
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol., 21 January 2021
Sec. Pediatric Oncology
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.614541
This article is part of the Research TopicRecent Advances in Pediatric Cancer Predisposition SyndromesView all 12 articles
 Anna Maria Caroleo1*
Anna Maria Caroleo1* Maria Antonietta De Ioris1
Maria Antonietta De Ioris1 Luigi Boccuto2,3Iside Alessi1
Luigi Boccuto2,3Iside Alessi1 Giada Del Baldo1
Giada Del Baldo1 Antonella Cacchione1
Antonella Cacchione1 Emanuele Agolini4
Emanuele Agolini4 Martina Rinelli4Annalisa Serra1
Martina Rinelli4Annalisa Serra1 Andrea Carai5
Andrea Carai5 Angela Mastronuzzi1
Angela Mastronuzzi1DICER1 syndrome is a rare genetic condition predisposing to hereditary cancer and caused by variants in the DICER1 gene. The risk to present a neoplasm before the age of 10 years is 5.3 and 31.5% before the age of 60. DICER1 variants have been associated with a syndrome involving familial pleuropulmonary blastoma (PPB), a rare malignant tumor of the lung, which occurs primarily in children under the age of 6 years and represents the most common life-threatening manifestation of DICER1 syndrome. Type I, II, III, and Ir (type I regressed) PPB are reported with a 5-year overall survival ranging from 53 to 100% (for type Ir). DICER1 gene should be screened in all patients with PPB and considered in other tumors mainly in thyroid neoplasms (multinodular goiter, thyroid cancer, adenomas), ovarian tumors (Sertoli-Leydig cell tumor, sarcoma, and gynandroblastoma), and cystic nephroma. A prompt identification of this syndrome is necessary to plan a correct follow-up and screening during lifetime.
DICER1 syndrome is a cancer-predisposing disorder caused by pathogenic variants in the DICER1 gene (OMIM 606241), which are known to confer a lifetime risks for a variety of neoplastic and dysplastic lesions (1).
Germline DICER1 variants have been detected in individuals affected with familial pleuropulmonary blastoma (PPB) (2–5), a rare malignant tumor of the lung, which occurs primarily in children under the age of 6 years (6). The International PPB Registry collected data from PPB patients and their families, reporting a variety of tumors in individuals with PPB and/or their relatives (6). A study on 207 carriers of DICER1 pathogenic variants reported that the risk to develop a neoplasm is 5.3% before the age of 10 years and of 31.5% before the age of 60, while in the American general population is estimated to be respectively 0.17 and 6.57% (1, 7). DICER1 syndrome occurs in children and young adults and its clinical presentation may include, beyond PPB, cystic nephroma, ovarian Sertoli-Leydig cell tumor (SLCT), multinodular goiter, cervix embryonal rhabdomyosarcoma, Wilms’ tumor, nasal chondromesenchymal hamartoma, ciliary body medulloepithelioma, differentiated thyroid carcinoma, pituitary blastoma, pineoblastoma, and sarcomas of different sites including, amongst others, the uterine cervix, kidney, and brain (8).
This syndrome shows an autosomal dominant inheritance pattern with reduced penetrance, which likely decreases the rate of familial cases. In cases with PPB, about 80% of the DICER1 germline pathogenic variants are inherited by a parent and nearly 20% are de novo (9).
This paper aims to review the clinical and genetic features of DICER1 syndrome, with particular focus on the description of the different types of cancer reported in this syndrome, grouped by systems.
The DICER1 gene, located on chromosome 14q32.13, encodes an RNA endonuclease (Dicer) that is involved in the post-transcriptional gene expression of over 30% of protein-coding genes by modulating microRNAs (miRNAs) (10, 11).
miRNAs are transcribed as pri-miRNAs, that are longer precursor, which are elaborated into pre-miRNAs in the nucleus. The pre-miRNAs, transported to the cytoplasm, are processed by Dicer to give a ∼21-bp RNA duplex intermediate. One strand of this RNA is incorporated into the RNA-induced silencing complex (RISC), and matched to complementary mRNA targets to regulate gene expression, inhibiting mRNA degradation (12).
In most syndrome’s neoplasms a biallelic pathogenic variant in DICER1 has been detected: usually a germline loss-of-function pathogenic variant in one allele and a tumor-specific somatic hotspot variant in the second allele. Several studies have shown that “monoallelic DICER1 inactivation promotes tumorigenesis, whereas biallelic loss is inhibitory, and although inactivation of one DICER1 allele is the initiating event in DICER1 syndrome”, leading “to dysregulation of miRNA levels, other events must be required for cancer to occur ” (13, 14). Only one third of DICER1 carriers present a neoplasm during the life, hinting that multiple additional events are required (13, 14).
This process suggests a predominant haploinsufficient tumor-suppressor function, where one copy of Dicer, albeit mutated, is functioning, rather than a more classical “two-hit” tumor suppressor model, which has been described in association with earlier diagnosis of DICER1-related conditions, where no function of the oncosuppressor gene is preserved (5, 15, 16).
Complete loss of Dicer is incompatible with life (4, 17, 18), while somatic mosaic mutations in the RNase IIIb domain have been associated with a more serious form of DICER1 syndrome, named GLOW syndrome from Global developmental delay, Lung cysts, Overgrowth, and Wilms tumor (19). Functional evidence links the hotspot mutations in the RNAse IIIb domain to specific dysregulation of certain miRNAs leading to activation of the PI3K/AKT/mTOR pathway (20). This mechanistic link to the PI3K/AKT/mTOR pathway may explain the fact that GLOW syndrome shares some clinical features with other conditions characterized by somatic gain-of-function mutations of genes of this pathway, such as lung cysts, reported in Proteus syndrome, and segmental overgrowth, a prominent feature of PROS (21).
