 Yan Tan
Yan Tan Mina Wang
Mina Wang Ke Yang
Ke Yang Tiange Chi
Tiange Chi Zehuan Liao
Zehuan Liao Peng Wei
Peng Wei
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 23 March 2021
Sec. Cancer Molecular Targets and Therapeutics
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.599995
Cancer is one of the leading causes of mortality worldwide. PPAR modulators may hold great potential for the management of cancer patients. Indeed, PPARs are critical sensors and regulators of lipid, and they are able to promote eNOS activation, regulate immunity and inflammation response, and affect proliferation and differentiation of cancer cells. Cancer, a name given to a group of diseases, is characterized by multiple distinctive biological behaviors, including angiogenesis, abnormal cell proliferation, aerobic glycolysis, inflammation, etc. In the last decade, emerging evidence has shown that PPAR-α, a nuclear hormone receptor, can modulate carcinogenesis via exerting effects on one or several characteristic pathological behaviors of cancer. Therefore, the multi-functional PPAR modulators have substantial promise in various types of cancer therapies. This review aims to consolidate the functions of PPAR-α, as well as discuss the current and potential applications of PPAR-α agonists and antagonists in tackling cancer.
Cancer is one of the worldwide health problems, and the well-established risk factors involving genetic susceptibility, ionizing radiation, infections, smoking, insobriety, unhealthy diet, sedentary lifestyle, obesity, and other carcinogenic environmental exposures, promote the prevalence of cancer (1–5). It is believed that more than 20 million new cancer cases annually are projected to occur by 2025. Lung cancer remains to be the leading cause of morbidity and mortality globally (6). Besides, the American Cancer Society has demonstrated that cancer is the second leading cause of death in the United States, with expected 1,762,450 new cancer cases and 606,880 cancer deaths in 2019 (7). In fact, the morbidity and mortality of multiple kinds of cancer declined over the past decade due to decreases in known risk factors, effective screening, early detection, and better treatments in high-income countries. In contrast, cancer rates increased in low- and meddle- income countries, resulting from increases in smoking, unhealthy diet, lack of physical activity and infections (8). For example, death rates in the poorest countries were 2-fold higher in terms of cervical cancer, and 40% higher for men with lung and liver cancers from 2012 to 2016, compared with the most developed countries (7). Moreover, since cancers are characterized by mutations, and cells in our body inevitably mutate as we age, some of the mutations trigger formation of malignancy (9, 10). Considering the characteristics of cancer, two major therapeutic schemes have been applied clinically. One is genotype-directed precision oncology, targeting specific genomic abnormalities of various types of cancer to provide individual treatment. Another is antitumor immunity, that is, therapies targeting at the components of tumor microenvironment, especially the immune system (11–16).
Peroxisome proliferator-activated receptors (PPARs) are members of three ligand-inducible transcription factors, which belong to the nuclear receptor super-family. PPARs play an important role in regulating the expression of a variety of genes regarding the metabolic homeostasis of glucose and lipid, adipogenesis, and inflammation (17, 18). In mammals, there are three subtypes of PPARs: PPAR-α, PPAR-γ, and PPAR-β/δ, possessing varying expression levels in different tissues, biological effects, and ligand affinities (19). PPAR-α is mainly expressed in brown adipose, skeletal muscle, heart, liver, and intestinal mucosa tissues, adjusting glucose and lipid metabolism and homeostasis, inflammation, immune response, and angiogenesis (20, 21). Therefore, due to the vital metabolic modulating roles and excellent druggability of PPARs, PPAR agonists and antagonists have been employed for the therapy of a number of diseases, such as dyslipidemia, type 2 diabetes, cardiovascular diseases, obesity, cancer, and other metabolic diseases (22). PPAR agonists promote the transcription of target genes, leading to structural adjustment in the heterodimerized PPAR (with retinoid X receptor). However, PPAR antagonists do not change receptor conformation, compete or not compete with other ligands (23).
Mounting evidence has been accumulating in effects of PPAR-α and PPAR-γ in carcinogenesis, which show overlapping functions in metabolism and inflammation modulation. Yet, respective distinctions in specificity and potency of PPAR-β/δ has been conflicting. Apart from the regulation of cancer cell proliferation and differentiation regulated by PPAR modulators (both agonists and antagonists), which have been widely investigated, their pleiotropic roles in cancer encompasses realms of metabolism and inflammation are highly associated with cancer types and specific microenvironment as well (24). To provide a more focused insight, this review aims to only discuss recent findings on the biological functions, as well as current and potential applications of the agonists and antagonists of PPAR-α in cancer treatments.
PPAR-α is universally expressed in an organism, whose RNA expression level lacks tissue specificity and that of protein is particularly significant in tissues that require fatty acid oxidation as a source of energy, primarily metabolic tissues like brown adipose tissue, liver, heart, and kidney (25). The anti- and pro-tumorigenesis properties of PPAR-α are intricately intertwined in the field of the metabolism of lipid, glucose, and amino acid, as well as inflammation, cell proliferation, and apoptosis (Table 1 and Figure 1).

Table 1. Summary of functions and mechanisms of PPAR-α.
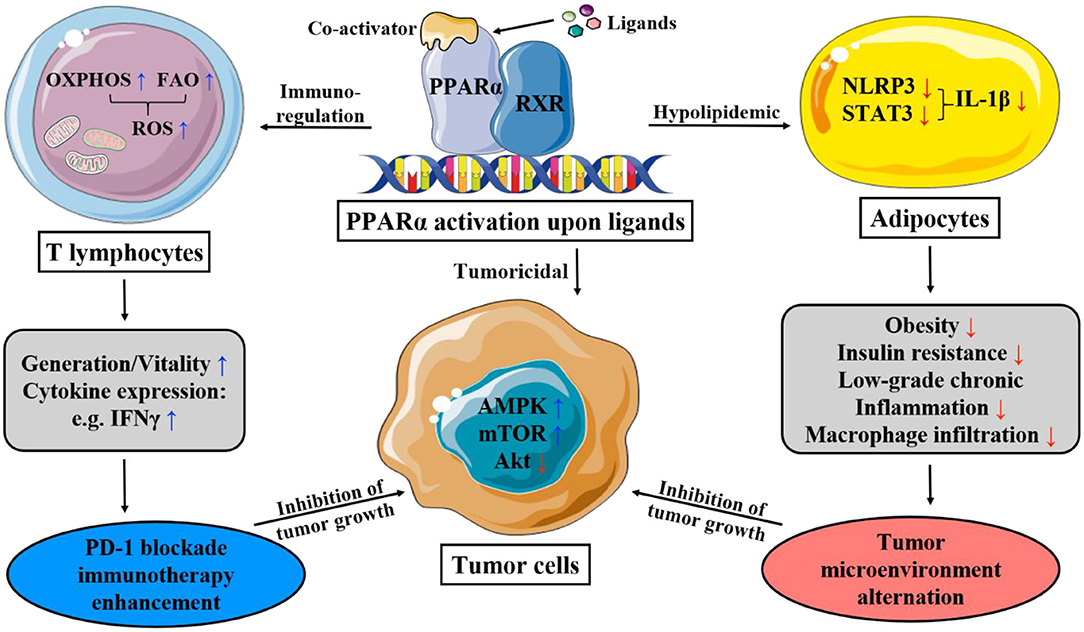
Figure 1. This figure illustrates the main mechanism of PPAR-α that lipid and immunity regulation lead to apoptosis of cancer cells. OXPHOS, oxidative phosphorylation; FAO, fatty acid oxidation; ROS, reactive oxygen species; PD-1, programmed death-1; AMPK, AMP-activated protein kinase; mTOR, mammalian target of rapamycin; STAT-3, signal transducer and activator of transcription-3; NLRP3, nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3.
PPAR-α maintains lipid metabolism and homeostasis via the modulation of genes of lipoprotein lipase, apolipoprotein (e.g. APOA1, APOA2, APOA5, and APOC3), as well as those involved in fatty acid transport and oxidation (e.g. FABP1, FABP3, ACS, ACO, CPT1, and CPT2), high-density lipoprotein (HDL) metabolism (e.g., PLTP), and ketone synthesis (e.g., HMGCS2), which take place in mitochondria, peroxisomes, and microsomes (22, 26, 27). Upon the activation of PPAR-α, the effect of substantial serum triglyceride clearance and HDL cholesterol increase is jointly yielded, as well as energy production. Moreover, PPAR-α also plays a key role in starvation response, which is underpinned by growing evidence on its regulation of carbohydrate and amino acid metabolism.
The catabolic action of PPAR-α on fatty acids causes an increased flux of fatty acids from peripheral tissues (e.g., skeletal muscle and adipose tissue), of which high content of triglyceride is significantly related to insulin resistance, to the liver. This effect might also consequently alleviate the FA-mediated inhibition of insulin-stimulated oxidative and non-oxidative glucose disposal in skeletal muscle, thus ameliorating insulin resistance. Additionally, the induction of hepatic lipogenesis of PPAR-α cooperates with insulin via regulating the sterol regulatory element-binding protein-1c (SREBP1c)-dependent pathway, a transcription factor modulating lipogenic enzyme expression and insulin action on both carbohydrates and lipids (28).
Cancer cells are characterized by their unique metabolic pathway, namely the Warburg effect, which in essence is glycolysis in oxygen-sufficient microenvironment. Under high energy demand and excessive oxidative stress, some cancer cells even adopt a hybrid metabolic method, employing both glycolysis and oxidative phosphorylation (OXPHOS). Upon activation, PPAR-α restricts mitochondrial OXPHOS, which non-tumor cells are not affected by; and increases the production of reactive oxygen species (ROS), causing the accumulation and exacerbation of the oxidative stress in cancer cell mitochondria. There is also a promotion of AMP-activated protein kinase (AMPK) signaling pathway witnessed alongside the activation of PPAR-α (29, 37), which increases the oxidation of fatty acids while potently inhibiting glycolysis, further cutting down ATP production. Synergistically, the mitochondria are impaired both structurally and functionally. Thus, the crippled energy production fails to meet the intensive energy demand of highly proliferative cancer cells, consequently impairing the viability, and even inducing cell cycle arrest, antagonizing the growth and metastasis of cancer (61).
Moreover, the prioritization of β-oxidation from PPAR-α as a starvation response has been illustrated. Upon agonist-binding, a decrease in urea concentration, an indicator of amino acid metabolism rate, is accompanied by an increase in ketone body concentration (30). Ketone bodies are of no energy-providing use for most cancer cells; however, for melanoma, the ketogenic gene expression is pro-tumorigenic, which is characterized by intracellular acetoacetate accumulation. Interestingly, acetoacetate is quite rapidly converted to beta-hydroxybutyrate in circulations and tissues within human body, and the latter suppresses inflammation, by the low-grade of which cancer microenvironment is always characterized, rather than immunosuppression. As the main regulator of physiological response to fasting and ketone body biosynthesis, PPAR-α has promising potential in the application in melanoma therapies (29).
