 Xiuzhen Zhang
Xiuzhen Zhang Tong Meng
Tong Meng Shuaishuai Cui
Shuaishuai Cui Ling Feng
Ling Feng Dongwu Liu1,3
Dongwu Liu1,3
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 11 February 2021
Sec. Cancer Molecular Targets and Therapeutics
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.621294
This article is part of the Research Topic Ubiquitin-modifying Enzymes as Drug Targets and Biomarkers in Cancer View all 10 articles
Ubiquitination, a crucial post-translation modification, regulates the localization and stability of the substrate proteins including nonhistone proteins. The ubiquitin-proteasome system (UPS) on nonhistone proteins plays a critical role in many cellular processes such as DNA repair, transcription, signal transduction, and apoptosis. Its dysregulation induces various diseases including cancer, and the identification of this process may provide potential therapeutic targets for cancer treatment. In this review, we summarize the regulatory roles of key UPS members on major nonhistone substrates in cancer-related processes, such as cell cycle, cell proliferation, apoptosis, DNA damage repair, inflammation, and T cell dysfunction in cancer. In addition, we also highlight novel therapeutic interventions targeting the UPS members (E1s, E2s, E3s, proteasomes, and deubiquitinating enzymes). Furthermore, we discuss the application of proteolysis-targeting chimeras (PROTACs) technology as a novel anticancer therapeutic strategy in modulating protein target levels with the aid of UPS.
Post-translational modification with ubiquitin plays an important role in the regulation of protein degradation and turnover. Ubiquitin, a small protein of 76 amino acids, can be covalently attached to target proteins to form mono- or polyubiquitinated types. This process occurs by a cascade of enzymatic reactions including E1-activating enzymes, E2-conjugating enzymes, and E3 ubiquitin ligases. Polyubinquitin with different chain topologies on specific lysine residues on substrates is related to different functional consequences (1). Generally, polyubiquitin chains linked at the 48 lysine site (K48) or K11 site lead to 26S proteasome-mediated proteolysis, which plays an essential role in maintaining protein homeostasis, regulating cell cycle, and apoptosis. On the other hand, chains with K63 site, as well as monoubiquitination, representing non-proteolytic ubiquitination, participate in diverse cellular processes, such as signal transduction, autophagy, and DNA damage repair (2, 3). As for most substrates, they are first covalently modified by ubiquitin and then directed to the proteasome to be degraded. Also, the function of ubiquitin ligases can be reversed by deubiquitinating enzymes (DUBs), which remove ubiquitin from substrate proteins and participate in the regulation of various cellular pathways (4).
Ubiquitination is ubiquitous, and second only to phosphorylation in abundance (5). Some reports have shown that histone ubiquitination regulating DNA-driven processes such as gene transcription and DNA damage repair (6, 7), and aberrant histone ubiquitination frequently occurs in cancers (8). Accumulating evidences indicate that ubiquitylation of nonhistone proteins plays an important role in many cellular processes, including DNA repair, transcription, signal transduction, autophagy, apoptosis, and so on (9). Nonhistone protein substrates for ubiquitination include general transcription factors, transcriptional activators or repressors, nonhistone chromatin-associated protein, and nuclear receptor coactivators. Dysregulation of nonhistone lysine ubiquitination is closely associated with various human cancers (10). Therefore, it is more important to study the role of nonhistone ubiquitination in tumorigenesis and tumor treatment. Moreover, interrogating the regulatory networks of UPS can offer a strategy for delineating the mechanism of cancer development and facilitate the identification of therapeutic targets. Meanwhile, the UPS exhibits high substrate specificity, which makes targeting it a promising strategy for cancer treatment. Nowadays, many UPS inhibitors such as bortezomib, carfilzomib and ixazomib, have been well applied in cancer treatment (11, 12). In this review, we summarize the regulatory roles of key UPS members on major nonhistone substrates in cancer-related processes.
Recently, a novel strategy named proteolysis-targeting chimeras (PROTACs) has been developed. PROTAC is a strategy that utilizes a hybrid molecule (a short peptide or a small molecule) to link a specific protein to an E3 ubiquitin ligase and induces the targeted protein degradation by the UPS in the cell (13). PROTACs link the target protein to an E3 ubiquitin ligase by a designed hybrid molecule, providing a path for ubiquitinating undruggable proteins such as transcription factors, scaffolding proteins and nonenzymatic proteins. Due to their high selectivities, low working concentrations, and less off-target toxicities, PROTACs may boost the development of drug discovery (14).
Considering the importance of UPS in the regulation of cancer development and treatment, we focus on the regulatory roles of key UPS members on nonhistone proteins in cancer development and highlight the novel therapeutic options targeting them. In addition, we also discuss and summarize the applications and recent advances of PROTAC technology focusing on nonhistone proteins.
The UPS contains a series of essential components: ubiquitin, E1s, E2s, E3s, DUBs, and the 26S proteasome. Until now, two E1s and about 40 E2s have been discovered, with more than 600 E3s conferring the diversity of protein substrates (15). Generally, E3 ligases are structurally classified into three subtypes: really interesting new gene (RING), homologous to E6-associated protein C-terminus (HECT) and RING-in-between-RING (RBR) E3s. RING E3 ligases are most abundant with more than 600 members in humans. About 30 HECT E3 ligases have been found in humans, including the NEDD4 family, the HERC family and other HECTs. RBR E3s have 14 members and work as hybrids of RING E3s and HECT E3s (16). In addition, there are approximate 100 DUBs and they are subdivided into 6 families based on sequence and structural similarity namely ubiquitin-specific proteases (USPs), ubiquitin carboxy-terminal hydrolases (UCHs), ovarian-tumor proteases (OTUs), Machado-Joseph disease protein proteases (MJD), JAB1/MPN/MOV34 metalloenzymes (JAMMs), and monocyte chemotactic protein-induced proteases (MCPIPs) (17). To date, more than 40 DUBs have been implicated in tumorigenesis (4).
The process of ubiquitylation contains three steps (Figure 1). Initially, the α-carboxyl group of the C-terminal glycine residue of ubiquitin links to a cysteine residue on E1 in an ATP-dependent manner, and a thioester bond is formed. Subsequently, E2 binds to the activated ubiquitin, and the complex of E1 and ubiquitin is transferred to the catalytic cysteine of E2 via a trans(thio)esterification reaction. Finally, E3 recognizes the substrate and catalyzes the linking of ubiquitin to a specific lysine residue on the substrate. The function of E3 ligases can be reversed by DUBs, which mediate the removal and processing of ubiquitin. DUBs regulate multiple biological processes including the cell cycle, DNA repair, apoptosis, inflammation, and signaling pathways.
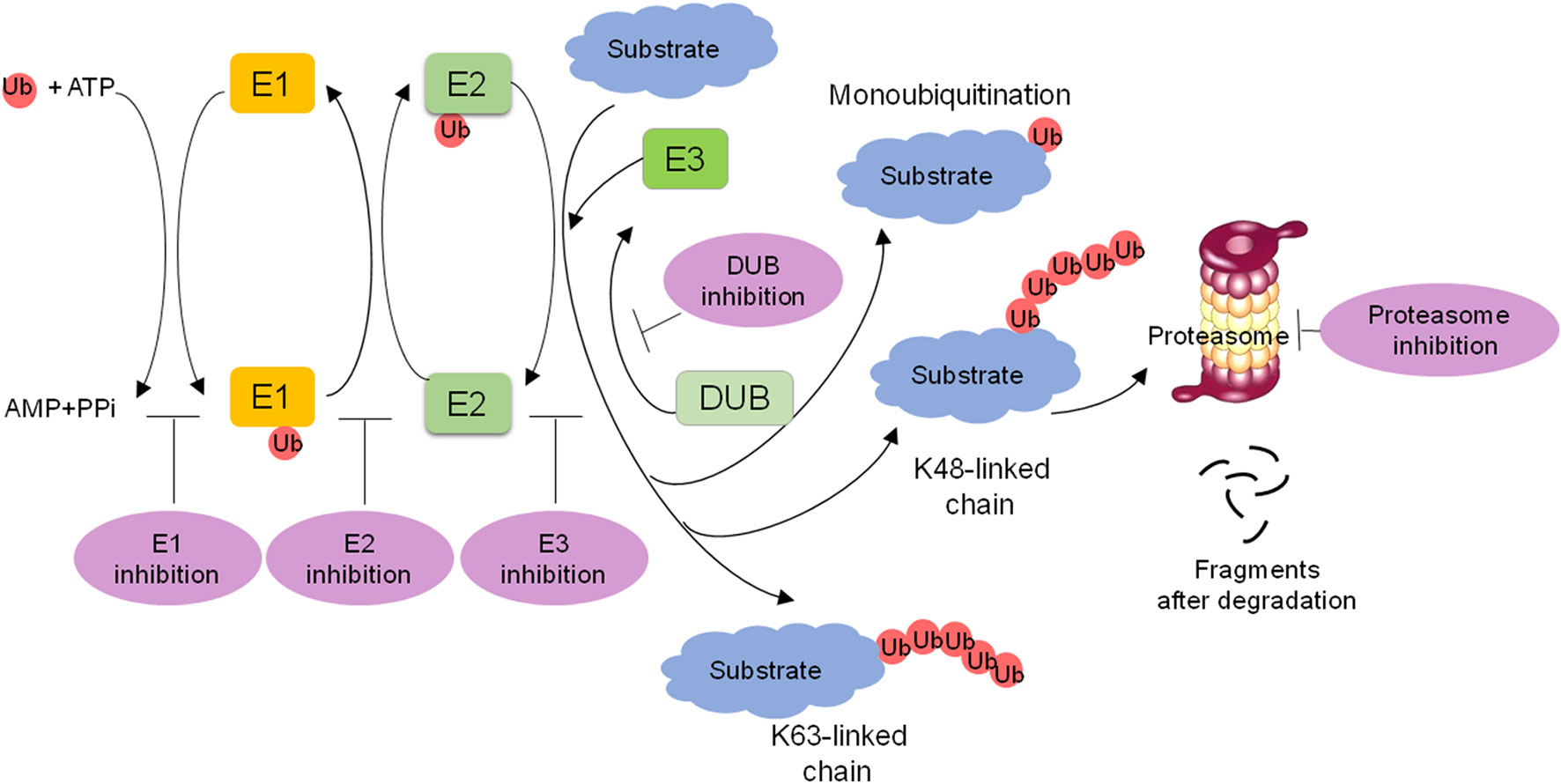
Figure 1 Overview of the ubiquitin-proteasome system (UPS) and targeting strategies for the UPS. The ubiquitin is activated with E1 in an ATP dependent manner, transferred to E2, and then transferred to the substrate through E3 ligase recognization, forming a mono- or polyubiquitinated protein. K48 or K11 polyubiquitin chains lead to 26S proteasome-mediated degradation. Monoubiquitination or K63 polyubiquitin chains are nonproteolytic ubiquitination signals and participate in many biological processes. DUBs remove or edit ubiquitins from substrate proteins. The targeting of E1s, E2s, E3s, proteasome and DUBs is a promising strategy for cancer treatment.
The UPS regulates diverse important cellular processes including cell cycle arrest, cell proliferation, and apoptosis. Thus, dysregulation of its key members and their regulatory network is often associated with human diseases, particularly cancer. Increasing studies have revealed that E3 ligases and DUBs are involved in cancer development through various biological processes, such as cell cycle, cell proliferation, apoptosis, DNA damage repair, inflammation, and T cell dysfunction in cancer and some of them are shown in Tables 1 and 2 (15).
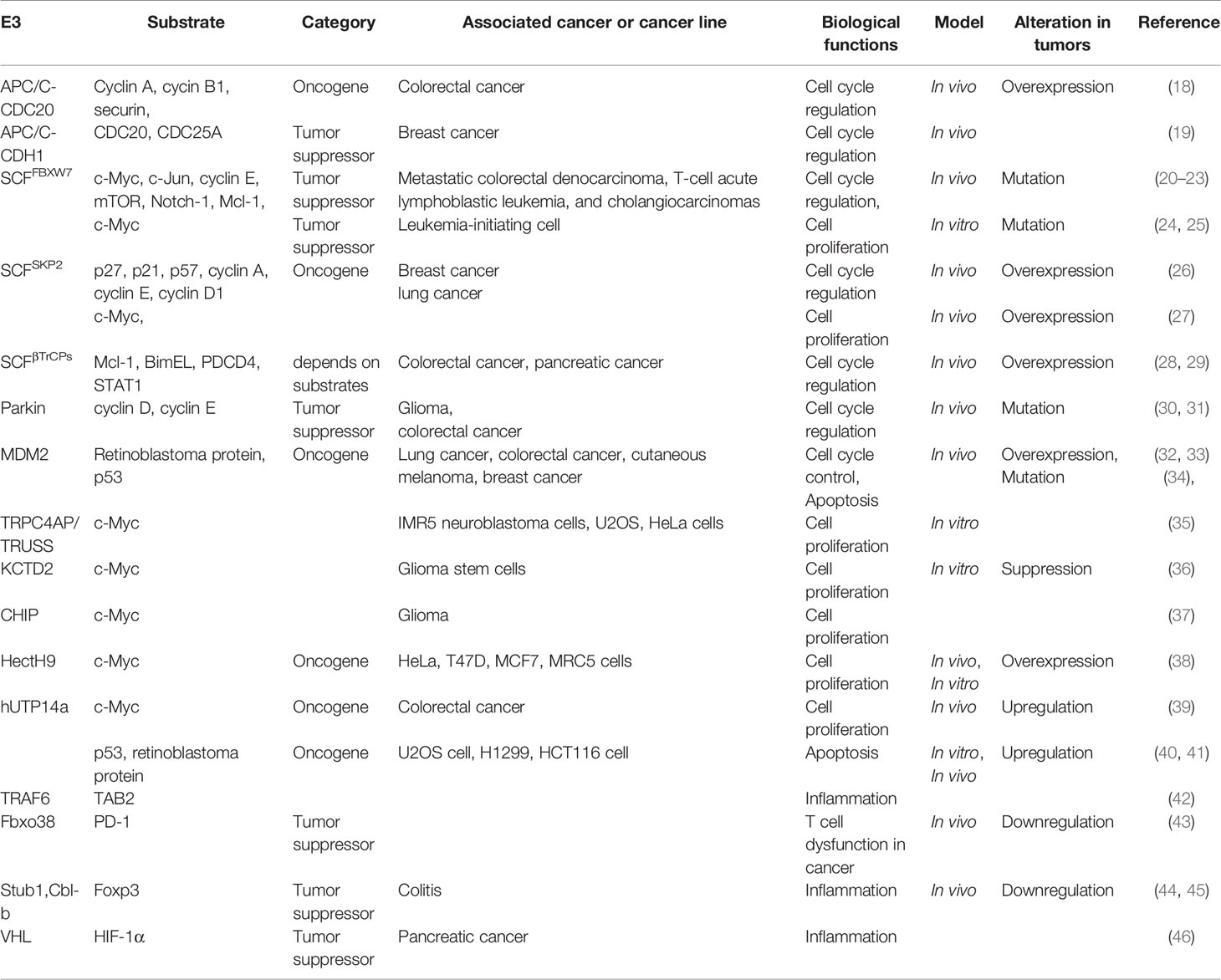
Table 1 Some E3s involved in cancers.
