 Alessandro Barbato1
Alessandro Barbato1 Grazia Scandura2
Grazia Scandura2 Fabrizio Puglisi1Daniela Cambria1Enrico La Spina2
Fabrizio Puglisi1Daniela Cambria1Enrico La Spina2 Giuseppe Alberto Palumbo3Giacomo Lazzarino4
Giuseppe Alberto Palumbo3Giacomo Lazzarino4 Daniele Tibullo5*
Daniele Tibullo5* Francesco Di Raimondo2
Francesco Di Raimondo2 Cesarina Giallongo3
Cesarina Giallongo3 Alessandra Romano6*
Alessandra Romano6*- 1Department of Clinical and Experimental Medicine, University of Catania, Catania, Italy
- 2Department of General Surgery and Medical-Surgical Specialties, University of Catania, Catania, Italy
- 3Department of Medical, Surgical Sciences and Advanced Technologies G.F. Ingrassia, University of Catania, Catania, Italy
- 4Saint Camillus International University of Health and Medical Sciences, Rome, Italy
- 5Department of Biotechnological and Biomedical Sciences, University of Catania, Catania, Italy
- 6Department of Surgery and Medical Specialties, University of Catania, Catania, Italy
The combined derangements in mitochondria network, function and dynamics can affect metabolism and ATP production, redox homeostasis and apoptosis triggering, contributing to cancer development in many different complex ways. In hematological malignancies, there is a strong relationship between cellular metabolism, mitochondrial bioenergetics, interconnections with supportive microenvironment and drug resistance. Lymphoma and chronic lymphocytic leukemia cells, e.g., adapt to intrinsic oxidative stress by increasing mitochondrial biogenesis. In other hematological disorders such as myeloma, on the contrary, bioenergetics changes, associated to increased mitochondrial fitness, derive from the adaptive response to drug-induced stress. In the bone marrow niche, a reverse Warburg effect has been recently described, consisting in metabolic changes occurring in stromal cells in the attempt to metabolically support adjacent cancer cells. Moreover, a physiological dynamic, based on mitochondria transfer, between tumor cells and their supporting stromal microenvironment has been described to sustain oxidative stress associated to proteostasis maintenance in multiple myeloma and leukemia. Increased mitochondrial biogenesis of tumor cells associated to acquisition of new mitochondria transferred by mesenchymal stromal cells results in augmented ATP production through increased oxidative phosphorylation (OX-PHOS), higher drug resistance, and resurgence after treatment. Accordingly, targeting mitochondrial biogenesis, electron transfer, mitochondrial DNA replication, or mitochondrial fatty acid transport increases therapy efficacy. In this review, we summarize selected examples of the mitochondrial derangements in hematological malignancies, which provide metabolic adaptation and apoptosis resistance, also supported by the crosstalk with tumor microenvironment. This field promises a rational design to improve target-therapy including the metabolic phenotype.
Introduction
Since the first description by Rudolf Albrecht von Kölliker in 1857, scientists have explored the essential roles of mitochondria in cell biology, as the powerhouse of the cells able to produce comparing weight to weight, thousands of times more energy per second as compared to sun production (1). Mitochondria can fuel cellular energy demands by using as substrate pyruvate, arising from glycolysis or lipolysis coupled to β-oxidation of fatty acids (FA), in the oxidative-phosphorylation (OX-PHOS) process coupled to the electron transport chain (ETC).
The combined derangements in mitochondria network function and dynamics can affect metabolism and ATP production, redox homeostasis and apoptosis triggering, contributing to cancer development in many different complex ways (2). In cancer, there is a gap in knowledge about the protein composition, structure and dynamics of lipid droplet–mitochondria structures and how bidirectional FAs exchange occur, even if the strong relationship between cellular metabolism, mitochondrial bioenergetics, and tumorigenesis development is an established emerging hallmark (2, 3).
There is a growing evidence that the metabolic reprogramming required in cancers to fuel the increased energy demand is coupled to the increased ability to evade apoptosis (2, 4). Modulating ATP availability might be an essential strategy in inducing cell resistance and sustaining cancer progression and growth (4). Recent findings have demonstrated that cancer cells take advantage of high OXPHOS, including leukemias, lymphomas, pancreatic ductal adenocarcinoma, melanoma, and endometrial carcinoma (5–16), while mitochondria can modulate their morphology regulating the intrinsic apoptotic pathway and participating in the resistance of cancer cells to apoptotic stimuli (17–27).
In this review, we will summarize the most advanced body of knowledge about the mitochondrial derangements in hematological malignancies which provide metabolic adaptation and apoptosis resistance, with a particular focus on the implication of novel relevant targets to reduce the risk of recurrence (Figure 1).
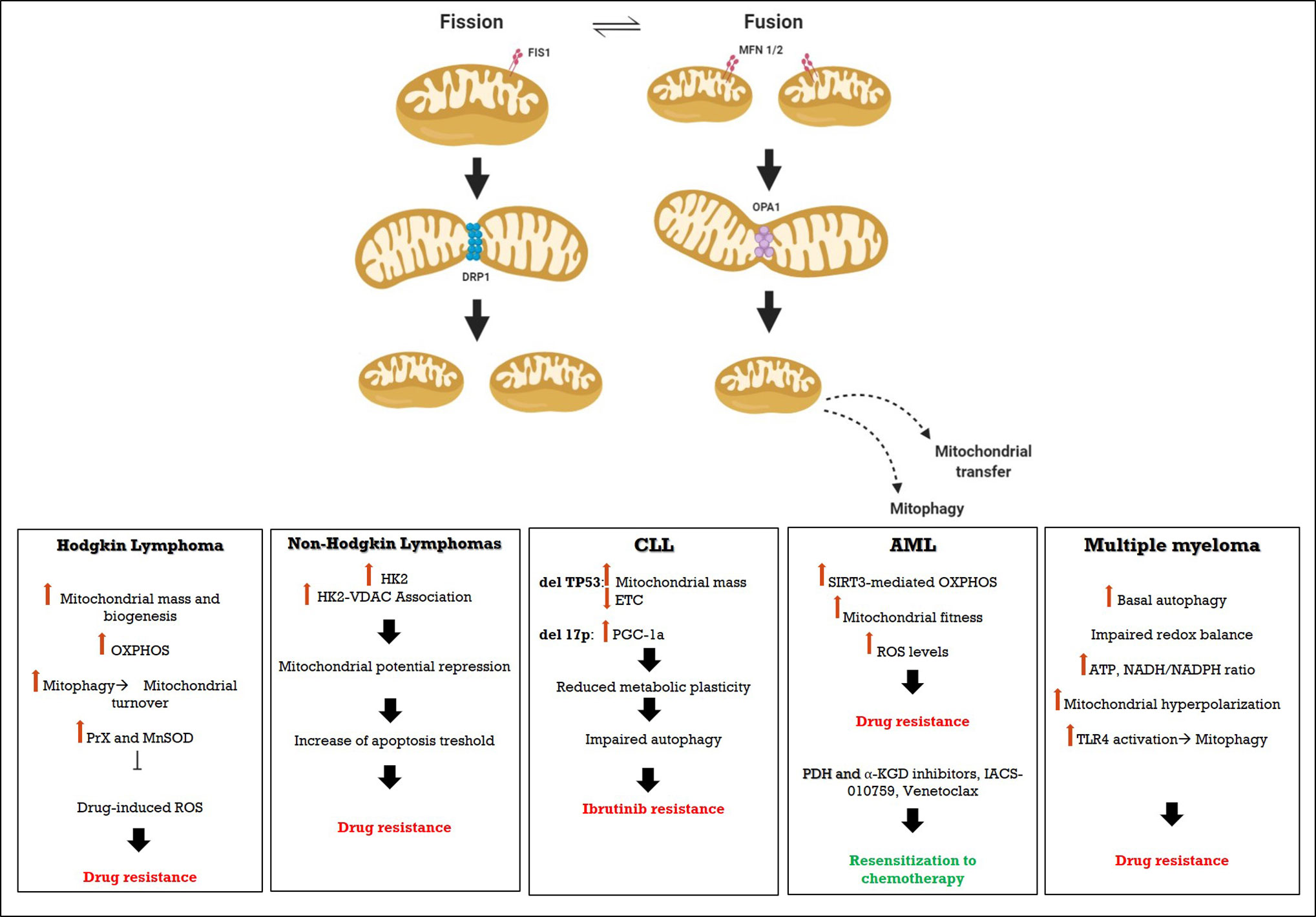
Figure 1 Schematic representation of Mitochondrial involvement in drug restistance in hematological malignancies.
