 Kristie Liu
Kristie Liu Kathleen Tsung2
Kathleen Tsung2 Frank J. Attenello
Frank J. Attenello
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 11 December 2020
Sec. Neuro-Oncology and Neurosurgical Oncology
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.608911
Glioblastoma (GBM) is the most common primary brain tumor, carrying a very poor prognosis, with median overall survival at about 12 to 15 months despite surgical resection, chemotherapy with temozolomide (TMZ), and radiation therapy. GBM recurs in the vast majority of patients, with recurrent tumors commonly displaying increase in resistance to standard of care chemotherapy, TMZ, as well as radiotherapy. One of the most commonly cited mechanisms of chemotherapeutic and radio-resistance occurs via the glucose-regulated protein 78 (GRP78), a well-studied mediator of the unfolded protein response (UPR), that has also demonstrated potential as a biomarker in GBM. Overexpression of GRP78 has been directly correlated with malignant tumor characteristics, including higher tumor grade, cellular proliferation, migration, invasion, poorer responses to TMZ and radiation therapy, and poorer patient outcomes. GRP78 expression is also higher in GBM tumor cells upon recurrence. Meanwhile, knockdown or suppression of GRP78 has been shown to sensitize cells to TMZ and radiation therapy. In light of these findings, various novel developing therapies are targeting GRP78 as monotherapies, combination therapies that enhance the effects of TMZ and radiation therapy, and as treatment delivery modalities. In this review, we delineate the mechanisms by which GRP78 has been noted to specifically modulate glioblastoma behavior and discuss current developing therapies involving GRP78 in GBM. While further research is necessary to translate these developing therapies into clinical settings, GRP78-based therapies hold promise in improving current standard-of-care GBM therapy and may ultimately lead to improved patient outcomes.
Gliomas are the most commonly diagnosed primary neoplasms in the central nervous system, constituting up to 81% of malignant brain tumors (1). Malignant gliomas include anaplastic astrocytoma (grade 3) and glioblastoma multiforme (GBM, grade 4). GBM is the most common histology, comprising 45% of gliomas (1), with a prognosis that remains dismal, with median overall survival for GBM at about 12 to 15 months (2) and a 5-year survival of only 5.5% despite therapy (3). The incidence of GBM also increases with age, and is associated with devastating neurological effects, including weakness, visual and sensory changes, headaches, seizures, and alterations in mood, memory, or executive function (4).
Standard of care for a newly suspected GBM involves surgical resection (or biopsy), followed by chemotherapy and radiation therapy. The standard chemotherapeutic regimen utilizes temozolomide (TMZ), a well-tolerated drug that has been shown to delay tumor progression and provide modest improvement in patient survival (5). Specifically, TMZ exerts anti-tumor effect via DNA methylation and substitution of cytosine by thymine (6). This repeated substitution activates the mismatch repair mechanism, which triggers cell stress and apoptosis in response to the detection of recurrent errors in DNA.
Despite surgery, chemotherapy, and radiotherapy, the majority of GBM patients experience tumor recurrence with increased chemo- and radio-resistance. Furthermore, there is currently no standard of care in second line management following initial adjuvant treatment (2). Because the mortality of GBM remains high and tumor chemo- and radio-resistance remain a critical challenge, new treatment modalities or approaches are needed to improve outcomes. Such treatment strategies have included multiple chemotherapeutic agents, anti-angiogenic therapy, and immunotherapy. Several promising treatments have focused on the unfolded protein response (UPR), a cellular stress response to accumulated proteins in the lumen of the endoplasmic reticulum (ER) (7). The UPR has emerged as one of the more promising targets due to its role in tumor survival and therapeutic resistance. Specifically, glucose-regulated protein 78 (GRP78) has emerged as a potential target in the majority of these studies due to its role as a central modulator of the UPR. Overexpression of GRP78 has been repeatedly demonstrated to modulate malignant and aggressive phenotypes in GBM tumor cells (8–12). In addition, GRP78 expression has been noted to promote propagation of glioma stem cells (GSCs), tumor-replenishing cells that form the pool of the highly proliferating transient cell population, while also driving GBM resistance and recurrence (13, 14). While UPR and GRP78 data is abundant in the literature, a focus on the role of this system in GBM is limited. Here, we have provided an updated review, including several years of novel studies evaluating and targeting endoplasmic reticulum proteostasis in GBM (15), with emphasis on the significance of GRP78 and targeted therapies for GRP78. We specifically aim to summarize the literature assessing the role of GRP78 and other mediators of the UPR within GBM, including novel studies exploring the role of the UPR in glioma stem cells. Furthermore, we review developing GBM therapies and treatment delivery methods to demonstrate how GRP78 is a compelling therapeutic target and biomarker that could potentially translate to improved GBM therapy and care.
The UPR is a cellular stress response that is activated when unfolded or misfolded proteins accumulate in the lumen of the endoplasmic reticulum. GRP78, also known as immunoglobulin heavy chain binding protein (BiP), is a well-studied chaperone heat shock protein that is central to the modulation of the UPR. The GRP78 protein primarily resides in the lumen of the ER but can be found on the ER membrane and on the cell surface (16, 17). As a molecular chaperone, GRP78 is important for protein folding and assembly, binding calcium in the ER, and export of misfolded proteins for degradation (18). GRP78 has two domains modulating UPR function, a nucleotide-binding domain (NBD) and a substrate binding domain (SBD) (19), described in further detail below. GRP78 expression is directly correlated with ER activity and regulated on the transcriptional level, based on an analysis of the human GRP78 gene demonstrating that the GRP78 promoter contains an ER stress-sensitive region located 170 nucleotides upstream from the transcription initiation site (20).
In the UPR, GRP78 functions as a molecular chaperone that binds to misfolded proteins and unassembled complexes and initiates ER-associated degradation (ERAD). ERAD targets these misfolded proteins for degradation via ubiquitination and proteasomal degradation. In normal homeostatic cell conditions, GRP78 is in an inactive form bound to (1) activating transcription factor 6 (ATF6), (2) protein kinase RNA-like endoplasmic reticulum kinase (PERK), and (3) inositol-requiring enzyme 1 (IRE1), all of which are UPR transmembrane sensors of cellular stress (Figure 1A, Table 1) (24). When unfolded proteins accumulate in the ER, GRP78 is released from these UPR stress sensors to exert its various aforementioned functions (Figure 1B, Table 1).
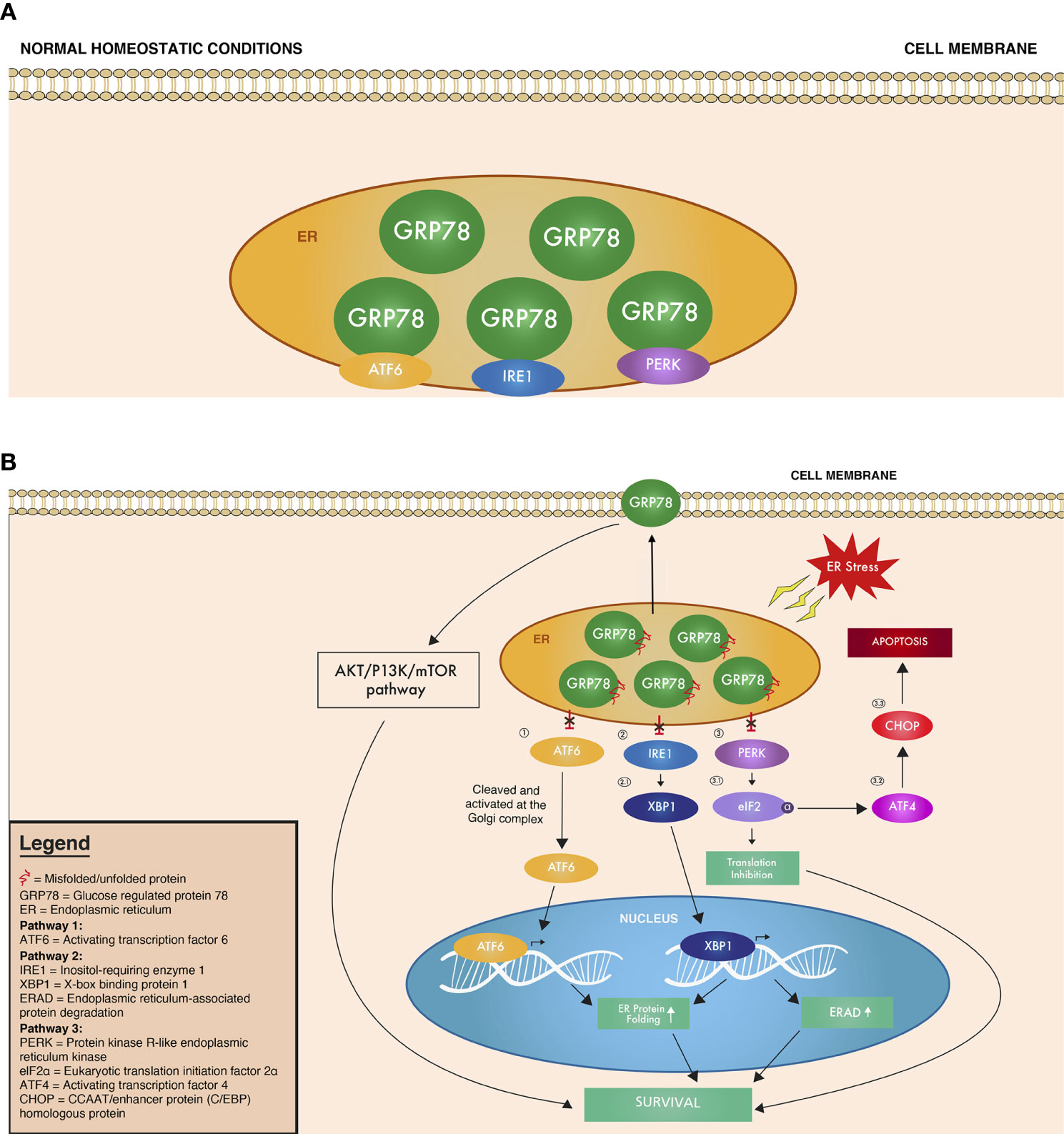
Figure 1 Overview of the UPR signaling cascade under (A) normal homeostatic conditions where GRP78 is bound to ATF6, IRE1, and PERK, inhibiting their function and (B) ER stress conditions, where ATF6, IRE1, and PERK are no longer bound, and thus no longer inhibited by GRP78. Circled numbers refer to the corresponding protein in the text and in Table 1.

Table 1 Overview of the UPR signaling cascade under normal homeostatic conditions and ER stress.
Upon release from GRP78, the UPR stress sensors (1) ATF6, (2) IRE1, and (3) PERK are also released to create a cascade of signaling down interacting UPR pathways (Figure 1B, Table 1). (1) ATF6 released from GRP78 then translocates to the Golgi apparatus, where it is cleaved into its active form. This active ATF6 then moves to the nucleus as a transcription factor that upregulates proteins involved in increasing the folding capacity of the ER (18). Meanwhile, after its release from GRP78, (2) the active form of IRE1 exerts its endoribonuclease activity, breaking a segment of mRNA intron and encoding X-box binding protein 1 (XBP1), which targets genes responsible for protein folding and ERAD (18, 27). Finally, when (3) PERK is released and activated, it phosphorylates the α subunit of eukaryotic translation initiation factor (eIF2α), which inhibits the initiation of translation, thus decreasing protein synthesis and the influx of proteins into the ER (18, 28). The phosphorylated eIF2α also activates activating transcription factor 4 (ATF4), which, in turn, activates the CCAAT/enhancer binding protein homologous transcription factor (CHOP/GADD153), which is associated with apoptosis (25, 29). All of these cascades continue to activate molecular targets to either promote survival of the cell or apoptosis, depending on the level of ER stress detected. If homeostasis cannot be restored, the pro-apoptotic arm of the UPR is induced, which is represented by the induction of CHOP (18, 24). CHOP then suppresses antiapoptotic outer mitochondrial membrane protein BCL2 and induces proapoptotic proteins, including death receptor 5 (DR5) and ER oxidoreductin protein 1α. All of the ATF6, IRE1, and PERK pathways overlap and with each pathway involved in both pro-survival and pro-apoptosis cellular responses. The mechanisms and nuances behind the coordinated activation of pro-survival or pro-apoptosis pathways still remain unclear, and further research is needed to pinpoint which ER stress signals can trigger apoptosis over survival and vice versa.