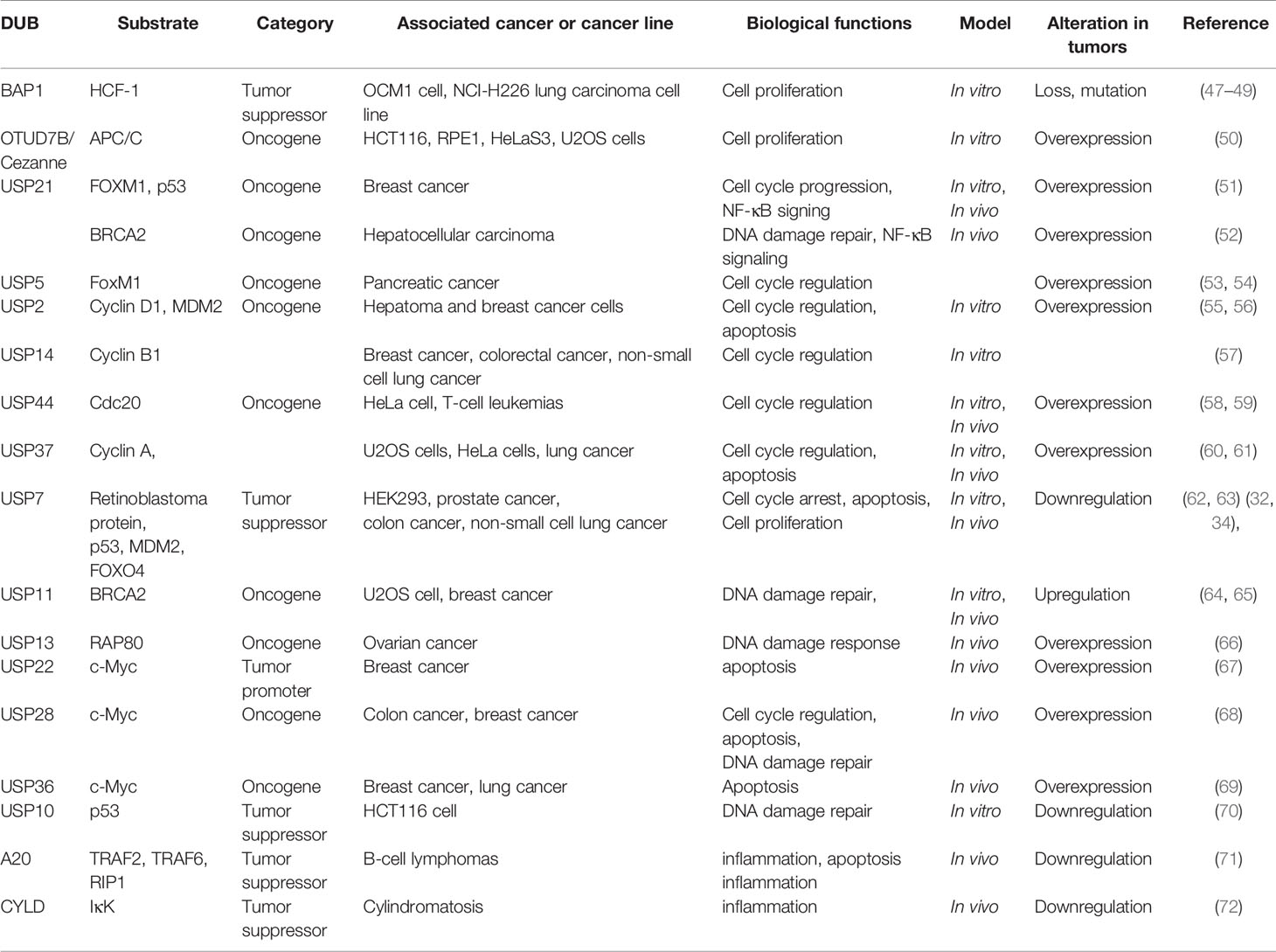
Table 2 Some DUBs involved in cancers.
Cell cycle progression and arrest are commonly deregulated in cancer (73). Increasing evidence indicates that multiple E3s participate in regulating cell cycle progression (Figure 2). Thus, the deregulation of E3s leads to the sustained proliferation and genomic instability of cancer cells. The anaphase-promoting complex named the cyclosome (APC/C) is the most sophisticated RING E3 ligase. It precisely governs cell cycle progression by recruiting cell division cycle 20 (CDC20) and CDC20-like protein 1 (CDH1) in turns. APC/C-CDC20 regulates cell cycle transition from metaphase to anaphase, while APC/C-CDH1 mediates mitotic exit and early G1 entry. Many studies indicate that Cdh1 functions as a tumor suppressor, whereas CDC20 may function as an oncoprotein to promote the development and progression of cancers (18, 74).
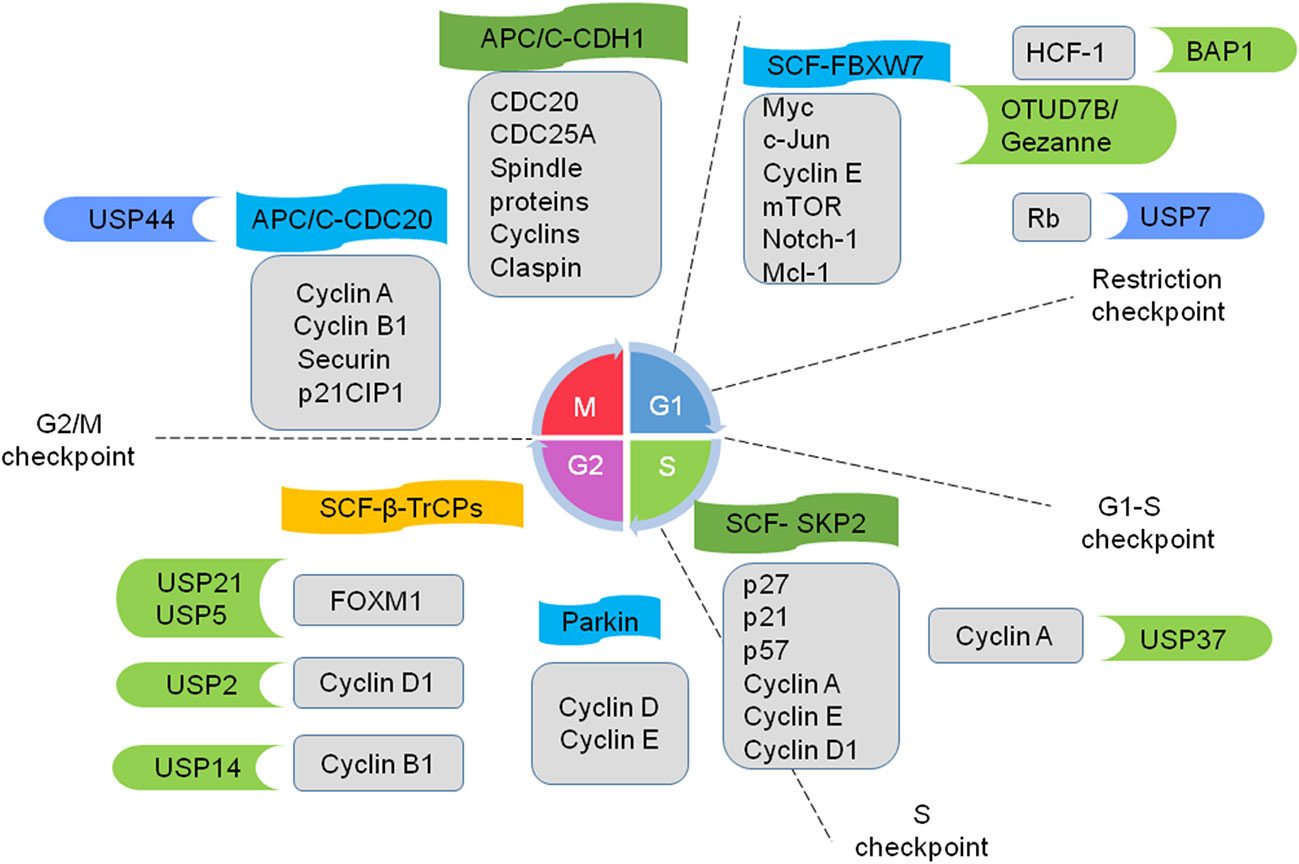
Figure 2 Ubiquitin ligases and DUBs coordinate to regulate cell cycle progression. E3 ligase APC/C (anaphase-promoting complex; also named as the cyclosome) recruits cell division cycle 20 (cdc20) and CDC20-like protein 1 (CDH1). APC/C-CDC20 promotes cell cycle transition from metaphase to anaphase, while APC/C-CDH1 mediates mitotic exit and early G1 entry. E3 ligases SCF (S-phase kinase-associated protein 1-cullin 1-F-box protein) complexes work on a subset of cyclins and CDK inhibitors and regulate progression from G1 to the onset of mitosis. FBXW7, SKP2, and β-TrCPs are well-studied F-box proteins. E3 Parkin downregulates some G1/S kinases. Several DUBs play crucial roles in cell-cycle progression in cancers. Some example substrates of E3 and DUBs are shown in the gray boxes. The E3 and DUBs in green are tumor promoters and the ones in blue are tumor suppressors.
Another representative example is SCF E3 ligases, which consist of four components: S-phase kinase-associated protein 1 (SKP1), cullin 1, Roc1/Rbx1/Hrt1 and an F-box protein (FBP). Commonly, FBPs serve for substrate recognition in the complexes and selectively regulate diverse biological processes (19). FBXW7, F-box/WD repeat-containing protein 7 (FBXW7), S-phase kinase associated protein2 (SKP2), and β-transducin repeat containing proteins (β-TrCPs) are well-studied FBPs. FBXW7 a tumor suppressor, works on many oncogenes including Myc, c-Jun, cyclin E, mTOR, Notch-1 and Mcl-1. It is often mutated or deleted in lots of cancers such as metastatic colorectal adenocarcinoma, T-cell acute lymphoblastic leukemia, and cholangiocarcinomas (20–22, 75). SKP2 plays a critical role during S and G2/M phases through regulating some cell cycle proteins, such as p21, p57, cyclin A, cyclin E, cyclin D1, and CDK inhibitors (e.g. p27). SKP2 is an important oncogene and is widely overexpressed in various cancers, such as breast cancer (23) and hepatocellular carcinoma (26). β-TrCPs-containing SCF complexes play a dual role in cell cycle checkpoint control: mediating and relieving cell cycle arrest via bonding different substrates (28, 76). Thus, the SCF complexes work on a subset of cyclins and CDK inhibitors to regulate the progression from G1 to the onset of mitosis. In addition, Parkin, a well-known RBR E3 ligase, controls the cell cycle by downregulating some G1/S kinases such as cyclin D and cyclin E (29, 30).
DUBs also participate in the regulation of cell-cycle progression (Figure 2) (31). For instance, E2F transcription factors play a key role in cell-cycle progression through G1 and into S-phase (77). The tumor suppressor retinoblastoma protein (Rb) maintains the cell in G1 through inhibiting E2F (78). However, hyperphosphorylated Rb dissociates from E2F, leading to the transcription of S-phase genes. The E3 ligase MDM2 promotes Rb degradation via ubiquitylation (79). On the contrary, the DUB USP7 directly reverses MDM2-mediated polyubiquitylation of Rb, stalling the cell cycle in G1 and inhibiting cell proliferation (32). Tumor suppressor BRCA1-associated protein 1 (BAP1), whose mutations can be seen in many cancers (62), has been found that it also could promote cell proliferation through deubiquitylating host cell factor 1 (HCF-1). HCF-1, an important transcriptional co-regulator of E2F, promotes cell cycle progression at the G1/S boundary by activating the E2F1 transcription factor. Therefore, BAP1 regulates cell proliferation at G1/S by co-regulating transcription from HCF-1/E2F-governed promoters. Moreover, BAP1 knockdown leads to G1 arrest and decreases the expression of S phase genes in OCM1 cells and NCI-H226 lung carcinoma cell line (47, 48, 80). It is well known that APC/C plays a crucial role in the completion of mitosis and maintenance of G1. Recently, OTUD7B/Cezanne has been reported to deubiquitinate and stabilize the APC/C substrates, as well as promote mitotic progression and cell proliferation. Cezanne is upregulated in multiple tumors, suggesting a potential role in cancer cell proliferation (49). Besides, the transcription factor FOXM1 participates in cell cycle progression and is upregulated in basal-like breast cancer. Arceci et al. reveal that USP21 directly binds to FOXM1, makes it deubiquitinate, and increases its expression level in vitro and in vivo. Suppression of USP21 causes a mitotic entry delay to slow proliferation and sensitivity to paclitaxel in cell culture and animal xenografts (50). The deubiquitinating enzyme USP5 is overexpressed in numerous malignancies, promoting tumor growth via modulating cell cycle regulators such as FoxM1. USP5 deficiency also induces DNA damage, cell cycle arrest and apoptosis in pancreatic ductal adenocarcinoma cells (51, 53). Besides, USP2 and USP14 regulate the cancer cell cycle via deubiquitinating cyclin D1 (54) and Cyclin B1 (55), respectively. Knocking down USP14 arrests cell cycle at the G2/M phase and inhibits the proliferation and migration of breast cancer cells (55), USP44 deubiquitinates the APC-inhibitory Mad2-Cdc20 complex, thereby preventing anaphase onset (57, 58). USP37 deubiquitinates and stabilizes Cyclin A and promotes S phase entry (59).
Many oncogenes can induce cancer cell proliferation, and UPS mediates their transcription by modulating general transcription factors, transcriptional activators and transcriptional coactivators via proteolytic and nonproteolytic ubiquitination (60). Here, we take the oncogene c-Myc as an example to show how ubiquitination regulates the transcription of oncogenes in cancer.
The overexpression of c-Myc is widely found in many cancers and is related to cell growth, proliferation, apoptosis and metabolic pathways (81). Its accumulation is also associated with poor cancer outcomes (82). Myc levels are controlled through targeted degradation by UPS (83). Multiple E3s are involved in modulating c-Myc activity in a tissue-specific manner. For instance, the ubiquitin ligase SCF-FBXW7 directly catalyzes c-Myc ubiquitination in a glycogen synthase kinase 3 phosphorylation-3-dependent manner and leads to c-Myc degradation in vitro (84). Furthermore, FBXW7 regulates the ubiquitylation of c-Myc protein and mediates leukemia-initiating cell activity (24). TRPC4AP (transient receptor potential cation channel, subfamily C, member 4-associated protein)/TRUSS (tumor necrosis factor receptor-associated ubiquitous scaffolding and signaling protein) binds to c-Myc and promotes its ubiquitination and degradation in multiple cancer cells (25). CRL3-potassium channel tetramerization domain-containing 2 (KCTD2) mediates c-Myc protein degradation by ubiquitination and suppresses gliomagenesis (35). E3 ligase CHIP interacts and degrades c-Myc by ubiquitination in glioma cells (36). In addition, 11S proteasomal activator REGγ has been reported to induce the degradation of c-Myc in cancer cells (37). On the other hand, SCF-SKP2 enhances c-Myc transcriptional activity by enabling the formation of c-Myc activator complexes (85). The E3 ligase HectH9 regulates the transcriptional activation of Myc through forming a lysine 63-linked polyubiquitin chain and promotes tumor cell proliferation in vivo and in vitro (27).
The deubiquitinating enzymes can prevent c-Myc degradation, maintain its stability, and then promote cancer progression. USP28 was the first DUB shown to regulate c-Myc stability. It is highly expressed in colon and breast carcinomas and binds to Myc through interacting with FBW7alpha to stabilize Myc in the nucleus (38). USP22 increased c-Myc stability via deubiquitination in breast cancer cells (68). We previously found that USP37 was significantly upregulated in human lung cancer tissues, and directly deubiquitinated and stabilized c-Myc independent of Fbw7 (67). USP36, a highly expressed USP in a subset of human breast and lung cancers, could interact with the nucleolar Fbw7γ and maintain c-Myc stability in the nucleolus (61). Recently, a novel E3 ligase, human U three protein 14a (hUTP14a) is upregulated in human colorectal cancer tissues, and it stabilizes c-Myc through forming a complex with USP36/Fbw7γ in the nucleolus and promote cancer progression (69).
Apoptosis could inhibit aberrant cell cycle progression and prevent tumorigenesis (39). If apoptotic pathways are abrogated, the cells may not appropriately induce apoptosis, which may lead to tumorigenesis. As a tumor suppressor protein, p53 is frequently mutated in most cancers and plays a pivotal role in apoptosis, genome instability and mutation. Ubiquitination has been found to play a key role in regulating p53 degradation as well as its activity and localization. For instance, MDM2 (murine double minute 2) has been found to negatively regulate p53 with diverse mechanisms. It can interact directly and degrade p53 via ubiquitination. Besides, it can connect p53 and pRb to form an Rb-Mdm2-p53 trimeric complex for the regulation of p53-induced apoptosis (86). Mdm2 can also form a heterodimer with MdmX (Mdm4) and participate in ubiquitin-mediated p53 degradation (33). Moreover, Mdm2 is upregulated in multiple cancers such as colorectal cancer, cutaneous melanoma and breast cancer (63). Therefore, the inhibition of p53-MDM2 interaction facilitates p53-mediated cell-cycle arrest or apoptosis in cancer cells.
Up to now, many DUBs are involved in the regulation of p53. For example, USP7 modulates the stability of both p53 and MDM2, and maintains the level of p53 ubiquitylation (34, 87); USP2 affects the stability of MDM2 (88); Otub1 inhibits p53 ubiquitination and activates p53 in cells (56); USP10 regulates the location and stability of p53, and stabilize both mutated and wild-type p53, thereby having a dual role in tumorigenesis (89).
