 Lucas V. dos Santos
Lucas V. dos Santos Carina M. Abrahão
Carina M. Abrahão William N. William Jr.
William N. William Jr.
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 05 March 2021
Sec. Head and Neck Cancer
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.596290
This article is part of the Research Topic Mechanisms of Resistance in Head and Neck Cancers View all 16 articles
Preclinical data suggest that head and neck squamous cell carcinomas (HNSCC) may evade immune surveillance and induce immunosuppression. One mechanism of immune evasion involves the expression of programmed death ligand-1 (PD-L1) in tumor and immune cells, which is, to date, the only biomarker routinely used in clinical practice to select patients with advanced HNSCCs more likely to benefit from anti-PD-1 therapy. Nonetheless, PD-L1 expression alone incompletely captures the degree of sensitivity of HNSCCs to PD-1 inhibitors. Most patients exposed to anti-PD-1 antibodies do not respond to therapy, suggesting the existence of mechanisms of de novo resistance to immunotherapy. Furthermore, patients that initially respond to PD-1 inhibitors will eventually develop acquired resistance to immunotherapy through mechanisms that have not yet been completely elucidated. In this article, we will provide an overview of the immune landscape of HNSCCs. We will briefly describe the clinical activity of inhibitors of the PD-1/PD-L1 axis in this disease, as well as biomarkers of benefit from these agents that have been identified so far. We will review pre-clinical and clinical work in cancers in general, and in HNSCCs specifically, that have characterized the mechanisms of de novo and acquired resistance to immunotherapy. Lastly, we will provide insights into novel strategies under investigation to overcome resistance to immune checkpoint inhibitors.
In recent years, our understanding of the importance of the immune system and its interaction with tumor cells and tumor microenvironment has allowed us to explore an increasing number of immune modulation strategies for cancer therapies (1). The identification of so-called checkpoints in T-cell immunity—namely, the molecules programmed death-1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), as well as the development of function-blocking antibodies against these molecules, have paved the way towards our understanding of the relevance of the immune system against cancer and its manipulation.
Immunologic checkpoints are a complex homeostatic system of signaling pathways that mediate the activation or selective tolerance of the immune system towards target cells (2). These pathways serve to establish an effector response to non-self-antigens while preventing the induction of autoimmune activity. Tumor cells, including head and neck squamous cell carcinomas (HNSCCs), hijack these mechanisms of immunologic surveillance and control to create an immunosuppressive and protumor microenvironment. As a result, immunotherapy with PD-1 blockade has emerged as the latest standard-of-care treatment strategy developed for advanced HNSCCs.
In this article, we will provide an overview of the immune landscape of HNSCCs. We will briefly describe the clinical activity of inhibitors of the PD-1/PD-L1 axis in this disease, as well as biomarkers of benefit from these agents that have been identified so far. We will review pre-clinical and clinical work in cancers in general, and in HNSCCs specifically, that have characterized the mechanisms of de novo and acquired resistance to immunotherapy. Lastly, we will provide insights into novel strategies under investigation to overcome resistance to immune checkpoint inhibitors.
HNSCC can evade immune surveillance through several crosslinked mechanisms that have now been recognized as being central to the development and progression of upper aerodigestive tract malignancies (3). The most clinically relevant mechanism of immune evasion identified so far is the modulation of cytotoxic T lymphocyte (CTL) activity.
A dual signal is mandatory for activation of CTLs against tumor antigens: the recognition of major histocompatibility complex (MHC)-antigen by the T cell receptor, and the interaction of B7 in the antigen presenting cell with CD28 in the CTL (4). This process primarily occurs in the lymph nodes and is regulated by immune checkpoint molecules. CTLA-4 is mostly expressed in CTLs, as well as in regulatory T lymphocytes (T regs). Upon binding to CTLA-4, the B7 protein induces CTL inhibition, and may cause CTL exhaustion (5). CTLA-4 expression is also upregulated by the immunosuppressive molecule transforming growth factor-β (TGF-β) produced by tumors cells (6). T regs are also one of the most important sources of TGF-β, thus contributing to T cell exhaustion (7, 8).
At the tissue level, T cell cytotoxicity is modulated by PD-1 and its ligands. PD-1 is a transmembrane protein belonging to the CD28 receptor family, which is highly expressed on T and B lymphocytes. The most important ligands for PD-1 include PD-ligand 1 and 2 (PD-L1 and PD-L2). These ligands are mostly expressed on antigen presenting cells, endothelial cells, as well as in CTLs (9, 10). Tregs inhibit CTLs by PD-1-PD-L1 interaction, thus promoting immunosuppression (11, 12). Hyperexpression of PD-1 in CTLs may also contribute to the immunosuppressive status due to enhanced sensitivity to PD-L1 (13). Like many other cancers, HNSCCs express PD-L1 on tumor cells, generating an immunosuppressive state and contributing to tumor progression and metastasis, with a negative impact on prognosis (14–16). Depending on the assay, PD-L1 is detected in about 50–70% of HNSCCs, and expression in Human Papilloma Virus (HPV)-related HNSCC is higher than in unrelated tumors (16). Indeed, HPV-related HNSCC are especially dependent on PD-L1 expression. HPV (+) tumors are characterized by more lymphocyte infiltration, with higher expression of PD-1 on CTLs when compared to HPV (−) tumors (17). In fact, at least three types of immune response in HPV (+) HNSCC have been described, rendering our understanding of the tumor host immune interactions more complex than previously thought (18).
In addition to CTLA-4 and PD-1 axes-mediated mechanisms, tumor immune evasion involves other related processes (19), selectively and briefly described as follows, some of which may represent therapeutic targets: (i) Neoangiogenesis induced by tumor associated macrophages exacerbates hypoxia and lowers the microenvironment pH, leading to PD-L1 upregulation and impairment of CTLs proliferation and efficiency (20–22); (ii) Chemokines and molecules, such as vascular endothelial growth factor (VEGF), interleukin 10 (IL-10), prostaglandin E2 and TGF-β, produced by Tregs and myeloid-derived suppressor cells (MDSCs), as well endothelial cells, reduce the attraction of CTLs (23). On the other hand, release of CXCL8, CCL2, CXCL5, and CXCL12, CCL22, and CCL28 attracts Tregs (24); (iii) Arrest of clonal expansion of CTLs, mediated by tumor cell, dendritic cell, and MDSCs secretion of indolamine-2,3-oxygenase (IDO), which induces degradation of tryptophan, an indispensable molecule for CTLs growth and production of Granzyme B (25); (iv) Impaired expression of human lymphocyte antigen-I (HLA-I) and other molecules involved in the antigen presentation machinery, leading to reduced tumor antigen recognition, impaired immune response, and worse prognosis (26, 27). Genetic alterations identified by The Cancer Genomic Atlas Network (TCGA), such as mutations in KMTD2 and HLA-A, contribute to this immunosuppressive behavior (28). Even though the complete loss of HLA-I could lead to T cell recognition evasion, activation of natural killer (NK) cells could take place, illustrating the potential for targeting multiple immune pathways for cancer therapy (29, 30).
Analyses of transcriptomics, genetic mutations, and copy number alterations in HNSCCs have revealed subtypes with common characteristics that may determine sensitivities to immunotherapies. Specifically, HPV (−) HNSCC may be subdivided into copy number high and low (28). Copy number low HPV (−) HNSCC, as well as HPV (+) HNSCC have been shown to have increased expression of immune signatures predictive of benefit from immune checkpoint inhibitors (31). Likewise, in a pan-TCGA analysis including HNSCC, lymphocyte infiltration correlated negatively with copy number variation segment burden, and positively with aneuploidy, loss of heterozygosity, homologous recombination deficiency, and tumor mutational burden (TMB) (32). In another pan-TCGA analysis, somatic copy number variation scores were positively correlated with mutations in driver genes involved in the DNA damage response pathway, as well as reduced cytotoxic immune infiltration—arm/chromosome somatic copy number variation scores were stronger predictors for decreased expression of immune signatures compared to focal copy number variation scores, including in HNSCCs with high TMB (33).
Recently, six immune subtypes across multiple tumor types were identified in an extensive pan-cancer TCGA immunogenomic analysis: wound healing, interferon-γ dominant, inflammatory, lymphocyte depleted, immunologically quiet, and TGF-β dominant. These tumors were characterized by differences in macrophage or lymphocyte signatures, Th1:Th2 cell ratio, intra-tumoral heterogeneity, copy number alterations, neoantigen load, cell proliferation, expression of immunomodulatory genes, and prognosis (34). The vast majority of squamous cell carcinomas were of the wound healing or interferon-γ dominant subtypes, with no significant differences in survival between these two groups. However, a more recent TCGA evaluation further stratified squamous cell carcinomas into six immune subtypes with distinct molecular characteristics and outcomes (35). The immune-cold subtype had the lowest level of T cell infiltration, the highest rate of aneuploidy, translating into worst survival. A subtype with M2-polarized macrophages, TGF-β signaling and reactive stroma also had a poor outcome compared to the other subtypes. The subgroup with the best survival rates was characterized by high CTLs and NK infiltration and elevated interferon-γ signature (35). In another study focusing specifically on HNSCCs, Chen et al. proposed three subgroups which were consistent with (albeit less granular than) the aforementioned analysis: immune active (enriched by proinflammatory M1 macrophage signature, with increased cytolytic activity and tumor infiltrating lymphocytes, and high incidence of HPV infection); immune exhausted (enriched by activated stroma and anti-inflammatory M2 macrophage signatures, with activation of the WNT/TGF-β signaling pathway activation and poor survival), and a non-immune class (36). In another TCGA comprehensive HNSCC immune landscape study, Mandal et al. demonstrated that both HPV (+) and HPV (−) HNSCCs were one of the most immune infiltrated tumors. However, most of the immune infiltrate was comprised of Tregs, which suppress immunological activities. NK population was also remarkably abundant in both subtypes of HNSCC (37). HPV-related HNSCC demonstrated the highest immune infiltration and increased cytolytic activity, which was counterbalanced by an increased Treg/CTLs ratio, whereas smoking related HNSCC had the lowest level of immune infiltration and interferon-γ signature. Patients with adaptative immune response cell infiltrates and mutations had improved survival when compared to those with innate immune response infiltrate and copy number alterations, suggesting a possible role for new immunotherapeutic approaches targeting Tregs and NK cells in improving efficacy of anti-PD-1 (37). Lastly, Cillo et al. assessed the transcriptional profiles of single cells from peripheral and intra-tumoral immune populations from patients with HPV (−) and HPV (+) HNSCCs and showed that helper CD4+ T cells and B cells were relatively divergent and CD8+ T cells and CD4+ regulatory T cells were relatively similar. They also identified a gene expression signature associated with CD4+ T follicular helper cells and longer progression-free survival (38).
The immune phenotype of HNSCC has also been characterized in terms of spatial distribution of tumor infiltrating lymphocytes. Troiano et al. classified tongue carcinomas into immune-inflamed (when lymphocytes were found next to tumor cells), immune-excluded (when lymphocytes were found in the stroma, outside the tumor), or immune-desert (absence of lymphocytes). Immune desert was the less frequent subgroup, but exhibited worse overall survival (39).
Taken together, these findings suggest a complex immune landscape, associated with (and possibly determined by) genomic alterations, with important implications to HNSCC prognosis. Interestingly, in one report, the transcriptomic variability of immunologic signatures seemed to be stable in both a spatially, and short-term, timely manner, minimizing the importance of tumor heterogeneity in selecting immunotherapeutic approaches, at least for untreated patients (40). The data provide rationale for development of PD-1 inhibitors for HNSCCs along with potential biomarkers of efficacy, and for development of combination immunotherapeutic approaches for management of patients harboring tumors with de novo and/or acquired resistance to such immunotherapies.
The identification that lymphocytes could take part in the immune response in cancers (including HNSCCs) was identified several years ago (41), and evolved to the development of cancer immunotherapy, initially for melanomas, later extended to HNSCCs and other tumor types. Simplistically, the ultimate goal of immunotherapy is to relieve immunosuppression, and thus induce responses in tumor, without auto-immune adverse events (42, 43). To date, the anti-PD-1 antibodies nivolumab and pembrolizumab have been investigated in phase 3 studies and are the only immunotherapies approved by regulatory agencies worldwide for treatment of advanced HNSCCs.
In patients with recurrent or metastatic HNSCC that failed platinum-based chemotherapy enrolled in the Checkmate-141 study, nivolumab, when compared to the investigators’ choice single agent therapy, improved overall survival (OS) and overall response rate (ORR). Importantly, the rate of grade 3–4 adverse events was lower with nivolumab than chemotherapy (44). In the KEYNOTE-040 study with analogous design, pembrolizumab demonstrated similar benefits against investigators’ choice standard therapy, although statistical significance for the primary OS endpoint was not reached (45).