Because the UPR manages cellular stress, it is a central component in promoting tumor survival. Solid tumors are, by definition, inherently stressed cells as a result of dysregulated cell proliferation, which leads to inadequate blood supply, hypoxia, nutrient deprivation, and immune reactions (30). To adapt to these conditions, tumors upregulate multiple pathways promoting cell survival, including those involved in protein folding and stabilization. These survival pathways subsequently upregulate molecular chaperone proteins and heat shock proteins in the UPR (30–32). This behavior is prevalent among multiple tumor types including GBM, as elevated GRP78 levels in gliomas compared to normal human astrocytes have been repeatedly demonstrated and confirmed via immunohistochemistry, immunoblotting, and proteomic approaches in patient samples (10, 33, 34). These data suggest a chronic activation of the UPR and GRP78 activity within GBM cells. While differential GRP78 expression has been seen between GBM tumors and normal astrocytes, very limited data exists on differential GRP78 expression across core and leading edge of GBM tumors as well as their surrounding parenchyma. While available data from tumor microarrays have included multiple samplings from single tumors, no clear data exist regarding differential expression from surrounding cortical tissue (35). A potential direction for future studies may potentially compare GRP78 expression in GBM tumor cores, leading edge, surrounding T2 positive signal, and surrounding cortex to better characterize the role of GRP78 in modulating tumor, invasion and microenvironment.
In addition to upregulation within tumor cells, several studies on clinical tumor samples have shown that GRP78 is associated with patient tumor phenotype and behavior. GRP78 expression is notably higher in more proliferative GBM cell lines, while both RNA and protein expression have been found to increase with tumor grades in patient astrocytoma specimens (8–12). When comparing GBM cell lines, cell lines with higher basal cell proliferation and migration have up to three-fold higher GRP78 expression than those cell lines with less aggressive phenotypes (36). Overexpression of GRP78 was also found to limit tumor cell apoptosis, with decreased activation of pro-apoptotic caspase 7 signaling (11). Modulation of GRP78 via siRNA knockdown has further resulted in decreased proliferation of GBM cell lines while attenuating pro-survival pathways, namely Akt and ERK1/2, suggesting these pathways may at least partially mediate the effects of GRP78 on GBM cell proliferation (8). Patient outcomes are linked to such tumor progression, with more rapidly preoperative tumor growth rates in glioblastoma resulting in increased tumor size and more limited extent of tumor resection (37). As noted above, increasing expression level of GRP78 is strongly correlated with increasing tumor grade and malignancy, with grade IV glioblastoma (and therefore those tumors with poorest prognosis) expressing the highest levels of GRP78. Patterns of GRP78 are also correlated with grade. GBM is associated with a scattered pattern, while grade I astrocytoma and non-neoplastic tissue are associated with a highly grouped pattern (36). Because cellular and genetic heterogeneity is a hallmark of glioblastoma, studies have suggested the scattered GRP78 pattern noted in high grade gliomas relative to normal glial tissue/low grade tumors may contribute to the heterogeneity of high grade astrocytomas.
Various studies on GRP78 have also demonstrated that GRP78 has an important role in recurrent GBM and tumor progression after initial treatment. Of particular importance is temozolomide (TMZ), the standard-of-care chemotherapeutic treatment for GBM. TMZ has been shown to result in activation of the UPR in GBM cells, inducing increased levels of UPR markers, GRP78 and CHOP (10). However, when repressing GRP78 in drug sensitivity analyses with colony survival assays, GBM cells are notably more sensitive to TMZ and multiple other chemotherapeutic agents including 5-FU, irinotecan, etoposide, and cisplatin (10, 11). Conversely, overexpression of GRP78 promotes GBM cell resistance to not only TMZ treatment (10), but also etoposide and cisplatin-induced apoptosis (11). Radiation therapy, also a critical component of first-line GBM adjuvant treatment, has shown induction of GRP78, with radiation-induced reactive oxygen species associated with induction of ER stress (26). Furthermore, the effects of radiation therapy are similarly modulated by GBM expression of GRP78. Radio-resistance of GSCs has been shown to increase with GRP78 expression (38), and knockdown of GRP78 sensitized cells to gamma radiation (11).
While GRP78 has been correlated with increasing glioma grade, as noted above, survival analyses among grade IV GBM patients have also noted direct correlations between aggressive clinical behavior and GRP expression. Specifically, GRP78 is elevated in patient GBM samples compared to non-neoplastic brain tissue, with the greatest levels of GRP78 in patients with shorter overall survival (11, 39). Furthermore, more rapid tumor recurrence (decreased time to progression) after initial tumor resection is associated with higher GRP78 levels (8). Importantly, direct clinical data evaluating primary and recurrent GBM tissue from patients following TMZ and radiation therapy demonstrates that GRP78 levels are elevated in resected GBM samples following these treatments (9). These studies may be of particular relevance to patient outcomes, with more aggressive phenotypes noted among patients with recurrent glioblastoma (40, 41). Taken together, clinical data reveal GRP78 expression not only increases tumor aggressiveness, but also shows upregulation in recurrent GBM in response to cytotoxic drugs, making GRP78 an attractive target for therapy as well as implicating it as a prognostic marker.
GRP78 predominantly resides in the ER lumen within normal cells, with the majority of GRP78 studies primarily focusing on cytosolic or total GRP78. However, in tumor microenvironments where GRP78 is overexpressed, GRP78 also localizes to the surface of GBM cell membranes (21). In studies across multiple tumor types, cell surface GRP78 (csGRP78) acts as a co-receptor and participates in various signaling processes, modulating tumor apoptosis, proliferation, and motility (22, 42). Furthermore, re-location of GRP78 to the cell surface is notably associated with drug resistance and cell transformation (23). Modulation of cell surface GRP78 in multiple tumor types has also shown effects on behaviors of cancer stem cell populations (43, 44). In one of the more well characterized pathways, surface GRP78 has been shown to interact with and promote activation of the tumorigenic PI3K/AKT pathway through complex formation with PI3K in prostate cancer (22).
More recently, cell surface GRP78 has been explored in the context of gliomas. Unsurprisingly, cell surface GRP78 has been detected in multiple high-grade glioma cell lines via immunocytochemistry and immunoblotting. Treatment of this surface GRP78 with a polyclonal antibody decreased high-grade glioma cell line survival and population growth (21). Notably, greater reductions in glioma growth are seen with antibody treatments of lower grade tumors. Combination treatment, with both radiotherapy and antibody to cell surface GRP78, also results in efficacy on tumor apoptosis relative to radiation alone (45). Targeting of cell surface GRP78 likely exerts apoptotic effects via the Akt and mTOR pathways, as antibody treatment of GBM cell lines revealed decreases in phosphorylated and total Akt and mTOR. Intravenous injection of antibody has proven a feasible delivery mechanism, as anti-GRP78 antibodies delivered via mouse tail vein are noted to extravasate cell from vasculature to tumor. Surface expression of GRP78 thus provides an accessible binding site for targeted GRP78 therapeutics discussed below. Although combination therapy of radiation and antibody to cell surface GRP78 has demonstrated promise in in vitro and in mouse xenograft models, Akt/mTOR pathway inhibitors have yet to demonstrate meaningful benefit in GBM clinical trials (46). While limitations have been attributed to the blood brain barrier, GBM tumor heterogeneity, and possible activation of alternative pathways that allow for immune escape, the lack of direct Akt/mTOR pathway inhibitors must be noted in subsequent studies. The expansion of studies to include in vivo intracranial GBM preclinical models, or evaluation of csGRP78 in clinical patient samples may shed further light on efficacy and mechanism of targeting csGRP78.
GRP78 ultimately modulates GBM phenotype via the well-characterized downstream elements in its pathway, with several studies showing direct correlation of these elements with GBM oncogenesis and resistance to treatments. As previously mentioned, these downstream elements include the UPR stress sensors (1), ATF6 (2), IRE1, and (3) PERK. The first of these downstream effector pathways, the (1) ATF6 pathway, was shown to contribute to GBM radiotherapy resistance, with ATF6 knockdown resulting in increased radiotherapy sensitivity. However, ATF6 activity does not entirely explain the above noted GRP78-modulated GBM phenotypes, as ATF6 expression was not shown to be correlated with astrocytoma tumor grade (25, 26). Meanwhile, in vitro knockdown and overexpression GBM studies have revealed the (2) IRE1 pathway to be involved in tumor growth, migration, invasion, and neovascularization in GBM (25, 47–50). Specifically, in vitro knockdown studies of IRE1 commonly modulate GBM expression of extracellular matrix proteins including SPARC/Osteonectin, a protein eliciting cell shape changes and modulating synthesis of extracellular matrix (47). IRE1 has specifically shown modulation of angiogenesis via studies showing that increased IRE1-XBP1 signaling stimulated angiogenesis, while decreased IRE1-XBP1 signaling suppressed angiogenesis (51). The (3) PERK pathway has been shown to initiate signaling that promotes protective metabolic processes such as glycolysis under cell stress (52), while also mediating autophagy responses to targeted GRP78 therapies such as OSU-03012, a treatment discussed in detail below (53). In evaluation of tissue microarrays from 148 glioblastoma patients, decreased expression of ATF4, a downstream element of the PERK pathway (as described above) is associated with prolonged overall survival (54). This was interpreted by the authors as clinical evidence, suggesting a link specifically between the PERK branch of the UPR and GBM patient prognosis. Interestingly, despite results implicating PERK and ATF4 in promotion of tumor survival, they have also shown upstream activation of pro-apoptotic CHOP, exemplifying how UPR pathways have roles in both pro-survival and pro-apoptotic responses.
Regulation of these pro- and anti-apoptotic pathways and their opposing consequences is complicated and continues to be studied. The pro-survival arm of endoplasmic reticulum stress, triggered by conditions of glucose starvation, hypoxia, Ca2+ depletion, and misfolding of proteins, initially activates GRPs and the UPR in an attempt to restore normal functioning (55). In this survival arm, GRP78 forms a complex with caspase-7, a mediator of apoptosis, inhibiting its activity. The pro-apoptotic arm seems to predominate when the protein aggregation is persistent and the stress is not resolved, which subsequently triggers signaling to become pro-apoptotic. CHOP, which is generally induced in ER stress, is thought to mediate the commitment to the pro-apoptotic response. When the stress is prolonged and severe, the PERK and IRE1 pathways converge on CHOP and increase its induction (56). Consequently, CHOP increases expression of pro-apoptotic genes and decreases expression of anti-apoptotic genes, including Bcl2. IRE1α also promotes both pro- and anti-apoptotic events including the activation of JNK and caspase pathways, as well as splicing of XBP-1 (57). Indeed, the interaction between UPR elements is very complex, with data from knockout of each individual UPR component suggesting that no single component is strictly necessary for ER induced apoptosis, underlining the need for further study (57).
