 Xian-Yang Qin
Xian-Yang Qin Luc Gailhouste
Luc Gailhouste- Liver Cancer Prevention Research Unit, RIKEN Cluster for Pioneering Research, Wako, Japan
Upregulated MYCN gene expression is restricted to specialized cell populations such as EpCAM+ cancer stem cells in liver cancer, regardless of DNA amplification and mutation. Here, we reviewed the role of MYCN gene expression in liver homeostasis, regeneration, and tumorigenesis, and discussed the potential non-genomic mechanisms involved in controlling MYCN gene expression in liver cancer, with a focus on inflammation-mediated signal transduction and microRNA-associated post-transcriptional regulation. We concluded that dynamic MYCN gene expression is an integrated consequence of multiple signals in the tumor microenvironment, including tumor growth-promoting signals, lipid desaturation-mediated endoplasmic reticulum stress adaptation signals, and tumor suppressive miRNAs, making it a potential predictive biomarker of tumor stemness and plasticity. Therefore, understanding and tracing the dynamic changes and functions of MYCN gene expression will shed light on the origin of liver tumorigenesis at the cellular level and the development of novel therapeutic and diagnostic strategies for liver cancer treatment.
Introduction
Liver cancer, mostly hepatocellular carcinoma (HCC), is a highly lethal cancer (>600,000 deaths per year worldwide) in which approximately 10% of patients survive the first 5 years after diagnosis (1). Liver cancer is recognized as an inflammation-related cancer, since more than 90% of HCC cases arise in the context of chronic liver injury and unresolved inflammatory microenvironment due to viral infection, alcohol consumption, or high-fat diet (HFD) hypernutrition (2–4). Advances in antiviral therapy have reduced the risk of developing hepatitis B virus- and hepatitis C virus-related HCC (5, 6). In contrast, non-alcoholic steatohepatitis (NASH) which is characterized by obesity-associated inflammation has attracted much attention, and is believed that it will soon be the leading etiology of HCC (7). Notably, mice fed with HFD alone did not develop liver injury and tumorigenesis. However, hyperresponsivity to lipopolysaccharide and endoplasmic reticulum (ER) stress were observed in fatty liver that contributed to the progression of NASH and HCC (8, 9). This suggested that a non-genomic mechanism was involved in the control of cellular responses such as adaptation to inflammatory stresses during hepatic tumorigenesis. In this line, whereas tumor initiation depends on somatic mutations, the mechanisms underlying tumor promotion are likely to involve epigenetic factors and environmental factors extrinsic to the cancer cell (10).
MYCN is a canonical proto-oncogene basic helix-loop-helix transcription factor that is mainly restricted to the migrating neural crest (11) and governs cell growth and differentiation during embryonic stages (12). Amplification of the MYCN locus was first observed in human neuroblastoma (13). MYCN amplification is observed in about 20% of neuroblastoma and represents one of the strongest clinical predictors of poor prognosis (14). MYCN amplicons are either organized as extrachromosomal double minutes or as homogeneously stained regions in addition to the single copy of MYCN on the short arm of chromosome 2, retained at 2p24, in neuroblastoma cells and other solid tumor cells (15). Notably, the MYCN gene is located in a non-fragile region of 2.8 Mbp between two common fragile sites, FRA2Ctel and FRA2Ccen, located at 2p24.3 and 2p24.4, respectively (16). A study by Blumrich and colleagues suggested that MYCN amplicons might arise from extra rounds of replication of unbroken DNA secondary structures that accumulate at FRA2C (16). Recent clinical studies have reported increased gene expression of MYCN in liver tumor tissues (17, 18). However, according to The Cancer Genome Atlas (TCGA) database, nine of the 371 HCC patients (2.4%) with upregulated MYCN mRNA expression but not the seven patients (1.9%) with MYCN amplification had a dramatically worse prognosis (Figure S1A). Data mining using the Cancer Cell Line Encyclopedia (CCLE) database identified a total of 65 MYCN mutations, but none of them was detected in HCC cell lines irrespective of their corresponding mRNA abundance (Table S1). This highlights the existence of non-genomic mechanisms potentially responsible for MYCN overexpression in liver cancer. Notably, data mining in TCGA showed that the expression of MYCN in human HCC was not correlated with that of c-MYC, another MYC family membranes known to be crucial for liver cancer maintenance (19) and oncogenic reprogramming of terminally differentiated hepatocytes into liver cancer stem cells (CSCs) (20) (Figure S1B). In addition, MYCN gene expression but not c-MYC gene expression was significantly correlated with the liver CSC marker EpCAM gene expression (Figure S1B). These data highlight the possibility that MYCN gene expression is restricted in CSC-like cells and serves as a more sensitive biomarker than c-MYC gene expression for the detection of tumor stemness during liver tumorigenesis. Here, we reviewed the role of dynamic MYCN gene expression in liver homeostasis, regeneration, and tumorigenesis, and discussed the potential non-genomic mechanisms involved in controlling MYCN gene expression in liver cancer, focusing on inflammation-mediated signal transduction and microRNA-associated (miRNA)-post-transcriptional regulation.
MYCN Gene Expression in Liver Homeostasis, Regeneration, and Tumorigenesis
Single-cell RNA sequencing provided a comprehensive view of MYCN gene expression in both human and mouse livers (21, 22). Under steady-state conditions, the expression of MYCN gene is low in hepatocytes (Figure S1C) (21). MYCN gene expression in the liver is significantly zonated, which is predominantly induced in the pericentral cells and progressively decreases along the liver lobule towards periportal cells (Figure S1D) (22). Metabolic liver zonation requires a Wnt/β-catenin signaling gradient (23). In the uninjured liver, diffusible Wnt ligands produced by the pericentral endothelial cells activate β-catenin signaling-induced target genes such as Axin2 and maintain a population of proliferating and self-renewing cells, surrounding the central vein, that contribute to homeostatic hepatocyte renewal (24). Wnt/β-catenin signaling is critical for organ development, homeostasis, and regeneration through governing stem cell pluripotency (25). During neocortical development, MYCN is a direct downstream target of the Wnt/β-catenin pathway and promotes neuronal fate commitment (26). Therefore, the basal expression of MYCN gene in the liver is a likely consequence of the activation of Wnt/β-catenin signaling during liver homeostasis.
Cap Analysis of Gene Expression(CAGE)-based transcriptional profiling of isolated primary mouse hepatocytes revealed that low level of MYCN gene expression was detected at 2 h and peaked at 48 h after 70% partial hepatectomy (Figure S1E) (27). Liver regeneration is a coordinated multistep process that is largely dependent on the re-entry of differentiated adult hepatocytes into the cell cycle and proliferation (28). In response to loss of hepatic tissue, hepatocyte DNA synthesis peaks at around 24 h, accompanied by the induction of gene expression of growth-regulated and cell-cycle-regulated genes at around 48 h (29). It is possible that the induction of MYCN gene expression is a mitogenic response of hepatocytes during liver regeneration. Indeed, a major direct mitogen of hepatocytes, the epidermal growth factor (EGF), stimulated MYCN gene expression in neuroblastoma cells via the recruitment of the transcription factor Sp1 to the MYCN promoter region (30).