These encouraging results led to the development of pembrolizumab in the first-line setting for recurrent/metastatic disease. In the KEYNOTE-048 study, with a complex statistical design and assumptions, pembrolizumab either as monotherapy or in combination with cisplatin or carboplatin and 5-fluorouracil (5-FU) was compared to platinum plus 5-FU combined with cetuximab (EXTREME regimen) in patients that had failed curative-intent therapies including surgery and/or radiation therapy (46). Compared to the EXTREME regimen, pembrolizumab monotherapy improved OS in the PD-L1 combined positive score (CPS) ≥ 20 or CPS ≥ 1 populations. The safety profile was improved in the pembrolizumab arm. Data on the chemotherapy plus pembrolizumab cohort is discussed below [see PD-(L)1 inhibitors plus chemotherapy subsection]. These results led to the FDA-approval of pembrolizumab as first-line monotherapy in patients with recurrent or metastatic disease with PD-L1 CPS ≥1, and in combination with chemotherapy independently of PD-L1 expression (47). However, the European regulatory agency recommended the approval of first-line pembrolizumab (whether monotherapy or in combination with platinum and 5-FU) only in patients with PD-L1 expressing tumors (CPS of 1 or above) (48). Nonetheless, PD-L1 expression alone incompletely captured the degree of sensitivity of HNSCCs to PD-1 inhibitors. Most patients exposed to anti-PD-1 antibodies did not respond to therapy, suggesting the existence of mechanisms of de novo resistance to immunotherapy. Furthermore, most patients that initially respond to PD-1 inhibitors eventually develop acquired resistance to immunotherapy through mechanisms that have not yet been completely elucidated. These data illustrate the need to discover more accurate biomarkers of sensitivity to PD-1 axis blockade, as well as strategies to enhance activity of and/or overcome resistance to these drugs.
Both PD-L1 and PD-L2 expression have been reported in many tumor types (including HNSCCs) and were amongst the first candidate biomarkers of immunotherapy efficacy investigated across several trials (49–51). Several PD-L1 assays are available in oncology, and they seem to be highly interchangeable in HNSCC (52), specially for assays evaluating tumor cells by the antibodies SP263, 22C3, and 28-8 (53). Concordance between staining scores that involve immune cells, and/or other antibodies (e.g., SP142) are more modest (53, 54) and require more careful interpretation. In the pivotal phase 3 Checkmate-141 trial, OS and ORR were improved by nivolumab across the entire study population. However, the magnitude of benefit seemed higher in patients with PD-L1 expression in at least 1% of the tumor cells using the 28-8 assay. HPV (+) and HPV (−) cancers derived the same benefit from nivolumab. No interaction between HPV and PD-L1 status was observed in this clinical trial (44, 55). Pembrolizumab was first explored in advanced HNSCC in the multi-cohort phase Ib KEYNOTE-012 trial (56). Anti-PD-L1 22C3 and anti-PD-L2 3G2 antibodies were used for PD-L1 and PD-L2 immunohistochemical assays, respectively. Overall, a 4% complete response (CR) and 14% partial response (PR) rate was observed, and 60% of patients experienced reductions in target lesions. PD-L1 CPS (which takes into account PD-L1 expression in both tumor and immune cells) performed better than the tumor proportion score (TPS) in predicting response to pembrolizumab, emerging as the most reliable biomarker for pembrolizumab. PD-L2 and PD-L1 expression were correlated, and PD-L2 expression was also associated with higher ORR. Patients with co-expression of PD-L1 and PD-L2 had higher ORR compared to PD-L1 positive patients alone. However, a 9% ORR was found in patients without the expression of any biomarker, underscoring the limitations of these strategies in selecting patients for pembrolizumab therapy (56). Similar results were found in the single arm, phase 2 KEYNOTE-055 study (57). These data supported the incorporation of PD-L1 expression (assessed by CPS) into the statistical design of the first-line KEYNOTE-048 study, as previously described.
TMB has been postulated as a possible biomarker of immunotherapy efficacy in cancers. Presumably, high TMB increases the abundance of neo-antigens (or neo-epitopes) resulting from non-synonymous mutations on cancer cells, allowing immune recognition and specific CTLs activation (58–60). However, only a small number of missense mutations produce neo-antigens, and a smaller part of those neo-antigens ultimately are recognizable by CTLs (61, 62). As such, specific immunogenic mutations, rather than total mutational burden, may be associated with improved prognosis, as it leads to increased expression of CD8A and hyperexpression of PD-1 and CTLA-4 (61). Despite these limitations, TMB has been associated with improved outcomes in clinical trials. In KEYNOTE-012, using a cut off ≥ 102 mutations per exome, TMB was associated with improved ORR (63–65). In a series of 126 patients treated at the Dana Farber Cancer Institute, TMB was higher in former smokers compared to non-smokers and HPV (+) patients, as well as in responders. Among HPV (−) responders, NOTCH1 and SMARCA4 were more frequently mutated, and frameshift events in tumor suppressor genes occurred more frequently. T cell immunoglobulin mucin-3 (TIM-3)/lymphocyte activated gene-3 (LAG-3) co-expression with PD-1 was higher on T cells among non-responders, suggesting a possible mechanism of adaptive de novo immune resistance (66). Consistent with the KEYNOTE-012 and Dana Farber data, a post hoc analysis of the EAGLE study revealed that a blood TMB of 16 mutations/Mb was associated with improved OS in the second-line setting for the anti-PD-L1 durvalumab alone, or in combination with the anti-CTLA4 tremelimumab versus single agent chemotherapy (67). Of note, blood TMB seemed also to be prognostic in the EAGLE study, since median OS in the standard of care arm was 4.0 months for high TMB versus 8.6 months for low TMB, raising the question whether this biomarker could be associated with poor outcomes to chemotherapy alone (67).
Interferon-γ and its co-stimulatory chemokines are implicated in tumor innate immune sensing, leading to immediate CTLs recruitment into the tumor micro ambient, a key step for an effective immune response (68–70). Interferon-γ gene expression was associated with clinical response in several cancer types treated with pembrolizumab (71). Likewise, in the KEYNOTE-012 study, a 6-gene interferon-γ signature including IDO1, CXCL10, CXCL9, HLA-DRA, STAT1, IFN-γ gene expression was found to be associated with improved ORR or progression-free survival (65).
A major role of fecal microbiome in determining response to immunotherapy has been increasingly recognized in recent years (72). Several mechanisms have been implicated in the dynamic interaction between microbiome and immunologic response, including T-cell activation, influence on recognition pattern of antigens (73). Some specific bacterial genera have been identified as predictors of response and toxicity in fecal microbiota transplant (FMT) experiments in mice. Akkermansia muciniphila was associated with increased response to anti-PD-1 (74). A. muciniphila, and Enterococcus hirae was able to reverse resistance to immunotherapy in mice. The mechanism implicated in such effects were related to increase in CCR9, CXCR3, and promotion of CTLs infiltration (75). Studies in melanoma suggested that the presence of certain genera, like Bifidobacterium longum, Collinsella aerofaciens, Enterococcus faecius, Faecalibacterium spp and Ruminococcaceae spp were associated with increased response to immunotherapy, and Bacteriodales were more common among non-responders. Responders were more likely to harbor greater microbiome diversity than non-responders (76, 77). Recently, data from phase 3 randomized trials comparing anti-PD-1 to chemotherapy showed that the use of antibiotics impaired the OS of patients receiving anti-PD-1 without compromising survival in the control group, suggesting a major role of microbiota in the benefit of immunotherapy in HNSCC (78). Oral cavity microbiome has also been implicated in HNSCC carcinogenesis and progression. Usually, Fusobacteria are abundant in primary and metastatic tissues, whereas Streptococcus have limited homing (79). Smoking and alcohol consumption are major risk factors for both HNSCC and periodontal disease, and are key modifiers of oral microbiota (80, 81). Abundant F. periodonticum and S. mitis and P. pasteri paucity are associated with late stage oral cancer (82). Despite these promising data, oral microbiome was not associated with outcomes in the Checkmate 141 study (83). Evaluation of fecal specimens may better reflect patients’ microbiome, but have not been assessed in anti-PD-1 trials in HNSCCs.
Taken together, these data demonstrate that a robust biomarker of sensitivity to PD-1/PD-L1 blockade has yet to be developed. It is likely that multiple mechanisms of resistance to immunotherapies are in place, leading to low response rates to single agent PD-1 inhibitors in all HNSCC clinical trials performed to date. Few comprehensive upfront and re-biopsy studies for biomarker evaluation (especially upon disease progression) have been completed. As will be discussed below, such investigations would be essential for the rational design of strategies aiming at mitigating resistance to treatment.
From a clinical perspective, resistance to immunotherapy may be divided into de novo or acquired. De novo (or primary) resistance may be defined as lack of benefit from upfront immunotherapy treatment, whereas acquired (or secondary) resistance is characterized by an initial period of benefit from immunotherapy followed by disease progression. Mechanistically, de novo and acquired resistance to immunotherapy may share common processes, including adaptive immune resistance (whereby the cancer is recognized by, but evades the immune system, by adapting to the immune attack). Additionally, acquired resistance to immunotherapy may emerge from adaptive resistance that occurs in a relatively homogenous fashion, and/or by selection of heterogenous clones over time that were already resistant to immunotherapy, even before treatment initiation (84).
Mechanisms of resistance to immunotherapy include tumor cell-intrinsic and tumor cell- extrinsic factors. Tumor cell-intrinsic resistance may stem from absence of antigenic proteins, absence of antigen presentation, genetic T cell exclusion, and/or insensitivity to CTLs. Tumor cell-extrinsic resistance may be a result of absence of CTLs, expression of inhibitory immune checkpoints, and/or presence of immunosuppressive cells (84–86). None of these mechanisms have been extensively studied in HNSCCs, limiting our understanding about the dynamic pressures on the immune system at play upon immunotherapy administration, and hindering our ability to rationally design combination and/or sequential approaches to mitigate resistance to PD-1 inhibitors. Knowledge gained from other cancers and pre-clinical work might prove to be relevant to HNSCC patients and is therefore described below.
Beta-2-microglobulin (B2M) has an important function on HLA class I transport to cell membrane, and inactivating mutations in B2M lead to loss of expression of HLA class I, impairing immune response (87, 88). This mechanism of resistance to anti-PD1 in metastatic melanoma has been detailed elsewhere (89). Other groups reported that B2M mutation in other clinical settings could also lead to acquired resistance to immunotherapy (90–92). Other causes of HLA class I loss of expression with intact B2M may also induce disease progression to anti-PD1 (90). Saloura et al. have demonstrated more diverse T-cell repertoire in HPV (+) versus (−) HNSCC, possibly due to impaired HLA class I expression induced by the virus (93). As such, strategies that could restore HLA class I expression could potentially be developed to augment immune response in this setting.
Release of IFN-γ by CTLs may induce PD-L1 and MHC class I expression in tumor cells through activation of the JAK-STAT pathway. Several mechanisms of tumor cell death derive from this pathway (94). Clinical evidence recognizes that mutations in JAK1 and JAK2 can be responsible for the progression of metastatic melanoma after initial response to anti-PD1 (89). It is unknown whether alterations in other molecules in the JAK-STAT pathway could be implicated in acquired resistance to immunotherapy, but their role in primary resistance has already been demonstrated (95).
The loss of mutations that preclude the expression of neoantigens recognized by the immune system through clonal selection, copy-number loss, or epigenetic mutation may lead to immune evasion and clinical progression (96). In a case series of four patients with non-small cell lung cancers, mutations encoding neoantigens were lost, and progressive disease occurred after initial response do anti-PD1 therapy (97) Clonal pressure has been implicated in immunoselection of tumor cells that respond to CTLs and adoptive cell transfer immunotherapy (98–100).
Phosphatase and tensin homolog on chromosome 10 (PTEN) inactivation and, consequently, phosphoinositide 3-kinase (PI3K) pathway activation is related to an immunosuppressive tumor microenvironment that may have implications in resistance to immunotherapy (101). A total of 55 isocitrate dehydrogenase 1 wild-type glioblastoma patients who received immunotherapy, including 13 long-term responders, were analyzed in one report, and PTEN mutations were identified in 23 out of 32 non-responders, but only in 3 responders (102). PTEN mutations were also associated with an immunosuppressive signature. Similar results were found in non-small cell lung cancer and melanoma patients, indicating a putative effect of PTEN-loss in acquired resistance to immunotherapy (103, 104). PTEN-loss may also be implicated in secondary resistance to immunotherapy in other distinct tumor types (105, 106). Patients with metastatic melanoma who initially responded to anti-PD-1 alone or in combination with anti-CTLA-4 and then progressed were analyzed, and PTEN-loss was identified in 5 cases in the post-progression biopsy out of 18 intact PTEN expression in pre-treatment biopsies (107). In surgically treated oral cavity squamous cell carcinomas, PTEN loss in tumor infiltrating immune cells has been associated with worse prognosis (108). Similarly to PTEN-loss, WNT-β-catenin promotes an immunosuppressive tumor microenvironment that may be responsible for secondary resistance to immunotherapy (106, 109).
