
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol., 27 October 2020
Sec. Women's Cancer
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.605633
This article is part of the Research TopicImmunotherapy as an Evolving Approach for the Treatment of Breast CancerView all 13 articles
 Jesús Fuentes-Antrás1,2*
Jesús Fuentes-Antrás1,2* Kissy Guevara-Hoyer3
Kissy Guevara-Hoyer3 Mariona Baliu-Piqué2
Mariona Baliu-Piqué2 José Ángel García-Sáenz1Pedro Pérez-Segura1,2
José Ángel García-Sáenz1Pedro Pérez-Segura1,2 Atanasio Pandiella4
Atanasio Pandiella4 Alberto Ocaña1,2
Alberto Ocaña1,2Immunotherapy has become a cornerstone in the treatment of cancer and changed the way clinicians and researchers approach tumor vulnerabilities. Durable responses are commonly observed with immune checkpoint inhibitors in highly immunogenic tumors, while the infusion of T cells genetically engineered to express chimeric antigen receptors (CARs) has shown impressive efficacy in certain types of blood cancer. Nevertheless, harnessing our own immunity has not proved successful for most breast cancer patients. In the era of genomic medicine, cellular immunotherapies may provide a more personalized and dynamic tool against tumors displaying heterogeneous mutational landscapes and antigenic pools. This approach encompasses multiple strategies including the adoptive transfer of tumor-infiltrating lymphocytes, dendritic cells, natural killer cells, and engineered immune components such as CAR constructs and engineered T cell receptors. Although far from permeating the clinical setting, technical advances have been overwhelming in recent years, with continuous improvement in traditional challenges such as toxicity, adoptive cell persistence, and intratumoral trafficking. Also, there is an avid search for neoantigens that can be targeted by these strategies, either alone or in combination. In this work, we aim to provide a clinically-oriented overview of preclinical and clinical data regarding the use of cellular immunotherapies in breast cancer.
Breast cancer (BC) is a leading cause of death worldwide and remains mostly incurable in advanced stages (1). Tumor initiation and progression is continuously controlled by innate and adaptive immune cells, which falter as cancer cells undergo mesenchymal dedifferentiation and/or evolve different mechanisms of tumor escape (2). In general, BC is not regarded as an inflamed tumor, triple negative BC (TNBC) and HER2+ tumors being more immunogenic than the most common luminal A-like subtype (3). Immunotherapeutic strategies against BC have traditionally been based on “passive immunotherapy” such as the HER2 blocking antibody trastuzumab. Encouraged by the success of immune checkpoint inhibitors (ICIs) in melanoma and lung cancer, numerous trials have tested the use of this “active immunotherapy” in BC with overall disappointing results (4). In the metastatic setting, the most significant achievement was observed in the IMpassion130 phase III trial, which demonstrated an increase in progression-free survival in TNBC patients receiving atezolizumab plus nab-paclitaxel compared to nab-paclitaxel alone (7.2 vs 5.5 months) (5). This humble benefit did not lead to a better overall survival and was not recapitulated when using paclitaxel as concomitant chemotherapy nor consistently associated to any predictive biomarker other than PD-L1 (6). Findings seem to be more clinically meaningful in the neoadjuvant setting, in which an increased pathological complete response rate has been reported in patients receiving atezolizumab (58 vs 41% for total population, 69 vs 49% in PD-L1 positive tumors) (7). This body of evidence underscores the need of a better understanding of the tumor-immune interaction, escape mechanisms, and the role of the microenvironment when a high tumor burden exists. Globally, the use of ICIs in BC would at best provide a nonspecific approach, guided by poorly understood biomarkers, to harnessing a debilitated immune system against a cold tumor. Instead, the development of omic-scale repositories and high-throughput technologies enable us to decode the genomic traits of each unique tumor and calls for the design of more specific and flexible immunotherapies, capable of targeting oncogenic addictions and overcoming temporal and spatial mutational heterogeneities. Thus, the aim of our work is to bridge the complex body of evidence on the different types of adoptive cell therapy (ACT) and the clinicians who everyday care for BC patients.
The adoptive transfer of lymphocytes to treat BC has been attempted in numerous occasions. Allogeneic stem cell transplants in addition to high-dose chemotherapy achieved successful long-term outcomes but arouse significant safety concerns, whereas ACT with autologous circulating lymphocytes conditioned in vitro was better tolerated but showed less efficacy (8–11). Tumor-infiltrating lymphocytes (TILs) include a subset of naturally occurring T cells capable of targeting neoantigens encoded by genes harboring nonsynonymus somatic mutations (12, 13). BC, particularly HER2+ and luminal-like tumors, have been traditionally considered as poorly immunogenic, with low numbers of TILs and a limited burden of neoantigens (3, 14, 15). However, a robust correlation exists between increased stromal TILs and a better prognosis in TNBC (16–19).
Adoptive transfer of autologous TILs was first described as a treatment modality by Rosenberg and colleagues in 1987 (Figure 1A) (20). Substantial objective responses have been observed in patients with tumors with high mutation rates such as melanoma, lung or bladder cancer (20–22). However, with few exceptions, the infusion of unselected heterogenous TILs appears mostly ineffective in epithelial malignancies (23–27). In order to boost tumor recognition and killing efficacy, TIL therapy has been refined by selecting TILs reactive for tumor antigens (TAs) identified by whole-exome sequencing and RNA sequencing. Zacharakis et al. recently described the case of a 49 year-old woman with ER+/HER2- metastatic BC refractory to multiple lines of chemotherapy, who exhibited a complete durable regression after ACT with TA-specific TILs in conjunction with IL-2 and an anti-PD1 agent (28). In this particular case, the genomic analysis of a right breast subcutaneous lesion revealed the presence of 62 nonsynonymous somatic mutations, of which the mutant versions of 4 proteins rendered the highest TIL reactivity. Further, a relevant impact of the concomitant anti-PD1 therapy was unlikely since no expression of PD-L1 was detected in tumor biopsies. A similar approach was used for a pulmonary metastasis of a TNBC patient, where an immunogenic mutation was found among 72 nonsynonymous mutations (29). However, outcome data from this tailored TIL therapy was not reported. Four clinical initiatives have been registered to date in ClinicalTrials.gov and are briefly displayed in Table 1. Notably, only two of them incorporate preconditioning with non-myeloablative chemotherapy regimens, and one of them will address the role of an anti-PD1 agent as concurrent medication. In sum, the transfer of selected autologous TILs primed against multiple MHC-restricted TAs may provide a safe and personalized option for patients with advanced BC.
