 Ying Fang
Ying Fang Chun-Kang Chang
Chun-Kang Chang- Department of Hematology, Shanghai Jiao Tong University Affiliated Sixth People’s Hospital, Shanghai, China
Despite the improvements in prognostication of the revised International Prognostic Scoring System (IPSS-R) in myelodysplastic syndrome (MDS), there remain a portion of patients with lower risk (low/intermediate risk, LR) but poor prognostics. This study aimed to evaluate the relative contribution of mutational status when added to the IPSS-R, for estimating overall survival (OS) and progression-free survival (PFS) in patients with LR-MDS. We retrospectively analyzed clinical and laboratory variables of 328 patients diagnosed with MDS according to the FAB criteria. Twenty-nine-gene NGS assay was applied to bone marrow samples obtained at diagnosis. 233 (71.04%) patients were classified as LR-MDS. Univariate analysis showed association between inferior outcome (OS and PFS) and presence of JAK2 (p = 0.0177, p = 0.0002), RUNX1 (p = 0.0250, p = 0.0387), and U2AF1 (p = 0.0227, p = 0.7995) mutations. Multivariable survival analysis revealed JAK2 (p < 0.0001) and RUNX1 (p = 0.0215) mutations were independently prognostic for PFS in LR-MDS. Interestingly, bone marrow blast >1.5% could further predict disease progression of patients with LR-MDS (HR 8.06, 95%CI 2.95–22.04, p < 0.0001). Incorporation of JAK2, RUNX1 mutation and bone marrow blast in the IPSS-R can improve risk stratification in patients with LR-MDS. In summary, our result provided new risk factors for LR-MDS prognostics to identify candidates for early therapeutic intervention.
Introduction
Myelodysplastic syndrome (MDS) represents a heterogeneous group of clonal hematopoietic disorders with diverse clinical manifestations such as ineffective bone marrow (BM) hematopoiesis, peripheral blood cytopenia and variable propensity to progress to acute myeloid leukemia (AML) (1).
30% of MDS patients may progress to acute myeloid leukemia (2, 3). This emphasizes the need to stratify MDS patients into low or high risk for progression to guide optimal MDS treatment. Currently, revised International Prognostic Scoring System (IPSS-R) was regarded as the dominant prognostic scoring system which is based on a comprehensive cytogenetic scoring system (MDS cytogenetic scoring system) and defines 5 risk groups with different clinical outcomes: very low risk, low risk, intermediated risk, high risk, and very high risk (4). Patients with lower-risk (low/intermediate risk according to IPSS-R) myelodysplastic syndrome (LR-MDS) account for approximately two-thirds of patients with MDS. There remains a proportion of patients with lower risk but poor prognosis (5). Meanwhile, with the advance of next generation sequencing (NGS), a number of driver mutations were found in MDS (6) with diverse prognosis under the standard immunochemotherapy (7, 8).
In this study, we analyzed the genetic and clinical characteristics of a large cohort of patients with LR-MDS according to IPSS-R, and identify new risk factors which could further segregate patients into different risk groups, thus, to provide rationale to refine risk stratification and improve the accuracy of current prognostic scoring system.
Materials and Methods
Patients and Clinical Methods
A total of 328 MDS patients analyzed in this study (Table 1). 233 (71.04%) patients were classified as LR-MDS (Table 2). Enrolment occurred between April 2016 and September 2019, and the median observation duration was approximately 19 months (range 0.3–168.0). All samples were obtained from patients treated at the Department of Hematology, Shanghai Jiao Tong University Affiliated Sixth People’s Hospital. Informed consent was obtained from all subjects according to institutional review board-approved protocols, which were carried out in accordance with the Declaration of Helsinki. All experiments were approved by the Ethics Committee of Shanghai Jiao Tong University Affiliated Sixth People’s Hospital. All methods used in this study were performed in accordance with approved guidelines. Response to treatment was assessed using the International Working Group (IWG) Response Criteria, which were revised in 2006 (9).

Table 1 Clinical characteristics of patients with MDS.

Table 2 Clinical characteristics of patients with LR-MDS (n = 233).
All patients with LR-MDS received disease‐modifying therapeutic strategies according to NCCN Guidelines (10), which included supportive care, such as red blood cell (RBC) transfusions, erythroid stimulating agents (ESAs), granulocyte-macrophage colony stimulating factors (GM-CSF), and lenalidomide. Patients especially those with hypocellular BM receive immunosuppressive therapy and iron chelation is indicated in patients with iron overload. More aggressive therapies such as hypomethylating agents are usually not administered to patients with LR-MDS. Bone marrow (BM) samples were harvested from patients (n = 328) at diagnosis before treatment. The percentage of BM blasts was assessed from total nucleated cells (TNCs). All samples were kept frozen at -80°C until mutational analysis performed.
Mutational Analysis
The mutational analysis by next‐generation sequencing (NGS) was carried out for all patients whose DNA was of adequate quality, following procedures as previously described. Twenty-nine-gene panel were listed in Table S1. All assays were performed blinded to the study end points, by pathologists who were not involved in patient management. Mutational data underlying the study is available in Table S2.
Statistical Analysis
All statistical analyses were conducted with SPSS software, version 23.0 (SPSS, Inc., Chicago, IL, USA). Progression-free survival (PFS) was measured from the date when diagnosed to the date when disease progression was recognized or the date of last follow-up. Overall survival (OS) was measured from the date when diagnosed to the date of death or last follow-up. Distributions of PFS and OS were estimated by the method of Kaplan-Meier, and compared between subgroups using the log rank test. Univariate-, multivariate hazard estimates were generated with Cox proportional hazards models. Categorical variables were compared using Fisher’s exact test or χ2 test as appropriate. A two-sided p value of <0.0500 was considered statistically significant.