Transcriptome profiling of frozen human liver tissues using microarray showed that MYCN gene expression was low in healthy livers, cirrhotic livers, and adjacent non-tumorous liver tissue, while it was dramatically increased in tumor tissues (17). Project HOPE (High-tech Omics-based Patient Evaluation), a clinical study aiming to provide multi-omics data of cancer patients, showed the upregulation of MYCN gene expression in tumor tissues compared to normal tissues in 22% of recruited HCC patients (18). Our previous cohort studies in Japan (n = 102) and Europe (n = 50) confirmed an increase in MYCN gene expression in HCC tumor regions as compared to non-tumor regions (17). Importantly, in a long-term (>10 years) follow-up study, MYCN gene expression in HCC tumors was significantly higher in patients with recurrence than in those without recurrence and was positively correlated with the de novo recurrence of HCC with a single tumor but not with multiple tumors (17). HCC recurrence at approximately 1–2 years after resection was considered to be mainly due to de novo carcinogenesis of liver CSCs or tumor-initiating cells (31). MYCN gene expression in HCC was positively correlated with the expression of liver CSC markers and Wnt/β-catenin signaling markers, suggesting that MYCN expression is restricted to CSC-like HCC (17). Consistently, MYCN expression marked an EpCAM+ CSC-like subpopulation, which was selectively depleted by acyclic retinoid (ACR), a promising chemopreventive agent against the recurrence of HCC after curative treatment (17, 32). EpCAM is a well-characterized liver CSC marker and is a direct transcriptional target of Wnt/β-catenin signaling (33). Similar to liver homeostasis, the restricted MYCN expression in liver CSCs is probably related to the activation of Wnt/β-catenin signaling. Furthermore, four out of six liver biopsies of HCC patients (66.7%) who had received 8 weeks of high-dose ACR treatment (600 mg/day), but not low-dose ACR treatment (300 mg/day), after definitive treatment showed decreased MYCN gene expression (< 0.5-fold) (17). In line with this, clinical studies showed that administration of ACR at 600 mg/day, but not 300 mg/day, reduced HCC recurrence after curative treatment (34). Collectively, MYCN expression marked CSC-like subpopulations in heterogeneous HCC and served as a potential therapeutic target and prognostic marker for HCC.
Regulation of Mycn Gene Expression by Tissue Repair Signals in the Inflammatory Microenvironment of Liver Cancer
Activation of inflammatory signal transduction in the tumor microenvironment is strongly linked to tumor initiation and progression based on two mechanisms: tissue repair and stress adaptation. Obesity-associated production of inflammatory cytokines, such as interleukin-6 (IL-6) and tumor necrosis factor-α (TNFα), induce repeated liver injury and compensatory proliferation, which might lead to aberrant stabilization and activation of “repair signals” such as signal transducer and activator of transcription 3 (STAT3)-dependent oncogenic signaling pathways and initiation and progression of HCC (35–37). The involvement of hyperactivated IL-6-STAT3 signaling axis as a driver oncogenic mechanism in promoting cell proliferation and suppressing antitumor immune response in the background of tumor microenvironment has been reported in several cancers (38). STAT3 directly mediates the initiation of MYCN transcription in neuroblastoma cells (39). Inhibition of STAT3 with antisense oligonucleotide or pharmacological inhibitors reduced MYCN gene expression and decreased neuroblastoma tumorigenicity in preclinical mouse models (39, 40). During early hepatocarcinogenesis, STAT3 activated by paracrine IL-6 produced by inflammatory cells, might directly bind to the promoter and upregulate the gene and protein expression of CD133, a well-defined liver CSC marker representing a specialized subpopulation of highly tumorigenic cells with high MYCN expression (17, 41, 42). Inhibition of STAT3 with sorafenib, the first-line recommended therapy for patients with advanced HCC, decreased CD133 levels and suppressed in vivo tumorigenicity by eradicating the liver tumor microenvironment (41). Of note, a recent proteomics-based pathway analysis showed that sorafenib inactivated downstream signaling of MYCN in HCC cells (43). In addition, growth factors such as EGF induced by inflammatory cytokines contribute to the upregulation of MYCN gene expression in an inflammatory microenvironment (30). Nerve growth factor (NGF) is expressed by hepatocytes during fibrotic liver injury (44). In MYCN-amplified neuroblastoma cells, NGF suppressed MYCN gene expression through mitogen-activated protein kinase signaling pathways (45). In contrast, a global transcriptome analysis showed reduced MYCN gene expression in NGF-deprived sympathetic neurons (46). It is unclear whether NGF directly regulates MYCN gene expression in normal livers and HCC cells.
Regulation of MYCN Gene Expression by Lipid Desaturation-Mediated Stress Adaptation Signals in the Inflammatory Microenvironment of Liver Cancer
The cell membrane serves as the barrier between life and death for individual cells and the first line of defense in response to environmental stress. In addition to their function as energy storage sources or as building blocks of membranes, membrane lipids have attracted much attention as biologically active molecules. They regulate the formation of membrane assembly of signal complexes by either binding to cognate receptors or recruiting proteins from the cytosol and coordinating signal transduction (47). Membrane lipids are highly diverse in chemical structures, varying in the desaturation and chain elongation of fatty acyl chains, backbones (such as glycerol, sphingoid base, and cholesterol), and head group substituents. Changes in membrane lipid composition affect membrane physical properties, as observed in mammalian cells in response to environmental stimuli (47). For example, macrophages rapidly reprogram their lipid metabolism, especially de novo cholesterol biosynthesis (48, 49) and desaturated fatty acid biosynthesis (50), for appropriate inflammatory and host defense functions in response to diverse inflammatory signals. Under inflammatory conditions, the fatty acid synthetic enzyme fatty acid synthase, reshapes macrophage lipid homeostasis for the assembly of cholesterol-dependent inflammatory signals such as Rho GTPase at the plasma membrane (51). In contrast, lipid desaturases such as stearoyl-CoA desaturase (SCD1) and fatty acid desaturase (FADS) were induced to inhibit the inflammatory responses through the production of anti-inflammatory omega-3 polyunsaturated fatty acids or disruption of membrane signaling complexes associated with lipid rafts, also known as membrane microdomains, which are enriched with saturated sphingolipids and cholesterol (52). Importantly, lipid reprograming, especially the upregulation of unsaturated fatty acids, has recently been recognized as a critical feature of stem cell maintenance under both physiological and abnormal conditions (53, 54). Our previous proteome and metabolome analyses demonstrated that high MYCN expression in liver CSCs was characterized by increased expression of lipid desaturases such as SCD1 and FADS and elevated levels of monounsaturated fatty acids such as palmitoleic acid and oleic acid in comparison to non-CSC HCC cells (55, 56). In addition to the upstream regulatory role of MYCN in lipid metabolism reprograming of cancer cells [reviewed in (57)], inhibition of lipid desaturation using both genetic and pharmacological approaches against SCD1 reduced MYCN gene expression and selectively suppressed the proliferation of high MYCN-expressing HCC cells, suggesting a direct regulatory role of lipid desaturation on MYCN transcription (56). Genome-wide transcriptome analysis using RNA-seq showed that ER stress-related signaling pathways were regulated upon siRNA knock-down of SCD1 but not MYCN in high MYCN-expressing HCC cells (56). Further, mechanistic studies showed that inhibition of lipid desaturation resulted in activation of ER stress signaling pathways, such as the expression of the transcription suppressor, cyclic AMP-dependent transcription factor 3 (ATF3), which reversibly regulates MYCN gene expression in high MYCN-expressing CSC-like HCC cells, CSC-rich spheroids, and in clinical HCC tissues (56).
