 Hainan Li1
Hainan Li1 Changguo Shan1Shengnan Wu1Baijie Cheng1Chongzu Fan1Linbo Cai1
Changguo Shan1Shengnan Wu1Baijie Cheng1Chongzu Fan1Linbo Cai1 Yedan Chen2Yuqian Shi2Kaihua Liu2Yang Shao2Dan Zhu1*Zhi Li3*
Yedan Chen2Yuqian Shi2Kaihua Liu2Yang Shao2Dan Zhu1*Zhi Li3*- 1Department of Pathology, Guangdong Sanjiu Brain Hospital, Guangzhou, China
- 2Medical Department, Nanjing Geneseeq Technology Inc., Nanjing, China
- 3Department of Pathology, Guangdong Provincial People’s Hospital, Guangzhou, China
Background: Molecular characteristics are essential for the classification and grading of gliomas. However, diagnostic classification of midline glioma is still debatable and substantial molecular and clinical heterogeneity within each subgroup suggested that they should be further stratified. Here, we studied the mutation landscape of Chinese midline glioma patients in hope to provide new insights for glioma prognosis and treatment.
Methods: Tissue samples from 112 midline glioma patients underwent next-generation sequencing targeting 425 cancer-relevant genes. Gene mutations and copy number variations were investigated for their somatic interactions and prognostic effect using overall survival data. Pathway-based survival analysis was performed for ten canonical oncogenic pathways.
Results: We identified several currently established diagnostic and prognostic biomarkers of glioma, including TP53 (33%), EGFR (26%), TERT (24%), PTEN (21%), PIK3CA (14%), ATRX (14%), BRAF (13%), and IDH1/2 (6%). Among all genetic aberrations with more than 5% occurrence rate, six mutations and three copy number gains were greatly associated with poor overall survival (univariate, P < 0.1). Of these, TERT mutations (hazard ratio [HR], 3.00; 95% confidence interval [CI], 1.37–6.61; P = 0.01) and PIK3CA mutations (HR, 2.04; 95% CI, 1.08–3.84; P = 0.02) remained significant in multivariate analyses. Additionally, we have also identified a novel MCL1 amplification (found in 31% patients) as a potential independent biomarker for glioma (multivariate HR, 2.78; 95% CI, 1.53–5.08; P < 0.001), which was seldom reported in public databases. Pathway analyses revealed significantly worse prognosis with abnormal PI3K (HR, 1.81; 95% CI, 1.12–2.95; P = 0.01) and cell cycle pathways (HR, 1.97; 95% CI, 1.15–3.37; P = 0.01), both of which stayed meaningful after multivariate adjustment.
Conclusions: In this study, we discovered shorter survival in midline glioma patients with PIK3CA and TERT mutations and with abnormal PI3K and cell cycle pathways. We also revealed a novel prognostic marker, MCL1 amplification that collectively provided new insights and opportunities in understanding and treating midline gliomas.
Introduction
Midline glioma is not a single disease but contains multiple histological and molecular subtypes. As they are situated along the pons in the brainstem, thalamus, cerebellum, or spinal cord, which are vital for regulating basic life functions, treatment and management of these gliomas are extremely crucial (1). In the recent decade, growing evidence of molecular biology has revolutionized the classification of glioma, where molecular features make the judgment call for any discordancy between histological and molecular classifications (1–3). Based on two hallmarks of glioma, mutation of IDH1 and IDH2 genes, and codeletion of chromosome arms 1p/19q, glioma is primarily distinguished into five principle molecular subtypes. However, the classification of midline glioma remained controversial and the intertwining histology and molecular classifications have brought great clinical challenges. In the 2016 WHO Guidelines and the 2018 cIMPACT-NOW update, diffused midline glioma with H3 K27M-mutant was introduced as a separate Grade IV entity, predominantly describing an astrocytic differentiation of glioma of different age groups with adverse overall prognosis (1, 4). This reserved diagnostic group has left lacuna for grading H3 K27-wildtype diffuse midline gliomas, lower-grade midline-crossing tumors, and midline gliomas of other histological and morphological categories, including pilocytic astrocytoma and ganglioglioma with various survival outcomes (5–8). Meanwhile, substantial molecular heterogeneity and non-universal prognosis within each subgroup also implied the necessity of further stratification (9–11).
H3 K27M is found in approximately 80% of pediatric diffuse intrinsic pontine gliomas (DIPG), as well as some adult midline glioma and pediatric ependymoma at a lower occurrence (9, 12–17). Molecular analyses usually found mutually exclusive distribution of H3 K27M with the characteristic IDH mutations. In addition to H3 K27M, ATRX, TP53, NF1, FGFR1, PDGFRA, PTPN11, and BRAF alterations have been found in various pediatric and adult midline glioma subtypes, while 1p/19q co-deletion were seldom reported (8, 12, 14, 18, 19). Many of these frequently observed genes are key members of PI3K/mTOR and RAS-MAPK pathways that could potentially provide novel therapeutic options for these otherwise devastating diseases (20–24). Nevertheless, the prognostic significance of genetic aberrations and oncogenic pathways was only weakly addressed for midline glioma with or without H3 K27M mutation (20) and comprehensive molecular characterization is still lacking.
In this study, we aim to explore the genomic makeup of midline glioma patients and to fill the gaps in the prognostic heterogeneity through comprehensive genetic and oncogenic pathways analyses. As mainstay treatment for tumors in the central nervous system remained to be surgery resection, radiotherapy, and chemotherapy, we also hope to provide fresh insights into future molecular stratification in midline glioma.
Methods
Patients and Tumor Specimen
Archived formalin-fixed and paraffin-embedded (FFPE) blocks of tumor tissues from 112 glioma patients were obtained from the Department of Pathology at Guangdong Sanjiu Brain Hospital. Clinical characteristics and overall survival data of patients were evaluated and obtained from March 2012 to September 2019. The study was approved by the research ethics board at the hospital. All patients provided written informed consent for participating in the study.
DNA Extraction and Sequencing Library Preparation
FFPE sections of tumor samples were sent to the CLIA/CAP-accredited central laboratory at Nanjing Geneseeq Technology Inc. (China, Nanjing) for genomic DNA extraction and hybridization capture-based targeted NGS of 425 cancer-relevant genes. Briefly, five to eight 10 µm FFPE sections were first de-paraffinized with xylene and then used for genomic DNA (gDNA) extraction by QIAamp DNA FFPE Tissue Kit (Qiagen) following the manufacturer’s instructions. The extracted gDNA samples were quantified on Qubit 3.0 fluorometer (Thermo Fisher Scientific) and its purity was measured on Nanodrop 2000 (Thermo Fisher Scientific). Purified gDNA was fragmented to a size of approximately 350 bp using a Covaris M220 sonication system (Covaris) and then purified by size selection with Agencourt AMPure XP beads (Beckman Coulter).