The recurrent involvement of specific organs (lungs, thyroid, kidneys, ovaries) in presence of DICER1 alterations may lead to infer that the effects of miRNAs on gene expression are tissue-specific (19). Nonetheless, the penetrance of each of the DICER1-associated neoplasms in inherited conditions is not fully understood. Individuals carrying germline loss-of-function mutations may present clinical features in few sites (0–2) of their body, while patients with mosaic “hotspot” mutations are more prone to manifestations in multiple site (6).
Different tumors are related to DICER1 syndrome as reported by Foulkes et al. and by Stewart et al. In Figure 1 we resumed the principal neoplasms according to the age of onset.
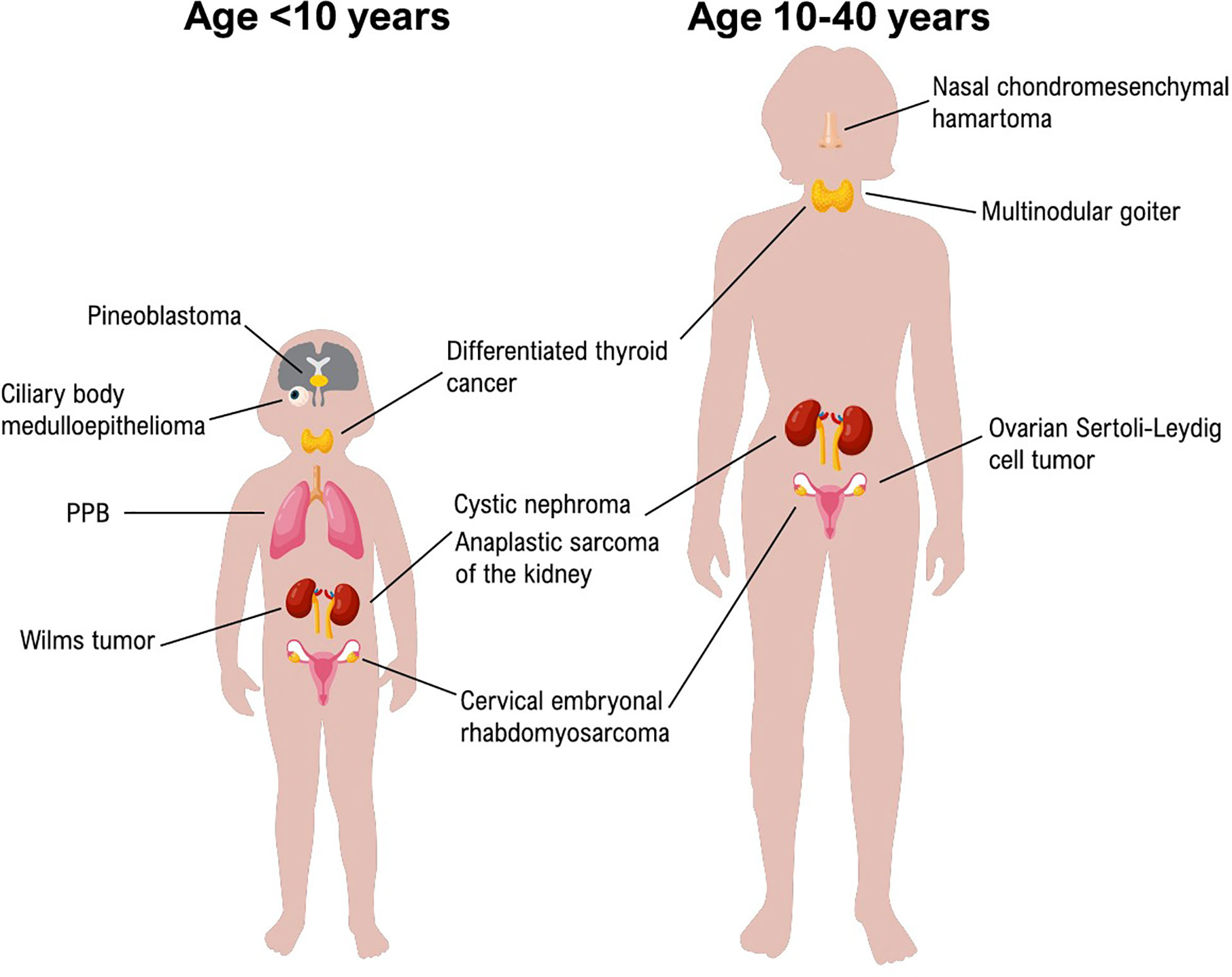
Figure 1 Principal DICER1-Syndrome neoplasms according to the age of onset.
Foulkes et al. in 2014 described the DICER1-associated features and their characteristics, as reported in Table 1 (5).
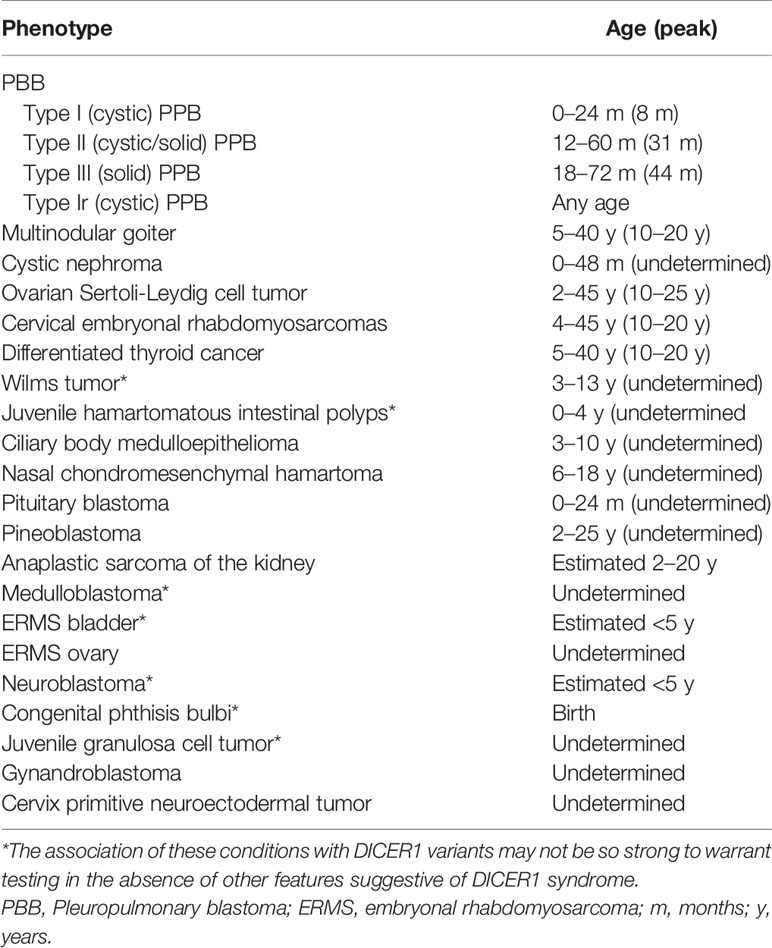
Table 1 Key clinical phenotypes (ordered by relative frequency) associated with germline DICER1 mutations.