ER stress response, also known as unfolded protein response (UPR), is activated as a cell-defensive mechanism triggered by multiple stress factors and plays a critical role in the switch between cell survival and cell death. Evading ER stress-induced apoptosis and differentiation is critical for the maintenance of long-living and self-renewing stem cells under both normal and malignant conditions (58–60). Therefore, modulation of ER stress-induced loss of stemness represents a potential therapeutic strategy for cancers and chronic inflammatory diseases (61, 62). In line with this, pharmacological targeting of SCD1 achieved remarkable therapeutic outcomes in glioblastoma and liver cancer by triggering ER stress-mediated apoptosis and differentiation of CSCs (63, 64). Mechanistically, enhanced levels of unsaturated fatty acids in CSCs could suppress ER stress by preventing saturated fatty acid-induced calcium accumulation, oxidative stress, or detrimental stiffening of the ER and plasma membrane (65–68). Collectively, under lipid-rich inflammatory conditions, both repair signals and stress adaptation signals contribute to the upregulation of MYCN gene expression (Figure 1). Inflammatory cytokine-induced chronic injury leads to the activation of repair signals, which triggers downstream MYCN gene expression and compensatory proliferation. In contrast, lipid desaturation-mediated membrane reprogramming reduces or counteracts the formation of membrane assembly of stress signal complexes and enables CSCs to survive and evade ER stress-induced apoptosis/differentiation. We propose that the stress adaption mechanism in long-living CSC-like cells contributes to tumorigenesis such as through accumulation of mutations in the survived cells, which is accompanied by the increase of MYCN gene expression.
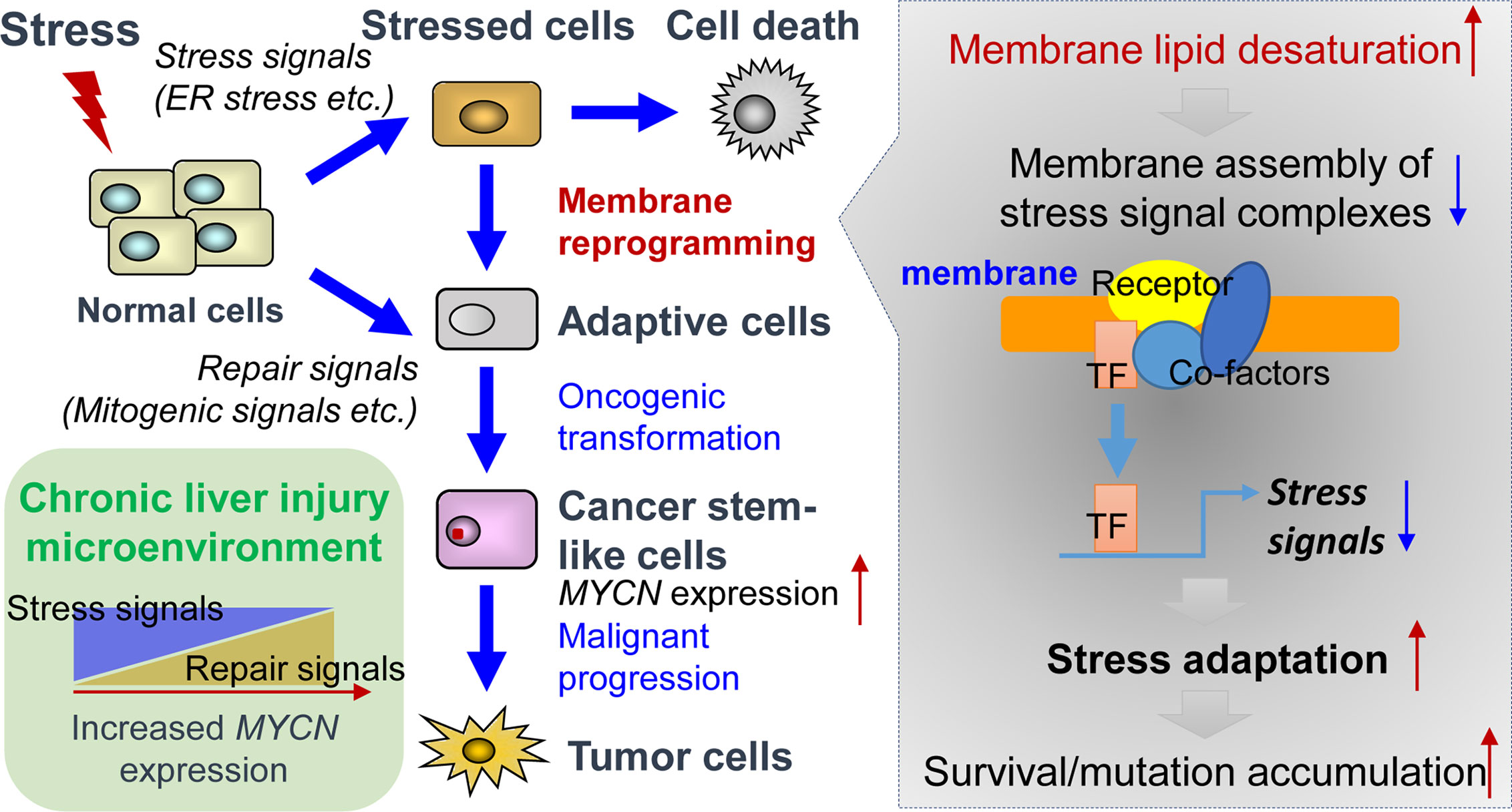
Figure 1 Tissue repair and stress adaptation signal-based control of MYCN gene expression in the inflammatory microenvironment of liver cancer. Under lipid-rich inflammatory conditions, inflammatory cytokines-induced chronic injury leads to the activation of repair signals such as mitogenic signals resulting in compensatory proliferation, thereby triggering downstream MYCN gene expression. In contrast, lipid desaturation-mediated membrane reprogramming reduces or counteracts the formation of membrane assembly of stress signal complexes and enables the CSCs to survive and evade ER stress-induced apoptosis/differentiation, which leads to the rescue of MYCN gene expression and initiates tumorigenesis by accumulation of mutations in long-living CSCs.