DNA libraries were prepared from purified gDNA with KAPA hyper library preparation kit (KAPA Biosystems) according to the manufacturer’s protocol. Libraries were then subjected to PCR amplification and purification with Agencourt AMPure XP beads before targeted enrichment.
Libraries with different sample indices were first pooled together to a total DNA amount of 2 µg and then enriched with IDT xGen Lockdown Reagents and a customized enrichment panel (Integrated DNA Technologies). The captured library was further amplified with Illumina p5 (5’ AAT GAT ACG GCG ACC ACC GA 3’) and p7 (5’ CAA GCA GAA GAC GGC ATA CGA GAT 3’) primers in KAPA Hifi HotStart ReadyMix (KAPA Biosystems, Wilmington, MA) and purified with Agencourt AMPure XP beads. Sequencing libraries were quantified by qPCR with KAPA Library Quantification kit (KAPA Biosystems), and its size distribution was examined on Bioanalyzer 2100 (Agilent Technologies). The final libraries were sequenced on Illumina Hiseq 4000 platform for 150 bp paired-end sequencing according to the manufacturer’s instructions.
Variant Filtering and Mutation Calling
Raw sequencing data were analyzed by a validated automation pipeline. In brief, bcl2fastq was used to demultiplex raw data and trimmomatic was used to trim adapters and remove low quality reads (quality reading below 20) or N bases from FASTQ files. Burrows-Wheeler Aligner (BWA) was then used to align clean paired-end reads to the reference human genome (hs37d5). PCR deduplication was performed using Picard and indel realignment and base quality score recalibration was performed using Genome Analysis Toolkit (GATK 3.4.0) (25). Cross sample contamination was estimated using ContEst (Broad Institute) by evaluating the likelihood of detecting alternate alleles of SNPs reported in the 1000g database. The resulted mutation lists were filtered through an internally collected list of recurrent sequencing errors on the same sequencing platform. Somatic SNV and insertion/deletions (INDELs) were called using Vardict (V 1.5.4). SNVs and INDELs were further filtered using previously reported criteria. 1) for mutations with more than 20 recurrences in COSMIC, minimum variant allele frequency (VAF) = 0.01 with at least 3 minimum variant supporting reads; 2) for others, minimum VAF = 0.02 with at least 5 minimum variant supporting reads; in addition, all variants also need to meet the standards of minimum read depth = 20, minimum base quality = 25, variant supporting reads mapped to both strands, and strand bias no greater than 10%. Final mutations were annotated using vcf2maf. H3K27M mutants were detected with high specificity and sensitivity via immunohistochemistry using H3K27M-specific antibodies (ABE419, EMD Millipore, Billerica, MA, 1:1,000 dilution). The H3 K27M immunostaining was run in a Ventana BenchMark XT immunostainer (Ventana Medical Systems, Tucson, AZ, USA) as previously reported (26–28).
Copy-Number Variants Analysis
Copy number (CN) analysis was performed using FACETS (Ver 0.5.13). Somatic CN alteration events were assigned based on sample-ploidy values calculated in the FACETS algorithm (29). Chromosome arm-level CN gain (>sample average ploidy +1) was defined if segments of amplification and deep amplification events account for more than 60% of total segments for the corresponding chromosome arm. Similarly, arm-level CN loss (<sample average ploidy -1) was identified if segments of deletion and deep deletion events account for more than 60% of total segments for the given chromosome.
Pathway Analysis and Independent Validation Dataset
For pathway analysis, sets of genetic aberrations were included according to the ten previously defined canonical oncogenic signaling pathways (30). Patients harboring one or more mutated pathway member genes were considered to be positive. To validate results from pathway analysis, sequencing data from a Memorial Sloan Kettering Cancer Center cohort that contains 923 glioma patients were obtained from cbioportal (31) and the same gene inclusion criteria were applied to assign patients. MCL1 gene expression data of 510 low-grade glioma patients were downloaded from OncoLnc.org (http://www.oncolnc.org/) (32), and survival rates were compared between high and low expression groups divided by the median RNA expression level.
Statistical Analysis
Overall survival was defined as the time between surgery and death or last follow-up time. The association of clinical features and biomarkers with overall survival was assessed using Cox proportional hazard model. Multivariate analyses included genetic biomarkers and clinical factors with univariate P ≤ 0.1. Kaplan-Meier estimates of overall survival were illustrated and statistically compared using the log-rank test. Two-sided P < 0.5 was considered significant. All statistical analyses were done in R (3.5.0).
Results
Patient Characteristics and Clinical Factors
A total of 112 glioma patients were enrolled in this retrospective study, with a median age of 31 (ranging from 1 to 71 years old) and comprising 61% of male patients (Table S1). Histologically, 76 were diagnosed with diffused glioma (68%), 15 with neuronal and mixed neuronal-glial tumors (13%), and nine with ependymoma (8%). All tumors were in the midline location, including the brainstem, thalamus, hypothalamus, basal ganglia, lateral ventricle, fourth ventricle, and the pineal gland. According to the WHO grading scheme (1), 11 of these patients were classified as WHO grade I glioma (10%), 45 as grade II glioma (40%), 27, and 29 patients with grade III and grade IV gliomas, respectively. All patients underwent surgical resection, in which more than half experienced gross total resection. The majority of patients (76%) received postoperative radiotherapy, and 60% of all patients received adjuvant chemotherapy. Median overall survival among this cohort was 17.1 months with a median follow-up of 20.5 months. Of all clinical factors, WHO grading, the extent of surgical resection and whether radiotherapy was administered were greatly associated with survival and were included in subsequent multivariate analyses (Table S1).
Identification of Prognostic Biomarkers
In this cohort, H3 K27M was observed in 32 patients (29%), majority of which were diffuse glioma located at the brainstem (12/32) or thalamus (11/32). Although the majority of H3 K27M-mutant tumors were diffuse glioma, we also found 1 neural-glial tumor and a few of undefined histological subtypes, with 14 pediatric and adolescent patients and 18 adults. The core glioma biomarkers, IDH1/2 were found at a lower prevalence (8/112, 7%) and the rest 104 patients had IDH wild-type tumors, while no 1p/19q deletions were identified. Other previously reported midline glioma biomarkers were observed at various frequencies, including TP53 (35/112,31%), TERT promoter mutations (27/112, 24%), PTEN (23/112, 21%), PDGFRA (18/112, 16%; recurrent point mutations at E227K, N659K, A810T, D842V/Y, and CN gain), ATRX (16/112, 14%), PIK3CA (16/112, 14%; at R81Q, R93W, R108H, E542K, E545K, H1047R, H1065Y), BRAF (14/112, 12%; primarily at V600E), NF1 (14/112, 12%; mainly frameshift and nonsense mutations) and FGFR1 (9/112, 8%; at G348E, N546K, V561L/M, and K656E) (Figure 1A, Figure S1, and Table S2). Of all the top altered genes in this cohort, we also noticed a high prevalence of MCL1 CN gain (35/112, 31%). Next, we examined somatic interactions among these molecular events. H3 K37M was often accompanied by mutations of ATRX and TP53, and CN gain of KIT, while being mutually exclusive with EGFR and TERT. In contrast, TERT was frequently found concurrently with PTEN mutations, gain of MDM4, and alterations of EGFR, but not with ATRX as expected. Gain of MCL1 was found to co-occurr with gain of STMN1 (Figure 1B).