Stewart et al. recently published the first quantitative analysis of site-specific neoplasm risk, analyzing the standardized incidence ratios of 207 individuals carrying DICER1 variants, selected combining data from three large cohorts of patients. The most remarkable rates were noted in PPB, in gynecologic tumors, especially SLCTs and rhabdomyosarcoma, and in cystic nephroma (1).
PPB is a rare tumor that develops during fetal life/infancy and constitutes the most common life-threatening manifestation of DICER1 syndrome (22). Type I PPB is typically a purely cystic mass occurring before age of 2 years, with a 5-year overall survival (OS) of 89% if it does not progress to type II or III PPB. Type II is a solid-cystic tumor while type III is purely solid; both types present from approximately 2 to 6 years of age and are malignant, although type III is generally more aggressive. If treated with chemotherapy and radiotherapy, OS rates may reach up to 74% in type II and 53% in type III. The fourth type, named type Ir, as “type I regressed”, is a cystic tumor lacking malignant cells and is supposed to represent regressed/non-progressed type I PPB. OS for this type of PPB is 100%. Cystic PPB is reported to be common in carriers of DICER1 variants, and only a limited number of cases had a type II II or III PPB progression (1).
The PPB begins as a cystic lung lesion, also defined as a Type I PPB, a well-defined pathology entity with a potential evolution in a more aggressive tumor. We need to underline that the imaging findings of Type I PPB is overlapped with congenital lung cyst; congenital lung cyst with congenital pulmonary airway malformation (CPAM) are almost diagnosed in prenatal period or over the first year and a surgical approach—with pathology study—was mandatory only in symptomatic cases. Indeed, in more than 70% of CPAM, a wait and see strategy is addressed (23); in this cases a DICER1 variants should always be considered in order to identify promptly with a strict follow-up and genetic screening patients at risk of more aggressive PBB. The pathology should always consider PPB evaluating a CPAM.
Shortness of breath and pneumothorax due to cyst rupture may be the presenting symptoms of PPB.
Multinodular goiter (MNG) is characterized by the development of thyroid nodular lesions. MNG is common in individuals with DICER1 pathogenic variants, as reported by Khan et al. (24). Germline DICER1 mutations have been reported in children with both MNG or familial MNG (25). The risk of DTC in carriers of DICER1 variants is elevated as compared to the general population and its occurrence is typically related to an indolent course (26).
Cystic nephroma is a benign multicystic kidney tumor that constitutes the most common neoplasm associated with PPB (3). It has a bimodal incidence: 65% of cases occur in the pediatric band, before the age of 4, while 35% of cases appear in adulthood and are usually seen between the fourth and the sixth decade (27, 28).
DICER1 syndrome also includes an elevated risk of Wilms’ tumor, an embryonal cancer of the kidney that affects children before the age of 6, without evidence to be a consequence of a prior cystic nephroma (29, 30).
Recent reports enumerate anaplastic sarcoma of the kidney in DICER1 syndrome, correlating the germline DICER1 mutations with the development of these tumors, and postulate that they may arise from pre-existing pediatric cystic nephromas (31–33).
The gynecologic tumors most frequently associated to DICER1 syndrome are ovarian SLCTs and embryonal rhabdomyosarcoma of the cervix. These neoplasms, as well as PPB and MNG, constitute key features leading to consider an underlying cancer predisposition syndrome, especially if found in children or adolescents (34).
Unlike PPB, the age range of increased risk for genital tract tumors is wide (2 to 40 years), even if some data suggest that ovarian SLCTs arising in patients carrying DICER1 variants occur mostly in the second decade (18, 35). Moderately differentiated SLCTs are most common, but juvenile granulosa cell tumor (JGCT), gynandroblastoma, and unclassified sex cord-stromal tumors have also been described. Most tumors are stage I, presenting with androgenic symptoms and a pelvic mass, that rarely may be bilateral (34). The prognosis of ovarian SLCT is generally favorable, but a recent report indicates that somatic DICER1 variants SLCTs may be linked to a higher relapse risk than others (36).
Even though rhabdomyosarcoma is the most common cervical sarcoma, it is still very rare (37). Most DICER1 cases are confined to the cervix at diagnosis, presenting with polypoid appearances (botryoides) and with vaginal bleeding. Even if ERMS is one of the more common sarcomas in childhood, approximately a third of DICER1-related ERMS arises in patients older than 20 years (38, 39). Studies report a quite favorable prognosis with an EFS over 50%, and an OS around 90% (34, 38–41).
Poorly differentiated ovarian sarcoma (42), retiform SLCT, and primitive neuroectodermal tumor (PNET) of the cervix (43) have also been reported in individuals with possible germline DICER1 variants.
Pituitary blastoma is an extremely rare tumor of the anterior pituitary. Genetic tests performed on 14 cases, on a total of 16 described to date, showed that all have at least one pathogenic variant in DICER1 (44–46). For such reason, pituitary blastoma may be considered pathognomonic for DICER1 syndrome (46).