Post-Transcriptional Control of MYCN Gene Expression by Mirnas in Liver Cancer
miRNAs are evolutionarily conserved small non-coding RNAs of approximately 22 nucleotides in length that modulate gene expression by complementary base pairing with the 3’-untranslated regions (3’-UTRs) of messenger RNAs [reviewed in (69)]. An essential feature of miRNA-based gene regulation is that a single miRNA can recognize numerous mRNAs and, conversely, a target mRNA can be recognized by several miRNAs. A large number of studies have reported the key role of these posttranscriptional regulators in the control of various cellular processes and human diseases (70). In cancer, aberrant expression of miRNAs has been well described and is associated with the deregulation of critical genes involved in tumor progression (71). Indeed, cancer-related miRNAs can act as oncogenes (called oncomirs) or tumor-suppressors, depending on their targets, and promote or negatively influence tumor growth, invasion, and/or drug resistance, respectively (72). Specific miRNA profiles have been identified in neuroblastoma, which reflect different subtypes of tumors and correlate with the advancement of the disease or its prognosis [reviewed in (73)]. In this malignancy, numerous MYCN-targeting miRNAs have been identified. Loss of miR-34a at chromosome band 1p36, a region frequently deleted due to loss of heterozygosity in neuroblastoma cells (74), is associated with MYCN amplification and promotion of tumor aggressiveness (75). Thus far, several additional miRNA/MYCN regulatory axes have been characterized. In a model of MYCN-amplified neuroblastoma cells, experimental overexpression of miR-101 and let-7e induced a decrease in MYCN protein levels and inhibited cell growth via the direct regulation of MYCN (76, 77). In another interesting study by Neviani and colleagues, the tumor-suppressor miR-186 was detected in natural killer cell-derived exosomes, which exhibited cytotoxicity against neuroblastoma cells with high MYCN levels (78). The authors showed that MYCN expression was directly inhibited by miR-186. In addition, the targeted delivery of miR-186 to MYCN-amplified neuroblastoma cells or natural killer cells resulted in significant tumor growth inhibition. A recent study based on the modeling of miRNA-mRNA interactions identified a regulatory loop between MYCN and miR-204 in neuroblastoma cells (79). The authors showed that miR-204 directly targeted MYCN mRNA and decreased its protein levels. In contrast, MYCN was able to bind to the promoter of miR-204 and inhibit the expression of the miRNA. Remarkably, the capability of MYCN to activate the expression of critical oncomirs, such as miR-221, miR-9, or the miR-17-92 cluster, has also been observed in neuroblastoma cells and other types of solid tumor cells (80).
A plethora of studies have described the functional interconnection between miRNAs and MYCN in neuroblastoma. However, little is known about the miRNAs involved in the posttranscriptional regulation of MYCN in liver cancer. The aberrant expression of miRNAs is a typical hallmark of hepatocarcinogenesis and tumor progression (81). We previously demonstrated that maternally expressed 3 (MEG3)-derived miR-493-5p tumor-suppressor was epigenetically silenced by CpG hypermethylation in HCC cells and tumor tissues from patients (82). Experimental overexpression of miR-493-5p promoted an anti-cancer response by inhibiting HCC cell growth and invasion, in part, through the negative regulation of insulin-like growth factor 2 (IGF2) and the IGF2-derived intronic oncomir miR-483-3p. More recently, our group highlighted MYCN as another major target of miR-493-5p using global gene expression analysis of liver cancer cells with restored expression of miR-493-5p (83). More precisely, real-time qPCR data showed an inverse and significant correlation between miR-493-5p and MYCN expression levels in the tumors of patients with advanced HCC. A dual-luciferase reporter activity assay validated miR-493-5p-mediated inhibition of MYCN via the targeting of two distinct regions in the MYCN 3’-UTR (Figure 2). To the best of our knowledge, no additional miRNA interacting directly with MYCN mRNA has been described in liver cancer thus far. However, in a study based on big data mining and connectivity map analysis, Xiong et al. uncovered the existence of a potential hsa_circRNA_104515/hsa-miR-142-5p/MYCN regulatory axis in HCC (84). In agreement with this finding, we found that TargetScanHuman predicted an exact consequential pairing of the MYCN 3’-UTR with positions 2-8 (7mer-m8) of mature miR-142-5p (Figure S1F). Interestingly, two reports described downregulation of miR-142-5p in liver cancer cells and showed that forced expression of miR-142-5p inhibited HCC cell growth and invasion (85, 86). Taken together, these data strongly suggest the tumor-suppressive role of miR-142-5p through post-transcriptional control of MYCN and its therapeutic potential in liver cancer. Finally, recent studies showed that all-trans retinoic acid (ATRA), which is an isomer of retinoic acid, was able to modulate the expression of more than 300 miRNAs and inhibit the growth of various types of tumor cells (87). Among the miRNAs upregulated after ATRA treatment, miR-34a-5p, miR-103a-3p, miR-200b/c-3p, miR-302-3p, and members of the let-7 family appeared appealing given their potential ability to target the MYCN 3’-UTR as predicted by TargetScanHuman 7.2 (Figure S1F). While the tumor-suppressor feature of the let-7 family members has been well-documented, further investigations will be required to evaluate the beneficial role of ATRA-stimulated miRNAs in HCC, since some of these miRNAs may also exhibit oncogenic activity.
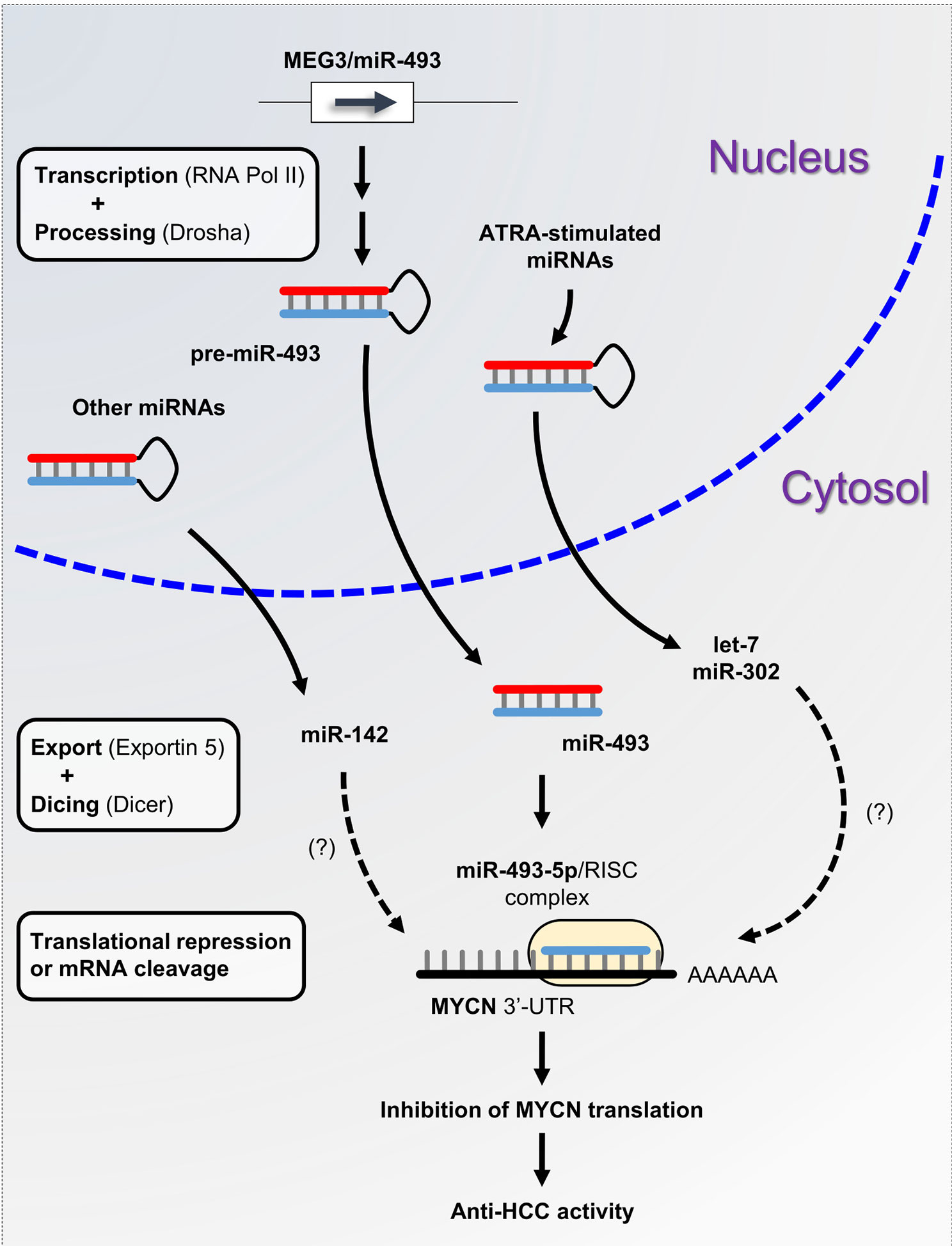
Figure 2 miRNA-based control of MYCN gene expression in liver cancer. miRNA biogenesis is a multistep process. Following transcription by RNA polymerase II, primary precursor miRNAs (pri-miRNAs) are cleaved into precursor miRNAs (pre-miRNAs) by the RNase III enzyme Drosha and exported out of the nucleus to produce mature miRNAs. Subsequently, mature miRNAs are loaded onto the RNA-induced silencing complex (RISC) and directed to the 3’-UTR of target mRNAs. Here, we propose the miRNA/MYCN regulatory network model, in which the tumor-suppressor miR-493-5p and the ATRA-stimulated miRNAs modulate MYCN expression and impede HCC progression.