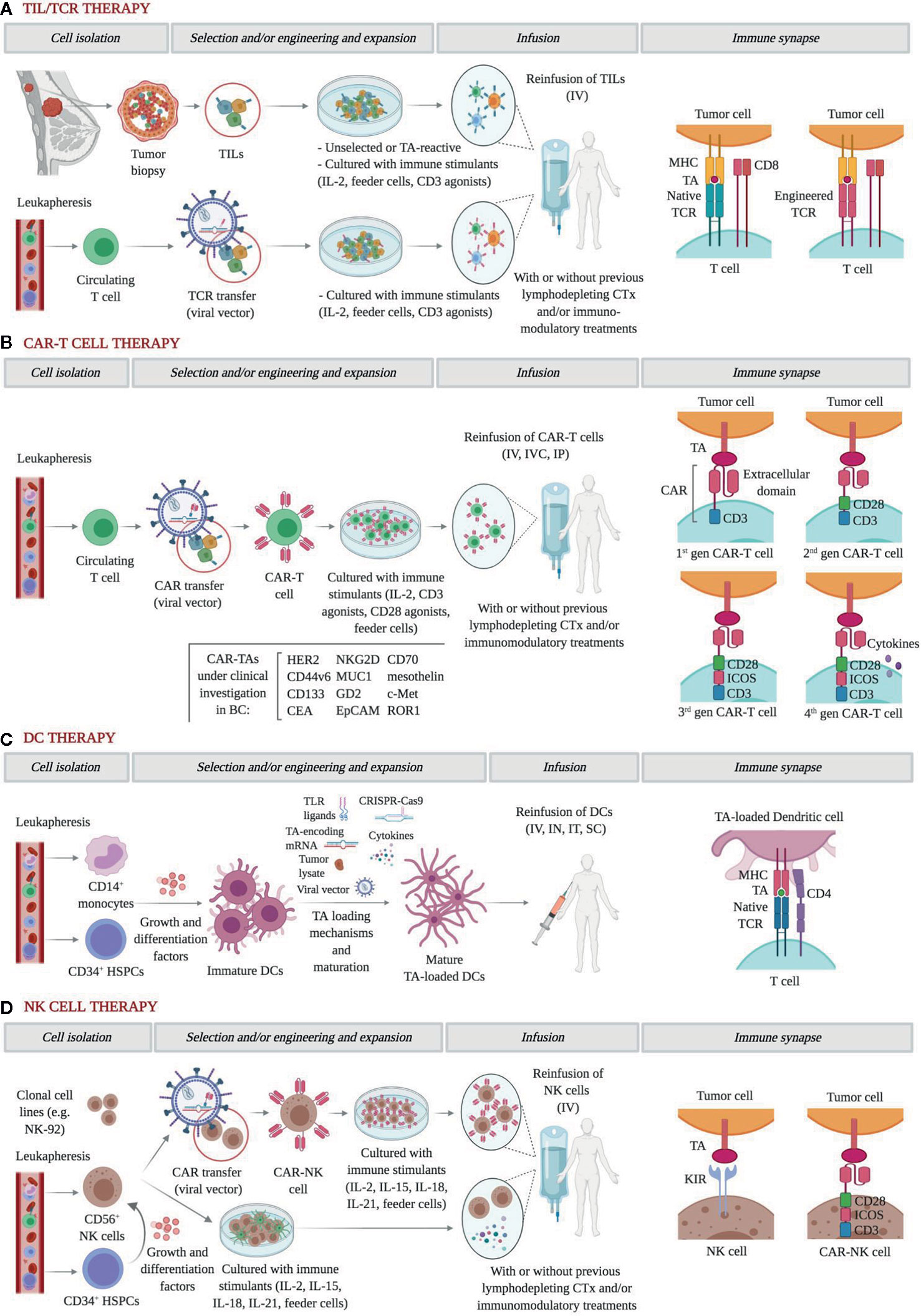
Figure 1 Graphical representation of the main approaches of adoptive cell therapy in breast cancer. (A) In general, TILs are enzymatically isolated, activated with high-dose IL-2, and eventually expanded for therapeutic use. More recently, they can also be screened for a high avidity for TAs. TCR transfer, usually using viral vectors on circulating T cells, endows T cells with TCRs with high affinity for TAs. Further, to help condition the body for the T cell transplant, patients often receive a non-myeloablative lymphodepleting chemotherapy regimen before IV infusion, which facilitates the access to growth-promoting cytokines and removes suppressor cells. The role of concomitant immunomodulatory therapies is yet to be elucidated. In both approaches, recognition of cognate TAs is MHC-restricted. (B) CAR engineering of circulating T cells has been progressively refined. First-generation CARs include only a CD3ζ chain as intracellular signaling domain; second-generation CARs add a single co-stimulatory domain, such as CD28, 4-1BB (CD137), CD27, or OX40; third-generation CARs add two or three co-stimulatory domains; fourth-generation CARs, also known as TRUCKs (T cells redirected for antigen‐unrestricted cytokine‐initiated killing) are further armored with potent antitumor cytokines and co-stimulatory ligands. CARs target a wide range of surface TAs in an MHC-independent manner, and multiple trials are currently testing the feasibility and efficacy of different administration routes. (C) DCs can be generated from PBMNC and HSPCs and become mature after being pulsed using a growing set of TA loading mechanisms. In trials, DCs are infused IV but also as IT or IN vaccines. (D) NK cells for ACT can be obtained from clonal cell lines, primary NK cells, or HSPCs. Whether they undergo CAR engineering or remain unmodified, NK cells ligate cognate TAs in an MHC-independent manner. After co-culture with immune stimulants and feeder cells, NK cells are infused IV with or without prior lymphodepleting chemotherapy and/or immunomodulatory treatments. TILs, tumor-infiltrating lymphocyte; IL, interleukin; TA, tumor antigen; TCR, T-cell receptor; MHC, major histocompatibility complex; CAR, chimeric antigen receptor; DC, dendritic cell; PBMC, peripheral blood mononuclear cell; HSPC, hemopoietic pluripotent stem cell; NK, natural killer. IV, intravenous; IVC, intraventricular; IP, intraperitoneal; IN, intranodal; IT, intratumoral; SC, subcutaneous. Figure created with BioRender.com.