Pineoblastoma is a rare primitive neuroectodermal grade IV tumor originating in the pineal gland (47). Only a few genes have been implicated in the pathogenesis of pineoblastomas, for instance, RB1 in the setting of “trilateral retinoblastoma” (48).
To date, in DICER1-related pineoblastomas loss of heterozygosity of the wild-type DICER1 allele seems to be the somatic event, in contrast from the typical missense hotspot mutations that usually lead to a factual germline heterozygosity (49–53). Moreover, somatic DROSHA and DGCR8 mutations, both related to the Dicer miRNA-regulating pathway, have been recently documented in pineoblastomas, in addition to germline and somatic DICER1 mutations (50), indicating that pineoblastoma development is influenced by disturbances of miRNA processes (46).
Other brain tumors associated to DICER1 alterations have also been reported but their genetic association has not been clearly demonstrated. These include medulloblastoma (6, 54), intracranial medulloepithelioma (55), anaplastic meningeal sarcoma (53), glioblastoma multiforme (56, 57), and embryonal tumor with multilayered rosettes (ETMR) (58).
Ciliary body medulloepithelioma is a rare embryonal ocular tumor, that arises from the eye’s ciliary body, which generally occurs during infancy and constitutes the second most common eye tumor of childhood, after retinoblastoma (59–61).
Some cases suspected to be DICER1-related have been documented but further studies are required to support their association with the syndrome (6, 15, 62–71).
Nasal chondromesenchymal hamartoma is a rare benign tumor of the sinus and nasal cavities that have been described in children with PPB. This peculiar association has led to the assumption that this hamartoma is also a manifestation of DICER1 syndrome (5, 72).
Molecular genetic testing methods, including single-gene or multigene panel testing, may be considered when clinical, imaging, and/or histopathological features evoke a DICER1 syndrome’s diagnosis. Heterozygosis is the most common condition through DICER1 syndrome’s patients, where commonly a germline loss‐of‐function gene variant (nonsense, frameshift, or splice-affected) generates a truncated protein. These variants can be identified by Sanger sequencing or next-generation sequencing (NGS). NGS has specific advantages over traditional Sanger sequencing, considered the gold standard for mutation analysis for many years, as multiple genes in several patients can be tested simultaneously. Indeed, when the phenotype is hard to distinguish from many other cancer predisposition syndromes, extensive genetic testing, based on multigene panels or exome analysis can be useful to identify the molecular defects underlying the condition.
Besides point mutations, other predisposing DICER1 alterations have also been documented, including deletion of the entire DICER1 locus (62), or intragenic deletions involving one or more exons (73). Methods used to detect these kinds of alterations may include quantitative polymerase chain reaction (PCR), multiplex ligation-dependent probe amplification (MLPA) and gene-targeted microarray. Finally, molecular genetic testing of tumor DNA may be necessary to identify somatic mosaicism, which is observed in 10% of individuals with DICER1 syndrome.
Although risks of malignancy are elevated, most patients with pathogenic germline DICER1 variants live healthy lives. Indeed, a tumor occurs in 19,3% of the patients who carry germline pathogenic variation by the age of 50 years old and the neoplastic risk rises with age, especially in females, that are exposed to the risk to present with gynecologic neoplasms (1).
Schultz et al. have defined the indications for DICER1 genetic counseling and testing, and they also provided specific screening strategies to manage risk in carriers of DICER1 pathogenic variants (2). Germline DICER1 genetic testing is to consider in individuals with one major or two minor criteria. “Major criteria are: PPB, lung cysts in childhood, thoracic embryonal rhabdomyosarcoma, cystic nephroma, genitourinary sarcomas including undifferentiated sarcoma, ovarian Sertoli−Leydig cell tumor, gynandroblastoma, uterine cervical or ovarian embryonal rhabdomyosarcoma, genitourinary/gynecologic neuroendocrine tumors, multinodular goiter or thyroid cancer in two or more first-degree relatives or in an index patient with a family history consistent with DICER1 syndrome, childhood-onset multinodular goiter or differentiated thyroid cancer, ciliary body medulloepithelioma, nasal chondromesenchymal hamartoma, pineoblastoma, pituitary blastoma. Minor criteria are: Lung cysts in adults, renal cysts, Wilms tumor, multinodular goiter or differentiated thyroid cancer, embryonal rhabdomyosarcoma other than thoracic or gynecologic, poorly differentiated neuroendocrine tumor, undifferentiated sarcoma, macrocephaly” (2).
Surveillance guidelines for individuals with a germline DICER1 pathogenic variant have been established. The current guidelines include “chest radiograph every 4–6 months until age 8 years, and every 12 months until 12 years; a chest computed tomography scan should be considered. Baseline chest radiograph or chest CT should be considered when the diagnosis is performed after age 12 years. Thyroid ultrasound is recommended by the age of eight years with subsequent ultrasounds every three to five years. Individuals with a history of chemotherapy exposure should begin thyroid ultrasound within three to five years from treatment. Pelvic ultrasounds for surveillance for gynecologic tumors in females are recommended every 6 to 12 months by the age of eight years and extending until at least age 40 years. Screening for cystic nephroma and other renal tumors includes abdominal ultrasounds every six months until age eight years and then annually until age 12 years. Visual acuity measurement and dilated ophthalmology examination for ciliary body medulloepithelioma is recommended annually from age three years until at least age ten years. Annual physical examination should be considered by an expert clinician” (2).
Some studies explored the use of metformin to upregulate DICER1 and linked proteins in mice, to counter the DICER1 syndrome’s effects (74–77). Despite patients affected by biallelic DICER1 mutations may not benefit from this treatment, metformin will be may proposed to patients with a single allele alteration, to try to augment DICER1 protein production and compensate the deficit, preventing the oncogenetic cascade.