Conclusions
Mature hepatocytes exhibit remarkable plasticity by direct dedifferentiation into an undifferentiated state in the tumor microenvironment, which are believed to represent the cells of origin for liver cancer (88). Since any cell has the potential to become a CSC, the stemness of liver CSCs could be considered as a dynamic state that can be acquired rather than a cell intrinsic property of specialized existing cells [reviewed in (89)]. MYCN gene is overexpressed in restricted cell populations such as EpCAM+ CSCs in liver cancer, regardless of DNA amplification and mutation. Dynamic MYCN gene expression is an integrated consequence of multiple signals in the tumor microenvironment, including tumor stemness/growth-promoting signals such as Wnt/β-catenin and IL-6-STAT3 signaling, lipid desaturation-mediated ER stress adaptation signals, and tumor suppressive miRNAs. We propose that MYCN gene expression might represent a potential predictive biomarker of tumor stemness and plasticity. Hence, understanding and tracing the dynamic changes and functions of MYCN gene expression during hepatic tumorigenesis will shed light on the origin of liver tumorigenesis at the cellular level and the development of novel therapeutic and diagnostic strategies for HCC treatment.
Author Contributions
X-YQ and LG performed the literature search and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan’s Grant-in-Aid for Scientific Research (C), JP20K07349 (to X-YQ), and JP20K07604 (to LG) and RIKEN Incentive Research Projects (to X-YQ and LG).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This manuscript is dedicated to the memory of Soichi Kojima (1961–2019).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.618515/full#supplementary-material
Supplementary Figure 1 | Supporting data of MYCN gene expression in the liver. (A) Overall survival Kaplan-Meier estimate of HCC patients with MYCN overexpression (left) or amplification (right) according to TCGA database (TCGA, PanCancer Atlas). (B) Correlation between MYCN gene expression and c-MYC gene expression (left), MYCN gene expression and EpCAM gene expression (middle), and c-MYC gene expression and EpCAM gene expression (right) in human HCC according to TCGA database (TCGA, PanCancer Atlas). (C) MYCN gene expression pattern in human liver visualized using a web interface (http://human-liver-cell-atlas.ie-freiburg.mpg.de/), which is based on the single cell RNA-seq data published in (21). (D) Mycn gene expression in mouse liver zonation according to the single cell RNA-seq data (GSE84498) published in (22). (E) Mycn gene expression in mouse primary hepatocyte isolated at 2, 30, 48 h or 1 w after partial hepatectomy and at 2 h from sham control during liver regeneration. The data was obtained from the CAGE-based transcriptome data published in Table S1 in (27). (F) Prediction of miR-142-5p, miR-34a-5p, miR-103a-3p, miR-200b/c-3p, miR-302-3p, and members of the let-7 family targeting MYCN 3’-UTR according to TargetScanHuman (http://www.targetscan.org, release 7.2).
References
1. Llovet JM, Villanueva A, Lachenmayer A, Finn RS. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat Rev Clin Oncol (2015) 12:408–24. doi: 10.1038/nrclinonc.2015.103
2. Bishayee A. The role of inflammation and liver cancer. Adv Exp Med Biol (2014) 816:401–35. doi: 10.1007/978-3-0348-0837-8_16
3. Perz JF, Armstrong GL, Farrington LA, Hutin YJ, Bell BP. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J Hepatol (2006) 45:529–38. doi: 10.1016/j.jhep.2006.05.013
4. Calvisi DF, Wang C, Ho C, Ladu S, Lee SA, Mattu S, et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology (2011) 140:1071–83. doi: 10.1053/j.gastro.2010.12.006
5. Hosaka T, Suzuki F, Kobayashi M, Seko Y, Kawamura Y, Sezaki H, et al. Long-term entecavir treatment reduces hepatocellular carcinoma incidence in patients with hepatitis B virus infection. Hepatology (2013) 58:98–107. doi: 10.1002/hep.26180
6. Ioannou GN, Green PK, Berry K. HCV eradication induced by direct-acting antiviral agents reduces the risk of hepatocellular carcinoma. J Hepatol (2017) 68:25–32. doi: 10.1016/j.jhep.2017.08.030
7. Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology (2010) 51:1820–32. doi: 10.1002/hep.23594
8. Imajo K, Fujita K, Yoneda M, Nozaki Y, Ogawa Y, Shinohara Y, et al. Hyperresponsivity to low-dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin-mediated signaling. Cell Metab (2012) 16:44–54. doi: 10.1016/j.cmet.2012.05.012
9. Nakagawa H, Umemura A, Taniguchi K, Font-Burgada J, Dhar D, Ogata H, et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell (2014) 26:331–43. doi: 10.1016/j.ccr.2014.07.001
10. Maeda S, Kamata H, Luo J-L, Leffert H, Karin M. IKKβ Couples Hepatocyte Death to Cytokine-Driven Compensatory Proliferation that Promotes Chemical Hepatocarcinogenesis. Cell (2005) 121:977–90. doi: 10.1016/j.cell.2005.04.014
11. Wakamatsu Y, Watanabe Y, Nakamura H, Kondoh H. Regulation of the neural crest cell fate by N-myc: promotion of ventral migration and neuronal differentiation. Development (1997) 124:1953–62.