Several E3s target anti-apoptotic protein myeloid cell leukemia 1 (MCL1) and sensitize cells to apoptosis. For example, DNA damage promotes HUWE1 bind to MCL1 and marks MCL1 for proteasomal degradation; the cell cycle regulators APC/C-CDC20 and SCF-FBXW7 degrade MCL1 and link apoptosis to prolonged mitotic arrest. Human UTP14a is upregulated in several types of tumors and involved in tumor progression via multiple mechanisms. It also exhibits an anti-apoptotic activity through the intrinsic apoptotic pathway, and protects tumor cells from chemotherapeutic drug-induced apoptosis (70). It binds p53 and induces p53 degradation through a ubiquitin-independent manner (40). Moreover, hUTP14a can also bind tumor suppressor pRb, and promote the polyubiquitination and degradation of pRb in vitro and in vivo (90). Thus hUTP14a might possess the potential as a target for anti-tumor therapy.
Errors in DNA replication and repair often cause genomic instability (73). DNA damage repair is critical to maintain genome integrity and prevent cancer. Many E3s including MDM2 and BRCA1 participate in regulating the DNA damage response and cell cycle checkpoints to cancer development. In brief, DNA double-strand breaks (DSBs) induce the activation of DNA damage sensors, which leads to the inactivation of MDM2, maintenance of p53 stability, promotion of SCF-β-TrCP mediated degradation of CDK phosphatase, and decrease of CDK activity. In the meantime, DNA repair machines are recruited to DNA damage sites under the control of ubiquitination. The inhibition of homologous recombination (HR) during G1 is also dependent on ubiquitylation mediated by APC/C-CDH1 and cullin 3-RING-E3 ligase (CRL3)-kelch-like ECH-associated protein 1 (KEAP1). USP11 is also involved in the regulation of DNA double-strand break repair, which is often up-regulated in cancer, resulting in resistance to poly ADP ribose polymerase 1 (PARP) inhibitors (41, 64). USP21 deubiquitinates and stabilizes BRCA2, promotes HR efficiency, and enhances homologous recombination efficiency and tumor cell growth (65). USP13 deubiquitinates receptor-associated protein 80 (RAP80) and promotes DNA damage response. Therefore, inhibiting USP13 makes ovarian cancer cells sensitive to cisplatin and olaparib (a PARP inhibitor) (52).
Cancer-related inflammation plays an important role in tumor development and progression. The transcription factor NF−κB regulates multiple biological processes including inflammation, immunity, cell proliferation and apoptosis. Abnormal activation of NF-κB has been involved in tumorigenesis. Ubiquitination regulates NF-κB pathways in proteasome-dependent and independent mechanisms (Figure 3) (66). For example, NF-κB is activated by the inflammatory cytokine interleukin-1 (IL-1). Without simulation, NF-κB is inactive in the cytoplasm binding to the inhibitory proteins of the κB family (IκB). IL-1β activates the ubiquitin E3 ligase tumor necrosis factor receptor-associated factor 6 (TRAF6). TRAF6 cooperates with the E2 enzyme Ubc13-Uev1A to synthesize K63 polyubiquitin chains and adds them to the TAB2 (TGFβ-activated kinase 1 binding protein 2) subunit of the TGF-β activated kinase 1 (TAK1) kinase complex, resulting in TAK1 activation. TAK1 then phosphorylates the IκB kinase β (IκKβ). Phosphorylated IκB is subsequently ubiquitinated and degraded by 26S proteasome, thereby allowing NF-κB to translocate to the nucleus and activate gene expression.
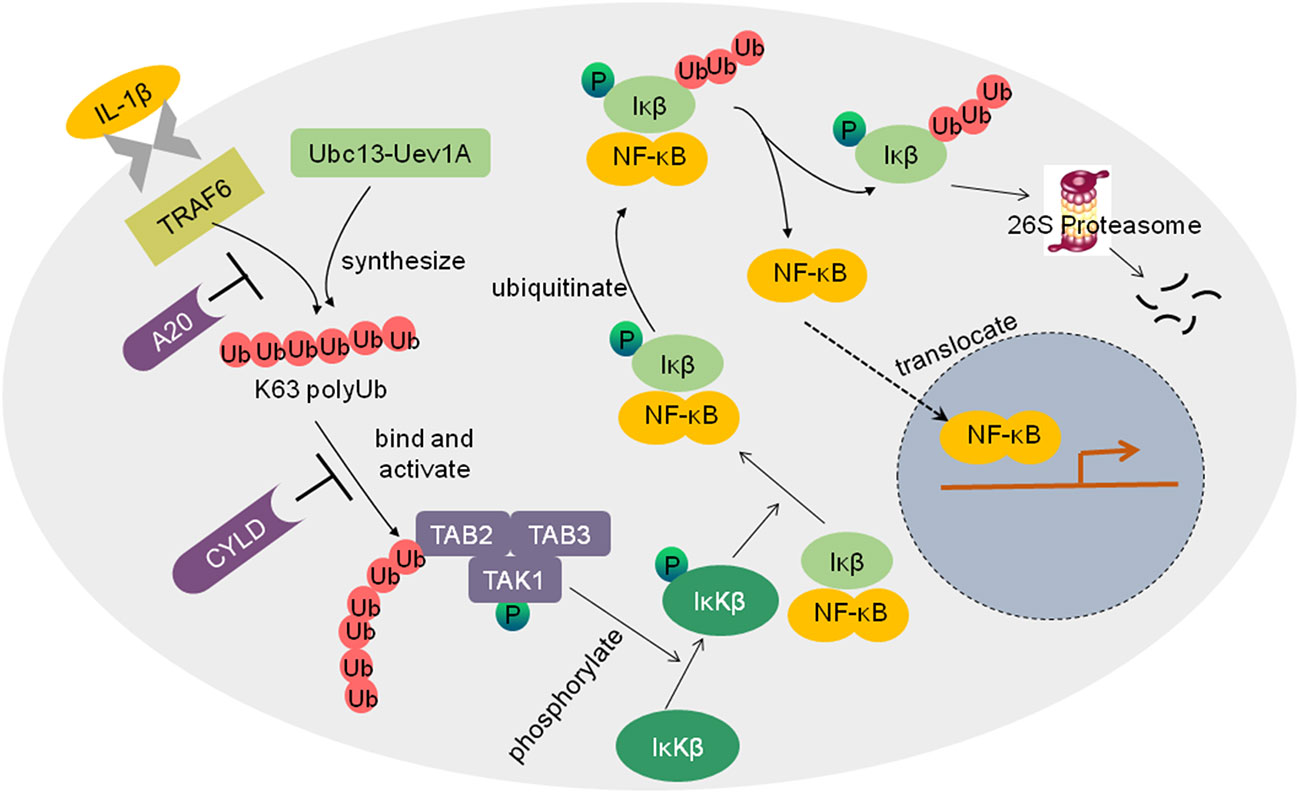
Figure 3 Schematic diagram of the regulation of NF-kB activation by ubiquitin ligases and DUBs. IL-1β activates the ubiquitin E3 ligase TRAF6, TRAF6 cooperated with the E2 enzyme Ubc13-Uev1A to synthesize K63 polyubiquitin chains and add them to the TAB2 subunit of the TGF-β activated kinase 1 (TAK1) kinase complex, which results in TAK1 activation. TAK1 then phosphorylates IκKβ. Phosphorylated IκB is subsequently ubiquitinated and degraded by 26S proteasome, thus allowing NF-κB to translocate to the nucleus, and the NF-κB pathway is activated. Deubiquitinases such as A20 and CYLD inhibit the activation of the NF-κB pathway.
Inappropriate activation of NF-κB has been linked to cancers. NF-κB activation could be tightly controlled by deubiquitinating enzymes as negative regulators of IκK. For example, DUB A20 inhibits IκK activation via three mechanisms, replacing K63 polyubiquitin chains from receptor-interacting protein 1(RIP1) with K48 polyubiquitin chains, blocking the interaction between Ubc13 and TRAFs, and inhibiting IκK phosphorylation by TAK1 (42). Another well-known DUB is the tumor suppressor CYLD, which inhibits NF-κB activation by cleaving K63 as well as linear polyubiquitin chains to inhibit IκK (71). Lack of CYLD in cells would elevate NF-κB activation, which likely contributes to tumor development.
T cell activation is critical for the initiation and regulation of the immune response in cancer immunotherapy. It requires at least two signals to become fully activated. One occurs after the engagement of the T cell receptor (TCR) and major histocompatibility complex (MHC). Another is provided when co-stimulator CD28 binds to CD80 and CD86 that are expressed on antigen-presenting cells (APCs). However, the multifaceted suppressive signals that existed in the tumor microenvironment make intratumoral T cells dysfunctional. The main traits of T cell dysfunction include some inhibitory receptors (e.g., PD-1), inhibitory cells (e.g., Treg cells), suppressive soluble mediators (e.g., TGFβ), transcriptional factors (e.g., T-bet), etc (72). UPS has been found to play a key regulatory role in maintaining T cell dysfunction with diverse mechanisms (91).
Dysfunctional T cells usually have abnormally high expression of multiple inhibitory receptors such as PD-1. Inhibitory receptors binding to their ligands negatively regulate an immune response. A recent study has identified that E3 ligase Fbxo38 ubiquitinates and degrades PD-1 in activated intratumoral T cells, which proves a novel mechanism for cancer immunotherapy. Fbxo38 can be activated by IL-2-induced STAT5 in activated T cells. In the dysfunctional T cells, Fbxo38 is downregulated, leading to an increased PD-1 abundance and impressive tumor immune response (92).
Regulatory T (Treg) cells are a subpopulation of CD4+ T cells that are crucial for maintaining immune tolerance. Treg cells usually produce immunosuppressive molecules such as TGFβ and inhibit the function of effector T cells. Treg cell development and function are determined by the transcription factor forkhead box protein 3 (Foxp3) and several E3s are involved in the process. For example, Stub1 and casitas B cell lymphoma protein b (Cbl-b) ubiquitinate Foxp3 and negatively regulate Treg cell development (43, 44). E3 ligase von Hippel-Lindau (VHL), Itchy homolog (Itch) and gene related to anergy in lymphocytes (Grail) participate in maintaining Treg cell repressive function (45, 46). Loss of VHL in Tregs leads to type 1 T helper (Th1)-like cell conversion and interferon-gamma (IFN-γ) production (45). Itch deficiency in Treg cells results in severe airway inflammation in mice, increasing TH2 cytokine production (46). Also, GRAIL-deficient Treg cells induce decreased suppressive function and increased Th17 cell-related gene expressions (93). Cbl-b and Grail have been found to play crucial roles in tumor immunosurveillance. Their loss inhibits tumor formation in mice. Cbl-b-/- and Grail-/- CD8+ T cells can be fully activated in the absence of costimulatory factors in vitro. They could promote tumor rejection and inhibit tumor formation when they are transferred into tumor-bearing mice (94, 95). These studies suggest that Cbl-b and Grail may serve as therapeutic targets to antitumor immunity.
TGFβ, a well-known immunosuppressor factor, plays an important role in immune tolerance (96). It not only promotes thymic Treg cell development by repressing T cell clonal deletion but also regulates peripheral Treg cell differentiation and maintains Treg cell function by inducing Foxp3 expression (96). Moreover, TGFβ inhibits T cell proliferation by decreasing IL-2 production and upregulating cell cycle inhibitors (97). It also blocks CD4+ T cell differentiation by modulating T-bet or GATA expression (97). Besides, TGFβ downregulates the expressions of cytolytic genes in cytotoxic T lymphocytes (98), costimulatory factors and MHC II molecules in dendritic cells and macrophages, reducing antigen resenting ability and regulating T cell function indirectly (99). In fact, as a versatile cytokine, TGFβ exerts pivotal functions in diverse processes of cancer development, such as proliferation, differentiation, apoptosis, and migration, depending on the target cells (100). Thus, TGFβ signaling has been regarded as a potential therapeutic target for the treatment of cancers.
Dynamic ubiquitination/deubiquitination plays a key role in the regulation of the TGFβ signaling pathway (Figure 4) (101). The TGFβ1-induced TGFβ pathway activation consists of receptors (TGF receptor I and II), receptor-SMADs (SMAD2 and SMAD3), co-SMAD (SMAD4), and inhibitor adaptor SMAD (SMAD7). TGFβ1 binding induces TGFRII to phosphate TGFRI, and then the activated-TGFRI phosphorylates SMAD2 and SMAD3. Subsequently, the phosphorylated SMAD2/3 dissociates from the receptor and oligomerizes with SMAD4. Following that, SMAD2/3/4 translocates to the nucleus and recruits other gene regulatory proteins and transcript specific genes. Many E3s and DUBs are reported to be involved in turning off the TGFβ pathway. For example, AIP4/Itch brings SMAD7 to TGFβRI and prevents the activation of SMAD2 (102). SMAD7 also serves as a scaffold to recruit E3 ligases SMURF1, SMURF2, Tuil1/WWP1 and NEDD4-2 to ubiquitinate and degrade the receptor complex (103–106). On the contrary, USP26 stabilizes SMAD7 via deubiquitination (107). As for SMADs, SMURF2 and NEDD4-2 target SMAD2 for degradation (106, 108) whereas SMAD3 is targeted by E3 ligases CHIP and ROC1-SCFFbw1a (109, 110). SMAD4 is indirectly regulated by E3 ligases SMURF1, SMURF2, Tuil1/WWP1, and NEDD4-2 through forming a complex with SMAD7, SMAD6 or activated SMAD2 (111). SMAD4 has a point mutation in many cancers. In this case, these protein variants are degraded by E3 ligases SCF-Skp2 and SCF-β-TrCP1 (112, 113) In addition, the R-SMAD/SMAD4 complex can be dissociated by SMURF2 monoubiquitinates SMAD3 or Ectodermin/Tif1γ monoubiquitinates SMAD4. Once the R-SMAD/SMAD4 complex enters the nucleus, the DNA-binding proteins SnoN and TGIF direct NEDD4-2 and Tiul/WWP1 to degrade SMAD2 and inhibit the signaling.