DICER1 syndrome is a rare condition caused by germline variants of DICER1; the occurrence of a second somatic tissue-specific mutation leads to different phenotypes ranging from benign lesions to malignant tumors. Screening for DICER1 variants should be performed in all patients with PPB and considered in few benign lesions and malignant tumors. A prompt identification of this syndrome is necessary to plan a correct follow-up and screening for tumor occurrence during the patient’s lifetime.
AC reviewed the literature and was a major contributor in writing the manuscript. MI reviewed the literature and wrote the manuscript. LB contributed to the concept and reviewed critically the manuscript. AM contributed to the concept, reviewed the literature, and reviewed critically the manuscript. All the authors state that no honorarium, grant, or other form of payment was given to anyone to produce the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Stewart DR, Best AF, Williams GM, Harney LA, Carr AG, Harris AK, et al. Neoplasm Risk Among Individuals With a Pathogenic Germline Variant in DICER1. J Clin Oncol (2019) 37(8):668–76. doi: 10.1200/JCO.2018.78.4678
2. Schultz KAP, Williams GM, Kamihara J, Stewart DR, Harris AK, Bauer AJ, et al. DICER1 and associated conditions: Identification of at-risk individuals and recommended surveillance strategies. Clin Cancer Res (2018) 24(10):2251–61. doi: 10.1158/1078-0432.CCR-17-3089
3. Boman F, Hill DA, Williams GM, Chauvenet A, Fournet J-C, Soglio DB-D, et al. Familial association of pleuropulmonary blastoma with cystic nephroma and other renal tumors: a report from the International Pleuropulmonary Blastoma Registry. J Pediatr (2006) 149(6):850–4. doi: 10.1016/j.jpeds.2006.08.068
4. Bahubeshi A, Bal N, Rio Frio T, Hamel N, Pouchet C, Yilmaz A, et al. Germline DICER1 mutations and familial cystic nephroma. J Med Genet (2010) 47(12):863–6. doi: 10.1136/jmg.2010.081216
5. Foulkes WD, Priest JR, Duchaine TF. DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer (2014) 14(10):662–72. doi: 10.1038/nrc3802
6. Slade I, Bacchelli C, Davies H, Murray A, Abbaszadeh F, Hanks S, et al. DICER1 syndrome: clarifying the diagnosis, clinical features and management implications of a pleiotropic tumour predisposition syndrome. J Med Genet (2011) 48(4):273–8. doi: 10.1136/jmg.2010.083790
7. White MC, Holman DM, Boehm JE, Peipins LA, Grossman M, Henley SJ. Age and cancer risk: a potentially modifiable relationship. Am J Prev Med (2014) 46(3 Suppl 1):S7–15. doi: 10.1016/j.amepre.2013.10.029
8. de Kock L, MK W, Foulkes WD. Ten years of DICER1 mutations: Provenance, distribution, and associated phenotypes. Hum Mutation (2019) 40(11):1939–53. doi: 10.1002/humu.23877
9. Schultz KAP, Stewart DR, Kamihara J, Bauer AJ, Merideth MA, Stratton P, et al. DICER1 Tumor Predisposition. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle (1993). Available from: http://www.ncbi.nlm.nih.gov/books/NBK196157/.
10. Matsuda S, Ichigotani Y, Okuda T, Irimura T, Nakatsugawa S, Hamaguchi M. Molecular cloning and characterization of a novel human gene (HERNA) which encodes a putative RNA-helicase. Biochim Biophys Acta (2000) 1490(1–2):163–9. doi: 10.1016/S0167-4781(99)00221-3
11. Carthew RW. Gene regulation by microRNAs. Curr Opin Genet Dev (2006) 16(2):203–8. doi: 10.1016/j.gde.2006.02.012
12. Cai X, Hagedorn CH, Cullen BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA (2004) 10(12):1957–66. doi: 10.1261/rna.7135204
13. Lambertz I, Nittner D, Mestdagh P, Denecker G, Vandesompele J, Dyer MA, et al. Monoallelic but not biallelic loss of Dicer1 promotes tumorigenesis in vivo. Cell Death Differ (2010) 17(4):633–41. doi: 10.1038/cdd.2009.202
14. Kumar MS, Pester RE, Chen CY, Lane K, Chin C, Lu J, et al. Dicer1 functions as a haploinsufficient tumor suppressor. Genes Dev (2009) 23(23):2700–4. doi: 10.1101/gad.1848209
15. de Kock L, Wang YC, Revil T, Badescu D, Rivera B, Sabbaghian N, et al. High-sensitivity sequencing reveals multi-organ somatic mosaicism causing DICER1 syndrome. J Med Genet (2016) 53(1):43–52. doi: 10.1136/jmedgenet-2015-103428
16. Brenneman M, Field A, Yang J, Williams G, Doros L, Rossi C, et al. Temporal order of RNase IIIb and loss-of-function mutations during development determines phenotype in pleuropulmonary blastoma / DICER1 syndrome: a unique variant of the two-hit tumor suppression model. F1000Res (2015) 4:214. doi: 10.12688/f1000research.6746.2
17. Bernstein E, Kim SY, Carmell MA, Murchison EP, Alcorn H, Li MZ, et al. Dicer is essential for mouse development. Nat Genet (2003) 35(3):215–7. doi: 10.1038/ng1253
18. Rio Frio T, Bahubeshi A, Kanellopoulou C, Hamel N, Niedziela M, Sabbaghian N, et al. DICER1 mutations in familial multinodular goiter with and without ovarian Sertoli-Leydig cell tumors. JAMA (2011) 305(1):68–77. doi: 10.1001/jama.2010.1910
19. Klein S, Lee H, Ghahremani S, Kempert P, Ischander M, Teitell MA, et al. Expanding the phenotype of mutations in DICER1: mosaic missense mutations in the RNase IIIb domain of DICER1 cause GLOW syndrome. J Med Genet (2014) 51(5):294–302. doi: 10.1136/jmedgenet-2013-101943
20. Klein SD, Martinez-Agosto JA. Hotspot Mutations in DICER1 Causing GLOW Syndrome-Associated Macrocephaly via Modulation of Specific microRNA Populations Result in the Activation of PI3K/ATK/mTOR Signaling. Microrna (2020) 9(1):70–80. doi: 10.2174/2211536608666190624114424
21. Neri G, Boccuto L, Stevenson R. Overgrowth Syndromes: A Clinical Guide. Overgrowth Syndromes. Oxford: Oxford University Press (2019). Available at: https://oxfordmedicine.com/view/10.1093/med/9780190944896.001.0001/med-9780190944896.