12. Knoepfler PS. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev (2002) 16:2699–712. doi: 10.1101/gad.1021202
13. Schwab M, Varmus HE, Bishop JM, Grzeschik K-H, Naylor SL, Sakaguchi AY, et al. Chromosome localization in normal human cells and neuroblastomas of a gene related to c-myc. Nature (1984) 308:288–91. doi: 10.1038/308288a0
14. Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet (2007) 369:2106–20. doi: 10.1016/S0140-6736(07)60983-0
15. Storlazzi CT, Lonoce A, Guastadisegni MC, Trombetta D, D’Addabbo P, Daniele G, et al. Gene amplification as double minutes or homogeneously staining regions in solid tumors: Origin and structure. Genome Res (2010) 20:1198–206. doi: 10.1101/gr.106252.110
16. Blumrich A, Zapatka M, Brueckner LM, Zheglo D, Schwab M, Savelyeva L. The FRA2C common fragile site maps to the borders of MYCN amplicons in neuroblastoma and is associated with gross chromosomal rearrangements in different cancers. Hum Mol Genet (2011) 20:1488–501. doi: 10.1093/hmg/ddr027
17. Qin XY, Suzuki H, Honda M, Okada H, Kaneko S, Inoue I, et al. Prevention of hepatocellular carcinoma by targeting MYCN-positive liver cancer stem cells with acyclic retinoid. Proc Natl Acad Sci USA. (2018) 115:4969–74. doi: 10.1073/pnas.1802279115
18. Okamura Y, Uesaka K, Sugiura T, Ito T, Yamamoto Y, Ashida R, et al. The Outcomes and Next Landscapes of Project HOPE (High-Tech Omics-Based Patient Evaluation) for Hepatocellular Carcinoma. Am J Gastroenterol (2015) 110:S851. doi: 10.14309/00000434-201510001-02018
19. Shachaf CM, Kopelman AM, Arvanitis C, Karlsson Å, Beer S, Mandl S, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature (2004) 431:1112–7. doi: 10.1038/nature03043
20. Holczbauer Á, Factor VM, Andersen JB, Marquardt JU, Kleiner DE, Raggi C, et al. Modeling Pathogenesis of Primary Liver Cancer in Lineage-Specific Mouse Cell Types. Gastroenterology (2013) 145:221–31. doi: 10.1053/j.gastro.2013.03.013
21. Aizarani N, Saviano A, Sagar, Mailly L, Durand S, Herman JS, et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature (2019) 572:199–204. doi: 10.1038/s41586-019-1373-2
22. Halpern KB, Shenhav R, Matcovitch-Natan O, Toth B, Lemze D, Golan M, et al. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature (2017) 542:352–6. doi: 10.1038/nature21065
23. Planas-Paz L, Orsini V, Boulter L, Calabrese D, Pikiolek M, Nigsch F, et al. The RSPO-LGR4/5-ZNRF3/RNF43 module controls liver zonation and size. Nat Cell Biol (2016) 18:467–79. doi: 10.1038/ncb3337
24. Wang B, Zhao L, Fish M, Logan CY, Nusse R. Self-renewing diploid Axin2+ cells fuel homeostatic renewal of the liver. Nature (2015) 524:180–5. doi: 10.1038/nature14863
25. Steinhart Z, Angers S. Wnt signaling in development and tissue homeostasis. Development (2018) 145:dev146589. doi: 10.1242/dev.146589
26. Kuwahara A, Hirabayashi Y, Knoepfler PS, Taketo MM, Sakai J, Kodama T, et al. Wnt signaling and its downstream target N-myc regulate basal progenitors in the developing neocortex. Development (2010) 137:1035–44. doi: 10.1242/dev.046417
27. Qin XY, Hara M, Arner E, Kawaguchi Y, Inoue I, Tatsukawa H, et al. Transcriptome Analysis Uncovers a Growth-Promoting Activity of Orosomucoid-1 on Hepatocytes. EBioMedicine (2017) 24:257–66. doi: 10.1016/j.ebiom.2017.09.008
28. Yanger K, Knigin D, Zong Y, Maggs L, Gu G, Akiyama H, et al. Adult hepatocytes are generated by self-duplication rather than stem cell differentiation. Cell Stem Cell (2014) 15:340–9. doi: 10.1016/j.stem.2014.06.003
29. Michalopoulos GK, DeFrances MC. Liver regeneration. Science (1997) 276:60–6. doi: 10.1126/science.276.5309.60
30. Hossain S, Takatori A, Nakamura Y, Suenaga Y, Kamijo T, Nakagawara A. NLRR1 enhances EGF-mediated MYCN induction in neuroblastoma and accelerates tumor growth in vivo. Cancer Res (2012) 72:4587–96. doi: 10.1158/0008-5472.CAN-12-0943
31. Mishra L, Banker T, Murray J, Byers S, Thenappan A, He AR, et al. Liver stem cells and hepatocellular carcinoma. Hepatology (2009) 49:318–29. doi: 10.1002/hep.22704
32. Muto Y, Moriwaki H, Ninomiya M, Adachi S, Saito A, Takasaki KT, et al. Prevention of Second Primary Tumors by an Acyclic Retinoid, Polyprenoic Acid, in Patients with Hepatocellular Carcinoma. New Engl J Med (1996) 334:1561–8. doi: 10.1056/NEJM199606133342402
33. Yamashita T, Ji J, Budhu A, Forgues M, Yang W, Wang HY, et al. EpCAM-Positive Hepatocellular Carcinoma Cells Are Tumor-Initiating Cells With Stem/Progenitor Cell Features. Gastroenterology (2009) 136:1012–1024.e4. doi: 10.1053/j.gastro.2008.12.004
34. Okita K, Izumi N, Ikeda K, Osaki Y, Numata K, Ikeda M, et al. Survey of survival among patients with hepatitis C virus-related hepatocellular carcinoma treated with peretinoin, an acyclic retinoid, after the completion of a randomized, placebo-controlled trial. J Gastroenterol (2014) 50:667–74. doi: 10.1007/s00535-014-0996-1
35. Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell (2010) 140:197–208. doi: 10.1016/j.cell.2009.12.052
36. Umemura A, Park EJ, Taniguchi K, Lee JH, Shalapour S, Valasek MA, et al. Liver damage, inflammation, and enhanced tumorigenesis after persistent mTORC1 inhibition. Cell Metab (2014) 20:133–44. doi: 10.1016/j.cmet.2014.05.001
37. Grohmann M, Wiede F, Dodd GT, Gurzov EN, Ooi GJ, Butt T, et al. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell (2018) 175:1289–1306 e20. doi: 10.1016/j.cell.2018.09.053
38. Johnson DE, O’Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol (2018) 15:234–48. doi: 10.1038/nrclinonc.2018.8
39. Sattu K, Hochgrafe F, Wu J, Umapathy G, Schonherr C, Ruuth K, et al. Phosphoproteomic analysis of anaplastic lymphoma kinase (ALK) downstream signaling pathways identifies signal transducer and activator of transcription 3 as a functional target of activated ALK in neuroblastoma cells. FEBS J (2013) 280:5269–82. doi: 10.1111/febs.12453
