 Zhiyuan Shi
Zhiyuan Shi Lei Zhang2
Lei Zhang2
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 01 April 2021
Sec. Cancer Metabolism
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.579286
The challenge of eradicating cancer is that cancer cells possess diverse mechanisms to protect themselves from clinical strategies. Recently, ferroptosis has been shown to exhibit appreciable anti-tumor activity that could be harnessed for cancer therapy in the future. Ferroptosis is an iron-dependent form of regulated cell death that is characterized by the oxidization of polyunsaturated fatty acids (PUFAs) and accumulation of lipid peroxides. Ferroptosis has been closely correlated with numerous biological processes, such as amino acid metabolism, glutathione metabolism, iron metabolism, and lipid metabolism, as well as key regulators including GPX4, FSP1, NRF2, and p53. Although ferroptosis could be involved in killing various cancer cells, multiple aspects of this phenomenon remain unresolved. In this review, we summarize the biochemistry and biology of ferroptosis in diverse cancers and discuss the potential mechanisms of ferroptosis, which might pave the way for guiding cancer therapeutics.
Cancer has been widely recognized as the second leading cause of death worldwide (1). According to recent statistics on cancers in 185 countries, there were 19.3 million new cancer cases (18.1 million excluding nonmelanoma skin cancer) and 10 million cancer deaths (9.9 million excluding nonmelanoma skin cancer) worldwide in 2020 (2). Although several therapeutic strategies such as surgery, radiotherapy, chemotherapy, ablation, and organ transplantation have been utilized to fight against cancer, many malignant cancers still do not show satisfactory results after treatment. The two major causes of cancer-related death are metastasis and recurrence of tumors; therefore, pinpointing mechanistic details of the pathways by which cancer cell death occurs is crucial for identifying treatment strategies and guiding clinical therapy.
Ferroptosis is defined as an iron-dependent form of nonapoptotic cell death (3), and is characterized by oxidization of polyunsaturated fatty acids (PUFAs) and accumulation of lipid peroxides. Cells undergoing ferroptosis cannot be rescued by chemical or genetic inhibitors of apoptosis or necroptosis, which indicates that ferroptosis is a distinct form of cell death (4). Ferroptosis has been investigated in many human diseases, such as neurodegenerative diseases [including Parkinson’s disease (5, 6), Alzheimer’s disease (7, 8), Friedreich’s ataxia (9), and Huntington’s disease (10)], organ injury [including hepatic damage (11, 12), brain injury (13–15), spinal cord injury (16, 17), and kidney injury (18–20)], organ fibrosis (21–23), cardiovascular diseases (24, 25), and gynecological diseases (26–28). Recently, an increasing number of studies on ferroptosis have been conducted in diverse cancers as ferroptosis is gradually being recognized as a potential form to eliminate cancer cells. In this review, we discuss the mechanism of ferroptosis and its molecular regulation in various cancers.
In 2012, the concept of ferroptosis was first described by Scott J. Dixon (a member of the Brent R. Stockwell group) and his collaborators who described the characteristics of ferroptosis (3). Ferroptosis, a novel type of regulated cell death (RCD), is a unique form of intracellular iron-dependent peroxidation of PUFA-containing phospholipids (PLs), and is morphologically, biochemically, and genetically distinct from other forms of RCD including apoptosis, autophagy, and necroptosis. Cells undergoing ferroptosis show unique hallmarks including rupture of cellular membranes, smaller mitochondria with increased mitochondrial membrane density, reduced/vanished mitochondria cristae, rupture of outer mitochondrial membranes, and a normal nucleus (29).
In 1955, Eagle et al. first found that human uterine carcinoma HeLa cells cultured without cystine exhibited a unique microscopic morphology that was different than that resulting from deprivation of other amino acids (30). They also found that cells cultured in cystine-free medium failed to grow but could be restored by supplementing them with glutathione (GSH) (31, 32). In 1977, Bannai et al. showed that cystine starvation of human lung fibroblasts resulted in rapid reduction of GSH and subsequent cell death; however, cell death could be rescued by the addition of the lipophilic antioxidant α-tocopherol (a component of vitamin E) (33). These results implied that cystine could sustain the intracellular level of GSH and that there might be an accumulation of reactive oxygen species (ROS) that could be prevented by lipophilic antioxidants.
In 1965, two separate research teams both identified lipid peroxidation as a prime cause of cellular damage in rat liver (34, 35). In the 1980s, lipid peroxidation was considered to be one of the main forms of oxidative damage via the destruction of unsaturated lipid components of cell membranes and lipoproteins in some pathologies (36, 37). Nonetheless, these discoveries were considered as mechanisms of cellular damage at that time.
Brent R. Stockwell and members tried to screen small molecules that could selectively kill cells overexpressing the oncogenic mutant HRAS. In 2003, they identified a novel compound that they named “erastin”, and explored the effect of erastin in engineering tumor cells. However, they found that no characteristics of apoptosis occurred, such as caspase activation, cleavage of caspase substrates, annexin V staining, and morphological changes in the nucleus (38). In 2007, they further reported that erastin induced the formation of oxidative species and subsequent death through an oxidative nonapoptotic mechanism, and that the cell death induced by erastin could be suppressed by α-tocopherol (39). In 2008, they reported yet another small compound, Ras selective lethal 3 (RSL3), which induced a similar iron-dependent non-apoptotic cell death in oncogenic RAS-harboring cancer cells, which could also be suppressed by both α-tocopherol and desferrioxamine mesylate (DFOM) (40). In 2011, the authors distinguished erastin- and RSL3-induced cell death from the mechanism of action of other cell death inducers (41). In 2012, they named this phenomenon of erastin-induced iron-dependent cell death as ferroptosis (3).
As an increasing number of groups actively investigate ferroptosis, numerous mechanisms have been found to be involved in ferroptosis, such as amino acid metabolism, GSH metabolism, iron metabolism, and lipid metabolism (Figure 1 and Table 1).
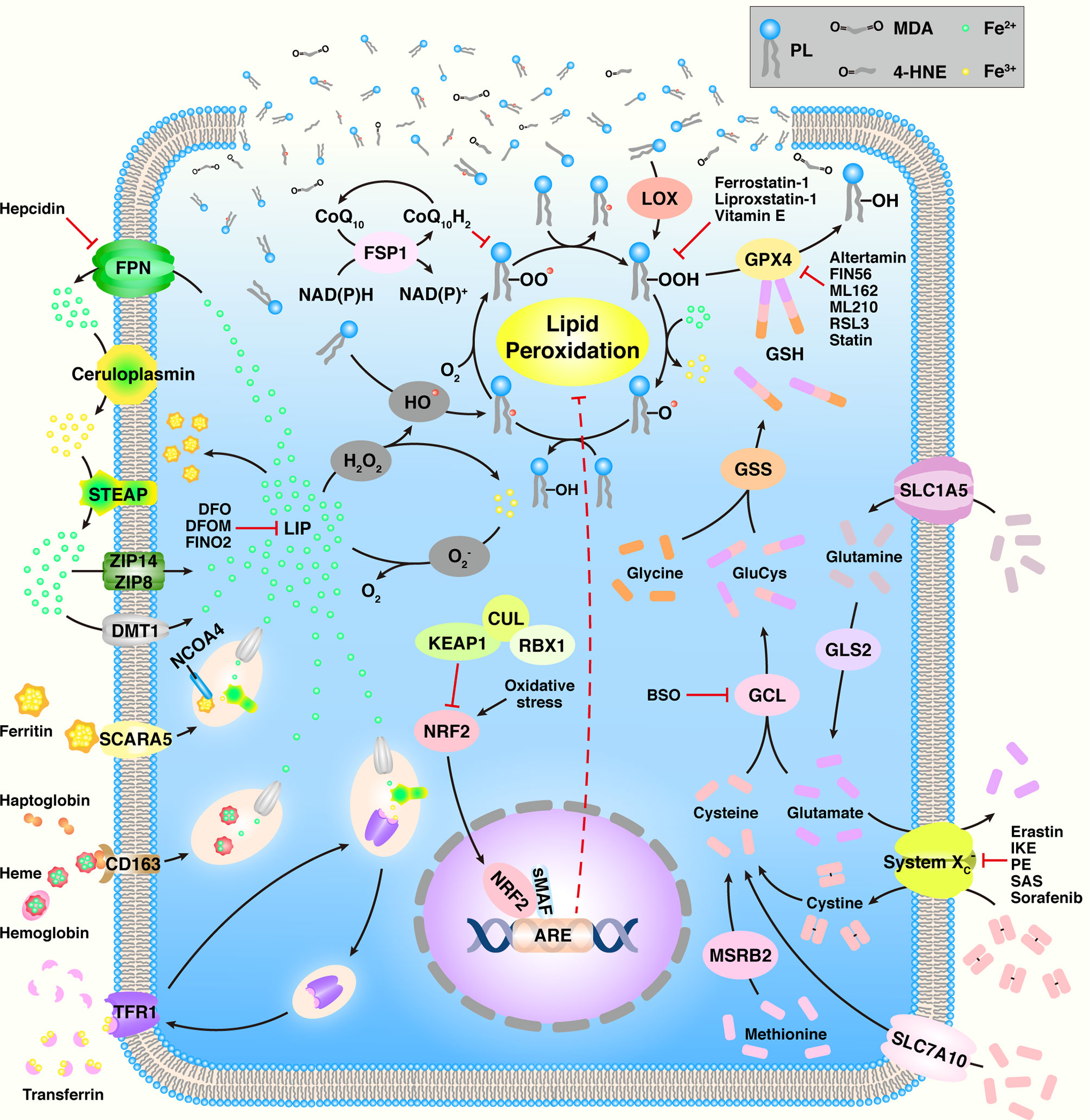
Figure 1 An overview of ferroptosis. ARE, antioxidant response element; BSO, buthionine sulfoximine; CoQ10, coenzyme Q10; CUL3, cullin3; DFO, deferoxamine; DFOM, desferrioxamine mesylate; DMT1, divalent metal transporter 1; FPN, ferroportin; FSP1, ferroptosis suppressor protein 1; GCL, glutamate-cysteine ligase; GLS2, glutaminase 2; GPX4, GSH peroxidase 4; GSH, glutathione; GSS, glutathione synthetase; 4-HNE, 4-hydroxynonenal; HO•, hydroxyl radical; IKE, imidazole ketone erastin; KEAP1, Kelch-like ECH-associated protein 1; LIP, labile iron pool; LOX, lipoxygenase; MDA, malondialdehyde; MSRB2, methionine-R-sulfide reductase B2; NCOA4, nuclear receptor coactivator 4; NRF2, nuclear factor erythroid 2-related factor 2; PE, piperazine erastin; PL, phospholipid; PL•, PL radical species; PLO•, PL alkoxyl radical; PLOH, PL alcohol; PLOO•, PL peroxyl radical; PLOOH, PL hydroperoxide; RBX1, ring box protein 1; RSL3, RAS synthetic lethal 3; SAS, sulfasalazine; SCARA5, scavenger receptor class A member 5; sMAF, small Maf; SLC1A5, solute carrier family member 1 member A5; STEAP, six-transmembrane epithelial antigen of prostate; TFR1, transferrin receptor 1; ZIP8, Zrt- and Irt-like protein 8.
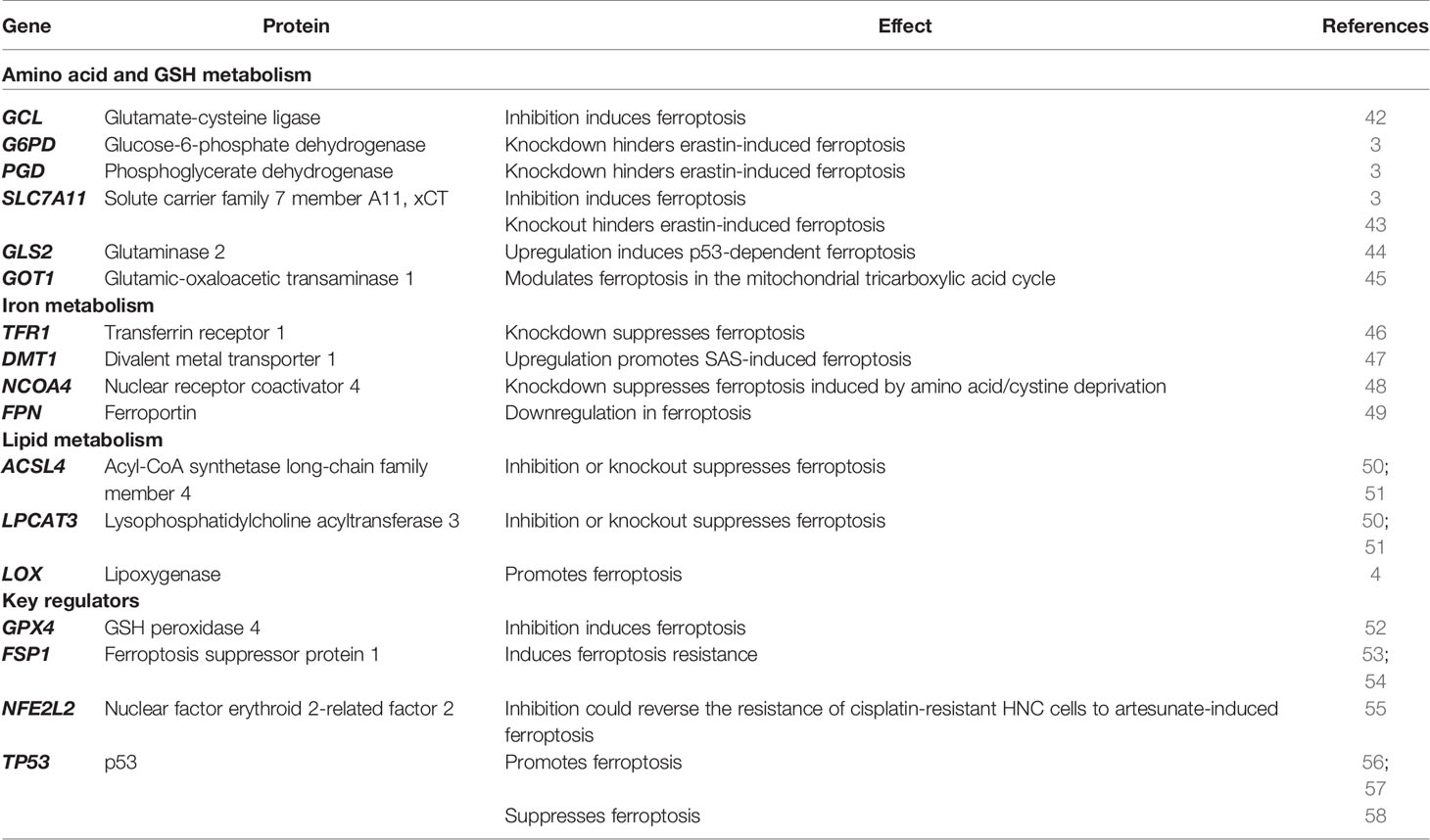
Table 1 Regulators of ferroptosis.