22. Messinger YH, Stewart DR, Priest JR, Williams GM, Harris AK, Schultz KAP, et al. Pleuropulmonary blastoma: a report on 350 central pathology-confirmed pleuropulmonary blastoma cases by the International Pleuropulmonary Blastoma Registry. Cancer (2015) 121(2):276–85. doi: 10.1002/cncr.29032
23. Kantor N, Wayne C, Nasr A. Symptom development in originally asymptomatic CPAM diagnosed prenatally: a systematic review. Pediatr Surg Int (2018) 34(6):613–20.
24. Khan NE, Bauer AJ, Schultz KAP, Doros L, Decastro RM, Ling A, et al. Quantification of Thyroid Cancer and Multinodular Goiter Risk in the DICER1 Syndrome: A Family-Based Cohort Study. J Clin Endocrinol Metab (2017) 102(5):1614–22. doi: 10.1210/jc.2016-2954
25. Bahubeshi A, Tischkowitz M, Foulkes WD. miRNA Processing and Human Cancer: DICER1 Cuts the Mustard. Sci Trans Med (2011) 3(111):111ps46–111ps46. doi: 10.1126/scitranslmed.3002493
26. Schultz KAP, Rednam SP, Kamihara J, Doros L, Achatz MI, Wasserman JD, et al. PTEN, DICER1, FH, and Their Associated Tumor Susceptibility Syndromes: Clinical Features, Genetics, and Surveillance Recommendations in Childhood. Clin Cancer Res (2017) 23(12):e76–82. doi: 10.1158/1078-0432.CCR-17-0629
27. Stamatiou K, Polizois K, Kollaitis G, Dahanis S, Zafeiropoulos G, Leventis C, et al. Cystic nephroma: a case report and review of the literature. Cases J (2008) 1:267. doi: 10.1186/1757-1626-1-267
28. Truong LD, Choi Y-J, Shen SS, Ayala G, Amato R, Krishnan B. Renal Cystic Neoplasms and Renal Neoplasms Associated With Cystic Renal Diseases: Pathogenetic and Molecular Links. Adv Anatomic Pathol (2003) 10(3):135–59. doi: 10.1097/00125480-200305000-00003
29. Breslow N, Beckwith JB, Ciol M, Sharples K. Age distribution of Wilms’ tumor: report from the National Wilms’ Tumor Study. Cancer Res (1988) 48(6):1653–7.
30. Stiller CA, Parkin DM. International variations in the incidence of childhood renal tumours. Br J Cancer (1990) 62(6):1026–30. doi: 10.1038/bjc.1990.432
31. Wu MK, Vujanic GM, Fahiminiya S, Watanabe N, Thorner PS, O’Sullivan MJ, et al. Anaplastic sarcomas of the kidney are characterized by DICER1 mutations. Mod Pathol (2018) 31(1):169–78. doi: 10.1038/modpathol.2017.100
32. Doros LA, Rossi CT, Yang J, Field A, Williams GM, Messinger Y, et al. DICER1 mutations in childhood cystic nephroma and its relationship to DICER1-renal sarcoma. Mod Pathol (2014) 27(9):1267–80. doi: 10.1038/modpathol.2013.242
33. Wu MK, Goudie C, Druker H, Thorner P, Traubici J, Grant R, et al. Evolution of Renal Cysts to Anaplastic Sarcoma of Kidney in a Child With DICER1 Syndrome. Pediatr Blood Cancer (2016) 63(7):1272–5. doi: 10.1002/pbc.25959
34. Stewart CJR, Charles A, Foulkes WD. Gynecologic Manifestations of the DICER1 Syndrome. Surg Pathol Clin (2016) 9(2):227–41. doi: 10.1016/j.path.2016.01.002
35. Schultz KAP, Pacheco MC, Yang J, Williams GM, Messinger Y, Hill DA, et al. Ovarian sex cord-stromal tumors, pleuropulmonary blastoma and DICER1 mutations: a report from the International Pleuropulmonary Blastoma Registry. Gynecol Oncol (2011) 122(2):246–50. doi: 10.1016/j.ygyno.2011.03.024
36. Goulvent T, Ray-Coquard I, Borel S, Haddad V, Devouassoux-Shisheboran M, Vacher-Lavenu M-C, et al. DICER1 and FOXL2 mutations in ovarian sex cord-stromal tumours: a GINECO Group study. Histopathology (2016) 68(2):279–85. doi: 10.1111/his.12747
37. Fadare O. Uncommon sarcomas of the uterine cervix: a review of selected entities. Diagn Pathol (2006) 1:30. doi: 10.1186/1746-1596-1-30
38. Daya DA, Scully RE. Sarcoma botryoides of the uterine cervix in young women: a clinicopathological study of 13 cases. Gynecol Oncol (1988) 29(3):290–304. doi: 10.1016/0090-8258(88)90228-4
39. Li RF, Gupta M, McCluggage WG, Ronnett BM. Embryonal rhabdomyosarcoma (botryoid type) of the uterine corpus and cervix in adult women: report of a case series and review of the literature. Am J Surg Pathol (2013) 37(3):344–55. doi: 10.1097/PAS.0b013e31826e0271
40. Dehner LP, Jarzembowski JA, Hill DA. Embryonal rhabdomyosarcoma of the uterine cervix: a report of 14 cases and a discussion of its unusual clinicopathological associations. Mod Pathol (2012) 25(4):602–14. doi: 10.1038/modpathol.2011.185
41. Kriseman ML, Wang W-L, Sullinger J, Schmeler KM, Ramirez PT, Herzog CE, et al. Rhabdomyosarcoma of the cervix in adult women and younger patients. Gynecol Oncol (2012) 126(3):351–6. doi: 10.1016/j.ygyno.2012.05.008