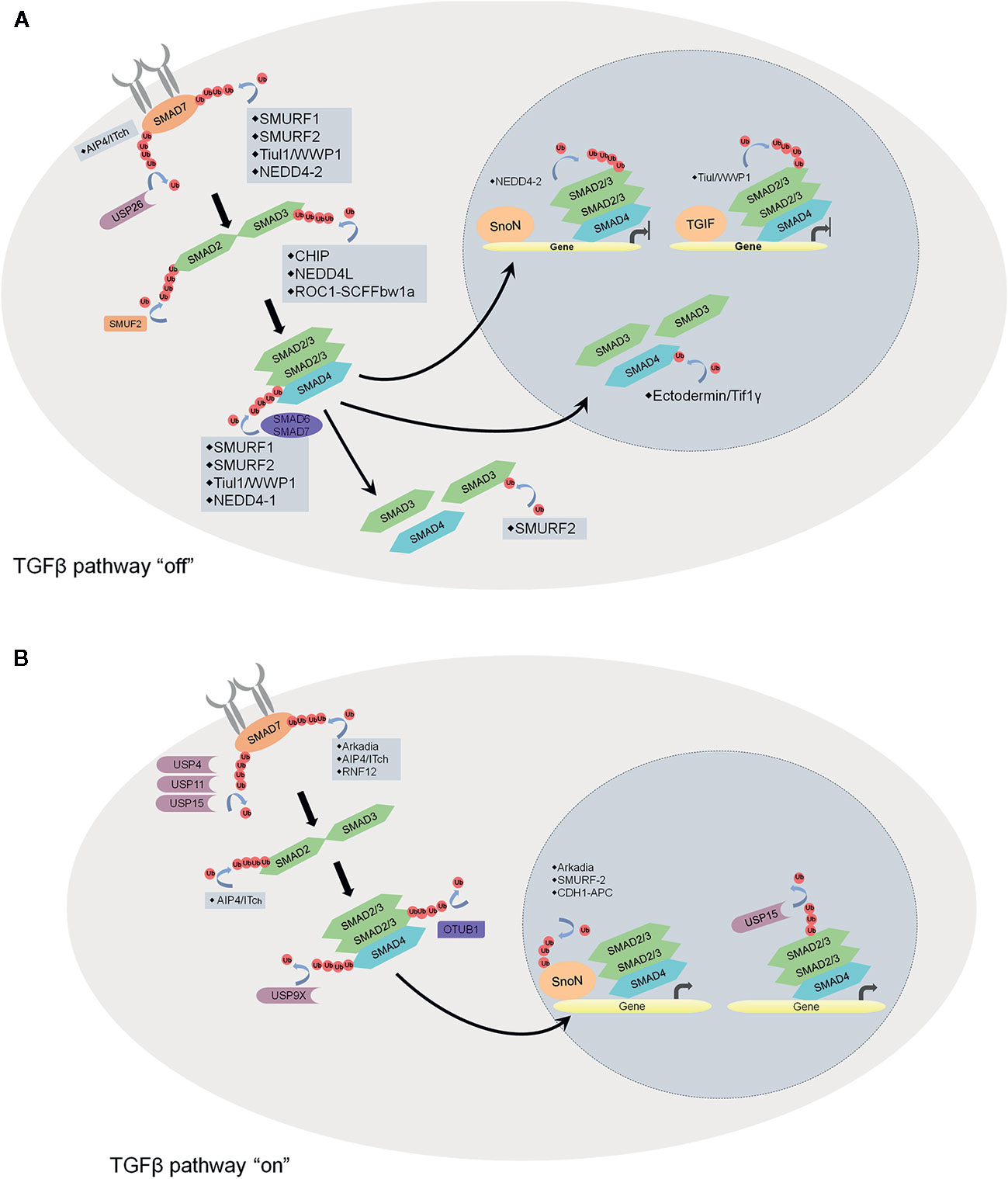
Figure 4 Schematic overview of the regulation of TGFβ pathway by ubiquitin ligases and DUBs. (A) factors that turn off the TGFβ pathway. AIP4/Itch brings SMAD7 to TGFβRI and prevents the activation of SMAD2. SMAD7 recruits E3 ligases SMURF1, SMURF2, Tuil1/WWP1, and NEDD4-2 to ubiquitinate and degrade the receptor complex. On the contrary, USP26 stabilizes SMAD7 via deubiquitination. As for SMADs, SMURF2, and NEDD4-2 target SMAD2 for degradation whereas SMAD3 is targeted by E3 ligases CHIP and ROC1-SCFFbw1a. SMAD4 is regulated by E3 ligases SMURF1, SMURF2, Tuil1/WWP1, and NEDD4-2 through forming a complex with SMAD7, SMAD6 or activated SMAD2. The R-SMAD/SMAD4 complex can be dissociated by SMURF2 monoubiquitinates SMAD3 or Ectodermin/Tif1γ monoubiquitinates SMAD4. Once the R-SMAD/SMAD4 complex enters the nucleus, the DNA-binding proteins SnoN and TGIF direct NEDD4-2 and Tiul/WWP1 to degrade SMAD2 and inhibit the signaling. (B) factors that turn on the TGFβ pathway. At the receptor level, USP4, USP11, and USP15 stabilize the receptor complex. E3s Arkadia, AIP4/Itch and RNF12 induce SMAD7 degradation. OTUB1 maintains the stability of SMAD2/3 and also promotes the R-SMAD/SMAD4 complex formation by preventing ubiquitination on R-SMAD. In the nucleus, transcriptional repressor SnoN can be degraded induced by E3s Arkadia, SMURF2 and CDH1-APC. Monoubiquitination of R-SMADs prevents the R-SMAD/SMAD4 complex binding with the DNA, while USP15 reverses the modification and promotes TGFβ-dependent transcription.
On the other hand, lots of E3s and DUBs participate in turning on the TGFβ pathway. At the receptor level, USP4 interacts directly with TGFβRI to maintain its stability (114). DUBs such as USP11 and USP15, stabilize the receptor complex by being associated with the scaffold protein SMAD7 (115, 116). SMAD7 can be degraded by E3 ligases Arkadia, AIP4/Itch and RNF12 mediated ubiquitination (117). OTUB1 maintains the stability of SMAD2/3 by reversing the ubiquitination of SMAD2 and USP9X, and also promotes the R-SMAD/SMAD4 complex formation by preventing ubiquitination on R-SMAD (118). In the nucleus, transcriptional repressor SnoN can be degraded by E3s Arkadia, SMURF2 and CDH1-APC mediated ubiquitination (119, 120). Monoubiquitination of R-SMADs prevents the R-SMAD/SMAD4 complex binding with the DNA, while USP15 reverses the modification and promotes TGFβ dependent transcription.
The T-box family transcription factor T-bet regulates the Th1 cell differentiation and induces the production of IFN-γ. Recently, it has been shown that it is expressed in Treg and participates in relevant immunosuppressive function (121). It has been suggested that T-bet is required in T cell dysfunction (72). Although the underlying mechanism of T-bet ubiquitination is unknown, USP10 has been found to stabilize T-bet via deubiquitination and enhance the secretion of IFN-γ (122).
Furthermore, UPS could regulate TCR activation. For instance, E3 ligases Cbl, Itch, and Grail degrade the TCR complex and inhibit T cell activation through proteolysis-dependent mechanisms (91, 123). In contrast, USP12 has been found to stabilize the TCR complex and promote TCR signaling through deubiquitylating TCR adaptor proteins LAT and Trat1 in primary mouse T lymphocytes (124). Naik et al. found that USP9X regulated TCR signaling and tolerance induction, and also the USP9X-deficient T cells were hyperproliferative (125). Therefore, E3 ligases and deubiquitinases keep the delicate balance between immunity and tolerance.
Numerous evidence indicates that every component of UPS can be regarded as valuable therapeutic targets in the development of anti-cancer drugs. Several drugs such as bortezomib (a proteasome inhibitor), have been approved by the FDA in cancer, and many other inhibitors are in development (Table 3) (138).
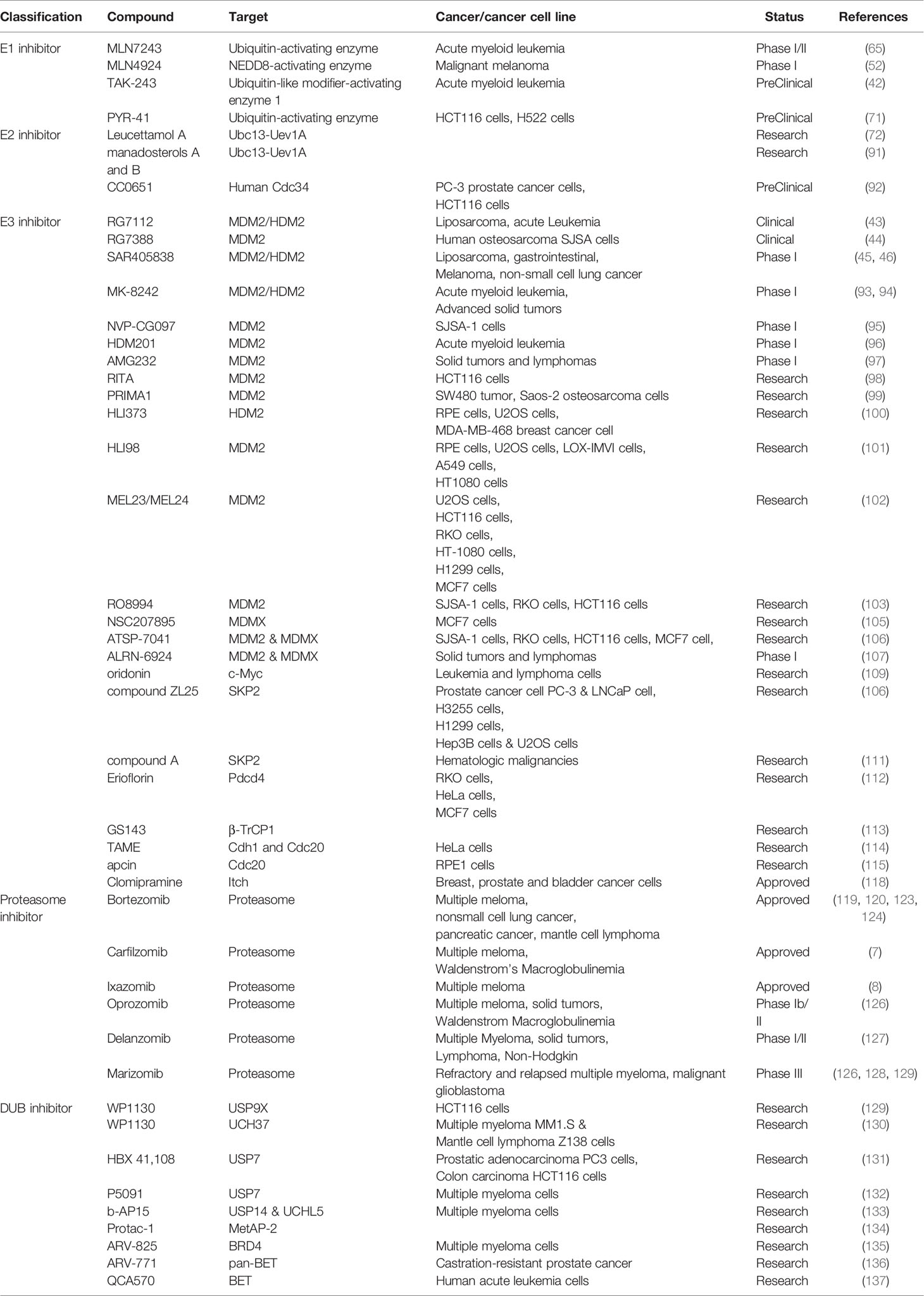
Table 3 Anti-cancer compounds in clinical trials targeting the ubiquitin-proteasome system and DUBs.
The E1 enzyme is responsible for activating ubiquitin molecules in the UPS, and several compounds have been identified to target E1. For example, adenosine sulfamate analogs, such as MLN7243 (126) and MLN4924, function as the ubiquitin-activating enzyme and NEDD8-activating enzyme inhibitors, respectively. They are currently undergoing Phase I/II and Phase I clinical trials (127, 139). Recently, TAK-243 was reported to induce leukemic cell death in preclinical models of acute myeloid leukemia cells through inhibition of the ubiquitin-like modifier-activating enzyme 1 (128). Experimental inhibitors of E1 have also been reported. For example, PYR-41, an irreversible inhibitor of ubiquitin E1, can inhibit the ubiquitylation of TRAF6 and decrease nuclear factor-kappa B activation. PYR-41 can also inhibit the degradation of p53 and activate its transcriptional activity (140). Due to lacking specificity, inhibition of E1 would cause remarkable side effects.
The E2 enzyme binds to E1, and then the activated ubiquitin is transferred to a cysteine of the E2 enzyme from the E1 enzyme. Thus, E2 enzymes mediate the conjugation of ubiquitin to substrates. Nowadays, several E2 inhibitors have been found to interfere with the process. For instance, Leucettamol A and manadosterols A and B, which are isolated from the sea sponges, inhibit the Ubc13-Uev1A interaction and block the E1-E2 complex formation (141, 142). Another example is CC0651, a small molecule inhibitor of the E2 enzyme hCdc34 (130). The E2 enzyme hCdc34 can ubiquitylate SCF (Skp2) substrate p27, and CC0651 decreases tumor cell growth by inhibiting p27 ubiquitylation and degradation.
E3 ligase recognizes substrate proteins and catalyzes the transfer of ubiquitin from E2 to target protein lysine. Therefore, E3 ligase has high substrate specificity which makes targeting E3 ligase become a promising tumor treatment strategy. So far, many studies have identified some compounds that could target specific E3 ligases and disturb UPS.
Due to the critical roles of p53 in regulating the genome, many efforts have been made to find the antagonists of E3 ligase MDM2/HDM2 to restore the function of p53. To date, a large number of inhibitors have been discovered based on MDM2-p53 interaction. Some of them are undergoing clinical assessment with different stages, such as RG7112 (129), RG7388 (131), SAR405838 (132, 143), MK-8242 (144, 145), NVP-CG097 (133), HDM201 (146), and AMG232 (147). Besides, more MDM2 inhibitors, such as RITA (134), PRIMA1 (135) HLI373 (148), HLI98 (149), MEL23 and MEL24 (150), and RO8994 (136) have been discovered to target MDM2 directly, thereby enhancing p53 activity and exhibiting anti-cancer ability.
MDMX/HDMX (murine/humans double minute X) shares significant homology with MDM2 and is also a negative regulator of p53. Though nutlin-3 has been found to inhibit MDM2-p53 but not MDMX-p53 interaction (151), NSC207895 targets MDMX specifically and acts addictively with nutlin-3a to activate p53 and induce apoptosis (137). Moreover, ATSP-7041 (152) and ALRN-6924 (153) decrease p53-dependent tumor growth as dual inhibitors of MDM2 and MDMX.
SCF (Skp1/cullin/F-box) E3 ligases are the largest family of E3 ubiquitin ligases. Their substrates play important roles in regulating the cell cycle, DNA replication, and signal transduction. Therefore, the dysregulation of these E3s often leads to cancer (154). Since FBPs are responsible for the specificity of SCFs, many small molecules are designed to target them. For instance, the natural compound oridonin enhances the ubiquitination and degradation of c-Myc mediated by FBW7, inducing apoptosis in leukemia and lymphoma cells (155). Furthermore, compound ZL25 inhibits SKP2 directly, resulting in the p53-independent cellular senescence in cancer cells (156). Another SKP2 inhibitor, compound A, induces p27-dependent cell cycle arrest and cell death by inhibition of SCF-SKP2 complexes formation (157). Erioflorin stabilizes the tumor suppressor Pdcd4 by blocking its interaction with β-TrCP1, suppresses the activity of AP-1 and NF-κB, and decelerates cancer cell proliferation (158). Another inhibitor, GS143, was shown to markedly decrease IκB ubiquitination by targeting β-TrCP1 and suppress the NF-κB signaling pathway (159).
Since Cdc20 is an oncogenic cofactor in the APC/C complex, many efforts have been made to find Cdc20 inhibitors to anti-cancer. TAME (tosyl-L-arginine methyl ester) was reported to bind to the APC complex. It could inhibit its activation by targeting both Cdh1 and Cdc20 and arrest cells in metaphase (160). Moreover, Apcin was found to bind directly to Cdc20, inhibiting the ubiquitylation of D-box-containing substrates, and subsequently inducing tumor cell death (161).
E3 ligase Cbl-b has been identified as a negative regulator of TCR signaling. When Cbl-b is inhibited, the T cell-mediated antitumor activity will be enhanced. Autologous peripheral blood mononuclear cells (PBMCs) from patients were collected and transfected with Cbl-b-siRNA, which were called APN401. The results of the Phase I clinical trial for APN401 revealed that its intravenous infusion in patients with refractory solid tumors was feasible and safe (162). Several small-molecule Cbl-b inhibitors have been discovered to decrease the ubiquitylation of TAM receptors and promote the activation of T cells as well as natural killer cells. They are expected to be utilized in combination with other approved agents in immunotherapy (163).
Itch, a HECT domain-containing E3 ligase, promotes the ubiquitylation of several proteins (e.g. p70, p63, c-Jun, JunB, Notch, and c-FLIP) and shows a potential target for cancer therapy. Rossi et al. identified that antidepressant drug clomipramine and its homologs could inhibit Itch auto-ubiquitylation and p73 ubiquitylation to reduce breast, prostate and bladder cancer cell growth by blocking autophagy (164).
Among all the UPS components, the proteasome has been successfully used as a target for cancer treatment. The proteasome is a large multi-protein complex containing multicatalytic proteases (e.g., chymotrypsin- and caspase-like enzyme) and is responsible for the degradation or processing of intracellular proteins. As such, it regulates the levels of some important mediators for cell-cycle progression and apoptosis in normal and malignant cells, such as cyclins, caspases, BCL2 and nuclear factor of κB (165). Bortezomib is the first proteasome inhibitor approved for recurrent refractory multiple meloma (MM) in 2003 (166, 167). It reversibly inhibits the activities of chymotrypsin- and caspase-like enzymes, leads to the apoptosis of MM cells, and suppresses the activation of NF-κB, production of cytokines (e.g., IL-6, IGF-1, and VEGF) in the tumor microenvironment, and adherence of myeloma cells to bone marrow stromal cells (165, 168). Later, it was extended to patients with non-small cell lung cancer, pancreatic cancer, and mantle cell lymphoma (169, 170). Although bortezomib has antitumor activity, it can cause side effects such as neuropathy and autophagy in some cases (171, 172). Besides, bortezomib resistance often occurred in about one year (173, 174). Carfilzomib, a second-in-class proteasome inhibitor drug, was approved in 2012 for MM by the US FDA (11). It irreversibly inhibits the chymotrypsin-like activities and shows improved safety in maintaining its cytotoxic potential in the bortezomib resistant cell lines (12). Carfilzomib treatment also causes adverse effects such as cardiovascular complications, hypertension, and heart failure, but they are reversible and manageable with careful monitoring. Both bortezomib and carfilzomib are not suitable for oral administration. Ixazomib is the first oral bioavailable proteasome inhibitor and was approved by the FDA in 2015. It reversibly inhibits the chymotrypsin-like activities and shows improved safety profiles over bortezomib, but its therapeutic advantages still need further investigation by randomized clinical trials (12).