GSH functions as a cofactor of GSH peroxidase 4 (GPX4, discussed in detail below) in lipid metabolism; thus, the synthesis of GSH regulates ferroptosis. GSH is synthesized with three substrates, namely glutamate, cysteine, and glycine, in two steps. The initial and limiting step is the combination of glutamate and cysteine to form GluCys in the presence of glutamate-cysteine ligase (GCL, previously known as γ-glutamyl-cysteine synthetase) utilizing ATP. Then, GluCys combines with glycine to yield GSH, which is catalyzed by GSH synthetase (GSS) in the presence of ATP. Buthionine sulfoximine (BSO) is a small molecular inhibitor of GCL known to indirectly inhibit the enzymatic activity of GPX4 (42). The pentose phosphate pathway (PPP) generates NADPH, which is essential for preserving the cellular levels of GSH. Inhibition of the PPP and silencing of two PPP enzymes, glucose-6-phosphate dehydrogenase (G6PD) and phosphoglycerate dehydrogenase (PGD), hinders erastin-induced ferroptosis in human lung cancer cells (3).
The heterodimeric amino acid antiporter system located on the cell surface consists of a twelve-pass transmembrane transporter protein solute carrier family 7 member A11 (SLC7A11, also known as xCT) and a single-pass transmembrane regulator protein SLC3A2 (4F2hc), both of which are linked by a disulfide bridge (59). It is a Na+-independent cystine/glutamate antiporter that imports one molecule of extracellular cystine by exporting the equivalent molecule of intracellular glutamate, followed by the conversion of the cystine to cysteine by disulfide bond breakage. Therefore, system nhibition will result in cysteine depletion in cells. In this context, several suitable compounds and their derivatives have been reported by researchers. Erastin, an oncogenic RAS-selective lethal small molecule, is the first compound reported to trigger ferroptosis by suppressing system (3). Although the precise mechanism underlying erastin-mediated inhibition of SLC7A11-mediated cysteine import remains unclear, it is agreed that erastin most likely inhibits SLC7A11 directly (60). The erastin derivatives imidazole ketone erastin (IKE) and piperazine erastin (PE) have been shown to induce ferroptosis (42, 61). Sulfasalazine (SAS), a medicine for arthritis, shows low potential inhibition of system at high concentrations (62). Sorafenib, an inhibitor of oncogenic kinases and approved by the Food and Drug Administration (FDA) as an anticancer drug, induces ferroptosis in different cancer cell lines (63–65). A high extracellular concentration of glutamate could initiate ferroptosis by preventing cystine import (3). Cysteine could also be imported into cells through the SLC7A10 transporter (66). Some cells utilize the trans-sulphuration pathway to biosynthesize cysteine from methionine through methionine-R-sulfide reductase B2 (MSRB2), that bypasses the requirement for cystine import via system (66, 67). Pharmacological and genetic inhibition of SLC7A11 induces ferroptosis and significantly sensitizes cisplatin-resistant head and neck cancer (HNC) cells to chemotherapy in vitro and in vivo (68). Deletion of SLC7A11 induces tumor-selective ferroptosis and inhibits pancreatic ductal adenocarcinoma (PDAC) growth (43).
Glutamine, transported into cells through SLC1A5, is also essential for the induction of ferroptotic cell death. Although glutaminase 1 (GLS1) and GLS2 are structurally and enzymatically similar and can catalyze the conversion of glutamine into glutamate, only GLS2 is required for ferroptosis (46). Upregulation of GLS2, a transcriptional target of the tumor suppressor p53, could induce p53-dependent ferroptosis (44). The glutamic-oxaloacetic transaminase 1 (GOT1) synthesizes α-ketoglutarate from glutamate, and glutaminolysis is supposed to modulate ferroptosis in the mitochondrial tricarboxylic acid (TCA) cycle (45).
The first observation that iron participates in the process of ferroptosis originated from reports that iron chelators could prevent cell death induced by cystine deprivation (considered as ferroptosis now) in 1996 (69). The biological function of iron is mainly dependent on its ability to accept and donate electrons while switching among the ferrous bivalent (Fe2+), ferric trivalent (Fe3+), and ferryl tetravalent (Fe4+) states by catalyzing various biochemical reactions (70). At the cellular level, three forms of ROS, including superoxide , hydrogen peroxide (H2O2), and hydroxyl radical (HO•), are related to iron in redox reactions. and H2O2 are respectively known as single and double univalent reductions of molecular oxygen (O2) respectively, while the most chemically active ROS is HO•, which can cause nonspecific oxidation that destroys biological molecules such as lipids. In the Fenton reaction, Fe2+ is oxidized to Fe3+ by a reaction with H2O2, while the electron transfers to H2O2 to form HO• (71). Conversely, Fe3+ could be reduced back to Fe2+ through reaction with while loses an electron to form O2, which is known as the Haber-Weiss reaction (72).
Sufficient levels of free intracellular iron is a necessity for triggering ferroptosis. There are four following routes for cells to import iron (73). The most crucial route is mediated by transferrin and its partner, the transferrin receptor 1 (TFR1). Transferrin, a type of globulin mainly synthesized by the liver and released into the serum, has an excellent ability to chelate two Fe3+ ions. When the transferrin reaches the cell membrane, it identifies and binds to TFR1 and the transferrin-TFR1 complex is internalized via clathrin-mediated endocytosis, whereas apo-transferrin (iron-free transferrin) is not identified by TFR1 and internalized. Fe3+ is liberated from the transferrin-TFR1 complex at the low pH conditions of endocytic vesicles and reduced to Fe2+ by the six-transmembrane epithelial antigen of prostate (STEAP) family (73, 74). Fe2+ is then released into the cytoplasm via the divalent metal transporter 1 (DMT1) and participates in establishing the intracellular labile iron pool (LIP) (75). Meanwhile, apo-transferrin and TFR1 remain bound until the complex is recycled back to the plasma membrane, followed by apo-transferrin releases (76). The second route is directly assimilating the free iron unbound by transferrin; however, each iron transporter only transports Fe2+. Therefore, Fe3+ is first reduced to Fe2+ by several types of ferrireductases (e.g., STEAP, cytochrome B reductase 1, and ferric chelate reductase 1), and the Fe2+ is directly transported into cells by cellular membrane transporters including DMT1, Zrt- and Irt-like protein 8 (ZIP8), and ZIP14 (73). The third pathway involves the uptake of hemoglobin-containing porphyrin-bound Fe2+, especially in macrophages (77). The fourth mechanism involves the assimilation of the iron-storage protein ferritin by its cellular membrane receptors such as the scavenger receptor class A member 5 (SCARA5) found in embryos and in the kidney (78).
After iron uptake, the intracellular iron can be utilized, stored, and exported. In terms of utilization, mitoferrin 1 and 2 specially transport iron into the mitochondria to assist with cellular respiration and synthesis of Fe-S clusters and heme (79). There are two forms of intracellular storage, LIP and ferritin. LIP causes active oxidative stress-related toxicity and is responsible for regulating cellular iron homeostasis through the iron regulatory protein-iron responsive element system. Ferritin is an iron-sequestering protein that contains up to 4500 iron atoms and possesses multiple functions in iron delivery, cell proliferation, angiogenesis, and immunosuppression (80). Ferritin can also be degraded to release free iron via the nuclear receptor coactivator 4 (NCOA4), a process termed as “ferritinophagy” (81). In terms of iron export, ferroportin (FPN, i.e., SLC40A1), once known as the only iron efflux pump that cooperates with ceruloplasmin or hephaestin, is mainly responsible for transporting Fe2+ out of cells (82). Ceruloplasmin suppresses ferroptosis by regulating iron homeostasis in HepG2 and Hep3B cells, and depletion of ceruloplasmin results in the accumulation of intracellular Fe2+ and lipid ROS and promotes erastin- and RSL3-induced ferroptotic cell death (83). In addition, prominin 2 promotes the formation of ferritin-containing multivesicular bodies and exosomes that transport iron out of the cell to facilitate ferroptosis resistance in breast cancer (84).
Treating cells without transferrin or with TFR RNAi could not lead to significant ferroptotic cell death, indicating that an external iron source is required for ferroptosis (46). STEAP1 and STEAP2 are highly expressed in various human cancer types, such as the colon, breast, cervix, prostate, pancreas, bladder, ovary, testis, and Ewing sarcoma (85–87). STEAP3 is overexpressed in malignant gliomas and induces cancer epithelial–mesenchymal transition (EMT) (88). STEAP4 is activated under hypoxic conditions, leading to mitochondrial iron imbalance and enhanced ROS production (89). FPN is dramatically suppressed in many cancer types (90), indicating that there might be abundant iron in cancer cells. Reduced FPN levels in triple-negative breast cancer (TNBC) cells stimulate proliferation and EMT (91). Hepcidin, synthesized by tumors or the liver, facilitates FPN degradation and contributes to cancer proliferation and progression (92). Proteins involved in raising the levels of intracellular iron (TFR1, DMT1, and hepcidin) are extensively upregulated in tumor cells, whereas proteins suppressing intracellular iron accumulation (FPN and hephaestin) are downregulated (93).
The fact that ferroptosis is driven by peroxidation of PUFAs, and not monounsaturated fatty acids (MUFAs) or deuterated PUFAs, was confirmed by Brent R. Stockwell and coworkers in 2016 (4). As is known to all, PUFAs, as well as essential fatty acids, could not be physiologically synthesized and must be obtained from food. PUFA is defined as a fatty acid containing at least two -CH=CH- groups, such as linoleic acid, linolenic acid, and arachidonic acid (AA). PLs, the basic components of the cellular membrane, are classified as glycerophospholipids and sphingomyelin. The predominant fatty acid moiety in sphingomyelin is saturated fatty acids or MUFAs, whereas glycerophospholipids contain an esterified C16 or C18 saturated fatty acid at C-1, an esterified C18 to C20 unsaturated fatty acid (e.g., AA) at C-2, and a phosphatidic ester group at C-3 of the glycerol backbone. The incorporation of de novo synthesized fatty acids into PLs requires acyl-CoA synthetases (ACSs) that convert long-chain fatty acids to acyl-CoA and lysophospholipid acyltransferases (LPLATs) that, in turn, catalyze the subsequent reacylation step to form PLs (94). For instance, lipidomic studies suggest that phosphatidylethanolamines containing AA or adrenic acid (AdA), are key PLs that undergo oxidation and drive the occurrence of ferroptosis (50, 51). ACS long-chain family member 4 (ACSL4) preferentially converts AA to acylated AA, and lysophosphatidylcholine acyltransferase 3 (LPCAT3) subsequently catalyzes the incorporation of acylated AA into the PL (51, 95).
There are two mechanisms of lipid peroxidation, the non-enzymatic free radical chain reaction and the enzymatic process. Compared with the tightly controlled enzymatic lipid peroxidation, the free radical chain reaction involving Fenton chemistry is poorly controlled. There are three types of lipid oxidation enzymes, namely cyclooxygenase (COX), cytochrome P450 (CYP), and lipoxygenase (LOX). COXs synthesize lipid endoperoxides and are partially responsible for the peroxidation of linoleic acid, while CYPs synthesize epoxyeicosatrienoic acids and LOXs contribute predominantly to the synthesis of lipid hydroperoxides.
Nonenzymatic lipid peroxidation involves the oxidation of PUFAs, initiated by ROS. In unsaturated acyl chains, the allylic hydrogen atoms, i.e., the hydrogen atoms on methylene groups adjacent to double bonds, exhibit low carbon-hydrogen (C-H) bond energies, while the hydrogen atoms located on methylene between two double bonds (bis-allylic hydrogen atoms) have even lower C-H bond energies, thus bis-allylic hydrogen atoms can be abstracted by ROS, forming PL radical species (PL•) with the radical centered on the allylic carbon atom (96). Therefore, PUFAs are highly susceptible to oxidative damage due to the existence of bis-allylic hydrogen atoms that saturated fatty acids and MUFAs do not possess. According to previous reports (96, 97), in the presence of HO• derived from the Fenton reaction, the PUFA of PL donates a hydrogen atom to HO• and becomes a carbon-centered PL• that further reacts with intracellular molecular O2 to form a PL peroxyl radical (PLOO•). Next, with the participation of PUFA moiety from another PL, PLOO• abstracts a hydrogen atom and subsequently converts to PL hydroperoxide (PLOOH) accompanied with a new PL•. PLOOH is cleaved in the presence of Fe2+ to form the PL alkoxyl radical (PLO•) that reacts with PUFA of another PL to form PL alcohol (PLOH) and a new PL•, followed by another lipid radical chain reaction.