Mitochondria and Cancer Cell Bioenergetics
Mitochondria and ATP Production: The Oxidative-Phosphorylation Process
The oxidative-phosphorylation (OX-PHOS) process is the electron transfer chain (ETC) driven by substrate oxidation (e.g pyruvate) coupled to the synthesis of ATP through an electrochemical transmembrane gradient. OX-PHOS is carried in the internal mitochondrial membrane (IMM) where four big multi-protein complexes, that contain flavins, iron-sulfur clusters, heme proteins, copper (Cu) structures, are organized to provide energy transformations in the ETC, namely Complex I, Complex II, Complex III Complex IV, as described in recent review for more details (20–22, 28–30), which operate together to generate water and a proton gradient, in presence of oxygen. The energy accumulated in the form of electrochemical proton gradient is used by F0-F1 ATP synthase (called Complex V also) to produce ATP from ADP and phosphate (4, 28, 31, 32).
Complex I (NADH–CoQ reductase), the largest respiratory complex, catalyzes the electron transfer from NADH to the CoQ (ubiquinone), thus to regenerate NAD+ levels oxidizing the NADH, which is formed both during the tricarboxylic acid (TCA) cycle and β-oxidation of fatty acids (31, 33). Complex moves four protons from the mitochondrial matrix to the intermembrane space generating a potential decrease of 360 mV (33).
Complex II (succinate dehydrogenase or succinate-ubiquinone oxidoreductase), a membrane-bound component of the Krebs cycle, permits the oxidation of the metabolite succinate to fumarate, transferring two protons to CoQ, trough the FAD/FADH2 coenzymes, thus to coupling the ETC and TCA cycle (34, 35).
Complex III (coenzyme Q: cytochrome c-oxidoreductase) catalyzes the reduction of cytochrome c, provides two protons to the CoQH2, originating from complex I and II to regenerate the CoQ, and pumps 4 protons from the mitochondrial matrix to the intermembrane space. The resulting so-called Q cycle allows the formation of a proton gradient across the membrane: oxidation of CoQ pumps four protons into the intermembrane space (positive side), two protons are taken up from the matrix (negative side) and two electrons are transferred from the ubiquinol to the ubiquinone, via two cytochrome c intermediates, to complete the cytochrome c reduction (36).
Complex IV (cytochrome c oxidase) transfers electrons from cytochrome c to oxygen to generate water and a proton gradient; its assembly and deregulation has been extensively reviewed recently (37).
Complex V consists of two main subunits: F0 is the transmembrane unit that works like a proton-driven turbine; F1 is the catalytic subunit located on the mitochondrial matrix side (38).
As a whole, OX-PHOS couples energy demands to the structural integrity of mitochondria within the cells. The membrane potential is essential for mitochondrial functions for cell survival and is well used by cancer cells to trigger molecular changes to make mitochondria network more efficient to provide energy requirements and resilience to changes in the redox status, induced by increased proliferation rate, and consequent increased nucleotide and lipid synthesis and/or drug exposure.
Mitochondria Network and Response to Dynamic Energy Demand: Fusion, Lipid Droplets (LDs) Trafficking, and Fission
Mitochondria are small organelles devoted to homeostasis and redox status maintenance within the cell, through cellular respiration, with consequent ATP production, heat production, biosynthesis of lipids and iron-containing prosthetic group heme enzymes, and apoptosis control (39, 40). These processes happen simultaneously in different compartments, identifiable by ultrastructural hallmarks, including double lipid membranes and inner membrane folds forming “cristae”. The outer mitochondrial membrane (OMM) is a double phospholipid membrane separating the inside of the organelle from the rest of the cell while the inner mitochondrial membrane (IMM), separates the inter-membrane space from the central matrix, the site of the electron transport chain (ETC). The IMM and the OMM enclose the intermembrane space (IMS) of mitochondria.
The adaptation of mitochondrial morphology to cellular bioenergetics occurs at IMM by remodeling of mitochondrial cristae (5, 17, 41). The number of mitochondria in the cell and their distribution in the mitochondrial networks is regulated by two interconnected and highly dynamic processes: the fusion, which allows the merge of two mitochondria into one, and the fission, which allows the division of one mitochondrion in two daughter mitochondria, in response to ATP request or to release of cytochrome C, leading to cell death (18, 20–22, 42).
In metabolic active cells, mitochondrial fusion is tightly regulated by three GTP-ase enzymes of the dynamin superfamily: Mfn1 (Mitofusin-1), Mfn2 (Mitofusin-2), localized on the OMM, and OPA1 (optic atrophy 1), associated with the IMM.
In highly-energy demanding cells not all mitochondria work with the same efficiency (43, 44) and a balance between fluctuating energy demands, energy storage in LDs and utilization in mitochondria is required. Indeed, “peridroplet” mitochondria, characterized by elevated Krebs cycle activity but low FA oxidation capacity, support LDs biogenesis and protect the cell from lipotoxicity, while non-lipid droplet-bound cytoplasmic mitochondria are addressed to FA oxidation and energy production (43). The presence of two kind of mitochondria networks become relevant when starvation occurs and autophagy, which leads to bulk release of FAs, is triggered. In these scenarios, LDs provide a lipid buffering system that sequesters FAs released during the autophagic degradation of membranous organelles, reducing mitochondria lipotoxicity. In turn, FAs are removed by cytoplasmatic lipases to enable their transfer to mitochondria to provide an “on-demand” source of fatty acids for bulk ATP production (44), only in highly fused peridroplet mitochondria. If mitochondrial fusion is prevented, FAs could not be efficiently metabolized and are re-associated with LDs and fluxed. Thus, relevant to cancer metabolism, mitochondrial fusion dynamics ensures maximum oxidative metabolism and avoids FA toxicity in starved cells (44). As shown in murine brown adipose cells, lack of Mfn2 induces LDs accumulation and mitochondrial dysfunction (45). Indeed, mitochondrial fusion is required for β-oxidation of fatty acids (FA) and mitochondrial respiration (46); otherwise, LDs accumulate and FA efflux into neighboring cells (44).
