 Tapas Pradhan1
Tapas Pradhan1 Vikas Kumar2†Evangeline Surya H1†R. Krishna1†
Vikas Kumar2†Evangeline Surya H1†R. Krishna1† Samu John1
Samu John1 V. T. Jissa3S. Anjana1
V. T. Jissa3S. Anjana1 K. Chandramohan4
K. Chandramohan4 S. Asha Nair1*
S. Asha Nair1*- 1Cancer Research Program 4, Rajiv Gandhi Centre for Biotechnology, Trivandrum, India
- 2Cardiovascular Diseases & Diabetes Biology, Rajiv Gandhi Centre for Biotechnology, Trivandrum, India
- 3Achutha Menon Centre for Health Science Studies (AMCHSS), Sree Chitra Tirunal Institute for Medical Sciences and Technology, Trivandrum, India
- 4Department of Surgical Oncology, Regional Cancer Centre, Trivandrum, India
The discovery of a potent gene regulating tumorigenesis and drug resistance is of high clinical importance. STIL is an oncogene; however, its molecular associations and role in colorectal oncogenesis are unknown. In this study, we have explored the role of STIL gene in tumorigenesis and studied its molecular targets in colorectal cancer (CRC). STIL silencing reduced proliferation and tumor growth in CRC. Further, STIL was found to regulate stemness markers CD133 and CD44 and drug resistant markers thymidylate synthase, ABCB1, and ABCG2 both in in-vitro and in-vivo CRC models. In addition, high expression of STIL mRNA was found to be associated with reduced disease-free survival in CRC cases. Interestingly, we observed that STIL-mediated regulation of stemness and drug resistant genes is not exclusively governed by Sonic hedgehog (Shh) signaling. Remarkably, we found STIL regulate β-catenin levels through p-AKT, independent of Shh pathway. This partially answers Shh independent regulatory mechanism of cancer stem cell (CSC) markers by STIL. Our study suggests an instrumental role of STIL in molecular manifestation of CRC and progression.
Introduction
SCL/TAL1 interrupting locus (STIL) gene is a crucial factor in centriole biogenesis, and dysfunction of this gene has been associated with abnormal brain development leading to microcephaly (1). Being a 1288 amino acid cytoplasmic protein, STIL functions as a cell cycle-regulatory protein specifically recruited at the mitotic centrosome to promote the duplication of centrioles in dividing cells. STIL has been shown to interact with CDK1, PLK4, and SAS-6,which are crucial in centriole duplication (2) and thus proven to have a crucial role in cell division (3). In lung cancer, upregulation of STIL has been reported to have significant effect on tumor mitotic activity (4). STIL also has been reported to be overexpressed in pancreatic ductal cell carcinoma (5) and altered in leukemia (6). Further, STIL overexpression has been reported to cause chromosomal instability in cancer cells (7). STIL has been studied as an integral part of Sonic hedgehog signaling (Shh) cascade. The C terminus of STIL can interact with conserved components of the Shh signaling such as suppressor-of-fused homolog (SUFU) and GLI1. Interaction of STIL with SUFU inhibits the repressor function of SUFU towards GLI1, resulting in activation of Shh-GLI1 cascades. Knockdown of STIL has been shown to increase nuclear accumulation of SUFU with GLI1 and repression of GLI1 mediated transcriptional activity (5). STIL−/− mouse embryos have been shown to have reduced expression of PTCH1 and GLI1 and have been reported to lack primary cilia, a structure present in almost all cell types mediating Shh signaling (8, 9). During the process of carcinogenesis, increased expression of STIL promotes the transcriptional activity of GLI1, leading to increased transcription of GLI1 targets that promote sustained proliferation, cell death resistance, stemness, angiogenesis, and genomic instability, which are the hallmarks of cancer (10). Therefore, increase in STIL expression likely represents a crucial step toward cancer progression. STIL depletion has been shown to enhance DNA double-strand breaks caused by DNA damaging agents in ovarian cancer (11). Another report has shown STIL to play a Shh-dependent role in drug susceptibility in PC12 cells (12). Further Shh signaling has been studied for its role in maintenance of CSC, metastasis, and disease recurrence in colorectal cancer (CRC) (13). GLI1-mediated Shh signaling has been found to have important role in CRC cell survival upon therapeutic insults (14). However, there have been hardly any studies deciphering the role of STIL in CRC tumorigenesis and drug resistance. Studies have shown regulation of ABCB1 and ABCG2 efflux pumps to be Shh dependent, which suggest that the Shh pathway could be a potent target to overcome drug resistance and surge chemotherapeutic response (15). Nevertheless, tumor fate against therapies is governed by various cues from tumor microenvironment, and thus a complex network of inter- and intracellular signaling decides therapy response. There has been reports suggesting the instrumental role of Wnt-Shh cross talk in basal cell carcinoma (16) and gastric cancer (17). In addition, cross talk between Wnt and Shh has been shown to be contributing towards CRC progression (18); however, core pathway components mediating cross talk still remain unexplored. Further studies to explore the mediators of this cross talk would provide an in-depth mechanistic perspective on tumor development and therapy resistance in CRC. In this study, we have explored the multifaceted role of STIL in CRC and also deciphered its role in mediating cross talk between Shh and Wnt signaling.
Methods
Patients’ Samples
Biopsies were collected from CRC patients undergoing curative surgery between 2014 and 2017 at the Regional Cancer Centre, Trivandrum, after human ethics committee approval and sanction from the Institutional Review Board. All subjects gave written informed consent in accordance with the Declaration of Helsinki. Patients’ details and clinical information were collected from medical records of the same institution.
Cell Culture
HCT116, HT-29, and HEK293T cells (ATCC, USA) were cultured with Dulbecco’s Eagle’s Medium (DMEM) (Invitrogen, USA) supplemented with 10% FBS (Invitrogen, USA) at 37°C and 5% CO2 in a humidified incubator (Thermo scientific, USA).
Chemical Inhibition of Shh Signaling
SANT1 [(4-Benzyl-piperazin-1-yl)-(3,5-dimethyl-1-phenyl-1H-pyrazol-4-ylmethylene)-amine], a chemical inhibitor of SMO receptor, was used at a concentration of 30 nM for 24 h to inhibit Shh signaling in HT29 cells. Post treatment, cells were processed for RNA and protein isolation.
RNA Isolation
Trizol reagent (Invitrogen, USA) was used to isolate total RNA from around 3×106 adherent cells or 50 mg tissues following manufacturer’s protocol. RNA quality and quantification were carried out on Nanodrop 1000 (Thermo Scientific, USA) after assessing its quality using gel electrophoresis.
Real-Time q-PCR
One µg of RNA was used for conversion of cDNA from each sample using PrimerScript cDNA conversion kit (TAKARA, Japan), following manufacturer’s protocol. Quantitative real-time PCR was performed using SYBR-Green-based fluorescence detection kit (TAKARA, Japan) and HT9700 detection system (AB, Life science, USA). Twenty-five ng of cDNA was used as template for each reaction. Analyzed genes and the primers used are shown in Supplementary Table 1. PCR data were analyzed using Data assist software (AB, Life science, USA).
Lentiviral-Mediated Gene Silencing
HEK293T cells (3×105) were seeded in a six-well plate (Nunc, Thermo, USA) with DMEM media (Invitrogen, USA) containing 10% FBS (Invitrogen, USA) and incubated till it reached 60% confluency. DNA constructs were mixed in a fixed ratio in a vial containing 75 µl OptiMEM media (Invitrogen, USA) as shRNA (0.75 µg/well), pREV (0.5 µg/well), pMDL (0.18 µg/well), and pVSVG (0.26 µg/well) (packaging plasmids were gifted from Vinay Tergaonkar, Department of Biochemistry, Yong Loo Lin School of Medicine, NUS). Details of shRNA employed are given in Supplementary Table 2. To the mixture of constructs, 2 mg/ml polyethylenimine (PEI) (Sigma-Aldrich, USA) was added followed by brisk vortex for 10 s and incubated for 30 min at RT. During incubation, HEK293T cells were rinsed with OptiMEM media, and 688 µl of OptiMEM was added to each well. After incubation, 75 µl DNA/PEI complex was added to cells drop-wise followed by gently shaking, and cells were kept in incubator overnight. Fresh 1.5 ml DMEM media with 10% FBS was added to the cells after overnight incubation, and media was collected every 24 h from cells and replenished by fresh media for 3 consecutive days. Collected media was filtered using 0.45 µm PES membrane (Minisart, Sartorius, Germany) to remove extracellular debris. Filtered media containing lentiviral particles were centrifuged at 24,000 rpm at 20°C for 2 h in 90Ti rotor in ultracentrifuge (Beckman Coulter Optima TM L-100K). Viral particle pellet was obtained after discarding supernatant post centrifugation and suspended in 100 µl OptiMEM media overnight at 4°C.The collected lentivirus suspension was used for transfection or stored for later use at −80°C. Then 3 × 105 number of HT29 and HCT116 cells were seeded in six-well plate in DMEM media containing 10% FBS and incubated till it reached 70% confluency. After reaching desired confluency, cells were infected with 100 µl of lentiviral particle suspension along with 10 µg/ml polybrene (Sigma-Aldrich, USA) and 1.5 ml of fresh DMEM media containing 10% FBS without antibiotics and incubated for a minimum of 24 h. Cells were selected using puromycin (Sigma-Aldrich, USA) antibiotic with a concentration of 2 µg/ml and 7.5 µg/ml for HCT116 and HT29, respectively, for 3 weeks for the development of stable transfected cells. Gene knockdown was evaluated using real-time qPCR and western blot.