The enzymatic pathway mainly involves iron-containing LOXs, which can catalyze the site-specific oxidation of PUFAs in a controlled manner. LOXs are classified based on their regioselectivity (e.g., the number of carbon atoms subjected to deoxygenation) and stereoselectivity (“S” or “R”) (98). There are six LOX isoforms discovered in humans, namely 5-LOX, 12-LOX, 12R-LOX, 15-LOX-1, 15-LOX-2, and e-LOX3, ranging from 662 to 711 amino acids and sharing 44% of their sequence identity (99). Encoded by the ALOX5 gene, 5-LOX could oxide AA at C-5 to form 5-hydroperoxyeicosatetraenoic acid (5-HpETE), which is subsequently converted to leukotriene A4 (LTA4) by 5-LOX (100, 101). Once 5-LOX is activated, it migrates to the nuclear membrane where it associates with two additional proteins: the 5-LOX activating protein (FLAP) and cytosolic phospholipase A2 (cPLA2) (102). cPLA2 is responsible for cleaving AA from PLs to increase substrate availability for 5-LOX. Although the exact function of FLAP is still unclear, pharmacological inhibition of FLAP function prevents oxidation of endogenous AA by 5-LOX, which demonstrates the necessary role of FLAP in lipid peroxidation (103). Compared with 5-LOX, which oxidizes hydrolyzed arachidonoyl groups of PLs by cPLA2, 12-LOXs and 15-LOXs, encoded by ALOX12/ALOX12B and ALOX15/ALOX15B, respectively, directly oxidize AA to synthesize 12-HpETE and 15-HpETE, which are rapidly reduced to their corresponding hydroxides and hepoxilins (104). Meanwhile, 12-LOXs and 15-LOXs can also oxidize linoleic acid to generate 13-hydroperoxyoctadecadienoic acid (13-HpODE), which is subsequently reduced to 13-HODE (101). Docosahexanoic acid (DHA) is also a substrate for 15-LOX-1, which metabolizes the conversion of ω-3 fatty acid to 17-hydroperoxydocosahexaenoic acid (17-HpDHA), which is rapidly transformed into resolvins and protectins (105).
Lipid peroxidation of membranes substantially alters the physical properties of lipid bilayers in terms of disrupted ion gradients, decreased membrane fluidity, slower lateral diffusion, and increased membrane permeability (106). There exist two mechanisms for eliminating lipid peroxides from cells without the generation of new radicals. GPXs, especially GPX4, are crucial regulators for reducing lipid peroxides to corresponding alcohols by utilizing GSH as a cofactor (107), thereby limiting the transition metal-dependent formation of toxic radicals (e.g., PLO•). Another mode of degradation involves the conversion of lipid peroxides to aldehydes such as malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE), which should undergo multiple oxidization steps. However, there is still no definitive consensus about the metabolic route that predominantly contributes to the degradation of lipid peroxides among multiple biosynthesis routes (101). MDA could react with primary amines on proteins or DNA to form crosslinks. Moreover, excessive MDA generation within cells is associated with major human diseases including cancer (108). Decomposition of AAs and longer PUFAs generates 4-HNE, which contains three functional groups: (i) an electrophilic C=C double bond that is a Michael acceptor and that forms covalent adducts with nucleophilic amino acids; (ii) an aldehyde that can form Schiff base adducts with primary amines; and (iii) a hydroxyl group that can be oxidized to an electrophilic ketone (109). The electrophile 4-HNE has been widely studied as a signaling molecule that stimulates the cell cycle and cellular proliferation, along with investigation into its cytotoxic nature known to inhibit gene expression (110).
According to phylogeny, the GPX family is divided into three groups: GPX1 and GPX2; GPX3, GPX5, and GPX6; GPX4, GPX7, and GPX8, and could also be classified as selenocysteine-containing GPXs and cysteine-containing GPXs depending on whether the active center contains Sec or Cys (111). GPX4, i.e., PL hydroperoxide GPX, is the second selenoperoxidase to be isolated and is regarded as a peroxidation-inhibiting protein after the identification of GPX1 (112). GPX4 is the only GPX for which PL hydroperoxides in membranes and protein-thiol groups can act as oxidizing and reducing substrates, respectively, under conditions of GSH deprivation (113). It was first isolated and purified from pig liver in 1982, and was identified to confer protection to liposomes and biomembranes from iron-catalyzed lipid peroxidation (114). The human GPX4 was isolated from human liver in 1994 (115). GPX4, containing 170 amino acids, has a typical thioredoxin motif consisting of four α-helices that are localized at the protein surface and seven β-strands, five of which form a central β-sheet (116). The catalytic triad, consisting of selenocysteine (U46), glutamine (Q81), and tryptophan (W136) residues, is localized at the protein surface, and mutations in any of these residues could lead to the inactivation of GPX4 (117). For instance, replacement of selenocysteine with cysteine could diminish the activity of GPX4 by 90% and strengthen its sensitivity to redox stress (118). GPX4 reduces lipid peroxides to the corresponding alcohols via oxidation of its active site selenol (Se-H) to selenenic acid (Se-OH), which is then reduced by two equivalents of GSH to the active Se-H (101).
RSL3 has four diastereomers, but only 1S,3R-RSL3 can induce ferroptosis by suppressing the enzymatic activity of GPX4 via covalent modifications without affecting intracellular GSH levels (42, 119). Conversely, overexpression of GPX4 could prevent RSL3-induced ferroptotic cell death (42). RSL3 induces ferroptosis in colorectal cancer cells due to decreased expression of GPX4 and both increased levels of ROS and cellular LIP (52). In addition to RSL3, ML162, ML210, and altretamine (an FDA-approved anticancer agent) can also induce ferroptosis by suppressing GPX4 (120). Inducible GPX4 inactivation could also lead to 12/15-LOX-derived lipid peroxidation as a specific downstream event, which indicates that GPX4 could prevent cell death by inhibiting 12/15-LOX (113). FIN56, derived from CIL56, exhibits greater potency and better oncogenic RAS selectivity than CIL56 (121). FIN56 induces ferroptosis by degrading GPX4 protein, instead of downregulating GPX4 mRNA transcription and subsequent synthesis of GPX4 protein, and simultaneously causes depletion of mevalonate-derived coenzyme Q10 (CoQ10), which is an electron carrier in the mitochondrial respiratory chain and an endogenous antioxidant. FINO2, an endoperoxide-containing 1,2-dioxolane, can initiate ferroptosis selectively in engineered cancer cells, such as renal cancer cell Caki-1 and fibrosarcoma cell HT-1080 (122). FINO2 does not inhibit the antiporter system ; instead, it directly represses the enzymatic activity of GPX4, or depletes GPX4 and CoQ10. However, it can directly oxidize iron and indirectly suppresses the enzymatic function of GPX4, ultimately causing widespread lipid peroxidation. The side effects of statin treatment could be attributed to reduced levels of GPX4 in some tissues (123), while the decreased incidence of several cancers on statin consumption (124) and the loss of GPX4 detected in some cancer cells with statin treatment could be ascribed to repression of GPX4 synthesis (125). Further, statin treatment has also been shown to deplete CoQ10 (126).
Ferroptosis suppressor protein 1 (FSP1), previously known as apoptosis-inducing factor mitochondrial-associated 2 (AIFM2), was recently identified as a ferroptosis resistance factor (54). Although GPX4 is essential for cancer cells to escape from ferroptosis, inhibition of GPX4 fails to trigger ferroptosis in some cancer cells regardless of ACSL4 expression, suggesting the existence of additional mechanisms of ferroptosis resistance. The anti-ferroptotic function of FSP1 is independent of cellular GSH level, GPX4 activity, ACSL4 expression, and oxidizable fatty acid content, showing that FSP1 does not interfere with canonical ferroptosis mechanisms. Moreover, p53 status does not affect FSP1 expression. FSP1 expression is positively correlated with ferroptosis resistance in many cancer cell lines (127). FSP1 can prevent PL peroxidation and its suppression has been shown to be enhanced in the presence of both CoQ10 and α-tocopherol (54). In the presence of FSP1, CoQ10 was reduced to CoQ10-H2 using NADPH to inhibit the propagation of PL peroxidation. Inhibition of FSP1 can robustly sensitize cells to RSL3-induced ferroptosis (53, 54).
Nuclear factor erythroid 2-related factor 2 (NRF2) encoded by NFE2L2 is a transcription factor that consists of 605 amino acids and contains seven conserved homology domains termed as Neh1-Neh7 (128, 129). Neh2 is the major regulatory domain that has two binding motifs known as DLG and ETGE, both of which help to regulate NRF2 stability by interacting with the Kelch domains of the E3 ubiquitin ligase Kelch-like ECH-associated protein 1 (KEAP1), which is the most notable negative regulator of NRF2 and contains a number of cysteine residues (including C151, C273, and C288) that act as electrophilic attack centers (130). Under physiological conditions, NRF2 is maintained at low levels in normal tissues because of KEAP1-dependent ubiquitination (130). KEAP1 acts as an adaptor for NRF2 binding to the KEAP1-cullin 3-ring box protein 1 (KEAP1-CUL3-RBX1) E3 ubiquitin ligase complex, which targets NRF2 for rapid proteasomal degradation (129). During increased oxidative stress, or if KEAP1, CUL3, or NRF2 are mutated, NRF2 can no longer be ubiquitylated and degraded, and the newly translated NRF2 transfers to the nucleus and activates transcription of anti-oxidant response element (ARE)-containing genes, many of which play a key role in preventing the initiation of ferroptosis (131). RSL3 or ML-162 treatment increases the expression of p62 and NRF2 in chemo-resistant HN3R and HN3-rslR cells, inactivates KEAP1, and increases the expression of the phospho-protein kinase R-like endoplasmic reticulum kinase (p-PERK)-activating transcription factor 4 (ATF4)-sertrin 2 (SESN2) (132). NRF2 inhibition could reverse the resistance of cisplatin-resistant HNC cells to artesunate-induced ferroptosis (55). With regard to other Neh domains, Neh1 known as a DNA binding domain enhances NRF2 transcriptional activation (133). The Neh3, Neh4, and Neh5 domains are known as trans-activation domains of NRF2 (134–136). Neh6, a serine-rich domain, negatively modulates NRF2 stability (137), and Neh7 has been shown to interact with the nuclear receptor retinoid X receptor α (RXRα), which inhibits the transcription of NRF2 target genes (138).
The genetic products of NRF2 activation can be categorized functionally with respect to their involvement in three cellular processes consisting of iron/metal metabolism, intermediate metabolism, and GSH synthesis/metabolism. Resistant melanoma cells can efficiently activate NRF2 (which upregulates the early ferroptotic marker GSH-specific γ-glutamylcyclotransferase 1 (CHAC1) in an endoplasmic reticulum (ER) stress-independent manner) and aldo-keto reductase AKR1C1-3 (which degrades lipid peroxides generated by 12/15-LOX), which could cumulatively result in resistance to ferroptotic cell death (139). NRF2 targets many genes involved in iron/metal metabolism, such as the light chain and heavy chain of ferritin (FTL/FTH1), FPN, heme oxygenase-1 (HO-1), biliverdin reductase A and B (BLVRA/B), ferrochelatase (FECH), ATP-binding cassette sub-family B member 6 (ABCB6), and SLC48A1, which are all regulated by NRF2 (140–145). In terms of the relationship between NRF2 and intermediate metabolism, NRF2 targets are involved in lipid metabolism (i.e., peroxisome proliferator-activated receptor γ, PPARG), the reduction of aldehydes and ketones to their alcohol forms (i.e., aldo-keto reductases, AKR1C1–3, AKR1B1, and AKR1B10), and glucose metabolism/NAPDH regeneration (i.e., G6PD) (146–149). Considering GSH synthesis/metabolism, numerous processes are under the control of NRF2, such as GSH metabolism (i.e., GCL, GSS, and SLC7A11), redox (i.e., GSH-S-transferases pi 1 and α 1, GSTP1 and GSTA1; peroxiredoxin 1 and 6, PRDX1 and PRDX6; and thioredoxin reductase, TXNRD), and the reduction of lipid peroxides (i.e., GPX4) (150–152). As mentioned above, some of these progresses have been confirmed to be involved in ferroptosis.
The tumor suppressor gene TP53, regarded as the “guardian of the genome”, was first discovered in 1979 in a complex with the simian virus 40 large T antigen and has been widely studied in cancers (153). Many studies have suggested that p53 could act synergistically with established oncogenes to promote the conversion of normal tissues to tumors and lead to accelerated metastasis. The first evidence that p53 might act as a tumor suppressor was reported in 1984 (154). Subsequently, it was concluded that these previous experiments were performed not with wild-type p53, but with its mutated version (155). The mutant p53 is highly abundant in cancers, and it promotes tumorigenesis by disabling the function of the wild-type p53, as well as by “gain-of-function” processes such as the accumulation of p53 mutations, that augments the oncogenic capacity of the mutated p53 and delivers a stronger tumoral resistance against anti-cancer treatments (156, 157). Under normal conditions, the levels of p53 are low because of the E3 ubiquitin-protein ligase MDM2, which can target and degrade p53, while oncogene activation could prevent MDM2 binding to p53 and stimulate p53 acetylation (158). Although 42% of cases across 12 tumor types occur due to TP53 mutation, the rate varies widely across diverse types of cancers (159).