Results
Patient Characteristics and Overall Predicted Outcome of International Prognostic Scoring System
A total of 328 de novo MDS patients, diagnosed according to the 2008 World Health Organization (WHO) classification, were included in our study. Their IPSS-R score was calculated based on their initial presentation to our institution. Among the total cohort, 11 patients (3.35%) had very low-risk disease, 115 patients (35.06%) had low-risk disease, 118 patients (35.98%) had intermediate-risk disease, 58 patients (17.68%) had high-risk disease and 26 patients (7.93%) had very high risk (Figure 1A). OS and PFS were shown in Figures 1B, C. Median OS was 19 months [95% confidence interval (CI) 26.2–33.0], PFS median follow-up was 18 months (range 0.3–168.0). However, as shown in Figure 1D, IPSS-R still has limitation to identify patients with lower risk but poor prognosis: 47/114 (41.23%) patients with LR-MDS were dead within 12 months and 18/114 (15.79%) patients had disease progression within 12 months.
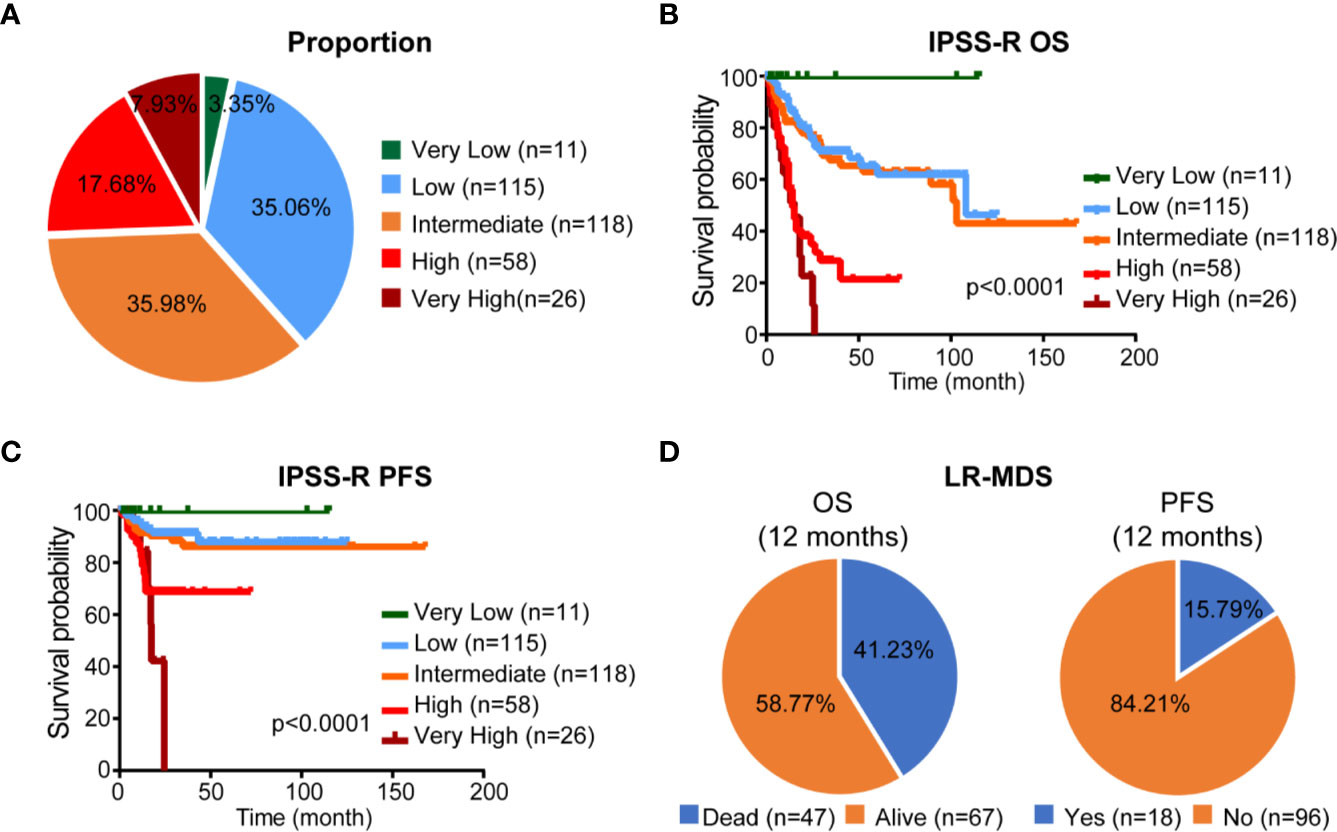
Figure 1 Proportion and prognosis of patients according to IPSS-R classification. (A) Proportion of patients according to IPSS-R classification. (B, C) Kaplan-Meier curves for OS and PFS according to IPSS-R classification. (D) Proportion of different clinical outcome of LR-MDS.
Mutational Analysis
We then focused on patients with LR-MDS (n = 233) based on IPSS-R, and investigated their genetic characteristics using 29-gene NGS assay (Figure 2A). One hundred ninety-five (83.69%) patients contained ≥1 mutation [median (IQR): 2 (1–3)]. Fifteen genes were recurrent mutated (mutation rate > 5%). The most frequently mutated gene was TET2 (15.45%) followed by SF3B1 (12.45%), SETBP1 (11.16%), ASXL1 (9.44%), RUNX1 (9.01%), ANKRD11 (9.01%), JAK2 (9.01%), DNMT3A (8.58%), U2AF1 (8.58%), ROBO1 (8.15%), MPL (7.30%), KIF20B (6.44%), IDH1 (5.58%), ROBO2 (5.58%), and ITIH3 (5.15%). We further investigate the prognostic influence of tumor mutation burden (TMB, ≥2 mutations) and recurrent mutations, univariate analysis indicated that TMB ≥2, RUNX1 and JAK2 mutations were associated with inferior OS and PFS, and U2AF1 mutations was associated with inferior OS [hazard ratio (HR) > 1.5, p < 0.0500, Figure 2B]. Kaplan-Meier curves of PFS and OS according to mutation status of RUNX1 and JAK2 were shown in Figure 2C. Multivariable survival analysis revealed JAK2 [HR 6.58, 95%CI 2.72–15.91, p < 0.0001] and RUNX1 [HR 3.25, 95%CI 1.19–8.89, p = 0.0215] mutations were independently prognostic for PFS in LR-MDS.