Modulation of other immune checkpoints has been identified in patients with secondary resistance to immunotherapy, including, but not limited to, TIM-3, LAG3, and V-domain immunoglobulin suppressor of T cell activation (VISTA), glucocorticoid induced TNFR family related gene (GITR), and T cell immunoglobulin and immunossupressor tyrosine kinase-based inhibitory motif (TIGIT) (90, 107, 110).
TIM-3 is a member of TIM family expressed on CD4(+) Th1 but not Th2 lymphocytes (111). It is also expressed in tumor cells and other immune cells (112). TIM-3 and its ligands, such as galectin-9, may regulate several biological functions of tumor cells, including aggregation, adhesion and apoptosis (113, 114). The binding of TIM-3 to galectin-9 leads to promotion of apoptosis of Th1 cells, impairs function of CTLs and induces major expansion of MDSCs, suppressing immune response. In early stages of disease, TIM-3 may have an immunostimulatory effect favoring CTLs secretion of interferon-γ, but TIM-3 expression in Tregs in late-stage tumors favor the suppression of CTLs and are important to create an immunosuppressive environment. Anti-TIM-3 monoclonal antibody may suppress the inhibition of CTLs and improve antitumor response (115). TIM-3 expression has been implicated in nodal metastasis and recurrence in HNSCC (116). TIM-3 may be related to the exhaustion of CTLs and ineffective immune response in HNSCC, favoring metastatic behavior (117) In a HNSCC mouse model, anti-TIM-3 antibody induced activation of CTLs and suppressed MDSCs, inhibiting carcinogenesis and improving antitumor responses (116).
LAG3, also known as CD223, is mainly expressed in activated T cells and, to a lesser extent, NK cells, B cells and dendritic cells. LAG-3 reduces T cell proliferation and activation (118, 119). LAG-3 is also an effector of Tregs inhibitory function (120). Tumor-infiltrating lymphocytes co-expressing PD-1 and LAG-3 may be susceptible to inhibition, leading to immune scape of cancer cells (121). LAG-3 also binds to liver and lymph node sinusoidal endothelial cell C-type lectin (LSECtin) and inhibit the secretion of interferon-γ by CTLs, therefore inhibiting immune response (122). Fibrinogen-like protein 1 (FGL1) is a liver secreted protein which inhibits the activation of T cells (123, 124). FGL1 is upregulated in several human cancers and it is associated with impaired outcome and blocking of FGL1-LAG-3 interaction enhances T cell response and improves antitumor immunity (123, 124). In HNSCCs, LAG-3 overexpression is associated with worse prognosis, and LAG-3 blockade retarded tumor growth in a HNSCC mouse model (125).
VISTA is another checkpoint similar in function to PD-L1 and capable of suppressing T effector cells. VISTA is expressed on myeloid APCs and Tregs (126). VISTA enhances Treg maturation and suppresses T cell activation (127). V-set and immunoglobulin domain-containing 3 (VSIG3) interacts with VISTA on activated T cells, suppress T cell proliferation and induces the production of immunosuppressive cytokines and chemokines. Data from several tumor types support blocking of VSIG3/VISTA pathway as a promising immunotherapy strategy (128). In HNSCC patients, overall survival was reduced when VISTA expression was high simultaneously with low CD8+ infiltration (129).
GITR is expressed on the surface of CD25+CD4+ Tregs, CTLS and NK cells (130). Binding of GITR to its ligand GITRL may impair the attraction of Tregs, weaken their suppression activity and activate the MAPK (mitogen-activated protein kinase)/ERK pathway and NF-κB signaling, which ultimately induces T cell proliferation and pro-inflammatory cytokines (131–133).
TIGIT is expressed mainly in effector lymphocytes and NK cells (134). CD155 is highly expressed in tumor cells and has high affinity to TIGIT, and induces IL-10 secretion, reduces the secretion of pro-inflammatory cytokines and inhibits antitumor response (135). TIGIT shares the same ligands with CD226, which, in part, counterbalances the TIGIT immunosuppressive effect (136). In mouse models of HNSCCs, TIGIT blockade delayed tumor progression through mechanisms involving CD8+ CTLs activation and Tregs inhibition. PD-1/PD-L1 inhibition increased expression of TIGIT on Tregs (137).
Some mechanisms of resistance may be induced by previous treatment. For example, in the Checkmate-064 study, patients with metastatic melanomas were randomized to ipilimumab followed by nivolumab after 12 weeks or the opposite order, and the immune landscape was analyzed at baseline and at week 13. Some immunophenotypes were more prone to show responses to ipilimumab and progression to nivolumab, and vice versa. Ipilimumab and nivolumab induced different patterns or immune landscape change after 12 weeks, and such patterns were related to patient outcomes. Furthermore, the alterations induced by ipilimumab favored progression in the nivolumab-ipilimumab cohort, whereas the alterations induced by nivolumab favored response in the nivolumab-ipilimumab cohort (138). These findings explain the superior survival outcomes in the nivolumab-ipilimumab arm (139).
There is an intricate interplay between the immune system, other components of the tumor microenvironment, and cancer cells that ultimately contribute to carcinogenesis and determine sensitivity and resistance to therapeutic strategies that have been developed so far to manage HNSCCs. The complexities of the microenvironment-cancer cell equilibrium outlined above in this review suggest that single-agent anti-PD-1/PD-L1 therapy would not be sufficient to promote long-term disease control. A natural evolution in the clinical development process of pharmacologic agents to treat HNSCCs would be the study of drug combinations, many of which have not been thoroughly investigated in pre-clinical systems specific to head and neck cancers but are already undergoing testing in human trials. This rapid pace of clinical investigations underscores a new model of information “cross pollination” from one cancer type to the next that, on the one hand, could reduce the likelihood of success of each individual study (given less robust rationale), but on the other hand may collectively result in identification of improved treatment options for patients with malignant diseases that were previously considered low priority for drug development, such as HNSCCs. Indeed, at the time of this writing, a search on clinicaltrials.gov using the terms “head and neck cancer” AND “nivolumab”, “pembrolizumab”, “durvalumab”, “atezolizumab”, “avelumab”, OR “cemiplimab” (i.e., PD-1/PD-L1 inhibitors already approved for at least one cancer type) resulted in 270 studies (Figure 1). Below, we briefly discuss all phase 3 drug combination trials identified, whether ongoing, completed or terminated.
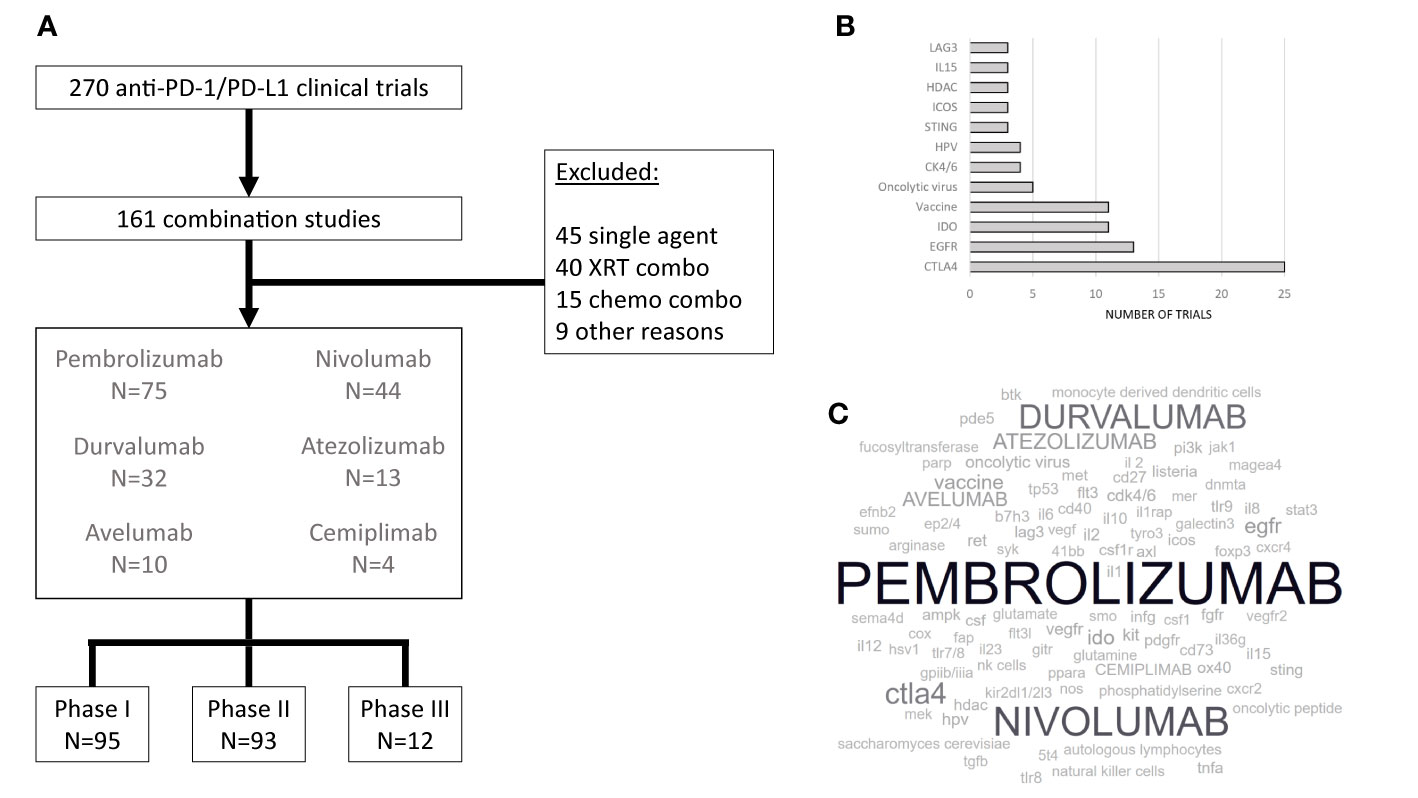
Figure 1 Clinical trials evaluating immunotherapy in HNSCC. (A) Completed, ongoing, or terminated HNSCC clinical trials involving pembrolizumab, nivolumab, durvalumab, atezolizumab, avelumab, or cemiplimab as single agents or in combination with drugs other than cytotoxic chemotherapy. (B). Most common co-targets (or mechanism of action, when appropriate – e.g. oncolytic virus) for anti-PD-1 or anti-PD-L1 combinations (C). A word cloud visual representation of PD-1 or PD-L1 inhibitors (in capital letters) and their co-targets (in small caps) under evaluation in combination studies. The font size is proportional to the number of studies employing the intervention.
Chemotherapy has been proposed as a combination strategy to enhance immunotherapy efficacy and bypass de novo and/or acquired resistance to PD-(L)1 inhibitors, through mechanisms that might involve increase of mutational load in cancer cells, depletion of suppressive regulatory T cells and myeloid-derived cells, normalization of neovasculature (thus facilitating T cell infiltration), upregulation of HLA class I expression and other components of antigen presentation machinery, induction of immunogenic cancer cell death (leading to neoantigen cross presentation), and modulation of cell signaling to increase sensitivity to interferon-gamma (140). In the KEYNOTE-048 phase 3 study, platinum, 5-FU plus pembrolizumab was compared to platinum, 5-FU plus cetuximab, and results demonstrated an improvement in overall survival for patients with PD-L1 expression CPS ≥ 20, CPS ≥ 1, or in the total population (regardless of PD-L1 expression) (46). Post-hoc analysis has demonstrated that efficacy improvement for the combination was primarily restricted to the PD-L1 positive group (141), raising the possibility that chemotherapy may not contribute to overcoming resistance to immunotherapy in patients without PD-L1 expression. A comparison between the pembrolizumab and chemotherapy plus pembrolizumab arms was not planned per trial design, and therefore it remains to be determined whether the long-term benefits from treatment in the pembrolizumab-containing arms can be attributed to the immunotherapy alone, or may be a result of a synergistic effect between chemotherapy and immunotherapy contributing to mitigation of resistance to treatment. Several chemotherapy plus immunotherapy trials are under way (Figure 1), combined or not with radiation therapy (see below).