42. Schultz KAP, Harris A, Messinger Y, Sencer S, Baldinger S, Dehner LP, et al. Ovarian tumors related to intronic mutations in DICER1: a report from the international ovarian and testicular stromal tumor registry. Fam Cancer (2016) 15(1):105–10. doi: 10.1007/s10689-015-9831-y
43. Panagiotou JP, Polychronopoulou S, Sofou K, Vanvliet-Constantinidou C, Papandreou E, Haidas S. Second and third malignant solid tumor in a girl with ovarian Sertoli-Leydig tumor. Pediatr Blood Cancer (2006) 46(5):654–6. doi: 10.1002/pbc.20486
44. de Kock L, Sabbaghian N, Plourde F, Srivastava A, Weber E, Bouron-Dal Soglio D, et al. Pituitary blastoma: a pathognomonic feature of germ-line DICER1 mutations. Acta Neuropathol (2014) 128(1):111–22. doi: 10.1007/s00401-014-1285-z
45. Sahakitrungruang T, Srichomthong C, Pornkunwilai S, Amornfa J, Shuangshoti S, Kulawonganunchai S, et al. Germline and somatic DICER1 mutations in a pituitary blastoma causing infantile-onset Cushing’s disease. J Clin Endocrinol Metab (2014) 99(8):E1487–1492. doi: 10.1210/jc.2014-1016
46. de Kock L, Priest JR, Foulkes WD, Alexandrescu S. An update on the central nervous system manifestations of DICER1 syndrome. Acta Neuropathol (2020) 139(4):689–701. doi: 10.1007/s00401-019-01997-y
47. Fauchon F, Jouvet A, Paquis P, Saint-Pierre G, Mottolese C, Ben Hassel M, et al. Parenchymal pineal tumors: a clinicopathological study of 76 cases. Int J Radiat Oncol Biol Phys (2000) 46(4):959–68. doi: 10.1016/S0360-3016(99)00389-2
48. Kivelä T. Trilateral retinoblastoma: a meta-analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma. J Clin Oncol (1999) 17(6):1829–37. doi: 10.1200/JCO.1999.17.6.1829
49. Sabbaghian N, Hamel N, Srivastava A, Albrecht S, Priest JR, Foulkes WD. Germline DICER1 mutation and associated loss of heterozygosity in a pineoblastoma. J Med Genet (2012) 49(7):417–9. doi: 10.1136/jmedgenet-2012-100898
50. Pfaff E, Aichmüller C, Sill M, Stichel D, Snuderl M, Karajannis MA, et al. Molecular subgrouping of primary pineal parenchymal tumors reveals distinct subtypes correlated with clinical parameters and genetic alterations. Acta Neuropathol (2020) 139(2):243–57. doi: 10.1007/s00401-019-02101-0
51. Heravi-Moussavi A, Anglesio MS, Cheng S-WG, Senz J, Yang W, Prentice L, et al. Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers. N Engl J Med (2012) 366(3):234–42. doi: 10.1056/NEJMoa1102903
52. Lee JC, Mazor T, Lao R, Wan E, Diallo AB, Hill NS, et al. Recurrent KBTBD4 small in-frame insertions and absence of DROSHA deletion or DICER1 mutation differentiate pineal parenchymal tumor of intermediate differentiation (PPTID) from pineoblastoma. Acta Neuropathol (2019) 137(5):851–4. doi: 10.1007/s00401-019-01990-5
53. de Kock L, Sabbaghian N, Druker H, Weber E, Hamel N, Miller S, et al. Germ-line and somatic DICER1 mutations in pineoblastoma. Acta Neuropathol (2014) 128(4):583–95. doi: 10.1007/s00401-014-1318-7
54. Priest JR, Watterson J, Strong L, Huff V, Woods WG, Byrd RL, et al. Pleuropulmonary blastoma: a marker for familial disease. J Pediatr (1996) 128(2):220–4. doi: 10.1016/S0022-3476(96)70393-1
55. Cross SF, Arbuckle S, Priest JR, Marshall G, Charles A, Dalla Pozza L. Familial pleuropulmonary blastoma in Australia. Pediatr Blood Cancer (2010) 55(7):1417–9. doi: 10.1002/pbc.22592
56. Priest JR, Williams GM, Hill DA, Dehner LP, Jaffé A. Pulmonary cysts in early childhood and the risk of malignancy. Pediatr Pulmonol (2009) 44(1):14–30. doi: 10.1002/ppul.20917
57. Vedanayagam J, Chatila WK, Aksoy BA, Majumdar S, Skanderup AJ, Demir E, et al. Cancer-associated mutations in DICER1 RNase IIIa and IIIb domains exert similar effects on miRNA biogenesis. Nat Commun (2019) 10(1):3682. doi: 10.1038/s41467-019-11610-1
58. Uro-Coste E, Masliah-Planchon J, Siegfried A, Blanluet M, Lambo S, Kool M, et al. ETMR-like infantile cerebellar embryonal tumors in the extended morphologic spectrum of DICER1-related tumors. Acta Neuropathol (2019) 137(1):175–7. doi: 10.1007/s00401-018-1935-7
59. Canning CR, McCartney AC, Hungerford J. Medulloepithelioma (diktyoma). Br J Ophthalmol (1988) 72(10):764–7. doi: 10.1136/bjo.72.10.764
60. Shields JA, Eagle RC, Shields CL, Singh AD, Robitaille J. Pigmented medulloepithelioma of the ciliary body. Arch Ophthalmol (2002) 120(2):207–10.