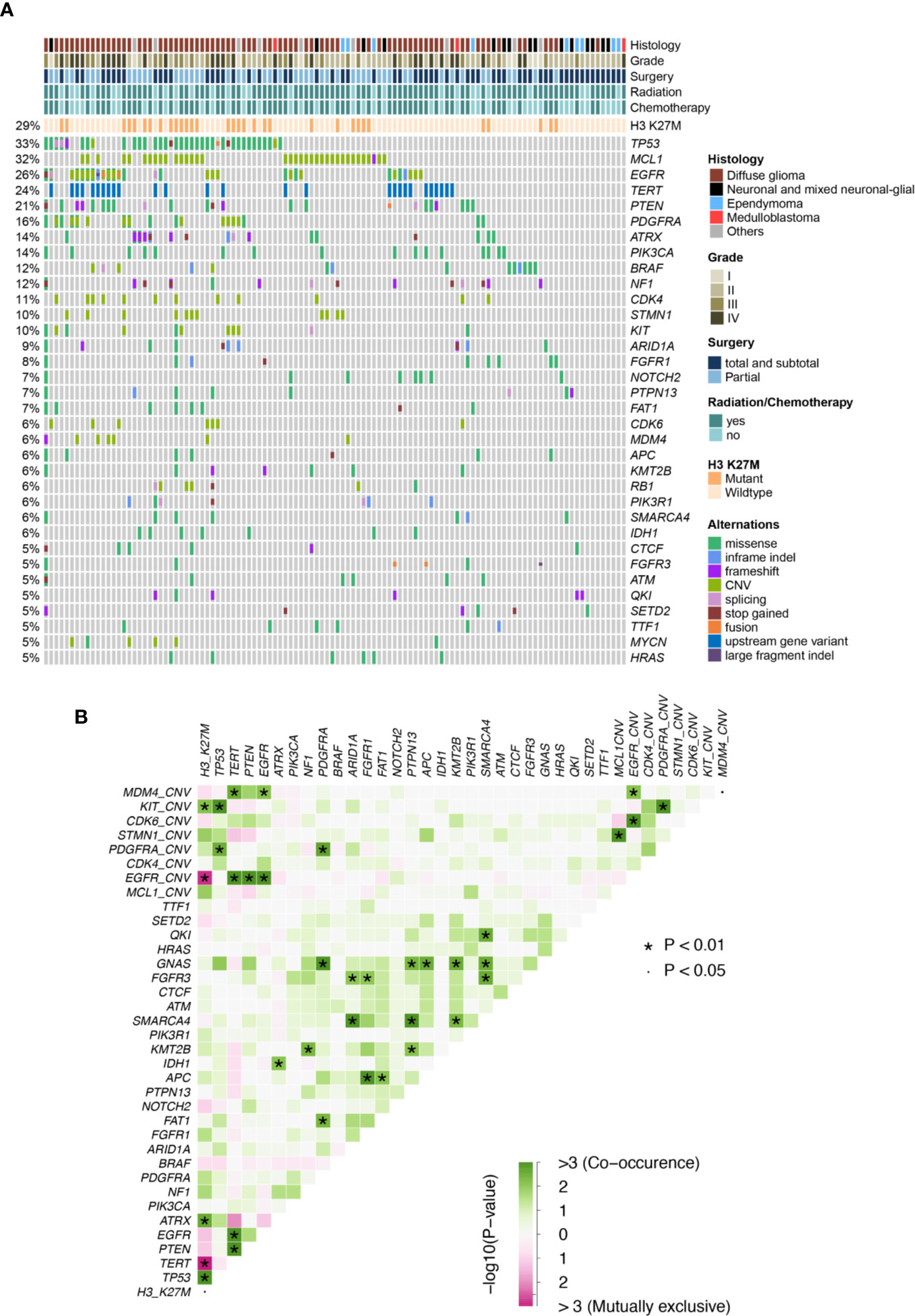
Figure 1 (A) Mutation plot of glioma patients. (B) Somatic interactions of midline glioma patients. Significant exclusive or co- occurrent genes were evaluated with Fisher’s exact test.
Only genes altered in more than 5% of patients (minimum 6 patients) were considered in the univariate analysis for their prognostic value. Clinical and genetic variables with P values ≤ 0.1 were considered for multivariate analysis, including WHO grading, the extent of resection, radiotherapy, H3 K27M, mutations of TP53, TERT, EGFR, PIK3CA, PIK3R1, and APC, as well as CN gain of MCL1, EGFR, and CDK6 (Table 1). After adjusting for the above factors in the multivariate analysis, significantly poor overall survival was found to associate with TERT promoter mutations (HR, 3.00 [95% CI, 1.37–6.61]; P = 0.01), PIK3CA mutations (HR, 3.01 [95% CI, 1.38–6.54]; P = 0.01) and CN gain of MCL1 (HR, 2.78 [95% CI, 1.53–5.08]; P < 0.001). In addition, marginal significance was observed in patients with CN gain of EGFR (HR, 2.30[95% CI, 0.97–5.48]; P = 0.06) and APC mutations (HR, 0.23 [95% CI, 0.05–1.05]; P = 0.06) after multivariate adjustments but predicted opposite prognostic outcomes.
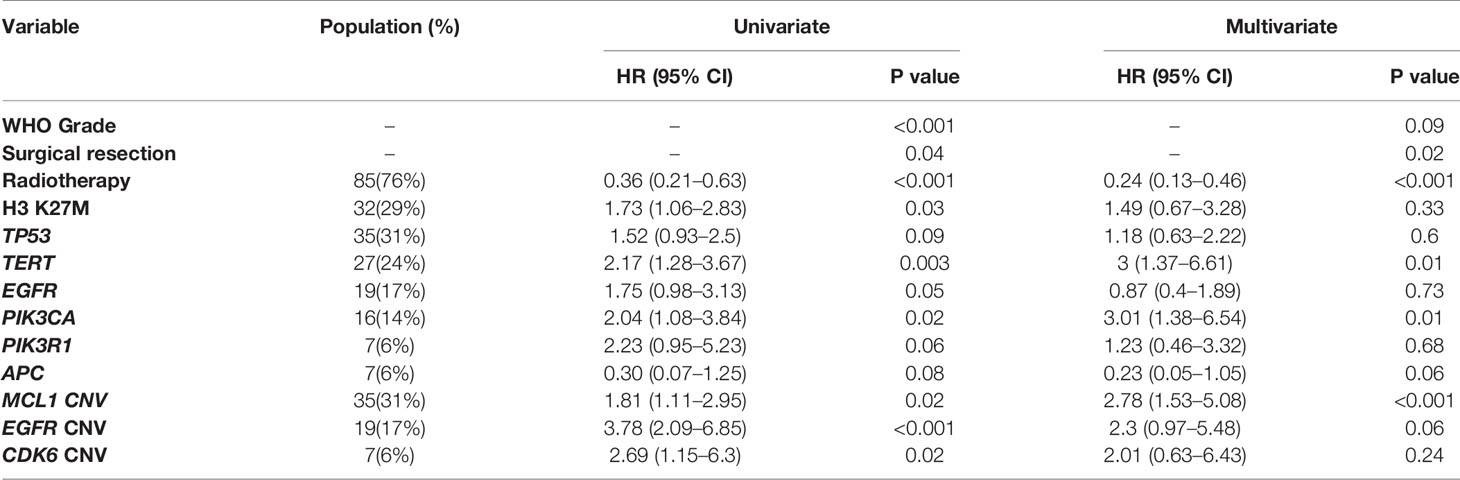
Table 1 Prognostic significance of genetic mutations and copy number variations by univariate and multivariate cox proportional hazard model.