The tumor immune microenvironment is dynamic and has been shown to be depleted of CD8+ T cells and B lymphocytes in recurrent versus primary tumors, with immune suppressive features apparent after receipt of chemoradiation therapy (142). Clonal expansion of tumor-infiltrating T cells has been identified in patients with untreated, locoregionally advanced SCCHN (93). These observations increase the enthusiasm for incorporating immunotherapy for earlier stages of HNSCC, in which immune function seems to be better preserved, and for which radiation therapy-based strategies are often used as a standard of care. Adiotherapy-induced immunosuppression has been well characterized (143). In animal models, PD-L1 blockade combined with radiation therapy reverses T cell exhaustion and leads to oligoclonal T cell expansion (144), suggesting a possible role of PD-(L)1 inhibitors in contributing to disease control in this setting. Radiation therapy may also synergize with immune checkpoint inhibitors through other nonredundant pathways that enhance antitumor activity, reviewed elsewhere (143, 145). Results of the first randomized studies combining PD-(L)1 inhibitors with radiation therapy for locally advanced HNSCC have recently been presented. In the GORTEC 2015-01 PembroRad trial, pembrolizumab plus radiation therapy failed to improve locoregional control compared to cetuximab plus radiation therapy in patients unfit for platinum (146). Likewise, in the phase 3 JAVELIN Head and Neck 100 trial, addition of avelumab to cisplatin/radiation therapy did not improve progression-free or overall survival (147). In advanced disease, addition of stereotactic radiation therapy to nivolumab has also been evaluated as a strategy to induce abscopal effect in a randomized phase 2 trial. Unfortunately, there were no improvements in overall response rates of nonirradiated lesions (primary endpoint), progression-free or overall survival (148). Despite these early negative results, several studies continue to evaluate immunotherapy in the context of radiation therapy (Figure 1) and will eventually determine whether PD-(L)1 inhibitors and radiation therapy can be combined to effectively circumvent resistance to treatment.
CTLA-4 was the first modern immunotherapy strategy to be widely explored in oncology. CTLA-4 can bind to B7, precluding the interaction between B7 and the co-stimulatory molecule CD28 and limiting the proliferation of T cells and the release of interleukin-2 (149). Blocking of CTLA-4 may limit the inhibitory effect on CTLs favoring host immune response. Due to its limited efficacy in other tumor types than melanoma, including HNSCC, anti-CTLA-4 has been developed mostly in combination with other agents (150–155). More recently, building on the results of the phase 2 CONDOR study (154), durvalumab, alone or in combination with tremelimumab, were compared to investigator’s choice chemotherapy (cetuximab, taxane, methotrexate, or fluoropyrimidine) in the phase 3 EAGLE study involving patients with HNSCC whose disease failed platinum-based chemotherapy. Durvalumab alone or in combination did not meet the primary outcome of OS benefit. Duration of response and 2-year survival were improved in the durvalumab monotherapy arm, suggesting that this drug is active in HNSCCs (156). PD-L1 expression, as assessed by SP263 assay, did not impact on any of the efficacy results, but a small benefit was found in patients with high TMB (≥16mutations/Mb) (67, 156). Despite these disappointing results, durvalumab alone or in combination with tremelimumab was evaluated in the first line setting in comparison with the EXTREME regimen in the KESTREL trial (NCT02551159), and results are pending. Nivolumab has also been tested in combination with ipilimumab versus nivolumab alone in the Checkmate-714 trial, and although the data have not yet been formally reported, a press release dated April 25, 2019 has indicated that the study did not meet its primary endpoint. In Checkmate-651 nivolumab plus ipilimumab has been compared against the EXTREME regimen (NCT02741570). Recruitment has already been completed, and the main data for these trials are expected in the following months. Nivolumab plus ipilimumab is also under development in the setting of locally advanced, potentially curable disease (NCT03700905). Taken together, the strategy of targeting PD-(L)1 plus CTLA-4 has not yielded promising results so far in phase 2/3 trials (154, 156). Unless the recently completed studies (NCT02551159, NCT02741570) report superior outcomes in the near future, other immunotherapy-immunotherapy combinations may need to be explored, as discussed below.
IDO is an enzyme that metabolizes tryptophan, limiting CTLs cytotoxicity. It is highly expressed in tumor-cells, macrophages and dendritic cells (157). Tryptophan depletion and its inhibitory metabolites has been implicated on how IDO is responsible for T cell anergy and suppression, as well as Treg activation and MDSCs infiltration (158, 159). IDO activity has been implicated on resistance to anti-PD-1 therapy (160, 161). Epacadostat, an IDO inhibitor, was evaluated in combination with pembrolizumab in advanced solid tumors, including HNSCCs. In phase I study ECHO-202/KEYNOTE-037, the ORR for this combination was 55%, including 13% complete responders. Two patients with refractory HNSCC were included, and one achieved a partial response and the other had stable disease with minor reduction in tumor burden (162). This study had a phase II part, including 36 additional HNSCC patients. The ORR was 30.5%, which was lower in patients with 3 or more lines of treatment (163). Nivolumab was also combined with epacadostat, with an ORR of 22.6% (164). Despite these results, its development as adjunctive therapy to anti-PD-1 in HNSCC (NCT03358472, NCT03342352) was halted due to the negative results of the combination IDO inhibitor and pembrolizumab in melanoma (165).
B7 constitutes a superfamily of inhibitory molecules in the cancer microenvironment was highly related to the immune evasion of cancer cells (4). B7-H3 (also known as CD276) is a newly identified member of the B7 family (166, 167), which is found in several human cancer cells and APCs. B7-H3 induces proliferation of both CD4+ and CD8+ T cells, enhances CTLs, and stimulates IFN-γ production in the presence of T cell receptor signaling (167). B7-H3 was later found to negatively regulate T cell function, affecting preferentially T helper type 1-mediated immune responses (168). Overexpression of B7-H3 was associated with larger tumor, advanced stage, and impaired survival in oral cancer patients (169). Retifanlimab, an anti-PD-1 antibody, and enoblituzumab, an anti-CD276, were evaluated in combination in multiple tumor types cohorts. In the anti-PD-1 naïve HNSCC cohort, 18 patients were treated, and ORR were 33.3%, with five partial responders and one complete responder (170). This led to the subsequent development of a phase II/III study (NCT04129320).
As described in Table 1, a number of phase 2 studies have been completed targeting the PD-1/PD-L1 axis and EGFR in recurrent/metastatic disease. A randomized, phase 3 study is currently ongoing evaluating the role of avelumab added to concurrent cetuximab/radiation therapy in locally advanced HNSCCs. GORTEC-2017-01 is a two-cohort prospective clinical trial enrolling treatment naïve patients with resectable stage III-IVa HNSCC. Cisplatin-eligible patients will be randomized to radiation therapy plus cisplatin or radiation therapy plus cetuximab plus avelumab. Those who are unfit for cisplatin therapy will be randomized to radiation therapy plus cetuximab or radiation therapy plus cetuximab plus avelumab (NCT02999087).
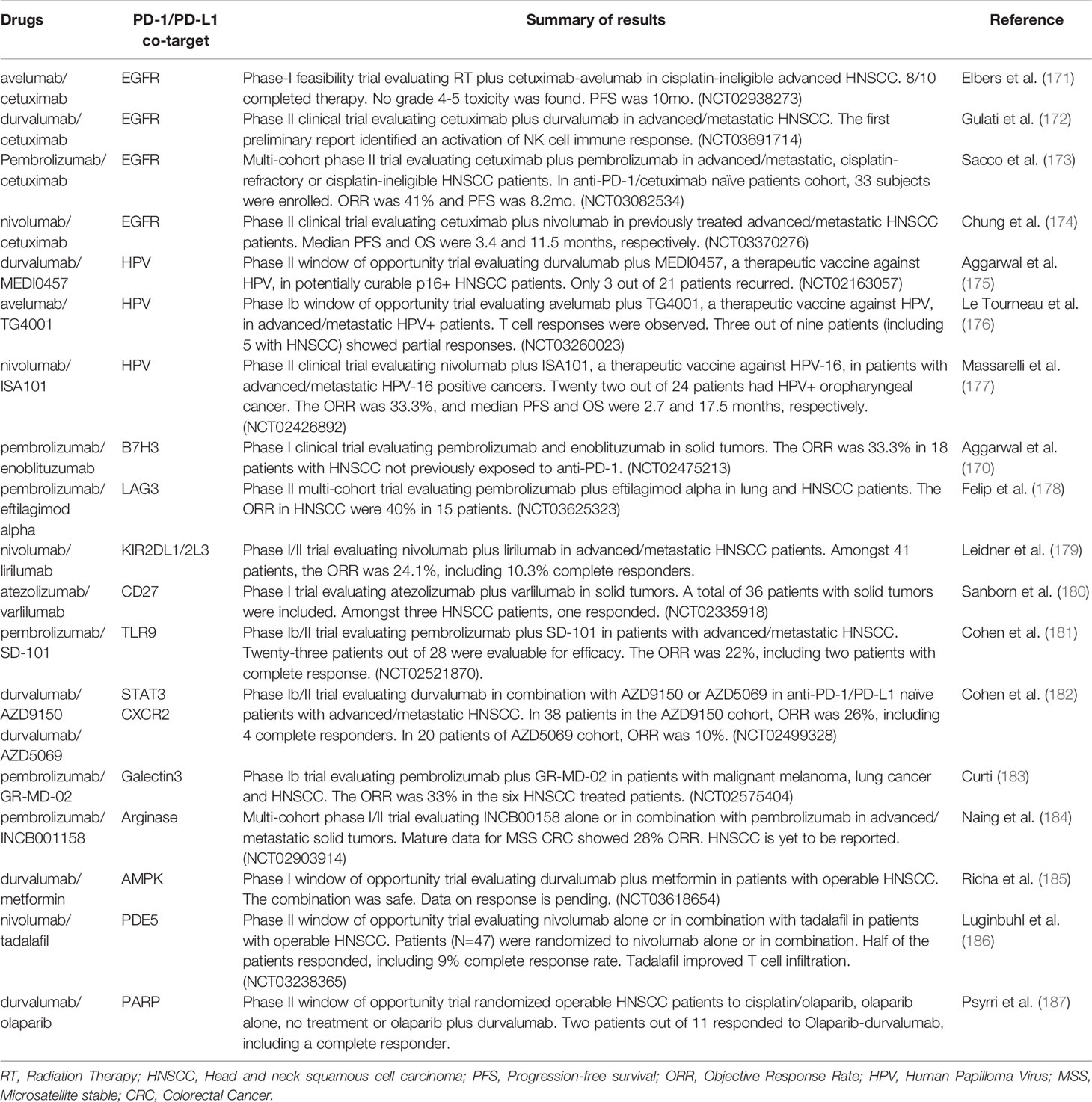
Table 1 Select Phase I and II drug combination clinical trials with PD-1/PD-L1 inhibitors.
As mentioned above, angiogenesis is closely related to immune response and may take part in the development of de novo or acquired resistance to immunotherapy. Lenvatinib is a multi-kinase inhibitor of vascular endothelial growth factor receptors 1–3, was combined with pembrolizumab in a phase Ib/II clinical trial. The ORR was 36.4% in 22 evaluated HNSCC patients (188). These results lead to the development of a placebo-controlled randomized phase 3 trial enrolling patients with HNSCC with no prior therapy for advanced or metastatic disease and CPS ≥ 1 to pembrolizumab plus Lenvatinib or pembrolizumab alone (NCT04199104).
The inducible co-stimulator of T cells (ICOS, or CD278) and its ligand (ICOSL) play important roles in memory and CTLs development and specific immune responses (189). ICOS and its pathway potentiates immunosuppression mediated by Tregs, but also induces antitumor responses when activated in CTLs (190, 191). Data on combination of anti-PD-1 with ICOS agonists are scarce, but synergy has been observed (192, 193). These data led to the rapid launching of a randomized phase 2 study evaluating the combination of ICOS agonists and anti-PD-1. Treatment naïve patients with advanced/metastatic HNSCC expressing PD-L1 (CPS ≥ 1) are randomized to receive pembrolizumab with GSK3359609 or placebo in the INDUCE-3 trial (NCT04128696). A second study will encompass HNSCC with or without PD-L1 expression. In this study, patients will be randomized to platinum-fluorouracil-pembrolizumab plus GSK3359609 or placebo (NCT04428333).