61. Peshtani A, Kaliki S, Eagle RC, Shields CL. Medulloepithelioma: A triad of clinical features. Oman J Ophthalmol (2014) 7(2):93–5. doi: 10.4103/0974-620X.137171
62. de Kock L, Geoffrion D, Rivera B, Wagener R, Sabbaghian N, Bens S, et al. Multiple DICER1-related tumors in a child with a large interstitial 14q32 deletion. Genes Chromosomes Cancer (2018) 57(5):223–30. doi: 10.1002/gcc.22523
63. de Kock L, Sabbaghian N, Soglio DB-D, Guillerman RP, Park B-K, Chami R, et al. Exploring the association Between DICER1 mutations and differentiated thyroid carcinoma. J Clin Endocrinol Metab (2014) 99(6):E1072–7. doi: 10.1210/jc.2013-4206
64. Durieux E, Descotes F, Nguyen A-M, Grange JD, Devouassoux-Shisheboran M. Somatic DICER1 gene mutation in sporadic intraocular medulloepithelioma without pleuropulmonary blastoma syndrome. Hum Pathol (2015) 46(5):783–7. doi: 10.1016/j.humpath.2015.01.020
65. Fremerey J, Balzer S, Brozou T, Schaper J, Borkhardt A, Kuhlen M. Embryonal rhabdomyosarcoma in a patient with a heterozygous frameshift variant in the DICER1 gene and additional manifestations of the DICER1 syndrome. Fam Cancer (2017) 16(3):401–5. doi: 10.1007/s10689-016-9958-5
66. Huryn LA, Turriff A, Harney LA, Carr AG, Chevez-Barrios P, Gombos DS, et al. DICER1 Syndrome: Characterization of the Ocular Phenotype in a Family-Based Cohort Study. Ophthalmology (2019) 126(2):296–304. doi: 10.1016/j.ophtha.2018.09.038
67. Sahm F, Jakobiec FA, Meyer J, Schrimpf D, Eberhart CG, Hovestadt V, et al. Somatic mutations of DICER1 and KMT2D are frequent in intraocular medulloepitheliomas. Genes Chromosomes Cancer (2016) 55(5):418–27. doi: 10.1002/gcc.22344
68. Kaliki S, Shields CL, Eagle RC, Vemuganti GK, Almeida A, Manjandavida FP, et al. Ciliary body medulloepithelioma: analysis of 41 cases. Ophthalmology (2013) 120(12):2552–9. doi: 10.1016/j.ophtha.2013.05.015
69. Kramer GD, Arepalli S, Shields CL, Shields JA. Ciliary body medulloepithelioma association with pleuropulmonary blastoma in a familial tumor predisposition syndrome. J Pediatr Ophthalmol Strabismus (2014) 51:Online:e48–50. doi: 10.3928/01913913-20140709-03
70. Laird PW, Grossniklaus HE, Hubbard GB. Ciliary body medulloepithelioma associated with pleuropulmonary blastoma. Br J Ophthalmol (2013) 97(8):1079, 1086–7. doi: 10.1136/bjophthalmol-2012-303019
71. Priest JR, Andic D, Arbuckle S, Gonzalez-Gomez I, Hill DA, Williams G. Great vessel/cardiac extension and tumor embolism in pleuropulmonary blastoma: a report from the International Pleuropulmonary Blastoma Registry. Pediatr Blood Cancer (2011) 56(4):604–9. doi: 10.1002/pbc.22583
72. Priest JR, Williams GM, Mize WA, Dehner LP, McDermott MB. Nasal chondromesenchymal hamartoma in children with pleuropulmonary blastoma–A report from the International Pleuropulmonary Blastoma Registry registry. Int J Pediatr Otorhinolaryngol (2010) 74(11):1240–4. doi: 10.1016/j.ijporl.2010.07.022
73. Apellaniz-Ruiz M, de Kock L, Sabbaghian N, Guaraldi F, Ghizzoni L, Beccuti G, et al. Familial multinodular goiter and Sertoli-Leydig cell tumors associated with a large intragenic in-frame DICER1 deletion. Eur J Endocrinol (2018) 178(2):K11–9. doi: 10.1530/EJE-17-0904
74. Robertson JC, Jorcyk CL, Oxford JT. DICER1 Syndrome: DICER1 Mutations in Rare Cancers. Cancers (Basel) (2018) 10(5). doi: 10.3390/cancers10050143
75. Blandino G, Valerio M, Cioce M, Mori F, Casadei L, Pulito C, et al. Metformin elicits anticancer effects through the sequential modulation of DICER and c-MYC. Nat Commun (2012) 3:865. doi: 10.1038/ncomms1859
76. Wu X, Yang Y, Huang Y, Chen Y, Wang T, Wu S, et al. RNA-binding protein AUF1 suppresses miR-122 biogenesis by down-regulating Dicer1 in hepatocellular carcinoma. Oncotarget (2018) 9(19):14815–27. doi: 10.18632/oncotarget.24079
Keywords: DICER1, cancer predisposition, pediatric, PPB, cystic nephroma
Citation: Caroleo AM, De Ioris MA, Boccuto L, Alessi I, Del Baldo G, Cacchione A, Agolini E, Rinelli M, Serra A, Carai A and Mastronuzzi A (2021) DICER1 Syndrome and Cancer Predisposition: From a Rare Pediatric Tumor to Lifetime Risk. Front. Oncol. 10:614541. doi: 10.3389/fonc.2020.614541
Received: 06 October 2020; Accepted: 24 November 2020;
Published: 21 January 2021.
Edited by:
Simone Cesaro, Integrated University Hospital Verona, ItalyReviewed by:
Katia Perruccio, University of Perugia, ItalyCopyright © 2021 Caroleo, De Ioris, Boccuto, Alessi, Del Baldo, Cacchione, Agolini, Rinelli, Serra, Carai and Mastronuzzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Maria Caroleo, YW5uYW1hcmlhLmNhcm9sZW9Ab3BiZy5uZXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.