The clinical successes of existing proteasome inhibitors encourage great efforts to discover more proteasome inhibitors with improved efficacy and safety. Thus, a lot of proteasome inhibitors have been identified including oprozomib, delanzomib and marizomib. Oprozomib is an orally available inhibitor with a homologous structure to carfilzomib. It is currently being studied in several clinical trials including a multicenter phase Ib/II trial for MM patients. Oprozomib can effectively decrease the viability of MM cells both in vitro and in vivo (175). Delanzomib, a reversible oral bioavailability of bortezomib analog, overcomes bortezomib’s resistance to peripheral neuropathy. But it causes severe skin toxicity to many patients (176). Marizomib, a novel proteasome inhibitor with a better therapeutic ratio, overcomes bortezomib resistance and exhibits broader anti-cancer activities (177). Moreover, marizomib has synergistic effects on refractory and recurrent MM patients with BTZ, linedoxamine, bormadoxamine and low dose dexamethasone (175, 178). In addition, marizomib can penetrate the blood-brain barrier and induces apoptosis in glioma cells with low toxicity on normal cells (179). Marizomib is currently being assessed in a phase III trial for the treatment of malignant glioblastoma in combination with temozolomide and radiotherapy.
Ubiquitination is a dynamic and reversible process and DUBs catalyze the removal of ubiquitin or polyubiquitin chains from the target protein. DUBs are actively involved in regulating tumorigenesis. Thus, DUB inhibitors are regarded as potential anti-cancer agents (180) To date, a number of DUB inhibitors have been identified to inhibit tumorigenesis (4, 10, 181).
WP1130, an inhibitor of DUBs, can suppress the activities of USP9X, USP5, USP14 and UCH37, deregulate anti-apoptotic protein MCL-1 and upregulate pro-apoptotic protein p53. It exhibits high anti-tumor activity (182). For example, the transcription factor E-twenty-six related gene (ERG) is overexpressed and promotes prostate carcinogenesis. Inhibition of USP9X by WP1130 leads to ERG degradation and inhibits tumor growth (183).
Recently, HBX 41,108, a small-molecule inhibitor of USP7, was reported to inhibit USP7-mediated p53 deubiquitination, stabilizing p53 and inducing p53-dependent apoptosis in cancer cells (184). Besides, P5091, a selective USP7 inhibitor, was found to induce apoptosis and overcome bortezomib resistance in MM cells. What’s more, it can inhibit tumor growth and exhibit synergistic anti-MM activity in combination with lenalidomide, HDAC inhibitor SAHA, or dexamethasone (185). A class of dual small molecule inhibitors of USP7 and USP47 has been identified to promote p53 activity and apoptosis in MM and B-cell leukemia cells in vitro and xenograft models (186).
Moreover, USP14 can inhibit the degradation of ubiquitin-protein conjugates in vitro and in vivo (187). The inhibitors of USP14 have been found to stimulate the proteasomal degradation of oxidized proteins, causing resistance to oxidative stress (188). Consistently, b-AP15 was shown to inhibit cell growth and overcome bortezomib resistance in MM cells by selectively blocking the deubiquitylating activity of USP14 and UCHL5 (189). These studies indicate that inhibiting specific oncogenic DUBs may be an effective anti-cancer approach.
Recently, emerging technologies based on PROTACs attract increasing attention in the pharmaceutical industry (190). PROTACs are heterobifunctional molecules that simultaneously bind a target protein and an E3 ubiquitin ligase, enabling ubiquitination and degradation of the target by the UPS in the cell (Figure 5) (13). PROTACs link the target protein to an E3 ubiquitin ligase by a designed hybrid molecule, providing a path for ubiquitinating undruggable proteins such as transcription factors, scaffolding proteins and nonenzymatic proteins. The first PROTACs were reported in 2001 by the Crews group and Ray Deshaies (191). They artificially synthesized a chimeric compound named Protac-1. Protac-1 has two domains: one domain contains the IκBa phosphopeptide that could recruit the F-box protein β-TrCP, and the other domain contains ovalicin which could bind to the target protein methionine aminopeptidase-2 (MetAP-2). As a result, MetAP-2 was ubiquitinated and degraded in a Protac-1-induced proteolysis manner.
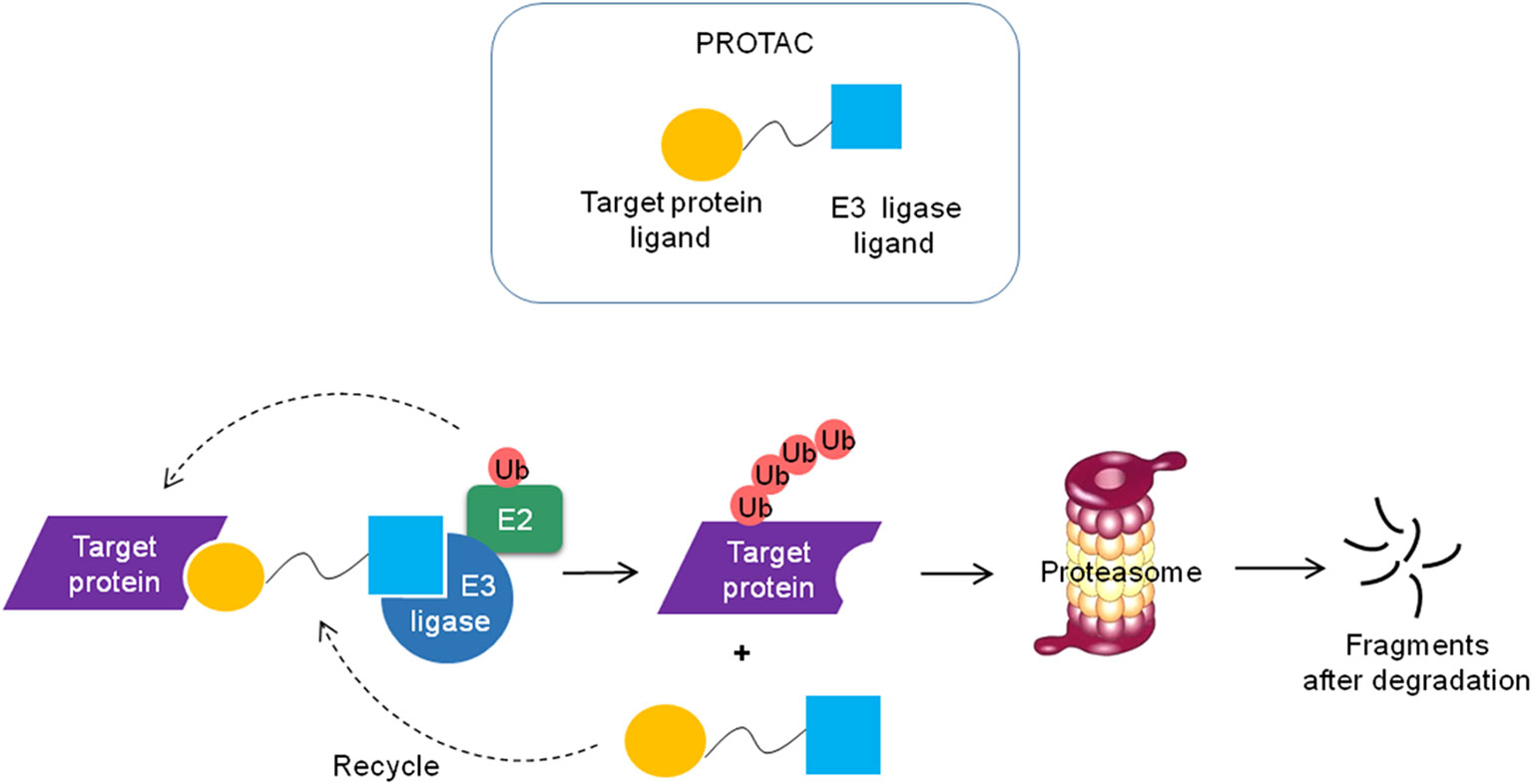
Figure 5 Schematic diagram of the PROTAC technology. The PROTC is a chimeric molecule that consists of two ligands, one is to interact with E3 ligase and the other is to bind the target protein. The target protein is polyubiquitinated and degraded by the proteasome and the PROTC molecule can be recycled.
Due to the excellent permeability and low working concentrations, small molecule-based PROTACs, which utilize small molecules to recruit E3 ubiquitin ligases, have more potential to be developed into drugs than peptide-based PROTACs (13). The PROTAC technology broadens the range of target proteins degraded by the UPS.
Recently, some transcriptional regulators (such as BRD4, TRIM24 and Smad3) have been reported to be targeted by PROTAC technologies (13). BRD4, a bromodomain and extraterminal domain (BET) family member, usually resides upstream of important oncogenes such as c-Myc, BCL-xL and BCL-6, and regulates their expressions. Therefore, BRD4 has become a promising therapeutic target in multiple cancer types. Preclinical studies of BRD4 inhibitors, JQ1 and OTX015, demonstrate their value in suppressing c-Myc expression and BL cell proliferation. However, owing to the reversible binding of inhibitors, the suppression is incomplete and requires high drug concentrations. Crews groups developed a bifunctional molecule, ARV-825, connecting the BRD4 inhibitor OTX015 to an E3 ligase cereblon binding moiety (pomalidomide) using PROTAC technology. As a result, ARV-825 actively recruits BRD4 to cereblon, leading to BRD4 efficient degradation via the proteasome in Burkitt’s Lymphoma cells. Moreover, ARV-825 treatment produces a more pronounced effect on the inhibition of c-Myc than that of the BRD4 inhibitors in five MM cell lines [SKO-007(J3), U266, RPMI-8226, ARP-1, JJN3] and an MM patient-derived CD138+ MM cells (192). In addition, Zengerle et al. designed another PROTAC, connecting JQ1 for BET family proteins and a ligand for VHL. Interestingly, the PROTAC not only triggered the degradation of BET family proteins particularly BRD4, but also regulated the transcription of BRD4 downstream genes such as Myc, p21 and AREG (193). In this way, it can also dampen the pro-inflammatory response in microglia, because BET proteins control the transcription of NF-κB-depended genes (194). These findings demonstrate that BRD4 PROTACs is a promising novel strategy to efficiently target BRD4 (195).
Raina and his colleagues reported that ARV-771 (another pan-BET inhibitor)-based PROTAC, dramatically suppressed androgen receptor (AR) protein level and AR signaling. It could lead to tumor regression in castration-resistant prostate cancer (CRPC) mouse xenograft model with more efficiency than BET inhibitors. This study provides evidence that small molecule-based PROTAC functions in a solid-tumor malignancy of CRPC (196). The results of BET-PROTACs ARV-825 and ARV-771 in the treatment of MCL cells demonstrate that they induce more apoptosis than BET inhibitors. Also, the results show that they can overcome the resistance of ibrutinib and exert a synergistic effect on apoptosis induction in the combination of other drugs such as ibrutinib, venetoclax (a BCL2-antagonist) and palbociclib (a CDK4/6 inhibitor) (197).
Recently, more BET-PROTACs have been designed. For instance, Qin et al. synthesized a BET-PROTAC called QCA570, utilizing a new class of BET inhibitors Oxazepines to recruit BET proteins. It could inhibit human acute leukemia cell proliferation at low picomolar concentrations, and abolish tumor growth in leukemia xenograft models in mice (198). Zhang and his colleagues demonstrated that BET-specific PROTACs were active against preclinical models of MM (199). Interestingly, the activity of BRD4-specific PROTACs can be improved over 100-fold through modification of hydroxylation of proline (200). In addition to the BET family, a functional PROTAC against TRIM24, another bromodomain-containing transcriptional regulator, has been designed and provides a path to find new undruggable targets (201). Wang et al. designed new PROTACs to prevent renal fibrosis by targeting SMAD3. They used hypoxia-inducible factor-1α to recruit VHL and screened compounds to bind SMAD3 from the Enamine library using the GLIDE molecular docking program. SMAD3 was degraded by PROTAC mediated ubiquitination (202). Thus, transcription factors can be targeted via PROTAC technology.
In addition, the undruggable transcription factors also can be degraded via alteration of the activity of an E3 ubiquitin ligase. For instance, Thalidomide and its derivatives Lenalidomide and Pomalidomide are effective drugs for the treatment of multiple myeloma and other B cell lymphomas. Thalidomide analogs bind Cereblon (CRBN), the substrate receptor of the CUL4-RBX1-DDB1-CRBN (CRL4CRBN) E3 ubiquitin ligase and alter its substrate selectivity to recruit, ubiquitinate and degrade unrelated transcription factors, such as Ikaros (IKZF1), Aiolos (IKZF3) and Casein kinase 1 alpha (CK1α) (203, 204). These findings provide a novel way to selective degrade specific targets through modulating the activity of an E3 ubiquitin ligase.
Ubiquitination of nonhistone proteins plays an important role in many cellular processes, including cell cycle, cell proliferation, DNA repair, apoptosis, inflammation, immune response, etc. Dysregulation of nonhistone lysine ubiquitylation is closely associated with the development of various human cancers. Therefore, UPS has been evolved as promising therapeutic targets for novel anti-cancer drugs. Nowadays, many proteasome inhibitors and E3 ligase modulators have been approved for anticancer treatment, whereas small-molecule inhibitor therapeutic strategies usually need high drug exposures and potentially increase the risk of off-target adverse effects. Fortunately, PROTAC technologies provide a path to target many undruggable proteins with UPS such as transcription factors.
To date, it remains an obstacle for the discovery of small molecule moiety to different targets. Another obstacle is specificity, how to get tissue-specific or disease-specific induced protein degradation? How to realize conditional triggered induced protein degradation? A deeper understanding of the tissue expression of E3 ligase and tumor microenvironment may provide a larger therapeutic window for appropriate PROTAC.
XZ wrote and drafted this article. TM and LF revised the manuscript critically. SC and DL drew the figures and prepared the table. QP and PW contributed to the drafting of the article and are responsible for the integrity of the work as a whole. All authors contributed to the article and approved the submitted version.