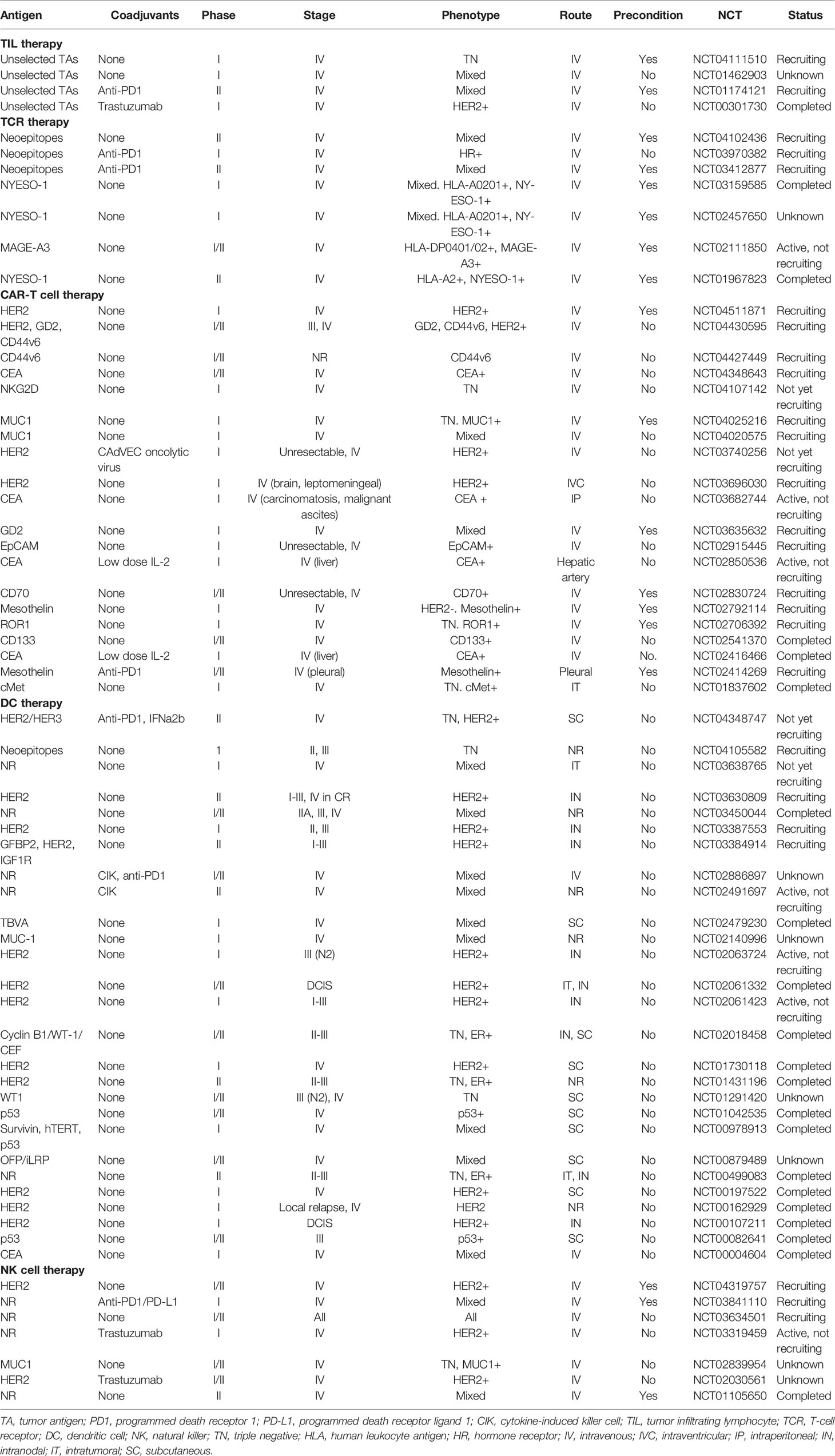
Table 1 Clinical trials of ACT in breast cancer.
Gene transfer-based strategies have been developed to overcome the main challenges of TIL therapy, including the low yield of TIL expansion, the low affinity of human TCRs for TAs, and the immune tolerance elicited by the downregulation of MHC molecules and TAs (30). Both TCR and chimeric antigen receptor (CAR) gene transfer endow polyclonal T cells with reactivities that are not naturally present against TAs of choice and thus provide an adaptable and highly subtle tool for personalized medicine (Figures 1A, B) (31).
The majority of engineered αβTCRs recognize epitopes presented by MHC molecules, thereby narrowing down the group of potential targets to those which are MHC-restricted, and exhibit an increased specificity recognition and affinity for tumor cells (Figure 1A) (32, 33). Mounting clinical evidence on several tumor types along with preclinical data on BC underscores the rationale for TCR use in BC patients (34–37). Of note, in both hormone-dependent and independent BC cell lines and in xenograft mice, Li et al. reported a notable enhancement of anti-tumor cytotoxicity by CD8+ T cells transduced with an MHC-A2-restricted placenta-specific 1 (PLAC1)-TCR molecule (38). However, to the best of our knowledge, evidence on humans is still lacking, with many ongoing clinical trials testing intravenous infusions of TCR-engineered T cells against TAs such as HER2, NYESO-1, and MAGE-A3 (Table 1). Interestingly, some of them will assess the value of adding anti-PD1 therapy to enhance immune reconstitution after lymphodepleting chemotherapy and cytotoxicity.
In order to bypass the limitations of MHC restriction of conventional αβTCRs, intensive research has focused on the development of CARs and, more recently, on the γδT cell compartment. γδT cells exhibit potent anti-tumor responses by bridging innate and adaptive immunities, since they incorporate both γδTCRs and killer cell immunoglobulin-like receptors (KIRs) (39, 40). Also, γδT cell ligand recognition requires the expression of accessory costimulatory molecules, which may prevent harmful self-reactivity. Infiltration by γδT cells has been associated with improved outcomes in a small cohort of TNBC patients (41). Consistently, ACT of γδT cells together with trastuzumab improved control of tumor growth as compared to trastuzumab alone in a mouse model of HER2+ BC (42). However, the function of the γδT cells may be extremely pleiotropic. In this regard, Peng et al. described a BC-infiltrating γδT cell subset with strong immunosuppressive effects on T cells and DCs regulated via the Toll-like receptor 8, thus suggesting that its depletion or reversal could enhance anti-tumor responses (43). ACT with unmodified or engineered γδT cells emerges as an appealing prospect for BC immunotherapy, but further functional characterization and data on clinical interventions are still required (44).
On the other hand, CAR-T cells are T cells engineered to express an artificial receptor with a modular design consisting of an extracellular ligand-binding domain, usually a single-chain antibody, a hinge, a transmembrane domain, and a cytoplasmic signaling domain, with increasing complexity and functionality across the four generations of CAR constructs (Figure 1B) (45–47). Compared to TILs, CAR-T cells are not as affected by the hurdles of isolation, expansion, and persistence limitation of natural tumor-specific T cells. Moreover, CAR recognition occurs in an MHC-independent manner, which helps overcome MHC downregulation as a mechanism of tumor escape, and can also recognize carbohydrate and glycolipid antigens (46). Yet, cognate antigens are consequently restricted to surface molecules. Numerous preclinical studies in vitro and in vivo have evaluated the use of CAR-T cells armed to specifically target TAs in BC, with HER2-CAR constructs attracting the most attention and achieving robust tumor regressions (48–60). To our knowledge, only one phase I trial has been published testing a HER2-CAR in BC patients. In the study by Lum et al., 23 metastatic BC patients independent of their HER status received 8 infusions of anti-CD3/HER2 bispecific antibody-armed T cells. In the evaluable patients at 14.5 weeks, 13 patients experienced clinical benefit, including 2 objective responses (61). Notwithstanding, serious adverse events have been reported following the use of HER2-CARs. The first evidence on the clinical use of HER2-CAR-T cells was a case report of a patient with metastatic HER2+ colon cancer in whom the administration of a 3rd generation HER2-CAR was followed by multiple cardiac arrests, respiratory distress, and multiorgan damage (62). This harm was attributed to an inflammatory cytokine release elicited by the immune-mediated recognition of HER2 in normal epithelial tissues, which is referred to as “on-target, off-tumor” toxicity.