Multiple UPR elements, including GRP78 and PERK, have been implicated in function of glioma stem cells (GSCs). GSCs are defined as a subpopulation of stem-like glioblastoma cells capable of self-renewal, differentiation into cortical lineages (astrocyte, oligodendrocyte, neuron), and have tumorigenic capacity. Not only are GSCs hypothesized to be the cell of origin for GBM, but the aggressive characteristics of GBM, including vascularization, invasion, chemo-resistance, radio-resistance, and recurrence, are often attributed to GSCs (58).
While few studies have evaluated UPR and GRP78 in GSCs, recent data suggests that GSCs show differential responses to cell stress, as UPR activation via a common cell stress agent, thapsigargin (Tg), results in variable results when comparing patient-derived GSCs (from mesenchymal and proneural subtypes) and their differentiated counterparts. GBM neurospheres show upregulation of all three branches of the UPR, with increased PERK phosphorylation, activation of IRE1α, and accumulation of ATF6. The contribution of apoptosis to cell death was seen with activation of caspases 3/7, while an apoptotic inhibitor suppressed cytotoxicity. Specifically, Tg-induced ER stress results in decreased survival of GSCs relative to effects on differentiated tumor cells (54). This GSC-specific sensitivity may be mediated by ER stress upregulation of the pro-apoptotic PERK signaling pathway of the UPR, as inhibition of PERK suppressed these GSC-specific cytotoxic effects. GSC self-renewal, in contrast to apoptosis, may be mediated by the PERK pathway. Specifically, cell stress was noted to decrease both as well as expression of Sox2, an essential protein in maintaining pluripotency and self-renewal properties of stem cells. These novel GSC effects are regulated by the PERK branch of the UPR during cell stress, as repression of the PERK branch (but not the IRE1/XBP1 branch) limits these effects (54). GSC self-renewal in association with PERK branch suppression has also been noted when BMI1, an epigenetic modifying factor, increases both GSC self-renewal and cell stress by suppressing ATF3, a downstream element of the PERK signaling pathway. Decreased ATF3 expression is also associated with increased stemness and poor prognoses (35, 59, 60). Finally, though the role of GSCs in GBM invasion has yet to be fully characterized, invasive GBM cells have been shown to have increased stemness, and future studies may explore whether invasive GBM cells beyond the tumor edge have more stem-like cells containing elevated GRP78 expression (57). Given the hypothesized central role of GSCs in GBM aggressiveness and resistance to therapy, induction of ER stress may offer selective targeting of the GSC subset. Ultimately, further studies are warranted to elucidate effects of UPR induction and GRP78 elevation on both GSC and differentiated GBM populations.
Although GRP78 is the more thoroughly investigated glucose-regulated protein in the UPR pathway, glucose-regulated protein 94 (GRP94) is another molecular chaperone, localized in the ER, that shares functions with GRP78. Similarly, GRP94 has been implicated in aggressive glioma behavior. Evaluation of GRP94 mRNA and protein levels reveal significant elevations in GRP94 in high-grade glioblastoma when compared with normal brain tissues (33). In addition, a gradual increase in GRP94 protein and RNA levels in patient samples of grade II to grade IV gliomas is also noted, with the highest levels of GRP94 in grade IV GBM. Just as with GRP78, high GRP94 levels when evaluated across 20 glioma patients were also associated with a significantly shorter overall patient survival. Functional evaluation of GRP94 in vitro reveals knockdown of GRP94 in GBM cell lines that suppressed cellular proliferation, impaired colony formation ability, and inhibited cell migration and invasion ability. However, microarray analysis of GRP94 knockdown cells suggests that, while similar to effects of GRP78, GRP94 may instead exert its effects via downstream dysregulation of the Wnt/β-catenin signaling pathway, which normally promotes the proliferation of GBM cells (61). Finally, there appears to be at least a partial interplay between GRP94 and GRP78 in GBM cells. Specifically, GRP94 expression appears to increase when GRP78 is suppressed, but not enough to completely compensate for its loss (18, 62). Thus, GRP94 may be a promising therapeutic target similar to GRP78, though it appears to function through different pathways. However, research on GRP94 is limited and more studies are warranted to further elucidate GRP94's role in GBM tumor malignancy.
GRP78 may influence not only GBM tumor cells, but the surrounding microenvironmental vasculature. GRP78 is notably significantly elevated in both in situ and in vitro primary cultures of human brain endothelial cells derived from blood vessels of malignant glioma tissues. In contrast, there appears to be minimal GRP78 expression in normal brain tissues and blood vessels. Functional studies further support a critical role for GRP78 in GBM endothelial cells, as GRP78 knockdown increases tumor endothelial cell susceptibility to not only TMZ, but multiple chemotherapeutic agents, including CPT-11 and etoposide. Conversely, GRP78 overexpression results in increased chemo-resistance of tumor endothelial cells (63). Though studies on GRP78 in tumor vasculature in the context of GBM is limited, there has also been evidence from studies on other solid tumors that suggest an important regulatory role for GRP78 in tumor vasculature. In a transgene-induced mammary tumor model, interference with GRP78 function exhibited dramatic reduction in the microvessel density of endogenous mammary tumors with no effect on normal organs, and a follow-up study demonstrated that interference with GRP78 function suppressed tumor growth and angiogenesis during the early phase of tumor growth (64). This suggests that GRP78 has a critical role in regulation of vasculature specific to the tumor microenvironment, while limiting injury to normal vasculature. While such data suggest GRP78 and the UPR may represent a therapeutic target for not only GBM tumor but associated microvasculature, further study is necessary both in vivo and at a from patient samples to clarify safety of such an approach.
An accumulating abundance of evidence thus continues to support a significant role for GRPs in modulating glioma and GSC behavior, as well as chemotherapy resistance. Therapeutic resistance of GBM cells to therapy remains a critical barrier to improving patient survival. Multiple groups have therefore focused on GRPs as therapeutic targets, developing several promising GRP-targeted agents to improve sensitivity to current conventional therapies and improve clinical outcomes.
In light of the potential that targeting GRPs have in improving glioblastoma therapy, various studies have sought to inhibit GRP78 using various agents and to examine their potential to be used in conjunction with current conventional therapies and in clinical settings. Other therapies modulate GRP78 expression without direct interaction or utilize GRP78 in treatment delivery modalities. While some efforts have directly targeted GRP78 via rational drug design or identified efficacious GRP targeting via screening, other targeted GBM therapies have incidentally been found to show effect in part through GRP effects. These therapies include natural products, fusion proteins, antibody-based treatments, phage-based treatment delivery methods, and more, all of which are shown in Table 2. More details on GRP78 interactions and downstream signaling effects of these GRP78-based therapies are also outlined in Table 3. The majority of these studies have not yet reached clinical trials, but in vitro and in vivo studies show promise in decreasing aggressive tumor phenotype upon treatment and reducing resistance to chemotherapy and radiation therapy.
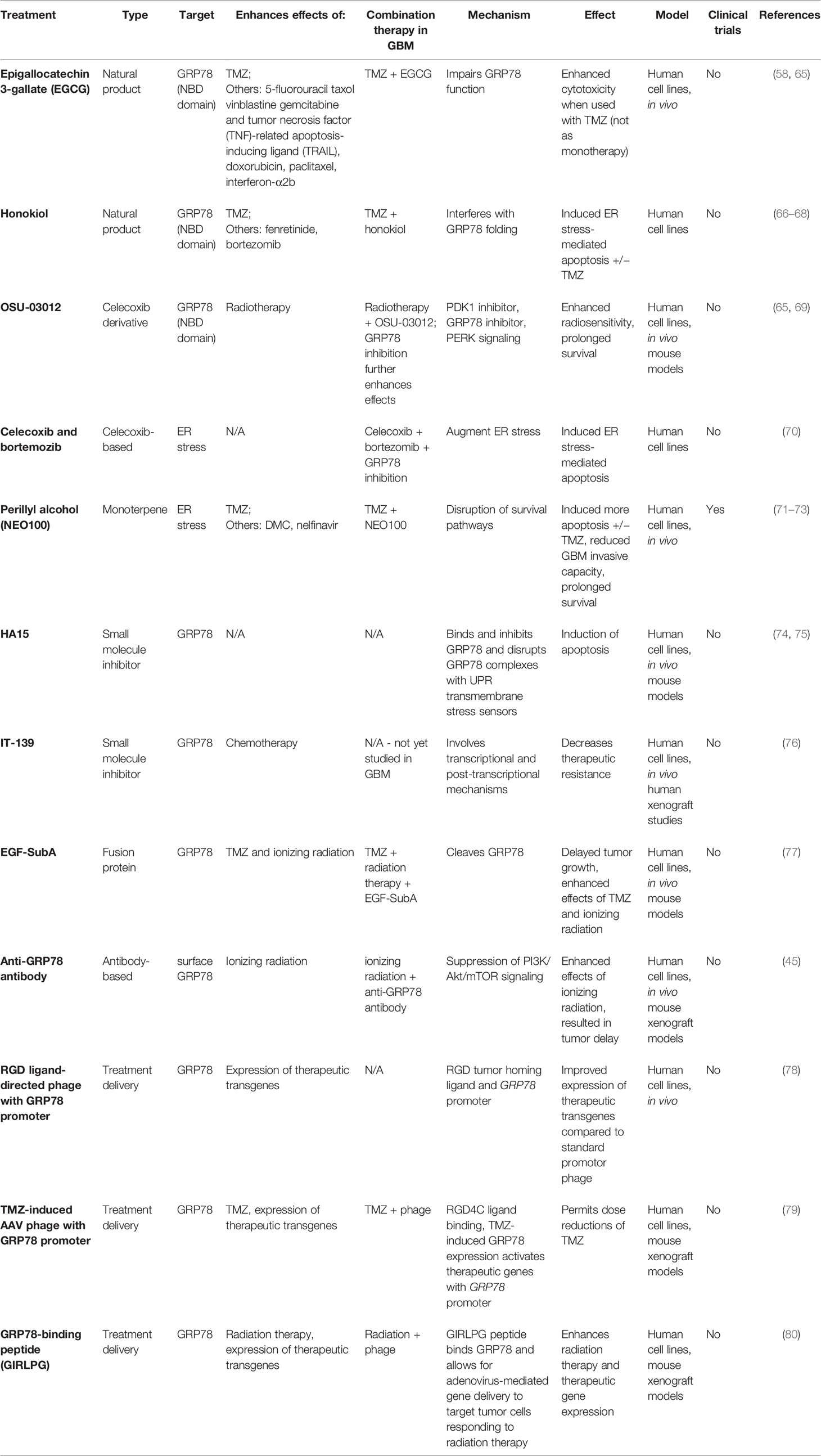
Table 2 General characteristics of GRP78-based treatments.