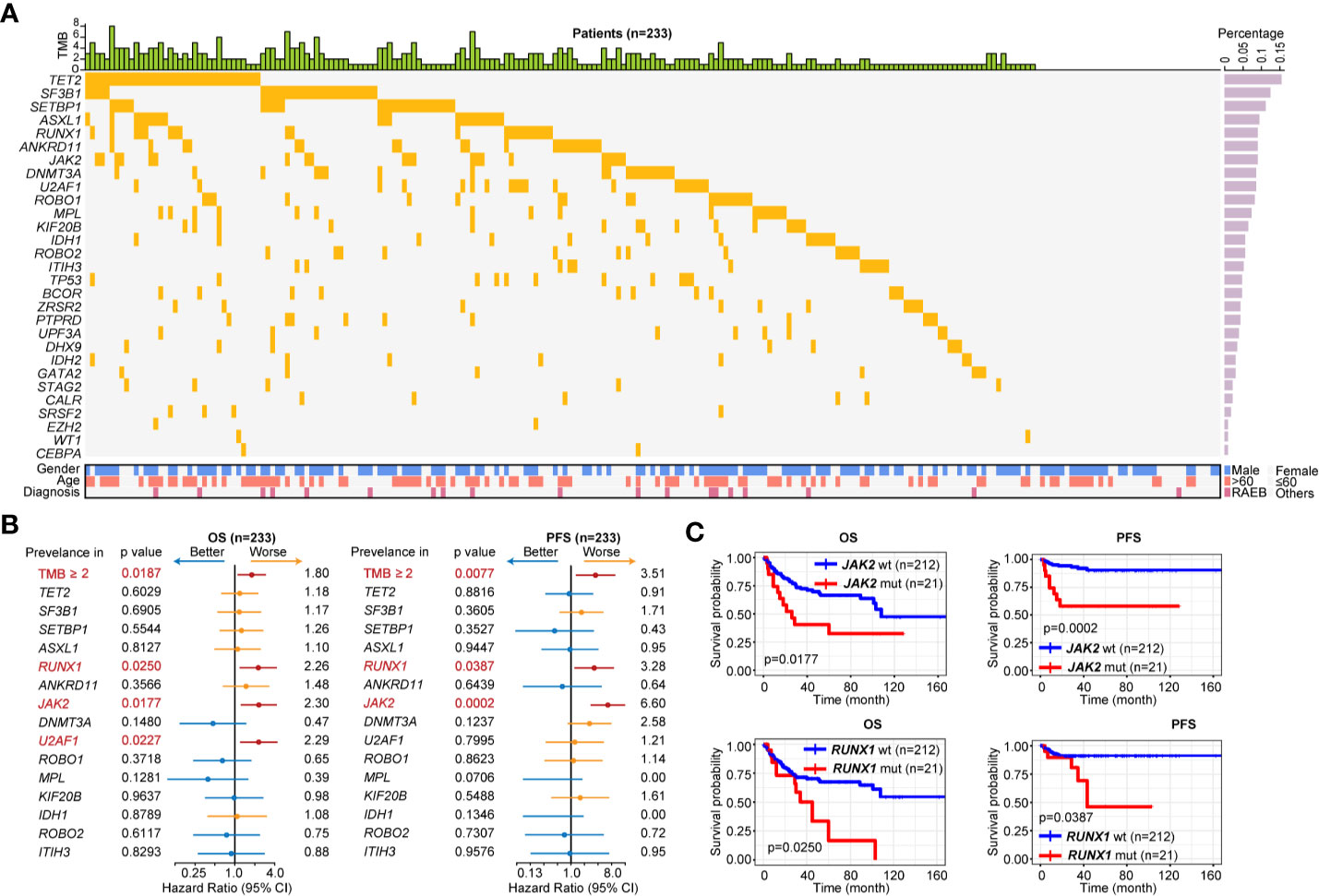
Figure 2 Gene mutation pattern of patients with LR-MDS. (A) Gene mutation pattern of patients with LR-MDS (n = 233). (B) Overall survival (OS) and progression-free survival (PFS) analysis of highly recurrent mutated genes among patients with LR-MDS (n = 233). (C) Kaplan-Meier curves for OS and PFS according to mutation status of JAK2 (top) and RUNX1 (bottom).
Clinical and Laboratory Characteristics
To further investigate the clinical implications of JAK2 and RUNX1 mutation, clinical and laboratory data were analyzed including age, gender, diagnosis, cytogenetics, percentage of bone marrow (BM) blast, absolute neutrophil count (ANC), hemoglobin (Hb), PLT and myelofibrosis (MF) status with Gomori staining positive (++~++++). As shown in Table 3, patients were reclassified according to their mutation status. JAK2-mutated patients had lower ANC (<0.8×10^9/L, 76.19 vs. 29.72%, p < 0.0001) than JAK2 wildtype patients and tended to be more responsible for MF (19.05 vs. 6.13%, p = 0.0129). Elderly patients (>60 y) tended to have recurrent RUNX1 mutation (p = 0.0098), and RUNX1-mutated patients had higher percentage of BM blast (>2%) when compared to those with RUNX1 wildtype (47.62 vs. 19.81%, p = 0.0452). We then evaluated the prognostic influence of BM blast (%) from 237 available LR-MDS patients, and tried to identify a suitable cutoff to improve PFS prediction.