Tissues highly involved in global metabolism (e.g., adipose tissue, liver, skeletal muscle, and vascular walls) are prone to inflammation when there is a metabolic disturbance (62). Chronic low-grade inflammation and metabolic disorders in lipid and glucose homeostasis often co-exist, which makes PPAR, the pleiotropic metabolism regulator, a strong candidate in the modulation of innate immunity in various metabolic diseases. PPAR-α directly or indirectly exerts an anti-inflammatory effect: the activation by leukotriene B4 limits its own activity and attenuates inflammatory response via negative feedback; stimulated by PPAR-α, the increased gene expression of IκB, an NF-κB inhibitor, together with the protein-protein interaction of PPAR-α with p65 and c-Jun, downregulates the activator protein-1 (AP-1) and NF-κB pathway and thus interferes with proinflammatory activity (22, 33); and the interaction of PPAR-α with glucocorticoid receptor α or estrogen receptor also trans-represses other proinflammatory transcription factors (22).
In addition, upon activation, PPAR-α plays a significant role in the regulation of endothelial nitric oxide synthase (eNOS) in a non-metabolic way (34). The sustainability of eNOS translates to stable production of nitric oxide, a vasodilator, and anti-thrombotic agent protecting epithelium, of which the activity is severely compromised in patients with cardiovascular diseases and arthrosclerosis (31).
Since ample blood supply is critical to tumor growth, angiogenesis is determinant in the progression of tumor, involving degradation of the surrounding matrix, cell proliferation, migration, differentiation, and tube formation. In the NADPH oxidase (NOX) family, as NOX2, NOX4, and NOX1 all expresses in endothelial cells, NOX2, and NOX4 are involved in cell proliferation, while NOX1 mediates endothelial cell migration and sprouting but not proliferation (35). PPAR-α upregulates the gene-expression of endostatin and thrombospondin 1, which are angiogenic inhibitors; and downregulates vascular endothelial growth factor (VEGF); and cytochrome P450-2C (CYP-2C), the neovascularization induced by the former in vivo and in vitro is reported to be dependent on NOX2 (22, 35). Interestingly, PPAR-α has also been found as a downstream modulator in NOX1-mediated angiogenesis, whose activity is repressed by the presence of NOX1; and in NOX1-deficient cells, the upregulated-expression of PPAR-α blocks angiogenic signaling needed in endothelial cell migration, sprouting, and angiogenesis (35).
Immunotherapy has been gaining significant momentum in cancer treatment, employing vaccines, antibodies, T cells, and cytokines to target the immune system to curb the growth of tumor cells. The valuable asset of metabolism-regulating of PPAR-α has established tight linkage to the generation, persistence, conversion, and apoptosis of T cell, of which the metabolic pathways play pivotal role in whose function and survival, greatly effecting the efficacy and outcome of the application. It is well-established that T-eff cells employ the classic metabolic mode—aerobic glycolysis—to sustain and recover effector function, which is the conversion from long-surviving memory cells to effectors (36); while T-memory cells majorly depend on fatty acid oxidation (FAO) and OXPHOS of mitochondria for energy.
According to studies, however, it is shown that in tumor microenvironment, with the metabolic constrains of hypoglycemia and hypoxia, due to the glucose depletion caused by tumor cells, which adopts glycolysis for energy production (63), T-effector cells perform better tumoricidal effect with increased mitochondrial metabolism, including OXPHOS, and FAO (64). It has been suggested that upon ligand-binding, PPAR-α, either working downstream in the activation of PPAR-δ by a PPAR-δ-specific ligand, GW501516 (65), or directly activated by co-activators (66), improves the efficacy of adoptive cell therapy by enhancing expression of carnitine palmitoyl transferase 1a, the rate-limiting enzyme of FAO, thereby enriching the uptake and oxidation of fatty acids. During which the expression of B-cell lymphoma-2 (Bcl2) is also upregulated, and the duo of the above two proteins can form a complex with the cytotoxic T lymphocytes (CTL) to exert an apoptosis-preventing effect (66). The activation of PPAR-α also improves anti-tumor immunity in PD-1 blockade cancer immunotherapy by reprogramming CD8+ T-cell metabolism from glycolysis to increased mitochondrial OXPHOS and FAO, supporting the extra energy demands of effector CTLs, thus lengthening the survival and potentiating activity (65, 66). Meanwhile, it also elevates cytokine expression (e.g., IFNγ) (64).
However, the pro-tumorigenesis effect of PPAR-α has also been contended with some solid evidence. With regard to its powerful oxidative property, contrary to the aforementioned anti-tumorigenesis effect with excessive oxidative stress on cancer cell mitochondria, other scientists argued that the inhibition of PPAR-α has yielded anti-proliferative effect on human paraganglioma, pancreatic and colorectal cancer cells in vitro with decreased antioxidant capacity and carnitine palmitoyl transferase-1A pattern expression (49–52). As suggested before, there exist interactions between PPAR-α and hormone metabolism. Upon activation, PPAR-α increases the expression and activity of CYP1B1, a subtype of Cytochromes P450. Through the biotransformation of endogenous estrogens and environmental carcinogens, it is critical in the initiation and progression of various hormone-dependent tumors, including breast cancer (53). Under long-term administration, the activation of PPAR-α is found to be hepatocarcinogenic in rodents, a mechanism related to the downregulation of let-7c micro RNA expression, which stabilizes MYC mRNA, contributing to increased mitogenic signaling and the consequent hepatocyte proliferation. This is an effect via both PPAR-α-dependent and -independent pathway, which has been testified to be absent in humans (54, 55). Cancer stem cell (CSC) is a subset of cancer cell population possessing self-renewal ability, and its sphere-formation rate is positively correlated with the advancement of malignancy. The higher number of CSCs population, the greater potential tumor possesses to advance. It has been found that maintenance of CSC properties of human hepatocellular carcinoma cells is upregulated by PPAR-α pathway activation, through activation of the PPAR-α- stearoyl-CoA desaturase-1 axis (56).
PPAR modulators including agonists and antagonists could represent a novel strategy for preventing and treating multiple types of cancer, regarding that dyslipidemias, obesity, glucose intolerance, and low-grade inflammation are strongly related to an increased risk of cancer, which PPAR modulators are able to directly or indirectly regulate. Thus, they are associated with cancer cell proliferation, differentiation, and apoptosis, supporting the potential of PPAR modulators as antitumor molecules. As far as PPAR-α agonists, they play an important role in the prevention of different cancers, including breast cancer, lung cancer, pancreatic cancer, and etc., by inhibiting the proliferation of cancer cells and affecting the Warburg effect. However, PPARs function as tumor suppressors or inducers is context-dependent, excessive expression of PPAR-α has been related to the progression of cell growth and survival in several cancer, suggesting that PPAR-α antagonists could be an effective therapeutic option for treating cancer (Table 2).
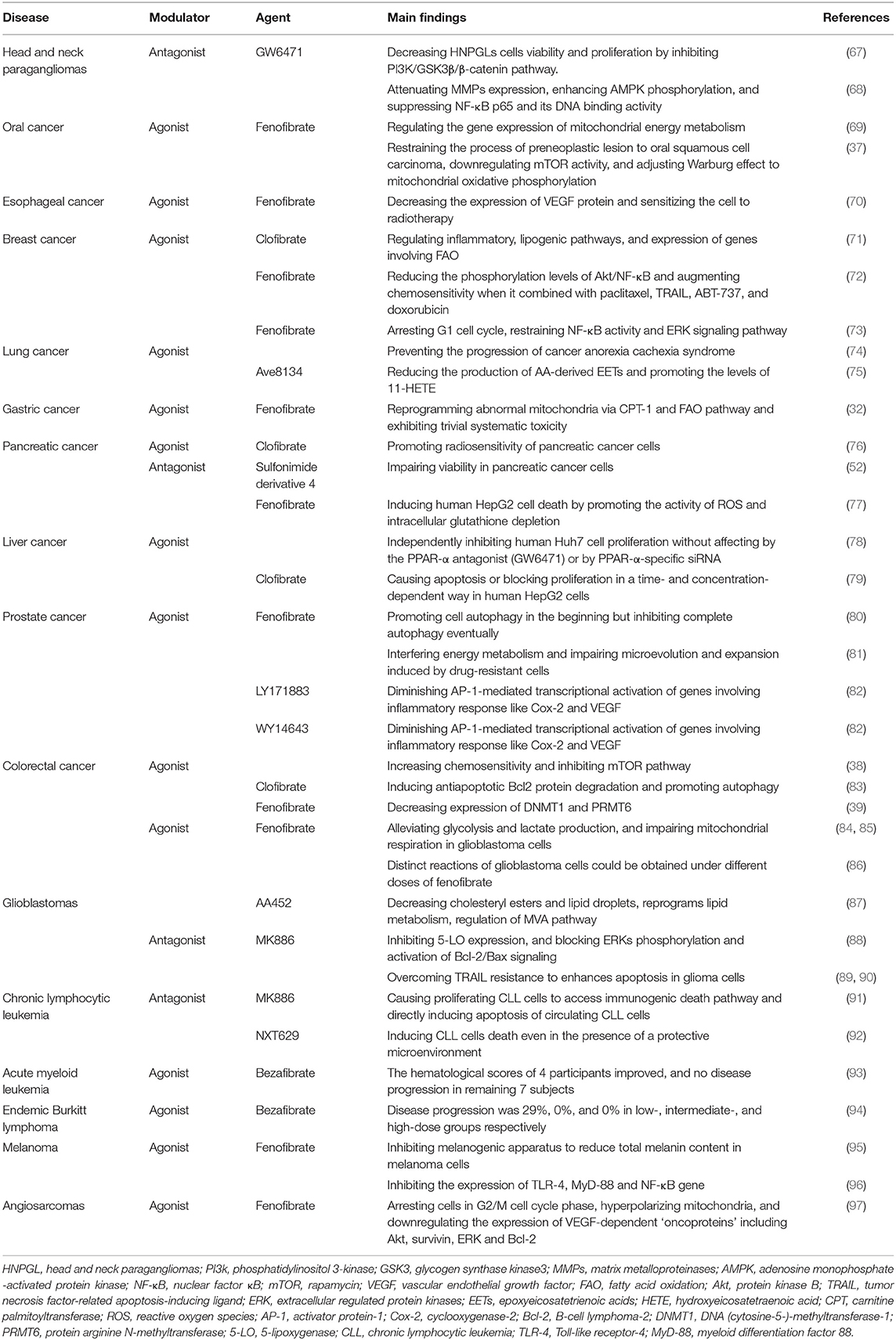
Table 2. Summary of the PPAR-α modulators in different types of cancer.
Head and neck paragangliomas (HNPGLs) are rare types of cancer that lead to significant morbidity regarding their ability to infiltrate the skull base (98). At present, surgery is the only option for patients, whereas, it is difficult to thoroughly remove the tumors (99). Therefore, other effective therapies are highly needed. Considering that PPAR-α is a candidate treatment for several types of cancer, a study has analyzed the expression of PPAR-α nuclear receptor in HNPGLs and assessed the functions of two PPAR-α modulators: PPAR-α agonist (WY14643) and PPAR-α antagonist (GW6471) in a unique model of HNPGLs cells, where the protein expression level of PPAR-α is remarkably high. The study has demonstrated that PPAR-α agonist (WY14643) fails to affect HNPGLs cells viability, while PPAR-α antagonist (GW6471) is associated with the decrease of HNPGLs cell viability and proliferation. Moreover, the underlying mechanisms involve the inhibition of the PI3K/GSK3β/β-catenin pathway, resulting in interfered cell cycles and induced apoptosis, resulting in the inhibition of clonogenicity and migration of HNPGL cells (67). Thus, PPAR-α antagonist (GW6471) could be considered as a potential therapy for HNPGLs.