Besides HER2, the single injection of accessible lesions with CAR-T cells targeting c-Met, a cell-surface protein tyrosine kinase aberrantly expressed in BC, in a group of 6 patients with metastatic BC comprised by two ER+ tumors and 4 TN tumors, did not render measurable responses but elicited extensive tumor necrosis and loss of c-Met immunoreactivity at the injection site, and also translated into detectable levels of c-Met-CAR-T cell mRNA in peripheral blood (63). Similarly, Specht et al. recently communicated preliminary safety results of a phase I trial targeting ROR-1, a tyrosine kinase protein expressed in TNBC and associated with a worse prognosis (64, 65). Interestingly, patients received a 2nd generation ROR1-CAR engineered with a truncated EGFR molecule to permit the elimination of infused cells in case of toxicity (64). Only 6 patients had been enrolled with no adverse events observed, but further update is expected to support this innovative approach.
A considerable number of trials are testing CAR constructs against multiple TAs in BC (Table 1). We expect that these studies also convey relevant information about on-target, off-tumor effects, and the benefits of the different administration routes, preconditioning or concomitant immunomodulatory therapies. In addition, it seems clear that a thorough genomic-scale understanding of molecular vulnerabilities and antigenic shifts will be of paramount importance in the design of CAR-based strategies.
Dendritic cells (DCs) are particularly well-suited for BC immunotherapy due to their ability to sensitize CD8+ T cells and also CD4+ T cells capable of generating memory T cells and contribute with additional cytotoxicity against tumors (66). DCs have been found infiltrating BC specimens in nearly half of the patients with either early or advanced disease, but are mostly relegated to the periphery, functionally compromised, and show a poor correlation with outcome (67–70).
Autologous DCs may be fused with tumor cells or pulsed with tumor lysates or TAs to activate T cells against tumors (Figure 1C) (71–74). Across these strategies, DCs may be either exposed to one particular neoantigen or to the entire repertoire of TAs, including those yet to be identified. In contrast to what was observed in TIL and TCR therapies, DCs can be obtained in large numbers from bone marrow precursors and monocyte-derived DCs from peripheral blood (75). The pioneering study by Brossart et al. evaluated the vaccination with autologous DCs pulsed with HER2 or MUC1-derived peptides in 7 BC patients. Although the clinical outcomes were disappointing, peptide-specific T cell responses could be detected even at 9 months after initiation of vaccinations, and T cell responses against epitopes not used for vaccination were identified as a result of cross priming (76). More encouraging objective responses were achieved by Avigan et al. in a phase I trial testing the vaccination with DCs fused with autologous tumor cells in 16 patients with metastatic BC (77). These included 2 patients attaining a partial response and 6 patients attaining a stable disease, although the anti-tumor effects were not maintained over time. In the neo/adjuvant setting, vaccination with autologous HER2-pulsed DCs achieved a modest rate of pathological complete responses in HER2+ BC patients, which yet correlated poorly with immune surrogates in peripheral blood (78). This study, however, demonstrated that intralesional and intranodal routes of administration may not substantially differ in terms of anti-tumor efficacy, thus facilitating vaccination when tumor locations are challenging. Likewise, the trial conducted by Qi et al. in stage II-IIIA ER-/PR- BC patients reported a 3-year relapse-free survival of 71% versus 31%, with and without vaccination, respectively (79). Other promising approaches consist of adding cytokine adjuvants, such as IL-2, or targeting both the innate and adaptive immune systems by complementing DCs with cytokine-induced killer cells, although the response to these strategies has so far been humble or confused by the effect of concurrent chemotherapies (80–82). More than 20 trials are registered to date testing DC vaccinations in BC patients of all major pathological and most of them are designed to pulse DCs with TAs of choice (Table 1). Although ACT with DCs has not yet materialized in a relevant clinical benefit, we believe that the role of DCs as stimulators of T-cell response and long-term memory, and their safety and ease of manufacture, may justify further development alone or in combination with other T cell therapies.
Different from the previous approaches, NK cells represent an attractive asset for cancer immunotherapy due to their innate ability to eliminate cancer cells in an MHC-independent and non-TA-restricted manner. The “loss of self” mediated by the downregulation of MHC molecules as a mechanism of tumor escape hinders the recognition of cancer cells by CD8+ T cells but unleashes the activity of NK cells, which are regulated by the interplay of activating and inhibitory receptors such as KIRs and natural killer group 2D (NKG2D) (83, 84).
Activated NK cells can be manufactured in large numbers ex vivo from primary NK cells, hemopoietic stem cells, and clonal cell lines, of which the NK-92 is approved by the US FDA for use in clinical trials (Figure 1D) (85–87). So far, adoptive transfer of autologous NK cells has been tested in a wide range of solid malignancies with poor clinical efficacy, which has been explained by the immunosuppressive state of the host and because the inhibitory receptors on autologous NK cells matched molecules exhibited on the tumor cell surface (87–89). Anecdotally, a report by Tian et al. described a partial response in a patient with progressing metastatic HER2+ BC who underwent treatment with trastuzumab-treated NK cells, which was consistent with an increased activation and expansion of NK cells mediated by trastuzumab in vitro (90). Allogeneic NK cells, however, have not proved to do much better in BC patients, with only one phase II trial published describing 4 patients with stabilized disease from a total of 6 patients evaluated at 4–6 weeks from infusion and after pre-conditioning with lymphodepleting chemotherapy and total body irradiation (91).
In order to enhance their cytotoxic properties, NK cells are also being modified with the addition of CARs against specific TAs. Compared to CAR-T cells, CAR-NK cells are theoretically less potent due to their lack of clonal expansion, relatively short lifespan, and less cytotoxic cytokines (87). Although CAR-T cells may mediate more incisive and long-term responses, the use of CAR-NK cells would minimize the risk of cytokine release syndrome and tumor-lysis syndrome, thereby increasing overall treatment safety (92). Importantly, CAR-NK therapy is expected to be much less expensive, considering that NK cells can be derived from multiple sources. Encouraging results have been reported in a phase I/IIa trial using cord blood-derived CAR-NK cells targeting CD19 in patients with relapsed or refractory non-Hodgkin’s lymphoma and chronic lymphocytic leukemia, with up to 64% of patients achieving a complete response (93). In BC, tissue factor (TF) was recently described by Hu as a novel and common yet selective molecule on TNBC, whose targeting by TF-CAR NK cells resulted in an increased cytotoxicity against TNBC cell lines and was effective and safe for the treatment of TNBC in an orthotopic mouse model (94). Chen et al. recapitulated these findings when investigating the effect of EGFR-CAR NK cells in TBNC cell lines and in mice pre-inoculated with brain metastases (95). To the best of our knowledge, there is not published data on human trials on BC to date, although several initiatives can be found registered in the Clinical Trials.gov repository including multiple trials evaluating the intravenous infusion of ex vivo expanded, autologous NK cells and also the administration of NK cells incorporating HER2- and MUC1-CAR constructs (Table 1).