Cell Proliferation Assay
MTT-3-(4, 5-Dimethylthiazol-2-yl)-2, 5-Diphenyltetrazolium Bromide) assay was used to determine cell survival. One hundred μl of culture medium containing 1×106 cells were seeded into a 96-well plate and incubated for 24 h. After that 0.5 mg/ml of MTT was added to each well and incubated for 3 h in the dark at 37°C. Culture medium from the wells was aspirated, and 100 µl of isopropanol was added to dissolve the formazan crystals formed inside cells. Absorbance was measured at 570 nm in a microtiter plate reader. A graph was plotted with concentration on the X axis and absorbance on the Y axis.
Cell Cycle Analysis
Cells were harvested after trypsinization by centrifugation at 1,200 g for 5 min at 4°C. Then 300 µl of ice-cold PBS was used for the suspension of cells, and 700 µl of 70% ethanol was added drop-wise with slight vortex followed by incubation for 1 h in ice. Post incubation, cells were centrifuged at 1,200 g for 5 min at 4°C, and the cell pellet was suspended in 1× PBS. Cells were treated with RNase A (Sigma-Aldrich, USA) at a concentration of 100 µg/ml for 1 h at 37°C in a heat block (Thermo mixer, Eppendorf, Germany). After incubation, 10 µg/ml of propidium iodide was added in the dark for 15 min. Cells were strained using 45 µm cell strainer and collected in FACS tube. Tubes were kept in ice till flow analysis.
Immunophenotyping
Cells were harvested after trypsinization by centrifugation at 400 g for 5 min and washed with 1 ml HBSS. Then 1×106 cells were used for antibody staining according to manufacturer’s protocol. Details of antibodies used are provided in Supplementary Table 3. After antibody staining, cells were washed with ice-cold HBSS and suspended in HBSS containing 2% FBS. Cells were kept on ice till flow cytometry analysis was carried out using BD FACS ARIA II (BD Bioscience, USA).
Side Population Analysis
Side population assay was performed according to the protocol developed by Goodell (1996) with slight modifications. Harvested cells were spun at 400 g for 5 min, and pellet was suspended at 1×106 cells per ml in prewarmed DMEM (supplemented with 2% serum) media (Gibco, Invitrogen) in two tubes. Hoechst 33342 (Sigma-Aldrich) was added to a final concentration of 5 µg/ml at dark in both tubes. In control tube, 100 µm verapamil was added to cells prior to addition of 5 µg/ml Hoechst. Cells were mixed well and placed in 37°C dry bath (Thermomixer, Eppendorf, Germany) with shaking at 500 rpm. After incubation, cells were spun down at 4°C and resuspended in cold HBSS (with 2% serum). Cells were suspended in ice-cold HBSS (2% serum) containing 2 µg/ml Propidium iodide (Sigma-Aldrich, USA) for dead cell discrimination. Cells were immediately taken for flow cytometry analysis (BD FACS Aria II), and results were analyzed using FACS Diva software.
Apoptosis Assay
Cells were seeded in a six-well plate and incubated for 24 h. Then 5-Flurouracil was added to wells at a sublethal concentration, keeping DMSO treatment as control for 24 h. Cells were washed with 1× PBS followed by trypsinization and pelleted by centrifugation at 400 g for 3 min at 4°C. Cells 100 µl were suspended in 1× binding buffer and 5 µl of Annexin reagent and incubated in the dark for 15 min at RT. After incubation, 5 µl propidium iodide was added to cells and kept for 5 min. Finally, 400 µl of 1× binding buffer was added, and cells were filtered using 40 µm strainer and analyzed by flow cytometry using BD FACS Aria II (BD Bioscience, USA).
Protein Isolation
The cells were washed with 1 ml 1× PBS. Then 1 ml 1× PBS was added, and the cells were scraped out using a cell scraper. The cells were collected into a vial and centrifuged at 5,000 rpm for 5 min at 4°C. The supernatant was discarded, and 50–200 µl of RIPA buffer was added based on the pellet size. Phosphatase and Protease inhibitors (10 µl/1 ml buffer) were added. The pellet was suspended well and vortexed and further mixed in thermomixer at 4°C for 1.40 h at 1,400 rpm and centrifuged at 14,000 rpm for 15 min at 4°C. The supernatant was collected and stored at −80°C.
Nuclear and Cytoplasmic Protein Fraction Isolation
Approximately 1 × 107 cells were washed with 1× PBS and harvested by centrifugation at 400 g for 5 min. The pellet was resuspended in 5 pellet volume of CE buffer (1× solution composed of 10 mM HEPES, 60 mM KCl, 1 mM EDTA, 0.075% (v/v) NP-40, 1 mM DTT, and 1 mM PMSF, pH 7.6), incubated on ice for 3 min, and spun at 1,500 rpm for 4 min. The supernatant (cytoplasmic extract) was removed into a clean tube, and the pellet was washed with 100 μl of CE buffer without NP-40 at 1,500 rpm for 4 min. One pellet volume of NE buffer (1× solution composed of 20 mM Tris Cl, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM PMSF, and 25% (v/v) glycerol, pH 8.0) was added to the pellet, salt concentration adjusted to 400 mM using 5 M NaCl (~35 μl), and an additional pellet volume of NE buffer added to it and resuspended by vortexing. It was incubated on ice for 10 min with periodic vortexing. Both CE and NE were then spun at maximum speed (14,000 rpm) for 10 min. The supernatants from all the tubes were then transferred separately to clean tubes and stored at −80°C.
Western Blot
After SDS-PAGE separation of proteins, transfer sandwich was prepared with PVDF membrane and placed in transfer buffer. PVDF membrane (Amersham) was activated with methanol for 15 s prior to use. The blot was allowed to run for 120 min at 100 volts. After the transfer of proteins, membrane was blocked using 5% skimmed milk powder as blocking buffer for 1 h at RT. Then membrane was washed with 1× TBST buffer and incubated with primary antibody overnight (details of antibody in Supplementary Table 3). Again, membrane was washed in 1× TBST buffer followed by secondary antibody (1:5,000 dilution) treatment, then incubated for 1 h at RT. After incubation, membrane was washed with 1× TBST buffer three times and detected with ECL reagent (TAKARA, Japan) using Versa Doc (BD Bioscience, USA) or X-ray film.
Development of Tumor Xenograft
Each experimental groups had five 8-week-old male NOD/SCID mice, which were housed in a specific pathogen-free environment. Four million cells/mice of each control shRNA and STIL-shRNA HT-29 cells were suspended in PBS and injected into flanks subcutaneously. Then mice were monitored every 2 days for tumor growth and health. Once visible tumor appeared, its volume was measured using calipers every 2 days for obtaining rate of tumor growth in both groups. Tumor volume was calculated using the formula V (mm3) = (Largest length) × (Shortest length)2/1 (19). At the end, mice were sacrificed using carbon dioxide inhalation, and tumors along with lungs and liver tissues were harvested and stored in RNAlater and 4% paraformaldehyde for molecular and histological analysis, respectively.
H&E Staining
Hematoxylin and Eosin (Merck-Millipore, USA) staining was performed according to manufacturer’s protocol for pathological examination of various tissues obtained after surgery.
Immunohistochemistry
Tissues received post-surgery were fixed with 4% paraformaldehyde (Sigma-Aldrich, USA) at 4°C for 16 h. They were then processed with alcohol gradient and xylene followed by embedding with paraffin to make paraffin blocks. Five µm thick sections were mounted on Starfrost glass slides (Leica, Germany) and were deparaffinized in xylene followed by rehydration with alcohol gradients. Sections were blocked for endogenous peroxidases, and antigen retrieval was performed using 10 mM citrate buffer (pH 6.0). After retrieval, BSA (3%) blocking was done for 30 min at RT followed by primary antibody staining overnight. Primary antibody details are given in Supplementary Table 3. After incubation, antigen-antibody binding was detected using ABC kit (Vecta stain, USA). DAB substrate (Sigma-Aldrich, USA) was used as a chromogen, and hematoxylin (Merck Millipore, Germany) was used as a counterstain. Sections without incubation of primary antibody served as negative control. Semiquantitative analysis was done by counting three independent microscopic fields (n=100–200 cells/field) for staining of specific antigen using upright microscope (Leica DM1000, Germany). Tissue array (US Biomax, USA) for STIL expression was also performed according to above protocol. Tissue array demographics has been detailed in Supplementary Table 4.
Cancer Datasets Analysis
STIL gene was used as an input in Oncomine (www.oncomine.org/resource/) and GEPIA (http://gepia.cancer-pku.cn/) online free cancer database to obtain gene expression pattern across multiple colorectal cancer transcriptomics studies globally, and survival curves for STIL-expressing patients were obtained from C-Bioportal (www.cbioportal.org) online tool.
Statistical Analysis
All statistical analysis was performed using SPSS version 25 and GraphPad Prism 5. Unpaired t-test was performed for mean comparison and p value determination for flow cytometry and immunohistochemistry data. Real-time PCR data analysis was carried out using one sample t-test to compare determined log2 fold change and determined p value among samples. Kaplan-Meier plots were obtained from C-Bioportal cancer genomic datasets. Multivariate analysis was performed using binary logistic regression model, and p-value and ODDS ratio were derived.
Results
Elevated Expression of STIL Gene in Tumors Correlates With Lower Disease-Free Survival In CRC Patients
We used Oncomine datasets to analyze differential mRNA expression of STIL between normal and cancerous colon tissues from three different studies (Hong, Notterman, and Kaiser), which revealed a significant enrichment of STIL gene in cancer tissues (Figure 1A). Similar trend in STIL mRNA expression was observed in TCGA datasets, using GEPIA web tool (20) (Supplementary Figure 1A). Further validation in our cohort CRC using real-time q-PCR showed a 2.1-fold high expression in tumor compared to matched normal tissues (p = 0.001) (Figure 1B). IHC of STIL protein in cancer tissues showed a very high cytoplasm-specific staining compared to normal tissue (Figure 1C). To assess the role of STIL enrichment in colon cancer with disease prognosis, we used c-Bioportal cancer database. Kaplan-Meier plots showed no significant differences for 5-year survival among STIL high- and low-expression groups; however, STIL high-expression group showed a very low disease-free survival rate among CRC patients with a p value 0.001 and 0.0007 in two different studies (Figure 1D). Put together, these results suggest that overexpression of STIL in CRC could have crucial unexplored role in tumor development and disease progression.