40. Odate S, Veschi V, Yan S, Lam N, Woessner R, Thiele CJ. Inhibition of STAT3 with the Generation 2.5 Antisense Oligonucleotide, AZD9150, Decreases Neuroblastoma Tumorigenicity and Increases Chemosensitivity. Clin Cancer Res (2017) 23:1771–84. doi: 10.1158/1078-0432.CCR-16-1317
41. Won C, Kim BH, Yi EH, Choi KJ, Kim EK, Jeong JM, et al. Signal transducer and activator of transcription 3-mediated CD133 up-regulation contributes to promotion of hepatocellular carcinoma. Hepatology (2015) 62:1160–73. doi: 10.1002/hep.27968
42. He G, Dhar D, Nakagawa H, Font-Burgada J, Ogata H, Jiang Y, et al. Identification of Liver Cancer Progenitors Whose Malignant Progression Depends on Autocrine IL-6 Signaling. Cell (2013) 155:384–96. doi: 10.1016/j.cell.2013.09.031
43. López-Grueso MJ, González R, Muntané J, Bárcena JA, Padilla CA. Thioredoxin Downregulation Enhances Sorafenib Effects in Hepatocarcinoma Cells. Antioxidants (2019) 8:501. doi: 10.3390/antiox8100501
44. Oakley F, Trim N, Constandinou CM, Ye W, Gray AM, Frantz G, et al. Hepatocytes Express Nerve Growth Factor during Liver Injury. Am J Pathol (2003) 163:1849–58. doi: 10.1016/S0002-9440(10)63544-4
45. Woo CW, Lucarelli E, Thiele CJ. NGF activation of TrkA decreases N-myc expression via MAPK path leading to a decrease in neuroblastoma cell number. Oncogene (2004) 23:1522–30. doi: 10.1038/sj.onc.1207267
46. Kristiansen M, Menghi F, Hughes R, Hubank M, Ham J. Global analysis of gene expression in NGF-deprived sympathetic neurons identifies molecular pathways associated with cell death. BMC Genomics (2011) 12:551. doi: 10.1186/1471-2164-12-551
47. Ibarguren M, López DJ, Escribá PV. The effect of natural and synthetic fatty acids on membrane structure, microdomain organization, cellular functions and human health. Biochim Biophys Acta (BBA) - Biomembr (2014) 1838:1518–28. doi: 10.1016/j.bbamem.2013.12.021
48. Autumn G. York, Williams KJ, Argus JP, Zhou QD, Brar G, Vergnes L, et al. Limiting Cholesterol Biosynthetic Flux Spontaneously Engages Type I IFN Signaling. Cell (2015) 163:1716–29. doi: 10.1016/j.cell.2015.11.045
49. Carroll RG, Zasłona Z, Galván-Peña S, Koppe EL, Sévin DC, Angiari S, et al. An unexpected link between fatty acid synthase and cholesterol synthesis in proinflammatory macrophage activation. J Biol Chem (2018) 293:5509–21. doi: 10.1074/jbc.RA118.001921
50. Hsieh WY, Zhou QD, York AG, Williams KJ, Scumpia PO, Kronenberger EB, et al. Toll-Like Receptors Induce Signal-Specific Reprogramming of the Macrophage Lipidome. Cell Metab (2020) 32:128–43 e5. doi: 10.1016/j.cmet.2020.05.003
51. Wei X, Song H, Yin L, Rizzo MG, Sidhu R, Covey DF, et al. Fatty acid synthesis configures the plasma membrane for inflammation in diabetes. Nature (2016) 539:294–8. doi: 10.1038/nature20117
52. Oishi Y, Spann NJ, Link VM, Muse ED, Strid T, Edillor C, et al. SREBP1 Contributes to Resolution of Pro-inflammatory TLR4 Signaling by Reprogramming Fatty Acid Metabolism. Cell Metab (2017) 25:412–27. doi: 10.1016/j.cmet.2016.11.009
53. Ben-David U, Gan Q-F, Golan-Lev T, Arora P, Yanuka O, Oren YS, et al. Selective Elimination of Human Pluripotent Stem Cells by an Oleate Synthesis Inhibitor Discovered in a High-Throughput Screen. Cell Stem Cell (2013) 12:167–79. doi: 10.1016/j.stem.2012.11.015
54. Li J, Condello S, Thomes-Pepin J, Ma X, Xia Y, Hurley TD, et al. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell (2017) 20:303–14.e5. doi: 10.1016/j.stem.2016.11.004
55. Qin X-Y, Dohmae N, Kojima S. Reply to Yoshida: Liver cancer stem cells: Identification and lipid metabolic reprogramming. Proc Natl Acad Sci (2018) 115:E6390–1. doi: 10.1073/pnas.1808740115
56. Qin X-Y, Su T, Yu W, Kojima S. Lipid desaturation-associated endoplasmic reticulum stress regulates MYCN gene expression in hepatocellular carcinoma cells. Cell Death Dis (2020) 11:66. doi: 10.1038/s41419-020-2257-y
57. Yoshida GJ. Beyond the Warburg Effect: N-Myc Contributes to Metabolic Reprogramming in Cancer Cells. Front Oncol (2020) 10:791. doi: 10.3389/fonc.2020.00791
58. Rouault-Pierre K, Lopez-Onieva L, Foster K, Anjos-Afonso F, Lamrissi-Garcia I, Serrano-Sanchez M, et al. HIF-2α Protects Human Hematopoietic Stem/Progenitors and Acute Myeloid Leukemic Cells from Apoptosis Induced by Endoplasmic Reticulum Stress. Cell Stem Cell (2013) 13:549–63. doi: 10.1016/j.stem.2013.08.011
59. Miharada K, Sigurdsson V, Karlsson S. Dppa5 Improves Hematopoietic Stem Cell Activity by Reducing Endoplasmic Reticulum Stress. Cell Rep (2014) 7:1381–92. doi: 10.1016/j.celrep.2014.04.056
60. Heijmans J, van Lidth de Jeude JF, Koo B-K, Rosekrans SL, Wielenga MCB, van de Wetering M, et al. ER Stress Causes Rapid Loss of Intestinal Epithelial Stemness through Activation of the Unfolded Protein Response. Cell Rep (2013) 3:1128–39. doi: 10.1016/j.celrep.2013.02.031
61. Wielenga MCB, Colak S, Heijmans J, van Lidth de Jeude JF, Rodermond HM, Paton JC, et al. ER-Stress-Induced Differentiation Sensitizes Colon Cancer Stem Cells to Chemotherapy. Cell Rep (2015) 13:489–94. doi: 10.1016/j.celrep.2015.09.016
62. Liu R, Li X, Huang Z, Zhao D, Ganesh BS, Lai G, et al. C/EBP homologous protein-induced loss of intestinal epithelial stemness contributes to bile duct ligation-induced cholestatic liver injury in mice. Hepatology (2018) 67:1441–57. doi: 10.1002/hep.29540
63. Ma MKF, Lau EYT, Leung DHW, Lo J, Ho NPY, Cheng LKW, et al. Stearoyl-CoA desaturase regulates sorafenib resistance via modulation of ER stress-induced differentiation. J Hepatol (2017) 67:979–90. doi: 10.1016/j.jhep.2017.06.015