This work was supported by the Natural Science Foundation of Shandong Province (No. ZR2016CM46).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Suryadinata R, Roesley SN, Yang G, Sarcevic B. Mechanisms of generating polyubiquitin chains of different topology. Cells (2014) 3:674–89. doi: 10.3390/cells3030674
2. Erpapazoglou Z, Walker O, Haguenauer-Tsapis R. Versatile roles of k63-linked ubiquitin chains in trafficking. Cells (2014) 3:1027–88. doi: 10.3390/cells3041027
3. Haglund K, Di Fiore PP, Dikic I. Distinct monoubiquitin signals in receptor endocytosis. Trends Biochem Sci (2003) 28:598–603. doi: 10.1016/j.tibs.2003.09.005
4. Wei R, Liu X, Yu W, Yang T, Cai W, Liu J, et al. Deubiquitinases in cancer. Oncotarget (2015) 6:12872–89. doi: 10.1007/s10555-016-9640-2
5. Gross S, Rahal R, Stransky N, Lengauer C, Hoeflich KP. Targeting cancer with kinase inhibitors. J Clin Invest (2015) 125:1780–9. doi: 10.1172/JCI76094
6. Uckelmann M, Sixma TK. Histone ubiquitination in the DNA damage response. DNA Repair (Amst) (2017) 56:92–101. doi: 10.1016/j.dnarep.2017.06.011
7. Osley MA, Fleming AB, Kao CF. Histone ubiquitylation and the regulation of transcription. Results Probl Cell Differ (2006) 41:47–75. doi: 10.1007/400_006
8. Jeusset LM, McManus KA-O. Developing Targeted Therapies That Exploit Aberrant Histone Ubiquitination in Cancer. Cells (2019) 8:165. doi: 10.3390/cells8020165
9. Deng L, Meng T, Chen L, Wei W, Wang P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct Target Ther (2020) 5. doi: 10.1038/s41392-020-0107-0
10. Morrow JK, Lin HK, Sun SC, Zhang S. Targeting ubiquitination for cancer therapies. Future Med Chem (2015) 7:2333–50. doi: 10.4155/fmc.15.148
11. Piva R, Ruggeri B, Williams M, Costa G, Tamagno I, Ferrero D, et al. CEP-18770: A novel, orally active proteasome inhibitor with a tumor-selective pharmacologic profile competitive with bortezomib. Blood (2008) 111:2765–75. doi: 10.1182/blood-2007-07-100651
12. Park JE, Miller Z, Jun Y, Lee W, Kim KB. Next-generation proteasome inhibitors for cancer therapy. Transl Res (2018) 198:1–16. doi: 10.1016/j.trsl.2018.03.002
13. Zou Y, Ma D, Wang Y. The PROTAC technology in drug development. Cell Biochem Funct (2019) 37:21–30. doi: 10.1002/cbf.3369
14. Neklesa TK, Winkler JD, Crews CM. Targeted protein degradation by PROTACs. Pharmacol Ther (2017) 174:138–44. doi: 10.1016/j.pharmthera.2017.02.027
15. Wang D, Ma L, Wang B, Liu J, Wei W. E3 ubiquitin ligases in cancer and implications for therapies. Cancer Metastasis Rev (2017) 36:683–702. doi: 10.1007/s10555-017-9703-z
16. Senft D, Qi J, Ronai ZA. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat Rev Cancer (2018) 18:69–88. doi: 10.1038/nrc.2017.105
17. Bhattacharya S, Ghosh MK. Cell death and deubiquitinases: perspectives in cancer. BioMed Res Int (2014) 2014:435197. doi: 10.1155/2014/435197
18. Fujita T, Liu W, Doihara H, Date H, Wan Y. Dissection of the APCCdh1-Skp2 cascade in breast cancer. Clin Cancer Res (2008) 14:1966–75. doi: 10.1158/1078-0432.CCR-07-1585
19. Wang Z, Liu P, Inuzuka H, Wei W. Roles of F-box proteins in cancer. Nat Rev Cancer (2014) 14:233–47. doi: 10.1038/nrc3700
20. Aydin IT, Melamed RD, Adams SJ, Castillo-Martin M, Demir A, Bryk D, et al. FBXW7 mutations in melanoma and a new therapeutic paradigm. J Natl Cancer Inst (2014) 106:dju107. doi: 10.1093/jnci/dju107
21. Xu J, Wu W, Wang J, Huang C, Wen W, Zhao F, et al. miR-367 promotes the proliferation and invasion of non-small cell lung cancer via targeting FBXW7. Oncol Rep (2017) 37:1052–8. doi: 10.3892/or.2016.5314
22. Gong J, Cui Z, Li L, Ma Q, Wang Q, Gao Y, et al. MicroRNA-25 promotes gastric cancer proliferation, invasion, and migration by directly targeting F-box and WD-40 Domain Protein 7, FBXW7. Tumour Biol (2015) 36(10):7831–40. doi: 10.1007/s13277-015-3510-3
23. Zhang W, Cao L, Sun Z, Xu J, Tang L, Chen W, et al. Skp2 is over-expressed in breast cancer and promotes breast cancer cell proliferation. Cell Cycle (2016) 15:1344–51. doi: 10.1080/15384101.2016.1160986
24. King B, Trimarchi T, Reavie L, Xu L, Mullenders J, Ntziachristos P, et al. The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell (2013) 153:1552–66. doi: 10.1016/j.cell.2013.05.041
25. Choi SH, Wright JB, Gerber SA, Cole MD. Myc protein is stabilized by suppression of a novel E3 ligase complex in cancer cells. Genes Dev (2010) 24:1236–41. doi: 10.1101/gad.1920310
26. Hung WC, Tseng WL, Shiea J, Chang HC. Skp2 overexpression increases the expression of MMP-2 and MMP-9 and invasion of lung cancer cells. Cancer Lett (2010) 288:156–61. doi: 10.1016/j.canlet.2009.06.032
27. Adhikary S, Marinoni F, Hock A, Hulleman E, Popov N, Beier R, et al. The ubiquitin ligase HectH9 regulates transcriptional activation by Myc and is essential for tumor cell proliferation. Cell (2005) 123:409–21. doi: 10.1016/j.cell.2005.08.016
28. Müerköster S, Arlt A, Sipos B, Witt M, Grossmann M, Klöppel G, et al. Increased expression of the E3-ubiquitin ligase receptor subunit betaTRCP1 relates to constitutive nuclear factor-kappaB activation and chemoresistance in pancreatic carcinoma cells. Cancer Res (2005) 65:1316–24. doi: 10.1158/0008-5472.CAN-04-1626
29. Veeriah S, Taylor BS, Meng S, Fang F, Yilmaz E, Vivanco I, et al. Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat Genet (2010) 42:77–82. doi: 10.1038/ng.491
30. Poulogiannis G, McIntyre RE, Dimitriadi M, Apps JR, Wilson CH, Ichimura K, et al. PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proc Natl Acad Sci USA (2010) 107:15145–50. doi: 10.1073/pnas.1009941107
31. Darling S, Fielding AB, Sabat-Pospiech D, Prior IA, Coulson JM. Regulation of the cell cycle and centrosome biology by deubiquitylases. Biochem Soc Trans (2017) 45:1125–36. doi: 10.1042/BST2017008
32. Bhattacharya S, Ghosh MK. HAUSP, a novel deubiquitinase for Rb - MDM2 the critical regulator. FEBS J (2014) 281(13):3061–78. doi: 10.1111/febs.12843
33. Marine JC. MDM2 and MDMX in cancer and development. Curr Top Dev Biol (2011) 94:45–75. doi: 10.1016/B978-0-12-380916-2.00003-6
34. Brooks CL, Li M, Hu M, Shi Y, Gu W. The p53–Mdm2–HAUSP complex is involved in p53 stabilization by HAUSP. Oncogene (2007) 26:7262–6. doi: 10.1038/sj.onc.1210531
35. Kim EJ, Kim SH, Jin X, Kim H. KCTD2, an adaptor of Cullin3 E3 ubiquitin ligase, suppresses gliomagenesis by destabilizing c-Myc. Cell Death Differ (2017) 24:649–59. doi: 10.1038/cdd.2016.151
36. Paul I, Ahmed SF, Bhowmik A, Deb S, Ghosh MK. The ubiquitin ligase CHIP regulates c-Myc stability and transcriptional activity. Oncogene (2013) 32:1284–95. doi: 10.1038/onc.2012.144
37. Li S, Jiang C, Pan J, Wang X, Jin J, Zhao L, et al. Regulation of c-Myc protein stability by proteasome activator REGγ. Cell Death Differ (2015) 22:1000–11. doi: 10.1038/cdd.2014.188
38. Popov N, Wanzel M, Madiredjo M, Zhang D, Beijersbergen R, Bernards R, et al. The ubiquitin-specific protease USP28 is required for MYC stability. Nat Cell Biol (2007) 9:765–74. doi: 10.1038/ncb1601
39. Hassan M, Watari H, AbuAlmaaty A, Ohba Y, Sakuragi N. Apoptosis and molecular targeting therapy in cancer. BioMed Res Int (2014) 2014:150845. doi: 10.1155/2014/150845
40. Hu L, Wang J, Liu Y, Zhang Y, Zhang L, Kong R, et al. A small ribosomal subunit (SSU) processome component, the human U3 protein 14A (hUTP14A) binds p53 and promotes p53 degradation. J Biol Chem (2011) 286:3119–28. doi: 10.1074/jbc.M110.157842
41. Wiltshire TD, Lovejoy CA, Wang T, Xia F, O’Connor MJ, Cortez D. Sensitivity to poly(ADP-ribose) polymerase (PARP) inhibition identifies ubiquitin-specific peptidase 11 (USP11) as a regulator of DNA double-strand break repair. J Biol Chem (2010) 285:14565–71. doi: 10.1074/jbc.M110.104745
42. da Silva CG, Minussi DC, Ferran C, Bredel M. A20 expressing tumors and anticancer drug resistance. Adv Exp Med Biol (2014) 809:65–81. doi: 10.1007/978-1-4939-0398-6_5
43. Chen Z, Barbi J, Bu S, Yang H-Y, Li Z, Gao Y, et al. The ubiquitin ligase Stub1 negatively modulates regulatory T cell suppressive activity by promoting degradation of the transcription factor Foxp3. Immunity (2013) 39:272–85. doi: 10.1016/j.immuni.2013.08.006
44. Zhao Y, Guo H, Qiao G, Zucker M, Langdon WY, Zhang J. E3 Ubiquitin Ligase Cbl-b Regulates Thymic-Derived CD4+CD25+ Regulatory T Cell Development by Targeting Foxp3 for Ubiquitination. J Immunol (Baltimore Md 1950) (2015) 194:1639–45. doi: 10.4049/jimmunol.1402434
45. Lee JH, Elly C, Park Y, Liu Y-C. E3 Ubiquitin Ligase VHL Regulates Hypoxia-Inducible Factor-1α to Maintain Regulatory T Cell Stability and Suppressive Capacity. Immunity (2015) 42:1062–74. doi: 10.1016/j.immuni.2015.05.016
46. Jin H-S, Park Y, Elly C, Liu Y-C. Itch expression by Treg cells controls Th2 inflammatory responses. J Clin Invest (2013) 123:4923–34. doi: 10.1172/JCI69355
47. Eletr ZM, Wilkinson KD. An emerging model for BAP1’s role in regulating cell cycle progression. Cell Biochem Biophys (2011) 60:3–11. doi: 10.1007/s12013-011-9184-6
48. Machida YJ, Machida Y, Vashisht AA, Wohlschlegel JA, Dutta A. The deubiquitinating enzyme BAP1 regulates cell growth via interaction with HCF-1. J Biol Chem (2009) 284:34179–88. doi: 10.1074/jbc.M109.046755
49. Bonacci T, Suzuki A, Grant GD, Stanley N, Cook JG, Brown NG, et al. Cezanne/OTUD7B is a cell cycle-regulated deubiquitinase that antagonizes the degradation of APC/C substrates. EMBOJ (2018) 37. doi: 10.15252/embj.201798701
50. Arceci A, Bonacci T, Wang X, Stewart K, Damrauer JS, Hoadley KA, et al. FOXM1 Deubiquitination by USP21 Regulates Cell Cycle Progression and Paclitaxel Sensitivity in Basal-like Breast Cancer. Cell Rep (2019) 26:3076–86.e3076. doi: 10.1016/j.celrep.2019.02.054
51. Kaistha BP, Krattenmacher A, Fredebohm J, Schmidt H, Behrens D, Widder M, et al. The deubiquitinating enzyme USP5 promotes pancreatic cancer via modulating cell cycle regulators. Oncotarget (2017) 8:66215–25. doi: 10.18632/oncotarget.19882
52. Li Y, Luo K, Yin Y, Wu C, Deng M, Li L, et al. USP13 regulates the RAP80-BRCA1 complex dependent DNA damage response. Nat Commun (2017) 8:15752. doi: 10.1038/ncomms15752
53. Li XY, Wu HY, Mao XF, Jiang LX, Wang YX. USP5 promotes tumorigenesis and progression of pancreatic cancer by stabilizing FoxM1 protein. Biochem Biophys Res Commun (2017) 492:48–54. doi: 10.1016/j.bbrc.2017.08.040
54. Nepal S, Shrestha A, Park PH. Ubiquitin specific protease 2 acts as a key modulator for the regulation of cell cycle by adiponectin and leptin in cancer cells. Mol Cell Endocrinol (2015) 412:44–55. doi: 10.1016/j.mce.2015.05.029
55. Liu B, Liu Y, Wang Y, Xie C, Gan M, Han T, et al. CyclinB1 deubiquitination by USP14 regulates cell cycle progression in breast cancer. Pathol Res Pract (2019) 215:152592. doi: 10.1016/j.prp.2019.152592
56. Sun X-X, Challagundla KB, Dai M-S. Positive regulation of p53 stability and activity by the deubiquitinating enzyme Otubain 1. EMBO J (2012) 31:576–92. doi: 10.1038/emboj.2011.434
57. Stegmeier F, Rape M, Draviam VM, Nalepa G, Sowa ME, Ang XL, et al. Anaphase initiation is regulated by antagonistic ubiquitination and deubiquitination activities. Nature (2007) 446:876–81. doi: 10.1038/nature05694
58. Zhang Y, van Deursen J, Galardy PJ. Overexpression of ubiquitin specific protease 44 (USP44) induces chromosomal instability and is frequently observed in human T-cell leukemia. PloS One (2011) 6:e23389. doi: 10.1371/journal.pone.0023389
59. Huang X, Summers MK, Pham V, Lill JR, Liu J, Lee G, et al. Deubiquitinase USP37 is activated by CDK2 to antagonize APC(CDH1) and promote S phase entry. Mol Cell (2011) 42:511–23. doi: 10.1016/j.molcel.2011.03.027
60. Geng F, Wenzel S, Tansey WP. Ubiquitin and proteasomes in transcription. Annu Rev Biochem (2012) 81:177–201. doi: 10.1146/annurev-biochem-052110-120012
61. Sun XX, He X, Yin L, Komada M, Sears RC, Dai MS. The nucleolar ubiquitin-specific protease USP36 deubiquitinates and stabilizes c-Myc. Proc Natl Acad Sci USA (2015) 112:3734–9. doi: 10.1073/pnas.1411713112
62. Murali R, Wiesner T, Scolyer RA. Tumours associated with BAP1 mutations. Pathology (2013) 45:116–26. doi: 10.1097/PAT.0b013e32835d0efb
63. Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer (2013) 13:83–96. doi: 10.1038/nrc3430
64. Schoenfeld AR, Apgar S, Dolios G, Wang R, Aaronson SA. BRCA2 is ubiquitinated in vivo and interacts with USP11, a deubiquitinating enzyme that exhibits prosurvival function in the cellular response to DNA damage. Mol Cell Biol (2004) 24:7444–55. doi: 10.1128/MCB.24.17.7444-7455.2004
65. Liu J, Kruswick A, Dang H, Tran AD, Kwon SM, Wang XW, et al. Ubiquitin-specific protease 21 stabilizes BRCA2 to control DNA repair and tumor growth. Nat Commun (2017) 8:137. doi: 10.1038/s41467-017-00206-2
66. Chen J, Chen ZJ. Regulation of NF-κB by ubiquitination. Curr Opin Immunol (2013) 25:4–12. doi: 10.1016/j.coi.2012.12.005
67. Pan J, Deng Q, Jiang C, Wang X, Niu T, Li H, et al. USP37 directly deubiquitinates and stabilizes c-Myc in lung cancer. Oncogene (2015) 34:3957–67. doi: 10.1038/onc.2014.327
68. Kim D, Hong A, Park HI, Shin WH, Yoo L, Jeon SJ, et al. Deubiquitinating enzyme USP22 positively regulates c-Myc stability and tumorigenic activity in mammalian and breast cancer cells. J Cell Physiol (2017) 232:3664–76. doi: 10.1002/jcp.25841
69. Zhang J, Ren P, Xu D, Liu X, Liu Z, Zhang C, et al. Human UTP14a promotes colorectal cancer progression by forming a positive regulation loop with c-Myc. Cancer Lett (2019) 440-441:106–15. doi: 10.1016/j.canlet.2018.10.010
70. Ma T, Lu C, Guo Y, Zhang C, Du X. Human U3 protein 14a plays an anti-apoptotic role in cancer cells. Biol Chem (2017) 398:1247–57. doi: 10.1515/hsz-2017-0121
71. Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D, Courtois G. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature (2003) 424:801–5. doi: 10.1038/nature01802
72. Xia A, Zhang Y, Xu J, Yin T, Lu X-J. T Cell Dysfunction in Cancer Immunity and Immunotherapy. Front Immunol (2019) 10:1719. doi: 10.3389/fimmu.2019.01719