The above results revealed MCL1 CN gain as an interesting independent biomarker of poor prognosis in midline glioma (Figure 2A). As the occurrence of MCL1 CN gain was relatively low in public datasets, we further validated this finding using RNA expression data in 510 TCGA low-grade glioma patients. High MCL1 expression was also associated with significantly worse overall outcome compared with patients with low MCL1 expression (HR, 1.44 [95% CI, 1.01–2.06]; P = 0.045) (Figure 2B).
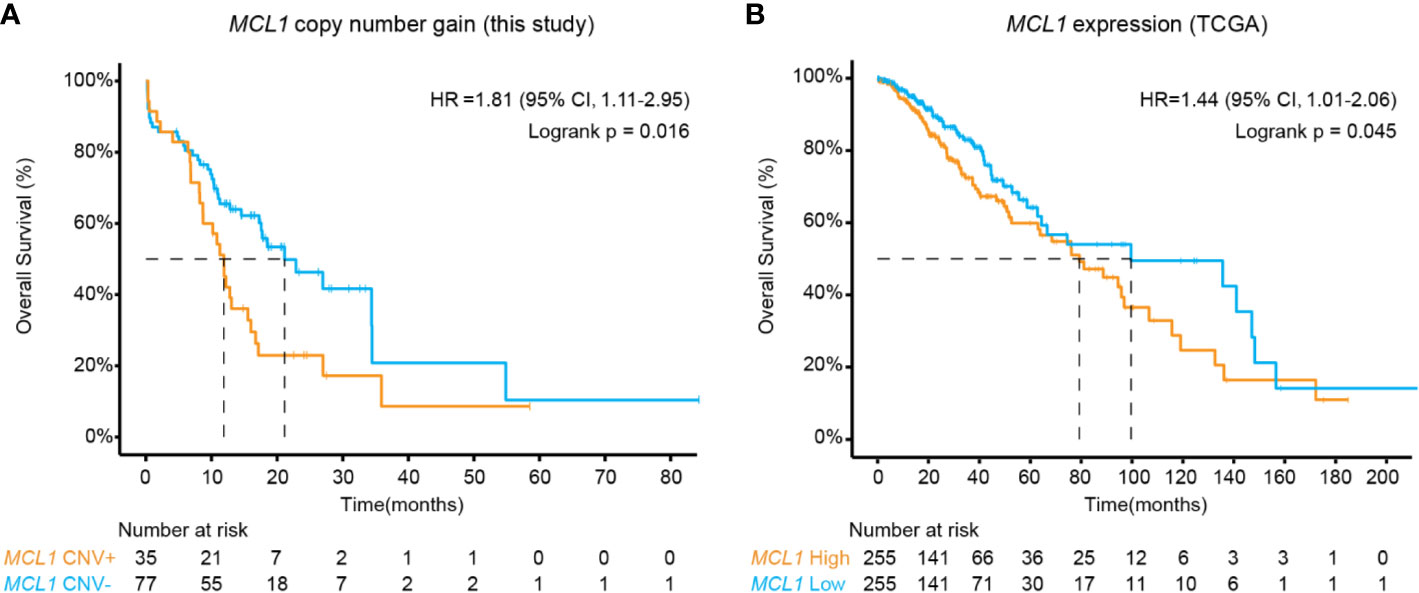
Figure 2 Kaplan-Meier estimates compared overall survival of patients (A) with or without MCL1 copy number gain in this study and (B) of high and low MCL1 expression in the TCGA cohort. P values were derived from the logrank test.
Mutations in the Oncogenic Signaling Pathways
PIK3CA is an emerging biomarker in glioma as reported by several previous studies (33, 34). Our data above again supported PIK3CA’s role to independently predict poor prognosis in the midline cohort (Table 1). Because previous conclusions on the PI3K pathway in glioma were primarily restricted to the scope of PI3K kinases, PTEN, and mTOR mutations, we sought to elaborate on other PI3K pathway genes. Meanwhile, we also explored the other nine canonical oncogenic signaling pathways to evaluate whether altered pathway genes could collectively influence prognosis.
In concert with previous descriptions of CNS tumors (30), we have shown that the RTK/RAS (75%), PI3K (44%), TP53 (43%), cell cycle (21%), and NOTCH (21%) pathways were frequently altered in midline glioma. Of these, patients harboring at least one of the PI3K pathway mutations, including PIK3CA, PTEN, TSC1, TSC2, AKT1, AKT2, AKT3, INPP4B, MTOR, PIK3R2, RICTOR, and RPTOR, were significantly associated with inferior survival comparing to those without these mutations (HR, 1.81 [95% CI, 1.12–2.95]; P = 0.01) (Figure 3A and Figure S2). Importantly, a similar difference in survival was seen after considering potential bias from other clinical and genetic factors (multivariate HR, 1.85 [95% CI, 1.07–3.18]; P = 0.03) (Table S3). We further validated this finding in an MSKCC glioma cohort, in which 51% of patients carried one or more mutations in the PI3K pathway, comparable to our study. Again, PI3K pathway mutations were associated with shorter overall survival (HR, 2.48 [95% CI, 1.99–3.09]; P < 0.001) (Figure 3B).