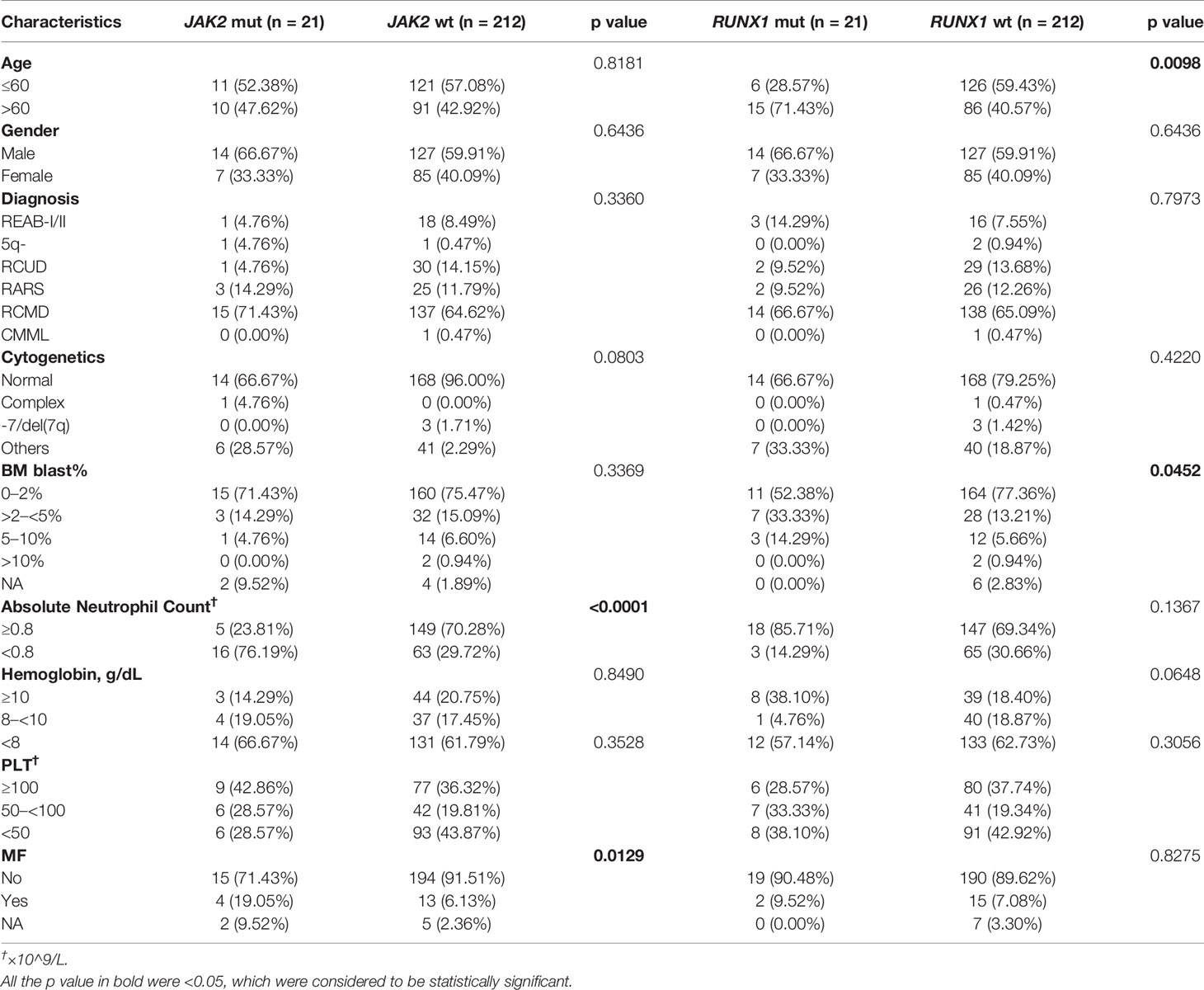
Table 3 Clinical characteristics of patients with LR-MDS (n = 233).
Subsequently, we classified patients according to their PFS status (progression or not) and conducted receiver operating curve (ROC) analyses to assess how the BM blast (%) could behave in predicting prognosis (Figure 3A). After detection, it was found that the area under the BM blast (%) curve was 0.7878 and the 95% CI was 0.6857–0.8900. When the cutoff value was 1.50%, the optimal specificity was 72.46%, the sensitivity 84.21% and the Youden index 0.5667 which revealed that at the cutoff (BM blast > 1.5%) could further improve risk stratification in patients with LR-MDS. As shown in Figures 3B, C, LR-MDS patients with BM blast >1.5% has an inferior OS and PFS when compared with those with BM blast ≤1.5% (median OS 20.33 months, 95%CI 14.00–27.00 vs. median OS 26.00 months, 95%CI 23.00–30.43, p < 0.0001; median PFS 17.50 months, 95%CI 12.47–27.00 vs. median PFS 26.00 months, 95%CI 23.00–30.43, p < 0.0001).
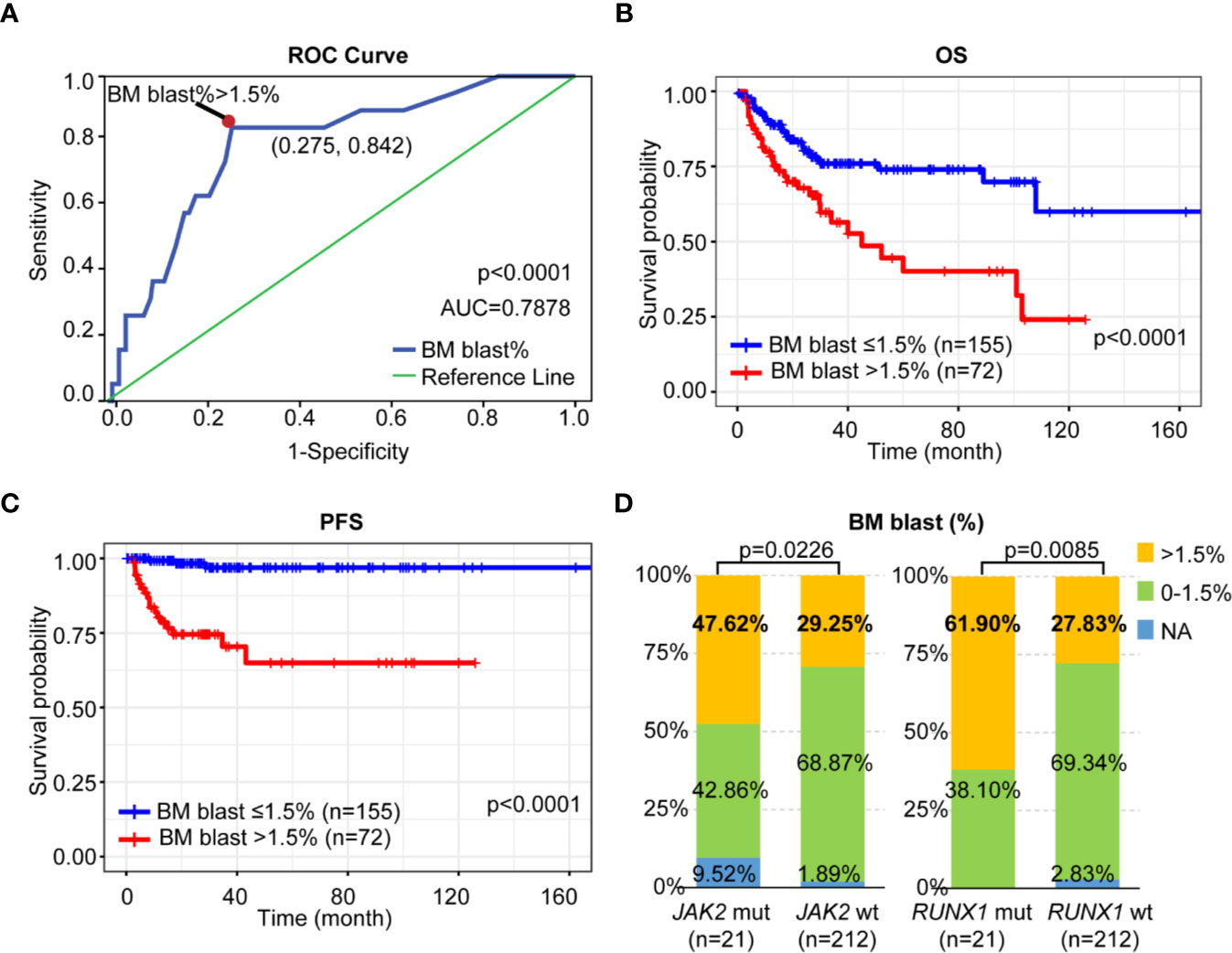
Figure 3 Prognostic impact of BM blast on LR-MDS. (A) ROC curve of BM blast-based model. (B) Kaplan-Meier curves for OS according to the percentage of BM blast. (C) Kaplan-Meier curves for PFS according to the percentage of BM blast. (D) Distribution of BM blast (%) among LR-MDS patients with or without JAK2 or RUNX1 mutations.
Interestingly, we found both JAK2 and RUNX1-mutated patients had high percentage of BM blast (>1.5%) as when compared to those with wildtype respectively (47.62 vs. 29.25%, p = 0.0226; 61.90 vs. 27.83%, p = 0.0085) (Figure 3D).
Prediction of Prognosis of Low/Intermediate Risk-Myelodysplastic Syndrome
According to BM blast (%) and mutation status of JAK2 and RUNX1, patients with LR-MDS could be further stratified into following three subgroups: low-risk subgroup (BM blast ≤1.5% without JAK2 and RUNX1 mutation), intermediate-risk subgroup (BM blast ≤1.5% with JAK2 or RUNX1 mutation, BM blast >1.5% without JAK2 and RUNX1 mutation), and high-risk subgroup (BM blast >1.5% with JAK2 and/or RUNX1 mutation) (Figure 4A). Finally, when incorporated JAK2, RUNX1 mutation and bone marrow blast, we further segregate patients with LR-MDS into following three risk groups: low-risk subgroup (60.79%, 138 of 227 patients) with median OS of 25.93 months (95%CI 23.00–30.33) and median PFS 25.93 months (95%CI 23.00–30.33); intermediate-risk subgroup (29.96%, 68 of 227 patients) with