Oral cancer is the one of the fatal malignancies with frequent lymph node metastasis and local invasion, thus, the prognosis of patients is poor even after targeted and chemotherapeutic drugs (100). Fenofibrate, a PPAR-α agonist, has been widely used to treat hyperlipidemia with its effects of anti-inflammatory and anti-atherosclerotic in humans. Meanwhile, the anticancer potential of fenofibrate has been report in several studies, involving induction of cancer cell apoptosis, decrease of cell proliferation, suppression of tumor angiogenesis, and inhibition of oxidative stress (101, 102). A study has investigated the anticancer activities of fenofibrate on the invasion and migration of human oral cancer Cal27 cells, which is associated with the attenuation of MMPs expression, enhancement of AMPK phosphorylation, and suppression of NF-κB p65 and its DNA binding activity, while such effect was observed to present in a AMPK-dependent manner, rather than PPARα signaling (68). The same research team has also found that in oral cancer mouse model, the expression level of PPAR-α protein was negatively correlated to cancer advancement; and the activation of PPAR-α by fenofibrate induced decreased migration ability in oral cancer cells in vitro, assumably via reprogramming ATP pathway, interfering with the characteristic Warburg effect of cancer cells (69). Moreover, in both animal and cell culture models, fenofibrate shows anti-oral cancer effects in restraining the process of preneoplastic lesion to oral squamous cell carcinoma, downregulating mTOR activity via TSC1/2-dependent signaling by triggering AMPK and suppressing Akt, and adjusting Warburg effect to mitochondrial oxidative phosphorylation to control energy production approach so as to repress proliferation of oral cancer cells and induction of metabolic reprogramming (37).
Esophageal cancer ranks as the sixth leading cause of cancer-related death worldwide. According to studies, there is a negative correlation between VEGF expression and cancer cell radiosensitivity, and the inhibition of VEGF expression promotes radiosensitivity of esophageal cancer cells, while the administration of fenofibrate can effectively diminish hypoxia-induced VEGF secretion in MCF-7 cells, a mechanism whose association with PPAR-α expression level remains unidentified. Either the combination of fenofibrate and radiation or fenofibrate alone is able to significantly decrease the expression of VEGF protein (70). Furthermore, a synergistic effect was observed in the combined administration of fenofibrate and radiation, which induced higher ratio of cells in G2/M phase and suppressed the growth of esophageal cancer cells (70).
Breast cancer is the second cause of cancer-related death in women (103), chemotherapy functions in preventing the progression of the primary breast cancer in neoadjuvant setting, but it may lead to several side effects and apoptosis resistance in breast cancer patients (104). Among fat and triglyceride lowering drugs, clofibrate, fenofibrate and WY14643 (a 2-aryl-thioacetic acid analog of clofibrate), PPAR-α agonists, present high chemosensitivity toward breast cancer cells. Clofibrate is the first lipid-lowering fibric acid derivative. According to Chandran et al. (71) the administration of clofibrate to breast cancer cells, the expression level of PPAR-α in which was abnormally high, showed significant cytotoxicity. The anti-carcinogenic effect was achieved possibly via the induction of PPAR-α DNA binding activity, causing cell cycle arrest, and the reduction of signaling, lipogenic, and inflammatory pathways, causing the suppressed proliferation of breast cancer cells (105). More specifically, researchers have the supposition that clofibrate suppresses the growth of breast cancer cells by repressing NF-κB and extracellular regulated protein kinases1/2 (ERK1/2) activation, inhibiting cyclinD1, cyclinA, cyclinE, and inducing pro-apoptotic P21 levels. Also, clofibrate significantly reduces Cox-2/PGE2(phenyl glycidyl ether-2) and 5-lipoxygenase/LTB4 (leukotriene B4) inflammatory pathways. Besides, clofibrate effectively mediates the expression of lipogenic and FAO pathways genes including SREBP-1c, SREBP-2, HMG-CoA synthase 2, Acyl-CoA oxidase, and CPT-1a (carnitine palmitoyltransferase 1a) (71). These findings provide new insight into our understating of clofibrate as a therapeutic anticancer agent.
In the process of cell apoptosis, the activation of Akt/NF-κB pathway plays a pivotal role in inhibiting the major apoptotic regulators, which hinders the activity of pro-apoptosis, are Bax, Bok, and Bim, leading to resistance of drug-induced cell death (106, 107). Fenofibrate is capable to potentiating chemosensitivity in breast cancer treatment by regulating Akt/NF-κB pathway, which is responsible for apoptosis resistance in some breast cancer patients. It is reported that fenofibrate promotes chemosensitivity by remarkably reducing the phosphorylation levels of Akt/NF-κB, as well as the downregulation of Mcl-1 and Bcl-xl and the upregulation of Bok and Bax at transcription level. Meanwhile, the study has indicated that the activation of caspase-9 and caspase-3 and the permeabilization of mitochondrial outer membrane affect fenofibrate-elevated chemosensitivity. Moreover, the synergistic effects of fenofibrate with paclitaxel, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), ABT-737, and doxorubicin significantly augment chemosensitivity to enhance the apoptosis of breast cancer cells (72). Basing on the emerging evidence, fenofibrate is a promising candidate in breast cancer treatments.
Lung cancer remains the most common cancer in the world, with a high mortality and a poor 5-year survival rate, even after radiation and immunotherapy (108). However, there is promising evidence that the PPAR-α agonist, fenofibrate, and the glucocorticoid, budesonide have been found to be beneficial to lung cancer (109, 110). A study used two types of lung adenocarcinoma cells: A549 (wild type TP53) and SK-MES-1 (TP53 deficient) to evaluate the effects of budesonide and fenofibrate, alone and in combination, which showed differential effects on the growth of lung cancer cells, in a PPAR-α-independent fashion. Budesonide inhibits cell proliferation in wild type TP53 A549 cells, whereas, this anti-proliferation effect is abrogated in TP53 deficient SK-MES-1 lung cancer cells. However, fenofibrate shows anti-proliferation effect in both A549 cells and SK-MES-1 cells. Furthermore, in A549 lung cancer cells, there is an additive effect against cell growth when using the combination of budesonide and fenofibrate, indicating a better therapeutic effect could be obtained with the combination of two compounds. The inhibition of cell growth induced by budesonide and fenofibrate is associated with G1 cell cycle arrest and the restraint of NF-κB activity and ERK signaling pathway (73). Besides, fenofibrate is able to prevent the progression of cancer anorexia cachexia syndrome (CACS) which is characterized by body weight loss, reduced food intake, and the catabolism of stored nutrients in muscle and adipose tissue. In lungs of CACS mice, the expression level of PPAR-α was reduced both in the nucleus and cytoplasm, compared to non-CACS counterparts. After the treatment of fenofibrate, the upregulation of the expression levels of several PPAR-α target genes, including Hmgcs2, Acadm, Cyp4a14, Acox1, and Ehhadh, has been observed. This effect may be related to improved FAO by facilitating peroxisome proliferator activity in the lungs (74).
In addition, a novel PPAR-α agonist, Ave8134, is well-tolerated in humans and shows advantages in cancer treatment. Ave813 suppresses the expression of CYP2c44, a functional homolog of human CYP2c9 that composes the major CYP epoxygenases for epoxyeicosatrienoic acids (EETs) biosynthesis in endothelial cells. Previous studies have confirmed that inhibiting the expression of CYP2c44 gene decreases endothelial proliferation and tumor growth. With the reduction of CYP2c44 expression, EET synthesis weakens, leading to reduced angiogenesis, and declined development of tumor (111, 112). However, there is conflicting evidence that Ave813 prominently promotes the levels of 11-hydroxyeicosatetraenoic acid (11-HETE), a bioactive lipid mediator converted by arachidonic acid (AA), which stimulates angiogenesis and tumor progression. Nevertheless, a Cox inhibitor indomethacin can effectively block the production of 11-HETE (75), thus the combination of indomethacin and AVE8134 may have promise in treatments for lung cancer.
Gastric cancer has a low rate of early diagnosis, and advanced gastric carcinoma is mainly treated by systematic chemotherapy, leading to serious adverse effects (113). PPAR-α is overexpressed and inversely related to prognosis of gastric cancer. Fenofibrate was shown to be capable of reprogramming abnormal mitochondria via CPT-1 and FAO pathway, as well as stimulating the AMPK signaling and suppressing the hexokinase-2 (HK-2) signaling. As a result, it is able to regulate the metabolism of glucose and lipid, prevent the growth of gastric cancer cells, and trigger the apoptosis of gastric cancer cells. Besides, fenofibrate also exhibits trivial toxicity in gastric tumor mouse model (32). It is expected that more investigations should be carried out on the effect of fenofibrate or other PPAR-α modulators in gastric cancer given the scarce existing evidence.
Pancreatic cancer remains as one of the most intractable and devastating types of cancer in the world, and few clinical development has been achieved for pancreatic cancer in the past decade, with surgery and chemotherapy being major curative treatments (114). A study used pancreatic cancer tissue samples from human to assess the level of PPAR-α in different pancreatic cancer tissues. The expression of PPAR-α is considerably lower in normal adjacent tissues than in pancreatic cancer tissues, which is strongly related to the prognosis of pancreatic cancer patients. Also, the activation of PPAR-α by its agonist, clofibrate, promotes radiosensitivity of pancreatic cancer cells via downregulating PTPRZ1 and Wnt8a transcription (two crucial components of Wnt/β-catenin pathway). This effect is abrogated by PPAR-α antagonist GW6471 and PPAR-α silencing, indicating a PPAR-dependent manner (76). Additionally, another study has examined the effects of a novel sulfonimide derivative 4 with PPAR-α antagonistic feature and a weaker PPAR-γ antagonism, and the novel compound potently impairs the viability of pancreatic cancer cell lines, indicating an inhibition of PPAR-α and PPAR-γ could be a therapeutic option for pancreatic cancer (52).
Hepatocellular carcinoma is one of the leading causes of cancer death in Asian countries. Unlike advanced breast cancer, PPAR-α expression is reduced in hepatocellular carcinoma, and PPAR-α agonists show anticancer property in liver tumor (115). In hepatocarcinoma HepG2 cells, fenofibrate and clofibrate induce cell apoptosis in a dose-related manner. Due to the different level of PPAR-α in human and rodent liver that human is significantly less than rodent, chronic application of fenofibrate causes liver cancer in rodent, however, high concentration of fenofibrate induces human HepG2 cells death by promoting the activity of ROS and intracellular glutathione depletion (77). Furthermore, clofibrate causes apoptosis or blocks proliferation in a time- and concentration- dependent way in human HepG2 cells by increasing protein phosphatase-2A expression and the pro-apoptotic BAD (79).