ACT offers a growing toolkit to overcome antigenic heterogeneity and the broad repertoire of immune escape mechanisms occurring in advanced BC. To fully capitalize these set of highly personalized treatments, we must address both approach-specific and cross-cutting challenges. ACT with autologous TILs may benefit from the standardization of TIL assessment in routine biopsies and the effective expansion of those TILs with the highest anti-tumor reactivity. Gene transfer-based TCR therapies increase antigen specificity but still fail to target those not presented by the MHC, whereas CAR engineering may provide additional versatility but entails elevated costs and significant on-target, off-tumor toxicity. Additionally, although DC and NK cell therapies may have not achieved relevant tumor responses, their better safety profile and reduced costs make them suitable companions for multimodal strategies. The successful transition of the different ACTs to the clinic poses a number of common considerations. The discovery of TAs that can guide ACT against BC is critically linked to its success and relies on comprehensive strategies integrating genomic sequencing, in silico prediction, and immunogenicity evaluation. Methodological refinement is also required to improve our ability to isolate immune components and modify them ex vivo and in vivo, and to enhance cell persistence and intratumor trafficking. Finally, clinical trials testing ACTs will progressively need to be more adaptable, explore the reliability of predictive biomarkers, and generate quality data from small sample sizes. Both puzzling and fascinating, this is the path ahead to materialize ACT and transform the therapeutic landscape of BC patients.
JF-A, KG-H, and AO contributed to the conception and scope of the study. JF-A and AO wrote the first draft. JF-A and KG-H composed the figures/tables. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank our colleagues from the Department of Medical Oncology at the San Carlos University Hospital for critical reading of the manuscript
1. Lord SJ, Kiely BE, Pearson S-A, Daniels B, O’Connell DL, Beith J, et al. Metastatic breast cancer incidence, site and survival in Australia, 2001–2016: a population-based health record linkage study protocol. BMJ Open (2019) 9. doi: 10.1136/bmjopen-2018-026414
2. Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev (2018) 32:1267–84. doi: 10.1101/gad.314617.118
3. Nagarajan D, McArdle SEB. Immune Landscape of Breast Cancers. Biomedicines (2018) 6. doi: 10.3390/biomedicines6010020
4. Nathan MR, Schmid P. The emerging world of breast cancer immunotherapy. Breast (2018) 37:200–6. doi: 10.1016/j.breast.2017.05.013
5. Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. New Engl J Med (2018) 379:2108–21. doi: 10.1056/NEJMoa1809615
6. ESMO. Encouraging Long-Term Survival Benefits with Atezolizumab Plus Nab-Paclitaxel in Metastatic Triple-Negative Breast Cancer . Available at: https://www.esmo.org/meetings/esmo-virtual-congress-2020/daily-reporter/daily-reporter-news/encouraging-long-term-survival-benefits-with-atezolizumab-plus-nab-paclitaxel-in-metastatic-triple-negative-breast-cancer (Accessed October 1, 2020).
7. Mittendorf EA, Zhang H, Barrios CH, Saji S, Jung KH, Hegg R, et al. Neoadjuvant atezolizumab in combination with sequential nab-paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy in patients with early-stage triple-negative breast cancer (IMpassion031): a randomised, double-blind, phase 3 trial. Lancet (2020) 396:1090–100. doi: 10.1016/S0140-6736(20)31953-X
8. Wright SE. Immunotherapy of breast cancer. Expert Opin Biol Ther (2012) 12:479–90. doi: 10.1517/14712598.2012.665445
9. Ueno NT, Rizzo JD, Demirer T, Cheng YC, Hegenbart U, Zhang M-J, et al. Allogeneic hematopoietic cell transplantation for metastatic breast cancer. Bone Marrow Transplant (2008) 41:537–45. doi: 10.1038/sj.bmt.1705940
10. Sparano JA, Fisher RI, Weiss GR, Margolin K, Aronson FR, Hawkins MJ, et al. Phase II trials of high-dose interleukin-2 and lymphokine-activated killer cells in advanced breast carcinoma and carcinoma of the lung, ovary, and pancreas and other tumors. J Immunother Emphasis Tumor Immunol (1994) 16:216–23. doi: 10.1097/00002371-199410000-00006
11. Bernhard H, Neudorfer J, Gebhard K, Conrad H, Hermann C, Nährig J, et al. Adoptive transfer of autologous, HER2-specific, cytotoxic T lymphocytes for the treatment of HER2-overexpressing breast cancer. Cancer Immunol Immunother (2008) 57:271–80. doi: 10.1007/s00262-007-0355-7
12. Wang K, Xu J, Zhang T, Xue D. Tumor-infiltrating lymphocytes in breast cancer predict the response to chemotherapy and survival outcome: A meta-analysis. Oncotarget (2016) 7:44288–98. doi: 10.18632/oncotarget.9988
13. Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell (2018) 174:1293–1308.e36. doi: 10.1016/j.cell.2018.05.060
14. Miyoshi Y, Shien T, Ogiya A, Ishida N, Yamazaki K, Horii R, et al. Associations in tumor infiltrating lymphocytes between clinicopathological factors and clinical outcomes in estrogen receptor−positive/human epidermal growth factor receptor type 2 negative breast cancer. Oncol Lett (2019) 17:2177–86. doi: 10.3892/ol.2018.9853
15. Luen SJ, Salgado R, Fox S, Savas P, Eng-Wong J, Clark E, et al. Tumour-infiltrating lymphocytes in advanced HER2-positive breast cancer treated with pertuzumab or placebo in addition to trastuzumab and docetaxel: a retrospective analysis of the CLEOPATRA study. Lancet Oncol (2017) 18:52–62. doi: 10.1016/S1470-2045(16)30631-3