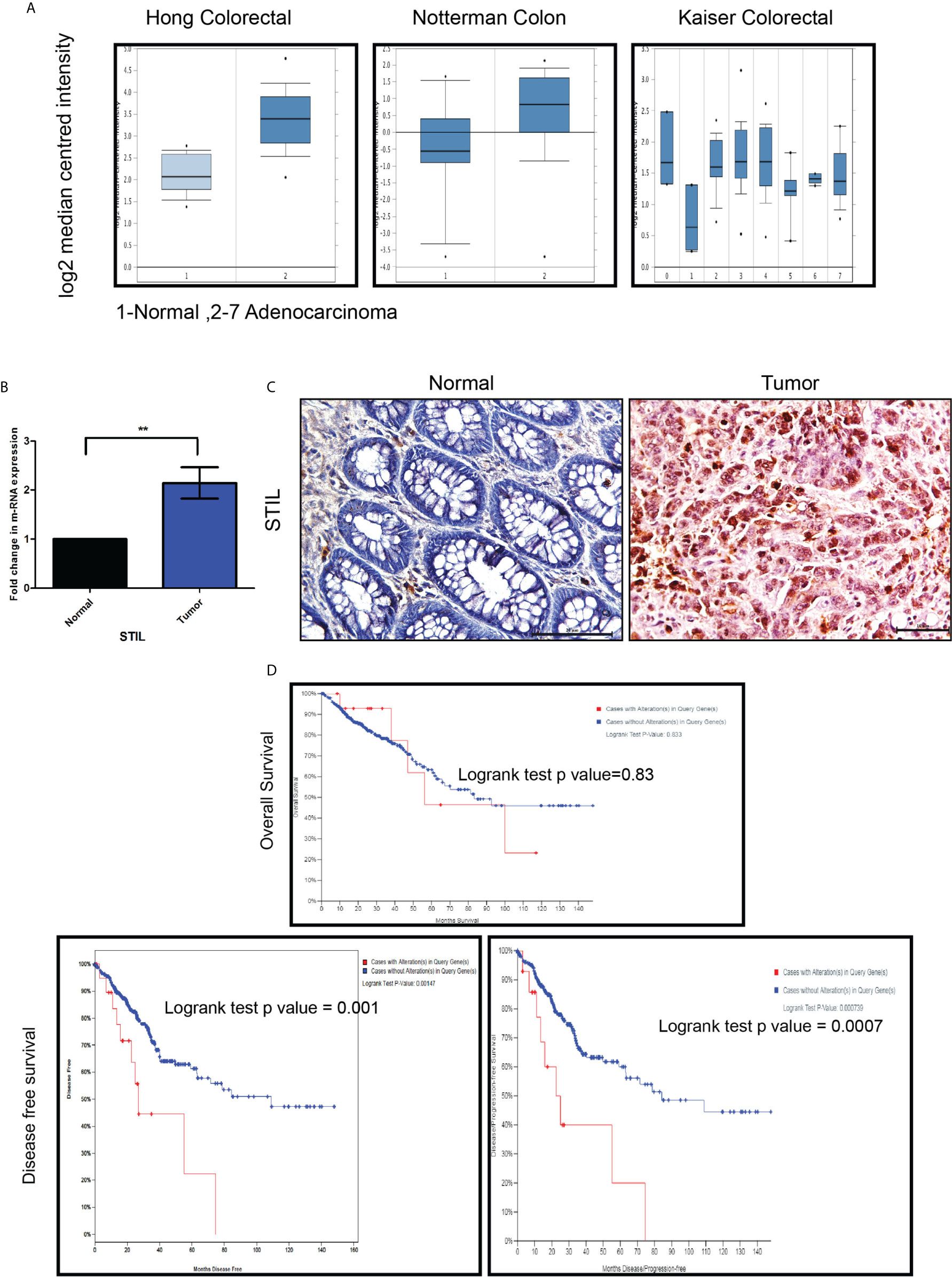
Figure 1 Expression of STIL gene in CRC and its role in prognosis. (A) STIL m-RNA expression in normal colon and tumor obtained from Oncomine datasets. (B, C) Differential expression of STIL gene mRNA (N=22) and protein in CRC. (D) Kaplan-Meier plot showing association of high mRNA expression with patients’ survival and disease free survival in CRC cases. One Sample t-test and log rank test results showing p value ≤0.01 is represented by **, Scale bar is 20µm. Note: In Kaplan-Meier plot, red and blue line in indicates high and low m-RNA expression respectively.
High STIL Protein Expression Is Found to Have Association With Early Tumor Stage in Rectal Cancer
To evaluate STIL protein expression in different tumor stages and grades of CRC, we performed IHC for STIL in rectal cancer tissue array (array details given in Supplementary Table 4). STIL was found to have intense cytoplasmic staining in cancer tissues compared to normal (p value 0.001). Early-stage (T1-T2) tumors were stained positively with a high IHC score compared to late-stage (T3-T4) tumors. (Figure 2A and Table 1). Both high- and low-grade tumor tissues showed moderate to strong STIL staining except signet-ring carcinoma (Figure 2B). Chi-square analysis revealed that females (69.4%) and early-stage tumor (73.3%) correlated well with higher expression of STIL with a p value of 0.04 and 0.03, respectively (Table 1). However, sex-adjusted binary regression analysis revealed that higher tumor stage has 71% lesser chance of high expression of STIL compared to early stage (p value 0.02), and earlier association of female sex with STIL expression was influenced by tumor stage. In addition, late-disease stage was found to have 60% lesser chance of STIL expression compared to early stage (p value 0.07) (Table 2). In addition, role of STIL in cancer cell migration was evaluated using scratch wound healing assay, which showed no significant difference between control and STIL-silenced HCT116 cells (Supplementary Figure 2). Further, we also analyzed lung and liver tissues for presence of secondary tumor in control and STIL-silenced xenograft mice using Ep-CAM protein expression and found no tumor deposits (Supplementary Figure 3). This further suggests that STIL may not play a role in cancer cell migration and thus metastasis in concordance to tissue array observation. Put together, this study suggests that STIL may not play a significant role in invasion of tumor; however, it could have a crucial role in early tumorigenesis.
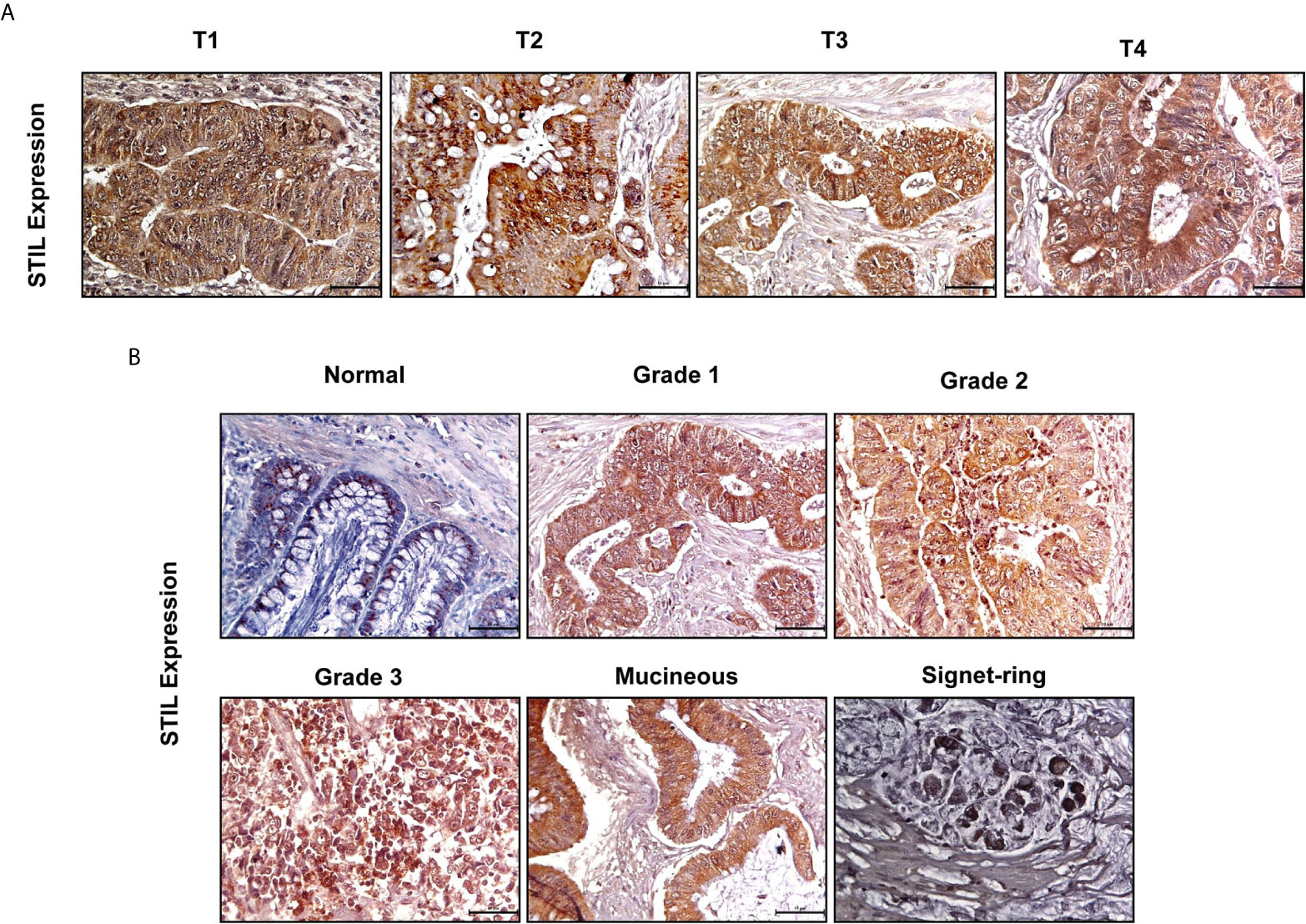
Figure 2 IHC of rectal cancer tissue array showing expression of STIL protein. (A, B) Representative images showing expression of STIL protein in different tumor stages and grades.