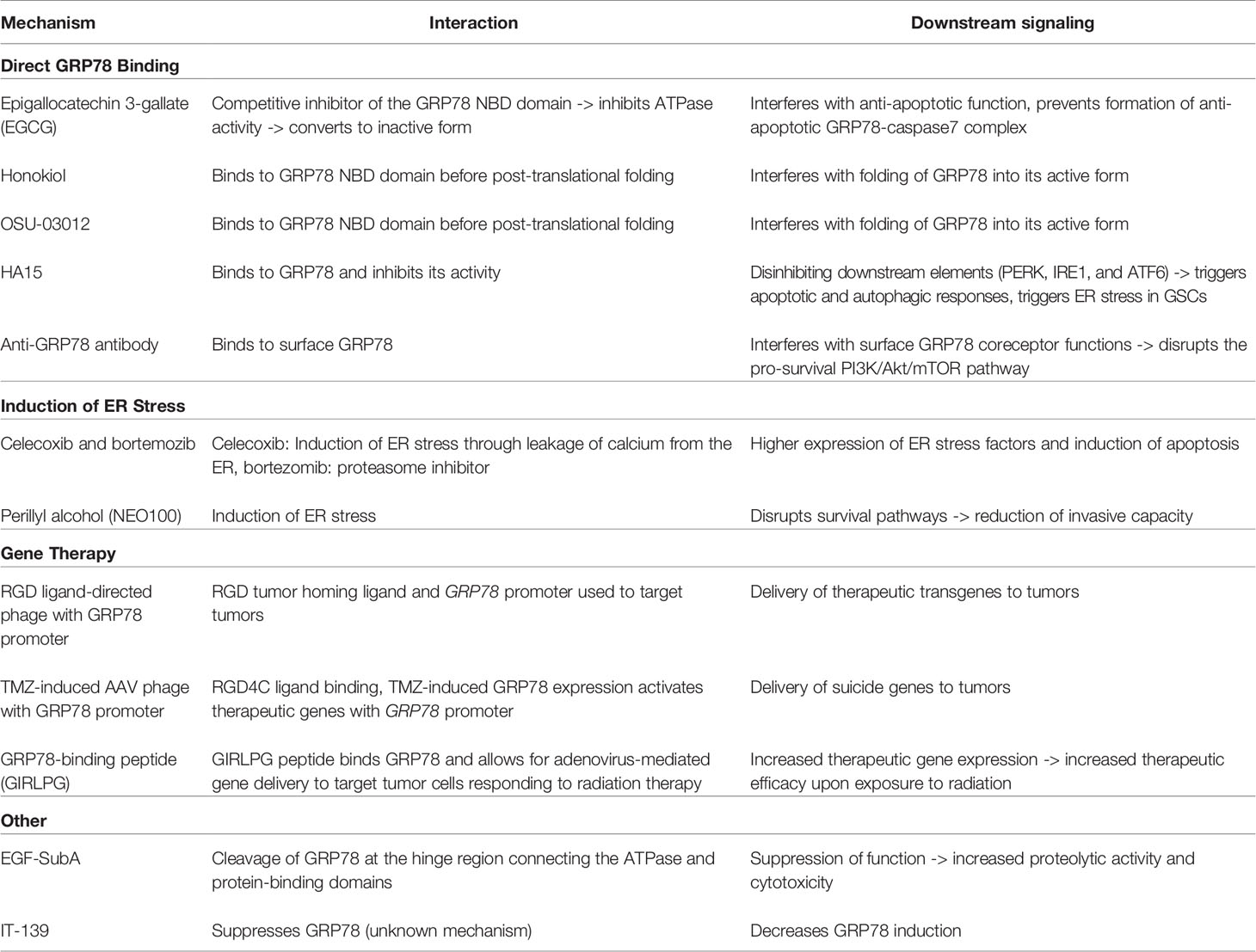
Table 3 Interactions and signaling of GRP78-based treatments.
One natural product that has garnered particular attention is epigallocatechin 3-gallate (EGCG), a polyphenolic bioflavinoid from green tea extract (65). This compound is currently being investigated for its potential in enhancing chemotherapy. EGCG has been shown to increase sensitivity of GBM and multiple other tumor cells to various cytotoxic agents including 5-fluorouracil, taxol, vinblastine, gemcitabine, and tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) in vitro, as well as doxorubicin, paclitaxel, or interferon-α2b in vivo, using a preclinical model of intracranially injected GBM cells (58). UPR components are critical in mediating the effects of EGCG. EGCG binds and inactivates GRP78, interfering with its anti-apoptotic function (58). More specifically, EGCG acts as a competitive inhibitor, inhibiting ATPase activity and impairing GRP78 function by binding to the nucleotide-binding domain (NBD), an ATPase binding domain. Upon binding, EGCG also converts the GRP78 NBD domain from its active unfolded form to its inactive folded form. In addition, EGCG can prevent the formation of the anti-apoptotic GRP78-caspase7 complex (65). In vitro and in vivo studies demonstrated that EGCG enhanced the cytotoxic effects of TMZ when they were used simultaneously. However, for unclear reasons, when used as a monotherapy, EGCG failed to demonstrate significant antitumor activity (58). These results indicate that both GRP78 are involved in mediating the cytotoxic effects of EGCG and that this compound holds significant promise in improving response to TMZ.
Honokiol (HNK), a Magnolia grandiflora cell wall derivative, is another natural product of interest (66). Like EGCG, HNK was shown to preferentially bind to the unfolded form of the NBD of GRP78, with studies suggesting that HNK binds with a greater affinity than EGCG. Studies in neuroectodermal tumor cell lines, including GBM cell lines, showed that HNK induced apoptosis through ER stress with twice the efficacy of EGCG. In addition, HNK has shown efficacy in augmenting TMZ-induced damage in GBM tumor cells when used in combination (67). Furthermore, when used in TMZ-resistant GBM cell lines, honokiol alone successfully induced tumor cell death (68). HNK has also demonstrated synergistic effects when used in combination with fenretinide or bortezomib, which are ER stress inducers and antitumor agents (66). It has been suggested that HNK may interact with GRP78 before post-translational folding of newly synthesized GRP78, thus interfering with translation and reducing the ability for GRP78 to fold into its active form (66). Ultimately, studies have been limited to in vitro conditions, with future studies requiring more data from preclinical xenograft models.
Another agent of interest is OSU-03012, which was developed with a chemical backbone of celecoxib, a cyclooxygenase (COX2) inhibitor (69). OSU-03012 was shown to suppress GRP78 expression in GBM cells and to bind to the NBD of GRP78. Molecular docking and molecular dynamics studies demonstrated that, similarly to EGCG, OSU-03012 binding induced conformational changes, converting it from its active unfolded form to a more inactive folded form (65). However, EGCG binds with higher specificity to GRP78 than OSU-03012, and thus EGCG is thought to be a more effective inhibitor for GRP78 (81). Nevertheless, OSU-03012 has shown promise in sensitization of GBM tumor cells to radiotherapy. In vitro, pre-treatment of GBM cells with OSU-03012 enhanced radiosensitivity (69). In vivo, OSU-03012 sensitized tumor cells to radiotherapy and prolonged survival in GBM tumor mouse models (69). Variable expression of GRP78 can also affect cytotoxicity effects. Knockdown of GRP78 enhanced OSU-03012 lethality, while overexpression of GRP78 essentially eliminated its toxicity (69). In terms of mechanisms behind OSU-03012, other knockdown studies have determined that PERK signaling may mediate these effects. OSU-03012 is also an inhibitor of phosphoinositide-dependent kinase 1 (PDK1), a kinase that is important in signaling for growth factors (69).
An emerging alternative combination therapy based on celecoxib (Celebrex), which causes ER stress through leakage of calcium from the ER into the cytosol, combines it with GRP78 inhibition and the proteasome inhibitor bortezomib, which is known to trigger ER stress through accumulation of proteins (70). When celecoxib and bortezomib were used in combination, elevated expression of ER stress factors was detected and apoptotic cell death was greatly increased (70). Importantly, when celecoxib and bortezomib were used in conjunction with siRNA-mediated knockdown of GRP78, tumor cells were further sensitized to the treatment (70). A novel compound structurally similar to celecoxib, 2,5-dimethyl-celecoxib (DMC), when used with bortezomib instead of celecoxib, demonstrated the same effects but with higher potency (70). Furthermore, DMC alone has been shown to induce tumor-associated brain endothelial cell death through GRP78 and CHOP induction (82), suggesting that DMC is better drug of choice than celecoxib to be used in conjunction with bortezomib. Notably, clinical trials combining celecoxib with temozolomide have not demonstrated additional benefit (83). However, the above combination therapies, in conjunction with a GRP78-inhibiting agent, may hold promise for alternative future clinical trials.
HA15 is a thiazole benzenesulfonamide small molecule inhibitor that directly interacts and targets GRP78, and has shown potential as a therapeutic agent in GBM. In melanoma cells, HA15 was shown to directly bind to GRP78, dissociating it from PERK, IRE1, and ATF6 and inhibiting its activity, which subsequently resulted in apoptotic and autophagic responses (74). In GBM, HA15 has similarly been shown to trigger ER stress in GSCs through GRP78-specific targeting (75). Another small molecular inhibitor, IT-139, has been shown to suppress GRP78 induction in therapy-resistant lung, prostate, liver, colon, pancreatic, gastric, and breast cancer cell lines, but not in non-cancerous cell lines, and decreased GRP78 in in vivo xenograft studies (76). Considering its efficacy in other cancer types, IT-139 could be a promising drug for GBM as well.
A novel fusion protein called EGF-SubA has also demonstrated promise as a novel form of therapy that targets GRP78. This fusion protein was created by engineering epidermal growth factor (EGF) and the bacterial toxin SubA, which selectively cleaves GRP78 at a single site in the hinge region connecting the ATPase and protein-binding domains (77). EGF-SubA demonstrated tumor-specific proteolytic activity and cytotoxicity in GBM cell lines and enhanced sensitivity of cells to therapeutic doses of TMZ and ionizing radiation (77). In in vivo mouse models, EGF-SubA was also shown to delay tumor growth (77). Thus, this fusion protein also holds promise as monotherapy or combination therapy with TMZ and ionizing radiation.
Antibody-based treatments have also emerged as therapeutic strategies. Studies have noted that antibodies targeting GRP78 obtain cytotoxic effect by interfering specifically with surface GRP78 coreceptor functions, disrupting the PI3K/Akt/mTOR signaling pro-survival pathway. Antibody binding therefore results in decreased tumor cell proliferation and colony formation, as well as enhanced apoptosis both in vitro and in vivo (45). Combining this anti-GRP78 antibody treatment with ionizing radiation therapy may have a sensitizing effect to radiation, with results showing more significant tumor growth delay with combination therapy (45). Importantly, it appears the GRP78 antibodies studied demonstrated specificity, binding specifically to cancer cells. Notably, antibody-based therapies must overcome challenges of chemotherapy delivery through the blood-brain barrier (BBB) in considerations of preclinical and clinical studies. In attempts to guide anti-GRP78 antibodies across the blood-brain barrier, antibody-conjugated nanoparticles have been shown to improve the accumulation of drugs in pathological sites and decrease side effects in normal tissue when utilized in neurodegenerative disorders (84). Future studies could also utilize this strategy with anti-GRP78 antibodies in GBM.
Another antibody based treatment, a micelle-based therapeutic delivery system, targets cell surface GRP78 limiting proliferation of GBM and GSC subpopulations (85). Micelles, “nanocarriers” for chemotherapeutics, were modified with two peptides. The first peptide, DVAP, had high-affinity for GRP78 while the second, DWSW, allowed blood-brain-barrier penetration necessary for in vivo access to tumors. Micelles were noted to co-localize with GRP78 on tumor cells. Subsequently, targeted micelles loaded with paclitaxel or parthenolide were noted to have potent anti-tumor activity, with increased survival of xenograft-bearing mice relative to free drug or non-targeted micelles. While such GRP78 targeted systems will require further study to ensure non-specific binding to csGRP in other systemic regions, results are promising.