The trafficking of FA between mitochondria and LDs is bidirectional, regulated by the lipid droplet-coating proteins PLIN1 and PLIN5 that, if overexpressed, promote clustering of mitochondria around LDs, by binding Mfn2 (43). In basal state, Plin1 and Plin5 participate to accumulate palmitate into triglycerides to limit its utilization by the mitochondria, by inhibiting hydrolysis and stabilizing the lipid droplet. In starvation, as consequence of protein kinase A-stimulated triggering, LD hydrolysis inhibition is lifted, and FAs are released from LDs to undergo β-oxidation in mitochondria (47, 48). This is associated to a transcriptional signaling, as well. Indeed, in response to starvation-induced lipolysis and protein kinase A-dependent translocation and enrichment of PLIN5 in the nucleus, transcriptional complexes with sirtuin 1 (SIRT1) and peroxisome proliferator-activated receptor γ co-activator 1α (PGC1α) can activate Nrf2-ARE system and promote transcription of target genes involved in mitochondrial biogenesis and oxidative metabolism (49). Finally, during starvation and autophagy activation, LDs can remove damaged lipids and proteins from mitochondria, to delay mitochondria fission and apoptosis triggering (43, 44, 49–52).
In resting cells, when metabolic requirements are reduced or when there is an insult leading to increased oxidative stress (e.g., oxidative damage, overcharge of ROS levels), mitochondrial fission is carried out by multi proteins machineries, such as the GTP-molecules Fis1 (Mitochondrial fission protein 1), which is integrated into the OMM, and Drp1 (Dynamin-related protein 1). Fis1 allows Drp1 to shuttle from the cytosol to the OMM, where it forms a ring that drives the division of the organelle, changing its function and structure, including mitochondrial outer membrane permeabilization, calcium influx and cytochrome c release. The resulting modifications occurring in both proteins and lipids damage mitochondria structures and induce the collapse of the mitochondrial membrane potential. The increased number of fragmented organelles in the mitochondrial network can then to be removed by a specialized form of autophagy, called mitophagy (13, 22, 26, 42, 53). Thus, mitophagy It plays a major role in maintaining a proper mitochondrial turnover by degrading damaged organelles and promoting a stable cellular pool of excellent working organelles (20, 27, 54), leading to ineffectiveness of drugs impairing mitochondrial function and consequently to chemotherapy resistance in cancer.
Warburg Effect and its Relationship to Mitochondrial Metabolism
In cancer cells, the rate of glucose uptake is dramatically increased with consequent high secretion of lactate to support malignant cell proliferation. This process, known as Warburg Effect, occurs in the presence of oxygen and performing mitochondria (55). Indeed, in the recent decade it has been demonstrated that while glycolysis is drastically increased in tumor cells, mitochondria fitness continues to operate normally (PMID: 25277420). This upregulation of glycolysis is not just for ATP production, but also for synthesis of biomass and the production of NADPH to reduce ROS and oxidative stress.
A symbiotic relationship exists among tumor and cancer-associated fibroblasts (CAFs). In this “two-compartment” model, cancer cells and CAFs become metabolically coupled (reverse Warburg effect). High production of ROS in tumor cells promotes the oxidative stress in CAFs inducing their metabolic reprogramming associated to increased aerobic glycolysis and production of energy-rich fuels such as pyruvate, lactate and fatty acids, which in turn support the OX-PHOS in cancer cells (56). Conversely, cancer cells take up these energy-rich metabolites, which in turn enter in the tricarboxylic acid (TCA) pathway, sustain ATP production by OX-PHOS, and in overall increase cell fitness for cell growth and migration (57–63).
Relevant for hematological malignancies, in the bone marrow there is a physiological dynamic, inverse metabolic state, based on mitochondria transfer, between hematopoietic stem and progenitor cells and their supporting stromal microenvironment during quiescence, proliferation and differentiation of these two populations (64, 65), in response to lactate in the extracellular space (66).
This mitochondrial transfer has been described to sustain oxidative stress associated to proteostasis maintenance in multiple myeloma (67, 68) and acute myeloid leukemia, associated to drug resistance and disease recurrence (39, 69–74).
Increased OX-PHOS in Hodgkin Lymphoma is Associated to Reverse Warburg Effect Promoting Drug Resistance
Hodgkin lymphoma (HL) is a hematopoietic neoplasm generated from B-cells, affecting secondary lymphoid tissues such as lymph nodes and spleen. Despite recent advances in the biological knowledge and targeted therapy, almost a quarter of lymphoma patients relapse, due to the complex interactions between residual neoplastic cells and the microenvironment, and the emergence of metabolic adaptive responses which mediate drug resistance and contribute to clonal selection, at different times and in the different sites of the same patient (75–79).
In classic HL (cHL) a few neoplastic cells (Hodgkin and Reed-Stenberg HRS cells) are surrounded by an inflammatory microenvironment of accessory myeloid and lymphoid cells (80–86). Compared to the normal counterpart of B cells deriving from (post-)germinal center (GC) cells, HRS have more mitochondrial mass, e.g., more TOMM20 (Transporter of the outer mitochondrial membrane 20) and mitochondrial biogenesis proteins, upregulate some OX-PHOS key proteins (11), have dismal lactate production (87), but increased expression of lactate importer MCT1 (monocarboxylate transporter 1) (11). As consequence, HRS metabolic profile is characterized by high ATP production, as consequence of high OX-PHOS (87).
Increased OX-PHOS is associated to increased basal autophagy flux and increased mitochondrial turnover via mitophagy, which provides to maintain high quality mitochondria and metabolic intermediates conveying drug resistance (88). Carbon skeletons can be further provided by the supportive microenvironment, since the surrounding cells in HL exhibit high-glycolytic activity, associated to increased lactate dehydrogenase activity and lactate release, with increased lactate exporter MCT4 expression (11). The induction of Warburg effect in the microenvironment and the reverse Warburg effect in HRS cells could mediate drug resistance to chemotherapy drugs that disrupt OX-PHOS function such as doxorubicin or affect the cellular redox state (11, 89).
Drugs commonly used in the first-line treatment, such as bleomycin, doxorubicine, and vinblastine, part of ABVD regimen (85, 90–92), generate large amount of reactive oxygen species, causing oxidative stress and apoptosis (93), but increasing at the same time the expression of antioxidant enzymes that could contribute to chemoresistance in HRS cells. Bur et al., assessed expression of peroxiredoxin (Prx) II, Prx III, Prx V, Prx VI, and manganese superoxide dismutase (MnSOD) in 99 cases of uniformly treated HL. Prxs I–VI participate in cellular antioxidant defense by reducing alkyl hydroperoxides and hydrogen peroxide to the corresponding alcohol and water, while MnSOD catalyzes the dismutation of superoxide to hydrogen peroxide and oxygen and is the most important antioxidant enzyme in mitochondria, where oxidative stress is most evident under physiological circumstances, owing to oxidative phosphorylation (94). Data reported by Bur et al., suggest that the induction of mitochondrial located antioxidant enzymes (MnSOD and Prx III) in both HRS cells and in reactive cellular infiltrate is significantly induced in the most aggressive cases. The evidence of a low rate of complete response to ABVD treatment in patients with low Prx V expression is therefore in line with the role of oxidative stress in the mechanism of action of these drugs. More precisely, all patients with low cytoplasmic Prx V expression in RS cells achieved CR, whereas the CR rate was highly low in those with high cytoplasmic Prx V expression in RS cells (95). Thanks to their mitochondria, HRS can sustain substantial amount of oxidative stress, and, in line with this, mitochondrial Prx V expression is related to a poor response to ABVD chemotherapy.