Table 1 Association of clinical and pathological parameters with STIL protein expression.
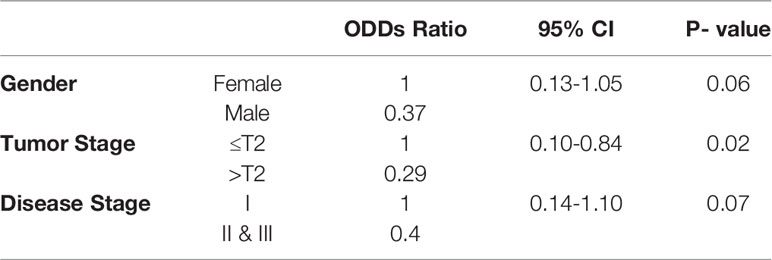
Table 2 Association of tumor and disease stage with STIL expression.
STIL Silencing Reduced Cell Proliferation and Tumor Growth in CRC
To understand the role of STIL in proliferation and tumor growth of CRC, we stably silenced STIL gene in HT-29 CRC cells using shRNA (Figures 3A, B). Cell proliferation was evaluated by MTT assay, where STIL silencing reduced the proliferation by 34% compared to control shRNA cells (p=0.05) (Figure 3C). STIL has been well studied for its role in cell cycle regulation, as it plays a crucial role in centrosome assembly. Thus, we analyzed cell cycle pattern, which revealed a two-fold accumulation of cells in G2/M phase in STIL-silenced cells (Figure 3D). These results led us to investigate the tumorigenic potential of STIL in-vivo, for which we developed a tumor xenograft using STIL-silenced HT-29 cells and compared it with control shRNA xenograft. We observed a higher tumor volume in control group, which became more significant with time compared to STIL-silenced tumor (Figures 3E, F). Dry weight of control tumor was found to be threefold more than STIL-silenced tumor (p=0.001) (Figure 3G). Further, we analyzed nuclear expression of the proliferation marker Ki-67 in tumor xenograft tissue and found a significantly reduced expression in STIL-silenced group compared to control (p=0.01) (Figure 3H). These results confirm a significant role of STIL in regulating tumor proliferation and growth in CRC.
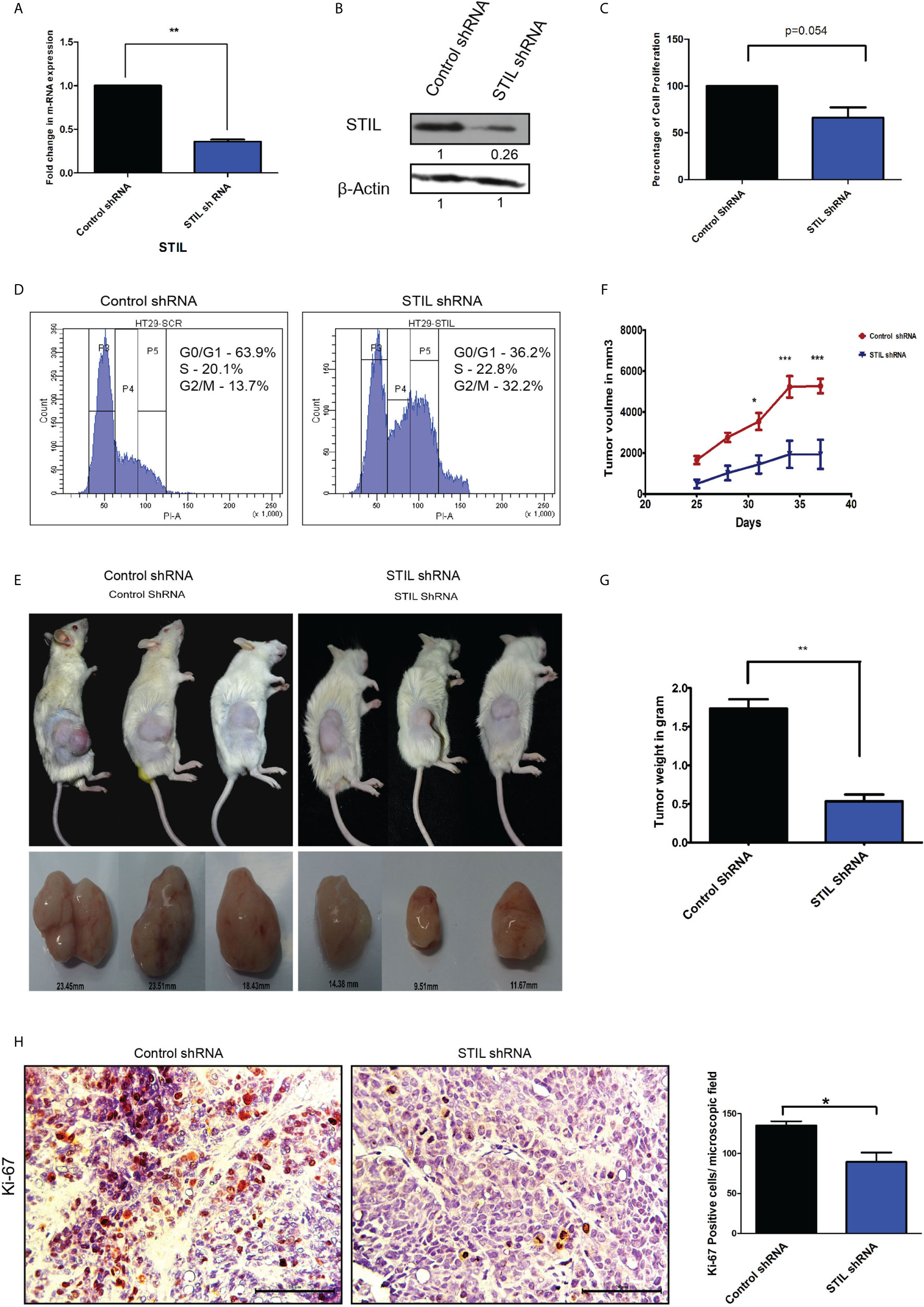
Figure 3 Role of STIL in CRC tumorigenesis. (A) Bar graph showing fold change in STIL mRNA expression upon STIL silencing. (B) Immunoblot validating STIL silencing at protein level. (C) Bar graph showing cell proliferation of HT-29 cells upon STIL silencing. (D) Histogram showing effect of STIL silencing on cell cycle. (E, F) Representative images of tumor xenograft and graph showing growth of tumor upon time (n = 3). (G) Bar graph showing mean dry weight of tumor in both groups (n = 3). (H) IHC showing nuclear expression and quantification of Ki-67 protein in xenograft derived tumor tissues. Unpaired t-test results showing p value ≤ 0.05, ≤ 0.01, ≤ 0.001 are represented by *, **, ***, respectively, Scale bar is 20 µm.
STIL Regulates Expression of Cancer Stem Cell–Associated Genes in CRC Independent of Shh Signaling
We have observed STIL overexpression to be associated with lower disease-free survival in CRC patients from our initial results. Disease recurrence has mostly been contributed by residual cells, which are drug resistant in nature. These cells are known to have cancer stem cell (CSC) properties including expression of established surface markers. However, the role of STIL in CRC stem cells is still unknown. We assessed the expression of established CSC markers in STIL-silenced cells and observed a significant reduction in CD133 (fold change 0.4, p=0.03) and CD44 (fold change0.7, p=0.02) expression in mRNA and protein (Figures 4A, B). STIL is a known positive regulator of Shh signaling, and thus, we analyzed expression of CSC markers CD133 and CD44 upon Shh inhibition by SANT1 treatment and found no significant reduction in their mRNA levels. Interestingly, CD44 protein expression was found elevated, suggesting STIL-mediated regulation of CSC markers may not be exclusively through Shh signaling (Figures 4C, D). We further validated these findings in xenograft tumor tissues using real-time q-PCR, which showed significant reduction in CD133 (fold change 0.41, p=0.002) and CD44 (fold change 0.77, p=0.05) mRNA levels in STIL-silenced tumor, which was further confirmed by IHC (Figures 4E–G), respectively. To examine if this stands true across other CRC cells, we analyzed CD133 and CD44 expression in HCT116 cells upon STIL silencing and SANT1 treatment, which also showed similar trend (Supplementary Figures 4A–E). Additionally, we also analyzed expression of stem cell maintenance factors oct4 and nanog in STIL-silenced cells and found a reduction (fold change 0.69 and 0.78) compared to control, respectively; however, statistical significance was not observed (Supplementary Figures 4F–H). Membranous expression of CD133 and CD44 are known to be characteristics of CSC; thus, we also analyzed their surface expression using flow cytometry. In concordance with our gene expression data, a significant reduction in CD133 and CD44-positive cells were also observed upon STIL silencing (Figure 4H). These results confirm a STIL-mediated regulation of CSC signatures associated with CRC.