Apart from the effects of p53 on apoptosis, autophagy, and the cell cycle, it also regulates ferroptosis via transcriptional or post-transcriptional mechanisms. Intriguingly, p53 can enhance or inhibit ferroptosis via different pathways. As to pro-ferroptosis, p53 promotes ferroptosis of MCF7 and U2OS cells due to SLC7A11 repression without affecting the expression of other p53 target genes involved in the cell cycle and apoptosis (56). p53 targets spermidine/spermine N1-acetyltransferase 1 (SAT1), which correlates with the expression of 15-LOX, and promotes the expression of SAT1 to induce ferroptosis (57). Depletion of p53 prevents nuclear accumulation of dipeptidyl peptidase-4 (DPP4) and triggers membrane-associated DPP4-mediated lipid peroxidation by binding to NADPH oxidase 1 (NOX1), which culminates in the ferroptosis of colorectal cancer cells. On the other hand, p53 mediates expression of the tumor suppressor cyclin-dependent kinase inhibitor 1A (CDKN1A/p21), which is a key mediator of p53-dependent cell cycle arrest after DNA damage, delays the onset of ferroptosis in response to cystine deprivation in cancer cells, and increases p53 expression by using the MDM2 inhibitor nutlin-3 that blocks erastin-induced ferroptosis in HT-1080 cells (58).
Here, different cancer groups are discussed in the context of ferroptosis, and the numerous inducers and inhibitors of ferroptosis, including drugs, genes, and RNA are listed in Table 2.
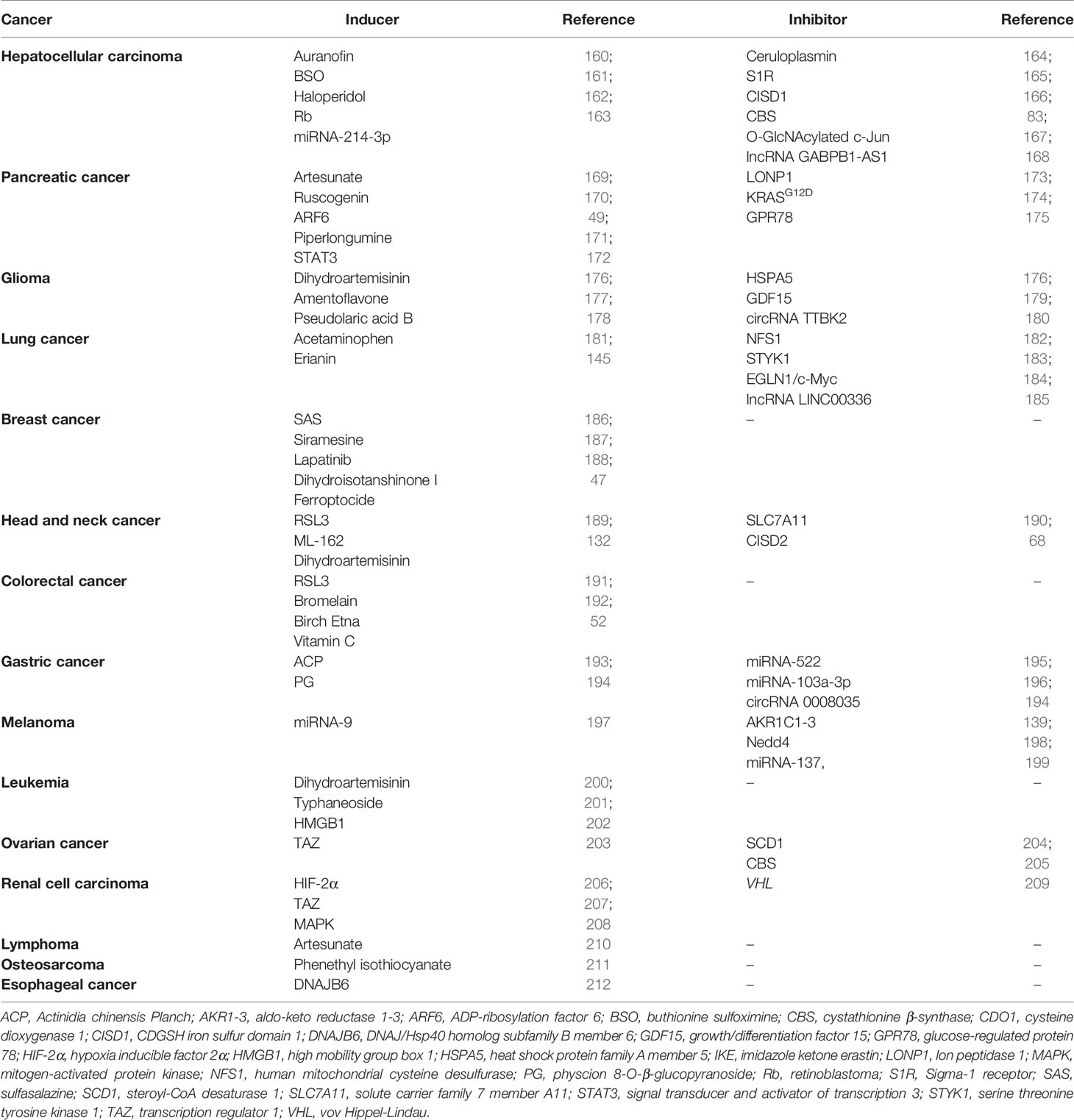
Table 2 Inducers and inhibitors of ferroptosis in different cancers.
Many clinical drugs have the ability to induce ferroptosis in cancer cells, which means that researchers need to comprehensively explore the potential mechanisms of these drugs. Artesunate, an anti-malarial drug, can specifically induce ferroptosis in pancreatic cancer cells without affecting pancreatic ductal epithelial cells (169). Artesunate also activates the ATF4-CHOP-CHAC1 cascade in DAUDI and CA-46 cells, and CHAC1 enhances artesunate-induced ferroptosis in Burkitt’s lymphoma cells (210). Dihydroartemisinin, a semisynthetic derivative of artemisinin, increases ROS levels in a dose-dependent manner and decreases the levels of both GPX4 and RAS in HEP-2 and CNE-1 cells (189). Dihydroartemisinin can also regulate the activity of the AMP-activated protein kinase (AMPK)/mTOR/p70S6k signaling pathway, thereby accelerating ferritin degradation, increasing LIP, promoting cellular ROS accumulation, and eventually triggering ferroptosis in acute myeloid leukemia cells (200). Several anti-inflammatory drugs have shown the potential to induce ferroptosis. Combinatorial treatment of auranofin and BSO can downregulate GPX4 and accumulate NRF2 and HO-1 (162). Ibuprofen induces ferroptosis in glioblastoma cells by downregulating the NRF2 signaling pathway (213). Acetaminophen can strengthen the sensitivity of erastin-induced ferroptosis by modulating the NRF2/HO-1 signaling pathway in non-small-cell lung cancer (NSCLC) (145). SAS can induce ferroptosis in breast cancer cells by upregulating TFR1 and DMT1, especially in cells with low expression of estrogen receptors (47). The tranquilizer haloperidol promotes ferroptosis by accelerating iron accumulation, lipid peroxidation, and GSH depletion under erastin or sorafenib treatment in HepG2 and Huh-7 cells (160). The lysosome disruptor siramesine and the tyrosine kinase inhibitor lapatinib synergistically induce ferroptosis by increasing the expression of transferrin and decreasing the expression of FPN (188).
Many extracts from plants and herbs also exhibit anti-tumor effects by inducing ferroptosis. Ruscogenin can induce ferroptosis by improving ROS generation and trigger intracellular iron accumulation by upregulating transferrin and downregulating FPN (49). Piperlongumine can also induce ferroptosis in pancreatic cancer cells (171). Amentoflavone represses the expression of FTH by autophagy via activation of the AMPK/mTOR/p70S6K signaling pathway to trigger in vitro and in vivo ferroptosis in an autophagy-dependent manner (177). Typhaneoside, a major flavonoid found in the extract of Pollen Typhae, promotes the activation of the AMPK signaling pathway to contribute to ferritin degradation, ROS accumulation, and ferroptosis in Kas-1, HL-60, and NB4 cells (202). Pseudolaric acid B isolated from cortex pseudolaricis triggers ferroptosis in vivo and in vitro by upregulating TFR1, activating NOX4, and inhibiting SLC7A11 (178). Actinidia chinensis Planch (ACP) has been shown to increase the accumulation of ROS by inhibiting GPX4 and SLC7A11 to induce ferroptosis in HGC-27 cells (193). Dihydroisotanshinone I, a pure compound present in danshen, can induce ferroptosis by downregulating the expression of GPX4 both in vitro and in vivo (186). The extract of Betula etnensis Raf. (Birch Etna) promotes an oxidative cellular microenvironment resulting in HO-1-mediated ferroptosis of CaCo-2 cells (191). Erianin, a natural product isolated from Dendrobium chrysotoxum Lindl, induces ferroptotic cell death in lung cancer cells via Ca2+/calmodulin signaling (181).
Some new compounds are still being found by researchers. The novel compound ferroptocide, identified as an inhibitor of thioredoxin, could rapidly and robustly induce ferroptotic cell death and positively modulate the immune system in a breast cancer model (187). Phenethyl isothiocyanate, present in cruciferous vegetables, induces ferroptosis, autophagy, and apoptosis in K7M2 cells by activating the ROS-related mitogen-activated protein kinase (MAPK) signaling pathway (211).
Overexpression of some genes and proteins can promote ferroptosis in cancer cells. The level of DNAJ/Hsp40 homolog subfamily B member 6 (DNAJB6) is negatively correlated with lymph node metastasis in esophageal squamous cell carcinoma (ESCC) patients, and the overexpression of its isoform DNAJB6a is accompanied by remarkable reduction in the protein levels of GPX4 and phospho-AKT (p-AKT), thus DNAJB6a plays an anti-oncogenic role in ESCC progression via ferroptosis (212). Signal transducer and activator of transcription-3 (STAT3) can promote ferroptosis through activation of cathepsin B-mediated lysosomal cell death in PANC-1 and CFPAC-1 cells (170).
Loss of function of genes and proteins could change the sensitivity of cancer cells to ferroptosis. The loss of function of retinoblastoma (Rb) protein is significant during liver carcinogenesis, and the Rb-negative status of hepatocellular carcinoma (HCC) cells promotes ferroptosis on sorafenib treatment (163). Knockdown of high mobility group box 1 (HMGB1) decreases erastin-induced ROS generation via an iron-mediated lysosomal pathway in HL-60 cells expressing NRASQ61L (201).
Some genes and proteins can affect lipid metabolism and subsequently induce ferroptosis. Bromelain induces ferroptosis by inhibiting the proliferation of KRAS mutant colorectal cancer cells via ACSL4 (192). ADP-ribosylation factor 6 (ARF6) can sensitize gemcitabine-resistance pancreatic cancer cells to RSL3-induced lipid peroxidation by affecting the ACSL4 protein levels (172). Hypoxia-inducible transcriptional factor 2α (HIF-2α) selectively enriches PUFAs by activating the expression of the hypoxia-inducible lipid droplet-associated (HILPDA) protein and induces a ferroptosis-susceptible cell state in 786-O cells (208).
Several cellular signaling pathways are also involved in ferroptosis. Removal of the Hippo pathway effector transcription regulator 1 (TAZ) confers ferroptosis resistance, whereas overexpression of TAZS89A sensitizes cells to ferroptosis. TAZ promotes ferroptosis by regulating epithelial membrane protein 1 (EMP1) and NOX4 in renal cell carcinoma (207). Low levels of TAZ in chemo-resistant recurrent ovarian cancer are responsible for reduced ferroptosis susceptibility (203). Blockade of MAPK signaling similarly protects cells from ferroptosis, whereas NSCLC with sustained MAPK activation is likely to respond to ferroptosis following cystine depletion (206).
Many genes and proteins can protect cancer cells from ferroptosis. The sigma-1 receptor (S1R) protects HCC cells against sorafenib, while knockdown of S1R could induce ferroptosis by blocking the expression of GPX4 (164). The human mitochondrial cysteine desulfurase (NFS1) is highly expressed in well-differentiated lung adenocarcinoma and protects cells from ferroptosis (182). CDGSH iron sulfur domain 1 (CISD1), an iron-containing outer mitochondrial membrane protein, negatively regulates erastin-induced ferroptosis in HepG2 and Hep3B cells by inhibition of mitochondrial lipid peroxidation in a GPX4-independent manner (168). Overexpression of the CISD2 gene has been shown to confer resistance to HN-6 and HN-10 cells against SAS-induced ferroptosis, while silencing CISD2 could reverse SAS-resistant cells to a ferroptotic state with increased levels of lipid ROS and mitochondrial Fe2+ (190).