The reverse Warburg effect in cHL could be overcome by drugs which target glycolysis in the microenvironment (e.g., arsenic or metformin) which become synergic with other agents directed against the crosstalk between neoplastic cells and the environment, like check-point inhibitors (96, 97). The high-glycolytic activity in HL microenvironment is clinically relevant, since and associated to prognostic meaning of 18-FDG-PET persistent positivity after first cycles of chemotherapy (81–83, 98).
Taken together these data reflect the high involvement of mitochondria in the resistance to the drugs commonly used for the treatment of cHL, making them a highly favorable target for therapeutic manipulation via biguanides and metformin (99).
OX-PHOS Identifies Metabolic Subtypes of B-Cell Non-Hodgkin Lymphomas
Non-Hodgkin lymphoma (NHL) includes quite heterogeneous group of blood neoplasms that differ for metabolism, cell of origin, clinical course, and response to treatment (79, 100–102). The relationship between metabolic pathway differences and drug resistance has led to an increasing interest in metabolic mechanisms important for lymphoma cells surviving.
A recent in vitro study about nine different B cell NHL cell lines has revealed their capacity to use glucose or glutamine as source of energy to sustain high proliferative rate. The capacity to use different substrates is related to differences in metabolic pathways. Particularly, glutamine-addicted cells use mitochondrial metabolism, while glucose-addicted cells have glycolytic metabolism also in presence of oxygen (Warburg effect), while cells that can use glutamine or glucose equally have a higher metabolic plasticity that allows them to use one or another pathway depending on the substrates availability (103).
In diffuse large B cell lymphoma (DLBCL), metabolic diversity is related to different expression profiles identified as a three-consensus cluster today used for DLBCL classification: B cell receptor (BCR)/proliferation cluster (BRC-DLBCL), oxidative phosphorylation cluster (OX-PHOS DLBCL), and host response (HR) cluster (102). Indeed, BCR-DLBCLs shows a higher expression of many component of the BCR signaling (102, 104, 105) and it is linked to the downstream PI3K/AKT/mTORC1 pathway involved in regulation of pro-survival factors, glucose acquisition, and glycolysis flux activation (14, 106, 107). Conversely, OX-PHOS DLBCLs shows an increase in mitochondrial activity and mitochondrial fatty acid oxidation (14). However, lymphoma cells show an important metabolic plasticity, and acute inhibition of BCR signaling increases glutamine catabolism fuelling the TCA cycle and palmitate-induced mitochondrial oxygen consumption (14). Increased contribution of mitochondria for energy production is related to differential activity or efficiency of mitochondrial ETC complexes which are encoded by nuclear and mitochondrial independently transcribed and translated genomes, with the exception of complex II (9, 14). The high expression of components of mitochondrial translation machinery is fundamental for OX-PHOS cells maintenance, so inhibition of mitochondrial translation machinery causes ROS production responsible of cell death (9). Moreover, lymphoma cell lines are able to regulate their metabolic activity in relation of oxygen availability. Indeed, OX-PHOS DLBCL can become resistant to hypoxic stress thanks to an increasing of Hexokinase II (HK2) expression upon eIF4E1 and HIF1α regulation, related to a high involvement of glycolysis pathway (108, 109). Gu and colleagues found that rituximab resistance in cell lines of lymphoma (110, 111) was associated to the impaired glucose metabolism, due to overexpression of HK2, which interacts with the protein of the mitochondrial outer membrane VDAC (voltage-dependent anion channel), to repress the mitochondrial membrane potential and increase the mitochondrial apoptosis threshold (112). Targeting HK2 resulted in decreased mitochondrial membrane potential, ATP production, cell viability, and re-sensitization to chemotherapy agents, suggesting that overexpression of HK2 could be a novel potential therapeutic target in rituximab-refractory lymphomas (111). Analysis of expression profile of newly diagnosed DLBCL has showed that glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is related to metabolic profile of lymphoma cells. Particularly, GAPDH expression is significantly correlated with the percentage of ATP generated from glycolysis, so low GAPDH level is related to oxygen consumption in OX-PHOS -DLBCL while high GAPDH level is related to lactate production in BCR-DLBCLs. Moreover, the increased activation of mTORC1 activity in OX-PHOS-DLBCL is associated with the increase in glutamine transport rate, reduction of intracellular metabolites involved in glycolysis (G6P, G3P, and lactate) and non-oxidative arm of the pentose phosphate pathway (113).
The not-uniform metabolic behavior of DLBLC has clinical implications: first, the limited role of early PET positivity during treatment, that could reflect residual glycolytic activity of neoplastic versus microenvironment cells, based on cell of origin; second, the suboptimal results of lymphoma treatment, as shown by the association between relation between gene expression related to mitochondrial energetic function and R-CHOP resistance (113); third, the metabolic rewiring associated to the residual activity of the B-cell receptor. Casola and colleagues demonstrated that the two lymphoma clusters have a different fitness. Indeed, BCR+ cell lines have a higher competitive fitness than BCR- counterparts thanks to BCR/PI3Kδ axis activation, which induces glycogen synthase kinase-3β phosphorylation (114) and regulates the transcriptional program, under MYC control, for the expression of OX-PHOS genes, which use carbon-skeleton of glutamine to fuel TCA cycle. Indeed, the competitive advantage of BCR- clones on BCR+ clones was due to increased glutamine catabolism, that could be observed also in absence of BCR/PI3Kδ axis through a constitutive activation of RAS/MAPK pathway (115).
Metabolic Rewiring in Chronic Lymphatic Leukemia
Chronic Lymphocytic leukemia (CLL) is due to the clonal expansion and accumulation of malignant B-cell lymphocytes in the blood stream and in homing tissues, such as bone marrow and lymphoid organs. Circulating CLL lymphocytes are quiescent and dependent on intrinsic survival factors and proliferate when they enter homing tissues, revealing a challenge for the design of therapeutic interventions that target intrinsic survival pathways (116). Circulating and homing require a plastic metabolic rewiring that allows cells to modify their metabolism and fulfill the requirements needed to sustain survival, differentiation or proliferation (117, 118). To this end, the number of mitochondria, the total mitochondrial mass, biogenesis, bioenergetics (basal, maximal, and ATP-linked respiration rates), membrane potential and ROS are increased in CLL cells compared to naïve B-lymphocytes (119). As discussed above, also CLL cells use preferentially the reverse Warburg effect, relying primarily on OX-PHOS for generating energy (120). Clinically relevant, 18-FDG-PET is not always informative to evaluate disease burden and it is indicated only when Richter’s Syndrome or transformation to another aggressive B-cell lymphoma is suspected (121).