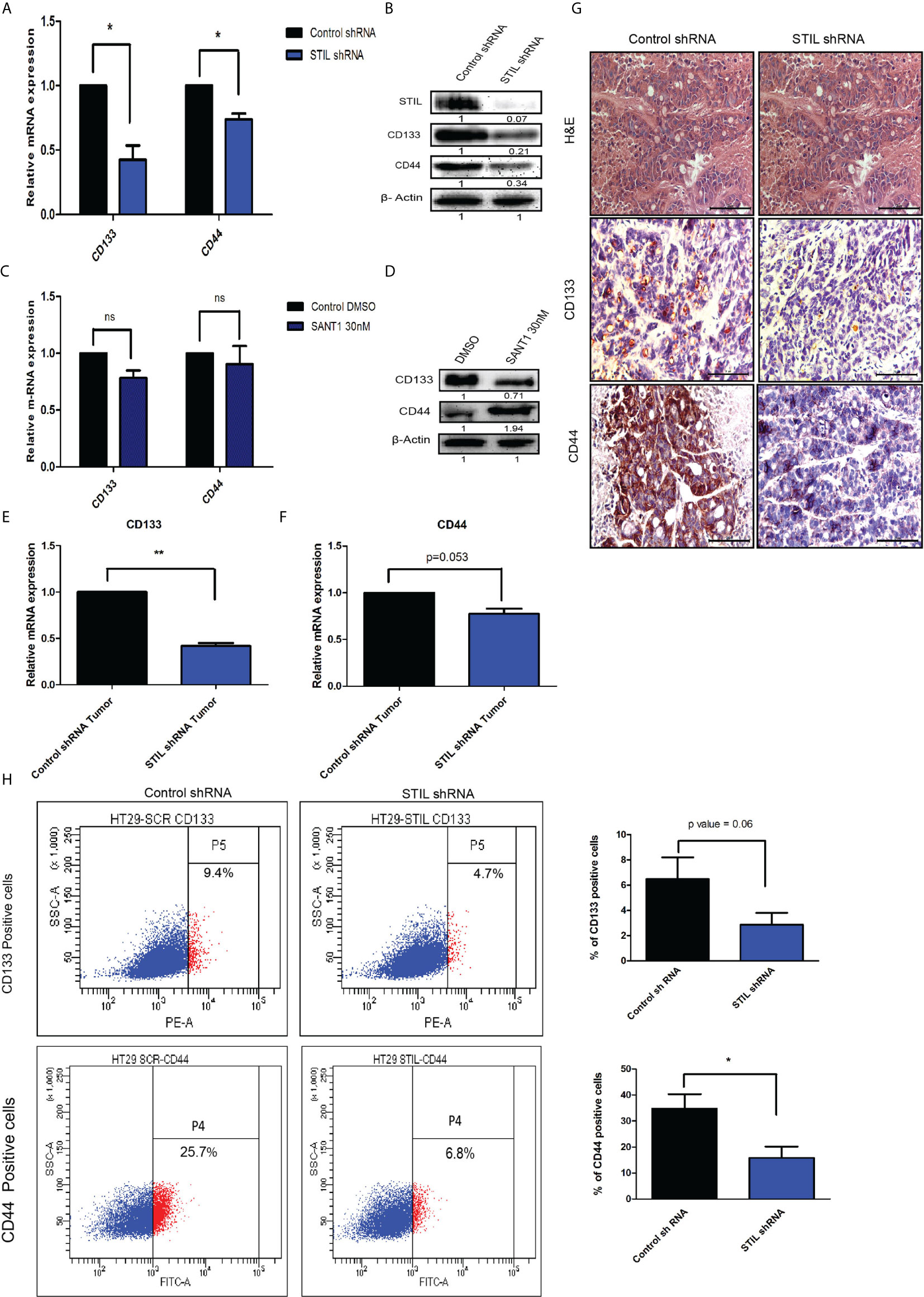
Figure 4 Effect of STIL silencing on CSC markers expression in CRC. (A, B) mRNA and protein level expression of CSC markers upon STIL silencing. (C, D) mRNA and protein level expression of CSC markers upon SANT1 treatment. (E–G) Showing CD133 and CD44 expression in xenograft tumors at mRNA and protein levels, respectively. (H) Dot plot representing % of CD133 and CD44-positive cells. Unpaired t-test results showing p value ≤ 0.05, ≤ 0.01, ≤ 0.001 are represented by *, **, ***, respectively. Scale bar is 20 µm.
STIL Silencing Reduced Drug Efflux Activity Via Depletion Of Drug Transporter Proteins in CRC Cells
Another hallmark of CSC is the ability to efflux drugs via active expression of drug transporter proteins. Here, we performed side population (SP) assay, which potentially identifies drug effluxing cells and CSCs. STIL silencing resulted in reduced SP in HT-29 cells with mean % population of 0.7 compared to control with 3.7 (p=0.051) (Figures 5A, B). We noted similar reduction in SP in HCT116 cells upon STIL silencing (Supplementary Figure 5A). Moreover, ABCG2, which is known to be the crucial transporter associated with stem cells and SP, was found to have reduced levels of mRNA and protein in both STIL-silenced HT29 cells and xenograft tumor tissue (Supplementary Figures 5B, C and Figures 5C, D). Our data further revealed depletion of ABCB1 and ABCG2 protein upon STIL silencing in HT29 cells and HCT 116 cells (Figure 5C, Supplementary Figure 5D). In addition, STIL silencing was also found to deplete expression of thymidylate synthase (TS) enzyme, known for its role in 5-fu resistance in CRC (Figure 5C and Supplementary Figure 5D). However, we observed no reduction in ABCG2 and TS protein expression upon SANT1 treatment, revealing a Shh-independent regulation of these proteins by STIL (Figure 5E and Supplementary Figure 5E). However, ABCB1 expression reduced significantly upon Shh inhibition (Figure 5E). Further, we analyzed the effect of STIL silencing on 5-fu drug treatment in HT29 cells. To our surprise, a slightly reduced cell death in STIL-silenced cells was observed compared to controls (Figures 5F, G). To investigate the cause behind reduced cell death, we looked for expression of antiapoptotic protein Bcl2 upon STIL silencing and observed an increased Bcl2 protein levels compared to control (Figure 5H). Put together, these data suggest that STIL may regulate drug efflux and resistance markers not solely through Shh pathway in CRC. However, the role of STIL in 5-fu sensitization for CRC cells demands further investigation.
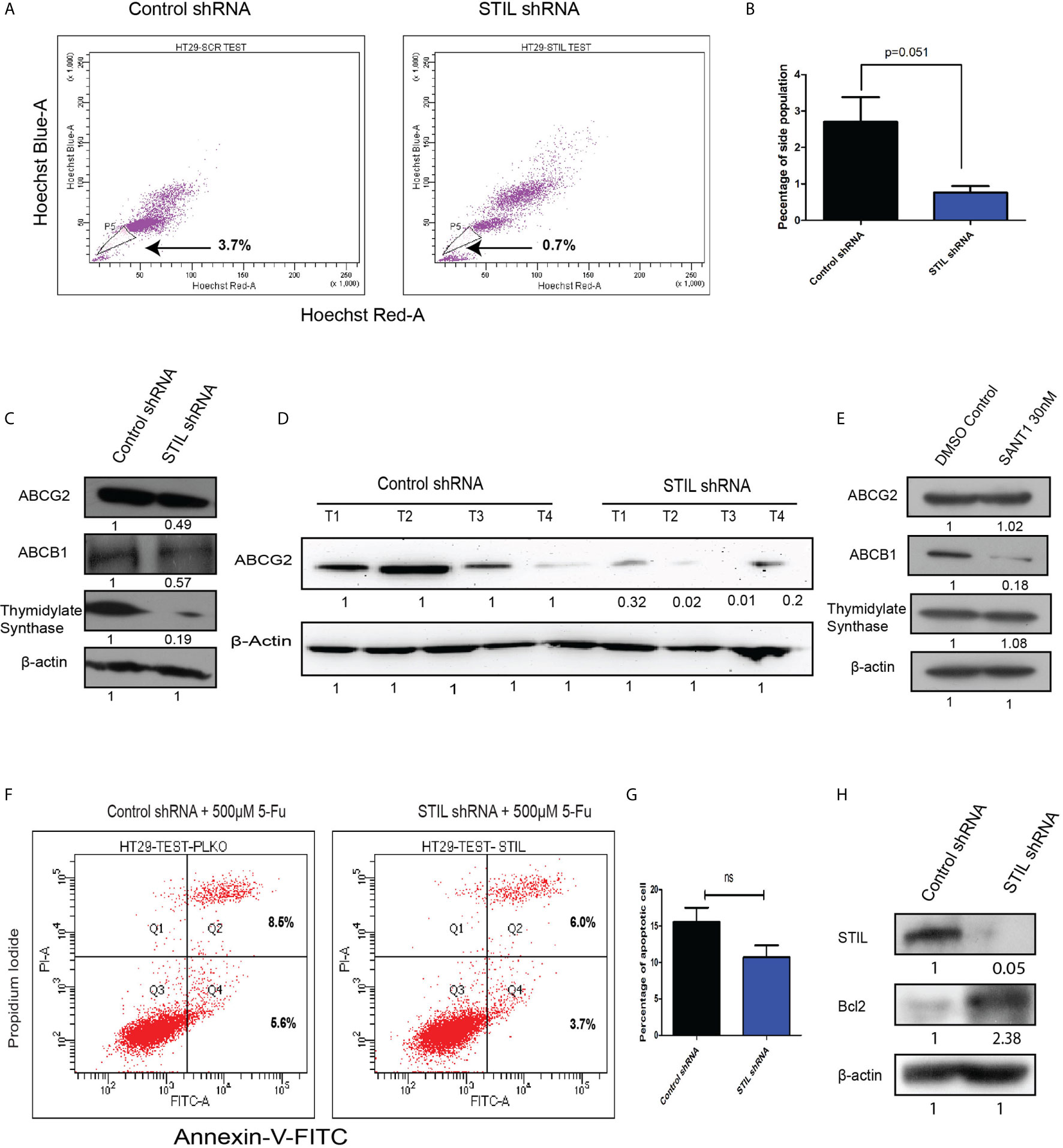
Figure 5 Effect of STIL silencing on drug effluxing cells and associated genes in CRC. (A, B) Dot plot showing side population cells and bar plot representing quantification of the same. (C) Western blot showing expression of ABCG2, ABCB1, and thymidylate synthase protein expression, upon STIL silencing. (D) Western blot showing ABCG2 protein expression in control and STIL-silenced tumor xenograft. (E) Western blot showing expression of ABCG2, ABCB1, and thymidylate synthase protein expression, upon SANT1 treatment. (F, G) Annexin-V assay showing percentage of cell death upon 5-FU treatment. (H) Western blot showing Bcl2 expression upon STIL silencing. Unpaired t-test results showing p value ≥ 0.05, is represented by ns, not significant.