Some genes and proteins hinder ferroptosis; however, ferroptosis can occur when these are suppressed. NRF2 can upregulate cystathionine β-synthase (CBS) to confer resistance to erastin-induced ferroptosis in ovarian cancer cells (204), however inhibition of CBS can trigger ferroptosis in HCC (167). Inhibition of Lon peptidase 1 (LONP1) could contribute to ferroptosis of PANC-1 cells by activating the NRF2/KEAP1 signaling pathway and upregulating GPX4 expression (175). Heat shock protein family A member 5 (HSPA5) can upregulate the expression and activity of GPX4 to prevent dihydroartemisinin-induced ferroptosis in glioma (176). Inhibition of glucose-regulated protein 78 (GRP78), one of the most active molecular chaperone components in the ER of cancer cells, could enhance the effect of artesunate-induced ferroptosis in KRAS mutant pancreatic cancer cells (174). Growth/differentiation factor 15 (GDF15) knockdown has been shown to promote erastin-induced ferroptosis in MGC803 cells by attenuating SLC7A11 expression and subsequently decreasing intracellular GSH levels (179).
Some genes and proteins can prevent ferroptosis via affecting lipid metabolism. EGLN1/c-Myc directly activates the expression of lymphoid-specific helicase (LSH) by inhibiting HIF-1α, then LSH as an oncogene epigenetically increases the expression level of lipid metabolic genes that inhibit ferroptosis in lung cancer (183). Reconstitution of the functional vov Hippel-Lindau (VHL) gene prevents ferroptosis by reverting cells back to an oxidative metabolism and increasing fatty acid degradation through β-oxidation (209). Steroyl-CoA desaturase 1 (SCD1), the rate-limiting enzyme of MUFA synthesis, is highly expressed in ovarian cancer tissues and cell lines, and overexpression of SCD1 could protect cells from ferroptosis (205).
Posttranslational modifications can also regulate ferroptosis. O-linked β-N-acetylglucosamine glycosylation of c-JUN antagonizes ferroptosis by inhibiting GSH synthesis in BEL-7402 and SMMC-7721 cells (165). Nedd4 ubiquitylates voltage-dependent anion channels 2/3 (VDAC2/3) and degrades them to suppress erastin-induced ferroptosis in melanoma cells (199).
Several genes and proteins related to ferroptosis can be utilized as prognostic markers. G12D is the most frequent mutation in KRAS (referred as KRASG12D), and it is confirmed that KRASG12D could be released from PDAC cells succumbing to autophagy-dependent ferroptosis, and then KRASG12D causes macrophages to switch to an M2-like pro-tumor phenotype via STAT3-dependent fatty acid oxidation and it is also found that high KRASG12D expression in macrophages is correlated with poor survival in PDAC patients (173). High expression of Serine Threonine Tyrosine kinase 1 (STYK1) predicts poorer prognosis and is related to high levels of GPX4 in NSCLC (184).
Many microRNAs (miRNAs) can protect cancer cells from ferroptosis. miRNA-522, regulated by heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1), is secreted from cancer-associated fibroblasts and packed into exosomes, leading to chemo-resistance through targeting 15-LOX and decreasing ROS accumulation in gastric cancer cells (196). The inhibitory effect of miRNA-103a-3p on GLS2 is downregulated to promote ferroptosis and anti-tumorigenesis on physcion 8-O-β-glucopyranoside (PG) treatment (194). miRNA-9 can suppress GOT1 by directly binding to its 3’-untranslated region to reduce erastin- or RSL3-induced ferroptosis (197). miRNA-137 negatively regulates ferroptosis by directly targeting SLC1A5 to decrease glutamine uptake and MDA accumulation (198). Nonetheless, there are also some miRNAs that promote ferroptosis; for instance, miRNA-214-3p could promote ferroptosis by inhibiting ATF4 in vitro and in vivo (161).
A few long non-coding RNAs (lncRNAs) may also regulate ferroptosis. Erastin could upregulate the lncRNA GABPB1-AS1, which could downregulate the level of GABPB1 by blocking GABPB1 translation, leading to the downregulation of peroxiredoxin-5 and suppression of the cellular anti-oxidant capacity, and high expression of GABPB1 has been correlated with poor prognosis in HCC patients (166). The lncRNA LINC00336 is upregulated in lung cancer and functions as an inhibitor of ferroptosis in carcinogenesis by interacting with ELAV-like RNA-binding protein 1, which acts as a novel regulator of ferroptosis (185).
Several circular RNAs (circRNAs) might participate in the ferroptosis of cancer cells. Levels of circRNA TTBK2 and integrin subunit β 8 (ITGB8) are upregulated in glioma tissues and cells with downregulated miRNA-761 levels, and circRNA TTBK2 regulates cell proliferation, invasion, and ferroptosis via the miRNA-761/ITGB8 axis (180). circRNA 0008035, which is upregulated in gastric cancer tissues and cells, promotes the growth of gastric cancer cells and represses ferroptosis by upregulating eukaryotic initiation factor 4A1 (EIF4A1) through sponging miRNA-599 (195).
In recent years, numerous studies have been conducted to study ferroptosis in cancers, and several anti-tumor drugs (such as sorafenib, SAS, altertamin) have been approved by the FDA. However, many unsolved issues regarding ferroptosis still exist. What is the executor of ferroptosis in cancer? Which is the main pathway of lipid peroxidation in ferroptosis, the nonenzymatic pathway or enzymatic one? Does there exist another mechanism of ferroptosis resistance? What is the likelihood that the ferroptosis-related anti-tumor drugs could be used in clinical settings, such as post-operation, chemo-resistance, or radio-resistance instances? What are the potential adverse effects of ferroptosis-related anti-tumor drugs? Could the administration of ferroptosis-related anti-tumor drugs be combined with immunotherapy? The research on ferroptosis is still in its infancy, and more studies are needed to comprehensively explore the mechanism of ferroptosis in various cancers.
ZS and CS designed and conceptualized the review. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Thanks to editor Maria Ida Amabile for her help. This work was supported by the National Natural Science Foundation of China (NSFC) (no. 81572529, 81972373) and the Scientific Research Foundation for Advanced Talents, Xiang’an Hospital of Xiamen University (no. PM201809170001). In addition, ZS especially wishes to thank his wife Meiying Huang and his parents who have given him strong spiritual and financial support during his hard times.
1. Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell (2019) 35:830–49. doi: 10.1016/j.ccell.2019.04.002
2. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71:1–41. doi: 10.3322/caac.21660
3. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
4. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA (2016) 113:E4966–4975. doi: 10.1073/pnas.1603244113
5. Do Van B, Gouel F, Jonneaux A, Timmerman K, Gele P, Petrault M, et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol Dis (2016) 94:169–78. doi: 10.1016/j.nbd.2016.05.011
6. Guiney SJ, Adlard PA, Bush AI, Finkelstein DI, Ayton S. Ferroptosis and cell death mechanisms in Parkinson’s disease. Neurochem Int (2017) 104:34–48. doi: 10.1016/j.neuint.2017.01.004
7. Cong L, Dong X, Wang Y, Deng Y, Li B, Dai R. On the role of synthesized hydroxylated chalcones as dual functional amyloid-beta aggregation and ferroptosis inhibitors for potential treatment of Alzheimer’s disease. Eur J Med Chem (2019) 166:11–21. doi: 10.1016/j.ejmech.2019.01.039
8. Yan N, Zhang J. Iron Metabolism, Ferroptosis, and the Links With Alzheimer’s Disease. Front Neurosci (2019) 13:1443. doi: 10.3389/fnins.2019.01443
9. Cotticelli MG, Xia S, Lin D, Lee T, Terrab L, Wipf P, et al. Ferroptosis as a Novel Therapeutic Target for Friedreich’s Ataxia. J Pharmacol Exp Ther (2019) 369:47–54. doi: 10.1124/jpet.118.252759
10. Mi Y, Gao X, Xu H, Cui Y, Zhang Y, Gou X. The Emerging Roles of Ferroptosis in Huntington’s Disease. Neuromolecular Med (2019) 21:110–9. doi: 10.1007/s12017-018-8518-6
11. Park SJ, Cho SS, Kim KM, Yang JH, Kim JH, Jeong EH, et al. Protective effect of sestrin2 against iron overload and ferroptosis-induced liver injury. Toxicol Appl Pharmacol (2019) 379:114665. doi: 10.1016/j.taap.2019.114665
12. Deng G, Li Y, Ma S, Gao Z, Zeng T, Chen L, et al. Caveolin-1 dictates ferroptosis in the execution of acute immune-mediated hepatic damage by attenuating nitrogen stress. Free Radic Biol Med (2020) 148:151–61. doi: 10.1016/j.freeradbiomed.2019.12.026
13. Magtanong L, Dixon SJ. Ferroptosis and Brain Injury. Dev Neurosci (2018) 40:382–95. doi: 10.1159/000496922
14. Zhang Z, Wu Y, Yuan S, Zhang P, Zhang J, Li H, et al. Glutathione peroxidase 4 participates in secondary brain injury through mediating ferroptosis in a rat model of intracerebral hemorrhage. Brain Res (2018b) 1701:112–25. doi: 10.1016/j.brainres.2018.09.012
15. Kenny EM, Fidan E, Yang Q, Anthonymuthu TS, New LA, Meyer EA, et al. Ferroptosis Contributes to Neuronal Death and Functional Outcome After Traumatic Brain Injury. Crit Care Med (2019) 47:410–8. doi: 10.1097/CCM.0000000000003555
16. Yao X, Zhang Y, Hao J, Duan HQ, Zhao CX, Sun C, et al. Deferoxamine promotes recovery of traumatic spinal cord injury by inhibiting ferroptosis. Neural Regener Res (2019) 14:532–41. doi: 10.4103/1673-5374.245480
17. Zhang Y, Sun C, Zhao C, Hao J, Zhang Y, Fan B, et al. Ferroptosis inhibitor SRS 16-86 attenuates ferroptosis and promotes functional recovery in contusion spinal cord injury. Brain Res (2019a) 1706:48–57. doi: 10.1016/j.brainres.2018.10.023
18. Martin-Sanchez D, Ruiz-Andres O, Poveda J, Carrasco S, Cannata-Ortiz P, Sanchez-Nino MD, et al. Ferroptosis, but Not Necroptosis, Is Important in Nephrotoxic Folic Acid-Induced AKI. J Am Soc Nephrol (2017) 28:218–29. doi: 10.1681/ASN.2015121376
19. Hu Z, Zhang H, Yang SK, Wu X, He D, Cao K, et al. Emerging Role of Ferroptosis in Acute Kidney Injury. Oxid Med Cell Longev (2019) 2019:8010614. doi: 10.1155/2019/8010614
20. Hu Z, Zhang H, Yi B, Yang S, Liu J, Hu J, et al. VDR activation attenuate cisplatin induced AKI by inhibiting ferroptosis. Cell Death Dis (2020) 11:73. doi: 10.1038/s41419-020-2256-z
21. Sui M, Jiang X, Chen J, Yang H, Zhu Y. Magnesium isoglycyrrhizinate ameliorates liver fibrosis and hepatic stellate cell activation by regulating ferroptosis signaling pathway. BioMed Pharmacother (2018a) 106:125–33. doi: 10.1016/j.biopha.2018.06.060
22. Kong Z, Liu R, Cheng Y. Artesunate alleviates liver fibrosis by regulating ferroptosis signaling pathway. BioMed Pharmacother (2019) 109:2043–53. doi: 10.1016/j.biopha.2018.11.030
23. Li X, Duan L, Yuan S, Zhuang X, Qiao T, He J. Ferroptosis inhibitor alleviates Radiation-induced lung fibrosis (RILF) via down-regulation of TGF-beta1. J Inflammation (Lond) (2019) 16:11. doi: 10.1186/s12950-019-0216-0
24. Liu B, Zhao C, Li H, Chen X, Ding Y, Xu S. Puerarin protects against heart failure induced by pressure overload through mitigation of ferroptosis. Biochem Biophys Res Commun (2018) 497:233–40. doi: 10.1016/j.bbrc.2018.02.061
25. Chen X, Xu S, Zhao C, Liu B. Role of TLR4/NADPH oxidase 4 pathway in promoting cell death through autophagy and ferroptosis during heart failure. Biochem Biophys Res Commun (2019a) 516:37–43. doi: 10.1016/j.bbrc.2019.06.015
26. Ng SW, Norwitz SG, Norwitz ER. The Impact of Iron Overload and Ferroptosis on Reproductive Disorders in Humans: Implications for Preeclampsia. Int J Mol Sci (2019) 20:3283. doi: 10.3390/ijms20133283
27. Ng SW, Norwitz SG, Taylor HS, Norwitz ER. Endometriosis: The Role of Iron Overload and Ferroptosis. Reprod Sci (2020) 27:1383–90. doi: 10.1007/s43032-020-00164-z
28. Zhang H, He Y, Wang JX, Chen MH, Xu JJ, Jiang MH, et al. miR-30-5p-mediated ferroptosis of trophoblasts is implicated in the pathogenesis of preeclampsia. Redox Biol (2020b) 29:101402. doi: 10.1016/j.redox.2019.101402
29. Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis (2020b) 11:88. doi: 10.1038/s41419-020-2298-2
30. Eagle H. The specific amino acid requirements of a human carcinoma cell (Stain HeLa) in tissue culture. J Exp Med (1955) 102:37–48. doi: 10.1084/jem.102.1.37
31. Eagle H. Amino acid metabolism in mammalian cell cultures. Science (1959) 130:432–7. doi: 10.1126/science.130.3373.432
32. Eagle H, Piez KA, Oyama VI. The biosynthesis of cystine in human cell cultures. J Biol Chem (1961) 236:1425–8. doi: 10.1016/S0021-9258(18)64190-0
33. Bannai S, Tsukeda H, Okumura H. Effect of antioxidants on cultured human diploid fibroblasts exposed to cystine-free medium. Biochem Biophys Res Commun (1977) 74:1582–8. doi: 10.1016/0006-291X(77)90623-4
34. Comporti M, Saccocci C, Dianzani MU. Effect of CCl-4 in vitro and in vivo on lipid peroxidation of rat liver homogenates and subcellular fractions. Enzymologia (1965) 29:185–204.