median OS of 24.00 months [95%CI 18.00–29.63, HR 1.73 (0.96–3.12), p = 0.0495] and median PFS 24.00 months [95%CI 17.00–29.63, HR 20.16 (5.75–70.72), p < 0.0001] and high-risk subgroup (9.25%, 21 of 227 patients) with median OS of 13.00 months [95%CI 7.00–33.07, HR 4.50 (1.65–12.29), p < 0.0001] and median PFS 12.47 months [95%CI 6.60–33.07, HR 81.89 (12.28–546.20), p < 0.0001] as shown in Figure 4B.
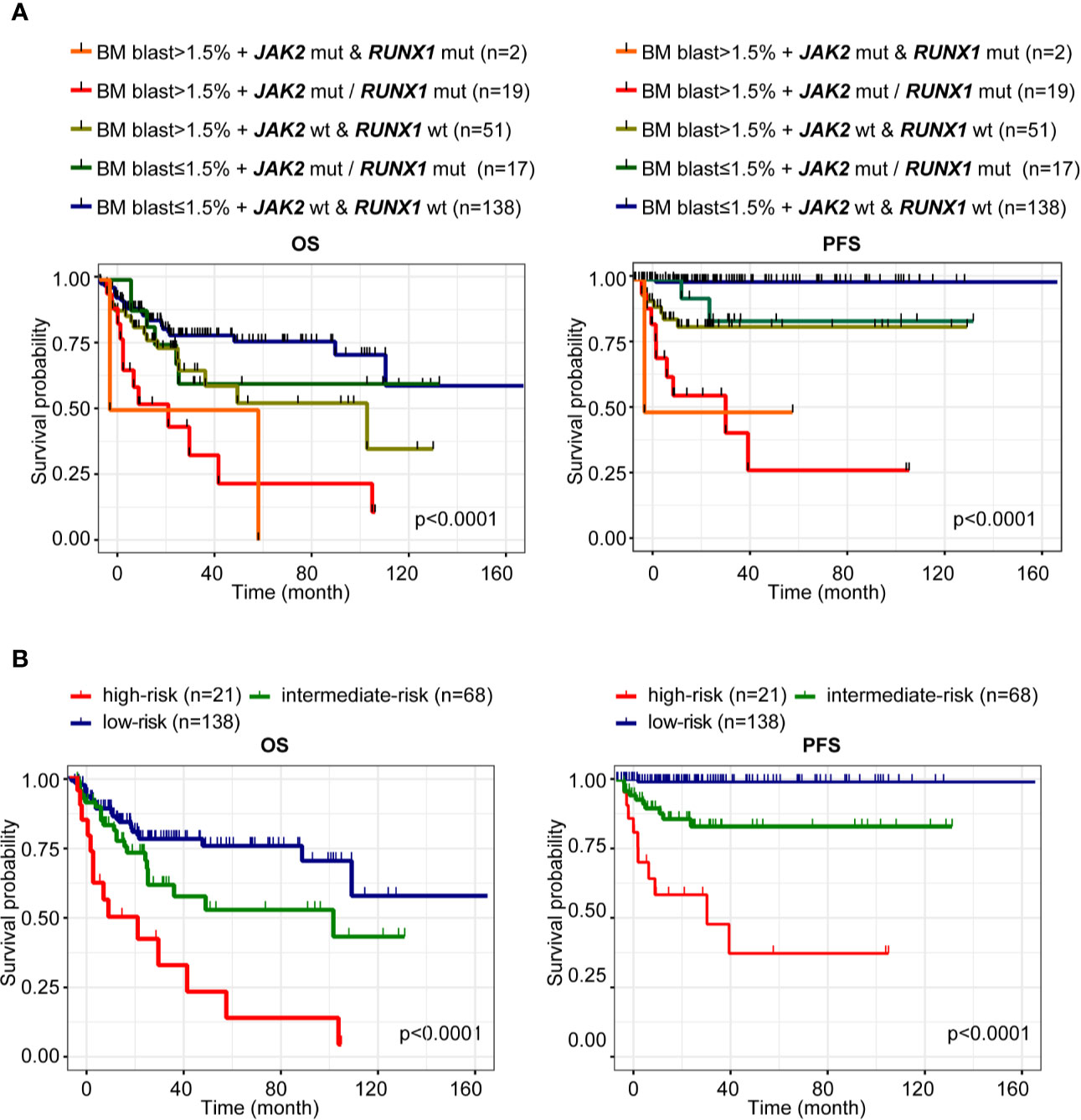
Figure 4 Prediction of prognosis of LR-MDS. (A) Kaplan-Meier curves for OS and PFS according to BM blast (%) and mutation status of JAK2 and RUNX1 of patients with LR-MDS. (B) Kaplan-Meier curves for OS and PFS of the following three subgroups: low-risk subgroup (BM blast ≤1.5% without JAK2 and RUNX1 mutation), intermediate-risk subgroup (BM blast ≤1.5% with JAK2 or RUNX1 mutation, BM blast >1.5% without JAK2 and RUNX1 mutation), and high-risk subgroup (BM blast >1.5% with JAK2 and/or RUNX1 mutation).
Discussion
MDS is a heterogeneous disease with various clinicopathological and molecular features (11, 12). Thus, accurate prognostication and risk stratification is important for patients with MDS to further guide risk-adapted therapy (13). IPSS (14), IPSS-R, MD Anderson Risk Model Score (MDAS) (15), WHO-classification based Prognostic Scoring System (WPSS) (16) and refined WPSS (WPSS-R) (17) are the most widely adopted models in clinical practice for risk stratification of MDS in the latest years. IPSS-R has been recognized as the score with the best prognostic capability in MDS (18–21). However, some concerns still exist about the real prognostic significance of the lower risk category, patients classified into intermediate risk group showed an outcome closer to the expected in higher risk MDS patients (22). Patients with lower risk still carry a significant excess mortality (23).
We investigated genetic characteristics of patients with LR-MDS using NGS assay and examined the prognostic utility of 15 recurrent mutated genes (mutation rate > 5%). We found RUNX1 (9.01%) and JAK2 (9.01%) mutations were associated with inferior OS and PFS among patients with LR-MDS. RUNX1, a member of the core-binding factor family of transcription factors, is indispensable for the establishment of definitive hematopoiesis (24). RUNX1 is one of the most frequently mutated genes in MDS, accounting for roughly 10% of the cases as previous reported (25, 26). It has been reported that MDS patients with RUNX1 mutations