In hepatocarcinoma Huh7 cells, fenofibrate is able to independently inhibit human Huh7 cells proliferation without affecting by the PPAR-α antagonist (GW6471) or by PPAR-α-specific siRNA. Fenofibrate triggers the expression of C-terminal modulator protein, leading to decreased Akt phosphorylation which stimulates the nuclear accumulation of cyclin-dependent kinase inhibitor p27. More accumulation of p27 and reduction of cyclin A and E2F transcription factor 1 cause G1 arrest, eventually contributing to human Huh7 cells death (78). Thus, the antiproliferative property of fenofibrate and clofibrate makes them potential for anti-hepatoma therapy.
The most common cancer of genital system among males is prostate cancer (116), and based on the existing evidence, fenofibrate may plays a crucial role in the management of prostate cancer. A study has investigated the effects of fenofibrate prostate cancer cells, where higher rate of apoptosis was observed after the administration of fenofibrate compared to the control group. Also, the study has found that fenofibrate promotes autophagy in the beginning but inhibits complete autophagy eventually by adjusting AMPK-mTOR pathway, which leads to increased ER stress via PERK and IRE1 pathways, and the cumulation of ER stress accelerates cell death. Meanwhile, it has been observed that fenofibrate significantly inhibits the growth of prostate tumor in vivo mice model (80).
In order to examine the effect of fenofibrate on the drug-resistance of prostate cancer cells, another study applied the combined treatment of docetaxel/mitoxantrone and fenofibrate to naïve and drug-resistant cells. As expected, fenofibrate increases the chemosensitivity of prostate cancer cells by interfering energy metabolism and impairing microevolution and expansion induced by drug-resistant cells (81). Noteworthily, these effects can be obtained in the presence of <25 μM fenofibrate, i.e., within the range of its tolerable serum concentrations (up to 100 μM) (117).
The morbidity of colorectal cancer is showing the tendency of increasing, with one of the important risk factors being dietary fat. Therefore, the regulation of systemic lipid homeostasis plays a key role in controlling the development of colorectal cancer, which may be achieved by the application of PPAR-α modulators (118). In colorectal carcinoma SW620 cells which express low levels of PPAR-α mRNA, two PPAR-α agonists, LY171883, and WY14643 attenuate early stages of colon tumorigenesis by diminishing AP-1-mediated transcriptional activation of genes involving inflammatory response like Cox-2 and VEGF via PPRE-driven transcription in a PPAR-α-dependent fashion (82). Moreover, WY14643 is able to increase chemosensitivity via affecting the transcriptional activity of glucose transporter-1, and inhibit mTOR pathway, leading to apoptosis of cancer cells (38). With regards to colorectal carcinoma SW480 cells, another PPAR-α agonist, clofibrate, significantly suppresses tumor proliferation and sensitizes SW480 cells to chemotherapy drugs in a PPAR-α-dependent manner that induces antiapoptotic Bcl2 protein degradation and promotes autophagy (83). Furthermore, PPAR-α transgenic mice with increased expression of DNMT1 and PRMT6 have higher susceptibility to the development of colorectal cancer, which can be reduced by the activation of PPAR-α with the application of fenofibrate (39). Based on the evidence available, PPAR-α agonists are posed as potential drugs in the treatment for colorectal cancer.
Glioblastomas are the most malignant and incurable brain tumors characterized by rapid proliferation, resistance to radio- and chemotherapy, and persistent invasion of the central nervous system. The rapid growing cancer cells require both large amounts of ATP generated by mitochondrial respiration and glucose carbons produced by glycolysis (119, 120). Thus, disturbing those pathways could be a therapeutic strategy for glioblastomas, and several studies have demonstrated fenofibrate's capability of selectively promoting metabolic catastrophe in glioblastoma cells through interfering with mitochondrial function and glucose uptake. A study has detected the effects of fenofibrate in two settings: in a PPAR-α-independent manner, the unprocessed fenofibrate represses mitochondrial respiration; and in a PPAR-α-dependent manner, fenofibric acid converted by blood and tissue esterases stimulate glioblastoma cells to switch from glycolysis to FAO. These effects attenuate intracellular ATP and promote the AMP-activated protein kinase, which leads to extensive glioblastoma cells death. Interestingly, the study has also found that autophagy activators decrease the cytotoxicity of fenofibrate, while autophagy inhibitors increase the fenofibrate-induced glioblastoma cytotoxicity including phosphorylation of AMPK and suppression of mTOR- dependent phosphorylation of p70S6K (84). Similar findings were acknowledged in another study where fenofibrate not only alleviates glycolysis and lactate production but also impairs mitochondrial respiration in glioblastoma cells by inhibiting the transcriptional activity of NF-κB/ RelA and disrupting its association with HIF1α (85). The role of NF-κB/ RelA has been intensively studied, which is related to cancer cell growth, angiogenesis, and metastasis (121), as well as triggers aerobic glycolysis through transcriptional activation of pyruvate kinase isozyme type M2 (PKM2) (122). Moreover, the high ratio of PKM1/PKM2 enhances glycolysis and inhibits oxidative phosphorylation, whereas, fenofibrate is able to reduce the PKM2/PKM1 ratio and result in mitochondrial dysfunction (85).
In addition, distinctively differential reactions from glioblastoma cells could be observed with different doses of fenofibrate applied. Compared to a dose of 25 μM fenofibrate, 50 μM fenofibrate induces massive apoptosis of glioblastoma cells, while 25 μM fenofibrate inhibits monolayer and clonogenic growth of the tumor, and merely leads to trivial cell death, which is associated with the accumulation and phosphorylation of forkhead box O-3A (FoxO-3A), as well as the increase of FoxO-dependent apoptotic protein, Bim (86). Although the anticancer property of fenofibrate has been widely acknowledged, a dilemma that fenofibrate hardly crosses blood brain barrier makes it less effective in promoting glioblastoma cells death. According to one study, a novel synthesized compound PP1 with similar chemical structure of fenofibrate, but more stable, water soluble, and tissue penetrable, causes extensive glioblastoma cells death without showing major signs of distress (123).
AA452 is another PPAR-α antagonist tested for the treatment of glioblastoma, which showed encouraging affects in regulating lipid metabolism and radiosensitivity of glioblastoma cells. It decreases cholesteryl esters and lipid droplets and reprograms lipid metabolism, in parallel with the regulation of mevalonate (MVA) pathway, consequently limiting cancer cells proliferation and migration, as well as decreasing the invasiveness of glioblastoma cells. This inhibition of cholesteryl esters, from AA452, is truly promising since cholesteryl esters play an important role in malignancy. Moreover, AA452 also sensitizes glioblastoma cells to radiotherapy, leading to more cell death, whose mechanism is associated with the downregulation of cyclinD1 protein and c-myc gene (87).
5-LO is the key enzyme that catalyzes the first two steps in the synthesis of all leukotrienes by AA dioxygenation, and leukotrienes have significantly higher expression levels in brain tumor tissues than in normal brain tissues. Also, 5-LO plays an essential role in the signaling via Ras-ERKs pathway which is associated with the growth and progression of brain tumor. Therefore, the inhibition of 5-LO is beneficial in brain tumor treatment (124). A study used human glioma cells U-87MG and A172 with strong expression of 5-LO, and 5-LO non-expressing cells U373 to detect the anticancer effects of a PPAR-α antagonist—MK886. The study has found that MK886 is a specific 5-LO inhibitor with a high dependence of 5-LO expression level, and that MK886 induced cytotoxicity in cells U-87MG and A172 but not in cells U373, with its antiproliferative property possibly linked to the blocking of ERKs phosphorylation and activation of Bcl-2/Bax signaling (88).
The TRAIL (TNF-related apoptosis-inducing ligand) selectively induces apoptosis in transformed cells without killing most normal cells, rendering it a promising candidate in brain tumor treatment. However, the resistance of TRAIL impedes its application in cancer therapy. Thrillingly, a study showed that MK886 may possess the ability to overcome TRAIL resistance, enhancing its apoptotic effect on glioma cells and suggesting a combination strategy, which may exert more potent effect than each agent alone (89). Moreover, the research team has also revealed the mechanisms of sensitization of TRAIL-induced apoptosis by MK886: it was found that MK886 reduced the expression of an antiapoptotic protein, namely survivin, which is related to tumor cell resistance to TRAIL, and MK886 activates p38 mitogen-activated protein kinase (MAPK) pathway, leading to the overexpression of the death receptor 5 (DR5), eventually causing intensive apoptosis in glioma cells (90).
Chronic lymphocytic leukemia (CLL) is the most common adult leukemia in the western countries awaiting advanced treatment (125). Unlike acute leukemias, CLL cells mainly generate energy through FAO, rather than Warburg effect and glycolysis. According to Spaner et al. (91), measured by real-time PCR, the mRNA expression levels of PPAR-α in circulating CLL cells from two patients were present and almost 3-fold higher than that of peripheral blood mononuclear cells from a normal donor. As a major transcriptional regulator of FAO, the PPAR-α antagonists may be promising for CLL treatment. It can be observed in the study that, MK886 not only causes proliferating CLL cells to access immunogenic death pathway, but also directly induces apoptosis of circulating CLL cells, which is related to the reduction of interleukin-10 (IL-10) and phospho-STAT3 (91). Compared with MK886, NXT629 is more selective in killing CLL cells, and it is able to induce CLL cells death even in the presence of a protective microenvironment (92). Although NXT629 shows curative effects of CLL in vitro and in vivo, its pharmacokinetic properties for further clinical development have not been identified to date (126).
Current cytotoxic chemotherapy deteriorates the situation of reduced hemopoiesis in patients with acute myeloid leukemia (AML). Nevertheless, a small scale of clinical study has examined the safety and efficacy of the combinational therapy of bezafibrate (a PPAR-α agonist) and medroxyprogesterone acetate in 20 AML patients, and it was found that the low-dose combinational therapy showed no hematological toxicity and could be applied persistently in treating AML (93). However, the latter study has demonstrated that no extra benefit has been found with the same regime at a higher dosage in improving hemopoiesis or treating AML (127).
Children living in malaria-prone place are susceptible to endemic Burkitt lymphoma, and limited treatments restrict the cure rates (128). A clinical trial recruited 95 children with endemic Burkitt lymphoma to investigate the anti-endemic Burkitt lymphoma activity of three differential doses (low, intermediate, and high) of the combination of bezafibrate and medroxyprogesterone acetate. It was observed that the CCR (complete clinical response) of the high BaP (Bezafibrate and Medroxyprogesterone acetate) dose cohort was significantly higher at 68%, while that of the intermediate and low BaP cohorts showed relatively unremarkable difference, at 18 and 24%, respectively (129). Additionally, another study has verified that clofibrate reduces cell proliferation in multiple lymphoma and myeloma cells, which may be associated with Wnt pathway (94).