35. Ghoshal AK, Recknagel RO. Positive Evidence of Acceleration of Lipoperoxidation in Rat Liver by Carbon Tetrachloride: In Vitro Experiments. Life Sci (1962) (1965) 4:1521–30. doi: 10.1016/0024-3205(65)90173-6
36. Halliwell B, Gutteridge JM. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem J (1984) 219:1–14. doi: 10.1042/bj2190001
37. Girotti AW. Mechanisms of lipid peroxidation. J Free Radic Biol Med (1985) 1:87–95. doi: 10.1016/0748-5514(85)90011-X
38. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell (2003) 3:285–96. doi: 10.1016/S1535-6108(03)00050-3
39. Yagoda N, Von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature (2007) 447:864–8. doi: 10.1038/nature05859
40. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol (2008) 15:234–45. doi: 10.1016/j.chembiol.2008.02.010
41. Wolpaw AJ, Shimada K, Skouta R, Welsch ME, Akavia UD, Pe’er D, et al. Modulatory profiling identifies mechanisms of small molecule-induced cell death. Proc Natl Acad Sci USA (2011) 108:E771–80. doi: 10.1073/pnas.1106149108
42. Yang WS, Sriramaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell (2014) 156:317–31. doi: 10.1016/j.cell.2013.12.010
43. Badgley MA, Kremer DM, Maurer HC, Delgiorno KE, Lee HJ, Purohit V, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science (2020) 368:85–9. doi: 10.1126/science.aaw9872
44. Jennis M, Kung CP, Basu S, Budina-Kolomets A, Leu JI, Khaku S, et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev (2016) 30:918–30. doi: 10.1101/gad.275891.115
45. Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of Mitochondria in Ferroptosis. Mol Cell (2019) 73:354–363 e353. doi: 10.1016/j.molcel.2018.10.042
46. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell (2015) 59:298–308. doi: 10.1016/j.molcel.2015.06.011
47. Yu H, Yang C, Jian L, Guo S, Chen R, Li K, et al. Sulfasalazineinduced ferroptosis in breast cancer cells is reduced by the inhibitory effect of estrogen receptor on the transferrin receptor. Oncol Rep (2019) 42:826–38. doi: 10.3892/or.2019.7189
48. Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res (2016) 26:1021–32. doi: 10.1038/cr.2016.95
49. Song Z, Xiang X, Li J, Deng J, Fang Z, Zhang L, et al. Ruscogenin induces ferroptosis in pancreatic cancer cells. Oncol Rep (2020) 43:516–24. doi: 10.3892/or.2019.7425
50. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol (2017) 13:91–8. doi: 10.1038/nchembio.2239
51. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol (2017) 13:81–90. doi: 10.1038/nchembio.2238
52. Sui X, Zhang R, Liu S, Duan T, Zhai L, Zhang M, et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front Pharmacol (2018b) 9:1371. doi: 10.3389/fphar.2018.01371
53. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature (2019) 575:688–92. doi: 10.1038/s41586-019-1705-2
54. Doll S, Freitas FP, Shah R, Aldrovandi M, Da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature (2019) 575:693–8. doi: 10.1038/s41586-019-1707-0
55. Roh JL, Kim EH, Jang H, Shin D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol (2017) 11:254–62. doi: 10.1016/j.redox.2016.12.010
56. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature (2015) 520:57–62. doi: 10.1038/nature14344
57. Ou Y, Wang SJ, Li D, Chu B, Gu W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci USA (2016) 113:E6806–12. doi: 10.1073/pnas.1607152113
58. Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD, et al. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep (2018) 22:569–75. doi: 10.1016/j.celrep.2017.12.077
59. Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem (1999) 274:11455–8. doi: 10.1074/jbc.274.17.11455
60. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife (2014) 3:e02523. doi: 10.7554/eLife.02523
61. Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, et al. Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem Biol (2019b) 26:623–633 e629. doi: 10.1016/j.chembiol.2019.01.008
62. Gout PW, Buckley AR, Simms CR, Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia (2001) 15:1633–40. doi: 10.1038/sj.leu.2402238
63. Louandre C, Ezzoukhry Z, Godin C, Barbare JC, Maziere JC, Chauffert B, et al. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int J Cancer (2013) 133:1732–42. doi: 10.1002/ijc.28159
64. Lachaier E, Louandre C, Godin C, Saidak Z, Baert M, Diouf M, et al. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res (2014) 34:6417–22.
65. Li Y, Yan H, Xu X, Liu H, Wu C, Zhao L. Erastin/sorafenib induces cisplatin-resistant non-small cell lung cancer cell ferroptosis through inhibition of the Nrf2/xCT pathway. Oncol Lett (2020c) 19:323–33. doi: 10.3892/ol.2019.11066
66. Felley-Bosco E, Gray SG. Mesothelioma Driver Genes, Ferroptosis, and Therapy. Front Oncol (2019) 9:1318. doi: 10.3389/fonc.2019.01318
67. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell (2017) 171:273–85. doi: 10.1016/j.cell.2017.09.021
68. Roh JL, Kim EH, Jang HJ, Park JY, Shin D. Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett (2016) 381:96–103. doi: 10.1016/j.canlet.2016.07.035
69. Yonezawa M, Back SA, Gan X, Rosenberg PA, Volpe JJ. Cystine deprivation induces oligodendroglial death: rescue by free radical scavengers and by a diffusible glial factor. J Neurochem (1996) 67:566–73. doi: 10.1046/j.1471-4159.1996.67020566.x
70. Hohenberger J, Ray K, Meyer K. The biology and chemistry of high-valent iron-oxo and iron-nitrido complexes. Nat Commun (2012) 3:720. doi: 10.1038/ncomms1718
71. Fenton HJH. Oxidation of tartaric acid in presence of iron. J Chem Soc Trans (1894) 65:899–910. doi: 10.1039/CT8946500899
72. Haber F, Weiss J. Uber die Katalyse des Hydroperoxydes. Naturwissenschaften (1932) 20:948–50. doi: 10.1007/BF01504715
73. Crielaard BJ, Lammers T, Rivella S. Targeting iron metabolism in drug discovery and delivery. Nat Rev Drug Discovery (2017) 16:400–23. doi: 10.1038/nrd.2016.248
74. Zhou L, Zhao B, Zhang L, Wang S, Dong D, Lv H, et al. Alterations in Cellular Iron Metabolism Provide More Therapeutic Opportunities for Cancer. Int J Mol Sci (2018) 19:1545. doi: 10.3390/ijms19051545
75. Hubert N, Hentze MW. Previously uncharacterized isoforms of divalent metal transporter (DMT)-1: implications for regulation and cellular function. Proc Natl Acad Sci USA (2002) 99:12345–50. doi: 10.1073/pnas.192423399
76. Dautry-Varsat A, Ciechanover A, Lodish HF. pH and the recycling of transferrin during receptor-mediated endocytosis. Proc Natl Acad Sci USA (1983) 80:2258–62. doi: 10.1073/pnas.80.8.2258
77. Kristiansen M, Graversen JH, Jacobsen C, Sonne O, Hoffman HJ, Law SK, et al. Identification of the haemoglobin scavenger receptor. Nature (2001) 409:198–201. doi: 10.1038/35051594
78. Li JY, Paragas N, Ned RM, Qiu A, Viltard M, Leete T, et al. Scara5 is a ferritin receptor mediating non-transferrin iron delivery. Dev Cell (2009) 16:35–46. doi: 10.1016/j.devcel.2008.12.002
79. Shaw GC, Cope JJ, Li L, Corson K, Hersey C, Ackermann GE, et al. Mitoferrin is essential for erythroid iron assimilation. Nature (2006) 440:96–100. doi: 10.1038/nature04512
80. Bou-Abdallah F. The iron redox and hydrolysis chemistry of the ferritins. Biochim Biophys Acta (2010) 1800:719–31. doi: 10.1016/j.bbagen.2010.03.021
81. Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature (2014) 509:105–9. doi: 10.1038/nature13148
82. Lei P, Bai T, Sun Y. Mechanisms of Ferroptosis and Relations With Regulated Cell Death: A Review. Front Physiol (2019) 10:139. doi: 10.3389/fphys.2019.00139
83. Shang Y, Luo M, Yao F, Wang S, Yuan Z, Yang Y. Ceruloplasmin suppresses ferroptosis by regulating iron homeostasis in hepatocellular carcinoma cells. Cell Signal (2020) 72:109633. doi: 10.1016/j.cellsig.2020.109633
84. Brown CW, Amante JJ, Chhoy P, Elaimy AL, Liu H, Zhu LJ, et al. Prominin2 Drives Ferroptosis Resistance by Stimulating Iron Export. Dev Cell (2019) 51:575–586 e574. doi: 10.1016/j.devcel.2019.10.007
85. Whiteland H, Spencer-Harty S, Morgan C, Kynaston H, Thomas DH, Bose P, et al. A role for STEAP2 in prostate cancer progression. Clin Exp Metastasis (2014) 31:909–20. doi: 10.1007/s10585-014-9679-9
86. Burnell SEA, Spencer-Harty S, Howarth S, Bodger O, Kynaston H, Morgan C, et al. STEAP2 Knockdown Reduces the Invasive Potential of Prostate Cancer Cells. Sci Rep (2018) 8:6252. doi: 10.1038/s41598-018-24655-x
87. Wu YY, Jiang JN, Fang XD, Ji FJ. STEAP1 Regulates Tumorigenesis and Chemoresistance During Peritoneal Metastasis of Gastric Cancer. Front Physiol (2018) 9:1132. doi: 10.3389/fphys.2018.01132
88. Han M, Xu R, Wang S, Yang N, Ni S, Zhang Q, et al. Six-Transmembrane Epithelial Antigen of Prostate 3 Predicts Poor Prognosis and Promotes Glioblastoma Growth and Invasion. Neoplasia (2018) 20:543–54. doi: 10.1016/j.neo.2018.04.002
89. Xue X, Bredell BX, Anderson ER, Martin A, Mays C, Nagao-Kitamoto H, et al. Quantitative proteomics identifies STEAP4 as a critical regulator of mitochondrial dysfunction linking inflammation and colon cancer. Proc Natl Acad Sci USA (2017) 114:E9608–17. doi: 10.1073/pnas.1712946114
90. Guo W, Zhang S, Chen Y, Zhang D, Yuan L, Cong H, et al. An important role of the hepcidin-ferroportin signaling in affecting tumor growth and metastasis. Acta Biochim Biophys Sin (Shanghai) (2015) 47:703–15. doi: 10.1093/abbs/gmv063
91. Shan Z, Wei Z, Shaikh ZA. Suppression of ferroportin expression by cadmium stimulates proliferation, EMT, and migration in triple-negative breast cancer cells. Toxicol Appl Pharmacol (2018) 356:36–43. doi: 10.1016/j.taap.2018.07.017
92. Wang Y, Yu L, Ding J, Chen Y. Iron Metabolism in Cancer. Int J Mol Sci (2018b) 20:95. doi: 10.3390/ijms20010095
93. Zhang C, Zhang F. Iron homeostasis and tumorigenesis: molecular mechanisms and therapeutic opportunities. Protein Cell (2015) 6:88–100. doi: 10.1007/s13238-014-0119-z
94. Hishikawa D, Hashidate T, Shimizu T, Shindou H. Diversity and function of membrane glycerophospholipids generated by the remodeling pathway in mammalian cells. J Lipid Res (2014) 55:799–807. doi: 10.1194/jlr.R046094
95. Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, et al. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem Biol (2015) 10:1604–9. doi: 10.1021/acschembio.5b00245
96. Reis A, Spickett CM. Chemistry of phospholipid oxidation. Biochim Biophys Acta (2012) 1818:2374–87. doi: 10.1016/j.bbamem.2012.02.002
97. Cheng Z, Li Y. What is responsible for the initiating chemistry of iron-mediated lipid peroxidation: an update. Chem Rev (2007) 107:748–66. doi: 10.1021/cr040077w
98. Haeggstrom JZ, Funk CD. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem Rev (2011) 111:5866–98. doi: 10.1021/cr200246d
99. Skrzypczak-Jankun E, Jankun J, Al-Senaidy A. Human lipoxygenase: developments in its structure, function, relevance to diseases and challenges in drug development. Curr Med Chem (2012) 19:5122–7. doi: 10.2174/092986712803530520
100. Rouzer CA, Matsumoto T, Samuelsson B. Single protein from human leukocytes possesses 5-lipoxygenase and leukotriene A4 synthase activities. Proc Natl Acad Sci USA (1986) 83:857–61. doi: 10.1073/pnas.83.4.857
101. Gaschler MM, Stockwell BR. Lipid peroxidation in cell death. Biochem Biophys Res Commun (2017) 482:419–25. doi: 10.1016/j.bbrc.2016.10.086
102. Radmark O, Werz O, Steinhilber D, Samuelsson B. 5-Lipoxygenase: regulation of expression and enzyme activity. Trends Biochem Sci (2007) 32:332–41. doi: 10.1016/j.tibs.2007.06.002
103. Peters-Golden M, Brock TG. 5-lipoxygenase and FLAP. Prostaglandins Leukot Essent Fatty Acids (2003) 69:99–109. doi: 10.1016/S0952-3278(03)00070-X
104. Ackermann JA, Hofheinz K, Zaiss MM, Kronke G. The double-edged role of 12/15-lipoxygenase during inflammation and immunity. Biochim Biophys Acta Mol Cell Biol Lipids (2017) 1862:371–81. doi: 10.1016/j.bbalip.2016.07.014
105. Dobrian AD, Lieb DC, Cole BK, Taylor-Fishwick DA, Chakrabarti SK, Nadler JL. Functional and pathological roles of the 12- and 15-lipoxygenases. Prog Lipid Res (2011) 50:115–31. doi: 10.1016/j.plipres.2010.10.005
106. Catala A, Diaz M. Editorial: Impact of Lipid Peroxidation on the Physiology and Pathophysiology of Cell Membranes. Front Physiol (2016) 7:423. doi: 10.3389/fphys.2016.00423