STIL Regulates β-Catenin Expression Seemingly via AKT Contrarily to Shh Signaling
STIL has been studied to be a positive regulator of hedgehog signaling; however, STIL-mediated hedgehog regulation in CRC remains unknown. To study its role in mediating regulation of hedgehog signaling in CRC, we screened for expression of hedgehog-associated genes (SHH, PTCH2, SMO, SUFU, and STIL) as well as effector genes (GLI1 and GLI2) after STIL silencing and treatment with SANT1. SANT1 treatment resulted in significant downregulation of the pathway-associated genes as well as effectors molecules GLI1&2 (Figure 6A). Conversely, STIL repression did not show similar trend in gene expression except for GLI1 (Figure 6B). These observations suggest the negative regulatory role of STIL on SUFU, SMO, and SHH genes in CRC. Although GLI2 showed an increase in mRNA level upon STIL silencing, it showed a significant reduction at protein level upon Shh inhibition and STIL repression (Figures 6C, D). There are studies that advocate a reciprocal relationship between Wnt and Shh signaling in CRC development (21). We further looked into probable STIL-mediated cross talk between Shh and Wnt signaling and observed STIL-mediated regulation of Wnt effector β-catenin, which was not altered upon Shh inhibition (Figures 6C, D and Supplementary Figures 6A, B). Further, when we checked the cytoplasmic and nuclear β-catenin levels upon STIL silencing, we observed a reduction in cytoplasmic as well as nuclear cleaved fragment (~56 kDa), which is reported to be the transcriptionally active fragment in CRC, suggestive of inhibited Wnt signaling (Supplementary Figure 7). However, further studies are warranted to delineate the STIL-β-catenin molecular axis in CRC. There has been a recent report suggesting STIL-mediated regulation of AKT protein in gastric cancer (22) and another one suggesting AKT-mediated regulation of β-catenin in CRC (23). Thus, we hypothesized that STIL regulates β-catenin via regulating AKT in CRC. STIL silencing resulted in a significant decrease in AKT and p-AKT protein, suggesting STIL-mediated β-catenin regulation is via AKT/pAKT regulation in CRC, unlike Shh signaling (Figures 6C, D). These results suggest that STIL functions in a Shh-independent manner for regulation of β-catenin in CRC.
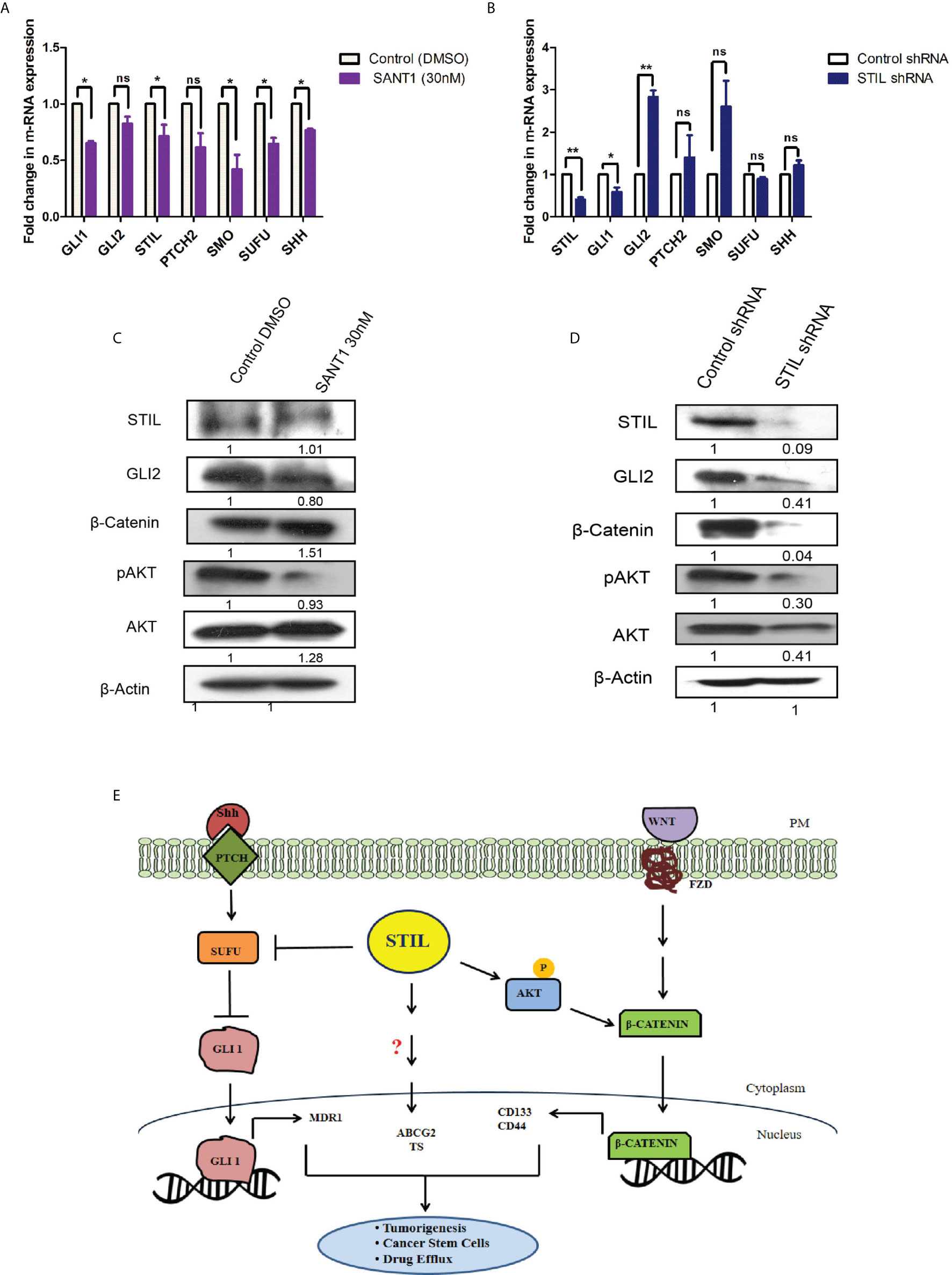
Figure 6 Effect of SANT1 treatment and STIL inhibition on Shh and Wnt signaling. (A, B) Bar graph showing expression of Shh signaling components upon SANT1 treatment and STIL silencing. (C, D) Western blot showing effect of SANT1 treatment and STIL silencing on GLI2, β-catenin, AKT, and p-AKT protein expression. (E) Schematic representation showing possible mechanistic regulation of multifaceted role by STIL in CRC. Unpaired t-test results showing p value ≤ 0.05, ≤ 0.01 are represented by *, **, respectively; ns, not significant.