Using NanoString technology, it was shown that CLL lymphocytes display heightened expression of mitochondrial IDH3 and citrate transporter (SLC25A1) which yield α-ketoglutarate from isocitrate and cytoplasmic export of citrate respectively (122). ZAP-70+ CLL cells exhibited significantly higher bioenergetics than B lymphocytes or ZAP-70- CLL cells and were more sensitive to the uncoupler, carbonyl cyanide-p-trifluoro-methoxyphenylhydrazone (FCCP). Univariable and multivariable linear regression analysis demonstrated that ZAP-70+ predicted increased maximal respiration. ZAP-70+ is a surrogate for B cell receptor (BCR) activation and can be targeted by ibrutinib, which is a clinically approved Bruton’s tyrosine kinase (BTK) inhibitor. Ibrutinib-treated patients exhibit decreased oxygen consumption rates (OCR) of CLL cells. similar to control B lymphocytes, suggesting that drug treatment resets the mitochondrial bioenergetics (117). Increased OX-PHOS is a resistance mechanism to BCL-2 inhibitor venetoclax, suggesting that the implementation of combinatorial therapy with metabolic modulators may overcome drug resistance (123).
Mitochondrial Fitness Mediates Resistance to Bortezomib in Multiple Myeloma
Multiple Myeloma (MM) is a neoplastic plasma cell disorder characterized by a complex array of clinical manifestations, including hypercalcemia, renal dysfunction, anemia, and bone lesions (collectively known as CRAB symptoms), in a wide spectrum of clinical variants ranging from benign MGUS and smoldering/indolent MM, to more aggressive, disseminated forms of MM and plasma cell leukemia (124). There is no a unique driver genetic event in MM onset, but a complex variety of chromosomal and genomic rearrangements (125), occurring at different timepoints in response to external driving forces (e.g., exposure to microbes, chronic antigen stimulation, oxidative stress). Among the most frequent mutated genes, FAM46C has been involved in both mitochondrial and bioenergetics dysfunction associated to drug resistance (126), proteostasis (127), and disease onset.
Physiologically, in the plasma cell (PCs) ontogeny, immunoglobulin synthesis and survival of competitive clones relies on preserved cell bioenergetics. In response to increased poli-ubiquitinated proteins requiring autophagy triggering (128), long-lived PCs robustly engage pyruvate-dependent respiration and rely on OX-PHOS whereas their short-lived counterparts could not (129). Thus, the transition from plasmablast to short-lived and long-lived PCs is associated to increased autophagy fluxes to allow removal of damaged mitochondria and lipid droplets accumulation to maintain protein and lipid homeostasis (44). Several groups have recently showed that integrity of mitochondrial function relies on p62 to limit oxidative stress, and conversely, lack of p62 is associated with inhibited complex I mitochondrial respiration. As consequence, reduced efficiency of electron transport chain (ETC) is associated to metabolic derangement inducing pentose phosphate pathway and increased cytosolic reduced glutathione (GSH) levels. Conversely, complex I inhibition resulted in lower mitochondrial membrane potential and higher cytosolic ROS production. Pharmacological activation of transcription factor Nrf2 increased mitochondrial NADH levels and restored mitochondrial membrane potential in p62-deficient cells (130).
Relying on the detoxifying mitochondrial function is consequence of other two aberrant metabolic changes occurring in MM: the oxidative stress, consequence of the aberrant protein synthesis of incomplete immunoglobulins with defective glycosylation (131), and the lack of glutamine synthetase which confers increased ammonium production and requires nitrates detoxification (132).
In relapsed and refractory patients MM PCs overexpress mitochondrial biogenesis signatures regulated by the cellular iron content (133). The consequent loss of integrity of redox balance, with the nuclear compartmentalization of heme-oxygenase 1 is associated to drug resistance and genomic instability (134). MM can intake iron to increase their scavenger antioxidant-related genes and mitochondrial mass. Iron trafficking, by modifying energetic metabolism of cancer cells and impairing inflammatory status of macrophages in the microenvironment, is a critical regulator to reshape the MM tumor niche (135). However, to make the picture more complex, MM cell lines are characterized by distinct ferritin levels, which directly correlate with bortezomib resistance and pre-treatment with ferric ammonium citrate (FAC) decreased bortezomib sensitivity in vitro (135).
Bioenergetics changes, associated to increased mitochondrial biomass and function, can be elicited as part of the adaptive response to treatment (135–138). In vitro, human MM cell lines resistant to bortezomib or dexamethasone have higher concentrations of ATP, NADH/NADPH ratio, associated to hyperpolarization of mitochondrial membrane leading to impaired drug response (137, 139). In vitro, pre-treament with the OX-PHOS inhibitor tigecycline can increase bortezomib sensitivity (Alejandra Ortiz-Ruiz, ASH 2019, poster 4408) (140). Similarly, inhibition of PGC-1α (SR18292), relevant for OX-PHOS, significantly impaired the proliferation and survival of MM cells due to the energy exhaustion and oxidative damage (141). These and other preclinical studies confirm OX-PHOS as possible targets for sensitization to chemotherapy treatment in MM (142), including the efficacy of Venetoclax (BCL-2 inhibitor) that could be used independently from the genetic lesion (143). Further steps could include a metabolic classification of MM subtypes based on mitochondria number and OX-PHOS quantification (143).
Our group has recently disclosed that TLR4 acts as a mitochondria protective factor against bortezomib-induced mitochondria damage and apoptosis (144). Targeting TLR4 signaling in bortezomib resistant cells damages mitochondrial fitness and increases mitophagy leading to apoptosis. As TLR4 pathway is also activated in MM mesenchymal stromal cells (MSCs) driving their commitment toward a pro-inflammatory and pro-tumor behavior (145), TLR4 inhibition could be an adjuvant therapy to interrupt the self-reinforcing stromal changes in MM microenvironment. Taken together, changes in microenvironment composition (146) and REDOX status can affect sensitivity to novel agents (147) and should be taken in account in designing novel combinations.
Mitochondrial Metabolism Dependency in Acute Myeloid Leukemia and Novel Therapeutic Targets
Acute myeloid leukemia (AML) is a heterogeneous disease characterized by a blockade in differentiation of hematopoietic stem cells with a clonal proliferation of myeloid blast in BM and peripheral blood. Due to the high relapse rate and poor clinical outcome, overcoming chemoresistance remains the most important goal in AML patients. Changes in cell metabolism and metabolic adaptation are a hallmark of many cancers, including AML, supporting tumor initiation, growth, and response to therapeutics. The discovery of enzymes deficiency and mutations in key metabolic enzymes has highlighted the importance of metabolism in cancer biology and how these changes might constitute a weakness for cancer treatment.
A study carried out by Chen et al., reported that some metabolites such as pyruvate and lactate were specifically enriched in the serum of patients at diagnosis compared to healthy controls and demonstrated prognostic value in cytogenetically normal AML (CN-AML) patients as it could predict poor survival for these patients (148). Interestingly, deletions of the two glycolytic enzymes PKM2 and LDHA, which catalyze the production of cytosolic pyruvate and lactate, respectively, inhibit leukemia initiation in vivo in AML mice models.
It has also been reported that a wide percentage of AML patients are deficient in arginosuccinate synthetase-1 (ASS1), an enzyme that allows the conversion of citrulline and aspartate into argininosuccinate (149). The loss of ASS1 has also been found in other tumor types where it is required to support cell proliferation and nucleotide synthesis by sustaining the intracellular aspartate level (150). A decrease in ASS1 can also lead to a dependence on arginine, which has been explored as a potential vulnerability in different cancer types, including AML (151).