16. Gao G, Wang Z, Qu X, Zhang Z. Prognostic value of tumor-infiltrating lymphocytes in patients with triple-negative breast cancer: a systematic review and meta-analysis. BMC Cancer (2020) 20:179. doi: 10.1186/s12885-020-6668-z
17. Denkert C, von Minckwitz G, Darb-Esfahani S, Lederer B, Heppner BI, Weber KE, et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: a pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol (2018) 19:40–50. doi: 10.1016/S1470-2045(17)30904-X
18. Ali HR, Provenzano E, Dawson S-J, Blows FM, Liu B, Shah M, et al. Association between CD8+ T-cell infiltration and breast cancer survival in 12 439 patients. Ann Oncol (2014) 25:1536–43. doi: 10.1093/annonc/mdu191
19. West NR, Milne K, Truong PT, Macpherson N, Nelson BH, Watson PH. Tumor-infiltrating lymphocytes predict response to anthracycline-based chemotherapy in estrogen receptor-negative breast cancer. Breast Cancer Res (2011) 13:R126. doi: 10.1186/bcr3072
20. Topalian SL, Muul LM, Solomon D, Rosenberg SA. Expansion of human tumor infiltrating lymphocytes for use in immunotherapy trials. J Immunol Methods (1987) 102:127–41. doi: 10.1016/S0022-1759(87)80018-2
21. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature (2013) 500:415–21. doi: 10.1038/nature12477
22. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable Complete Responses in Heavily Pretreated Patients with Metastatic Melanoma Using T-Cell Transfer Immunotherapy. Clin Cancer Res (2011) 17:4550–7. doi: 10.1158/1078-0432.CCR-11-0116
23. Tran E, Ahmadzadeh M, Lu Y-C, Gros A, Turcotte S, Robbins PF, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science (2015) 350:1387–90. doi: 10.1126/science.aad1253
24. Tran E, Robbins PF, Lu Y-C, Prickett TD, Gartner JJ, Jia L, et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med (2016) 375:2255–62. doi: 10.1056/NEJMoa1609279
25. Tran E, Turcotte S, Gros A, Robbins PF, Lu Y-C, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science (2014) 344:641–5. doi: 10.1126/science.1251102
26. Stevanović S, Pasetto A, Helman SR, Gartner JJ, Prickett TD, Howie B, et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science (2017) 356:200–5. doi: 10.1126/science.aak9510
27. Tran E, Robbins PF, Rosenberg SA. “Final common pathway” of human cancer immunotherapy: targeting random somatic mutations. Nat Immunol (2017) 18:255–62. doi: 10.1038/ni.3682
28. Zacharakis N, Chinnasamy H, Black M, Xu H, Lu Y-C, Zheng Z, et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med (2018) 24:724–30. doi: 10.1038/s41591-018-0040-8
29. Assadipour Y, Zacharakis N, Crystal JS, Prickett TD, Gartner JJ, Somerville RPT, et al. Characterization of an Immunogenic Mutation in a Patient with Metastatic Triple-Negative Breast Cancer. Clin Cancer Res (2017) 23:4347–53. doi: 10.1158/1078-0432.CCR-16-1423
30. Pilipow K, Darwich A, Losurdo A. T-cell-based breast cancer immunotherapy. Semin Cancer Biol (2020). doi: 10.1016/j.semcancer.2020.05.019
31. Karpanen T, Olweus J. T-cell receptor gene therapy – ready to go viral? Mol Oncol (2015) 9:2019–42. doi: 10.1016/j.molonc.2015.10.006
32. Zhao L, Cao YJ. Engineered T Cell Therapy for Cancer in the Clinic. Front Immunol (2019) 10:2250. doi: 10.3389/fimmu.2019.02250
33. Huang J, Brameshuber M, Zeng X, Xie J, Li Q, Chien Y, et al. A single peptide-major histocompatibility complex ligand triggers digital cytokine secretion in CD4(+) T cells. Immunity (2013) 39:846–57. doi: 10.1016/j.immuni.2013.08.036
34. Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood (2009) 114:535–46. doi: 10.1182/blood-2009-03-211714
35. Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan D-AN, Feldman SA, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther (2011) 19:620–6. doi: 10.1038/mt.2010.272
36. Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol (2011) 29:917–24. doi: 10.1200/JCO.2010.32.2537
37. Kageyama S, Ikeda H, Miyahara Y, Imai N, Ishihara M, Saito K, et al. Adoptive Transfer of MAGE-A4 T-cell Receptor Gene-Transduced Lymphocytes in Patients with Recurrent Esophageal Cancer. Clin Cancer Res (2015) 21:2268–77. doi: 10.1158/1078-0432.CCR-14-1559
38. Li Q, Liu M, Wu M, Zhou X, Wang S, Hu Y, et al. PLAC1-specific TCR-engineered T cells mediate antigen-specific antitumor effects in breast cancer. Oncol Lett (2018) 15:5924–32. doi: 10.3892/ol.2018.8075
39. Janssen A, Villacorta Hidalgo J, Beringer DX, van Dooremalen S, Fernando F, van Diest E, et al. γδ T-cell Receptors Derived from Breast Cancer-Infiltrating T Lymphocytes Mediate Antitumor Reactivity. Cancer Immunol Res (2020) 8:530–43. doi: 10.1158/2326-6066.CIR-19-0513
40. Yazdanifar M, Barbarito G, Bertaina A, Airoldi I. γδ T Cells: The Ideal Tool for Cancer Immunotherapy. Cells (2020) 9. doi: 10.3390/cells9051305
41. Wu Y, Kyle-Cezar F, Woolf RT, Naceur-Lombardelli C, Owen J, Biswas D, et al. An innate-like Vδ1+ γδ T cell compartment in the human breast is associated with remission in triple-negative breast cancer. Sci Trans Med (2019) 11. doi: 10.1126/scitranslmed.aax9364
42. Capietto A-H, Martinet L, Fournié J-J. Stimulated γδ T cells increase the in vivo efficacy of trastuzumab in HER-2+ breast cancer. J Immunol (2011) 187:1031–8. doi: 10.4049/jimmunol.1100681