In addition to the aforementioned phase 3 studies, multiple phase 1/2 clinical trials with PD-1/PD-L1 inhibitors combined with a second drug targeting a variety of pathways are ongoing or have been completed (Table 1) (170–187). Studies for which data have been reported are summarized in Table 1. These clinical trials include patients that are immunotherapy-naïve (thus potentially addressing de novo resistance) and/or patients who have developed acquired resistance to anti-PD-1. While it is premature to elect a dominant combination strategy that will move forward to become a new standard of care, preliminary results for many of these studies are encouraging. Nonetheless, it is expected that resistance mechanisms will not be uniform in all patients, and biomarker-informed approaches will likely be needed to maximize the chances of achieving long-term successful outcomes, thus leading, in the future, to the development of precision immunotherapy for recurrent/metastatic HNSCCs, and ultimately earlier stage disease as well.
All authors contributed to the article and approved the submitted version.
LS is a paid advisor for Merck and reports receiving speaker’s bureau honoraria from Bristol-Myers-Squib and Merck. WW is a paid advisor for Pfizer, AstraZeneca, Merck, Bristol-Myers Squibb, and Roche/Genentech and reports receiving speaker’s bureau honoraria from AstraZeneca, Boehringer-Ingelheim, Roche/Genentech, Bristol-Myers Squibb, and Merck.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
2. Waldman AD, Fritz JM. Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol (2020) 20:651–68. doi: 10.1038/s41577-020-0306-5
3. Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer (2012) 12(4):298–306. doi: 10.1038/nrc3245
4. Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol (2008) 8(6):467–77. doi: 10.1038/nri2326
5. Wing JB, Ise W, Kurosaki T, Sakaguchi S. Regulatory T cells control antigen-specific expansion of Tfh cell number and humoral immune responses via the coreceptor CTLA-4. Immunity (2014) 41(6):1013–25. doi: 10.1016/j.immuni.2014.12.006
6. Sullivan TJ, Letterio JJ, van Elsas A, Mamura M, van Amelsfort J, Sharpe S, et al. Lack of a role for transforming growth factor-beta in cytotoxic T lymphocyte antigen-4-mediated inhibition of T cell activation. Proc Natl Acad Sci U S A (2001) 98(5):2587–92. doi: 10.1073/pnas.051632398
7. Kuiper HM, Brouwer M, Linsley PS, van Lier RA. Activated T cells can induce high levels of CTLA-4 expression on B cells. J Immunol (1995) 155(4):1776–83.
8. Jie HB, Schuler PJ, Lee SC, Srivastava RM, Argiris A, Ferrone S, et al. CTLA-4(+) Regulatory T Cells Increased in Cetuximab-Treated Head and Neck Cancer Patients Suppress NK Cell Cytotoxicity and Correlate with Poor Prognosis. Cancer Res (2015) 75(11):2200–10. doi: 10.1158/0008-5472
9. Lenouvel D, Gonzalez-Moles MA, Ruiz-Avila I, Gonzalez-Ruiz L, Gonzalez-Ruiz I, Ramos-Garcia P. Prognostic and clinicopathological significance of PD-L1 overexpression in oral squamous cell carcinoma: A systematic review and comprehensive meta-analysis. Oral Oncol (2020) 106:104722. doi: 10.1016/j.oraloncology.2020.104722
10. Maruse Y, Kawano S, Jinno T, Matsubara R, Goto Y, Kaneko N, et al. Significant association of increased PD-L1 and PD-1 expression with nodal metastasis and a poor prognosis in oral squamous cell carcinoma. Int J Oral Maxillofac Surg (2018) 47(7):836–45. doi: 10.1016/j.ijom.2018.01.004
11. Wang W, Lau R, Yu D, Zhu W, Korman A, Weber J. PD1 blockade reverses the suppression of melanoma antigen-specific CTL by CD4+ CD25(Hi) regulatory T cells. Int Immunol (2009) 21(9):1065–77. doi: 10.1093/intimm/dxp072
12. Wong RM, Scotland RR, Lau RL, Wang C, Korman AJ, Kast WM, et al. Programmed death-1 blockade enhances expansion and functional capacity of human melanoma antigen-specific CTLs. Int Immunol (2007) 19(10):1223–34. doi: 10.1093/intimm/dxm091
13. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med (2000) 192(7):1027–34. doi: 10.1084/jem.192.7.1027
14. De Meulenaere A, Vermassen T, Aspeslagh S, Huvenne W, Van Dorpe J, Ferdinande L, et al. Turning the tide: Clinical utility of PD-L1 expression in squamous cell carcinoma of the head and neck. Oral Oncol (2017) 70:34–42. doi: 10.1016/j.oraloncology.2017.05.002
15. de Vicente JC, Rodriguez-Santamarta T, Rodrigo JP, Blanco-Lorenzo V, Allonca E, Garcia-Pedrero JM. PD-L1 Expression in Tumor Cells Is an Independent Unfavorable Prognostic Factor in Oral Squamous Cell Carcinoma. Cancer Epidemiol Biomarkers Prev (2018) 28(3):546–54. doi: 10.1158/1055-9965.EPI-18-0779
16. Zandberg DP, Strome SE. The role of the PD-L1:PD-1 pathway in squamous cell carcinoma of the head and neck. Oral Oncol (2014) 50(7):627–32. doi: 10.1016/j.oraloncology.2014.04.003
17. Lyford-Pike S, Peng S, Young GD, Taube JM, Westra WH, Akpeng B, et al. Evidence for a role of the PD-1:PD-L1 pathway in immune resistance of HPV-associated head and neck squamous cell carcinoma. Cancer Res (2013) 73(6):1733–41. doi: 10.1158/0008-5472.CAN-12-2384
18. Kim MH, Kim JH, Lee JM, Choi JW, Jung D, Cho H, et al. Molecular subtypes of oropharyngeal cancer show distinct immune microenvironment related with immune checkpoint blockade response. Br J Cancer (2020) 122(11):1649–60. doi: 10.1038/s41416-020-0796-8
19. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39(1):1–10. doi: 10.1016/j.immuni.2013.07.012
20. Huber V, Camisaschi C, Berzi A, Ferro S, Lugini L, Triulzi T, et al. Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin Cancer Biol (2017) 43:74–89. doi: 10.1016/j.semcancer.2017.03.001
21. Lugano R, Ramachandran M, Dimberg A. Tumor angiogenesis: causes, consequences, challenges and opportunities. Cell Mol Life Sci (2019) 77(9):1745–70. doi: 10.1007/s00018-019-03351-7
22. Schaaf MB, Garg AD, Agostinis P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis (2018) 9(2):115. doi: 10.1038/s41419-017-0061-0
23. Mauge L, Terme M, Tartour E, Helley D. Control of the adaptive immune response by tumor vasculature. Front Oncol (2014) 4:61. doi: 10.3389/fonc.2014.00061
24. Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell (2014) 26(5):605–22. doi: 10.1016/j.ccell.2014.10.006
25. Sukumar M, Kishton RJ, Restifo NP. Metabolic reprograming of anti-tumor immunity. Curr Opin Immunol (2017) 46:14–22. doi: 10.1016/j.coi.2017.03.011
26. Lopez-Albaitero A, Nayak JV, Ogino T, Machandia A, Gooding W, DeLeo AB, et al. Role of antigen-processing machinery in the in vitro resistance of squamous cell carcinoma of the head and neck cells to recognition by CTL. J Immunol (2006) 176(6):3402–9. doi: 10.4049/jimmunol.176.6.3402
27. Mizukami Y, Kono K, Maruyama T, Watanabe M, Kawaguchi Y, Kamimura K, et al. Downregulation of HLA Class I molecules in the tumour is associated with a poor prognosis in patients with oesophageal squamous cell carcinoma. Br J Cancer (2008) 99(9):1462–7. doi: 10.1038/sj.bjc.6604715
28. Lawrence M, Sougnez C, Lichtenstein L, Cibulskis K, Lander E, Chin L, et al. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature (2015) 517(7536):576–82. doi: 10.1038/nature14129
29. Ferris RL. Immunology and Immunotherapy of Head and Neck Cancer. J Clin Oncol (2015) 33(29):3293–304. doi: 10.1200/JCO.2015.61.1509
30. Ferris RL, Whiteside TL, Ferrone S. Immune escape associated with functional defects in antigen-processing machinery in head and neck cancer. Clin Cancer Res (2006) 12(13):3890–5. doi: 10.1158/1078-0432.CCR-05-2750
31. Ock CY, Hwang JE, Keam B, Kim SB, Shim JJ, Jang HJ, et al. Genomic landscape associated with potential response to anti-CTLA-4 treatment in cancers. Nat Commun (2017) 8(1):1050. doi: 10.1038/s41467-017-01018-0
32. Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The Immune Landscape of Cancer. Immunity (2018) 48(4):812–30.e14. doi: 10.1016/j.immuni.2018.03.023
33. Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science (2017) 355(6322). doi: 10.1126/science.aaf8399
34. Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The Immune Landscape of Cancer. Immunity (2019) 51(2):411–2. doi: 10.1016/j.immuni.2019.08.004
35. Li B, Cui Y, Nambiar DK, Sunwoo JB, Li R. The Immune Subtypes and Landscape of Squamous Cell Carcinoma. Clin Cancer Res (2019) 25(12):3528–37. doi: 10.1158/1078-0432.CCR-18-4085
36. Chen YP, Wang YQ, Lv JW, Li YQ, Chua MLK, Le QT, et al. Identification and validation of novel microenvironment-based immune molecular subgroups of head and neck squamous cell carcinoma: implications for immunotherapy. Ann Oncol (2019) 30(1):68–75. doi: 10.1093/annonc/mdy470
37. Mandal R, Senbabaoglu Y, Desrichard A, Havel JJ, Dalin MG, Riaz N, et al. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight (2016) 1(17):e89829. doi: 10.1172/jci.insight.89829
38. Cillo AR, Kurten CHL, Tabib T, Qi Z, Onkar S, Wang T, et al. Immune Landscape of Viral- and Carcinogen-Driven Head and Neck Cancer. Immunity (2020) 52(1):183–99 e9. doi: 10.1016/j.immuni.2019.11.014
39. Troiano G, Rubini C, Togni L, Caponio VCA, Zhurakivska K, Santarelli A, et al. The immune phenotype of tongue squamous cell carcinoma predicts early relapse and poor prognosis. Cancer Med (2020) 9(22):8333–44. doi: 10.1002/cam4.3440
40. Wood O, Clarke J, Woo J, Mirza AH, Woelk CH, Thomas GJ, et al. Head and Neck Squamous Cell Carcinomas Are Characterized by a Stable Immune Signature Within the Primary Tumor Over Time and Space. Clin Cancer Res (2017) 23(24):7641–9. doi: 10.1158/1078-0432.CCR-17-0373
41. Letessier EM, Heo DS, Okarma T, Johnson JT, Herberman RB, Whiteside TL. Enrichment in tumor-reactive CD8+ T-lymphocytes by positive selection from the blood and lymph nodes of patients with head and neck cancer. Cancer Res (1991) 51(15):3891–9.
42. Varilla V, Atienza J, Dasanu CA. Immune alterations and immunotherapy prospects in head and neck cancer. Expert Opin Biol Ther (2013) 13(9):1241–56. doi: 10.1517/14712598.2013.810716
43. Freiser ME, Serafini P, Weed DT. The immune system and head and neck squamous cell carcinoma: from carcinogenesis to new therapeutic opportunities. Immunol Res (2013) 57(1-3):52–69. doi: 10.1007/s12026-013-8462-3
44. Ferris RL, Blumenschein G Jr, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med (2016) 375(19):1856–67. doi: 10.1056/NEJMoa1602252
45. Cohen EEW, Soulieres D, Le Tourneau C, Dinis J, Licitra L, Ahn MJ, et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): a randomised, open-label, phase 3 study. Lancet (2019) 393(10167):156–67. doi: 10.1016/S0140-6736(18)31999-8
46. Burtness B, Harrington KJ, Greil R, Soulieres D, Tahara M, de Castro G Jr, et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet (2019) 394(10212):1915–28. doi: 10.1016/S0140-6736(19)32591-7
47. HIGHLIGHTS OF PRESCRIBING INFORMATION. KEYTRUDA® (pembrolizumab) injection, for intravenous use. Merck & Co, Inc (2020).
48. Keytruda - Assessment Report. European Medicines Agency. (2019) 17 October 2019. Contract No.: EMA/CHMP/591139/2019.
49. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature (2014) 515(7528):563–7. doi: 10.1038/nature14011
50. Strome SE, Dong H, Tamura H, Voss SG, Flies DB, Tamada K, et al. B7-H1 blockade augments adoptive T-cell immunotherapy for squamous cell carcinoma. Cancer Res (2003) 63(19):6501–5.