Phage-directed targeting of GRP78 for treatment delivery has generated significant interest from various labs due to the well-documented overexpression of GRP78 in aggressive tumors. A dual tumor-targeted phage, containing both a tumor homing ligand (the tripeptide Arg-Gly-Asp) and GRP78 promoter, leverages tumor specificity of the bacteriophage with introduction of highly expressed GRP78 promoter within tumor cells. This was shown to be more effective in GRP78-guided expression of therapeutic transgenes compared to the standard cytomegalovirus promotor phage both in vitro and in vivo (78). Other attempts for delivery of therapeutic genes to GBM have utilized a hybrid AAV/phage to deliver a suicide genes under the control of a TMZ-induced promoter of GRP78 (79). Dual tumor targeting is first accomplished when the phage capsid displays the RGD4C ligand that binds to an integrin receptor. The virus plasmid thus infects tumor xenografts in mice incorporating the viral plasmid within tumor cells. Subsequently, when TMZ is administered and GRP78 expression is upregulated in tumor tissue, the GRP78 promoter is induced on the viral plasmid, which activates therapeutic gene expression (79). This method offers a compelling mode of combination therapy using TMZ and targeted suicide gene therapy that may potentially permit dose reductions of TMZ. Finally, another glioma-specific gene therapy study focused on direct targeting of cell surface GRP78. Specifically, radiation-induction of plasma membrane GRP78 on both tumor cells and associated endothelial cells was targeted by adenovirus. The authors utilized a GIRLPG peptide that specifically binds to GRP78 (80). The study found that using the GRP78-binding peptide resulted in increased gene expression in irradiated tumors after infection with the adenoviruses, demonstrating its increased efficacy in recognizing tumor cells that are responding to radiation therapy (80). While studies leveraging GRP78 for mediating treatment delivery in gene therapy are ongoing and must address targeting specificity and safety profiles in clinical delivery, ultimately, these therapies hold promise at supplementing current therapies.
Perillyl alcohol (POH), or NEO100, is another promising anti-cancer agent that has been shown to induce cytotoxicity through ER stress, as demonstrated by elevated expression of GRP78 (71). NEO100 is a monoterpene initially used as an oral treatment for systemic cancer (71). Clinical administration via intranasal delivery in patients has been successful in GBM, as a phase II trial in Brazil for intranasal NEO100 treatment of TMZ-resistant malignant gliomas was well-tolerated (72). NEO100 induces effects in GBM via disruption of survival pathways, with a reduction of invasive capacity of both chemosensitive and resistant glioma cell lines (71). These effects are likely mediated in part via ER stress and the UPR pathway, with administration of NEO 100 resulting in elevated levels of GRP78, CHOP, PARP, and ATF3 (71). Further, functional assessment via knockdown of GRP78 results in a significant decrease in tumor cell viability, with a corresponding increase in chemosensitivity. In parallel with the effects of GRP78 on GSCs noted above, NEO100 was also shown to be cytotoxic for different subtypes of GSCs (73). NEO100 may therefore offer a GRP-mediated treatment modality, currently in the clinical trial stage, that offers promising monotherapy or combination therapy with TMZ.
Various therapeutic strategies currently being investigated for efficacy in GBM tumors have notable effects on GRP78 expression without directly binding or interacting with the protein. The first of these therapies of interest is pterostilbene (PT). Pterostilbene is an analogue of resveratrol, a phenol compound found in plants (86). Studies have shown that when treated with PT, GSCs in GBM tumors demonstrated increased radiosensitivity (86). An increase of tumor suppressor miR-205 and negative modulation of GRP78 signaling suggested that these effects were mediated through the GRP78/miR-205 axis (86). Tubastatin A, a novel HDAC6 inhibitor, also functions through modulation of GRP78, resulting in reduced cell viability and induced apoptosis in TMZ-resistant glioma cells (87). Tubulin was noted to induce hyperacetylation of GRP78, resulting in dissociation of GRP78 from target proteins. Coupled with TMZ exposure, Tubulin A HDAC6 inhibition resulted in downstream effects that favored pro-apoptotic mechanisms (87).
GRP78 has been further indicated as a promising anti-angiogenic target, with studies noting that recombinant plasminogen kringle 5 (rK5) can induce apoptosis of brain derived dermal microvessel endothelial cells (MvEC) through mediation by GRP78 (88). Using knockdown studies, authors demonstrated that this apoptosis requires both GRP78 and lipoprotein receptor-related protein 1 (LRP1) (88). Of note, this study focused primarily on brain-derived MyECs, suggesting efficacy in GBM given a demonstration of increased GRP78 in GBM. Ultimately, further study would require direct evaluation of this treatment paradigm in GBM-derived endothelial cells. Finally, another treatment that involves GRP78 is the antimalarial agent chloroquine, which has also been shown to sensitize GBM cells to TMZ and whose effects were further enhanced by GRP78 knockdown (89).
While GRP78 has been leveraged as a therapeutic target as described above, a large number of studies assess the efficacy of GBM treatments using GRP78 overexpression as an indicator of therapy-inducted ER stress. These treatments do not specifically upregulate GRP78, but rather overexpress GRP78 as a result of the ER stress that is caused upon treatment. While this GRP78 overexpression may mediate function of these treatments, or simply act an indicator of a naturally stressed tumor cell environment, further GRP78 specific studies would be required to clarify GRP78’s role in each of these therapies. These novel therapies, demonstrating GRP78 overexpression, include tumor necrosis factor-related apoptosis inducing ligand (TRAIL) (90), endothelial-monocyte activating polypeptide II (EMAP-II) (91), asiatic acid (92), ellagic acid (93), canavanine treatment under lack of arginine (94), C-150 (Mannich-type curcumin derivative) (95), gamitrinib with bromodomain and extraterminal (BET)-inhibitors (96), dihydroartemisinin (97), lysine demethylase KDM1A inhibitor (98), and withaferin A (99). More of these novel therapies are listed in Table 4. Perhaps the most compelling and widely implemented treatment modality demonstrating GRP78 elevation following treatment is tumor-treating fields (TTFields). TTFields is a recently FDA-approved antimitotic GBM treatment that acts via disruption GBM cell division and organelle assembly through low-intensity alternating electric fields (100). GRP78 was found to be elevated in cell lines following TTFields, indicating the induction of ER stress (101). Considering GRP78 overexpression and its role in tumor survival, GRP78 suppression in conjunction with these novel therapies may potentially improve and enhance their effects.

Table 4 Table of treatments that cause ER stress and result in GRP78 overexpression.
Overall, GRP78 can be leveraged in many diverse ways and through various approaches to improve GBM therapy, demonstrating great potential as a therapeutic target in improving first-line therapies as well as in developing alternative therapies. Ultimately, further study of GRP78 and UPR targeting in GBM is warranted. In addition to the future studies suggested throughout this review, studies must clarify GRP-specific and GBM-specific targeting that crosses the blood brain barrier, minimize effects on normal non-cancerous cells within the host, and maximize an acceptable safety profile for a very promising target.
GRP78 and other components of the UPR have important roles in mediating GBM-specific survival, therapeutic resistance, and tumor progression. Many treatment modalities targeting the UPR and GRP78 are under investigation, with some at the clinical trial stage. Many of these therapies sensitize tumor cells to the current standard-of-care therapies, TMZ and radiation therapy, suggesting potential for combination therapy with GRP78-suppressing agents and current therapy. Further investigation is warranted to evaluate the efficacy of these treatment modalities within the clinic, as well as synergistic effects in the cases involving combination therapies. While GBM remains a devastating disease, GRP78-based therapies may hold significant promise in prolonging overall survival, delaying tumor progression, and decreasing treatment resistance and recurrence in GBM, ultimately providing hope that future treatments might convert GBM from a malignant disease to a chronic one.
KL did the majority of the literature search and wrote the majority of the manuscript. KL also designed the figures and made the tables. KT made edits to the manuscript, gave crucial feedback for the text, and offered significant guidance in the design of the figures. FA is the principal investigator. FA gave guidance on the focus and general direction of the paper, provided detailed edits, added specific data based on additional literature search, and did much of the rewriting. All authors contributed to the article and approved the submitted version.
Laboratory studies evaluating noncoding RNAs modulating the glioblastoma cell stress response is funded by the Margaret E. Early Medical Research Trust. Laboratory work and publication fees are also supported by the USC Department of Neurosurgery Research Startup Fund.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Dr. Amy S. Lee from the University of Southern California Norris Comprehensive Cancer Center for her valuable discussion and helpful feedback with the manuscript. In addition, FA is funded by the Margaret E. Early Research Trust.
1. Ostrom QT, Bauchet L, Davis FG, Deltour I, Fisher JL, Langer CE, et al. The epidemiology of glioma in adults: a “state of the science” review. Neuro Oncol (2014) 16(7):896–913. doi: 10.1093/neuonc/nou087
2. Stupp R, Hegi ME, van den Bent MJ, Mason WP, Weller M, Mirimanoff RO, et al. Changing paradigms–an update on the multidisciplinary management of malignant glioma. Oncologist (2006) 11(2):165–80. doi: 10.1634/theoncologist.11-2-165