Developing from melanocytes present in the epidermis, dermis, hair follicle of the skin, and other parts of the body, melanoma has an increasing morbidity worldwide in the past decades (130). In a study investigating the association of PPAR-α expression level and pigmentation of melanoma cells, which showed a inverse correlation, fenofibrate was observed to act as a depigmentation agent and suppresses the melanogenic apparatus in a PPAR-α-independent manner, which reduces total melanin content in B16 F10-derived cell lines (95). Additionally, fenofibrate can also exert anti-melanoma effects as a result of its significant anti-inflammatory effect, through inhibiting the expression of TLR-4, myeloid differentiation factor 88 (MyD-88), and NF-κB gene and the generation of TNF-α. However, the effect showed no dose-dependent pattern (96).
Angiosarcomas are lethal and aggressive soft tissue malignancies originating from endothelia. According to Majeed et al. (97) fenofibrate was observed to exhibit prominent anti-proliferative property independent of PPAR-α in VEGF-dependent angiosarcomas cells. It induced cell arrest in G2/M phase, hyperpolarizes mitochondria, and downregulates the expression of VEGF-dependent “oncoproteins” including Akt, survivin, ERK, and Bcl-2, without reducing viability or inducing apoptosis of angiosarcomas cells. Noteworthily, the effects are observed to be not abrogated by PPAR-α and NF-κB inhibitors, and their combination with fenofibrate was cytotoxic. Whereas, other PPAR-α agonists including bezafibrate, WY14643, and fenofibric acid fail to replicate the effects of fenofibrate (97).
Cancer remains one of the leading causes of morbidity and mortality globally, but PPAR-α modulators shows great capability to manage the process of cancer. Indeed, by analyzing research evidence from a number of studies, we have discussed the functions of PPAR-α to clarify the underlying mechanisms of PPAR-α modulators in treating different malignancies. PPAR-α regulates energy metabolism of lipid, glucose and protein, affects inflammation and eNOS, modulates immune response, and adjusts proliferation, differentiation and apoptosis of cancer cell. Simultaneously, possible PPAR agonists and antagonists that may hold potential treatment for cancer patients have been comprehended and provided in this review for comparison across the clinical strategies. However, PPAR-α presents pro-tumorigenesis property as well, indicating that PPAR-α may promote the progression of tumor, which has been demonstrated in several studies that PPAR-α agonists induced hepatocarcinoma or peroxisome proliferation in rodents (131). Also, studies have reported that exposures of weeks to several years of PPAR-α agonists treatment may induce hepatocellular chronic active and cholestatic hepatitis, as well as increase the level of serum transaminase, which may be dose related (132). Besides, fibrates have a risk of muscle astrophy, and the combination of fibrates and stains may also increase the risk of rhabdomyolysis (133). Other adverse effects include renal damage that fibrates may impair the generation of vasodilatory prostaglandins, leading to a reversible decrease in glomerular filtration rate and a elevation of serum creatinine (134, 135), and gallbladder disease such as gall stone, but the incidence varies remarkably between compounds (132). Therefore, routine monitoring of liver and renal function should be administrated in patients with PPAR-α modulators therapy. By establishing this thorough review on PPAR-α modulators, we hope to provide valuable insight to how we can better tackle cancer in the future.
ZL and PW: conceptualization and supervision. YT and PW: resources. KY, TC, and MW: writing—original draft preparation. TC, MW, YT, and ZL: writing—review and editing. PW: project administration. PW and YT: funding acquisition. All authors have read and agreed to the published version of the manuscript.
This work was funded by the National Natural Science Foundation of China (Grant No. 31800652), Young Scientist Program by Beijing University of Chinese Medicine (Grant No. BUCM-2019-QNKXJ-C014), the Double First Class Construction Funds of Discipline of Integrated Traditional Chinese and Western Medicine of Beijing University of Chinese Medicine, the Young Elite Scientists Sponsorship Program by CAST (Project No: CACM-2018-QNRC2-C06).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
5-LO, 5-lipoxygenase; AA, arachidonic acid; ACO, acyl-CoA oxidase; ACS, acyl-CoA synthetase; Akt, protein kinase B; AMP, adenosine monophosphate; AMPK, AMP-activated protein kinase; AML, acute myeloid leukemia; AP-1, activator protein-1; APOA1, apolipoprotein A1; APOA2, apolipoprotein A2; APOA5, apolipoprotein A5; APOC3, apolipoprotein C3; Bcl2, B-cell lymphoma-2; CACS, cancer anorexia cachexia syndromes; CKD4, cyclin-dependent kinases4; CLL, chronic lymphocytic leukemia; Cox, cyclooxygenase; CPT, carnitine palmitoyltransferase; CSC, cancer stem cell; CTL, cytotoxic T lymphocyte; CYP, cytochrome P450; DNMT1, DNA (cytosine-5-)-methyltransferase-1; DR5, death receptor5; EETs, epoxyeicosatetrienoic acids; eNOS, endothelial nitric oxide synthase; ER stress, endoplasmic reticulum stress; ERK, extracellular regulated protein kinases; FABP1, fatty ccid-binding protein 1; FABP3, fatty ccid-binding protein 3; FA, fatty acid; FAO, fatty acid oxidation; FLAP, 5-lipoxygenase activating protein; FoxO, forkhead box O; FASN, fatty acid synthase; GSK3, glycogen synthase kinase3; HEI-OC1, house ear institute organ of corti1; HETE, hydroxyeicosatetraenoic acid; HK-2, hexokinase-2; HIF1α, hypoxia inducible factor-1α; HDL, high-density lipoprotein; HMGCS2, hydroxymethylglutaryl CoA synthase 2; HNPGLs, head and neck paragangliomas; IFNγ, interferon gamma; IκB, inhibitor of NF-κB; JNK, c-Jun N-terminal kinase; LTB4, leukotriene B4; MAPK, mitogen-activated protein kinase; MMPs, matrix metalloproteinases; mTOR, mammalian target of rapamycin; MSC, mesenchymal stem cells; MVA, mevalonate; MYC, avian myelocytomatosis virus oncogene homolog; MyD-88, myeloid differentiation factor 88; NADPH, nicotinamide adenine dinucleotide phosphate; NF-κB, nuclear factor κB; NLRP3, nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3; NOX, NADPH oxidase; OXPHOS, oxidative phosphorylation; PI3k, phosphatidylinositol 3-kinase; PKM1, Pyruvate kinase isozyme type M1; PKM2, Pyruvate kinase isozyme type M2; PRMT6, protein arginine N-methyltransferase; PD-1, programmed cell death protein 1; PGE, phenyl glycidyl ether; PLTP, phospholipid transfer protein; PPAR, peroxisome proliferator-activated receptors; ROS, reactive oxygen species; RXR, retinoid X receptor; SREBP, sterol regulatory element-binding transcription factor; STAT3, signal transducer and activator of transcription 3; TLR-4, Toll-like receptor-4; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; VEGF, vascular endothelial growth factor.
1. Avgerinos KI, Spyrou N, Mantzoros CS, Dalamaga M. Obesity and cancer risk: emerging biological mechanisms and perspectives. Metabolism. (2019) 92:121–35. doi: 10.1016/j.metabol.2018.11.001
2. Yang Y, Yang T, Liu S, Cao Z, Zhao Y, Su X, et al. Concentrated ambient PM2. 5 exposure affects mice sperm quality and testosterone biosynthesis. PeerJ. (2019) 7:e8109. doi: 10.7717/peerj.8109
3. Khan RMM, Chua ZJY, Tan JC, Yang Y, Liao Z, Zhao Y. From pre-diabetes to diabetes:diagnosis, treatments and translational research. Medicina. (2019) 55:546. doi: 10.3390/medicina55090546
4. Wang M, Yang Y, Liao Z. Diabetes and cancer:epidemiological and biological links. World J Diabetes. (2020) 11:227. doi: 10.4239/wjd.v11.i6.227
5. Wang M, Tan Y, Shi Y, Wang X, Liao Z, Wei P. Diabetes and sarcopenic obesity: pathogenesis, diagnosis, and treatments. Front Endocrinol. (2020) 11:568. doi: 10.3389/fendo.2020.00568
6. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide:sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. (2015) 136:E359–86. doi: 10.1002/ijc.29210
7. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. (2019) 69:7–34. doi: 10.3322/caac.21551
8. Torre LA, Siegel RL, Ward EM, Jemal A. Global cancer incidence and mortality rates and trends–an update. Cancer Epidemiol Biomarkers Prev. (2016) 25:16–27. doi: 10.1158/1055-9965.EPI-15-0578
9. Curtius K, Wright NA, Graham TA. An evolutionary perspective on field cancerization. Nat Rev Cancer. (2018) 18:19–32. doi: 10.1038/nrc.2017.102
10. Liao Z, Chua D, Tan NS. Reactive oxygen species:a volatile driver of field cancerization and metastasis. Mol Cancer. (2019) 18:1–10. doi: 10.1186/s12943-019-0961-y
11. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas Primary A. Adaptive, and acquired resistance to cancer immunotherapy. Cell. (2017) 168:707–23. doi: 10.1016/j.cell.2017.01.017
12. Zugazagoitia J, Guedes C, Ponce S, Ferrer I, Molina-Pinelo S, Paz-Ares L. Current challenges in cancer treatment. Clin Ther. (2016) 38:1551–66. doi: 10.1016/j.clinthera.2016.03.026
13. Gonzalez-Molina J, Gramolelli S, Liao Z, Carlson JW, Ojala PM, Lehti K. MMP14 in sarcoma:a regulator of tumor microenvironment communication in connective tissues. Cells. (2019) 8:991. doi: 10.3390/cells8090991
14. Liao Z, Tan ZW, Zhu P, Tan NS. Cancer-associated fibroblasts in tumor microenvironment–Accomplices in tumor malignancy. Cell Immunol. (2019) 343:103729. doi: 10.1016/j.cellimm.2017.12.003
15. Dong R, Tan Y, Fan A, Liao Z, Liu H, Wei P. Molecular dynamics of the recruitment of immunoreceptor signaling module DAP12 homodimer to lipid raft boundary regulated by PIP2. J Phys Chem. (2019) 124:504–10. doi: 10.1021/acs.jpcb.9b11095
16. Liao Z, Wong SW, Yeo HL, Zhao Y. Smart nanocarriers for cancer treatment: clinical impact and safety. NanoImpact. (2020) 11:100253. doi: 10.1016/j.impact.2020.100253
17. Botta M, Audano M, Sahebkar A, Sirtori CR, Mitro N, Ruscica M. PPAR agonists and metabolic syndrome: an established role? Int J Mol Sci. (2018) 19:1197. doi: 10.3390/ijms19041197