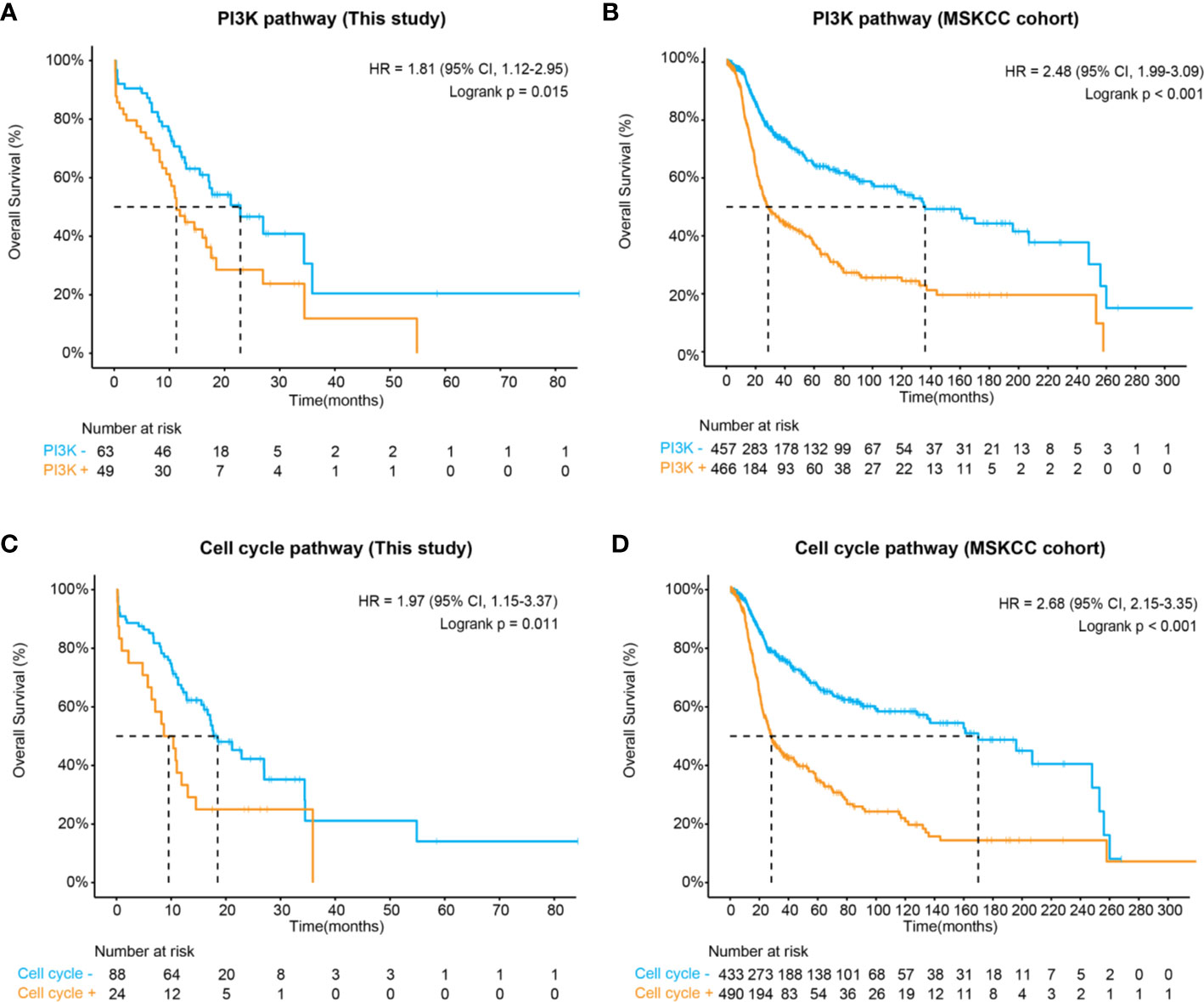
Figure 3 Kaplan-Meier estimates compared overall survival of patients with and without (A) altered PI3K pathway in this study, (B) altered PI3K pathway in the MSKCC cohort, (C) altered cell cycle pathways in this study, and (D) altered cell cycle pathway in the MSKCC cohort.
Apart from the PI3K pathway, our data also demonstrated that the altered cell cycle pathway was significantly associated with poor prognosis (HR, 1.97 [95% CI, 1.15–3.37]; P = 0.011) (Figure 3C), which could be considered as an independent biomarker (multivariate HR, 0.29 [95% CI, 0.15–0.53]; P = 0.05) (Table S3). In the MSKCC cohort, although a higher frequency of cell cycle pathway mutations was observed (53% of patients), patients carrying these mutations also showed remarkably inferior overall survival (HR, 2.68 [95% CI, 2.15–3.68]; P < 0.001) (Figure 3D). Additionally, both the RTK/RAS and p53 pathways demonstrated marginal significance associated with poor prognosis in our cohort (Table S3). In the MSKCC cohort, the RTK/RAS pathway remained to be a significant biomarker for poor prognosis. However, similar results were not observed for the p53 pathway (Table S4), probably due to heterogeneity between cohorts, which might need further exploration.
Discussion
The 2016 WHO Classification has marked the start of the molecular classification era of glioma. For midline gliomas, multiple histological and molecular subtypes have led to emerging debates in defining diagnostic entities, while the current guideline covered only one particular subtype. This unmet clinical need calls for the exploration of additional biomarkers to help stratify and clarify. In this study, we examined 425 cancer-related genes and evaluated how genetic alterations and abnormal oncogenic pathways correlated with survival outcomes in midline glioma patients. In this cohort, H3 K27M was associated with poor survival by univariate, but not multivariate analysis, probably masked by other clinical and genetic factors. Prognostication by H3 K27M could be affected by histology of the tumor. For example, in diffuse gliomas, H3 K27M was found to be a negative prognostic factor in diffuse glioma while in circumscribed and non-diffuse subtypes, it was not associated with prognosis (12, 35–37). We also found a mutually exclusive pattern of H3 K27M and TERT. In line with this, we identified that TERT promoter, PIK3CA, and CN gain of EGFR were of great prognostic significance for midline glioma despite histological subtypes. TERT promoter mutations and ATRX alterations have been described across glioma subtypes in a mutually exclusive pattern (10). In IDH-wildtype low-grade tumors, up to 60% of patients are TERT mutated, while only approximately 6% of IDH-mutant oligodendrogliomas are positive for TERT. TERT-mutant tumors are less responsive to surgical resection with poor survival outcomes similar to that of malignant grade IV glioblastoma across glioma subtypes, implying its prognostic role in other midline gliomas (38). Other frequently altered genes, such as FGFR1, PDGFRA, were also discovered in previous reports of DIPG and adult midline gliomas with shared recurrent mutation sites (39–42). Here, we also revealed a few novel sites including PDGFRA A810T and FGFR1 V561L/M, both located within their tyrosine kinase domains.
Our comprehensive pathway analysis further elucidated that mutations in the PI3K as well as the cell cycle pathways also conferred poor patient survival. The PI3K pathway is one of the most altered pathways in IDH-wildtype gliomas, in consequence of PI3K kinase mutations, loss of PTEN suppressor functions, or activated mTOR (43–46) and resulted in poor prognosis (33, 34) as shown by previous researches. Here, we expanded beyond these star members of the PI3K pathway and considered a collection of pathway genes with a low abundance that were often excluded in single-gene survival analyses. Among these, TSC1 or TSC2 characterizes rare subependymal giant-cell astrocytomas in patients with tuberous sclerosis, causing aberrant activation of mTOR (47). By compiling a broader spectrum of genes, our data revealed that patients with any aberrant PI3K pathway mutations were greatly associated with poor survival regardless of their clinical classification. The RTK/RAS pathway was found to have a prognostic impact on pediatric thalamic glioma (20). However, in this study, this pathway showed only marginal significance univariately and no effect after multivariate adjustments, probably due to differences in the number of genes considered for pathway analysis and heterogeneity among cohorts.