64. Pinkham K, Park DJ, Hashemiaghdam A, Kirov AB, Adam I, Rosiak K, et al. Stearoyl CoA Desaturase Is Essential for Regulation of Endoplasmic Reticulum Homeostasis and Tumor Growth in Glioblastoma Cancer Stem Cells. Stem Cell Rep (2019) 12:712–27. doi: 10.1016/j.stemcr.2019.02.012
65. Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol-Endocrinol Metab (2006) 291:E275–81. doi: 10.1152/ajpendo.00644.2005
66. Borradaile NM, Han X, Harp JD, Gale SE, Ory DS, Schaffer JE. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J Lipid Res (2006) 47:2726–37. doi: 10.1194/jlr.M600299-JLR200
67. Wei Y, Wang D, Gentile CL, Pagliassotti MJ. Reduced endoplasmic reticulum luminal calcium links saturated fatty acid-mediated endoplasmic reticulum stress and cell death in liver cells. Mol Cell Biochem (2009) 331:31–40. doi: 10.1007/s11010-009-0142-1
68. Ariyama H, Kono N, Matsuda S, Inoue T, Arai H. Decrease in Membrane Phospholipid Unsaturation Induces Unfolded Protein Response. J Biol Chem (2010) 285:22027–35. doi: 10.1074/jbc.M110.126870
69. Gebert LFR, MacRae IJ. Regulation of microRNA function in animals. Nat Rev Mol Cell Biol (2019) 20:21–37. doi: 10.1038/s41580-018-0045-7
70. Mendell JT, Olson EN. MicroRNAs in stress signaling and human disease. Cell (2012) 148:1172–87. doi: 10.1016/j.cell.2012.02.005
71. Lujambio A, Lowe SW. The microcosmos of cancer. Nature (2012) 482:347–55. doi: 10.1038/nature10888
72. Gailhouste L, Ochiya T. Cancer-related microRNAs and their role as tumor suppressors and oncogenes in hepatocellular carcinoma. Histol Histopathol (2013) 28:437–51. doi: 10.14670/HH-28.437
73. Domingo-Fernandez R, Watters K, Piskareva O, Stallings RL, Bray I. The role of genetic and epigenetic alterations in neuroblastoma disease pathogenesis. Pediatr Surg Int (2013) 29:101–19. doi: 10.1007/s00383-012-3239-7
74. Attiyeh EF, London WB, Mosse YP, Wang Q, Winter C, Khazi D, et al. Chromosome 1p and 11q deletions and outcome in neuroblastoma. N Engl J Med (2005) 353:2243–53. doi: 10.1056/NEJMoa052399
75. Wei JS, Song YK, Durinck S, Chen QR, Cheuk AT, Tsang P, et al. The MYCN oncogene is a direct target of miR-34a. Oncogene (2008) 27:5204–13. doi: 10.1038/onc.2008.154
76. Molenaar JJ, Domingo-Fernandez R, Ebus ME, Lindner S, Koster J, Drabek K, et al. LIN28B induces neuroblastoma and enhances MYCN levels via let-7 suppression. Nat Genet (2012) 44:1199–206. doi: 10.1038/ng.2436
77. Buechner J, Tomte E, Haug BH, Henriksen JR, Lokke C, Flaegstad T, et al. Tumour-suppressor microRNAs let-7 and mir-101 target the proto-oncogene MYCN and inhibit cell proliferation in MYCN-amplified neuroblastoma. Br J Cancer (2011) 105:296–303. doi: 10.1038/bjc.2011.220
78. Neviani P, Wise PM, Murtadha M, Liu CW, Wu CH, Jong AY, et al. Natural Killer-Derived Exosomal miR-186 Inhibits Neuroblastoma Growth and Immune Escape Mechanisms. Cancer Res (2018) 79:1151–64. doi: 10.1158/0008-5472.CAN-18-0779
79. Ooi CY, Carter DR, Liu B, Mayoh C, Beckers A, Lalwani A, et al. Network Modeling of microRNA-mRNA Interactions in Neuroblastoma Tumorigenesis Identifies miR-204 as a Direct Inhibitor of MYCN. Cancer Res (2018) 78:3122–34. doi: 10.1158/0008-5472.CAN-17-3034
80. Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol (2010) 12:247–56. doi: 10.1038/ncb2024
81. Gailhouste L, Gomez-Santos L, Ochiya T. Potential applications of miRNAs as diagnostic and prognostic markers in liver cancer. Front Biosci (Landmark Ed) (2013) 18:199–223. doi: 10.2741/4096
82. Gailhouste L, Liew LC, Yasukawa K, Hatada I, Tanaka Y, Kato T, et al. MEG3-derived miR-493-5p overcomes the oncogenic feature of IGF2-miR-483 loss of imprinting in hepatic cancer cells. Cell Death Dis (2019) 10:553. doi: 10.1038/s41419-019-1788-6
83. Yasukawa K, Liew LC, Hagiwara K, Hironaka-Mitsuhashi A, Qin XY, Furutani Y, et al. MicroRNA-493-5p-mediated repression of the MYCN oncogene inhibits hepatic cancer cell growth and invasion. Cancer Sci (2020) 111:869–80. doi: 10.1111/cas.14292
84. Xiong DD, Dang YW, Lin P, Wen DY, He RQ, Luo DZ, et al. A circRNA-miRNA-mRNA network identification for exploring underlying pathogenesis and therapy strategy of hepatocellular carcinoma. J Transl Med (2018) 16:220. doi: 10.1186/s12967-018-1593-5
85. Lou K, Chen N, Li Z, Zhang B, Wang X, Chen Y, et al. MicroRNA-142-5p Overexpression Inhibits Cell Growth and Induces Apoptosis by Regulating FOXO in Hepatocellular Carcinoma Cells. Oncol Res (2017) 25:65–73. doi: 10.3727/096504016X14719078133366
86. Tsang FH, Au SL, Wei L, Fan DN, Lee JM, Wong CC, et al. MicroRNA-142-3p and microRNA-142-5p are downregulated in hepatocellular carcinoma and exhibit synergistic effects on cell motility. Front Med (2015) 9:331–43. doi: 10.1007/s11684-015-0409-8
87. Lima L, de Melo TCT, Marques D, de Araujo JNG, Leite ISF, Alves CX, et al. Modulation of all-trans retinoic acid-induced MiRNA expression in neoplastic cell lines: a systematic review. BMC Cancer (2019) 19:866. doi: 10.1186/s12885-019-6081-7
88. Mu X, Español-Suñer R, Mederacke I, Affò S, Manco R, Sempoux C, et al. Hepatocellular carcinoma originates from hepatocytes and not from the progenitor/biliary compartment. J Clin Invest (2015) 125:3891–903. doi: 10.1172/JCI77995
Keywords: MYCN, liver cancer, microenvironment, inflammation, plasticity, lipid desaturation, endoplasmic reticulum stress, miRNA
Citation: Qin X-Y and Gailhouste L (2021) Non-Genomic Control of Dynamic MYCN Gene Expression in Liver Cancer. Front. Oncol. 10:618515. doi: 10.3389/fonc.2020.618515
Received: 17 October 2020; Accepted: 23 December 2020;
Published: 16 April 2021.
Edited by:
Yusuke Suenaga, Chiba Cancer Center, JapanReviewed by:
Shoma Tsubota, Nagoya University, JapanAlexander Schramm, Essen University Hospital, Germany
Copyright © 2021 Qin and Gailhouste. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xian-Yang Qin, eHlxaW5AcmlrZW4uanA=; Luc Gailhouste, bHVjLmdhaWxob3VzdGVAcmlrZW4uanA=