3. Ostrom QT, Gittleman H, Xu J, Kromer C, Wolinsky Y, Kruchko C, et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2009-2013. Neuro Oncol (2016) 18(suppl_5):v1–v75. doi: 10.1093/neuonc/now207
4. Alexander BM, Cloughesy TF. Adult Glioblastoma. J Clin Oncol (2017) 35(21):2402–9. doi: 10.1200/JCO.2017.73.0119
5. Dehdashti AR, Hegi ME, Regli L, Pica A, Stupp R. New trends in the medical management of glioblastoma multiforme: the role of temozolomide chemotherapy. Neurosurg Focus (2006) 20(4):E6. doi: 10.3171/foc.2006.20.4.3
6. Nagasubramanian R, Dolan ME. Temozolomide: realizing the promise and potential. Curr Opin Oncol (2003) 15(6):412–8. doi: 10.1097/00001622-200311000-00002
7. Lee AS. The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem Sci (2001) 26(8):504–10. doi: 10.1016/S0968-0004(01)01908-9
8. Zhang LH, Yang XL, Zhang X, Cheng JX, Zhang W. Association of elevated GRP78 expression with increased astrocytoma malignancy via Akt and ERK pathways. Brain Res (2011) 1371:23–31. doi: 10.1016/j.brainres.2010.11.063
9. Wen X, Chen X, Cheng X. Increased Expression of GRP78 Correlates with Adverse Outcome in Recurrent Glioblastoma Multiforme Patients. Turk Neurosurg (2020) 30(1):11–6. doi: 10.5137/1019-5149.JTN.21840-17.4
10. Pyrko P, Schonthal AH, Hofman FM, Chen TC, Lee AS. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res (2007) 67(20):9809–16. doi: 10.1158/0008-5472.CAN-07-0625
11. Lee HK, Xiang C, Cazacu S, Finniss S, Kazimirsky G, Lemke N, et al. GRP78 is overexpressed in glioblastomas and regulates glioma cell growth and apoptosis. Neuro Oncol (2008) 10(3):236–43. doi: 10.1215/15228517-2008-006
12. Gimenez M, Marie SK, Oba-Shinjo S, Uno M, Izumi C, Oliveira JB, et al. Quantitative proteomic analysis shows differentially expressed HSPB1 in glioblastoma as a discriminating short from long survival factor and NOVA1 as a differentiation factor between low-grade astrocytoma and oligodendroglioma. BMC Cancer (2015) 15:481. doi: 10.1186/s12885-015-1473-9
13. Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature (2012) 488(7412):522–6. doi: 10.1038/nature11287
14. Hombach-Klonisch S, Mehrpour M, Shojaei S, Harlos C, Pitz M, Hamai A, et al. Glioblastoma and chemoresistance to alkylating agents: Involvement of apoptosis, autophagy, and unfolded protein response. Pharmacol Ther (2018) 184:13–41. doi: 10.1016/j.pharmthera.2017.10.017
15. Obacz J, Avril T, Le Reste PJ, Urra H, Quillien V, Hetz C, et al. Endoplasmic reticulum proteostasis in glioblastoma-From molecular mechanisms to therapeutic perspectives. Sci Signal (2017) 10(470):eaal2323. doi: 10.1126/scisignal.aal2323
16. Lee AS. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer (2014) 14(4):263–76. doi: 10.1038/nrc3701
17. Zhang Y, Liu R, Ni M, Gill P, Lee AS. Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. J Biol Chem (2010) 285(20):15065–75. doi: 10.1074/jbc.M109.087445
18. Ibrahim IM, Abdelmalek DH, Elfiky AA. GRP78: A cell’s response to stress. Life Sci (2019) 226:156–63. doi: 10.1016/j.lfs.2019.04.022
19. Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet (1988) 22:631–77. doi: 10.1146/annurev.ge.22.120188.003215
20. Chang SC, Wooden SK, Nakaki T, Kim YK, Lin AY, Kung L, et al. Rat gene encoding the 78-kDa glucose-regulated protein GRP78: its regulatory sequences and the effect of protein glycosylation on its expression. Proc Natl Acad Sci U S A (1987) 84(3):680–4. doi: 10.1073/pnas.84.3.680
21. Kang BR, Yang SH, Chung BR, Kim W, Kim Y. Cell surface GRP78 as a biomarker and target for suppressing glioma cells. Sci Rep (2016) 6:34922. doi: 10.1038/srep34922
22. Zhang Y, Tseng CC, Tsai YL, Fu X, Schiff R, Lee AS. Cancer cells resistant to therapy promote cell surface relocalization of GRP78 which complexes with PI3K and enhances PI(3,4,5)P3 production. PLoS One (2013) 8(11):e80071. doi: 10.1371/journal.pone.0080071
23. Roller C, Maddalo D. The Molecular Chaperone GRP78/BiP in the Development of Chemoresistance: Mechanism and Possible Treatment. Front Pharmacol (2013) 4:10. doi: 10.3389/fphar.2013.00010
24. Pfaffenbach KT, Lee AS. The critical role of GRP78 in physiologic and pathologic stress. Curr Opin Cell Biol (2011) 23(2):150–6. doi: 10.1016/j.ceb.2010.09.007
25. Le Reste PJ, Avril T, Quillien V, Morandi X, Chevet E. Signaling the Unfolded Protein Response in primary brain cancers. Brain Res (2016) 1642:59–69. doi: 10.1016/j.brainres.2016.03.015
26. Dadey DY, Kapoor V, Khudanyan A, Urano F, Kim AH, Thotala D, et al. The ATF6 pathway of the ER stress response contributes to enhanced viability in glioblastoma. Oncotarget (2016) 7(2):2080–92. doi: 10.18632/oncotarget.6712
27. Hendershot LM, Valentine VA, Lee AS, Morris SW, Shapiro DN. Localization of the gene encoding human BiP/GRP78, the endoplasmic reticulum cognate of the HSP70 family, to chromosome 9q34. Genomics (1994) 20(2):281–4. doi: 10.1006/geno.1994.1166
28. Li J, Ni M, Lee B, Barron E, Hinton DR, Lee AS. The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death Differ (2008) 15(9):1460–71. doi: 10.1038/cdd.2008.81
29. Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol (2013) 15(5):481–90. doi: 10.1038/ncb2738
30. Graner MW. The unfolded protein response in glioblastomas: targetable or trouble? Future Sci OA (2015) 1(2):FSO45. doi: 10.4155/fso.15.45
31. Ma Y, Hendershot LM. The role of the unfolded protein response in tumour development: friend or foe? Nat Rev Cancer (2004) 4(12):966–77. doi: 10.1038/nrc1505
32. Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol (2012) 197(7):857–67. doi: 10.1083/jcb.201110131
33. Graner MW, Cumming RI, Bigner DD. The heat shock response and chaperones/heat shock proteins in brain tumors: surface expression, release, and possible immune consequences. J Neurosci (2007) 27(42):11214–27. doi: 10.1523/JNEUROSCI.3588-07.2007
34. Banerjee HN, Hyman G, Evans S, Manglik V, Gwebu E, Banerjee A, et al. Identification of the Transmembrane Glucose Regulated Protein 78 as a Biomarker for the Brain Cancer Glioblastoma Multiforme by Gene Expression and Proteomic Studies. J Membr Sci Technol (2014) 4(1):1000126. doi: 10.4172/2155-9589.1000126
35. Penaranda Fajardo NM, Meijer C, Kruyt FA. The endoplasmic reticulum stress/unfolded protein response in gliomagenesis, tumor progression and as a therapeutic target in glioblastoma. Biochem Pharmacol (2016) 118:1–8. doi: 10.1016/j.bcp.2016.04.008
36. Ramao A, Gimenez M, Laure HJ, Izumi C, Vida RC, Oba-Shinjo S, et al. Changes in the expression of proteins associated with aerobic glycolysis and cell migration are involved in tumorigenic ability of two glioma cell lines. Proteome Sci (2012) 10(1):53. doi: 10.1186/1477-5956-10-53
37. Stensjoen AL, Solheim O, Kvistad KA, Haberg AK, Salvesen O, Berntsen EM. Growth dynamics of untreated glioblastomas in vivo. Neuro Oncol (2015) 17(10):1402–11. doi: 10.1093/neuonc/nov029
38. Shah SS, Rodriguez GA, Musick A, Walters WM, de Cordoba N, Barbarite E, et al. Targeting Glioblastoma Stem Cells with 2-Deoxy-D-Glucose (2-DG) Potentiates Radiation-Induced Unfolded Protein Response (UPR). Cancers (Basel) (2019) 11(2):159. doi: 10.3390/cancers11020159
39. Gimenez M, Marie SK, Oba-Shinjo SM, Uno M, da Silva R, Laure HJ, et al. Quantitative proteomic analysis and functional studies reveal that nucleophosmin is involved in cell death in glioblastoma cell line transfected with siRNA. Proteomics (2012) 12(17):2632–40. doi: 10.1002/pmic.201200034
40. Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol (2010) 5:99–118. doi: 10.1146/annurev-pathol-121808-102144
41. Pavlyukov MS, Yu H, Bastola S, Minata M, Shender VO, Lee Y, et al. Apoptotic Cell-Derived Extracellular Vesicles Promote Malignancy of Glioblastoma Via Intercellular Transfer of Splicing Factors. Cancer Cell (2018) 34(1):119–35.e10. doi: 10.1016/j.ccell.2018.05.012
42. Misra UK, Deedwania R, Pizzo SV. Binding of activated alpha2-macroglobulin to its cell surface receptor GRP78 in 1-LN prostate cancer cells regulates PAK-2-dependent activation of LIMK. J Biol Chem (2005) 280(28):26278–86. doi: 10.1074/jbc.M414467200
43. Chen HY, Chang JT, Chien KY, Lee YS, You GR, Cheng AJ. The Endogenous GRP78 Interactome in Human Head and Neck Cancers: A Deterministic Role of Cell Surface GRP78 in Cancer Stemness. Sci Rep (2018) 8(1):536. doi: 10.1038/s41598-017-14604-5
44. Miharada K, Karlsson G, Rehn M, Rorby E, Siva K, Cammenga J, et al. Cripto regulates hematopoietic stem cells as a hypoxic-niche-related factor through cell surface receptor GRP78. Cell Stem Cell (2011) 9(4):330–44. doi: 10.1016/j.stem.2011.07.016
45. Dadey DYA, Kapoor V, Hoye K, Khudanyan A, Collins A, Thotala D, et al. Antibody Targeting GRP78 Enhances the Efficacy of Radiation Therapy in Human Glioblastoma and Non-Small Cell Lung Cancer Cell Lines and Tumor Models. Clin Cancer Res (2017) 23(10):2556–64. doi: 10.1158/1078-0432.CCR-16-1935
46. Li X, Wu C, Chen N, Gu H, Yen A, Cao L, et al. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget (2016) 7(22):33440–50. doi: 10.18632/oncotarget.7961
47. Dejeans N, Pluquet O, Lhomond S, Grise F, Bouchecareilh M, Juin A, et al. Autocrine control of glioma cells adhesion and migration through IRE1alpha-mediated cleavage of SPARC mRNA. J Cell Sci (2012) 125(Pt 18):4278–87. doi: 10.1242/jcs.099291
48. Auf G, Jabouille A, Delugin M, Guerit S, Pineau R, North S, et al. High epiregulin expression in human U87 glioma cells relies on IRE1alpha and promotes autocrine growth through EGF receptor. BMC Cancer (2013) 13:597. doi: 10.1186/1471-2407-13-597
49. Chevet E, Hetz C, Samali A. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov (2015) 5(6):586–97. doi: 10.1158/2159-8290.CD-14-1490
50. Jabouille A, Delugin M, Pineau R, Dubrac A, Soulet F, Lhomond S, et al. Glioblastoma invasion and cooption depend on IRE1alpha endoribonuclease activity. Oncotarget (2015) 6(28):24922–34. doi: 10.18632/oncotarget.4679
51. Madden E, Logue SE, Healy SJ, Manie S, Samali A. The role of the unfolded protein response in cancer progression: From oncogenesis to chemoresistance. Biol Cell (2019) 111(1):1–17. doi: 10.1111/boc.201800050
52. Hou X, Liu Y, Liu H, Chen X, Liu M, Che H, et al. PERK silence inhibits glioma cell growth under low glucose stress by blockage of p-AKT and subsequent HK2’s mitochondria translocation. Sci Rep (2015) 5:9065. doi: 10.1038/srep09065