43. Peng G, Wang HY, Peng W, Kiniwa Y, Seo KH, Wang R-F. Tumor-Infiltrating γδ T Cells Suppress T and Dendritic Cell Function via Mechanisms Controlled by a Unique Toll-like Receptor Signaling Pathway. Immunity (2007) 27:334–48. doi: 10.1016/j.immuni.2007.05.020
44. Meraviglia S, Eberl M, Vermijlen D, Todaro M, Buccheri S, Cicero G, et al. In vivo manipulation of Vgamma9Vdelta2 T cells with zoledronate and low-dose interleukin-2 for immunotherapy of advanced breast cancer patients. Clin Exp Immunol (2010) 161:290–7. doi: 10.1111/j.1365-2249.2010.04167.x
45. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol (2020) 17:147–67. doi: 10.1038/s41571-019-0297-y
46. Mirzaei HR, Rodriguez A, Shepphird J, Brown CE, Badie B. Chimeric Antigen Receptors T Cell Therapy in Solid Tumor: Challenges and Clinical Applications. Front Immunol (2017) 8:1850. doi: 10.3389/fimmu.2017.01850
47. Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther (2015) 15:1145–54. doi: 10.1517/14712598.2015.1046430
48. Sun M, Shi H, Liu C, Liu J, Liu X, Sun Y. Construction and evaluation of a novel humanized HER2-specific chimeric receptor. Breast Cancer Res (2014) 16:R61. doi: 10.1186/bcr3674
49. Globerson-Levin A, Waks T, Eshhar Z. Elimination of Progressive Mammary Cancer by Repeated Administrations of Chimeric Antigen Receptor-Modified T Cells. Mol Ther (2014) 22:1029–38. doi: 10.1038/mt.2014.28
50. Tóth G, Szöllősi J, Abken H, Vereb G, Szöőr Á. A Small Number of HER2 Redirected CAR T Cells Significantly Improves Immune Response of Adoptively Transferred Mouse Lymphocytes against Human Breast Cancer Xenografts. Int J Mol Sci (2020) 21. doi: 10.3390/ijms21031039
51. Wilkie S, van Schalkwyk MCI, Hobbs S, Davies DM, van der Stegen SJC, Pereira ACP, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol (2012) 32:1059–70. doi: 10.1007/s10875-012-9689-9
52. Bajgain P, Tawinwung S, D’Elia L, Sukumaran S, Watanabe N, Hoyos V, et al. CAR T cell therapy for breast cancer: harnessing the tumor milieu to drive T cell activation. J Immunother Cancer (2018) 6:34. doi: 10.1186/s40425-018-0347-5
53. Petrovic K, Robinson J, Whitworth K, Jinks E, Shaaban A, Lee SP. TEM8/ANTXR1-specific CAR T cells mediate toxicity in vivo. PLoS One (2019) 14:e0224015. doi: 10.1371/journal.pone.0224015
54. Zhou R, Yazdanifar M, Roy LD, Whilding LM, Gavrill A, Maher J, et al. CAR T Cells Targeting the Tumor MUC1 Glycoprotein Reduce Triple-Negative Breast Cancer Growth. Front Immunol (2019) 10:1149. doi: 10.3389/fimmu.2019.01149
55. Byrd TT, Fousek K, Pignata A, Szot C, Samaha H, Seaman S, et al. TEM8/ANTXR1-Specific CAR T Cells as a Targeted Therapy for Triple-Negative Breast Cancer. Cancer Res (2018) 78:489–500. doi: 10.1158/0008-5472.CAN-16-1911
56. Han Y, Xie W, Song D-G, Powell DJ. Control of triple-negative breast cancer using ex vivo self-enriched, costimulated NKG2D CAR T cells. J Hematol Oncol (2018) 11:92. doi: 10.1186/s13045-018-0635-z
57. VanSeggelen H, Hammill JA, Dvorkin-Gheva A, Tantalo DGM, Kwiecien JM, Denisova GF, et al. T Cells Engineered With Chimeric Antigen Receptors Targeting NKG2D Ligands Display Lethal Toxicity in Mice. Mol Ther (2015) 23:1600–10. doi: 10.1038/mt.2015.119
58. Tchou J, Wang L-C, Selven B, Zhang H, Conejo-Garcia J, Borghaei H, et al. Mesothelin, a novel immunotherapy target for triple negative breast cancer. Breast Cancer Res Treat (2012) 133:799–804. doi: 10.1007/s10549-012-2018-4
59. Song D-G, Ye Q, Poussin M, Chacon JA, Figini M, Powell DJ. Effective adoptive immunotherapy of triple-negative breast cancer by folate receptor-alpha redirected CAR T cells is influenced by surface antigen expression level. J Hematol Oncol (2016) 9:56. doi: 10.1186/s13045-016-0285-y
60. Wei J, Sun H, Zhang A, Wu X, Li Y, Liu J, et al. A novel AXL chimeric antigen receptor endows T cells with anti-tumor effects against triple negative breast cancers. Cell Immunol (2018) 331:49–58. doi: 10.1016/j.cellimm.2018.05.004
61. Lum LG, Thakur A, Al-Kadhimi Z, Colvin GA, Cummings FJ, Legare RD, et al. Targeted T-cell Therapy in Stage IV Breast Cancer: A Phase I Clinical Trial. Clin Cancer Res (2015) 21:2305–14. doi: 10.1158/1078-0432.CCR-14-2280
62. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther (2010) 18:843–51. doi: 10.1038/mt.2010.24
63. Tchou J, Zhao Y, Levine BL, Zhang PJ, Davis MM, Melenhorst JJ, et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol Res (2017) 5:1152–61. doi: 10.1158/2326-6066.CIR-17-0189
64. Specht JM, Lee S, Turtle C, Berger C, Veatch J, Gooley T, et al. Phase I study of immunotherapy for advanced ROR1+ malignancies with autologous ROR1-specific chimeric antigen receptor-modified (CAR)-T cells. JCO (2018) 36:TPS79–9. doi: 10.1200/JCO.2018.36.5_suppl.TPS79
65. Saleh RR, Antrás JF, Peinado P, Pérez-Segura P, Pandiella A, Amir E, et al. Prognostic value of receptor tyrosine kinase-like orphan receptor (ROR) family in cancer: A meta-analysis. Cancer Treat Rev (2019) 77:11–9. doi: 10.1016/j.ctrv.2019.05.006
66. Wylie B, Macri C, Mintern JD, Waithman J. Dendritic Cells and Cancer: From Biology to Therapeutic Intervention. Cancers (Basel) (2019) 11. doi: 10.3390/cancers11040521
67. Lissoni P, Vigore L, Ferranti R, Bukovec R, Meregalli S, Mandala M, et al. Circulating dendritic cells in early and advanced cancer patients: diminished percent in the metastatic disease. J Biol Regul Homeost Agents (1999) 13:216–9.