18. Phua WWT, Wong MXY, Liao Z, Tan NS. An aPPARent functional consequence in skeletal muscle physiology via peroxisome proliferator-activated receptors. Int J Mol Sci. (2018) 19:1425. doi: 10.3390/ijms19051425
19. Mirza AZ, Althagafi II, Shamshad H. Role of PPAR receptor in different diseases and their ligands:Physiological importance and clinical implications. Eur J Med Chem. (2019) 166:502–13. doi: 10.1016/j.ejmech.2019.01.067
20. Yousefnia S, Momenzadeh S, Seyed Forootan FK. Ghaedi, Nasr Esfahani MH. The influence of peroxisome proliferator-activated receptor γ. (PPARγ) ligands on cancer cell tumorigenicity. Gene. (2018) 649:14–22. doi: 10.1016/j.gene.2018.01.018
21. Han L, Shen W-J, Bittner S, Kraemer FB, Azhar S. PPARs:regulators of metabolism and as therapeutic targets in cardiovascular disease. Part II:PPAR-β/δ and PPAR-γ. Future Cardiol. (2017) 13:279–96. doi: 10.2217/fca-2017-0019
22. Cheng HS, Tan WR, Low ZS, Marvalim C, Lee JYH, Tan NS. Exploration and development of PPAR modulators in health and disease: an update of clinical evidence. Int J Mol Sci. (2019) 20:5055. doi: 10.3390/ijms20205055
23. De Lellis L, Cimini A, Veschi S, Benedetti E, Amoroso R, Cama A, et al. The anticancer potential of peroxisome proliferator-activated receptor antagonists. Chem Med Chem. (2018) 13:209–19. doi: 10.1002/cmdc.201700703
24. Heudobler D, Rechenmacher M, Lüke F, Vogelhuber M, Pukrop T, Herr W, et al. Peroxisome proliferator-activated receptors (PPAR)γ agonists as master modulators of tumor tissue. Int J Mol Sci. (2018) 19:3540. doi: 10.3390/ijms19113540
25. Escher P, Braissant O, Basu-Modak S, Michalik L, Wahli W, Desvergne Ba. Rat PPARs: quantitative analysis in adult rat tissues and regulation in fasting and refeeding. Endocrinology. (2001) 142:4195–202. doi: 10.1210/endo.142.10.8458
26. Auwerx J, Schoonjans K, Fruchart JC, Staels B. Regulation of triglyceride metabolism by PPARs:fibrates and thiazolidinediones have distinct effects. J Atheroscler Thromb. (1996) 3:81–9. doi: 10.5551/jat1994.3.81
27. Pyper SR, Viswakarma N, Yu S, Reddy JK. PPARalpha: energy combustion, hypolipidemia, inflammation and cancer. Nucl Recept Signal. (2010) 8:e002–2. doi: 10.1621/nrs.08002
28. Fernández-Alvarez A, Alvarez MS, Gonzalez R, Cucarella C, Muntané J, Casado M. Human SREBP1c expression in liver is directly regulated by peroxisome proliferator-activated receptor alpha (PPARalpha). J Biol Chem. (2011) 286:21466–77. doi: 10.1074/jbc.M110.209973
29. Grabacka M, Plonka PM, Reiss K. Melanoma—time to fast or time to feast? An interplay between PPARs, metabolism and immunity. Exp Dermatol. (2020) 29:436–45. doi: 10.1111/exd.14072
30. Kersten S, Mandard S, Escher P, Gonzalez FJ, Tafuri S, Desvergne B, Wahli W. The peroxisome proliferator-activated receptor α regulates amino acid metabolism. FASEB J. (2001) 15:1971–8. doi: 10.1096/fj.01-0147com
31. Guerre-Millo M, Gervois P, Raspé E, Madsen L, Poulain P, Derudas B, et al. Peroxisome proliferator-activated receptor alpha activators improve insulin sensitivity and reduce adiposity. J Biol Chem. (2000) 275:16638–42. doi: 10.1074/jbc.275.22.16638
32. Chen L, Peng J, Wang Y, Jiang H, Wang W, Dai J, et al. Fenofibrate-induced mitochondrial dysfunction and metabolic reprogramming reversal:the anti-tumor effects in gastric carcinoma cells mediated by the PPAR pathway. Am J Transl Res. (2020) 12:428–46.
33. Contreras AV, Torres N, Tovar AR. PPAR-α as a key nutritional and environmental sensor for metabolic adaptation. Adv Nutr. (2013) 4:439–52. doi: 10.3945/an.113.003798
34. Maccallini C, Mollica A, Amoroso R. The positive regulation of eNOS signaling by PPAR agonists in cardiovascular diseases. Am J Cardiovascular Drugs. (2017) 17:273–81. doi: 10.1007/s40256-017-0220-9
35. Garrido-Urbani S, Jemelin S, Deffert C, Carnesecchi S, Basset O, Szyndralewiez C, et al. Targeting vascular NADPH oxidase 1 blocks tumor angiogenesis through a PPARα mediated mechanism. PLoS ONE. (2011) 6:e14665. doi: 10.1371/journal.pone.0014665
36. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun. (2015) 6:6692. doi: 10.1038/ncomms7692
37. Chang N-W, Huang Y-P. The RNA degradation pathway is involved in PPARα-modulated anti-oral tumorigenesis. BioMedicine. (2019) 9:27. doi: 10.1051/bmdcn/2019090427
38. Gou Q, Dong C, Jin J, Liu Q, Lu W, Shi J, et al. PPARα agonist alleviates tumor growth and chemo-resistance associated with the inhibition of glucose metabolic pathway. Eur J Pharmacol. (2019) 863:172664. doi: 10.1016/j.ejphar.2019.172664
39. Luo Y, Xie C, Brocker CN, Fan J, Wu X, Feng L, et al. Intestinal PPARα protects against colon carcinogenesis via regulation of methyltransferases DNMT1 and PRMT6. Gastroenterology. (2019) 157:744–59.e4. doi: 10.1053/j.gastro.2019.05.057
40. Mou Y, Wang J, Wu J, He D, Zhang C, Duan C, et al. Ferroptosis, a new form of cell death:opportunities and challenges in cancer. J Hematol Oncol. (2019) 12:34. doi: 10.1186/s13045-019-0720-y
41. Li J, Cao F, Yin H-l, Huang Z-j, Lin Z-t, Mao N, et al. Ferroptosis:past, present and future. Cell Death Dis. (2020) 11:88. doi: 10.1038/s41419-020-2298-2
42. Venkatesh D, O'Brien N, Zandkarimi F, Tong D, Stokes M, Dunn D, et al. MDM2 and MDMX promote ferroptosis by PPARα-mediated lipid remodeling. Genes Dev. (2020) 34:526–43. doi: 10.1101/gad.334219.119
43. Xu L, Xia H, Ni D, Hu Y, Liu J, Qin Y, et al. High-dose dexamethasone manipulates the tumor microenvironment and internal metabolic pathways in anti-tumor progression. Int J Mol Sci. (2020) 21:E1846. doi: 10.3390/ijms21051846
44. Kwong S, Jamil A, Rhodes A, Mohd Taib NA, Chung I. Metabolic role of fatty acid binding protein 7 in mediating triple negative breast cancer cell death via PPAR-alpha signalling. J Lipid Res. (2019) 60:jlr.M092379. doi: 10.1194/jlr.M092379
45. Hentze JL, Høgdall CK, Høgdall EV. Methylation and ovarian cancer: can DNA methylation be of diagnostic use? Mol Clin Oncol. (2019) 10:323–30. doi: 10.3892/mco.2019.1800
46. Koch A, Joosten SC, Feng Z, de Ruijter TC, Draht MX, Melotte V, et al. Analysis of DNA methylation in cancer: location revisited. Nat Rev Clin Oncol. (2018) 15:459–66. doi: 10.1038/s41571-018-0004-4
47. Mahmoud AM, Ali MM. Methyl donor micronutrients that modify DNA methylation and cancer outcome. Nutrients. (2019) 11:608. doi: 10.3390/nu11030608
48. Brunetti L, Loiodice F, Piemontese L, Tortorella P, Laghezza A. New approaches to cancer therapy: combining fatty acid amide hydrolase (FAAH) inhibition with peroxisome proliferator-activated receptors (PPARs) activation. J Med Chem. (2019) 62:10995–1003. doi: 10.1021/acs.jmedchem.9b00885
49. Ammazzalorso A, De Lellis L, Florio R, Laghezza A, De Filippis B, Fantacuzzi M, et al. Synthesis of novel benzothiazole amides:Evaluation of PPAR activity and anti-proliferative effects in paraganglioma, pancreatic and colorectal cancer cell lines. Bioorganic Med Chem Lett. (2019) 29:2302–6. doi: 10.1016/j.bmcl.2019.06.020
50. Ammazzalorso A, Giancristofaro A, D'Angelo A, Filippis BD, Fantacuzzi M, Giampietro L, et al. Benzothiazole-based N-(phenylsulfonyl)amides as a novel family of PPARα antagonists. Bioorganic Med Chem Lett. (2011) 21:4869–72. doi: 10.1016/j.bmcl.2011.06.028
51. Uremis N, Uremis MM, Tolun FI, Ceylan M, Doganer A, Kurt AH. Synthesis of 2-substituted benzothiazole derivatives and their in vitro anticancer effects and antioxidant activities against pancreatic cancer cells. Anticancer Res. (2017) 37:6381–9. doi: 10.21873/anticanres.12091
52. Ammazzalorso A, De Lellis L, Florio R, Bruno I, De Filippis B, Fantacuzzi M, et al. Cytotoxic effect of a family of peroxisome proliferator-activated receptor antagonists in colorectal and pancreatic cancer cell lines. Chem Biol Drug Design. (2017) 90:1029–35. doi: 10.1111/cbdd.13026
53. Hwang YP, Won SS, Jin SW, Lee GH, Pham TH, Choi JH, et al. WY-14643 Regulates CYP1B1 expression through peroxisome proliferator-activated receptor α-mediated signaling in human breast cancer cells. Int J Mol Sci. (2019) 20:5928. doi: 10.3390/ijms20235928
54. Pauley CJ, Ledwith BJ, Kaplanski C. Peroxisome proliferators activate growth regulatory pathways largely via peroxisome proliferator-activated receptor alpha-independent mechanisms. Cell Signal. (2002) 14:351–8. doi: 10.1016/S0898-6568(01)00260-1
55. Peters JM, Shah YM, Gonzalez FJ. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nat Rev Cancer. (2012) 12:181–95. doi: 10.1038/nrc3214
56. Ma X-L, Sun Y-F, Wang B-L, Shen M-N, Zhou Y, Chen J-W, et al. Sphere-forming culture enriches liver cancer stem cells and reveals Stearoyl-CoA desaturase 1 as a potential therapeutic target. BMC Cancer. (2019) 19:760. doi: 10.1186/s12885-019-5963-z
57. Brown ZJ, Fu Q, Ma C, Kruhlak M, Zhang H, Luo J, et al. Carnitine palmitoyltransferase gene upregulation by linoleic acid induces CD4(+) T cell apoptosis promoting HCC development. Cell Death Dis. (2018) 9:620. doi: 10.1038/s41419-018-0687-6
58. Hichami A, Yessoufou A, Ghiringhelli F, Salvadori F, Moutairou K, Zwetyenga N, et al. Peroxisome proliferator-activated receptor alpha deficiency impairs regulatory T cell functions: possible application in the inhibition of melanoma tumor growth in mice. Biochimie. (2016) 131:1–10. doi: 10.1016/j.biochi.2016.09.001
59. Morse E, Selim E, Cunard R. PPARα ligands cause lymphocyte depletion and cell cycle block and this is associated with augmented TRB3 and reduced Cyclin B1 expression. Mol Immunol. (2009) 46:3454–61. doi: 10.1016/j.molimm.2009.08.008
60. Selim E, Frkanec JT, Cunard R. Fibrates upregulate TRB3 in lymphocytes independent of PPARα by augmenting CCAAT/enhancer-binding proteinβ (C/EBPβ) expression. Mol Immunol. (2007) 44:1218–29. doi: 10.1016/j.molimm.2006.06.006
61. Chen L, Peng J, Wang Y, Jiang H, Wang W, Dai J, et al. Fenofibrate-induced mitochondrial dysfunction and metabolic reprogramming reversal:the anti-tumor effects in gastric carcinoma cells mediated by the PPAR pathway. Am J Transl Res. (2020) 12:428–46.