had a higher risk and shorter latency for progression to AML (27). However, the exact role of RUNX1 in progression of MDS to AML is not known. In our study, among patients with LR-MDS, cytogenetic changes such as deletion of the entire chromosome 7 or its long arm (-7/7q-) was not common in mutated RUNX1 patients. It’s worth noting that patients with RUNX1 mutation tended to had higher percentage of BM blast (> 2%) when compared to those without RUNX1 mutation which further confirmed the pathogenic role of RUNX1 mutation in hematopoietic defects and propensity to leukemogenesis.
Recently, JAK2 mutations have been reported to not only be associated with inferior survival in MDS receiving hematopoietic stem cell transplantation (HSCT) (28), but also in LR-MDS (29). JAK2 mutation leads to constitutive activation of the JAK2/STAT3 pathway which further resulted in growth factor independence, increased proliferation, and differentiation failure (30). JAK2 mutations are mainly found in patients with myeloproliferative characteristics (31). It has been reported that JAK2 mutation patients had significantly higher hemoglobin levels, white blood cell and platelet counts compared to unmutated patients (32, 33). In our study, we found JAK2-mutated patients had lower ANC (<0.8×10^9/L, 76.19 vs. 29.72%, p < 0.0001) than JAK2 wildtype patients and tended to be more responsible for MF (19.05% vs. 6.13%, p=0.0129) which further confirmed JAK2 mutations reinforced the pathogenesis of MDS, may be involved in disease progression in LR-MDS.
TP53 has also had a poor prognosis in LR-MDS with a relative low mutation rate (4.72%) than frequent RUNX1 or JAK2 mutation (9.01%). Kaplan-Meier curves for OS and PFS according to TP53 status were shown in Supplementary Figure S1. However, other mutations which are known to confer poor prognosis including ASXL1 and SRSF2 were not shown to affect prognosis in our cohort. Mutation of ASXL1 gene has been reported to be a molecular marker of disease progression in MDS, in particular its high frequency in REAB-I/II (31.00%) (34). However, only 19 patients with REAB-I/II were included in our cohort with a relatively low ASXL1 mutation rate 5.26% in REAB-I/II. The mutation rate of SRSF2 were only 1.72% in our cohort of LR-MDS. Accordingly, ASXL1 and SRSF2 mutation did not show significant correlation with the prognosis in our cohort may due to the different proportion of disease classification of enrolled patients.