107. Ursini F, Maiorino M, Gregolin C. The selenoenzyme phospholipid hydroperoxide glutathione peroxidase. Biochim Biophys Acta (1985) 839:62–70. doi: 10.1016/0304-4165(85)90182-5
108. Garcia SC, Grotto D, Bulcao RP, Moro AM, Roehrs M, Valentini J, et al. Evaluation of lipid damage related to pathological and physiological conditions. Drug Chem Toxicol (2013) 36:306–12. doi: 10.3109/01480545.2012.720989
109. Feng H, Stockwell BR. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PloS Biol (2018) 16:e2006203. doi: 10.1371/journal.pbio.2006203
110. Ayala A, Munoz MF, Arguelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev (2014) 2014:360438. doi: 10.1155/2014/360438
111. Brigelius-Flohe R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta (2013) 1830:3289–303. doi: 10.1016/j.bbagen.2012.11.020
112. Hirschhorn T, Stockwell BR. The development of the concept of ferroptosis. Free Radic Biol Med (2019) 133:130–43. doi: 10.1016/j.freeradbiomed.2018.09.043
113. Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab (2008) 8:237–48. doi: 10.1016/j.cmet.2008.07.005
114. Ursini F, Maiorino M, Valente M, Ferri L, Gregolin C. Purification from Pig-Liver of a Protein Which Protects Liposomes and Biomembranes from Peroxidative Degradation and Exhibits Glutathione-Peroxidase Activity on Phosphatidylcholine Hydroperoxides. Biochim Et Biophys Acta (1982) 710:197–211. doi: 10.1016/0005-2760(82)90150-3
115. Chambers SJ, Lambert N, Williamson G. Purification of a cytosolic enzyme from human liver with phospholipid hydroperoxide glutathione peroxidase activity. Int J Biochem (1994) 26:1279–86. doi: 10.1016/0020-711X(94)90097-3
116. Scheerer P, Borchert A, Krauss N, Wessner H, Gerth C, Hohne W, et al. Structural basis for catalytic activity and enzyme polymerization of phospholipid hydroperoxide glutathione peroxidase-4 (GPx4). Biochemistry (2007) 46:9041–9. doi: 10.1021/bi700840d
117. Forcina GC, Dixon SJ. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics (2019) 19:e1800311. doi: 10.1002/pmic.201800311
118. Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell (2018) 172:409–422 e421. doi: 10.1016/j.cell.2017.11.048
119. Weiwer M, Bittker JA, Lewis TA, Shimada K, Yang WS, Macpherson L, et al. Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorg Med Chem Lett (2012) 22:1822–6. doi: 10.1016/j.bmcl.2011.09.047
120. Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci (2016) 73:2195–209. doi: 10.1007/s00018-016-2194-1
121. Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol (2016) 12:497–503. doi: 10.1038/nchembio.2079
122. Gaschler MM, Andia AA, Liu H, Csuka JM, Hurlocker B, Vaiana CA, et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat Chem Biol (2018) 14:507–15. doi: 10.1038/s41589-018-0031-6
123. Moosmann B, Behl C. Selenoprotein synthesis and side-effects of statins. Lancet (2004) 363:892–4. doi: 10.1016/S0140-6736(04)15739-5
124. Demierre MF, Higgins PD, Gruber SB, Hawk E, Lippman SM. Statins and cancer prevention. Nat Rev Cancer (2005) 5:930–42. doi: 10.1038/nrc1751
125. Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature (2017) 547:453–7. doi: 10.1038/nature23007
126. Qi XF, Kim DH, Yoon YS, Kim SK, Cai DQ, Teng YC, et al. Involvement of oxidative stress in simvastatin-induced apoptosis of murine CT26 colon carcinoma cells. Toxicol Lett (2010) 199:277–87. doi: 10.1016/j.toxlet.2010.09.010
127. Hadian K. Ferroptosis Suppressor Protein 1 (FSP1) and Coenzyme Q10 Cooperatively Suppress Ferroptosis. Biochemistry (2020) 59:637–8. doi: 10.1021/acs.biochem.0c00030
128. Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci USA (1994) 91:9926–30. doi: 10.1073/pnas.91.21.9926
129. Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev (2013) 27:2179–91. doi: 10.1101/gad.225680.113
130. Tong KI, Katoh Y, Kusunoki H, Itoh K, Tanaka T, Yamamoto M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol Cell Biol (2006) 26:2887–900. doi: 10.1128/MCB.26.8.2887-2900.2006
131. Anandhan A, Dodson M, Schmidlin CJ, Liu P, Zhang DD. Breakdown of an Ironclad Defense System: The Critical Role of NRF2 in Mediating Ferroptosis. Cell Chem Biol (2020) 27:436–47. doi: 10.1016/j.chembiol.2020.03.011
132. Shin D, Kim EH, Lee J, Roh JL. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic Biol Med (2018) 129:454–62. doi: 10.1016/j.freeradbiomed.2018.10.426
133. Itoh K, Igarashi K, Hayashi N, Nishizawa M, Yamamoto M. Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. Mol Cell Biol (1995) 15:4184–93. doi: 10.1128/MCB.15.8.4184
134. Katoh Y, Itoh K, Yoshida E, Miyagishi M, Fukamizu A, Yamamoto M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells (2001) 6:857–68. doi: 10.1046/j.1365-2443.2001.00469.x
135. Zhu M, Fahl WE. Functional characterization of transcription regulators that interact with the electrophile response element. Biochem Biophys Res Commun (2001) 289:212–9. doi: 10.1006/bbrc.2001.5944
136. Nioi P, Nguyen T, Sherratt PJ, Pickett CB. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol Cell Biol (2005) 25:10895–906. doi: 10.1128/MCB.25.24.10895-10906.2005
137. Chowdhry S, Zhang Y, Mcmahon M, Sutherland C, Cuadrado A, Hayes JD. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene (2013) 32:3765–81. doi: 10.1038/onc.2012.388
138. Wang H, Liu K, Geng M, Gao P, Wu X, Hai Y, et al. RXRalpha inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res (2013) 73:3097–108. doi: 10.1158/0008-5472.CAN-12-3386
139. Gagliardi M, Cotella D, Santoro C, Cora D, Barlev NA, Piacentini M, et al. Aldo-keto reductases protect metastatic melanoma from ER stress-independent ferroptosis. Cell Death Dis (2019) 10:902. doi: 10.1038/s41419-019-2143-7
140. Hubner RH, Schwartz JD, De Bishnu P, Ferris B, Omberg L, Mezey JG, et al. Coordinate control of expression of Nrf2-modulated genes in the human small airway epithelium is highly responsive to cigarette smoking. Mol Med (2009) 15:203–19. doi: 10.2119/molmed.2008.00130
141. Harada N, Kanayama M, Maruyama A, Yoshida A, Tazumi K, Hosoya T, et al. Nrf2 regulates ferroportin 1-mediated iron efflux and counteracts lipopolysaccharide-induced ferroportin 1 mRNA suppression in macrophages. Arch Biochem Biophys (2011) 508:101–9. doi: 10.1016/j.abb.2011.02.001
142. Agyeman AS, Chaerkady R, Shaw PG, Davidson NE, Visvanathan K, Pandey A, et al. Transcriptomic and proteomic profiling of KEAP1 disrupted and sulforaphane-treated human breast epithelial cells reveals common expression profiles. Breast Cancer Res Treat (2012) 132:175–87. doi: 10.1007/s10549-011-1536-9
143. Chorley BN, Campbell MR, Wang X, Karaca M, Sambandan D, Bangura F, et al. Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor alpha. Nucleic Acids Res (2012) 40:7416–29. doi: 10.1093/nar/gks409
144. Campbell MR, Karaca M, Adamski KN, Chorley BN, Wang X, Bell DA. Novel hematopoietic target genes in the NRF2-mediated transcriptional pathway. Oxid Med Cell Longev (2013) 2013:120305. doi: 10.1155/2013/120305
145. Gai C, Yu M, Li Z, Wang Y, Ding D, Zheng J, et al. Acetaminophen sensitizing erastin-induced ferroptosis via modulation of Nrf2/heme oxygenase-1 signaling pathway in non-small-cell lung cancer. J Cell Physiol (2020) 235:3329–39. doi: 10.1002/jcp.29221
146. Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res (2002) 62:5196–203.
147. Cho HY, Gladwell W, Wang X, Chorley B, Bell D, Reddy SP, et al. Nrf2-regulated PPAR{gamma} expression is critical to protection against acute lung injury in mice. Am J Respir Crit Care Med (2010) 182:170–82. doi: 10.1164/rccm.200907-1047OC
148. Huang J, Tabbi-Anneni I, Gunda V, Wang L. Transcription factor Nrf2 regulates SHP and lipogenic gene expression in hepatic lipid metabolism. Am J Physiol Gastrointest Liver Physiol (2010) 299:G1211–1221. doi: 10.1152/ajpgi.00322.2010
149. Jung KA, Choi BH, Nam CW, Song M, Kim ST, Lee JY, et al. Identification of aldo-keto reductases as NRF2-target marker genes in human cells. Toxicol Lett (2013) 218:39–49. doi: 10.1016/j.toxlet.2012.12.026
150. Salazar M, Rojo AI, Velasco D, De Sagarra RM, Cuadrado A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J Biol Chem (2006) 281:14841–51. doi: 10.1074/jbc.M513737200
151. Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci (2014) 39:199–218. doi: 10.1016/j.tibs.2014.02.002
152. Abdalkader M, Lampinen R, Kanninen KM, Malm TM, Liddell JR. Targeting Nrf2 to Suppress Ferroptosis and Mitochondrial Dysfunction in Neurodegeneration. Front Neurosci (2018) 12:466. doi: 10.3389/fnins.2018.00466
153. Barabutis N, Schally AV, Siejka A. P53, GHRH, inflammation and cancer. EBioMedicine (2018) 37:557–62. doi: 10.1016/j.ebiom.2018.10.034
154. Levine AJ. The road to the discovery of the p53 protein. The Steiner Cancer Prize Award Lecture. Int J Cancer (1994) 56:775–6. doi: 10.1002/ijc.2910560602
155. Hinds P, Finlay C, Levine AJ. Mutation is required to activate the p53 gene for cooperation with the ras oncogene and transformation. J Virol (1989) 63:739–46. doi: 10.1128/JVI.63.2.739-746.1989
156. Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol (2010) 2:a001107. doi: 10.1101/cshperspect.a001107
157. Yue X, Zhao Y, Xu Y, Zheng M, Feng Z, Hu W. Mutant p53 in Cancer: Accumulation, Gain-of-Function, and Therapy. J Mol Biol (2017) 429:1595–606. doi: 10.1016/j.jmb.2017.03.030
158. Bykov VJN, Eriksson SE, Bianchi J, Wiman KG. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer (2018) 18:89–102. doi: 10.1038/nrc.2017.109
159. Kandoth C, Mclellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature (2013) 502:333–9. doi: 10.1038/nature12634
160. Bai T, Wang S, Zhao Y, Zhu R, Wang W, Sun Y. Haloperidol, a sigma receptor 1 antagonist, promotes ferroptosis in hepatocellular carcinoma cells. Biochem Biophys Res Commun (2017) 491:919–25. doi: 10.1016/j.bbrc.2017.07.136
161. Bai T, Liang R, Zhu R, Wang W, Zhou L, Sun Y. MicroRNA-214-3p enhances erastin-induced ferroptosis by targeting ATF4 in hepatoma cells. J Cell Physiol (2020) 235:5637–48. doi: 10.1002/jcp.29496
162. Lippmann J, Petri K, Fulda S, Liese J. Redox Modulation and Induction of Ferroptosis as a New Therapeutic Strategy in Hepatocellular Carcinoma. Transl Oncol (2020) 13:100785. doi: 10.1016/j.tranon.2020.100785
163. Louandre C, Marcq I, Bouhlal H, Lachaier E, Godin C, Saidak Z, et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett (2015) 356:971–7. doi: 10.1016/j.canlet.2014.11.014
164. Bai T, Lei P, Zhou H, Liang R, Zhu R, Wang W, et al. Sigma-1 receptor protects against ferroptosis in hepatocellular carcinoma cells. J Cell Mol Med (2019) 23:7349–59. doi: 10.1111/jcmm.14594
165. Chen Y, Zhu G, Liu Y, Wu Q, Zhang X, Bian Z, et al. O-GlcNAcylated c-Jun antagonizes ferroptosis via inhibiting GSH synthesis in liver cancer. Cell Signal (2019c) 63:109384. doi: 10.1016/j.cellsig.2019.109384
166. Qi W, Li Z, Xia L, Dai J, Zhang Q, Wu C, et al. LncRNA GABPB1-AS1 and GABPB1 regulate oxidative stress during erastin-induced ferroptosis in HepG2 hepatocellular carcinoma cells. Sci Rep (2019) 9:16185. doi: 10.1038/s41598-019-52837-8
167. Wang L, Cai H, Hu Y, Liu F, Huang S, Zhou Y, et al. A pharmacological probe identifies cystathionine beta-synthase as a new negative regulator for ferroptosis. Cell Death Dis (2018a) 9:1005. doi: 10.1038/s41419-018-1063-2
168. Yuan H, Li X, Zhang X, Kang R, Tang D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem Biophys Res Commun (2016) 478:838–44. doi: 10.1016/j.bbrc.2016.08.034
169. Eling N, Reuter L, Hazin J, Hamacher-Brady A, Brady NR. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience (2015) 2:517–32. doi: 10.18632/oncoscience.160
170. Gao H, Bai Y, Jia Y, Zhao Y, Kang R, Tang D, et al. Ferroptosis is a lysosomal cell death process. Biochem Biophys Res Commun (2018) 503:1550–6. doi: 10.1016/j.bbrc.2018.07.078
171. Yamaguchi Y, Kasukabe T, Kumakura S. Piperlongumine rapidly induces the death of human pancreatic cancer cells mainly through the induction of ferroptosis. Int J Oncol (2018) 52:1011–22. doi: 10.3892/ijo.2018.4259
172. Ye Z, Hu Q, Zhuo Q, Zhu Y, Fan G, Liu M, et al. Abrogation of ARF6 promotes RSL3-induced ferroptosis and mitigates gemcitabine resistance in pancreatic cancer cells. Am J Cancer Res (2020) 10:1182–93.