62. Wahli W, Michalik L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol Metabol. (2012) 23:351–63. doi: 10.1016/j.tem.2012.05.001
63. Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, et al. Enhancing CD8(+) T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell. (2017) 32:377–91.e9. doi: 10.1016/j.ccell.2017.08.004
64. Saibil SD, St Paul M, Laister RC, Garcia-Batres CR, Israni-Winger K, Elford AR, et al. Activation of peroxisome proliferator-activated receptors α and δ synergizes with inflammatory signals to enhance adoptive cell therapy. Cancer Res. (2019) 79:445–51. doi: 10.1158/0008-5472.CAN-17-3053
65. Garbacz WG, Huang JTJ, Higgins LG, Wahli W, Palmer CNA. PPARα is required for PPARδ action in regulation of body weight and hepatic steatosis in mice. PPAR Res. (2015) 2015:927057. doi: 10.1155/2015/927057
66. Chowdhury PS, Chamoto K, Kumar A, Honjo T. PPAR-induced fatty acid oxidation in t cells increases the number of tumor-reactive CD8 T cells and facilitates anti–PD-1 therapy. Cancer Immunol Res. (2018) 6:1375. doi: 10.1158/2326-6066.CIR-18-0095
67. Florio R, De Lellis L, di Giacomo V, Di Marcantonio MC, Cristiano L, Basile M, et al. Effects of PPARα inhibition in head and neck paraganglioma cells. PLoS ONE. (2017) 12:e0178995. doi: 10.1371/journal.pone.0178995
68. Tsai SC, Tsai MH, Chiu CF, Lu CC, Kuo SC, Chang NW, et al. AMPK-dependent signaling modulates the suppression of invasion and migration by fenofibrate in CAL 27 oral cancer cells through NF-κB pathway. Environ Toxicol. (2016) 31:866–76. doi: 10.1002/tox.22097
69. Huang YP, Chang NW. PPARα modulates gene expression profiles of mitochondrial energy metabolism in oral tumorigenesis. Biomedicine. (2016) 6:3. doi: 10.7603/s40681-016-0003-7
70. Li XQ, Zhou JD, Zou ST, Yu J, Meng XJ, Wu JC. Enhancement of radiosensitivity in human esophageal carcinoma cells by fenofibrate and its potential mechanism. Tumori. (2015) 101:123–30. doi: 10.5301/tj.5000228
71. Chandran K, Goswami S, Sharma-Walia N. Implications of a peroxisome proliferator-activated receptor alpha (PPARα) ligand clofibrate in breast cancer. Oncotarget. (2016) 7:15577–99. doi: 10.18632/oncotarget.6402
72. Sun J, Zheng Z, Chen Q, Pan Y, Quan M, Dai Y. Fenofibrate potentiates chemosensitivity to human breast cancer cells by modulating apoptosis via AKT/NF-κB pathway. Onco Targets Ther. (2019) 12:773–83. doi: 10.2147/OTT.S191239
73. Liang H, Kowalczyk P, Junco JJ, Klug-De Santiago HL, Malik G, Wei SJ, et al. Differential effects on lung cancer cell proliferation by agonists of glucocorticoid and PPARα receptors. Mol Carcinog. (2014) 53:753–63. doi: 10.1002/mc.22029
74. Goncalves MD, Hwang SK, Pauli C, Murphy CJ, Cheng Z, Hopkins BD, et al. Fenofibrate prevents skeletal muscle loss in mice with lung cancer. Proc Natl Acad Sci USA. (2018) 115:E743–52. doi: 10.1073/pnas.1714703115
75. Wu L, Wang W, Dai M, Li H, Chen C, Wang D. PPARα ligand, AVE8134, and cyclooxygenase inhibitor therapy synergistically suppress lung cancer growth and metastasis. BMC Cancer. (2019) 19:1166. doi: 10.1186/s12885-019-6379-5
76. Xue J, Zhu W, Song J, Jiao Y, Luo J, Yu C, et al. Activation of PPARα by clofibrate sensitizes pancreatic cancer cells to radiation through the Wnt/β-catenin pathway. Oncogene. (2018) 37:953–62. doi: 10.1038/onc.2017.401
77. Jiao HL, Zhao BL. Cytotoxic effect of peroxisome proliferator fenofibrate on human HepG2 hepatoma cell line and relevant mechanisms. Toxicol Appl Pharmacol. (2002) 185:172–9. doi: 10.1006/taap.2002.9538
78. Yamasaki D, Kawabe N, Nakamura H, Tachibana K, Ishimoto K, Tanaka T, et al. Fenofibrate suppresses growth of the human hepatocellular carcinoma cell via PPARα-independent mechanisms. Eur J Cell Biol. (2011) 90:657–64. doi: 10.1016/j.ejcb.2011.02.005
79. Maggiora M, Oraldi M, Muzio G, Canuto RA. Involvement of PPARα and PPARγ in apoptosis and proliferation of human hepatocarcinoma HepG2 cells. Cell Biochem Funct. (2010) 28:571–7. doi: 10.1002/cbf.1691
80. Tao T, Zhao F, Xuan Q, Shen Z, Xiao J, Shen Q. Fenofibrate inhibits the growth of prostate cancer through regulating autophagy and endoplasmic reticulum stress. Biochem Biophys Res Commun. (2018) 503:2685–9. doi: 10.1016/j.bbrc.2018.08.024
81. Luty M, Piwowarczyk K, Łabedz-Masłowska A, Wróbel T, Szczygieł M, Catapano J, et al. Fenofibrate augments the sensitivity of drug-resistant prostate cancer cells to docetaxel. Cancers. (2019) 11:E77. doi: 10.3390/cancers11010077
82. Grau R, Punzón C, Fresno M, Iñiguez MA. Peroxisome-proliferator-activated receptor alpha agonists inhibit cyclo-oxygenase 2 and vascular endothelial growth factor transcriptional activation in human colorectal carcinoma cells via inhibition of activator protein-1. Biochem J. (2006) 395:81–8. doi: 10.1042/BJ20050964
83. You M, Gao J, Jin J, Hou Y. PPARα enhances cancer cell chemotherapy sensitivity by autophagy induction. J Oncol. (2018) 2018:6458537. doi: 10.1155/2018/6458537
84. Wilk A, Wyczechowska D, Zapata A, Dean M, Mullinax J, Marrero L, et al. Molecular mechanisms of fenofibrate-induced metabolic catastrophe and glioblastoma cell death. Mol Cell Biol. (2015) 35:182–98. doi: 10.1128/MCB.00562-14
85. Han D, Wei W, Chen X, Zhang Y, Wang Y, Zhang J, et al. NF-κB/RelA-PKM2 mediates inhibition of glycolysis by fenofibrate in glioblastoma cells. Oncotarget. (2015) 6:26119–28. doi: 10.18632/oncotarget.4444
86. Wilk A, Urbanska K, Grabacka M, Mullinax J, Marcinkiewicz C, Impastato D, et al. Fenofibrate-induced nuclear translocation of FoxO3A triggers Bim-mediated apoptosis in glioblastoma cells in vitro. Cell Cycle. (2012) 11:2660–71. doi: 10.4161/cc.21015
87. Benedetti E, d'Angelo M, Ammazzalorso A, Gravina GL, Laezza C, Antonosante A, et al. PPARα antagonist AA452 triggers metabolic reprogramming and increases sensitivity to radiation therapy in human glioblastoma primary cells. J Cell Physiol. (2017) 232:1458–66. doi: 10.1002/jcp.25648
88. Lim JY, Oh JH, Jung JR, Kim SM, Ryu CH, Kim HT, et al. MK886-induced apoptosis depends on the 5-LO expression level in human malignant glioma cells. J Neuro-Oncol. (2010) 97:339–46. doi: 10.1007/s11060-009-0036-9
89. Kim SM, Woo JS, Jeong CH, Ryu CH, Lim JY, Jeun SS. Effective combination therapy for malignant glioma with TRAIL-secreting mesenchymal stem cells and lipoxygenase inhibitor MK886. Cancer Res. (2012) 72:4807–17. doi: 10.1158/0008-5472.CAN-12-0123
90. Woo JS, Kim SM, Jeong CH, Ryu CH, Jeun SS. Lipoxygenase inhibitor MK886 potentiates TRAIL-induced apoptosis through CHOP- and p38 MAPK-mediated up-regulation of death receptor 5 in malignant glioma. Biochem Biophys Res Commun. (2013) 431:354–9. doi: 10.1016/j.bbrc.2012.11.134
91. Spaner DE, Lee E, Shi Y, Wen F, Li Y, Tung S, et al. PPAR-alpha is a therapeutic target for chronic lymphocytic leukemia. Leukemia. (2013) 27:1090–9. doi: 10.1038/leu.2012.329
92. Messmer D, Lorrain K, Stebbins K, Bravo Y, Stock N, Cabrera G, et al. A Selective novel peroxisome proliferator-activated receptor (PPAR)-α antagonist induces apoptosis and inhibits proliferation of CLL cells in vitro and in vivo. Mol Med. (2015) 21:410–9. doi: 10.2119/molmed.2015.00139
93. Murray JA, Khanim FL, Hayden RE, Craddock CF, Holyoake TL, Jackson N, et al. Combined bezafibrate and medroxyprogesterone acetate have efficacy without haematological toxicity in elderly and relapsed acute myeloid leukaemia. (AML). Br J Haematol. (2010) 149:65–9. doi: 10.1111/j.1365-2141.2009.08055.x
94. Schmeel LC, Schmeel FC, Schmidt-Wolf IG. Clofibrate demonstrates efficacy in in vitro treatment of lymphoma and multiple myeloma. Anticancer Res. (2016) 36:3395–400. Available online at: https://ar.iiarjournals.org/content/36/7/3395.long
95. Grabacka M, Wieczorek J, Michalczyk-Wetula D, Malinowski M, Wolan N, et al. Peroxisome proliferator-activated receptor α (PPARα) contributes to control of melanogenesis in B16 F10 melanoma cells. Arch Dermatol Res. (2017) 309:141–57. doi: 10.1007/s00403-016-1711-2
96. Dana N, Haghjooy JS, Vaseghi G. The effect of fenofibrate, a PPARalpha activator on toll-like receptor-4 signal transduction in melanoma both in vitro and in vivo. Clin Translat Oncol. (2020) 22:486–94. doi: 10.1007/s12094-019-02150-7
97. Majeed Y, Upadhyay R, Alhousseiny S, Taha T, Musthak A, Shaheen Y, et al. Potent and PPARα-independent anti-proliferative action of the hypolipidemic drug fenofibrate in VEGF-dependent angiosarcomas in vitro. Sci Rep. (2019) 9:6316. doi: 10.1038/s41598-019-42838-y