Interestingly, we also unraveled CN gain of MCL1 as a potential novel biomarker for poor survival in midline glioma. MCL1 is a BCL-2 family of anti-apoptotic genes. Focal amplification surrounding the MCL1 chromosome region was observed in 10% of various human cancers, promoting pro-survival signals, and preventing cell death (48). The poor prognosis associated with MCL1 amplification or overexpression has been described in a spectrum of cancers, including non-small cell lung cancer, breast cancer, esophageal squamous cell carcinoma, and acute myeloid leukemia, but not in glioma (49–52). After a thorough literature search, we noticed that the prognostic significance of MCL1 in glioma might be neglected as many cohort studies did not specifically cover or focus on MCL1 gene-level amplification (9, 33, 53, 54). For instance, in the MSKCC validation dataset, only 3 patients reported CN gain of MCL1. In contrast, MCL1 was relatively prevalent in roughly one-third of the entire cohort in this study, including 3 neuronal-glial tumors and 2 ependymoma. As speculated, patients with gain of MCL1 displayed significantly shorter overall survival irrespective of their clinical status, providing the first evidence of its prognostic value in glioma. This observation was also supported by the TCGA datasets that higher MCL1 expression was markedly associated with inferior outcomes in low-grade glioma patients. MCL1 controls cell survival signals downstream of multiple oncogenic pathways. Co-inhibition of MCL1 with key pathway regulators induces synergistic toxicity through different mechanisms. Of note, several clues have pointed to the canonical PI3K/AKT/mTOR pathway. In IDH-mutant tumors, an elevated level of the oncometabolite, 2-R-2-hydroxyglutarate, stimulates mTOR signaling that makes cells susceptible to inhibition of Bcl-xL (46, 55). Another group further demonstrated that the silencing of MCL1 compromised cell proliferation and cell cycle signals through inhibition of the PI3K/Akt pathway (56). Such synthetic lethal interaction has not yet been studied specifically in IDH-wildtype gliomas. Due to the mutual exclusivity between IDH and PTEN mutations, a proposed mechanism of increased Mcl-1 in these tumors is mTOR activation by the absence of negative PTEN regulation. Indeed, an earlier in vitro study examined PTEN-mutated glioblastoma cell lines and demonstrated that co-inhibition of PI3K and Bcl-1 significantly depleted pAKT and Mcl-1 protein levels and enhanced cell apoptosis (57). Also, depletion of Mcl-1 by mTOR inhibitors increase vulnerability to PARP inhibitors, providing additional therapeutic options for glioma (58).
A potential limitation of this study is that a relatively small sample size of mixed histological subtypes was collected for retrospective analysis. To address potential sampling bias, the prognostic significance of genetic alterations and oncogenic pathways were examined by multivariate analyses and verified with public datasets.
In conclusion, we performed comprehensive molecular profiling of midline glioma and revealed that not only the PIK3CA mutations alone but aberrations on the entire PI3K pathway would together predict poor prognosis for midline gliomas. We also discovered CN gain of MCL1 as a novel prognostic biomarker of poor survival that potentially interplays with the PI3K pathway. Collectively, our findings provided new insights in understanding and stratifying midline glioma and developing potential therapeutic targets of glioma in the future.
Data Availability Statement
The data presented in the study are deposited in the Genome Sequence Archive for Human repository, accession number HDAC000322.
Ethics Statement
The studies involving human participants were reviewed and approved by Guangdong Sanjiu Brain Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
HL, DZ, and ZL conceptualized and designed the study. CS, SW, BC, CF, and LC managed patient information and collected samples. HL, YC, YQS, and KL performed data analysis and interpretation. HL and YC wrote the manuscript. YS, DZ, and ZL revised the manuscript. The study was supervised by ZL. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by the Guangdong Natural Science Foundation (No.2017A030313779) and the Guangdong Medical Science and Technology Research Foundation (No. A2019315).
Conflict of Interest
YC, YQS, KL, and YS are employees of Nanjing Geneseeq Technology Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.607429/full#supplementary-material
Supplementary Figure 1 | Lollipop graphs showing the distribution of recurrent mutation sites in the frequently mutated genes across their protein functional domains.
Supplementary Figure 2 | Distribution of genetic alterations in the PI3K pathway.
References
1. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol (2016) 131(6):803–20. doi: 10.1007/s00401-016-1545-1
2. Cancer Genome Atlas Research N, Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N Engl J Med (2015) 372(26):2481–98. doi: 10.1056/NEJMoa1402121
3. Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N Engl J Med (2015) 372(26):2499–508. doi: 10.1056/NEJMoa1407279
4. Louis DN, Giannini C, Capper D, Paulus W, Figarella-Branger D, Lopes MB, et al. cIMPACT-NOW update 2: diagnostic clarifications for diffuse midline glioma, H3 K27M-mutant and diffuse astrocytoma/anaplastic astrocytoma, IDH-mutant. Acta Neuropathol (2018) 135(4):639–42. doi: 10.1007/s00401-018-1826-y
5. von Bueren AO, Karremann M, Gielen GH, Benesch M, Fouladi M, van Vuurden DG, et al. A suggestion to introduce the diagnosis of “diffuse midline glioma of the pons, H3 K27 wildtype (WHO grade IV)”. Acta Neuropathol (2018) 136(1):171–3. doi: 10.1007/s00401-018-1863-6
6. Yekula A, Gupta M, Coley N, HS U. Adult H3K27M-mutant diffuse midline glioma with gliomatosis cerebri growth pattern: Case report and review of the literature. Int J Surg Case Rep (2020) 68:124–8. doi: 10.1016/j.ijscr.2020.02.046