Proportion of BM blasts has been demonstrated as an independent prognostic factor in MDS. It has been reported that reconsidering the methods of enumerating bone marrow blasts could improve outcome prediction (35). In our study, we found both JAK2 and RUNX1-mutated patients had high percentage of BM blast (> 1.5%), BM blast (> 1.5%) adequately predicted OS and PFS respectively (p < 0.0001, p < 0.0001) among LR-MDS patients.
In conclusion, integrative mutational and clinical analyses reveal that considering JAK2, RUNX1 mutation status and BM blast percentage (> 1.5%) can further improve risk stratification of disease progression and over survival among the patients with LR-MDS in the context of IPSS-R.
Data Availability Statement
The mutational data underlying the study is available in Table S2.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Shanghai Jiao Tong University Affiliated Sixth People’s Hospital. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
C-KC and Y-SZ contributed to the conception and design of the study; DW, L-YW, L-XS and ZZ recruited the patients and collected the data; YF was responsible for the clinical and mutational analysis; JG assisted with biological sample preparation; YF and Y-SZ wrote the manuscript. C-KC and Y-SZ supervised the study. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by research funding from the National Natural Science Foundation of China (81770121).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.610525/full#supplementary-material
Supplementary Table 1 | Genes for targeted sequencing.
Supplementary Table 2 | Somatic mutations of hypermutated genes in LR-MDS.
Supplementary Figure 1 | Kaplan-Meier curves for OS and PFS according to TP53 status.
References
1. Adès L, Itzykson R, Fenaux P. Myelodysplastic syndromes. Lancet (London England) (2014) 383(9936):2239–52. doi: 10.1016/S0140-6736(13)61901-7
2. Nachtkamp K, Stark R, Strupp C, Kündgen A, Giagounidis A, Aul C, et al. Causes of death in 2877 patients with myelodysplastic syndromes. Ann Hematol (2016) 95(6):937–44. doi: 10.1007/s00277-016-2649-3
3. Chen J, Kao YR, Sun D, Todorova TI, Reynolds D, Narayanagari SR, et al. Myelodysplastic syndrome progression to acute myeloid leukemia at the stem cell level. Nat Med (2019) 25(1):103–10. doi: 10.1038/s41591-018-0267-4
4. Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Solé F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood (2012) 120(12):2454–65. doi: 10.1182/blood-2012-03-420489
5. Zeidan AM, Sekeres MA, Wang XF, Al Ali N, Garcia-Manero G, Steensma DP, et al. Comparing the prognostic value of risk stratifying models for patients with lower-risk myelodysplastic syndromes: Is one model better? Am J Hematol (2015) 90(11):1036–40. doi: 10.1002/ajh.24173
6. Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood (2013) 122(22):3616–27. doi: 10.1182/blood-2013-08-518886
7. Li B, Liu J, Xu Z, Qin T, Shi Z, Song Z, et al. The usefulness of mutational data on prognosis of myelodysplastic syndromes: alone or incorporated into the IPSS-R? Br J Haematol (2018) 183(5):815–9. doi: 10.1111/bjh.15036
8. Nazha A, Bejar R. Molecular Data and the IPSS-R: How Mutational Burden Can Affect Prognostication in MDS. Curr Hematol Malignancy Rep (2017) 12(5):461–7. doi: 10.1007/s11899-017-0407-9
9. Cheson BD, Greenberg PL, Bennett JM, Lowenberg B, Wijermans PW, Nimer SD, et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood (2006) 108(2):419–25. doi: 10.1182/blood-2005-10-4149
10. Greenberg PL, Stone RM, Bejar R, Bennett JM, Bloomfield CD, Borate U, et al. Myelodysplastic syndromes, version 2.2015. J Natl Compr Cancer Netw: JNCCN (2015) 13(3):261–72. doi: 10.6004/jnccn.2015.0038
11. Zeidan AM, Komrokji RS. There’s risk, and then there’s risk: The latest clinical prognostic risk stratification models in myelodysplastic syndromes. Curr Hematol Malignancy Rep (2013) 8(4):351–60. doi: 10.1007/s11899-013-0172-3
12. Raza A, Galili N. The genetic basis of phenotypic heterogeneity in myelodysplastic syndromes. Nat Rev Cancer (2012) 12(12):849–59. doi: 10.1038/nrc3321
13. Faltas B, Zeidan A, Gergis U. Myelodysplastic syndromes: toward a risk-adapted treatment approach. Expert Rev Hematol (2013) 6(5):611–24. doi: 10.1586/17474086.2013.840997
14. Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood (1997) 89(6):2079–88. doi: 10.1182/blood.V89.6.2079
15. Kantarjian H, O’Brien S, Ravandi F, Cortes J, Shan J, Bennett JM, et al. Proposal for a new risk model in myelodysplastic syndrome that accounts for events not considered in the original International Prognostic Scoring System. Cancer (2008) 113(6):1351–61. doi: 10.1002/cncr.23697
16. Malcovati L, Germing U, Kuendgen A, Della Porta MG, Pascutto C, Invernizzi R, et al. Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J Clin Oncol Off J Am Soc Clin Oncol (2007) 25(23):3503–10. doi: 10.1200/JCO.2006.08.5696
17. Malcovati L, Della Porta MG, Strupp C, Ambaglio I, Kuendgen A, Nachtkamp K, et al. Impact of the degree of anemia on the outcome of patients with myelodysplastic syndrome and its integration into the WHO classification-based Prognostic Scoring System (WPSS). Haematologica (2011) 96(10):1433–40. doi: 10.3324/haematol.2011.044602