98. Taïeb D, Kaliski A, Boedeker CC, Martucci V, Fojo T, Adler JR Jr, Pacak K. Current approaches and recent developments in the management of head and neck paragangliomas. Endocrine Rev. (2014) 35:795–819. doi: 10.1210/er.2014-1026
99. Lack EE, Lloyd RV, Carney JA, Woodruff JM. Recommendations for reporting of extra-adrenal paragangliomas. Mod Pathol. (2003) 16:833–5. doi: 10.1097/01.MP.0000081050.89276.CA
100. Seema S, Krishnan M, Harith AK, Sahai K, Iyer SR, Arora V, et al. Laser ionization mass spectrometry in oral squamous cell carcinoma. J Oral Pathol Med. (2014) 43:471–83. doi: 10.1111/jop.12117
101. Varet J, Vincent L, Mirshahi P, Pille JV, Legrand E, Opolon P, et al. Fenofibrate inhibits angiogenesis in vitro and in vivo. Cell Mol Life Sci. (2003) 60:810–9. doi: 10.1007/s00018-003-2322-6
102. Fruchart JC. Peroxisome proliferator-activated receptor-alpha activation and high-density lipoprotein metabolism. Am J Cardiol. (2001) 88:24–29n. doi: 10.1016/S0002-9149(01)02149-X
103. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018:GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
104. Pan Y, Zhang F, Zhao Y, Shao D, Zheng X, Chen Y, et al. Berberine enhances chemosensitivity and induces apoptosis through dose-orchestrated AMPK signaling in breast cancer. J Cancer. (2017) 8:1679–89. doi: 10.7150/jca.19106
105. Rakhshandehroo M, Knoch B, Müller M, Kersten S. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. (2010) 2010:393–416. doi: 10.1155/2010/612089
106. Bentires-Alj M, Dejardin E, Viatour P, Van Lint C, Froesch B, Reed JC, et al. Inhibition of the NF-kappa B transcription factor increases Bax expression in cancer cell lines. Oncogene. (2001) 20:2805–13. doi: 10.1038/sj.onc.1204343
107. Inta I, Paxian S, Maegele I, Zhang W, Pizzi M, Spano P, et al. Bim and Noxa are candidates to mediate the deleterious effect of the NF-kappa B subunit RelA in cerebral ischemia. J Neurosci. (2006) 26:12896–903. doi: 10.1523/JNEUROSCI.3670-06.2006
108. Duruisseaux M, Esteller M. Lung cancer epigenetics:From knowledge to applications. Semin Cancer Biol. (2018) 51:116–28. doi: 10.1016/j.semcancer.2017.09.005
109. Yao R, Wang Y, Lemon WJ, Lubet RA, You M. Budesonide exerts its chemopreventive efficacy during mouse lung tumorigenesis by modulating gene expressions. Oncogene. (2004) 23:7746–52. doi: 10.1038/sj.onc.1207985
110. Bougarne N, Paumelle R, Caron S, Hennuyer N, Mansouri R, Gervois P, et al. PPARalpha blocks glucocorticoid receptor alpha-mediated transactivation but cooperates with the activated glucocorticoid receptor alpha for transrepression on NF-kappaB. Proc Natl Acad Sci USA. (2009) 106:7397–402. doi: 10.1073/pnas.0806742106
111. Pozzi A, Popescu V, Yang S, Mei S, Shi M, Puolitaival SM, et al. The anti-tumorigenic properties of peroxisomal proliferator-activated receptor alpha are arachidonic acid epoxygenase-mediated. J Biol Chem. (2010) 285:12840–50. doi: 10.1074/jbc.M109.081554
112. Skrypnyk N, Chen X, Hu W, Su Y, Mont S, Yang S, et al. PPARα activation can help prevent and treat non-small cell lung cancer. Cancer Res. (2014) 74:621–31. doi: 10.1158/0008-5472.CAN-13-1928
113. Ajani JA, Lee J, Sano T, Janjigian YY, Fan D, Song S. Gastric adenocarcinoma. Nat Rev Dis Primers. (2017) 3:17036. doi: 10.1038/nrdp.2017.36
114. Changela K, Patil R, Duddempudi S, Gaduputi V. Endoscopic ultrasound-guided radiofrequency ablation of the pancreatic tumors:a promising tool in management of pancreatic tumors. Can J Gastroenterol Hepatol. (2016) 2016:4189358. doi: 10.1155/2016/4189358
115. Chen H, Shen ZY, Xu W, Fan TY, Li J, Lu YF, et al. Expression of P450 and nuclear receptors in normal and end-stage Chinese livers. World J Gastroenterol. (2014) 20:8681–90. doi: 10.3748/wjg.v20.i26.8681
116. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. (2018) 68:7–30. doi: 10.3322/caac.21442
117. Doser K, Guserle R, Nitsche V, Arnold G. Comparative steady state study with 2 fenofibrate 250 mg slow release capsules. An example of bioequivalence assessment with a highly variable drug. Int J Clin Pharmacol Ther. (1996) 34:345–8.
118. Park JI, Kwak JY. The role of peroxisome proliferator-activated receptors in colorectal cancer. PPAR Res. (2012) 2012:876418. doi: 10.1155/2012/876418
119. Nakada M, Nakada S, Demuth T, Tran NL, Hoelzinger DB, Berens ME. Molecular targets of glioma invasion. Cell Mol Life Sci. (2007) 64:458–78. doi: 10.1007/s00018-007-6342-5
120. Hsu PP, Sabatini DM. Cancer cell metabolism:Warburg and beyond. Cell. (2008) 134:703–7. doi: 10.1016/j.cell.2008.08.021
121. Tabata S, Ikeda R, Yamamoto M, Shimaoka S, Mukaida N, Takeda Y, et al. Thymidine phosphorylase activates NFκB and stimulates the expression of angiogenic and metastatic factors in human cancer cells. Oncotarget. (2014) 5:10473–85. doi: 10.18632/oncotarget.2242
122. Yang W, Xia Y, Cao Y, Zheng Y, Bu W, Zhang L, et al. EGFR-Induced and PKCε monoubiquitylation-dependent NF-κB activation upregulates PKM2 expression and promotes tumorigenesis. Mol Cell. (2018) 69:347. doi: 10.1016/j.molcel.2017.12.034
123. Stalinska J, Zimolag E, Pianovich NA, Zapata A, Lassak A, Rak M, et al. Chemically modified variants of fenofibrate with antiglioblastoma potential. Transl Oncol. (2019) 12:895–907. doi: 10.1016/j.tranon.2019.04.006
124. Fischer AS, Metzner J, Steinbrink SD, Ulrich S, Angioni C, Geisslinger G, et al. 5-Lipoxygenase inhibitors induce potent anti-proliferative and cytotoxic effects in human tumour cells independently of suppression of 5-lipoxygenase activity. Br J Pharmacol. (2010) 161:936–49. doi: 10.1111/j.1476-5381.2010.00915.x
125. Tsimberidou AM, Keating MJ. Treatment of fludarabine-refractory chronic lymphocytic leukemia. Cancer. (2009) 115:2824–36. doi: 10.1002/cncr.24329
126. Stebbins KJ, Broadhead AR, Cabrera G, Correa LD, Messmer D, Bundey R, et al. In vitro and in vivo pharmacology of NXT629, a novel and selective PPARα antagonist. Eur J Pharmacol. (2017) 809:130–40. doi: 10.1016/j.ejphar.2017.05.008
127. Murray J, Pratt G, Jacob A, Clark F, Blundred R, Fox S, et al. Single arm phase II trial assessing the safety, compliance with and activity of Bezafibrate and medroxyProgesterone acetate. (BaP) therapy against myeloid and lymphoid cancers. Contemp Clin Trials Commun. (2019) 14:100361. doi: 10.1016/j.conctc.2019.100361
128. Molyneux EM, Rochford R, Griffin B, Newton R, Jackson G, Menon G, et al. Burkitt's lymphoma. Lancet. (2012) 379:1234–44. doi: 10.1016/S0140-6736(11)61177-X
129. Molyneux E, Merrick B, Khanim FL, Banda K, Dunn JA, Iqbal G, et al. Bezafibrate and medroxyprogesterone acetate in resistant and relapsed endemic Burkitt lymphoma in Malawi; an open-label, single-arm, phase 2 study. (ISRCTN34303497). Br J Haematol. (2014) 164:888–90. doi: 10.1111/bjh.12681
130. Wilking MJ, Singh C, Nihal M, Zhong W, Ahmad N. SIRT1 deacetylase is overexpressed in human melanoma and its small molecule inhibition imparts anti-proliferative response via p53 activation. Arch Biochem Biophys. (2014) 563:94–100. doi: 10.1016/j.abb.2014.04.001
131. Hedrington MS, Davis SN. Peroxisome proliferator-activated receptor alpha-mediated drug toxicity in the liver. Exp Opin Drug Metab Toxicol. (2018) 14:671–7. doi: 10.1080/17425255.2018.1483337
132. Bortolini M, Wright MB, Bopst M, Balas B. Examining the safety of PPAR agonists - current trends and future prospects. Exp Opin Drug Safety. (2013) 12:65–79. doi: 10.1517/14740338.2013.741585
133. Ajima H, Kai Y, Fujimaki J, Akashi S, Morita A, Ezaki O, et al. Effects of fenofibrate and its combination with lovastatin on the expression of genes involved in skeletal muscle atrophy, including FoxO1 and its targets. J Toxicol Sci. (2021) 46:11–24. doi: 10.2131/jts.46.11
134. Tsimihodimos V, Miltiadous G, Bairaktari E, Elisaf M. Possible mechanisms of the fibrate-induced increase in serum creatinine. Clin Nephrol. (2002) 57:407–8. doi: 10.1046/j.1464-410X.2002.02735.x
Keywords: PPAR-α, PPAR-α modulators, cancer, cancer treatment, PPAR-α agonists, PPAR-α antagonists
Citation: Tan Y, Wang M, Yang K, Chi T, Liao Z and Wei P (2021) PPAR-α Modulators as Current and Potential Cancer Treatments. Front. Oncol. 11:599995. doi: 10.3389/fonc.2021.599995
Received: 28 August 2020; Accepted: 22 February 2021;
Published: 23 March 2021.
Edited by:
Kwong Tsang, Precision Biologics, Inc., United StatesReviewed by:
Massimo Fantini, Precision Biologics, Inc., United StatesCopyright © 2021 Tan, Wang, Yang, Chi, Liao and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peng Wei, d2VpcGVuZ0BidWNtLmVkdS5jbg==; Zehuan Liao, bGlhbzAwNThAZS5udHUuZWR1LnNn
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.