7. Kraus TFJ, Machegger L, Poppe J, Zellinger B, Dovjak E, Schlicker HU, et al. Diffuse midline glioma of the cervical spinal cord with H3 K27M genotype phenotypically mimicking anaplastic ganglioglioma: a case report and review of the literature. Brain Tumor Pathol (2020) 37(3):89–94. doi: 10.1007/s10014-020-00365-z
8. Di Carlo DT, Duffau H, Cagnazzo F, Benedetto N, Morganti R, Perrini P. IDH wild-type WHO grade II diffuse low-grade gliomas. A heterogeneous family with different outcomes. Systematic review and meta-analysis. Neurosurg Rev (2020) 43(2):383–95. doi: 10.1007/s10143-018-0996-3
9. Aibaidula A, Chan AK, Shi Z, Li Y, Zhang R, Yang R, et al. Adult IDH wild-type lower-grade gliomas should be further stratified. Neuro Oncol (2017) 19(10):1327–37. doi: 10.1093/neuonc/nox078
10. Pekmezci M, Rice T, Molinaro AM, Walsh KM, Decker PA, Hansen H, et al. Adult infiltrating gliomas with WHO 2016 integrated diagnosis: additional prognostic roles of ATRX and TERT. Acta Neuropathol (2017) 133(6):1001–16. doi: 10.1007/s00401-017-1690-1
11. Shirahata M, Ono T, Stichel D, Schrimpf D, Reuss DE, Sahm F, et al. Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol (2018) 136(1):153–66. doi: 10.1007/s00401-018-1849-4
12. Kleinschmidt-DeMasters BK, Mulcahy Levy JM. H3 K27M-mutant gliomas in adults vs. children share similar histological features and adverse prognosis. Clin Neuropathol (2018) 37(2018):53–63. doi: 10.5414/NP301085
13. Daoud EV, Rajaram V, Cai C, Oberle RJ, Martin GR, Raisanen JM, et al. Adult Brainstem Gliomas With H3K27M Mutation: Radiology, Pathology, and Prognosis. J Neuropathol Exp Neurol (2018) 77(4):302–11. doi: 10.1093/jnen/nly006
14. Enomoto T, Aoki M, Hamasaki M, Abe H, Nonaka M, Inoue T, et al. Midline Glioma in Adults: Clinicopathological, Genetic, and Epigenetic Analysis. Neurol Med Chir (Tokyo) (2020) 60(3):136–46. doi: 10.2176/nmc.oa.2019-0168
15. Yao K, Duan Z, Wang Y, Zhang M, Fan T, Wu B, et al. Detection of H3K27M mutation in cases of brain stem subependymoma. Hum Pathol (2019) 84:262–9. doi: 10.1016/j.humpath.2018.10.011
16. Gessi M, Capper D, Sahm F, Huang K, von Deimling A, Tippelt S, et al. Evidence of H3 K27M mutations in posterior fossa ependymomas. Acta Neuropathol (2016) 132(4):635–7. doi: 10.1007/s00401-016-1608-3
17. Tanrikulu B, Danyeli AE, Ozek MM. Is H3K27me3 status really a strong prognostic indicator for pediatric posterior fossa ependymomas? A single surgeon, single center experience. Childs Nerv Syst (2020) 36(5):941–9. doi: 10.1007/s00381-020-04518-5
18. Chen LH, Pan C, Diplas BH, Xu C, Hansen LJ, Wu Y, et al. The integrated genomic and epigenomic landscape of brainstem glioma. Nat Commun (2020) 11(1):3077. doi: 10.1038/s41467-020-16682-y
19. Jones DT, Hutter B, Jäger N, Korshunov A, Kool M, Hans-Jörg Warnatz HJ, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet (2013) 45(8):927–32. doi: 10.1038/ng.268
20. Ryall S, Krishnatry R, Arnoldo A, Buczkowicz P, Mistry M, Siddaway R, et al. Targeted detection of genetic alterations reveal the prognostic impact of H3K27M and MAPK pathway aberrations in paediatric thalamic glioma. Acta Neuropathol Commun (2016) 4(1):93. doi: 10.1186/s40478-016-0353-0
21. Georgescu MM, Islam MZ, Li Y, Circu ML, Traylor J, Notarianni CM, et al. Global activation of oncogenic pathways underlies therapy resistance in diffuse midline glioma. Acta Neuropathol Commun (2020) 8(1):111. doi: 10.1186/s40478-020-00992-9
22. Chang R, Tosi U, Voronina J, Adeuyan O, Wu LY, Schweitzer ME, et al. Combined targeting of PI3K and MEK effector pathways via CED for DIPG therapy. Neuro-Oncol Adv (2019) 1(1):vdz004. doi: 10.1093/noajnl/vdz004
23. Dufour C, Perbet R, Leblond P, Vasseur R, Stechly L, Pierache A, et al. Identification of prognostic markers in diffuse midline gliomas H3K27M-mutant. Brain Pathol (2020) 30(1):179–90. doi: 10.1111/bpa.12768
24. Gojo J, Pavelka Z, Zapletalova D, Schmook MT, Mayr L, Madlener S, et al. Personalized Treatment of H3K27M-Mutant Pediatric Diffuse Gliomas Provides Improved Therapeutic Opportunities. Front Oncol (2020) 9:1436. doi: 10.3389/fonc.2019.01436
25. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res (2010) 20(9):1297–303. doi: 10.1101/gr.107524.110
26. Tanboon J, Williams EA, Louis DN. The Diagnostic Use of Immunohistochemical Surrogates for Signature Molecular Genetic Alterations in Gliomas. J Neuropathol Exp Neurol (2016) 75(1):4–18. doi: 10.1093/jnen/nlv009
27. Lopez GY, Oberheim Bush NA, Phillips JJ, Bouffard JP, Moshel YA, Jaeckle K, et al. Diffuse midline gliomas with subclonal H3F3A K27M mutation and mosaic H3.3 K27M mutant protein expression. Acta Neuropathol (2017) 134(6):961–3. doi: 10.1007/s00401-017-1780-0
28. Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M, et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet (2014) 46(5):451–6. doi: 10.1038/ng.2936
29. Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell (2017) 171(4):934–49 e16. doi: 10.1016/j.cell.2017.09.028
30. Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell (2018) 173(2):321–37.10. doi: 10.1016/j.cell.2018.03.035
31. Jonsson P, Lin AL, Young RJ, DiStefano NM, Hyman DM, Li BT, et al. Genomic Correlates of Disease Progression and Treatment Response in Prospectively Characterized Gliomas. Clin Cancer Res (2019) 25(18):5537–47. doi: 10.1158/1078-0432.Ccr-19-0032
32. Anaya J. OncoLnc: linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. PeerJ Comput Sci (2016) 2:e67. doi: 10.7717/peerj-cs.67
33. Draaisma K, Wijnenga MM, Weenink B, Gao Y, Smid M, Robe P, et al. PI3 kinase mutations and mutational load as poor prognostic markers in diffuse glioma patients. Acta Neuropathol Commun (2015) 3:88. doi: 10.1186/s40478-015-0265-4
34. Tanaka S, Batchelor TT, Iafrate AJ, Dias-Santagata D, Borger DR, Ellisen LW, et al. PIK3CA activating mutations are associated with more disseminated disease at presentation and earlier recurrence in glioblastoma. Acta Neuropathol Commun (2019) 7(1):66. doi: 10.1186/s40478-019-0720-8