Recent advances in cancer genetics have found mutations in the isocitrate dehydrogenase 1 (IDH1) and 2 (IDH2) genes occur frequently in a variety of human cancers, including AML. Wild-type IDH1 and IDH2 are important metabolic enzymes catalyzing the oxidative decarboxylation of isocitrate to generate α-ketoglutarate (αKG) and CO2. IDH1 represents the peroxisomes and cytosol isoform, while IDH2 is localized in mitochondria. The common function of IDH1/2 active-site mutations is a new enzyme activity that catalyzes the conversion of αKG to D-2-hydroxyglutarate (D2HG). Under physiological conditions, cellular D2HG accumulation is limited due to the actions of the endogenous D2HG dehydrogenase (D2HGDH), which catalyzes the reverse reaction from D2HG to αKG.
D2HG has been demonstrated to inhibit αKG-dependent dioxygenases that are involved in the regulation of epigenetics and differentiation and is thought to induce epigenetic dysfunction inhibiting normal cellular differentiation. Specifically, elevated D2HG levels competitively inhibit αKG-dependent lysine demethylases, resulting in elevated levels of histone methylation in a variety of cell line models (152, 153). Consequently, inhibition of cellular differentiation by D2HG is thought to promote the pathological self-renewal of stem-like progenitor cells, which may create a cellular state prone to malignant transformation.
Evidence from AML patients and preclinical models strongly suggests that IDH1 and IDH2 mutations are oncogenic drivers of AML and myelodysplastic syndrome and that targeting IDH mutant neomorphic activity in this context may provide therapeutic benefit by promoting the differentiation of malignant myeloid cells. Research attempts have been made to identify small molecule inhibitors of mutant IDH enzymes and to develop these molecules as drugs for anti-cancer therapy (153).
In addition to targeting metabolic enzymes, targeting OX-PHOS turned out to be a promising strategy to improve the treatment outcomes of AML. Indeed, leukemic cells have higher copy number of mitochondrial DNA, more mitochondria and increased oxygen consumption in comparison to normal hematopoietic stem cells, without a concomitant increase in respiratory chain complex activity, which confers increased susceptibility to oxidative stress (154). Acquisition of chemoresistance is associated to a shift toward a high OX-PHOS status characterized by increased mitochondrial fitness and high levels of ROS (155). Mechanistically, this can be due to increased SIRT3 expression, which significantly decreased nicotinamide adenine dinucleotide phosphate (NADP)/reduced NADP ratio and increased reduced glutathione/oxidized glutathione ratio, associated to OX-PHOS induction (156).
For those cancers, like AML, that rely on OX-PHOS, its inhibition could represent an effective therapeutic strategy. In solid cancers OH-PHOS inhibitors, including biguanides and metformin, are currently under investigation in several trials, designed to evaluate the combination of metformin with chemotherapy, as recently reviewed (157, 158). Several drugs, including biguanides, metformin, atovaquone, and arsenic trioxide, are used at clinical level for non-oncologic indications, but growing evidences indicate their potential use as OX-PHOS inhibitors (8). The detrimental effect of metformin is emerging in AML, alone (159) or in combination with cytarabine (160), and Venetoclax induced cell-cycle arrest leading to clinical trials.
CPI-613, designated as orphan drug for the treatment of peripheral T-cell lymphoma, is a lipoate analog that blocks pyruvate dehydrogenase (PDH) and α-ketoglutarate dehydrogenase (KGDH), induces collapse of mitochondrial function associated to large, tumor-specific production of mitochondrial ROS (161). Based on encouraging results in phase I clinical studies (162, 163), a phase III randomized study is ongoing in the setting of refractory/relapsing AML to compare the efficacy of standard chemotherapy supplemented or not with CPI-613. Similarly, phase I trials disclosed promising results using IACS-010759, is an ECT inhibitor, acting against complex I in AML and NHL (164).
Taken together, ongoing trials in AML and other hematological malignancies show that targeting OX-PHOS is a promising strategy to induce a metabolic rewiring leading to chemo-sensitization.
Conclusions
Mitochondria play many important roles in cell functions and homeostasis, including the production of ATP, the release of death-promoting factors upon apoptotic stimuli and a variety of metabolic pathways. Contrary to conventional wisdom, functional mitochondria are essential for cancer cells. Although mutations in mitochondrial genes are common in cancer cells, they do not inactivate mitochondrial energy metabolism but rather alter the mitochondrial bioenergetic and biosynthetic state. These alterations activate out-of-context programs that are important in the onset and the development of malignancies. However, different cancer cell types undergo different bioenergetic alterations, some to more glycolytic and others to more oxidative, depending in part on the developmental state of the cell undergoing neoplastic transformation (165).
In most hematological malignancies, cancer cells show greater basal mitochondrial activity compared to healthy counterparts leading to higher levels of oxidative stress. ROS, adaptation to ROS, and mitochondrial biogenesis appear to form a self-amplifying feedback loop to sustain recurrence, as shown in CLL (119). However, in other settings, like MM, bioenergetics of tumor cells can change, as consequence of increased mitochondrial biomass and function, as part of the adaptive response to drug-induced stress (139).
In particular, interactions between cancer cells and surrounding microenvironment highly affect the growth, metabolism, metastasis and progression of cancer. The so-called reverse Warburg effect has been proposed to reconsider bioenergetics of cancer cells and stromal cells become metabolically coupled (56, 58, 59). In a vicious circle, neoplastic cells induce oxidative stress in neighboring microenvironment to undergo aerobic glycolysis and generate high level of energy-rich fuels (such as pyruvate, ketone bodies, fatty acids, and lactate) that fuel mitochondrial OX-PHOS in cancer cells and are utilized for efficient ATP production (56). In addition, microenvironment can contribute through horizontal mitochondrial transfer, when neoplastic cells become incapable of aerobic respiration due to defective or deleted mtDNA (166), Taking up functional mitochondria derived from the microenvironment can increase mitochondrial mass to improve metabolic fitness of neoplastic cells and conferring drug resistance.
It is probably that in next years “mitochondrial medicine” will play an active role to design effective therapeutic strategies to target the interplay between microenvironment and neoplastic cells to tailor the metabolic phenotype and not only the genomic aberrancies, with novel targets for selective anti-cancer therapy.
Author Contributions
AB, GS, CG, and AR designed the paper and wrote the manuscript. FP and FR reviewed literature about myeloma. DC and ES reviewed literature about lymphoma. GP reviewed literature about AML. GL and DT reviewed literature about OX-PHOS targeting. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with one of the authors FR.