73. Abbas T, Keaton MA, Dutta A. Genomic instability in cancer. Cold Spring Harb Perspect Biol (2013) 5:a012914. doi: 10.1101/cshperspect.a012914
74. Wu WJ, Hu KS, Wang DS, Zeng ZL, Zhang DS, Chen DL, et al. CDC20 overexpression predicts a poor prognosis for patients with colorectal cancer. J Transl Med (2013) 11:142. doi: 10.1186/1479-5876-11-142
75. Korphaisarn K, Morris VK, Overman MJ, Fogelman DR, Kee BK, Raghav KPS, et al. FBXW7 missense mutation: a novel negative prognostic factor in metastatic colorectal adenocarcinoma. Oncotarget (2017) 8:39268–79. doi: 10.18632/oncotarget.16848
76. Ougolkov A, Zhang B, Yamashita K, Bilim V, Mai M, Fuchs SY, et al. Associations among beta-TrCP, an E3 ubiquitin ligase receptor, beta-catenin, and NF-kappaB in colorectal cancer. J Natl Cancer Inst (2004) 96:1161–70. doi: 10.1093/jnci/djh219
77. Johnson DG, Schneider-Broussard R. Role of E2F in cell cycle control and cancer. Front Biosci (1998) 3:d447–8. doi: 10.2741/a291
78. Helin K, Harlow E, Fattaey A. Inhibition of E2F-1 transactivation by direct binding of the retinoblastoma protein. Mol Cell Biol (1993) 13:6501–8. doi: 10.1128/mcb.13.10.6501
79. Miwa S, Uchida C, Kitagawa K, Hattori T, Oda T, Sugimura H, et al. Mdm2-mediated pRB downregulation is involved in carcinogenesis in a p53-independent manner. Biochem Biophys Res Commun (2006) 340:54–61. doi: 10.1016/j.bbrc.2005.11.148
80. Pan H, Jia R, Zhang L, Xu S, Wu Q, Song X, et al. BAP1 regulates cell cycle progression through E2F1 target genes and mediates transcriptional silencing via H2A monoubiquitination in uveal melanoma cells. Int J Biochem Cell Biol (2015) 60:176–84. doi: 10.1016/j.biocel.2015.01.001
81. Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol (1999) 19:1–11. doi: 10.1128/mcb.19.1.1
83. Farrell AS, Sears RC. MYC degradation. Cold Spring Harb Perspect Med (2014) 4:a014365. doi: 10.1101/cshperspect.a014365
84. Welcker M, Orian A, Jin J, Grim JE, Grim JA, Harper JW, et al. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci USA (2004) 101:9085–90. doi: 10.1073/pnas.0402770101
85. Kim SY, Herbst A, Tworkowski KA, Salghetti SE, Tansey WP. Skp2 regulates Myc protein stability and activity. Mol Cell (2003) 11:1177–88. doi: 10.1016/S1097-2765(03)00173-4
86. Yap DB, Hsieh JK, Chan FS, Lu X. mdm2: a bridge over the two tumour suppressors, p53 and Rb. Oncogene (1999) 18:7681–9. doi: 10.1038/sj.onc.1202954
87. Kon N, Kobayashi Y, Li M, Brooks CL, Ludwig T, Gu W. Inactivation of HAUSP in vivo modulates p53 function. Oncogene (2010) 29:1270–9. doi: 10.1038/onc.2009.427
88. Stevenson LF, Sparks A, Allende-Vega N, Xirodimas DP, Lane DP, Fau - Saville MK, Saville MK. The deubiquitinating enzyme USP2a regulates the p53 pathway by targeting Mdm2. EMBO J (2007) 26:976–86. doi: 10.1038/sj.emboj.7601567
89. Yuan J, Luo K, Zhang L, Cheville JC, Lou Z. USP10 regulates p53 localization and stability by deubiquitinating p53. Cell (2010) 140:384–96. doi: 10.1016/j.cell.2009.12.032
90. Liu H, Wang J, Liu Y, Hu L, Zhang C, Xing B, et al. Human U3 protein14a is a novel type ubiquitin ligase that binds RB and promotes RB degradation depending on a leucine-rich region. Biochim Biophys Acta Mol Cell Res (2018) 1865:1611–20. doi: 10.1016/j.bbamcr.2018.08.016
91. Fujita Y, Tinoco R, Li Y, Senft D, Ronai Z. Ubiquitin Ligases in Cancer Immunotherapy - Balancing Antitumor and Autoimmunity. Trends Mol Med (2019) 25:428–43. doi: 10.1016/j.molmed.2019.02.002
92. Meng X, Liu X, Guo X, Jiang S, Chen T, Hu Z, et al. FBXO38 mediates PD-1 ubiquitination and regulates anti-tumour immunity of T cells. Nature (2018) 564:130–5. doi: 10.1038/s41586-018-0756-0
93. Nurieva RI, Zheng S, Jin W, Chung Y, Zhang Y, Martinez GJ, et al. The E3 ubiquitin ligase GRAIL regulates T cell tolerance and regulatory T cell function by mediating T cell receptor-CD3 degradation. Immunity (2010) 32:670–80. doi: 10.1016/j.immuni.2010.05.002
94. Chiang JY, Jang IK, Hodes R, Gu H. Ablation of Cbl-b provides protection against transplanted and spontaneous tumors. J Clin Invest (2007) 117:1029–36. doi: 10.1172/JCI29472
95. Haymaker C, Yang Y, Wang J, Zou Q, Sahoo A, Alekseev A, et al. Absence of Grail promotes CD8 T cell anti-tumour activity. Nat Commun (2017) 8:239. doi: 10.1172/JCI29472
96. Zhang L, Yi H, Xia X-P, Zhao Y. Transforming growth factor-beta: an important role in CD4+CD25+ regulatory T cells and immune tolerance. Autoimmunity (2006) 39:269–76. doi: 10.1080/08916930600753903
97. Gorelik L, Flavell RA. Transforming growth factor-beta in T-cell biology. Nat Rev Immunol (2002) 2:46–53. doi: 10.1038/nri704
98. Thomas DA, Massagué J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell (2005) 8:369–80. doi: 10.1016/j.ccr.2005.10.012
99. Li MO, Wan YY, Sanjabi S, Robertson A-KL, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol (2006) 24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737
101. Kumari N, Jaynes PW, Saei A, Iyengar PV, Richard JLC, Eichhorn PJA. The roles of ubiquitin modifying enzymes in neoplastic disease. Biochim Biophys Acta Rev Cancer (2017) 1868:456–83. doi: 10.1016/j.bbcan.2017.09.002
102. Lallemand F, Seo SR, Ferrand N, Pessah M, L’Hoste S, Rawadi G, et al. AIP4 restricts transforming growth factor-beta signaling through a ubiquitination-independent mechanism. J Biol Chem (2005) 280:27645–53. doi: 10.1074/jbc.M500188200
103. Seo SR, Lallemand F, Ferrand N, Pessah M, L’Hoste S, Camonis J, et al. The novel E3 ubiquitin ligase Tiul1 associates with TGIF to target Smad2 for degradation. EMBO J (2004) 23:3780–92. doi: 10.1038/sj.emboj.7600398
104. Komuro A, Imamura T, Saitoh M, Yoshida Y, Yamori T, Miyazono K, et al. Negative regulation of transforming growth factor-beta (TGF-beta) signaling by WW domain-containing protein 1 (WWP1). Oncogene (2004) 23:6914–23. doi: 10.1038/sj.onc.1207885
105. Ebisawa T, Fukuchi M, Murakami G, Chiba T, Tanaka K, Imamura T, et al. Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J Biol Chem (2001) 276:12477–80. doi: 10.1074/jbc.C100008200
106. Kuratomi G, Komuro A, Goto K, Shinozaki M, Miyazawa K, Miyazono K, et al. NEDD4-2 (neural precursor cell expressed, developmentally down-regulated 4-2) negatively regulates TGF-beta (transforming growth factor-beta) signalling by inducing ubiquitin-mediated degradation of Smad2 and TGF-beta type I receptor. Biochem J (2005) 86:461–70. doi: 10.1042/BJ20040738
107. Kit Leng Lui S, Iyengar PV, Jaynes P, Isa Z, Pang B, Tan TZ. Eichhorn PA-OX: USP26 regulates TGF-β signaling by deubiquitinating and stabilizing SMAD7. EMBO Rep (2017) 18:797–808. doi: 10.15252/embr.201643270
108. Lin X, Liang M Fau - Feng XH, Feng XH. Smurf2 is a ubiquitin E3 ligase mediating proteasome-dependent degradation of Smad2 in transforming growth factor-beta signaling. J Biol Chem (2000) 275:36818–22. doi: 10.1074/jbc.C000580200
109. Fukuchi M, Imamura T, Chiba T, Ebisawa T, Kawabata M, Tanaka K, et al. Ligand-dependent degradation of Smad3 by a ubiquitin ligase complex of ROC1 and associated proteins. Mol Biol Cell (2001) 12:1431–43. doi: 10.1091/mbc.12.5.1431
110. Xin H, Xu X, Li L, Ning H, Rong Y, Shang Y, et al. CHIP controls the sensitivity of transforming growth factor-beta signaling by modulating the basal level of Smad3 through ubiquitin-mediated degradation. J Biol Chem (2005) 280:20842–50. doi: 10.1074/jbc.M412275200
111. Morén A, Hellman U, Inada Y, Imamura T, Heldin CH, Moustakas A. Differential ubiquitination defines the functional status of the tumor suppressor Smad4. J Biol Chem (2003) 278:33571–82. doi: 10.1074/jbc.M300159200
112. Liang M, Liang YY, Wrighton K, Ungermannova D, Wang XP, Brunicardi FC, et al. Ubiquitination and proteolysis of cancer-derived Smad4 mutants by SCFSkp2. Mol Cell Biol (2004) 24:7524–37. doi: 10.1128/MCB.24.17.7524-7537.2004
113. Wan M, Tang Y, Tytler EM, Lu C, Jin B, Vickers SM, et al. Smad4 protein stability is regulated by ubiquitin ligase SCF beta-TrCP1. J Biol Chem (2004) 279:14484–7. doi: 10.1074/jbc.C400005200
114. Zhang L, Zhou F, Drabsch Y, Gao R, Snaar-Jagalska BE, Mickanin C, et al. USP4 is regulated by AKT phosphorylation and directly deubiquitylates TGF-β type I receptor. Nat Cell Biol (2012) 14:717–26. doi: 10.1038/ncb2522
115. Al-Salihi MA, Herhaus L, Macartney T, Sapkota GP. USP11 augments TGFβ signalling by deubiquitylating ALK5. Open Biol (2012) 2:120063. doi: 10.1098/rsob.120063
116. Eichhorn PJ, Rodón L, Gonzàlez-Juncà A, Dirac A, Gili M, Martínez-Sáez E, et al. USP15 stabilizes TGF-β receptor I and promotes oncogenesis through the activation of TGF-β signaling in glioblastoma. Nat Med (2012) 18:429–35. doi: 10.1038/nm.2619
117. Briones-Orta MA, Levy L, Madsen CD, Das D, Erker Y, Sahai E, et al. Arkadia regulates tumor metastasis by modulation of the TGF-β pathway. Cancer Res (2013) 73:1800–10. doi: 10.1158/0008-5472.CAN-12-1916
118. Herhaus L, Al-Salihi M, Macartney T, Weidlich S, Sapkota GP. OTUB1 enhances TGFβ signalling by inhibiting the ubiquitylation and degradation of active SMAD2/3. Nat Commun (2013) 4:2519. doi: 10.1038/ncomms3519
119. Nagano Y, Mavrakis KJ, Lee KL, Fujii T, Koinuma D, Sase H, et al. Arkadia induces degradation of SnoN and c-Ski to enhance transforming growth factor-beta signaling. J Biol Chem (2007) 282:20492–501. doi: 10.1074/jbc.M701294200
120. Bonni S, Wang HR, Causing CG, Kavsak P, Stroschein SL, Luo K, et al. TGF-beta induces assembly of a Smad2-Smurf2 ubiquitin ligase complex that targets SnoN for degradation. Nat Cell Biol (2001) 3:587–95. doi: 10.1038/35078562
121. Levine AG, Mendoza A, Hemmers S, Moltedo B, Niec RE, Schizas M, et al. Stability and function of regulatory T cells expressing the transcription factor T-bet. Nature (2017) 546:421–5. doi: 10.1038/nature22360
122. Pan L, Chen Z, Wang L, Chen C, Li D, Wan H, et al. Deubiquitination and stabilization of T-bet by USP10. Biochem Biophys Res Commun (2014) 449:289–94. doi: 10.1016/j.bbrc.2014.05.037
123. Sitaram P, Uyemura B, Malarkannan S, Riese MJ. Beyond the Cell Surface: Targeting Intracellular Negative Regulators to Enhance T cell Anti-Tumor Activity. Int J Mol Sci (2019) 20:5821. doi: 10.3390/ijms20235821
124. Jahan AS, Lestra M, Swee LK, Fan Y, Lamers MM, Tafesse FG, et al. Usp12 stabilizes the T-cell receptor complex at the cell surface during signaling. Proc Natl Acad Sci U S A (2016) 113:E705–14. doi: 10.1073/pnas.1521763113
125. Naik E, Webster JD, DeVoss J, Liu J, Suriben R, Dixit VM. Regulation of proximal T cell receptor signaling and tolerance induction by deubiquitinase Usp9X. J Exp Med (2014) 211:1947–55. doi: 10.1084/jem.20140860
126. Hyer ML, Milhollen MA, Ciavarri J, Fleming P, Traore T, Sappal D, et al. A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment. Nat Med (2018) 24:186–93. doi: 10.1038/nm.4474
127. Nawrocki ST, Griffin P, Kelly KR, Carew JS. MLN4924: a novel first-in-class inhibitor of NEDD8-activating enzyme for cancer therapy. Expert Opin Invest Drugs (2012) 21:1563–73. doi: 10.1517/13543784.2012.707192
128. Barghout SH, Patel PS, Wang X, Xu GW, Kavanagh S, Halgas O, et al. Preclinical evaluation of the selective small-molecule UBA1 inhibitor, TAK-243, in acute myeloid leukemia. Leukemia (2019) 33:37–51. doi: 10.1038/s41375-018-0167-0
129. Vu B, Wovkulich P, Pizzolato G, Lovey A, Ding Q, Jiang N, et al. Discovery of RG7112: A Small-Molecule MDM2 Inhibitor in Clinical Development. ACS Med Chem Lett (2013) 4:466–9. doi: 10.1021/ml4000657
130. Ceccarelli DF, Tang X, Pelletier B, Orlicky S, Xie W, Plantevin V, et al. An allosteric inhibitor of the human Cdc34 ubiquitin-conjugating enzyme. Cell (2011) 145:1075–87. doi: 10.1016/j.cell.2011.05.039
131. Ding Q, Zhang Z, Liu J-J, Jiang N, Zhang J, Ross TM, et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J Med Chem (2013) 56:5979–83. doi: 10.1021/jm400487c
132. Wang S, Sun W, Zhao Y, McEachern D, Meaux I, Barrière C, et al. SAR405838: an optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res (2014) 74:5855–65. doi: 10.1158/0008-5472.CAN-14-0799
133. Holzer P, Masuya K, Furet P, Kallen J, Valat-Stachyra T, Ferretti S, et al. Discovery of a Dihydroisoquinolinone Derivative (NVP-CGM097): A Highly Potent and Selective MDM2 Inhibitor Undergoing Phase 1 Clinical Trials in p53wt Tumors. J Med Chem (2015) 58:6348–58. doi: 10.1021/acs.jmedchem.5b00810
134. Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, Masucci M, et al. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med (2004) 10:1321–8. doi: 10.1038/nm1146
135. Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med (2002) 8:282–8. doi: 10.1038/nm0302-282
136. Zhang Z, Ding Q, Liu J-J, Zhang J, Jiang N, Chu X-J, et al. Discovery of potent and selective spiroindolinone MDM2 inhibitor, RO8994, for cancer therapy. Bioorg Med Chem (2014) 22:4001–9. doi: 10.1016/j.bmc.2014.05.072
137. Wang H, Ma X, Ren S, Buolamwini JK, Yan C. A small-molecule inhibitor of MDMX activates p53 and induces apoptosis. Mol Cancer Ther (2011) 10:69–79. doi: 10.4161/cc.9.6.11067
138. Nalepa G, Rolfe M, Harper JW. Drug discovery in the ubiquitin-proteasome system. Nat Rev Drug Discovery (2006) 5:596–613. doi: 10.1038/nrd2056
139. Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature (2009) 458:732–6. doi: 10.1038/nature07884
140. Yang Y, Kitagaki J, Dai RM, Tsai YC, Lorick KL, Ludwig RL, et al. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res (2007) 67:9472–81. doi: 10.1158/0008-5472.CAN-07-0568
141. Tsukamoto S, Takeuchi T, Rotinsulu H, Mangindaan RE, van Soest RW, Ukai K, et al. Leucettamol A: a new inhibitor of Ubc13-Uev1A interaction isolated from a marine sponge, Leucetta aff. microrhaphis. Bioorg Med Chem Lett (2008) 18:6319–20. doi: 10.1016/j.bmcl.2008.10.110
142. Ushiyama S, Umaoka H, Kato H, Suwa Y, Morioka H, Rotinsulu H, et al. Manadosterols A and B, sulfonated sterol dimers inhibiting the Ubc13-Uev1A interaction, isolated from the marine sponge Lissodendryx fibrosa. J Nat Prod (2012) 75:1495–9. doi: 10.1021/np300352u