51. Tsushima F, Tanaka K, Otsuki N, Youngnak P, Iwai H, Omura K, et al. Predominant expression of B7-H1 and its immunoregulatory roles in oral squamous cell carcinoma. Oral Oncol (2006) 42(3):268–74. doi: 10.1016/j.oraloncology.2005.07.013
52. Ratcliffe MJ, Sharpe A, Rebelatto M, Scott M, Barker C, Scorer P, et al. A comparative study of PD-L1 diagnostic assays in squamous cell carcinoma of the head and neck (SCCHN). Ann Oncol (2016) 27(Supplement 6):vi328–50. doi: 10.1093/annonc/mdw376.07
53. Scott M, Wildsmith S, Ratcliffe M, Al-Masri H, Scorer PW, Barker C, et al. Comparison of Patient Populations Identified by Different PD-L1 Assays in Head and Neck Squamous Cell Carcinoma (HNSCC). Ann Oncol (2018) 29(Supplement 8):viii372–99. doi: 10.1093/annonc/mdy287
54. Kintsler S, Cassataro MA, Drosch M, Holenya P, Knuechel R, Braunschweig T. Expression of programmed death ligand (PD-L1) in different tumors. Comparison of several current available antibody clones and antibody profiling. Ann Diagn Pathol (2019) 41:24–37. doi: 10.1016/j.anndiagpath.2019.05.005
55. Ferris RL, Blumenschein G Jr, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab vs investigator’s choice in recurrent or metastatic squamous cell carcinoma of the head and neck: 2-year long-term survival update of CheckMate 141 with analyses by tumor PD-L1 expression. Oral Oncol (2018) 81:45–51. doi: 10.1016/j.oraloncology.2018.04.008
56. Mehra R, Seiwert TY, Gupta S, Weiss J, Gluck I, Eder JP, et al. Efficacy and safety of pembrolizumab in recurrent/metastatic head and neck squamous cell carcinoma: pooled analyses after long-term follow-up in KEYNOTE-012. Br J Cancer (2018) 119(2):153–9. doi: 10.1038/s41416-018-0131-9
57. Bauml J, Seiwert TY, Pfister DG, Worden F, Liu SV, Gilbert J, et al. Pembrolizumab for Platinum- and Cetuximab-Refractory Head and Neck Cancer: Results From a Single-Arm, Phase II Study. J Clin Oncol (2017) 35(14):1542–9. doi: 10.1200/JCO.2016.70.1524
58. Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med (2015) 21(1):81–5. doi: 10.1038/nm.3773
59. Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ, et al. Corrigendum: High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4(+) T cells in human melanoma. Nat Med (2016) 22(10):1192. doi: 10.1038/nm1016-1192d
60. Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med (2013) 19(6):747–52. doi: 10.1038/nm.3161
61. Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res (2014) 24(5):743–50. doi: 10.1101/gr.165985.113
62. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science (2015) 348(6230):69–74. doi: 10.1126/science.aaa4971
63. Mogg R, Ayers M, Albright A, Murphy E, Yearley J, Sher X, et al. Erratum for the Research Article “Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy” by R. Cristescu. Science (2019) 363(6430). doi: 10.1126/science.aax1384
64. Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science (2018) 362(6411). doi: 10.1126/science.aar3593
65. Seiwert TY, Burtness B, Mehra R, Weiss J, Berger R, Eder JP, et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol (2016) 17(7):956–65. doi: 10.1016/S1470-2045(16)30066-3
66. Hanna GJ, Lizotte P, Cavanaugh M, Kuo FC, Shivdasani P, Frieden A, et al. Frameshift events predict anti-PD-1/L1 response in head and neck cancer. JCI Insight (2018) 3(4). doi: 10.1172/jci.insight.98811
67. Li W, Wildsmith S, Ye J, Si H, Morsli N, He P, et al. Plasma-based tumor mutational burden (bTMB) as predictor for survival in phase III EAGLE study: Durvalumab (D) ± tremelimumab (T) versus chemotherapy (CT) in recurrent/metastatic head and neck squamous cell carcinoma (R/M HNSCC) after platinum failure. J Clin Oncol (2020) 38(Supplement 15):6511. doi: 10.1200/jco.2020.38.15_suppl.6511
68. Corrales L, Gajewski TF. Molecular Pathways: Targeting the Stimulator of Interferon Genes (STING) in the Immunotherapy of Cancer. Clin Cancer Res (2015) 21(21):4774–9. doi: 10.1158/1078-0432.CCR-15-1362
69. Gajewski TF, Fuertes MB, Woo SR. Innate immune sensing of cancer: clues from an identified role for type I IFNs. Cancer Immunol Immunother (2012) 61(8):1343–7. doi: 10.1007/s00262-012-1305-6
70. Woo SR, Corrales L, Gajewski TF. Innate immune recognition of cancer. Annu Rev Immunol (2015) 33:445–74. doi: 10.1146/annurev-immunol-032414-112043
71. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest (2017) 127(8):2930–40. doi: 10.1172/JCI91190
72. Roy S, Trinchieri G. Microbiota: a key orchestrator of cancer therapy. Nat Rev Cancer (2017) 17(5):271–85. doi: 10.1038/nrc.2017.13
73. Zitvogel L, Ma Y, Raoult D, Kroemer G, Gajewski TF. The microbiome in cancer immunotherapy: Diagnostic tools and therapeutic strategies. Science (2018) 359(6382):1366–70. doi: 10.1126/science.aar6918
74. Gopalakrishnan V, Helmink BA, Spencer CN, Reuben A, Wargo JA. The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell (2018) 33(4):570–80. doi: 10.1016/j.ccell.2018.03.015
75. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science (2018) 359(6371):91–7. doi: 10.1126/science.aan3706
76. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science (2018) 359(6371):97–103. doi: 10.1126/science.aan4236
77. Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science (2018) 359(6371):104–8. doi: 10.1126/science.aao3290
78. Vellanki P, Marur S, Bandaru P, Mishra-Kalyani P, By K, Girvin A, et al. Evaluation of the correlation between antibiotic use and survival in patients with recurrent or metastatic head and neck squamous cell carcinoma (R/M HNSCC) treated with immune checkpoint inhibitors (ICIs). J Clin Oncol (2020) 38(15 Supplement):6509. doi: 10.1200/JCO.2020.38.15_suppl.6509
79. Shin JM, Luo T, Kamarajan P, Fenno JC, Rickard AH, Kapila YL. Microbial Communities Associated with Primary and Metastatic Head and Neck Squamous Cell Carcinoma - A High Fusobacterial and Low Streptococcal Signature. Sci Rep (2017) 7(1):9934. doi: 10.1038/s41598-017-09786-x
80. Chen J, Domingue JC, Sears CL. Microbiota dysbiosis in select human cancers: Evidence of association and causality. Semin Immunol (2017) 32:25–34. doi: 10.1016/j.smim.2017.08.001
81. Fan X, Peters BA, Jacobs EJ, Gapstur SM, Purdue MP, Freedman ND, et al. Drinking alcohol is associated with variation in the human oral microbiome in a large study of American adults. Microbiome (2018) 6(1):59. doi: 10.1186/s40168-018-0448-x
82. Yang CY, Yeh YM, Yu HY, Chin CY, Hsu CW, Liu H, et al. Oral Microbiota Community Dynamics Associated With Oral Squamous Cell Carcinoma Staging. Front Microbiol (2018) 9:862. doi: 10.3389/fmicb.2018.00862
83. Ferris R, Blumenschein G, Harrington K, Fayette J, Guigay J, Dimitrios Colevas A, et al. Evaluation of oral microbiome profiling as a response biomarker in squamous cell carcinoma of the head and neck: Analyses from CheckMate 141. Cancer Res (2017) 77(13 Supplement):CT022. doi: 10.1158/1538-7445.AM2017-CT022
84. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell (2017) 168(4):707–23. doi: 10.1016/j.cell.2017.01.017
85. Ma C, Cheung AF, Chodon T, Koya RC, Wu Z, Ng C, et al. Multifunctional T-cell analyses to study response and progression in adoptive cell transfer immunotherapy. Cancer Discovery (2013) 3(4):418–29. doi: 10.1158/2159-8290.CD-12-0383
86. Ma C, Fan R, Ahmad H, Shi Q, Comin-Anduix B, Chodon T, et al. A clinical microchip for evaluation of single immune cells reveals high functional heterogeneity in phenotypically similar T cells. Nat Med (2011) 17(6):738–43. doi: 10.1038/nm.2375
87. D’Urso CM, Wang ZG, Cao Y, Tatake R, Zeff RA, Ferrone S. Lack of HLA class I antigen expression by cultured melanoma cells FO-1 due to a defect in B2m gene expression. J Clin Invest (1991) 87(1):284–92. doi: 10.1172/JCI114984
88. Restifo NP, Marincola FM, Kawakami Y, Taubenberger J, Yannelli JR, Rosenberg SA. Loss of functional beta 2-microglobulin in metastatic melanomas from five patients receiving immunotherapy. J Natl Cancer Inst (1996) 88(2):100–8. doi: 10.1093/jnci/88.2.100
89. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med (2016) 375(9):819–29. doi: 10.1056/NEJMoa1604958
90. Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discovery (2017) 7(12):1420–35. doi: 10.1158/2159-8290.CD-17-0593
91. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science (2017) 357(6349):409–13. doi: 10.1126/science.aan6733
92. Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun (2017) 8(1):1136. doi: 10.1038/s41467-017-01062-w
93. Saloura V, Fatima A, Zewde M, Kiyotani K, Brisson R, Park JH, et al. Characterization of the T-Cell Receptor Repertoire and Immune Microenvironment in Patients with Locoregionally Advanced Squamous Cell Carcinoma of the Head and Neck. Clin Cancer Res (2017) 23(16):4897–907. doi: 10.1158/1078-0432.CCR-17-0103
94. Bach EA, Aguet M, Schreiber RD. The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu Rev Immunol (1997) 15:563–91. doi: 10.1146/annurev.immunol.15.1.563
95. Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell (2016) 167(2):397–404.e9. doi: 10.1016/j.cell.2016.08.069
96. Rosenthal R, Cadieux EL, Salgado R, Bakir MA, Moore DA, Hiley CT, et al. Neoantigen-directed immune escape in lung cancer evolution. Nature (2019) 567(7749):479–85. doi: 10.1038/s41586-019-1032-7
97. Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discovery (2017) 7(3):264–76. doi: 10.1158/1538-7445.AM2017-NG01
98. Ruella M, Barrett DM, Kenderian SS, Shestova O, Hofmann TJ, Perazzelli J, et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Invest (2016) 126(10):3814–26. doi: 10.1172/JCI87366
99. Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov (2015) 5(12):1282–95. doi: 10.1158/2159-8290.CD-15-1020
100. Verdegaal EM, de Miranda NF, Visser M, Harryvan T, van Buuren MM, Andersen RS, et al. Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature (2016) 536(7614):91–5. doi: 10.1038/nature18945
101. Conciatori F, Bazzichetto C, Falcone I, Ciuffreda L, Ferretti G, Vari S, et al. PTEN Function at the Interface between Cancer and Tumor Microenvironment: Implications for Response to Immunotherapy. Int J Mol Sci (2020) 21(15). doi: 10.3390/ijms21155337
102. Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med (2019) 25(3):462–9. doi: 10.1038/s41591-019-0349-y
103. Chen H, Chong W, Teng C, Yao Y, Wang X, Li X. The immune response-related mutational signatures and driver genes in non-small-cell lung cancer. Cancer Sci (2019) 110(8):2348–56. doi: 10.1111/cas.14113
104. Roh W, Chen PL, Reuben A, Spencer CN, Prieto PA, Miller JP, et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci Transl Med (2017) 9(379). doi: 10.1126/scitranslmed.aah3560
105. George S, Miao D, Demetri GD, Adeegbe D, Rodig SJ, Shukla S, et al. Loss of PTEN Is Associated with Resistance to Anti-PD-1 Checkpoint Blockade Therapy in Metastatic Uterine Leiomyosarcoma. Immunity (2017) 46(2):197–204. doi: 10.1016/j.immuni.2017.02.001
106. Trujillo JA, Luke JJ, Zha Y, Segal JP, Ritterhouse LL, Spranger S, et al. Secondary resistance to immunotherapy associated with beta-catenin pathway activation or PTEN loss in metastatic melanoma. J Immunother Cancer (2019) 7(1):295. doi: 10.1186/s40425-019-0780-0
107. Kakavand H, Jackett LA, Menzies AM, Gide TN, Carlino MS, Saw RPM, et al. Negative immune checkpoint regulation by VISTA: a mechanism of acquired resistance to anti-PD-1 therapy in metastatic melanoma patients. Mod Pathol (2017) 30(12):1666–76. doi: 10.1038/modpathol.2017.89
108. Ibrahim MY, Nunez MI, Harun N, Lee JJ, El-Naggar AK, Ferrarotto R, et al. PI3-kinase pathway biomarkers in oral cancer and tumor immune cells. Head Neck (2019) 41(3):615–22. doi: 10.1002/hed.25350
109. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature (2015) 523(7559):231–5. doi: 10.1038/nature14404
110. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun (2016) 7:10501. doi: 10.1038/ncomms10501
111. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature (2002) 415(6871):536–41. doi: 10.1038/415536a
112. Anderson AC, Anderson DE, Bregoli L, Hastings WD, Kassam N, Lei C, et al. Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science (2007) 318(5853):1141–3. doi: 10.1126/science.1148536
113. Golden-Mason L, Rosen HR. Galectin-9: Diverse roles in hepatic immune homeostasis and inflammation. Hepatology (2019) 66(1):271–9. doi: 10.1002/hep.29106
114. Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood (2011) 117(17):4501–10. doi: 10.1182/blood-2010-10-310425
115. Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer Res (2011) 71(10):3540–51. doi: 10.1158/0008-5472.CAN-11-0096
116. Liu JF, Ma SR, Mao L, Bu LL, Yu GT, Li YC, et al. T-cell immunoglobulin mucin 3 blockade drives an antitumor immune response in head and neck cancer. Mol Oncol (2017) 11(2):235–47. doi: 10.1002/1878-0261.12029
117. Finn OJ. Immuno-oncology: understanding the function and dysfunction of the immune system in cancer. Ann Oncol (2012) 23 Suppl 8:viii6–9. doi: 10.1093/annonc/mds256
118. Huard B, Tournier M, Hercend T, Triebel F, Faure F. Lymphocyte-activation gene 3/major histocompatibility complex class II interaction modulates the antigenic response of CD4+ T lymphocytes. Eur J Immunol (1994) 24(12):3216–21. doi: 10.1002/eji.1830241246
119. Workman CJ, Vignali DA. The CD4-related molecule, LAG-3 (CD223), regulates the expansion of activated T cells. Eur J Immunol (2003) 33(4):970–9. doi: 10.1002/eji.200323382
120. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in regulatory T cells. Immunity (2004) 21(4):503–13. doi: 10.1016/j.immuni.2004.08.010
121. Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res (2012) 72(4):917–27. doi: 10.1158/0008-5472.CAN-11-1620
122. Xu F, Liu J, Liu D, Liu B, Wang M, Hu Z, et al. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res (2014) 74(13):3418–28. doi: 10.1158/0008-5472.CAN-13-2690
123. Demchev V, Malana G, Vangala D, Stoll J, Desai A, Kang HW, et al. Targeted deletion of fibrinogen like protein 1 reveals a novel role in energy substrate utilization. PloS One (2013) 8(3):e58084. doi: 10.1371/journal.pone.0058084
124. Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, et al. Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell (2019) 176(1-2):334–47 e12. doi: 10.1016/j.cell.2018.11.010
125. Deng WW, Mao L, Yu GT, Bu LL, Ma SR, Liu B, et al. LAG-3 confers poor prognosis and its blockade reshapes antitumor response in head and neck squamous cell carcinoma. Oncoimmunology (2016) 5(11):e1239005. doi: 10.1080/2162402X.2016.1239005
126. Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med (2011) 208(3):577–92. doi: 10.1084/jem.20100619
127. Liu J, Yuan Y, Chen W, Putra J, Suriawinata AA, Schenk AD, et al. Immune-checkpoint proteins VISTA and PD-1 nonredundantly regulate murine T-cell responses. Proc Natl Acad Sci U S A (2015) 112(21):6682–7. doi: 10.1073/pnas.1420370112
128. Wang J, Wu G, Manick B, Hernandez V, Renelt M, Erickson C, et al. VSIG-3 as a ligand of VISTA inhibits human T-cell function. Immunology (2019) 156(1):74–85. doi: 10.1111/imm.13001
129. Wu L, Deng WW, Huang CF, Bu LL, Yu GT, Mao L, et al. Expression of VISTA correlated with immunosuppression and synergized with CD8 to predict survival in human oral squamous cell carcinoma. Cancer Immunol Immunother (2017) 66(5):627–36. doi: 10.1007/s00262-017-1968-0
130. Stephens GL, McHugh RS, Whitters MJ, Young DA, Luxenberg D, Carreno BM, et al. Engagement of glucocorticoid-induced TNFR family-related receptor on effector T cells by its ligand mediates resistance to suppression by CD4+CD25+ T cells. J Immunol (2004) 173(8):5008–20. doi: 10.4049/jimmunol.173.8.5008
131. Esparza EM, Arch RH. Glucocorticoid-induced TNF receptor functions as a costimulatory receptor that promotes survival in early phases of T cell activation. J Immunol (2005) 174(12):7869–74. doi: 10.4049/jimmunol.174.12.7869
132. Schaer DA, Budhu S, Liu C, Bryson C, Malandro N, Cohen A, et al. GITR pathway activation abrogates tumor immune suppression through loss of regulatory T cell lineage stability. Cancer Immunol Res (2013) 1(5):320–31. doi: 10.1158/2326-6066.CIR-13-0086
133. Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol (2002) 3(2):135–42. doi: 10.1038/ni759
134. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol (2009) 10(1):48–57. doi: 10.1038/ni.1674
135. Fuchs A, Colonna M. The role of NK cell recognition of nectin and nectin-like proteins in tumor immunosurveillance. Semin Cancer Biol (2006) 16(5):359–66. doi: 10.1016/j.semcancer.2006.07.002
136. Lozano E, Dominguez-Villar M, Kuchroo V, Hafler DA. The TIGIT/CD226 axis regulates human T cell function. J Immunol (2012) 188(8):3869–75. doi: 10.4049/jimmunol.1103627
137. Wu L, Mao L, Liu JF, Chen L, Yu GT, Yang LL, et al. Blockade of TIGIT/CD155 Signaling Reverses T-cell Exhaustion and Enhances Antitumor Capability in Head and Neck Squamous Cell Carcinoma. Cancer Immunol Res (2019) 7(10):1700–13. doi: 10.1158/2326-6066.CIR-18-0725
138. Woods DM, Laino AS, Winters A, Alexandre J, Freeman D, Rao V, et al. Nivolumab and ipilimumab are associated with distinct immune landscape changes and response-associated immunophenotypes. JCI Insight (2020) 5(11):e137066. doi: 10.1172/jci.insight.137066
139. Weber JS, Gibney G, Sullivan RJ, Sosman JA, Slingluff CL Jr, Lawrence DP, et al. Sequential administration of nivolumab and ipilimumab with a planned switch in patients with advanced melanoma (CheckMate 064): an open-label, randomised, phase 2 trial. Lancet Oncol (2016) 17(7):943–55. doi: 10.1016/s1470-2045(16)30126-7
140. Syn N, Roudi R, Wang L, Wang L, Loh M, Huang Y, et al. Immune checkpoint inhibitors plus chemotherapy versus chemotherapy or immunotherapy for first-line treatment of advanced non-small cell lung cancer: a generic protocol. Cochrane Database System Rev (2018) 4:CD013009. doi: 10.1002/14651858.CD013009
141. Burtness B, Rischin D, Greil R, Soulières D, Tahara M, de Castro G, et al. Efficacy of first-line (1L) pembrolizumab by PD-L1 combined positive score <1, 1-19, and ≥20 in recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC): KEYNOTE-048 subgroup analysis, in: (2020). (Accessed 2020 27-28 April).
142. Watermann C, Pasternack H, Idel C, Ribbat-Idel J, Bragelmann J, Kuppler P, et al. Recurrent HNSCC Harbor an Immunosuppressive Tumor Immune Microenvironment Suggesting Successful Tumor Immune Evasion. Clin Cancer Res (2020) 27:632–44. doi: 10.1007/s12026-012-8306-6
143. Sato H, Okonogi N, Nakano T. Rationale of combination of anti-PD-1/PD-L1 antibody therapy and radiotherapy for cancer treatment. Int J Clin Oncol (2020) 25(5):801–9. doi: 10.1007/s10147-020-01666-1
144. Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature (2015) 520(7547):373–7. doi: 10.1038/nature14292
145. Gong J, Le TQ, Massarelli E, Hendifar AE, Tuli R. Radiation therapy and PD-1/PD-L1 blockade: the clinical development of an evolving anticancer combination. J Immunother Cancer (2018) 6(1):46. doi: 10.1186/s40425-018-0361-7
146. Bourhis J, Sire C, Tao Y, Martin L, Alfonsi M, Prevost J, et al. Pembrolizumab versus cetuximab, concomitant with radiotherapy (RT) in locally advanced head and neck squamous cell carcinoma (LA-HNSCC): Results of the GORTEC 2015-01 “PembroRad” randomized trial. Ann Oncol (2020) 314:S1168. doi: 10.1016/j.annonc.2020.08.2268
147. Cohen E, Ferris R, Psyrri A, Haddad R, Tahara M, Bourhis J, et al. Primary results of the phase III JAVELIN head & neck 100 trial: Avelumab plus chemoradiotherapy (CRT) followed by avelumab maintenance vs CRT in patients with locally advanced squamous cell carcinoma of the head and neck (LA SCCHN). Ann Oncol (2020) 31(Supplement 4):S658. doi: 10.1016/j.annonc.2020.08.1025
148. McBride S, Sherman E, Tsai CJ, Baxi S, Aghalar J, Eng J, et al. Randomized Phase II Trial of Nivolumab With Stereotactic Body Radiotherapy Versus Nivolumab Alone in Metastatic Head and Neck Squamous Cell Carcinoma. J Clin Oncol (2020) 39(1):30–1. doi: 10.1200/JCO.20.00290
149. Blank CU, Enk A. Therapeutic use of anti-CTLA-4 antibodies. Int Immunol (2015) 27(1):3–10. doi: 10.1093/intimm/dxu076
150. Camacho LH, Antonia S, Sosman J, Kirkwood JM, Gajewski TF, Redman B, et al. Phase I/II trial of tremelimumab in patients with metastatic melanoma. J Clin Oncol (2009) 27(7):1075–81. doi: 10.1200/JCO.2008.19.2435
151. Hamid O, Schmidt H, Nissan A, Ridolfi L, Aamdal S, Hansson J, et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J Transl Med (2011) 9:204. doi: 10.1186/1479-5876-9-204
152. Lynch TJ, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol (2012) 30(17):2046–54. doi: 10.1200/JCO.2011.38.4032
153. Reck M, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: results from a randomized, double-blind, multicenter phase 2 trial. Ann Oncol (2013) 24(1):75–83. doi: 10.1093/annonc/mds213
154. Siu LL, Even C, Mesia R, Remenar E, Daste A, Delord JP, et al. Safety and Efficacy of Durvalumab With or Without Tremelimumab in Patients With PD-L1-Low/Negative Recurrent or Metastatic HNSCC: The Phase 2 CONDOR Randomized Clinical Trial. JAMA Oncol (2019) 5(2):195–203. doi: 10.1001/jamaoncol.2018.4628
155. Zandberg DP, Algazi AP, Jimeno A, Good JS, Fayette J, Bouganim N, et al. Durvalumab for recurrent or metastatic head and neck squamous cell carcinoma: Results from a single-arm, phase II study in patients with >/=25% tumour cell PD-L1 expression who have progressed on platinum-based chemotherapy. Eur J Cancer (2019) 107:142–52. doi: 10.1016/j.ejca.2018.11.015
156. Ferris RL, Haddad R, Even C, Tahara M, Dvorkin M, Ciuleanu TE, et al. Durvalumab with or without tremelimumab in patients with recurrent or metastatic head and neck squamous cell carcinoma: EAGLE, a randomized, open-label phase III study. Ann Oncol (2020) 31(7):942–50. doi: 10.1016/j.annonc.2020.04.001
157. Munn DH, Mellor AL. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol (2016) 37(3):193–207. doi: 10.1016/j.it.2016.01.002
158. Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest (2007) 117(5):1147–54. doi: 10.1172/JCI31178
159. Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, et al. Tumor-Expressed IDO Recruits and Activates MDSCs in a Treg-Dependent Manner. Cell Rep (2015) 13(2):412–24. doi: 10.1016/j.celrep.2015.08.077
160. Botticelli A, Cerbelli B, Lionetto L, Zizzari I, Salati M, Pisano A, et al. Can IDO activity predict primary resistance to anti-PD-1 treatment in NSCLC? J Transl Med (2018) 16(1):219. doi: 10.1186/s12967-018-1595-3
161. Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med (2013) 210(7):1389–402. doi: 10.1084/jem.20130066
162. Mitchell TC, Hamid O, Smith DC, Bauer TM, Wasser JS, Olszanski AJ, et al. Epacadostat Plus Pembrolizumab in Patients With Advanced Solid Tumors: Phase I Results From a Multicenter, Open-Label Phase I/II Trial (ECHO-202/KEYNOTE-037). J Clin Oncol (2018) 36(32):3223–30. doi: 10.1200/JCO.2018.78.9602
163. Hamid O, Bauer T, Spira A, Olszanski AJ, Patel S, Wasser JS, et al. Epacadostat plus pembrolizumab in patients with SCCHN: Preliminary phase I/II results from ECHO-202/KEYNOTE-037. J Clin Oncol (2017) 35(15 Supplement):6010. doi: 10.1200/JCO.2017.35.15_suppl.6010
164. Perez R, Riese M, Lewis K, Saleh M, Daud A, Berlin J, et al. Epacadostat plus nivolumab in patients with advanced solid tumors: Preliminary phase I/II results of ECHO-204. J Clin Oncol (2017) 35(15 Supplement):3003. doi: 10.1200/JCO.2017.35.15_suppl.3003
165. Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol (2019) 20(8):1083–97. doi: 10.1016/S1470-2045(19)30274-8
166. Castriconi R, Dondero A, Augugliaro R, Cantoni C, Carnemolla B, Sementa AR, et al. Identification of 4Ig-B7-H3 as a neuroblastoma-associated molecule that exerts a protective role from an NK cell-mediated lysis. Proc Natl Acad Sci U S A (2004) 101(34):12640–5. doi: 10.1073/pnas.0405025101
167. Chapoval AI, Ni J, Lau JS, Wilcox RA, Flies DB, Liu D, et al. B7-H3: a costimulatory molecule for T cell activation and IFN-gamma production. Nat Immunol (2001) 2(3):269–74. doi: 10.1038/85339
168. Suh WK, Gajewska BU, Okada H, Gronski MA, Bertram EM, Dawicki W, et al. The B7 family member B7-H3 preferentially down-regulates T helper type 1-mediated immune responses. Nat Immunol (2003) 4(9):899–906. doi: 10.1038/ni967
169. Chen JT, Chen CH, Ku KL, Hsiao M, Chiang CP, Hsu TL, et al. Glycoprotein B7-H3 overexpression and aberrant glycosylation in oral cancer and immune response. Proc Natl Acad Sci U S A (2015) 112(42):13057–62. doi: 10.1073/pnas.1516991112
170. Aggarwal C, Joshua A, Ferris R, Antonia S, Rahma E, Tolcher A, et al. A phase 1, open-label, dose-escalation study of enoblituzumab in combination with pembrolizumab in patients with select solid tumors. In: 33rd Society for Immunotherapy of Cancer Annual Meeting. Washington, DC: e Society for Immunotherapy of Cancer (SITC) (2018).