53. Hamed HA, Yacoub A, Park MA, Eulitt P, Sarkar D, Dimitrie IP, et al. OSU-03012 enhances Ad.7-induced GBM cell killing via ER stress and autophagy and by decreasing expression of mitochondrial protective proteins. Cancer Biol Ther (2010) 9(7):526–36. doi: 10.4161/cbt.9.7.11116
54. Penaranda-Fajardo NM, Meijer C, Liang Y, Dijkstra BM, Aguirre-Gamboa R, den Dunnen WFA, et al. ER stress and UPR activation in glioblastoma: identification of a noncanonical PERK mechanism regulating GBM stem cells through SOX2 modulation. Cell Death Dis (2019) 10(10):690. doi: 10.1038/s41419-019-1934-1
55. Wang M, Wey S, Zhang Y, Ye R, Lee AS. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid Redox Signal (2009) 11(9):2307–16. doi: 10.1089/ars.2009.2485
56. Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep (2006) 7(9):880–5. doi: 10.1038/sj.embor.7400779
57. Urra H, Dufey E, Lisbona F, Rojas-Rivera D, Hetz C. When ER stress reaches a dead end. Biochim Biophys Acta (2013) 1833(12):3507–17. doi: 10.1016/j.bbamcr.2013.07.024
58. Chen TC, Wang W, Golden EB, Thomas S, Sivakumar W, Hofman FM, et al. Green tea epigallocatechin gallate enhances therapeutic efficacy of temozolomide in orthotopic mouse glioblastoma models. Cancer Lett (2011) 302(2):100–8. doi: 10.1016/j.canlet.2010.11.008
59. Gargiulo G, Cesaroni M, Serresi M, de Vries N, Hulsman D, Bruggeman SW, et al. In vivo RNAi screen for BMI1 targets identifies TGF-beta/BMP-ER stress pathways as key regulators of neural- and malignant glioma-stem cell homeostasis. Cancer Cell (2013) 23(5):660–76. doi: 10.1016/j.ccr.2013.03.030
60. Jiang HY, Wek SA, McGrath BC, Lu D, Hai T, Harding HP, et al. Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol Cell Biol (2004) 24(3):1365–77. doi: 10.1128/MCB.24.3.1365-1377.2004
61. Hu T, Xie N, Qin C, Wang J, You Y. Glucose-regulated protein 94 is a novel glioma biomarker and promotes the aggressiveness of glioma via Wnt/beta-catenin signaling pathway. Tumour Biol (2015) 36(12):9357–64. doi: 10.1007/s13277-015-3635-4
62. Zhang LH, Zhang X. Roles of GRP78 in physiology and cancer. J Cell Biochem (2010) 110(6):1299–305. doi: 10.1002/jcb.22679
63. Virrey JJ, Dong D, Stiles C, Patterson JB, Pen L, Ni M, et al. Stress chaperone GRP78/BiP confers chemoresistance to tumor-associated endothelial cells. Mol Cancer Res (2008) 6(8):1268–75. doi: 10.1158/1541-7786.MCR-08-0060
64. Dong D, Stapleton C, Luo B, Xiong S, Ye W, Zhang Y, et al. A critical role for GRP78/BiP in the tumor microenvironment for neovascularization during tumor growth and metastasis. Cancer Res (2011) 71(8):2848–57. doi: 10.1158/0008-5472.CAN-10-3151
65. Gurusinghe K, Mishra A, Mishra S. Glucose-regulated protein 78 substrate-binding domain alters its conformation upon EGCG inhibitor binding to nucleotide-binding domain: Molecular dynamics studies. Sci Rep (2018) 8(1):5487. doi: 10.1038/s41598-018-22905-6
66. Martin S, Lamb HK, Brady C, Lefkove B, Bonner MY, Thompson P, et al. Inducing apoptosis of cancer cells using small-molecule plant compounds that bind to GRP78. Br J Cancer (2013) 109(2):433–43. doi: 10.1038/bjc.2013.325
67. Chio CC, Tai YT, Mohanraj M, Liu SH, Yang ST, Chen RM. Honokiol enhances temozolomide-induced apoptotic insults to malignant glioma cells via an intrinsic mitochondrion-dependent pathway. Phytomedicine (2018) 49:41–51. doi: 10.1016/j.phymed.2018.06.012
68. Chio CC, Chen KY, Chang CK, Chuang JY, Liu CC, Liu SH, et al. Improved effects of honokiol on temozolomide-induced autophagy and apoptosis of drug-sensitive and -tolerant glioma cells. BMC Cancer (2018) 18(1):379. doi: 10.1186/s12885-018-4267-z
69. Booth L, Cazanave SC, Hamed HA, Yacoub A, Ogretmen B, Chen CS, et al. OSU-03012 suppresses GRP78/BiP expression that causes PERK-dependent increases in tumor cell killing. Cancer Biol Ther (2012) 13(4):224–36. doi: 10.4161/cbt.13.4.18877
70. Kardosh A, Golden EB, Pyrko P, Uddin J, Hofman FM, Chen TC, et al. Aggravated endoplasmic reticulum stress as a basis for enhanced glioblastoma cell killing by bortezomib in combination with celecoxib or its non-coxib analogue, 2,5-dimethyl-celecoxib. Cancer Res (2008) 68(3):843–51. doi: 10.1158/0008-5472.CAN-07-5555
71. Cho HY, Wang W, Jhaveri N, Torres S, Tseng J, Leong MN, et al. Perillyl alcohol for the treatment of temozolomide-resistant gliomas. Mol Cancer Ther (2012) 11(11):2462–72. doi: 10.1158/1535-7163.MCT-12-0321
72. Da Fonseca CO, Masini M, Futuro D, Caetano R, Gattass CR, Quirico-Santos T. Anaplastic oligodendroglioma responding favorably to intranasal delivery of perillyl alcohol: a case report and literature review. Surg Neurol (2006) 66(6):611–5. doi: 10.1016/j.surneu.2006.02.034
73. Marin-Ramos NI, Perez-Hernandez M, Tam A, Swenson SD, Cho HY, Thein TZ, et al. Inhibition of motility by NEO100 through the calpain-1/RhoA pathway. J Neurosurg (2019) 133(4):1–12. doi: 10.3171/2019.5.JNS19798
74. Cerezo M, Lehraiki A, Millet A, Rouaud F, Plaisant M, Jaune E, et al. Compounds Triggering ER Stress Exert Anti-Melanoma Effects and Overcome BRAF Inhibitor Resistance. Cancer Cell (2016) 29(6):805–19. doi: 10.1016/j.ccell.2016.04.013
75. Pinkham K, Park DJ, Hashemiaghdam A, Kirov AB, Adam I, Rosiak K, et al. Stearoyl CoA Desaturase Is Essential for Regulation of Endoplasmic Reticulum Homeostasis and Tumor Growth in Glioblastoma Cancer Stem Cells. Stem Cell Rep (2019) 12(4):712–27. doi: 10.1016/j.stemcr.2019.02.012
76. Bakewell SJ, Rangel DF, Ha DP, Sethuraman J, Crouse R, Hadley E, et al. Suppression of stress induction of the 78-kilodalton glucose regulated protein (GRP78) in cancer by IT-139, an anti-tumor ruthenium small molecule inhibitor. Oncotarget (2018) 9(51):29698–714. doi: 10.18632/oncotarget.25679
77. Prabhu A, Sarcar B, Kahali S, Shan Y, Chinnaiyan P. Targeting the unfolded protein response in glioblastoma cells with the fusion protein EGF-SubA. PLoS One (2012) 7(12):e52265. doi: 10.1371/journal.pone.0052265
78. Kia A, Przystal JM, Nianiaris N, Mazarakis ND, Mintz PJ, Hajitou A. Dual systemic tumor targeting with ligand-directed phage and Grp78 promoter induces tumor regression. Mol Cancer Ther (2012) 11(12):2566–77. doi: 10.1158/1535-7163.MCT-12-0587
79. Przystal JM, Waramit S, Pranjol MZI, Yan W, Chu G, Chongchai A, et al. Efficacy of systemic temozolomide-activated phage-targeted gene therapy in human glioblastoma. EMBO Mol Med (2019) 11(4):e8492. doi: 10.15252/emmm.201708492
80. Kaliberov SA, Kaliberova LN, Yan H, Kapoor V, Hallahan DE. Retargeted adenoviruses for radiation-guided gene delivery. Cancer Gene Ther (2016) 23(9):303–14. doi: 10.1038/cgt.2016.32
81. Bhattacharjee R, Devi A, Mishra S. Molecular docking and molecular dynamics studies reveal structural basis of inhibition and selectivity of inhibitors EGCG and OSU-03012 toward glucose regulated protein-78 (GRP78) overexpressed in glioblastoma. J Mol Model (2015) 21(10):272. doi: 10.1007/s00894-015-2801-3
82. Virrey JJ, Liu Z, Cho HY, Kardosh A, Golden EB, Louie SG, et al. Antiangiogenic activities of 2,5-dimethyl-celecoxib on the tumor vasculature. Mol Cancer Ther (2010) 9(3):631–41. doi: 10.1158/1535-7163.MCT-09-0652
83. Penas-Prado M, Hess KR, Fisch MJ, Lagrone LW, Groves MD, Levin VA, et al. Randomized phase II adjuvant factorial study of dose-dense temozolomide alone and in combination with isotretinoin, celecoxib, and/or thalidomide for glioblastoma. Neuro Oncol (2015) 17(2):266–73. doi: 10.1093/neuonc/nou155
84. Loureiro JA, Gomes B, Coelho MA, do Carmo Pereira M, Rocha S. Targeting nanoparticles across the blood-brain barrier with monoclonal antibodies. Nanomed (Lond) (2014) 9(5):709–22. doi: 10.2217/nnm.14.27
85. Ran D, Zhou J, Chai Z, Li J, Xie C, Mao J, et al. All-stage precisional glioma targeted therapy enabled by a well-designed D-peptide. Theranostics (2020) 10(9):4073–87. doi: 10.7150/thno.41382
86. Huynh TT, Lin CM, Lee WH, Wu AT, Lin YK, Lin YF, et al. Pterostilbene suppressed irradiation-resistant glioma stem cells by modulating GRP78/miR-205 axis. J Nutr Biochem (2015) 26(5):466–75. doi: 10.1016/j.jnutbio.2014.11.015
87. Li ZY, Zhang C, Zhang Y, Chen L, Chen BD, Li QZ, et al. A novel HDAC6 inhibitor Tubastatin A: Controls HDAC6-p97/VCP-mediated ubiquitination-autophagy turnover and reverses Temozolomide-induced ER stress-tolerance in GBM cells. Cancer Lett (2017) 391:89–99. doi: 10.1016/j.canlet.2017.01.025
88. McFarland BC, Stewart J Jr., Hamza A, Nordal R, Davidson DJ, Henkin J, et al. Plasminogen kringle 5 induces apoptosis of brain microvessel endothelial cells: sensitization by radiation and requirement for GRP78 and LRP1. Cancer Res (2009) 69(13):5537–45. doi: 10.1158/0008-5472.CAN-08-4841
89. Golden EB, Cho HY, Jahanian A, Hofman FM, Louie SG, Schonthal AH, et al. Chloroquine enhances temozolomide cytotoxicity in malignant gliomas by blocking autophagy. Neurosurg Focus (2014) 37(6):E12. doi: 10.3171/2014.9.FOCUS14504
90. Siegelin MD. Utilization of the cellular stress response to sensitize cancer cells to TRAIL-mediated apoptosis. Expert Opin Ther Targets (2012) 16(8):801–17. doi: 10.1517/14728222.2012.703655
91. Li Z, Ma J, Liu L, Liu X, Wang P, Liu Y, et al. Endothelial-Monocyte Activating Polypeptide II Suppresses the In Vitro Glioblastoma-Induced Angiogenesis by Inducing Autophagy. Front Mol Neurosci (2017) 10:208. doi: 10.3389/fnmol.2017.00208
92. Kavitha CV, Jain AK, Agarwal C, Pierce A, Keating A, Huber KM, et al. Asiatic acid induces endoplasmic reticulum stress and apoptotic death in glioblastoma multiforme cells both in vitro and in vivo. Mol Carcinog (2015) 54(11):1417–29. doi: 10.1002/mc.22220
93. Wang D, Chen Q, Liu B, Li Y, Tan Y, Yang B. Ellagic acid inhibits proliferation and induces apoptosis in human glioblastoma cells. Acta Cir Bras (2016) 31(2):143–9. doi: 10.1590/S0102-865020160020000010
94. Bobak Y, Kurlishchuk Y, Vynnytska-Myronovska B, Grydzuk O, Shuvayeva G, Redowicz MJ, et al. Arginine deprivation induces endoplasmic reticulum stress in human solid cancer cells. Int J Biochem Cell Biol (2016) 70:29–38. doi: 10.1016/j.biocel.2015.10.027
95. Hackler L Jr., Ozsvari B, Gyuris M, Sipos P, Fabian G, Molnar E, et al. The Curcumin Analog C-150, Influencing NF-kappaB, UPR and Akt/Notch Pathways Has Potent Anticancer Activity In Vitro and In Vivo. PLoS One (2016) 11(3):e0149832. doi: 10.1371/journal.pone.0149832