143. de Jonge M, de Weger VA, Dickson MA, Langenberg M, Le Cesne A, Wagner AJ, et al. A phase I study of SAR405838, a novel human double minute 2 (HDM2) antagonist, in patients with solid tumours. Eur J Cancer (Oxford Engl 1990) (2017) 76:144–51. doi: 10.1016/j.ejca.2017.02.005
144. Ravandi F, Gojo I, Patnaik MM, Minden MD, Kantarjian H, Johnson-Levonas AO, et al. A phase I trial of the human double minute 2 inhibitor (MK-8242) in patients with refractory/recurrent acute myelogenous leukemia (AML). Leuk Res (2016) 48:92–100. doi: 10.1016/j.leukres.2016.07.004
145. Wagner AJ, Banerji U, Mahipal A, Somaxiah N, Hirsch H, Fancourt C, et al. Phase I Trial of the Human Double Minute 2 Inhibitor MK-8242 in Patients With Advanced Solid Tumors. J Clin Oncol (2017) 35:1304–11. doi: 10.1200/JCO.2016.70.7117
146. Seipel K, Marques MAT, Sidler C, Mueller BU, Pabst T. MDM2- and FLT3-inhibitors in the treatment of -ITD acute myeloid leukemia, specificity and efficacy of NVP-HDM201 and midostaurin. Haematologica (2018) 103:1862–72. doi: 10.3324/haematol.2018.191650
147. Sun D, Li Z, Rew Y, Gribble M, Bartberger MD, Beck HP, et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J Med Chem (2014) 57:1454–72. doi: 10.1021/jm401753e
148. Kitagaki J, Agama KK, Pommier Y, Yang Y, Weissman AM. Targeting tumor cells expressing p53 with a water-soluble inhibitor of Hdm2. Mol Cancer Ther (2008) 7:2445–54. doi: 10.1158/1535-7163.MCT-08-0063
149. Yang Y, Ludwig RL, Jensen JP, Pierre SA, Medaglia MV, Davydov IV, et al. : Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell (2005) 7:547–59. doi: 10.1016/j.ccr.2005.04.029
150. Herman AG, Hayano M, Poyurovsky MV, Shimada K, Skouta R, Prives C, et al. Discovery of Mdm2-MdmX E3 ligase inhibitors using a cell-based ubiquitination assay. Cancer Discovery (2011) 1:312–25. doi: 10.1158/2159-8290.CD-11-0104
151. Joseph TL, Madhumalar A, Brown CJ, Lane DP, Verma CS. Differential binding of p53 and nutlin to MDM2 and MDMX: computational studies. Cell Cycle (2010) 9:1167–81. doi: 10.4161/cc.9.6.11067
152. Chang YS, Graves B, Guerlavais V, Tovar C, Packman K, To KH, et al. Stapled alpha-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc Natl Acad Sci USA (2013) 110:E3445–3454. doi: 10.1073/pnas.1303002110
153. Carvajal LA, Neriah DB, Senecal A, Benard L, Thiruthuvanathan V, Yatsenko T, et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci Trans Med (2018) 10:eaao3003. doi: 10.1126/scitranslmed.aao3003
154. Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer (2006) 6:369–81. doi: 10.1038/nrc1881
155. Huang HL, Weng HY, Wang LQ, Yu CH, Huang QJ, Zhao PP, et al. Triggering Fbw7-mediated proteasomal degradation of c-Myc by oridonin induces cell growth inhibition and apoptosis. Mol Cancer Ther (2012) 11:1155–65. doi: 10.1158/1535-7163.MCT-12-0066
156. Chan CH, Morrow JK, Li CF, Gao Y, Jin G, Moten A, et al. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell (2013) 154:556–68. doi: 10.1016/j.cell.2013.06.048
157. Chen Q, Xie W, Kuhn DJ, Voorhees PM, Lopez-Girona A, Mendy D, et al. Targeting the p27 E3 ligase SCF(Skp2) results in p27- and Skp2-mediated cell-cycle arrest and activation of autophagy. Blood (2008) 111:4690–9. doi: 10.1182/blood-2007-09-112904
158. Blees JS, Bokesch HR, Rübsamen D, Schulz K, Milke L, Bajer MM, et al. Erioflorin stabilizes the tumor suppressor Pdcd4 by inhibiting its interaction with the E3-ligase β-TrCP1. PloS One (2012) 7:e46567. doi: 10.1371/journal.pone.0046567
159. Nakajima H, Fujiwara H, Furuichi Y, Tanaka K, Shimbara N. A novel small-molecule inhibitor of NF-kappaB signaling. Biochem Biophys Res Commun (2008) 368:1007–13. doi: 10.1016/j.bbrc.2008.01.166
160. Zeng X, Sigoillot F, Gaur S, Choi S, Pfaff KL, Oh DC, et al. Pharmacologic inhibition of the anaphase-promoting complex induces a spindle checkpoint-dependent mitotic arrest in the absence of spindle damage. Cancer Cell (2010) 18:382–95. doi: 10.1016/j.ccr.2010.08.010
161. Sackton KL, Dimova N, Zeng X, Tian W, Zhang M, Sackton TB, et al. Synergistic blockade of mitotic exit by two chemical inhibitors of the APC/C. Nature (2014) 514:646–9. doi: 10.1038/nature13660
162. Triozzi PK,M, Alistar A, Bitting R, Neal A, Lametschwandtner G, Loibner H. : Phase I clinical trial of adoptive cellular immunotherapy with APN401 in patients with solid tumors. J Immunother Cancer (2015) 3:175. doi: 10.1186/2051-1426-3-S2-P175
163. Riling CS, III., Cunnion B, Todd E, Mattern M, Wu J, Kambayashi T, et al. Small-molecule Cbl-b inhibitors as novel intracellular checkpoint inhibitors for cancer immunotherapy. Mol Targets Cancer Ther (2018) 17:A206. doi: 10.1158/1535-7163.TARG-17-A206
164. Rossi M RB, Ansell K, Amelio I, Caraglia M, Misso G, Bernassola F, et al. High throughput screening for inhibitors of the HECT ubiquitin E3 ligase ITCH identifies antidepressant drugs as regulators of autophagy. Cell Death Dis (2014) 5:e1203. doi: 10.1038/cddis.2014.113
165. Adams J. The proteasome: a suitable antineoplastic target. Nat Rev Cancer (2004) 4:349–60. doi: 10.1038/nrc1361
166. Richardson PG, Hideshima T, Anderson KC. Bortezomib (PS-341): a novel, first-in-class proteasome inhibitor for the treatment of multiple myeloma and other cancers. Cancer Control (2003) 10:361–9. doi: 10.1177/107327480301000502
167. Kouroukis TC, Baldassarre FG, Haynes AE, Imrie K, Reece DE, Cheung MC. Bortezomib in multiple myeloma: systematic review and clinical considerations. Curr Oncol (2014) 21:e573–603. doi: 10.3747/co.21.1798
168. Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targ (2011) 11:239–53. doi: 10.2174/156800911794519752
169. Frankland-Searby S, Bhaumik SR. The 26S proteasome complex: an attractive target for cancer therapy. Biochim Biophys Acta (2012) 1825:64–76. doi: 10.1016/j.bbcan.2011.10.003
170. Bonvini P, Zorzi E, Basso G, Rosolen A. Bortezomib-mediated 26S proteasome inhibition causes cell-cycle arrest and induces apoptosis in CD-30+ anaplastic large cell lymphoma. Leukemia (2007) 21:838–42. doi: 10.1038/sj.leu.2404528
171. Richardson PG, Briemberg H, Jagannath S, Wen PY, Barlogie B, Berenson J, et al. Frequency, characteristics, and reversibility of peripheral neuropathy during treatment of advanced multiple myeloma with bortezomib. J Clin Oncol (2006) 24:3113–20. doi: 10.1200/JCO.2005.04.7779
172. Gelman JS, Sironi J, Berezniuk I, Dasgupta S, Castro LM, Gozzo FC, et al. Alterations of the intracellular peptidome in response to the proteasome inhibitor bortezomib. PloS One (2013) 8:e53263. doi: 10.1371/journal.pone.0053263
173. Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med (2003) 348:2609–17. doi: 10.1056/NEJMoa030288
174. Suzuki E, Demo S, Deu E, Keats J, Arastu-Kapur S, Bergsagel PL, et al. Molecular mechanisms of bortezomib resistant adenocarcinoma cells. PloS One (2011) 6:e27996. doi: 10.1371/journal.pone.0027996
175. Chauhan D, Singh AV, Aujay M, Kirk CJ, Bandi M, Ciccarelli B, et al. A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma. Blood (2010) 116:4906–15. doi: 10.1182/blood-2010-04-276626
176. Vogl DT, Martin TG, Vij R, Hari P, Mikhael JR, Siegel D, et al. Phase I/II study of the novel proteasome inhibitor delanzomib (CEP-18770) for relapsed and refractory multiple myeloma. Leukemia Lymphoma (2017) 58:1872–9. doi: 10.1080/10428194.2016.1263842
177. Potts BC, Albitar MX, Anderson KC, Baritaki S, Berkers C, Bonavida B, et al. Marizomib, a proteasome inhibitor for all seasons: preclinical profile and a framework for clinical trials. Curr Cancer Drug Targ (2011) 11:254–84. doi: 10.2174/156800911794519716
178. Spencer A, Harrison S, Zonder J, Badros A, Laubach J, Bergin K, et al. A phase 1 clinical trial evaluating marizomib, pomalidomide and low-dose dexamethasone in relapsed and refractory multiple myeloma (NPI-0052-107): final study results. Br J Haematol (2018) 180:41–51. doi: 10.1111/bjh.14987
179. Di K, Lloyd GK, Abraham V, MacLaren A, Burrows FJ, Desjardins A, et al. Marizomib activity as a single agent in malignant gliomas: ability to cross the blood-brain barrier. Neuro-oncology (2016) 18:840–8. doi: 10.1093/neuonc/nov299
180. Fang Y, Fu D, Shen XZ. The potential role of ubiquitin c-terminal hydrolases in oncogenesis. Biochim Biophys Acta (2010) 1806:1–6. doi: 10.1016/j.bbcan.2010.03.001
181. Liu J, Shaik S, Dai X, Wu Q, Zhou X, Wang Z, et al. Targeting the ubiquitin pathway for cancer treatment. Biochim Biophys Acta (2015) 1855:50–60. doi: 10.1016/j.bbcan.2014.11.005
182. Kapuria V, Peterson LF, Fang D, Bornmann WG, Talpaz M, Donato NJ. Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Res (2010) 70:9265–76. doi: 10.1158/0008-5472.CAN-10-1530
183. Wang S, Kollipara RK, Srivastava N, Li R, Ravindranathan P, Hernandez E, et al. Ablation of the oncogenic transcription factor ERG by deubiquitinase inhibition in prostate cancer. Proc Natl Acad Sci U S A (2014) 111:4251–6. doi: 10.1073/pnas.1322198111
184. Colland F, Formstecher E, Jacq X, Reverdy C, Planquette C, Conrath S, et al. Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol Cancer Ther (2009) 8:2286–95. doi: 10.1158/1535-7163.MCT-09-0097
185. Chauhan D, Tian Z, Nicholson B, Kumar KG, Zhou B, Carrasco R, et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell (2012) 22:345–58. doi: 10.1016/j.ccr.2012.08.007
186. Weinstock J, Wu J, Cao P, Kingsbury WD, McDermott JL, Kodrasov MP, et al. Selective Dual Inhibitors of the Cancer-Related Deubiquitylating Proteases USP7 and USP47. ACS Med Chem Lett (2012) 3:789–92. doi: 10.1021/ml200276j
187. Lee BH, Lee MJ, Park S, Oh DC, Elsasser S, Chen PC, et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature (2010) 467:179–84. doi: 10.1038/nature09299
188. Lee BH, Finley D, King RW. A High-Throughput Screening Method for Identification of Inhibitors of the Deubiquitinating Enzyme USP14. Curr Protoc Chem Biol (2012) 4:311–30. doi: 10.1002/9780470559277.ch120078
189. Tian Z, D’Arcy P, Wang X, Ray A, Tai YT, Hu Y, et al. A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood (2014) 123:706–16. doi: 10.1182/blood-2013-05-500033
190. Lai AC, Crews CM. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discovery (2017) 16:101–14. doi: 10.1038/nrd.2016.211
191. Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci USA (2001) 98:8554–9. doi: 10.1073/pnas.141230798
192. Abruzzese MP, Bilotta MT, Fionda C, Zingoni A, Soriani A, Vulpis E, et al. Inhibition of bromodomain and extra-terminal (BET) proteins increases NKG2D ligand MICA expression and sensitivity to NK cell-mediated cytotoxicity in multiple myeloma cells: role of cMYC-IRF4-miR-125b interplay. J Hematol Oncol (2016) 9:134. doi: 10.1186/s13045-016-0362-2
193. Zengerle M, Chan KH, Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem Biol (2015) 10:1770–7. doi: 10.1021/acschembio.5b00216
194. DeMars KM, Yang C, Castro-Rivera CI, Candelario-Jalil E. Selective degradation of BET proteins with dBET1, a proteolysis-targeting chimera, potently reduces pro-inflammatory responses in lipopolysaccharide-activated microglia. Biochem Biophys Res Commun (2018) 497:410–5. doi: 10.1016/j.bbrc.2018.02.096
195. Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem Biol (2015) 22:755–63. doi: 10.1016/j.chembiol.2015.05.009
196. Raina K, Lu J, Qian Y, Altieri M, Gordon D, Rossi AM, et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc Natl Acad Sci USA (2016) 113:7124–9. doi: 10.1073/pnas.1521738113
197. Sun B, Fiskus W, Qian Y, Rajapakshe K, Raina K, Coleman KG, et al. BET protein proteolysis targeting chimera (PROTAC) exerts potent lethal activity against mantle cell lymphoma cells. Leukemia (2018) 32:343–52. doi: 10.1038/leu.2017.207
198. Qin C, Hu Y, Zhou B, Fernandez-Salas E, Yang CY, Liu L, et al. Discovery of QCA570 as an Exceptionally Potent and Efficacious Proteolysis Targeting Chimera (PROTAC) Degrader of the Bromodomain and Extra-Terminal (BET) Proteins Capable of Inducing Complete and Durable Tumor Regression. J Med Chem (2018) 61:6685–704. doi: 10.1021/acs.jmedchem.8b00506
199. Zhang X, Lee HC, Shirazi F, Baladandayuthapani V, Lin H, Kuiatse I, et al. Protein targeting chimeric molecules specific for bromodomain and extra-terminal motif family proteins are active against pre-clinical models of multiple myeloma. Leukemia (2018) 32:2224–39. doi: 10.1038/s41375-018-0044-x
200. Testa A, Lucas X, Castro GV, Chan KH, Wright JE, Runcie AC, et al. 3-Fluoro-4-hydroxyprolines: Synthesis, Conformational Analysis, and Stereoselective Recognition by the VHL E3 Ubiquitin Ligase for Targeted Protein Degradation. J Am Chem Soc (2018) 140:9299–313. doi: 10.1021/jacs.8b05807
201. Gechijian LN, Buckley DL, Lawlor MA, Reyes JM, Paulk J, Ott CJ, et al. Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands. Nat Chem Biol (2018) 14:405–12. doi: 10.1038/s41589-018-0010-y
202. Wang X, Feng S, Fan J, Li X, Wen Q, Luo N. New strategy for renal fibrosis: Targeting Smad3 proteins for ubiquitination and degradation. Biochem Pharmacol (2016) 116:200–9. doi: 10.1016/j.bcp.2016.07.017
203. Krönke J, Fink EC, Hollenbach PW, MacBeth KJ, Hurst SN, Udeshi ND, et al. Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature (2015) 523:183–8. doi: 10.1038/nature14610
Keywords: ubiquitination, E3 ligase, deubiquitinase, nonhistone protein, cancer, proteolysis-targeting chimeras
Citation: Zhang X, Meng T, Cui S, Feng L, Liu D, Pang Q and Wang P (2021) Ubiquitination of Nonhistone Proteins in Cancer Development and Treatment. Front. Oncol. 10:621294. doi: 10.3389/fonc.2020.621294
Received: 26 October 2020; Accepted: 24 December 2020;
Published: 11 February 2021.
Edited by:
Yi Sun, Zhejiang University, ChinaReviewed by:
Huabo Su, Augusta University, United StatesCopyright © 2021 Zhang, Meng, Cui, Feng, Liu, Pang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiuxiang Pang, cGFuZ3FpdXhpYW5nQHNkdXQuZWR1LmNu; Ping Wang, d2FuZ3BAdG9uZ2ppLmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.