Discussion
STIL alteration has been implicated in lymphoblastic leukemia and microcephaly (24, 25); however, the role of STIL in solid epithelial tumors stands least explored. Our study substantiates a novel role for STIL in regulation of tumor growth, drug resistance, and stem cells in CRC. STIL plays an instrumental role in centriole division and thus has been very critical for cell cycle progression and proliferation (26). Being a regulator of cell proliferation, its role in cancer becomes obvious, and many tumor types have been reported to have enriched expression of STIL (1, 4). In this study, we found an enriched expression of STIL at mRNA and protein level in CRC tissues, whereas adjacent normal tissue showed a basal-level expression (Figure 1). However, to date the role of STIL in CRC growth and proliferation has not been studied. STIL, being a critical factor in cell cycle machinery, its downregulation resulted in aberrant cell cycle and reduced proliferation in HT29 cancer cells (Figure 3). Further studies with STIL-silenced xenograft showed a remarkable difference in tumor growth in NOD/SCID mice with lesser tumor volume and weight compared to control shRNA group. Ki-67 antigen, an established proliferation marker, was also found to have less nuclear staining in STIL-silenced tumor (Figure 3). Put together, these data showed a critical role of STIL in CRC proliferation and growth. Another aspect of STIL, which has not been explored, is its role in Shh signaling-mediated stem cell maintenance in cancer. CSC are being studied for development of future therapeutic interventions in CRC as they are known to be the fuel behind therapy failure and recurrence (27). Although CSCs are well-known to be regulated by developmental signaling cascades, specific targets are lacking, which can be exploited for development of advanced therapeutics. CD133 and CD44 surface molecules have been studied well in CRC for their enriched expression in CSCs and responsible for drug resistance, recurrence, and metastasis in CRC (28–30). Our study has shown that repression of STIL in HT-29 cells results in a significant reduction of CD133 and CD44-positive cells (Figure 4). Interestingly, STIL was found to regulate expression of CSC markers (Figure 4) and regulators such as OCT4 and NANOG at the transcriptional level both in vitro and in vivo (Supplementary Figure 4). These data suggest a stem cell regulatory function of STIL in CRC. However, when Shh signaling was inhibited by SANT1, we did not observe any significant reduction in mRNA levels of CD133 and CD44 markers (Figure 4), along with OCT4 and NANOG genes (Supplementary Figure 4). Interestingly, CD44 protein level was found elevated upon SANT1 treatment, which signs a heterogenous mode of regulation of CSC markers by STIL may not be solely through Shh signaling in CRC. Another hallmark feature of CSCs and drug-resistant cells is the ability to efflux drugs by enhancing ABC pumps expression (31). SP cells show high ABC transporter activities, and CSCs are reported to be enriched in SP cells in multiple gastrointestinal cancer (32). Hedgehog signaling has been reported to regulate expression of ABC transporters in ovarian epithelial cancer (33), and STIL, being a positive regulator of GLI1-mediated hedgehog regulation, was found to be significantly regulating side population in CRC cells (Figure 5). ABCG2 remains as one of the major efflux pumps regulating side population as well as stem cells (34), and we observed downregulation of ABCG2 and partial reduction in ABCB1 expression upon STIL repression (Figure 5). Further, STIL silencing was found to repress expression of thymidylate synthase (Figure 5), a known target for 5-fu and a poor prognostic marker in CRC (35). To our concern we did not observe reduction of ABCG2 and Thymidylate synthase protein upon Shh inhibition, suggesting a critical role of STIL in regulation of drug-resistant cells and genes in CRC independent of Shh signaling (Figure 5). Previous studies have already shown that ABCG2 function and localization are regulated by AKT (36). Since we found that STIL modulates AKT expression (Figure 6), thus STIL-mediated regulation of ABCG2 in CRC could possibly be via AKT. Surprisingly, inhibition of STIL failed to sensitize HT29 cells for 5-fu treatment (Figure 5). Further, antiapoptotic protein Bcl2 showed enriched expression upon STIL silencing, partially explaining the reason behind reduced apoptosis in STIL-repressed cells after 5-fu treatment. Again, there are reports of increased thymidylate synthase levels upon 5-fu treatment (37), which in turn could desensitize cells to 5-fu treatment. In this study, 5-fu treatment might be elevating the TS levels, thus replenishing the reduced TS caused by STIL silencing resulting in 5-fu resistance. However, in a recent study, STIL repression was shown to have a synergistic effect with DNA-damaging agents in ovarian cancer (11). This suggests that function of STIL in therapy sensitization could be drug and tissue specific. Nevertheless, this heterogeneous mode of action of STIL in 5-fu and other chemotherapeutics needs further investigation.
STIL has been established to be a positive regulator of Shh signaling and known to regulate GLI1 (38). Shh inhibition by SANT1 treatment showed reduced gene expression of its pathway components including GLI1/2; however, STIL silencing was found to downregulate GLI1 and GLI2 but no other Shh components (Figure 6). These results advocate for a Shh-independent regulatory function of STIL, which demands further studies. Shh and Wnt signaling has been studied for their antagonistic mode of regulation (21), and Wnt being the most critical aberrant signaling in CRC (39), we further looked into STIL-mediated regulation of Wnt pathway. Interestingly, we found STIL to be regulating β-catenin protein. However, Shh inhibition did not show similar result, which signs at existence of a STIL-mediated molecular axis between Wnt and Shh signaling (Figure 6). A recent study has delineated that STIL regulates PI3K/AKT in gastric cancer. Reiteratively, AKT is known to regulate β-catenin stabilization (40, 41). These studies led us to explore an AKT mediated β-catenin regulation by STIL. One of the most interesting findings of our study remains to be STIL-mediated regulation of β-catenin via AKT and p-AKT independent of Shh signaling (Figure 6). Eventually, our study revealed a STIL-mediated cross talk between Wnt and Shh signaling in CRC, which opens new molecular insights on existing interplay between these developmental pathways during carcinogenesis and therapy failure in CRC. Despite few studies having shown the role of STIL in various cancers, its association with cancer prognosis stands elusive. Our rectal tissue array analysis showed no association of STIL protein expression with invasive stages of tumor but was found to be highly expressed in early stages (Figure 2 and Table 2). This again may be supported by our scratch assay experiment where we have observed that STIL silencing have no significant effect on HCT116 migration, and tumor xenograft also showed no tumor deposits in distant metastatic sites (Supplementary Figure 2). Further, overall survival (OS) and disease-free survival (DFS) analyses from c-Bioportal datasets revealed STIL overexpression to be significantly associated with lower DFS in CRC, but no association was found with OS (Figure 1). These observations show that STIL may not have a possible role in tumor invasion but could be critical in early tumorigenesis and therapy resistance, thus leading to lower DFS in CRC. However, more studies are warranted for elucidation of detailed molecular events mediated via STIL in CRC. Nonetheless, our study suggests Shh and Wnt regulation by STIL to be a possible mechanism governing its role in CRC (Figure 6E).
In conclusion, this report stands as the first study demonstrating an oncogenic role of STIL in CRC. Our study showed the role of STIL oncogene regulating CSC characteristics along with drug resistance properties, which had not been investigated to date in CRC. Further, STIL signs at a Shh-independent regulation of β-catenin, which we consider to be a novel finding, as regulation of Wnt by STIL opens a whole new prospective of research for Shh and Wnt cross talk in CRC. In addition, this study also sheds light on prognostic implications of STIL in CRC, which ensue to be very critical and imperative from a clinical perspective. Association of STIL with poor disease-free survival suggests its potency for a prognostic biomarker in CRC. Though the mechanistic proof of STIL function in details remains elusive, our study urges that STIL could be an important oncogene with an inherent ability to regulate multiple aspects of cancer progression in CRC, and it could be a warranted target for therapeutic intervention and prognostic marker in the future.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
All human biopsies were collected at Regional cancer center, Trivandrum, Kerala, India after approval of the Regional Cancer center Ethical committee (HEC No.43/2011) and from donors that signed written informed consent. The patients/participants provided their written informed consent to participate in this study. All the experiments on NOD-SCID mice were performed after approval from the Institute Animal Ethics Committee (IAEC/683/ASN/28).
Author Contributions