96. Ishida CT, Shu C, Halatsch ME, Westhoff MA, Altieri DC, Karpel-Massler G, et al. Mitochondrial matrix chaperone and c-myc inhibition causes enhanced lethality in glioblastoma. Oncotarget (2017) 8(23):37140–53. doi: 10.18632/oncotarget.16202
97. Qu C, Ma J, Liu X, Xue Y, Zheng J, Liu L, et al. Dihydroartemisinin Exerts Anti-Tumor Activity by Inducing Mitochondrion and Endoplasmic Reticulum Apoptosis and Autophagic Cell Death in Human Glioblastoma Cells. Front Cell Neurosci (2017) 11:310. doi: 10.3389/fncel.2017.00310
98. Sareddy GR, Viswanadhapalli S, Surapaneni P, Suzuki T, Brenner A, Vadlamudi RK. Novel KDM1A inhibitors induce differentiation and apoptosis of glioma stem cells via unfolded protein response pathway. Oncogene (2017) 36(17):2423–34. doi: 10.1038/onc.2016.395
99. Tang Q, Ren L, Liu J, Li W, Zheng X, Wang J, et al. Withaferin A triggers G2/M arrest and intrinsic apoptosis in glioblastoma cells via ATF4-ATF3-CHOP axis. Cell Prolif (2020) 53(1):e12706. doi: 10.1111/cpr.12706
100. Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA (2017) 318(23):2306–16. doi: 10.1001/jama.2017.18718
101. Shteingauz A, Porat Y, Voloshin T, Schneiderman RS, Munster M, Zeevi E, et al. AMPK-dependent autophagy upregulation serves as a survival mechanism in response to Tumor Treating Fields (TTFields). Cell Death Dis (2018) 9(11):1074. doi: 10.1038/s41419-018-1085-9
102. Meng X, Leyva ML, Jenny M, Gross I, Benosman S, Fricker B, et al. A ruthenium-containing organometallic compound reduces tumor growth through induction of the endoplasmic reticulum stress gene CHOP. Cancer Res (2009) 69(13):5458–66. doi: 10.1158/0008-5472.CAN-08-4408
103. Koncarevic S, Urig S, Steiner K, Rahlfs S, Herold-Mende C, Sueltmann H, et al. Differential genomic and proteomic profiling of glioblastoma cells exposed to terpyridineplatinum(II) complexes. Free Radic Biol Med (2009) 46(8):1096–108. doi: 10.1016/j.freeradbiomed.2009.01.013
104. Jalota A, Kumar M, Das BC, Yadav AK, Chosdol K, Sinha S. Synergistic increase in efficacy of a combination of 2-deoxy-D-glucose and cisplatin in normoxia and hypoxia: switch from autophagy to apoptosis. Tumour Biol (2016) 37(9):12347–58. doi: 10.1007/s13277-016-5089-8
105. Noack J, Choi J, Richter K, Kopp-Schneider A, Regnier-Vigouroux A. A sphingosine kinase inhibitor combined with temozolomide induces glioblastoma cell death through accumulation of dihydrosphingosine and dihydroceramide, endoplasmic reticulum stress and autophagy. Cell Death Dis (2014) 5:e1425. doi: 10.1038/cddis.2014.384
106. Jia W, Loria RM, Park MA, Yacoub A, Dent P, Graf MR. The neuro-steroid, 5-androstene 3beta,17alpha diol; induces endoplasmic reticulum stress and autophagy through PERK/eIF2alpha signaling in malignant glioma cells and transformed fibroblasts. Int J Biochem Cell Biol (2010) 42(12):2019–29. doi: 10.1016/j.biocel.2010.09.003
107. Chen TC, Lai KC, Yang JS, Liao CL, Hsia TC, Chen GW, et al. Involvement of reactive oxygen species and caspase-dependent pathway in berberine-induced cell cycle arrest and apoptosis in C6 rat glioma cells. Int J Oncol (2009) 34(6):1681–90. doi: 10.3892/ijo_00000299
108. Eom KS, Kim HJ, So HS, Park R, Kim TY. Berberine-induced apoptosis in human glioblastoma T98G cells is mediated by endoplasmic reticulum stress accompanying reactive oxygen species and mitochondrial dysfunction. Biol Pharm Bull (2010) 33(10):1644–9. doi: 10.1248/bpb.33.1644
109. Shen S, Zhang Y, Wang Z, Liu R, Gong X. Bufalin induces the interplay between apoptosis and autophagy in glioma cells through endoplasmic reticulum stress. Int J Biol Sci (2014) 10(2):212–24. doi: 10.7150/ijbs.8056
110. Qian Y, Zheng Y, Abraham L, Ramos KS, Tiffany-Castiglioni E. Differential profiles of copper-induced ROS generation in human neuroblastoma and astrocytoma cells. Brain Res Mol Brain Res (2005) 134(2):323–32. doi: 10.1016/j.molbrainres.2004.11.004
111. Hwang MS, Baek WK. Glucosamine induces autophagic cell death through the stimulation of ER stress in human glioma cancer cells. Biochem Biophys Res Commun (2010) 399(1):111–6. doi: 10.1016/j.bbrc.2010.07.050
112. Tian X, Ye J, Alonso-Basanta M, Hahn SM, Koumenis C, Dorsey JF. Modulation of CCAAT/enhancer binding protein homologous protein (CHOP)-dependent DR5 expression by nelfinavir sensitizes glioblastoma multiforme cells to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). J Biol Chem (2011) 286(33):29408–16. doi: 10.1074/jbc.M110.197665
113. Liu WT, Huang CY, Lu IC, Gean PW. Inhibition of glioma growth by minocycline is mediated through endoplasmic reticulum stress-induced apoptosis and autophagic cell death. Neuro Oncol (2013) 15(9):1127–41. doi: 10.1093/neuonc/not073
114. Chou YC, Chang MY, Wang MJ, Harnod T, Hung CH, Lee HT, et al. PEITC induces apoptosis of Human Brain Glioblastoma GBM8401 Cells through the extrinsic- and intrinsic -signaling pathways. Neurochem Int (2015) 81:32–40. doi: 10.1016/j.neuint.2015.01.001
115. Chou YC, Chang MY, Wang MJ, Liu HC, Chang SJ, Harnod T, et al. Phenethyl isothiocyanate alters the gene expression and the levels of protein associated with cell cycle regulation in human glioblastoma GBM 8401 cells. Environ Toxicol (2017) 32(1):176–87. doi: 10.1002/tox.22224
116. Chou YC, Chang MY, Wang MJ, Yu FS, Liu HC, Harnod T, et al. PEITC inhibits human brain glioblastoma GBM 8401 cell migration and invasion through the inhibition of uPA, Rho A, and Ras with inhibition of MMP-2, -7 and -9 gene expression. Oncol Rep (2015) 34(5):2489–96. doi: 10.3892/or.2015.4260
117. Lu DY, Chang CS, Yeh WL, Tang CH, Cheung CW, Leung YM, et al. The novel phloroglucinol derivative BFP induces apoptosis of glioma cancer through reactive oxygen species and endoplasmic reticulum stress pathways. Phytomedicine (2012) 19(12):1093–100. doi: 10.1016/j.phymed.2012.06.010
118. Zhong JT, Xu Y, Yi HW, Su J, Yu HM, Xiang XY, et al. The BH3 mimetic S1 induces autophagy through ER stress and disruption of Bcl-2/Beclin 1 interaction in human glioma U251 cells. Cancer Lett (2012) 323(2):180–7. doi: 10.1016/j.canlet.2012.04.009
119. Kuder CH, Sheehy RM, Neighbors JD, Wiemer DF, Hohl RJ. Functional evaluation of a fluorescent schweinfurthin: mechanism of cytotoxicity and intracellular quantification. Mol Pharmacol (2012) 82(1):9–16. doi: 10.1124/mol.111.077107
120. White MC, Johnson GG, Zhang W, Hobrath JV, Piazza GA, Grimaldi M. Sulindac sulfide inhibits sarcoendoplasmic reticulum Ca2+ ATPase, induces endoplasmic reticulum stress response, and exerts toxicity in glioma cells: relevant similarities to and important differences from celecoxib. J Neurosci Res (2013) 91(3):393–406. doi: 10.1002/jnr.23169
121. Antal O, Hackler L Jr., Shen J, Man I, Hideghety K, Kitajka K, et al. Combination of unsaturated fatty acids and ionizing radiation on human glioma cells: cellular, biochemical and gene expression analysis. Lipids Health Dis (2014) 13:142. doi: 10.1186/1476-511X-13-142
122. Bown CD, Wang JF, Young LT. Increased expression of endoplasmic reticulum stress proteins following chronic valproate treatment of rat C6 glioma cells. Neuropharmacology (2000) 39(11):2162–9. doi: 10.1016/S0028-3908(00)00029-0
123. Cattaneo M, Baronchelli S, Schiffer D, Mellai M, Caldera V, Saccani GJ, et al. Down-modulation of SEL1L, an unfolded protein response and endoplasmic reticulum-associated degradation protein, sensitizes glioma stem cells to the cytotoxic effect of valproic acid. J Biol Chem (2014) 289(5):2826–38. doi: 10.1074/jbc.M113.527754
124. Tsai CF, Yeh WL, Huang SM, Tan TW, Lu DY. Wogonin induces reactive oxygen species production and cell apoptosis in human glioma cancer cells. Int J Mol Sci (2012) 13(8):9877–92. doi: 10.3390/ijms13089877
125. Wang JF, Bown C, Young LT. Differential display PCR reveals novel targets for the mood-stabilizing drug valproate including the molecular chaperone GRP78. Mol Pharmacol (1999) 55(3):521–7.
126. Ciechomska IA, Gabrusiewicz K, Szczepankiewicz AA, Kaminska B. Endoplasmic reticulum stress triggers autophagy in malignant glioma cells undergoing cyclosporine a-induced cell death. Oncogene (2013) 32(12):1518–29. doi: 10.1038/onc.2012.174
127. Ciechomska IA, Kaminska B. ER stress and autophagy contribute to CsA-induced death of malignant glioma cells. Autophagy (2012) 8(10):1526–8. doi: 10.4161/auto.21155
128. Hsieh KP, Wilke N, Harris A, Miles MF. Interaction of ethanol with inducers of glucose-regulated stress proteins. Ethanol potentiates inducers of grp78 transcription. J Biol Chem (1996) 271(5):2709–16. doi: 10.1074/jbc.271.5.2709
129. Miles MF, Wilke N, Elliot M, Tanner W, Shah S. Ethanol-responsive genes in neural cells include the 78-kilodalton glucose-regulated protein (GRP78) and 94-kilodalton glucose-regulated protein (GRP94) molecular chaperones. Mol Pharmacol (1994) 46(5):873–9.
130. Qian Y, Falahatpisheh MH, Zheng Y, Ramos KS, Tiffany-Castiglioni E. Induction of 78 kD glucose-regulated protein (GRP78) expression and redox-regulated transcription factor activity by lead and mercury in C6 rat glioma cells. Neurotox Res (2001) 3(6):581–9. doi: 10.1007/BF03033212
131. Qian Y, Zheng Y, Ramos KS, Tiffany-Castiglioni E. GRP78 compartmentalized redistribution in Pb-treated glia: role of GRP78 in lead-induced oxidative stress. Neurotoxicology (2005) 26(2):267–75. doi: 10.1016/j.neuro.2004.09.002
132. Romero-Ramirez L, Garcia-Alvarez I, Casas J, Barreda-Manso MA, Yanguas-Casas N, Nieto-Sampedro M, et al. New oleyl glycoside as anti-cancer agent that targets on neutral sphingomyelinase. Biochem Pharmacol (2015) 97(2):158–72. doi: 10.1016/j.bcp.2015.07.009
Keywords: GBM—Glioblastoma multiforme, UPR—unfolded protein response, glioma, GRP94, ER stress, TMZ (Temozolomide), glucose regulated protein 78 (GRP78), GBM therapeutic target
Citation: Liu K, Tsung K and Attenello FJ (2020) Characterizing Cell Stress and GRP78 in Glioma to Enhance Tumor Treatment. Front. Oncol. 10:608911. doi: 10.3389/fonc.2020.608911
Received: 22 September 2020; Accepted: 11 November 2020;
Published: 11 December 2020.
Edited by:
Jose Ramon Pineda, University of the Basque Country, SpainReviewed by:
Jens Gempt, Technische Universität München, GermanyCopyright © 2020 Liu, Tsung and Attenello. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kristie Liu, a3FsaXVAdXNjLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.