171. Elbers JBW, Al-Mamgani A, Tesseslaar MET, van den Brekel MWM, Lange CAH, van der Wal JE, et al. Immuno-radiotherapy with cetuximab and avelumab for advanced stage head and neck squamous cell carcinoma: Results from a phase-I trial. Radiother Oncol (2019) 142:79–84. doi: 10.1016/j.radonc.2019.08.007
172. Gulati S, Vachon R, Desai S, Steele A, Palackdharry S, Takiar V, et al. Preliminary effects on the innate immune system with the combination of cetuximab and durvalumab in metastatic/ relapsed head and neck squamous cell carcinoma in a phase II trial. J Clin Oncol (2019) 37(15 Supplement):e14273. doi: 10.1200/JCO.2019.37.15_suppl.e14273
173. Sacco A, Chen R, Ghosh D, Worden F, Wong D, Adkins D, et al. An open-label, non-randomized, multi-arm, phase II trial evaluating pembrolizumab combined with cetuximab in patients (pts) with recurrent/metastatic (R/M) head and neck squamous cell carcinoma (HNSCC): updated results of cohort 1 analysis. Int J Rad Oncol Biol Phys (2020) 106(5):P1121–2. doi: 10.1016/j.ijrobp.2019.11.376
174. Chung C, Raul Bonomi M, Steuer C, Schell M, Li J, Johnson M, et al. Concurrent cetuximab (CTX) and nivolumab (NIVO) in patients with recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC): Results of phase II study. J Clin Oncol (2020) 38(15 Supplement):6515. doi: 10.1200/JCO.2020.38.15_suppl.6515
175. Aggarwal C, Cohen RB, Morrow MP, Kraynyak KA, Sylvester AJ, Knoblock DM, et al. Immunotherapy Targeting HPV16/18 Generates Potent Immune Responses in HPV-Associated Head and Neck Cancer. Clin Cancer Res (2019) 25(1):110–24. doi: 10.1158/1078-0432.CCR-18-1763
176. Le Tourneau C, Delord J, Cassier P, Loirat D, Tavernaro A, Bastien B, et al. Phase Ib/II trial of TG4001 (Tipapkinogene sovacivec), a therapeutic HPV-vaccine, and Avelumab in patients with recurrent/metastatic (R/M) HPV-16+ cancers. Ann Oncol (2019) 30(5 Supplement):v475. doi: 10.1093/annonc/mdz253.036
177. Massarelli E, William W, Johnson F, Kies M, Ferrarotto R, Guo M, et al. Combining Immune Checkpoint Blockade and Tumor-Specific Vaccine for Patients With Incurable Human Papillomavirus 16-Related Cancer: A Phase 2 Clinical Trial. JAMA Oncol (2019) 5(1):67–73. doi: 10.1001/jamaoncol.2018.4051
178. Felip E, Doger B, Majem M, Carcereny E, Krebs M, Peguero J, et al. Initial results from a phase II study (TACTI-002) in metastatic non-small cell lung or head and neck carcinoma patients receiving eftilagimod alpha (soluble LAG-3 protein) and pembrolizumab. J Clin Oncol (2020) 38(15 Supplement):3100. doi: 10.1200/JCO.2020.38.15_suppl.3100
179. Leidner R, Kang H, Haddad R, Segal N, Wirth L, Ferris R, et al. (2016). Preliminary efficacy from a phase 1/2 study of the natural killer cell–targeted antibody, lirilumab in combination with nivolumab in squamous cell carcinoma of the head and neck, in: 31st Society for Immunotherapy of Cancer Annual Meeting, National Harbor, MD: e Society for Immunotherapy of Cancer (SITC). 2016 9-13 November.
180. Sanborn RE, Pishvaian MJ, Kluger HM, Callahan MK, Weise AM, Lutzky J, et al. Clinical results with combination of anti-CD27 agonist antibody, varlilumab, with anti-PD1 antibody nivolumab in advanced cancer patients. J Clin Oncol (2017) 35(15 Supplement):3007. doi: 10.1200/JCO.2017.35.15_suppl.3007
181. Cohen EEW, Nabell L, Wong DJL, Day TA, Daniels GA, Milhem MM, et al. Phase 1b/2, open label, multicenter study of intratumoral SD-101 in combination with pembrolizumab in anti-PD-1 treatment naïve patients with recurrent or metastatic head and neck squamous cell carcinoma (HNSCC). J Clin Oncol (2019) 37(15 Supplement):6039. doi: 10.1158/1538-7445.AM2018-CT098
182. Cohen EEW, Harrington KJ, Hong DS, Mesia R, Brana I, Segura PP, et al. A phase Ib/II study (SCORES) of durvalumab (D) plus danvatirsen (DAN; AZD9150) or AZD5069 (CX2i) in advanced solid malignancies and recurrent/metastatic head and neck squamous cell carcinoma (RM-HNSCC): Updated results. Ann Oncol (2018) 29(8 Supplement):viii372. doi: 10.1093/annonc/mdy287
183. Curti B. Galectin Therapeutics, Inc. Announces Positive Preliminary Results from Phase 1b Clinical Trial of GR-MD-02 and KEYTRUDA in Advanced Melanoma and Expansion of the Trial NORCROSS. Ga: Galectin Therapeutics Inc (2018). Available at: https://www.sec.gov/Archives/edgar/data/1133416/000119312518278101/d626312dex991.htm.
184. Naing A, Bauer T, Papadopoulos KP, Rahma O, Tsai F, Garralda E, et al. PHASE 1 STUDY OF THE ARGINASE INHIBITOR INCB001158 (1158) ALONE AND IN COMBINATION WITH PEMBROLIZUMAB (PEM) IN PATIENTS (PTS) WITH ADVANCED/METASTATIC (ADV/MET) SOLID TUMORS. Ann Oncol (2019) 30(5 Supplement):V159. doi: 10.1093/annonc/mdz244.002
185. Richa T, Johnson JM, Cognetti DM, Argiris A, Luginbuhl A, Zinner R, et al. Window of opportunity for durvalumab (MEDI4736) plus metformin trial in squamous cell carcinoma of the head and neck (SCCHN): Interim safety analysis. Ann Oncol (2019) 30(5 Supplement):v458. doi: 10.1093/annonc/mdz252.021
186. Luginbuhl AJ, Johnson JM, Harshyne L, Tuluc M, Mardekian S, Leiby BE, et al. A window of opportunity trial of preoperative nivolumab with or without tadalafil in squamous cell carcinoma of the head and neck (SCCHN): Safety, clinical, and correlative outcomes. Ann Oncol (2019) 30(5 Supplement):v453. doi: 10.1093/annonc/mdz252.008
187. Psyrri A, Papaxoinis G, Gavrielatou N, Gkotzamanidou M, Economopoulou P, Kotsantis I, et al. Molecular correlates of response to preoperative olaparib alone or with cisplatin or with durvalumab in head and neck squamous cell carcinoma (HNSCC): A Hellenic Cooperative Oncology Group study. J Clin Oncol (2020) 38(15 Supplement):6556. doi: 10.1200/JCO.2020.38.15_suppl.6556
188. Taylor M, Rasco D, Brose M, Vogelzang N, Richey S, Cohn A, et al. A phase 1b/2 trial of lenvatinib plus pembrolizumab in patients with squamous cell carcinoma of the head and neck. J Clin Oncol (2018) 36(15 Supplement):6016. doi: 10.1200/JCO.2018.36.15_suppl.6016
189. Hutloff A, Dittrich AM, Beier KC, Eljaschewitsch B, Kraft R, Anagnostopoulos I, et al. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature (1999) 397(6716):263–6. doi: 10.1038/16717
190. Amatore F, Gorvel L, Olive D. Inducible Co-Stimulator (ICOS) as a potential therapeutic target for anti-cancer therapy. Expert Opin Ther Targets (2018) 22(4):343–51. doi: 10.1080/14728222.2018.1444753
191. Marinelli O, Nabissi M, Morelli MB, Torquati L, Amantini C, Santoni G. ICOS-L as a Potential Therapeutic Target for Cancer Immunotherapy. Curr Protein Pept Sci (2018) 19(11):1107–13. doi: 10.2174/1389203719666180608093913
192. Hansen A, Bauer T, Moreno V, Maio M, Groenland S, Martin-Liberal J, et al. First in human study with GSK3359609 [GSK609], inducible T cell costimulator (ICOS) receptor agonist in patients [Pts] with advanced, solid tumors: Preliminary results from INDUCE-1. Ann Oncol (2018) 29(8 Supplement):viii404. doi: 10.1093/annonc/mdy288.011
193. Yap T, Burris H, Kummar S, Falchook G, Pachynski R, LoRusso P, et al. ICONIC: Biologic and clinical activity of first in class ICOS agonist antibody JTX-2011 +/- nivolumab (nivo) in patients (pts) with advanced cancers. J Clin Oncol (2018) 36(15 Supplement):3000. doi: 10.1200/JCO.2018.36.15_suppl.3000
Keywords: head and neck (H&N) cancer, immunotherapy, programmed death-1 (PD-1), programmed death ligand-1 (PD-L1), programmed death-1 (PD-1)/programmed death ligand-1 (PD-L1) axis, resistance
Citation: dos Santos LV, Abrahão CM and William WN Jr (2021) Overcoming Resistance to Immune Checkpoint Inhibitors in Head and Neck Squamous Cell Carcinomas. Front. Oncol. 11:596290. doi: 10.3389/fonc.2021.596290
Received: 19 August 2020; Accepted: 08 January 2021;
Published: 05 March 2021.
Edited by:
Floriana Morgillo, Second University of Naples, ItalyReviewed by:
Thorsten Fuereder, Medical University of Vienna, AustriaCopyright © 2021 dos Santos, Abrahão and William. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: William N. William Jr., d2lsbGlhbXdpbGxpYW1qckBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.