35. Wang T, Qiu Y, Liang L, Zheng E, Gao T. H3 K27M-mutant glioma: clinical characteristics and outcomes. Neuro Oncol (2019) 21(11):1480–1. doi: 10.1093/neuonc/noz135
36. Pratt D, Natarajan SK, Banda A, Giannini C, Vats P, Koschmann C, et al. Circumscribed/non-diffuse histology confers a better prognosis in H3K27M-mutant gliomas. Acta Neuropathol (2018) 135(2):299–301. doi: 10.1007/s00401-018-1805-3
37. Orillac C, Thomas C, Dastagirzada Y, Hidalgo ET, Golfinos JG, Zagzag D, et al. Pilocytic astrocytoma and glioneuronal tumor with histone H3 K27M mutation. Acta Neuropathol Commun (2016) 4(1):84. doi: 10.1186/s40478-016-0361-0
38. Molinaro AM, Taylor JW, Wiencke JK, Wrensch MR. Genetic and molecular epidemiology of adult diffuse glioma. Nat Rev Neurol (2019) 15(7):405–17. doi: 10.1038/s41582-019-0220-2
39. Paugh BS, Zhu X, Qu C, Endersby R, Diaz AK, Zhang J, et al. Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res (2013) 73(20):6219–29. doi: 10.1158/0008-5472.CAN-13-1491
40. Hoffman LM, DeWire M, Ryall S, Buczkowicz P, Leach J, Miles L, et al. Spatial genomic heterogeneity in diffuse intrinsic pontine and midline high-grade glioma: implications for diagnostic biopsy and targeted therapeutics. Acta Neuropathol Commun (2016) 4:1. doi: 10.1186/s40478-015-0269-0
41. Picca A, Berzero G, Bielle F, Touat M, Savatovsky J, Polivka M, et al. FGFR1 actionable mutations, molecular specificities, and outcome of adult midline gliomas. Neurology (2018) 90(23):e2086–e94. doi: 10.1212/WNL.0000000000005658
42. Jones DT, Hutter B, Jager N, Korshunov A, Kool M, Warnatz HJ, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet (2013) 45(8):927–32. doi: 10.1038/ng.2682
43. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell (2013) 155(2):462–77. doi: 10.1016/j.cell.2013.09.034
44. McBride SM, Perez DA, Polley MY, Vandenberg SR, Smith JS, Zheng S, et al. Activation of PI3K/mTOR pathway occurs in most adult low-grade gliomas and predicts patient survival. J Neurooncol (2010) 97(1):33–40. doi: 10.1007/s11060-009-0004-4
45. Bai H, Harmanci AS, Erson-Omay EZ, Li J, Coskun S, Simon M, et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat Genet (2016) 48(1):59–66. doi: 10.1038/ng.3457
46. Carbonneau M, MG L, Lalonde ME, Germain MA, Motorina A, Guiot MC, et al. The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway. Nat Commun (2016) 7:12700. doi: 10.1038/ncomms12700
47. Weller M, Wick W, Aldape K, Brada M, Berger M, Pfister SM, et al. Glioma. Nat Rev Dis Primers (2015) 1(1):15017. doi: 10.1038/nrdp.2015.17
48. Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature (2010) 463(7283):899–905. doi: 10.1038/nature08822
49. Campbell KJ, Dhayade S, Ferrari N, Sims AH, Johnson E, Mason SM, et al. MCL-1 is a prognostic indicator and drug target in breast cancer. Cell Death Dis (2018) 9(2):19. doi: 10.1038/s41419-017-0035-2
50. Xu C, Liu Y, Huang J, Wang H, Tan L, Xu Y, et al. The prognostic significance of MCL1 copy number gain in esophageal squamous cell carcinoma. Oncotarget (2017) 8(50):87699–709. doi: 10.18632/oncotarget.21181
51. Yin J, Li Y, Zhao H, Qin Q, Li X, Huang J, et al. Copy-number variation of MCL1 predicts overall survival of non-small-cell lung cancer in a Southern Chinese population. Cancer Med (2016) 5(9):2171–9. doi: 10.1002/cam4.774
52. Li XX, Zhou JD, Wen XM, Zhang TJ, Wu DH, Deng ZQ, et al. Increased MCL-1 expression predicts poor prognosis and disease recurrence in acute myeloid leukemia. Onco Targets Ther (2019) 12:3295–304. doi: 10.2147/OTT.S194549
53. Wijnenga MMJ, Dubbink HJ, French PJ, Synhaeve NE, Dinjens WNM, Atmodimedjo PN, et al. Molecular and clinical heterogeneity of adult diffuse low-grade IDH wild-type gliomas: assessment of TERT promoter mutation and chromosome 7 and 10 copy number status allows superior prognostic stratification. Acta Neuropathol (2017) 134(6):957–9. doi: 10.1007/s00401-017-1781-z
54. Reinhardt A, Stichel D, Schrimpf D, Sahm F, Korshunov A, Reuss DE, et al. Anaplastic astrocytoma with piloid features, a novel molecular class of IDH wildtype glioma with recurrent MAPK pathway, CDKN2A/B and ATRX alterations. Acta Neuropathol (2018) 136(2):273–91. doi: 10.1007/s00401-018-1837-8
55. Karpel-Massler G, Ishida CT, Bianchetti E, Zhang Y, Shu C, Tsujiuchi T, et al. Induction of synthetic lethality in IDH1-mutated gliomas through inhibition of Bcl-xL. Nat Commun (2017) 8(1):1067. doi: 10.1038/s41467-017-00984-9
56. Wu DM, Hong XW, Wen X, Han XR, Wang S, Wang YJ, et al. MCL1 gene silencing promotes senescence and apoptosis of glioma cells via inhibition of the PI3K/Akt signaling pathway. IUBMB Life (2019) 71(1):81–92. doi: 10.1002/iub.1944
57. Pareja F, Macleod D, Shu C, Crary JF, Canoll PD, Ross AH, et al. PI3K and Bcl-2 inhibition primes glioblastoma cells to apoptosis through downregulation of Mcl-1 and Phospho-BAD. Mol Cancer Res (2014) 12(7):987–1001. doi: 10.1158/1541-7786.MCR-13-0650
Keywords: midline glioma, PI3K-AKT pathway, cell cycle pathway, prognostic genetic markers, next-generation sequencing
Citation: Li H, Shan C, Wu S, Cheng B, Fan C, Cai L, Chen Y, Shi Y, Liu K, Shao Y, Zhu D and Li Z (2021) Genomic Profiling Identified Novel Prognostic Biomarkers in Chinese Midline Glioma Patients. Front. Oncol. 10:607429. doi: 10.3389/fonc.2020.607429
Received: 17 September 2020; Accepted: 26 November 2020;
Published: 03 March 2021.
Edited by:
David M. Peereboom, Case Western Reserve University, United StatesReviewed by:
Antonio Silvani, Fondazione IRCCS Istituto Neurologio Carlo Besta, ItalyQuan Cheng, Central South University, China
Copyright © 2021 Li, Shan, Wu, Cheng, Fan, Cai, Chen, Shi, Liu, Shao, Zhu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dan Zhu, emh1ZGFuOTk5YnJhaW5AMTYzLmNvbQ==; Zhi Li, bGl6aGlAbWFpbC5zeXN1LmVkdS5jbg==