TP conceived, planned, and carried out the experiments, and lead in data analysis and manuscript writing. VK and SJ assisted in planning and execution of xenograft experiment. ES and KR performed immunoblots and gave critical comments to improve work and also helped in article corrections. JV helped with statistical analysis of IHC tissue array data. CK performed the surgery and helped in obtaining the human biopsy samples and their storage. AS helped in RNA isolation from biopsies and qPCR. SN conceived, designed the project, and supervised its execution, manuscript proofreading, and provided critical feedbacks in manuscript writing. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Department of Biotechnology, Government of India (grant no-BT/PR3223/BRB/10/964/2011) and research fellowship to TP from Department of Biotechnology, Government of India.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Dr. Arya, Dr. Vishnu, and Dr. Archana for their immense support in animal experiment. Dr. Debasree Dutta for HEK293T cells. Indu Ramachandran and Arya (Flow Cytometry facility, RGCB, Trivandrum) for her help and guidance in flow cytometry, Vijiand Hima (Histology facility, RGCB, Trivandrum) for their assistance in histology work, Rekha for her help in biopsy collection.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.581671/full#supplementary-material
References
1. Patwardhan D, Mani S, Passemard S, Gressens P, El Ghouzzi V. STIL Balancing Primary Microcephaly and Cancer. Cell Death Dis (2018) 9:65. doi: 10.1038/s41419-017-0101-9
2. Arquint C, Nigg EA. The PLK4-STIL-SAS-6 Module at the Core of Centriole Duplication. Biochem Soc Trans (2016) 44:1253–63. doi: 10.1042/BST20160116
3. D’Angiò M, Valsecchi MG, Testi AM, Conter V, Nunes V, Parasole R, et al. Clinical Features and Outcome of SIL/TAL1-Positive T-Cell Acute Lymphoblastic Leukemia in Children and Adolescents: A 10-Year Experience of the AIEOP Group. Haematologica (2015) 100:e10–3. doi: 10.3324/haematol.2014.112151
4. Erez A, Perelman M, Hewitt SM, Cojacaru G, Goldberg I, Shahar I, et al. Sil Overexpression in Lung Cancer Characterizes Tumors With Increased Mitotic Activity. Oncogene (2004) 23:5371–7. doi: 10.1038/sj.onc.1207685
5. Kasai K, Inaguma S, Yoneyama A, Yoshikawa K, Ikeda H. SCL/TAL1 Interrupting Locus Derepresses GLI1 From the Negative Control of Suppressor-of-Fused in Pancreatic Cancer Cell. Cancer Res (2008) 68:7723–9. doi: 10.1158/0008-5472.CAN-07-6661
6. Furness CL, Mansur MB, Weston VJ, Ermini L, van Delft FW, Jenkinson S, et al. The Subclonal Complexity of STIL-TAL1+ T-Cell Acute Lymphoblastic Leukaemia. Leukemia (2018) 32:1984–93. doi: 10.1038/s41375-018-0046-8
7. Nigg EA. Centrosome Aberrations: Cause or Consequence of Cancer Progression? Nat Rev Cancer (2002) 2:815–25. doi: 10.1038/nrc924
8. Izraeli S, Lowe LA, Bertness VL, Good DJ, Dorward DW, Kirsch IR, et al. The SIL Gene is Required for Mouse Embryonic Axial Development and Left–Right Specification. Nature (1999) 399:691–4. doi: 10.1038/21429
9. David A, Liu F, Tibelius A, Vulprecht J, Wald D, Rothermel U, et al. Lack of Centrioles and Primary Cilia in STIL –/– Mouse Embryos. Cell Cycle (2014) 13(18):2859–68. doi: 10.4161/15384101.2014.946830
10. Gulino A, Ferretti E, De Smaele E. Hedgehog Signalling in Colon Cancer and Stem Cells. EMBO Mol Med (2009) 1:300–2. doi: 10.1002/emmm.200900042
11. Rabinowicz N, Mangala LS, Brown KR, Checa-Rodriguez C, Castiel A, Moskovich O, et al. Targeting the Centriolar Replication Factor STIL Synergizes With DNA Damaging Agents for Treatment of Ovarian Cancer. Oncotarget (2017) 8:27380–92. doi: 10.18632/oncotarget.16068
12. Li L, Carr AL, Sun L, Drewing A, Lee J, Rao Z. A Novel Function of the Human Oncogene Stil: Regulation of PC12 Cell Toxic Susceptibility Through the Shh Pathway. Sci Rep (2015) 5:16513. doi: 10.1038/srep16513
13. Varnat F, Duquet A, Malerba M, Zbinden M, Mas C, Gervaz P, et al. Human Colon Cancer Epithelial Cells Harbour Active HEDGEHOG-GLI Signalling That is Essential for Tumour Growth, Recurrence, Metastasis and Stem Cell Survival and Expansion. EMBO Mol Med (2009) 1:338–51. doi: 10.1002/EMMM.200900039
14. Mazumdar T, DeVecchio J, Shi T, Jones J, Agyeman A, Houghton JA. Hedgehog Signaling Drives Cellular Survival in Human Colon Carcinoma Cells. Cancer Res (2011) 71:1092–102. doi: 10.1158/0008-5472.CAN-10-2315
15. Sims-Mourtada J, Izzo JG, Ajani J, Chao KSC. Sonic Hedgehog Promotes Multiple Drug Resistance by Regulation of Drug Transport. Oncogene (2007) 26:5674–9. doi: 10.1038/sj.onc.1210356
16. Noubissi FK, Yedjou CG, Spiegelman VS, Tchounwou PB. Cross-Talk Between Wnt and Hh Signaling Pathways in the Pathology of Basal Cell Carcinoma. Int J Environ Res Public Health (2018) 15:1442. doi: 10.3390/ijerph15071442
17. Yanai K, Nakamura M, Akiyoshi T, Nagai S, Wada J, Koga K, et al. Crosstalk of Hedgehog and Wnt Pathways in Gastric Cancer. Cancer Lett (2008) 263:145–56. doi: 10.1016/j.canlet.2007.12.030
18. Song L, Li Z-Y, Liu W-P, Zhao M-R. Crosstalk Between Wnt/β-Catenin and Hedgehog/Gli Signaling Pathways in Colon Cancer and Implications for Therapy. Cancer Biol Ther (2015) 16:1–7. doi: 10.4161/15384047.2014.972215
19. Jung J. Human Tumor Xenograft Models for Preclinical Assessment of Anticancer Drug Development. Toxicol Res (2014) 30:1–5. doi: 10.5487/TR.2014.30.1.001
20. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: A Web Server for Cancer and Normal Gene Expression Profiling and Interactive Analyses. Nucleic Acids Res (2017) 45:W98–W102. doi: 10.1093/NAR/GKX247
21. Ding M, Wang X. Antagonism Between Hedgehog and Wnt Signaling Pathways Regulates Tumorigenicity. Oncol Lett (2017) 14:6327–33. doi: 10.3892/ol.2017.7030
22. Wang J, Zhang Y, Dou Z, Jiang H, Wang Y, Gao X, et al. Knockdown of STIL Suppresses the Progression of Gastric Cancer by Down - Regulating the IGF - 1/PI3K/AKT Pathway. J Cell Mol Med (2019) 23:5566–75. doi: 10.1111/jcmm.14440
23. Ou B, Cheng X, Xu Z, Chen C, Shen X, Zhao J, et al. A Positive Feedback Loop of β -Catenin/CCR2 Axis Promotes Regorafenib Resistance in Colorectal Cancer. Cell Death Dis (2019) 10:1–12. doi: 10.1038/s41419-019-1906-5
24. Aplan PD, Lombardi DP, Kirsch IR. Structural Characterization of SIL, a Gene Frequently Disrupted in T-Cell Acute Lymphoblastic Leukemia. Mol Cell Biol (1991) 11:5462–9. doi: 10.1128/mcb.11.11.5462
25. Marthiens V, Basto R. Microcephaly: STIL(L) a Tale of Too Many Centrosomes. Curr Biol (2014) 24:R162–4. doi: 10.1016/J.CUB.2013.12.054
26. Vulprecht J, David A, Tibelius A, Castiel A, Konotop G, Liu F, et al. STIL is Required for Centriole Duplication in Human Cells. J Cell Sci (2012) 125:1353–62. doi: 10.1242/jcs.104109
27. Hu T, Li Z, Gao C-Y, Cho CH. Mechanisms of Drug Resistance in Colon Cancer and its Therapeutic Strategies. World J Gastroenterol (2016) 22:6876–89. doi: 10.3748/wjg.v22.i30.6876
28. Du L, Wang H, He L, Zhang J, Ni B, Wang X, et al. CD44 is of Functional Importance for Colorectal Cancer Stem Cells. Clin Cancer Res (2008) 14:6751–60. doi: 10.1158/1078-0432.CCR-08-1034
29. Li Z. CD133: A Stem Cell Biomarker and Beyond. Exp Hematol Oncol (2013) 2:17. doi: 10.1186/2162-3619-2-17
30. Wang K, Xu J, Zhang J, Huang J. Prognostic Role of CD133 Expression in Colorectal Cancer: A Meta-Analysis. BMC Cancer (2012) 12:573. doi: 10.1186/1471-2407-12-573
31. Moitra K, Lou H, Dean M. Multidrug Efflux Pumps and Cancer Stem Cells: Insights Into Multidrug Resistance and Therapeutic Development. Clin Pharmacol Ther (2011) 89:491–502. doi: 10.1038/clpt.2011.14
32. Haraguchi N, Utsunomiya T, Inoue H, Tanaka F, Mimori K, Barnard GF, et al. Characterization of a Side Population of Cancer Cells From Human Gastrointestinal System. Stem Cells (2006) 24:506–13. doi: 10.1634/stemcells.2005-0282
33. Chen Y, Bieber MM, Teng NNH. Hedgehog Signaling Regulates Drug Sensitivity by Targeting ABC Transporters ABCB1 and ABCG2 in Epithelial Ovarian Cancer. Mol Carcinog (2013) 53:n/a–a. doi: 10.1002/mc.22015
34. Zhou S, Schuetz JD, Bunting KD, Colapietro A-M, Sampath J, Morris JJ, et al. The ABC Transporter Bcrp1/ABCG2 is Expressed in a Wide Variety of Stem Cells and is a Molecular Determinant of the Side-Population Phenotype. Nat Med (2001) 7:1028–34. doi: 10.1038/nm0901-1028
35. Popat S, Matakidou A, Houlston RS. Thymidylate Synthase Expression and Prognosis in Colorectal Cancer: A Systematic Review and Meta-Analysis. J Clin Oncol (2004) 22:529–36. doi: 10.1200/JCO.2004.05.064
36. Lin J, Feng J, Yang H, Yan Z, Li Q, Wei L, et al. Scutellaria Barbata D. Don Inhibits 5-Fluorouracil Resistance in Colorectal Cancer by Regulating PI3K/AKT Pathway. Oncol Rep (2017) 38:2293–300. doi: 10.3892/or.2017.5892
37. Chu E, Allegra CJ. Regulation of Thymidylate Synthase in Human Colon Cancer Cells Treated With 5-Fluorouracil and Interferon-Gamma. In: Advances in Experimental Medicine and Biology. New York LLC: Springer (1993). p. 143–51. doi: 10.1007/978-1-4615-2488-5_16
38. Izraeli S, Lowe LA, Bertness VL, Campaner S, Hahn H, Kirsch IR, et al. Genetic Evidence Thatsil is Required for the Sonic Hedgehog Response Pathway. genesis (2001) 31:72–7. doi: 10.1002/gene.10004
39. Novellasdemunt L, Antas P, Li VSW. Targeting Wnt Signaling in Colorectal Cancer. A Review in the Theme: Cell Signaling: Proteins, Pathways and Mechanisms. Am J Physiol Physiol (2015) 309:C511–21. doi: 10.1152/ajpcell.00117.2015
40. Liu C, Kato Y, Zhang Z, Do VM, Yankner BA, He X. β-Trcp Couples β-Catenin Phosphorylation-Degradation and Regulates Xenopus Axis Formation. Proc Natl Acad Sci USA (1999) 96:6273–8. doi: 10.1073/pnas.96.11.6273
Keywords: colorectal cancer, STIL, hedgehog signaling, cancer stem cell, drug resistance, β-catenin, prognosis STIL Oncogene in CRC
Citation: Pradhan T, Kumar V, Surya H E, Krishna R, John S, Jissa VT, Anjana S, Chandramohan K and Nair SA (2021) STIL Endows Oncogenic and Stem-Like Attributes to Colorectal Cancer Plausibly by Shh and Wnt Signaling. Front. Oncol. 11:581671. doi: 10.3389/fonc.2021.581671
Received: 09 July 2020; Accepted: 21 July 2021;
Published: 12 August 2021.
Edited by:
Shihori Tanabe, National Institute of Health Sciences (NIHS), JapanReviewed by:
Fuming Li, University of Pennsylvania, United StatesClaudio Cantù, Linköping University, Sweden
Copyright © 2021 Pradhan, Kumar, Surya H, Krishna, John, Jissa, Anjana, Chandramohan and Nair. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: S. Asha Nair, c2FzaGFAcmdjYi5yZXMuaW4=
†These authors have contributed equally to this work