18. Voso MT, Fenu S, Latagliata R, Buccisano F, Piciocchi A, Aloe-Spiriti MA, et al. Revised International Prognostic Scoring System (IPSS) predicts survival and leukemic evolution of myelodysplastic syndromes significantly better than IPSS and WHO Prognostic Scoring System: validation by the Gruppo Romano Mielodisplasie Italian Regional Database. J Clin Oncol Off J Am Soc Clin Oncol (2013) 31(21):2671–7. doi: 10.1200/JCO.2012.48.0764
19. Della Porta MG, Tuechler H, Malcovati L, Schanz J, Sanz G, Garcia-Manero G, et al. Validation of WHO classification-based Prognostic Scoring System (WPSS) for myelodysplastic syndromes and comparison with the revised International Prognostic Scoring System (IPSS-R). A study of the International Working Group for Prognosis in Myelodysplasia (IWG-PM). Leukemia (2015) 29(7):1502–13. doi: 10.1038/leu.2015.55
20. van Spronsen MF, Ossenkoppele GJ, Holman R, van de Loosdrecht AA. Improved risk stratification by the integration of the revised international prognostic scoring system with the myelodysplastic syndromes comorbidity index. Eur J Cancer (Oxford England: 1990) (2014) 50(18):3198–205. doi: 10.1016/j.ejca.2014.09.016
21. Neukirchen J, Lauseker M, Blum S, Giagounidis A, Lübbert M, Martino S, et al. Validation of the revised international prognostic scoring system (IPSS-R) in patients with myelodysplastic syndrome: a multicenter study. Leukemia Res (2014) 38(1):57–64. doi: 10.1016/j.leukres.2013.10.013
22. Pfeilstöcker M, Tuechler H, Sanz G, Schanz J, Garcia-Manero G, Solé F, et al. Time-dependent changes in mortality and transformation risk in MDS. Blood (2016) 128(7):902–10. doi: 10.1182/blood-2016-02-700054
23. Nomdedeu M, Pereira A, Ramos F, Valcárcel D, Costa D, Arnan M, et al. Excess mortality in the myelodysplastic syndromes. Am J Hematol (2017) 92: (2):149–54. doi: 10.1002/ajh.24606
24. Sood R, Kamikubo Y, Liu P. Role of RUNX1 in hematological malignancies. Blood (2017) 129(15):2070–82. doi: 10.1182/blood-2016-10-687830
25. Chen CY, Lin LI, Tang JL, Ko BS, Tsay W, Chou WC, et al. RUNX1 gene mutation in primary myelodysplastic syndrome–the mutation can be detected early at diagnosis or acquired during disease progression and is associated with poor outcome. Br J Haematol (2007) 139(3):405–14. doi: 10.1111/j.1365-2141.2007.06811.x
26. Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia (2014) 28(2):241–7. doi: 10.1038/leu.2013.336
27. Tsai SC, Shih LY, Liang ST, Huang YJ, Kuo MC, Huang CF, et al. Biological Activities of RUNX1 Mutants Predict Secondary Acute Leukemia Transformation from Chronic Myelomonocytic Leukemia and Myelodysplastic Syndromes. Clin Cancer Res: Off J Am Assoc Cancer Res (2015) 21(15):3541–51. doi: 10.1158/1078-0432.CCR-14-2203
28. Lindsley RC, Saber W, Mar BG, Redd R, Wang T, Haagenson MD, et al. Prognostic Mutations in Myelodysplastic Syndrome after Stem-Cell Transplantation. New Engl J Med (2017) 376(6):536–47. doi: 10.1056/NEJMoa1611604
29. Jiang L, Luo Y, Zhu S, Wang L, Ma L, Zhang H, et al. Mutation status and burden can improve prognostic prediction of patients with lower-risk myelodysplastic syndromes. Cancer Sci (2020) 111(2):580–91. doi: 10.1111/cas.14270
30. James C, Ugo V, Le Couédic JP, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature (2005) 434(7037):1144–8. doi: 10.1038/nature03546
31. Jädersten M, Hellström-Lindberg E. New clues to the molecular pathogenesis of myelodysplastic syndromes. Exp Cell Res (2010) 316(8):1390–6. doi: 10.1016/j.yexcr.2010.02.043
32. Raya JM, Arenillas L, Domingo A, Bellosillo B, Gutiérrez G, Luño E, et al. Refractory anemia with ringed sideroblasts associated with thrombocytosis: comparative analysis of marked with non-marked thrombocytosis, and relationship with JAK2 V617F mutational status. Int J Hematol (2008) 88(4):387–95. doi: 10.1007/s12185-008-0169-1
33. Ganguly BB, Kadam NN. Mutations of myelodysplastic syndromes (MDS): An update. Mutat Res Rev Mutat Res (2016) 769:47–62. doi: 10.1016/j.mrrev.2016.04.009
34. Boultwood J, Perry J, Pellagatti A, Fernandez-Mercado M, Fernandez-Santamaria C, Calasanz MJ, et al. Frequent mutation of the polycomb-associated gene ASXL1 in the myelodysplastic syndromes and in acute myeloid leukemia. Leukemia (2010) 24(5):1062–5. doi: 10.1038/leu.2010.20
35. Calvo X, Arenillas L, Luño E, Senent L, Arnan M, Ramos F, et al. Enumerating bone marrow blasts from nonerythroid cellularity improves outcome prediction in myelodysplastic syndromes and permits a better definition of the intermediate risk category of the Revised International Prognostic Scoring System (IPSS-R). Am J Hematol (2017) 92(7):614–21. doi: 10.1002/ajh.24732
Keywords: lower-risk myelodysplastic syndrome, revised International Prognostic Scoring System, next-generation sequencing, bone marrow blast, risk factor
Citation: Fang Y, Guo J, Wu D, Wu L-Y, Song L-X, Zhang Z, Zhao Y-S and Chang C-K (2021) Integration Analysis of JAK2 or RUNX1 Mutation With Bone Marrow Blast Can Improve Risk Stratification in the Patients With Lower Risk Myelodysplastic Syndrome. Front. Oncol. 10:610525. doi: 10.3389/fonc.2020.610525
Received: 26 September 2020; Accepted: 10 November 2020;
Published: 13 January 2021.
Edited by:
Massimo Breccia, Sapienza University of Rome, ItalyReviewed by:
Antonella Poloni, Marche Polytechnic University, ItalyGang Zheng, Mayo Clinic, United States
Copyright © 2021 Fang, Guo, Wu, Wu, Song, Zhang, Zhao and Chang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chun-Kang Chang, Y2hhbmdjaHVua2FuZ0BzanR1LmVkdS5jbg==; You-Shan Zhao, emhhb3lvdXNoYW4xOTg1QDE2My5jb20=