68. Ma Y, Shurin GV, Peiyuan Z, Shurin MR. Dendritic cells in the cancer microenvironment. J Cancer (2013) 4:36–44. doi: 10.7150/jca.5046
69. Satthaporn S, Robins A, Vassanasiri W, El-Sheemy M, Jibril JA, Clark D, et al. Dendritic cells are dysfunctional in patients with operable breast cancer. Cancer Immunol Immunother (2004) 53:510–8. doi: 10.1007/s00262-003-0485-5
70. Engelhardt JJ, Boldajipour B, Beemiller P, Pandurangi P, Sorensen C, Werb Z, et al. Marginating dendritic cells of the tumor microenvironment cross-present tumor antigens and stably engage tumor-specific T cells. Cancer Cell (2012) 21:402–17. doi: 10.1016/j.ccr.2012.01.008
71. Gong J, Avigan D, Chen D, Wu Z, Koido S, Kashiwaba M, et al. Activation of antitumor cytotoxic T lymphocytes by fusions of human dendritic cells and breast carcinoma cells. Proc Natl Acad Sci U S A (2000) 97:2715–8. doi: 10.1073/pnas.050587197
72. Neidhardt-Berard E-M, Berard F, Banchereau J, Palucka AK. Dendritic cells loaded with killed breast cancer cells induce differentiation of tumor-specific cytotoxic T lymphocytes. Breast Cancer Res (2004) 6:R322–328. doi: 10.1186/bcr794
73. Wang B, Zaidi N, He L-Z, Zhang L, Kuroiwa JMY, Keler T, et al. Targeting of the non-mutated tumor antigen HER2/neu to mature dendritic cells induces an integrated immune response that protects against breast cancer in mice. Breast Cancer Res (2012) 14:R39. doi: 10.1186/bcr3135
74. Sakai S, Kauffman KD, Sallin MA, Sharpe AH, Young HA, Ganusov VV, et al. CD4 T Cell-Derived IFN-γ Plays a Minimal Role in Control of Pulmonary Mycobacterium tuberculosis Infection and Must Be Actively Repressed by PD-1 to Prevent Lethal Disease. PLoS Pathog (2016) 12:e1005667. doi: 10.1371/journal.ppat.1005667
75. Banchereau J, Schuler-Thurner B, Palucka AK, Schuler G. Dendritic cells as vectors for therapy. Cell (2001) 106:271–4. doi: 10.1016/s0092-8674(01)00448-2
76. Brossart P, Wirths S, Stuhler G, Reichardt VL, Kanz L, Brugger W. Induction of cytotoxic T-lymphocyte responses in vivo after vaccinations with peptide-pulsed dendritic cells. Blood (2000) 96:3102–8. doi: 10.1182/blood.V96.9.3102.h8003102_3102_3108
77. Avigan D, Vasir B, Gong J, Borges V, Wu Z, Uhl L, et al. Fusion cell vaccination of patients with metastatic breast and renal cancer induces immunological and clinical responses. Clin Cancer Res (2004) 10:4699–708. doi: 10.1158/1078-0432.CCR-04-0347
78. Lowenfeld L, Mick R, Datta J, Xu S, Fitzpatrick E, Fisher CS, et al. Dendritic Cell Vaccination Enhances Immune Responses and Induces Regression of HER2pos DCIS Independent of Route: Results of Randomized Selection Design Trial. Clin Cancer Res (2017) 23:2961–71. doi: 10.1158/1078-0432.CCR-16-1924
79. Qi C-J, Ning Y-L, Han Y-S, Min H-Y, Ye H, Zhu Y-L, et al. Autologous dendritic cell vaccine for estrogen receptor (ER)/progestin receptor (PR) double-negative breast cancer. Cancer Immunol Immunother (2012) 61:1415–24. doi: 10.1007/s00262-011-1192-2
80. Baek S, Kim C-S, Kim S-B, Kim Y-M, Kwon S-W, Kim Y, et al. Combination therapy of renal cell carcinoma or breast cancer patients with dendritic cell vaccine and IL-2: results from a phase I/II trial. J Transl Med (2011) 9:178. doi: 10.1186/1479-5876-9-178
81. Cui Y, Yang X, Zhu W, Li J, Wu X, Pang Y. Immune response, clinical outcome and safety of dendritic cell vaccine in combination with cytokine-induced killer cell therapy in cancer patients. Oncol Lett (2013) 6:537–41. doi: 10.3892/ol.2013.1376
82. Ren J, Di L, Song G, Yu J, Jia J, Zhu Y, et al. Selections of appropriate regimen of high-dose chemotherapy combined with adoptive cellular therapy with dendritic and cytokine-induced killer cells improved progression-free and overall survival in patients with metastatic breast cancer: reargument of such contentious therapeutic preferences. Clin Transl Oncol (2013) 15:780–8. doi: 10.1007/s12094-013-1001-9
83. Grossenbacher SK, Canter RJ, Murphy WJ. Natural killer cell immunotherapy to target stem-like tumor cells. J Immunother Cancer (2016) 4:19. doi: 10.1186/s40425-016-0124-2
84. Martinet L, Smyth MJ. Balancing natural killer cell activation through paired receptors. Nat Rev Immunol (2015) 15:243–54. doi: 10.1038/nri3799
85. Eguizabal C, Zenarruzabeitia O, Monge J, Santos S, Vesga MA, Maruri N, et al. Natural killer cells for cancer immunotherapy: pluripotent stem cells-derived NK cells as an immunotherapeutic perspective. Front Immunol (2014) 5:439. doi: 10.3389/fimmu.2014.00439
86. Klingemann H-G, Martinson J. Ex vivo expansion of natural killer cells for clinical applications. Cytotherapy (2004) 6:15–22. doi: 10.1080/14653240310004548
87. Hu W, Wang G, Huang D, Sui M, Xu Y. Cancer Immunotherapy Based on Natural Killer Cells: Current Progress and New Opportunities. Front Immunol (2019) 10:1205. doi: 10.3389/fimmu.2019.01205
88. Krause SW, Gastpar R, Andreesen R, Gross C, Ullrich H, Thonigs G, et al. Treatment of colon and lung cancer patients with ex vivo heat shock protein 70-peptide-activated, autologous natural killer cells: a clinical phase i trial. Clin Cancer Res (2004) 10:3699–707. doi: 10.1158/1078-0432.CCR-03-0683
89. Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin Cancer Res (2011) 17:6287–97. doi: 10.1158/1078-0432.CCR-11-1347
90. Tian X, Wei F, Wang L, Yu W, Zhang N, Zhang X, et al. Herceptin Enhances the Antitumor Effect of Natural Killer Cells on Breast Cancer Cells Expressing Human Epidermal Growth Factor Receptor-2. Front Immunol (2017) 8:1426. doi: 10.3389/fimmu.2017.01426
91. Geller MA, Cooley S, Judson PL, Ghebre R, Carson LF, Argenta PA, et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy (2011) 13:98–107. doi: 10.3109/14653249.2010.515582
92. Klingemann H. Are natural killer cells superior CAR drivers? Oncoimmunology (2014) 3. doi: 10.4161/onci.28147
93. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med (2020) 382:545–53. doi: 10.1056/NEJMoa1910607
94. Hu Z. Tissue factor as a new target for CAR-NK cell immunotherapy of triple-negative breast cancer. Sci Rep (2020) 10:2815. doi: 10.1038/s41598-020-59736-3
Keywords: adoptive cell therapy, breast cancer, TIL, TCR, CAR, dendritic cell, natural killer cell, tumor antigen
Citation: Fuentes-Antrás J, Guevara-Hoyer K, Baliu-Piqué M, García-Sáenz J́Á, Pérez-Segura P, Pandiella A and Ocaña A (2020) Adoptive Cell Therapy in Breast Cancer: A Current Perspective of Next-Generation Medicine. Front. Oncol. 10:605633. doi: 10.3389/fonc.2020.605633
Received: 12 September 2020; Accepted: 07 October 2020;
Published: 27 October 2020.
Edited by:
Cesar Augusto Santa-Maria, Johns Hopkins Medicine, United StatesReviewed by:
John Maher, King’s College London, United KingdomCopyright © 2020 Fuentes-Antrás, Guevara-Hoyer, Baliu-Piqué, García-Sáenz, Pérez-Segura, Pandiella and Ocaña. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jesús Fuentes-Antrás, amZ1ZW50ZXNhQHNhbHVkLm1hZHJpZC5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.