173. Dai E, Han L, Liu J, Xie Y, Kroemer G, Klionsky DJ, et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy (2020) 16:2069–83. doi: 10.1080/15548627.2020.1714209
174. Wang K, Zhang Z, Wang M, Cao X, Qi J, Wang D, et al. Role of GRP78 inhibiting artesunate-induced ferroptosis in KRAS mutant pancreatic cancer cells. Drug Des Devel Ther (2019a) 13:2135–44. doi: 10.2147/DDDT.S199459
175. Wang H, Liu C, Zhao Y, Zhang W, Xu K, Li D, et al. Inhibition of LONP1 protects against erastin-induced ferroptosis in Pancreatic ductal adenocarcinoma PANC1 cells. Biochem Biophys Res Commun (2020) 522:1063–8. doi: 10.1016/j.bbrc.2019.11.187
176. Chen Y, Mi Y, Zhang X, Ma Q, Song Y, Zhang L, et al. Dihydroartemisinin-induced unfolded protein response feedback attenuates ferroptosis via PERK/ATF4/HSPA5 pathway in glioma cells. J Exp Clin Cancer Res (2019b) 38:402. doi: 10.1186/s13046-019-1413-7
177. Chen Y, Li N, Wang H, Wang N, Peng H, Wang J, et al. Amentoflavone suppresses cell proliferation and induces cell death through triggering autophagy-dependent ferroptosis in human glioma. Life Sci (2020c) 247:117425. doi: 10.1016/j.lfs.2020.117425
178. Wang Z, Ding Y, Wang X, Lu S, Wang C, He C, et al. Pseudolaric acid B triggers ferroptosis in glioma cells via activation of Nox4 and inhibition of xCT. Cancer Lett (2018c) 428:21–33. doi: 10.1016/j.canlet.2018.04.021
179. Chen L, Qiao L, Bian Y, Sun X. GDF15 knockdown promotes erastin-induced ferroptosis by decreasing SLC7A11 expression. Biochem Biophys Res Commun (2020a) 526:293–9. doi: 10.1016/j.bbrc.2020.03.079
180. Zhang HY, Zhang BW, Zhang ZB, Deng QJ. Circular RNA TTBK2 regulates cell proliferation, invasion and ferroptosis via miR-761/ITGB8 axis in glioma. Eur Rev Med Pharmacol Sci (2020c) 24:2585–600. doi: 10.26355/eurrev_202003_20528
181. Chen P, Wu Q, Feng J, Yan L, Sun Y, Liu S, et al. Erianin, a novel dibenzyl compound in Dendrobium extract, inhibits lung cancer cell growth and migration via calcium/calmodulin-dependent ferroptosis. Signal Transduct Target Ther (2020b) 5:51. doi: 10.1038/s41392-020-0149-3
182. Alvarez SW, Sviderskiy VO, Terzi EM, Papagiannakopoulos T, Moreira AL, Adams S, et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature (2017) 551:639–43. doi: 10.1038/nature24637
183. Jiang Y, Mao C, Yang R, Yan B, Shi Y, Liu X, et al. EGLN1/c-Myc Induced Lymphoid-Specific Helicase Inhibits Ferroptosis through Lipid Metabolic Gene Expression Changes. Theranostics (2017) 7:3293–305. doi: 10.7150/thno.19988
184. Lai Y, Zhang Z, Li J, Li W, Huang Z, Zhang C, et al. STYK1/NOK correlates with ferroptosis in non-small cell lung carcinoma. Biochem Biophys Res Commun (2019) 519:659–66. doi: 10.1016/j.bbrc.2019.09.032
185. Wang M, Mao C, Ouyang L, Liu Y, Lai W, Liu N, et al. Long noncoding RNA LINC00336 inhibits ferroptosis in lung cancer by functioning as a competing endogenous RNA. Cell Death Differ (2019b) 26:2329–43. doi: 10.1038/s41418-019-0304-y
186. Lin YS, Shen YC, Wu CY, Tsai YY, Yang YH, Lin YY, et al. Danshen Improves Survival of Patients With Breast Cancer and Dihydroisotanshinone I Induces Ferroptosis and Apoptosis of Breast Cancer Cells. Front Pharmacol (2019) 10:1226. doi: 10.3389/fphar.2019.01226
187. Llabani E, Hicklin RW, Lee HY, Motika SE, Crawford LA, Weerapana E, et al. Diverse compounds from pleuromutilin lead to a thioredoxin inhibitor and inducer of ferroptosis. Nat Chem (2019) 11:521–32. doi: 10.1038/s41557-019-0261-6
188. Ma S, Henson ES, Chen Y, Gibson SB. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis (2016) 7:e2307. doi: 10.1038/cddis.2016.208
189. Lin R, Zhang Z, Chen L, Zhou Y, Zou P, Feng C, et al. Dihydroartemisinin (DHA) induces ferroptosis and causes cell cycle arrest in head and neck carcinoma cells. Cancer Lett (2016) 381:165–75. doi: 10.1016/j.canlet.2016.07.033
190. Kim EH, Shin D, Lee J, Jung AR, Roh JL. CISD2 inhibition overcomes resistance to sulfasalazine-induced ferroptotic cell death in head and neck cancer. Cancer Lett (2018) 432:180–90. doi: 10.1016/j.canlet.2018.06.018
191. Malfa GA, Tomasello B, Acquaviva R, Genovese C, La Mantia A, Cammarata FP, et al. Betula etnensis Raf. (Betulaceae) Extract Induced HO-1 Expression and Ferroptosis Cell Death in Human Colon Cancer Cells. Int J Mol Sci (2019) 20:2723. doi: 10.3390/ijms20112723
192. Park S, Oh J, Kim M, Jin EJ. Bromelain effectively suppresses Kras-mutant colorectal cancer by stimulating ferroptosis. Anim Cells Syst (Seoul) (2018) 22:334–40. doi: 10.1080/19768354.2018.1512521
193. Gao Z, Deng G, Li Y, Huang H, Sun X, Shi H, et al. Actinidia chinensis Planch prevents proliferation and migration of gastric cancer associated with apoptosis, ferroptosis activation and mesenchymal phenotype suppression. BioMed Pharmacother (2020b) 126:110092. doi: 10.1016/j.biopha.2020.110092
194. Niu Y, Zhang J, Tong Y, Li J, Liu B. Physcion 8-O-beta-glucopyranoside induced ferroptosis via regulating miR-103a-3p/GLS2 axis in gastric cancer. Life Sci (2019) 237:116893. doi: 10.1016/j.lfs.2019.116893
195. Li C, Tian Y, Liang Y, Li Q. Circ_0008035 contributes to cell proliferation and inhibits apoptosis and ferroptosis in gastric cancer via miR-599/EIF4A1 axis. Cancer Cell Int (2020a) 20:84. doi: 10.1186/s12935-020-01168-0
196. Zhang H, Deng T, Liu R, Ning T, Yang H, Liu D, et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol Cancer (2020a) 19:43. doi: 10.1186/s12943-020-01168-8
197. Zhang K, Wu L, Zhang P, Luo M, Du J, Gao T, et al. miR-9 regulates ferroptosis by targeting glutamic-oxaloacetic transaminase GOT1 in melanoma. Mol Carcinog (2018a) 57:1566–76. doi: 10.1002/mc.22878
198. Luo M, Wu L, Zhang K, Wang H, Zhang T, Gutierrez L, et al. miR-137 regulates ferroptosis by targeting glutamine transporter SLC1A5 in melanoma. Cell Death Differ (2018) 25:1457–72. doi: 10.1038/s41418-017-0053-8
199. Yang Y, Luo M, Zhang K, Zhang J, Gao T, Connell DO, et al. Nedd4 ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nat Commun (2020b) 11:433. doi: 10.1038/s41467-020-14324-x
200. Du J, Wang T, Li Y, Zhou Y, Wang X, Yu X, et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic Biol Med (2019) 131:356–69. doi: 10.1016/j.freeradbiomed.2018.12.011
201. Ye FH, Chai WW, Xie M, Yang MH, Yu Y, Cao LZ, et al. HMGB1 regulates erastin-induced ferroptosis via RAS-JNK/p38 signaling in HL-60/NRAS(Q)(61L) cells. Am J Cancer Res (2019) 9:730–+. doi: 10.1186/s13046-019-1328-3
202. Zhu HY, Huang ZX, Chen GQ, Sheng F, Zheng YS. Typhaneoside prevents acute myeloid leukemia (AML) through suppressing proliferation and inducing ferroptosis associated with autophagy. Biochem Biophys Res Commun (2019) 516:1265–71. doi: 10.1016/j.bbrc.2019.06.070
203. Yang WH, Huang Z, Wu J, Ding CC, Murphy SK, Chi JT. A TAZ-ANGPTL4-NOX2 Axis Regulates Ferroptotic Cell Death and Chemoresistance in Epithelial Ovarian Cancer. Mol Cancer Res (2020a) 18:79–90. doi: 10.1158/1541-7786.MCR-19-0691
204. Liu N, Lin X, Huang C. Activation of the reverse transsulfuration pathway through NRF2/CBS confers erastin-induced ferroptosis resistance. Br J Cancer (2020) 122:279–92. doi: 10.1038/s41416-019-0660-x
205. Tesfay L, Paul BT, Konstorum A, Deng Z, Cox AO, Lee J, et al. Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells from Ferroptotic Cell Death. Cancer Res (2019) 79:5355–66. doi: 10.1158/0008-5472.CAN-19-0369
206. Poursaitidis I, Wang X, Crighton T, Labuschagne C, Mason D, Cramer SL, et al. Oncogene-Selective Sensitivity to Synchronous Cell Death following Modulation of the Amino Acid Nutrient Cystine. Cell Rep (2017) 18:2547–56. doi: 10.1016/j.celrep.2017.02.054
207. Yang WH, Ding CC, Sun T, Rupprecht G, Lin CC, Hsu D, et al. The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma. Cell Rep (2019) 28:2501–2508 e2504. doi: 10.1016/j.celrep.2019.07.107
208. Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun (2019) 10:1617. doi: 10.1038/s41467-019-09277-9
209. Miess H, Dankworth B, Gouw AM, Rosenfeldt M, Schmitz W, Jiang M, et al. The glutathione redox system is essential to prevent ferroptosis caused by impaired lipid metabolism in clear cell renal cell carcinoma. Oncogene (2018) 37:5435–50. doi: 10.1038/s41388-018-0315-z
210. Wang N, Zeng GZ, Yin JL, Bian ZX. Artesunate activates the ATF4-CHOP-CHAC1 pathway and affects ferroptosis in Burkitt’s Lymphoma. Biochem Biophys Res Commun (2019c) 519:533–9. doi: 10.1016/j.bbrc.2019.09.023
211. Lv HH, Zhen CX, Liu JY, Shang P. PEITC triggers multiple forms of cell death by GSH-iron-ROS regulation in K7M2 murine osteosarcoma cells. Acta Pharmacol Sin (2020) 41:1119–32. doi: 10.1038/s41401-020-0376-8
212. Jiang B, Zhao Y, Shi M, Song L, Wang Q, Qin Q, et al. DNAJB6 Promotes Ferroptosis in Esophageal Squamous Cell Carcinoma. Dig Dis Sci (2020) 65:1999–2008. doi: 10.1007/s10620-019-05929-4
Keywords: ferroptosis, cancers, amino acid metabolism, iron metabolism, lipid metabolism
Citation: Shi Z, Zhang L, Zheng J, Sun H and Shao C (2021) Ferroptosis: Biochemistry and Biology in Cancers. Front. Oncol. 11:579286. doi: 10.3389/fonc.2021.579286
Received: 02 July 2020; Accepted: 15 March 2021;
Published: 01 April 2021.
Edited by:
Maria Ida Amabile, Sapienza University of Rome, ItalyReviewed by:
Jeff Liddell, The University of Melbourne, AustraliaCopyright © 2021 Shi, Zhang, Zheng, Sun and Shao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chen Shao, Y3NoYW9AeGFoLnhtdS5lZHUuY24=; Huimin Sun, aG1zdW5AeGFoLnhtdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.