References
1. Schatz G. The Magic Garden. Annu Rev Biochem (2007) 76:673–8. doi: 10.1146/annurev.biochem.76.060806.091141
2. Vakifahmetoglu-Norberg H, Ouchida AT, Norberg E. The role of mitochondria in metabolism and cell death. Biochem Biophys Res Commun (2017) 482:426–31. doi: 10.1016/j.bbrc.2016.11.088
3. Frezza C, Gottlieb E. Mitochondria in cancer: not just innocent bystanders. Semin Cancer Biol (2009) 19:4–11. doi: 10.1016/j.semcancer.2008.11.008
4. Moreno-Sánchez R, Rodríguez-Enríquez S, Marín-Hernández A, Saavedra E. Energy metabolism in tumor cells. FEBS J (2007) 274:1393–418. doi: 10.1111/j.1742-4658.2007.05686.x
5. Field CS, Baixauli F, Kyle RL, Puleston DJ, Cameron AM, Sanin DE, et al. Mitochondrial Integrity Regulated by Lipid Metabolism Is a Cell-Intrinsic Checkpoint for Treg Suppressive Function. Cell Metab (2020) 31:422–37.e5. doi: 10.1016/j.cmet.2019.11.021
6. Panuzzo C, Jovanovski A, Pergolizzi B, Pironi L, Stanga S, Fava C, et al. Mitochondria: A Galaxy in the Hematopoietic and Leukemic Stem Cell Universe. Int J Mol Sci (2020) 21:3298. doi: 10.3390/ijms21113928
7. Zhang L, Yao Y, Zhang S, Liu Y, Guo H, Ahmed M, et al. Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma. Sci Transl Med (2019) 11(491):eaau1667. doi: 10.1126/scitranslmed.aau1167
8. Ashton TM, McKenna WG, Kunz-Schughart LA, Higgins GS. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin Cancer Res (2018) 24:2482–90. doi: 10.1158/1078-0432.CCR-17-3070
9. Norberg E, Lako A, Chen PH, Stanley IA, Zhou F, Ficarro SB, et al. Differential contribution of the mitochondrial translation pathway to the survival of diffuse large B-cell lymphoma subsets. Cell Death Differ (2017) 24:251–62. doi: 10.1038/cdd.2016.116
10. Gooptu M, Whitaker-Menezes D, Sprandio J, Domingo-Vidal M, Lin Z, Uppal G, et al. Mitochondrial and glycolytic metabolic compartmentalization in diffuse large B-cell lymphoma. Semin Oncol (2017) 44:204–17. doi: 10.1053/j.seminoncol.2017.10.002
11. Mikkilineni L, Whitaker-Menezes D, Domingo-Vidal M, Sprandio J, Avena P, Cotzia P, et al. Hodgkin lymphoma: A complex metabolic ecosystem with glycolytic reprogramming of the tumor microenvironment. Semin Oncol (2017) 44:218–25. doi: 10.1053/j.seminoncol.2017.10.003
12. Sewastianik T, Szydlowski M, Jablonska E, Bialopiotrowicz E, Kiliszek P, Gorniak P, et al. FOXO1 is a TXN- and p300-dependent sensor and effector of oxidative stress in diffuse large B-cell lymphomas characterized by increased oxidative metabolism. Oncogene (2016) 35:5989–6000. doi: 10.1038/onc.2016.126
13. Giménez-Cassina A, Danial NN. Regulation of mitochondrial nutrient and energy metabolism by BCL-2 family proteins. Trends Endocrinol Metab (2015) 26:165–75. doi: 10.1016/j.tem.2015.02.004
14. Caro P, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB, et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell (2012) 22:547–60. doi: 10.1016/j.ccr.2012.08.014
15. Flis K, Irvine D, Copland M, Bhatia R, Skorski T. Chronic myeloid leukemia stem cells display alterations in expression of genes involved in oxidative phosphorylation. Leuk Lymphoma (2012) 53:2474–8. doi: 10.3109/10428194.2012.696313
16. Shahruzaman SH, Fakurazi S, Maniam S. Targeting energy metabolism to eliminate cancer cells. Cancer Manag Res (2018) 10:2325–35. doi: 10.2147/CMAR.S167424
17. Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, et al. Mitochondrial Cristae Shape Determines Respiratory Chain Supercomplexes Assembly and Respiratory Efficiency. Cell (2013) 155:160–71. doi: 10.1016/j.cell.2013.08.032
18. Maycotte P, Marín-Hernández A, Goyri-Aguirre M, Anaya-Ruiz M, Reyes-Leyva J, Cortés-Hernández P. Mitochondrial dynamics and cancer. Tumour Biol (2017) 39:1010428317698391. doi: 10.1177/1010428317698391
19. Humphries BA, Cutter AC, Buschhaus JM, Chen YC, Qyli T, Palagama DSW, et al. Enhanced mitochondrial fission suppresses signaling and metastasis in triple-negative breast cancer. Breast Cancer Res (2020) 22:60. doi: 10.1186/s13058-020-01301-x
20. Chan DC. Mitochondrial Dynamics and Its Involvement in Disease. Annu Rev Pathol (2020) 15:235–59. doi: 10.1146/annurev-pathmechdis-012419-032711
21. Farmer T, Naslavsky N, Caplan S. Tying trafficking to fusion and fission at the mighty mitochondria. Traffic (2018) 19:569–77. doi: 10.1111/tra.12573
22. Herst PM, Rowe MR, Carson GM, Berridge MV. Functional Mitochondria in Health and Disease. Front Endocrinol (Lausanne) (2017) 8:296. doi: 10.3389/fendo.2017.00296
23. Chen H, Chan DC. Mitochondrial Dynamics in Regulating the Unique Phenotypes of Cancer and Stem Cells. Cell Metab (2017) 26:39–48. doi: 10.1016/j.cmet.2017.05.016
24. Morciano G, Pedriali G, Sbano L, Iannitti T, Giorgi C, Pinton P. Intersection of mitochondrial fission and fusion machinery with apoptotic pathways: Role of Mcl-1. Biol Cell (2016) 108:279–93. doi: 10.1111/boc.201600019
25. Babbar M, Sheikh MS. Metabolic Stress and Disorders Related to Alterations in Mitochondrial Fission or Fusion. Mol Cell Pharmacol (2013) 5:109–33.
26. Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, et al. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J (2012) 26:2175–86. doi: 10.1096/fj.11-196543
27. Figge MT, Reichert AS, Meyer-Hermann M, Osiewacz HD. Deceleration of fusion-fission cycles improves mitochondrial quality control during aging. PLoS Comput Biol (2012) 8:e1002576. doi: 10.1371/journal.pcbi.1002576
28. Mills EL, Kelly B, O’Neill LAJ. Mitochondria are the powerhouses of immunity. Nat Immunol (2017) 18:488–98. doi: 10.1038/ni.3704
29. Vyas S, Zaganjor E, Haigis MC. Mitochondria and Cancer. Cell (2016) 166:555–66. doi: 10.1016/j.cell.2016.07.002
30. Ulivieri C. Cell death: insights into the ultrastructure of mitochondria. Tissue Cell (2010) 42:339–47. doi: 10.1016/j.tice.2010.10.004
31. Sazanov LA. The mechanism of coupling between electron transfer and proton translocation in respiratory complex I. J Bioenerg Biomembr (2014) 46:247–53. doi: 10.1007/s10863-014-9554-z
32. Buttgereit F, Brand MD. A hierarchy of ATP-consuming processes in mammalian cells. Biochem J (1995) 312( Pt 1):163–7. doi: 10.1042/bj3120163
33. Urra FA, Muñoz F, Lovy A, Cárdenas C. The Mitochondrial Complex(I)ty of Cancer. Front Oncol (2017) 7:118. doi: 10.3389/fonc.2017.00118
34. Kluckova K, Bezawork-Geleta A, Rohlena J, Dong L, Neuzil J. Mitochondrial complex II, a novel target for anti-cancer agents. Biochim Biophys Acta (2013) 1827:552–64. doi: 10.1016/j.bbabio.2012.10.015
35. Cecchini G. Function and structure of complex II of the respiratory chain. Annu Rev Biochem (2003) 72:77–109. doi: 10.1146/annurev.biochem.72.121801.161700
36. Bleier L, Dröse S. Superoxide generation by complex III: from mechanistic rationales to functional consequences. Biochim Biophys Acta (2013) 1827:1320–31. doi: 10.1016/j.bbabio.2012.12.002
37. Dennerlein S, Rehling P. Human mitochondrial COX1 assembly into cytochrome c oxidase at a glance. J Cell Sci (2015) 128:833–7. doi: 10.1242/jcs.161729
38. Jonckheere AI, Smeitink JA, Rodenburg RJ. Mitochondrial ATP synthase: architecture, function and pathology. J Inherit Metab Dis (2012) 35:211–25. doi: 10.1007/s10545-011-9382-9
39. Wang J, Liu X, Qiu Y, Shi Y, Cai J, Wang B, et al. Cell adhesion-mediated mitochondria transfer contributes to mesenchymal stem cell-induced chemoresistance on T cell acute lymphoblastic leukemia cells. J Hematol Oncol (2018) 11:11. doi: 10.1186/s13045-018-0554-z
40. Wang C, Youle RJ. The role of mitochondria in apoptosis*. Annu Rev Genet (2009) 43:95–118. doi: 10.1146/annurev-genet-102108-134850
41. Soultawi C, Fortier Y, Soundaramourty C, Estaquier J, Laforge M. Mitochondrial Bioenergetics and Dynamics During Infection. Exp Suppl (2018) 109:221–33. doi: 10.1007/978-3-319-74932-7_5
42. Xie LL, Shi F, Tan Z, Li Y, Bode AM, Cao Y. Mitochondrial network structure homeostasis and cell death. Cancer Sci (2018) 109:3686–94. doi: 10.1111/cas.13830
43. Benador IY, Veliova M, Mahdaviani K, Petcherski A, Wikstrom JD, Assali EA, et al. Mitochondria Bound to Lipid Droplets Have Unique Bioenergetics, Composition, and Dynamics that Support Lipid Droplet Expansion. Cell Metab (2018) 27:869–85.e6. doi: 10.1016/j.cmet.2018.03.003
44. Rambold AS, Cohen S, Lippincott-Schwartz J. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell (2015) 32:678–92. doi: 10.1016/j.devcel.2015.01.029
45. Boutant M, Kulkarni SS, Joffraud M, Ratajczak J, Valera-Alberni M, Combe R, et al. Mfn2 is critical for brown adipose tissue thermogenic function. EMBO J (2017) 36:1543–58. doi: 10.15252/embj.201694914
46. Koundouros N, Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J Cancer (2020) 122:4–22. doi: 10.1038/s41416-019-0650-z
47. Zhu Y, Ren C, Zhang M, Zhong Y. Perilipin 5 Reduces Oxidative Damage Associated With Lipotoxicity by Activating the PI3K/ERK-Mediated Nrf2-ARE Signaling Pathway in INS-1 Pancreatic β-Cells. Front Endocrinol (Lausanne) (2020) 11:166. doi: 10.3389/fendo.2020.00166
48. Wang H, Sreenivasan U, Hu H, Saladino A, Polster BM, Lund LM, et al. Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. J Lipid Res (2011) 52:2159–68. doi: 10.1194/jlr.M017939
49. Jarc E, Petan T. Lipid Droplets and the Management of Cellular Stress. Yale J Biol Med (2019) 92:435–52.
50. Voisin P, Bernard M, Bergès T, Regnacq M. Amino acid starvation inhibits autophagy in lipid droplet-deficient cells through mitochondrial dysfunction. Biochem J (2020) 477(18):3613–23. doi: 10.1042/BCJ20200551
51. Li Y, Zong WX, Ding WX. Recycling the danger via lipid droplet biogenesis after autophagy. Autophagy (2017) 13:1995–7. doi: 10.1080/15548627.2017.1371394
52. Klecker T, Braun RJ, Westermann B. Lipid Droplets Guard Mitochondria during Autophagy. Dev Cell (2017) 42:1–2. doi: 10.1016/j.devcel.2017.06.018
53. Simula L, Nazio F, Campello S. The mitochondrial dynamics in cancer and immune-surveillance. Semin Cancer Biol (2017) 47:29–42. doi: 10.1016/j.semcancer.2017.06.007
54. Mai S, Muster B, Bereiter-Hahn J, Jendrach M. Autophagy proteins LC3B, ATG5 and ATG12 participate in quality control after mitochondrial damage and influence lifespan. Autophagy (2012) 8:47–62. doi: 10.4161/auto.8.1.18174
55. Liberti MV, Locasale JW. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci (2016) 41:211–8. doi: 10.1016/j.tibs.2015.12.001
56. Fu Y, Liu S, Yin S, Niu W, Xiong W, Tan M, et al. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget (2017) 8:57813–25. doi: 10.18632/oncotarget.18175
57. Martinez-Outschoorn U, Sotgia F, Lisanti MP. Tumor microenvironment and metabolic synergy in breast cancers: critical importance of mitochondrial fuels and function. Semin Oncol (2014) 41:195–216. doi: 10.1053/j.seminoncol.2014.03.002
58. Witkiewicz AK, Whitaker-Menezes D, Dasgupta A, Philp NJ, Lin Z, Gandara R, et al. Using the “reverse Warburg effect” to identify high-risk breast cancer patients: stromal MCT4 predicts poor clinical outcome in triple-negative breast cancers. Cell Cycle (2012) 11:1108–17. doi: 10.4161/cc.11.6.19530
59. Sotgia F, Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, et al. Mitochondrial metabolism in cancer metastasis: visualizing tumor cell mitochondria and the “reverse Warburg effect” in positive lymph node tissue. Cell Cycle (2012) 11:1445–54. doi: 10.4161/cc.19841
60. Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Tanowitz HB, Sotgia F, et al. Stromal-epithelial metabolic coupling in cancer: integrating autophagy and metabolism in the tumor microenvironment. Int J Biochem Cell Biol (2011) 43:1045–51. doi: 10.1016/j.biocel.2011.01.023
61. Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Daumer KM, Milliman JN, Chiavarina B, et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle (2010) 9:2423–33. doi: 10.4161/cc.9.12.12048
62. Bonuccelli G, Whitaker-Menezes D, Castello-Cros R, Pavlides S, Pestell RG, Fatatis A, et al. The reverse Warburg effect: glycolysis inhibitors prevent the tumor promoting effects of caveolin-1 deficient cancer associated fibroblasts. Cell Cycle (2010) 9:1960–71. doi: 10.4161/cc.9.10.11601
63. Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle (2009) 8:3984–4001. doi: 10.4161/cc.8.23.10238
64. Golan K, Wellendorf A, Takihara Y, Kumari A, Khatib-Massalha E, Kollet O, et al. Mitochondria Transfer from Hematopoietic Stem and Progenitor Cells to Pdgfrα+/Sca-1-/CD48dim BM Stromal Cells Via CX43 Gap Junctions and AMPK Signaling Inversely Regulate ROS Generation in Both Cell Populations. Blood (2016) 128:5–5. doi: 10.1182/blood.V128.22.5.5
65. Maryanovich M, Zaltsman Y, Ruggiero A, Goldman A, Shachnai L, Zaidman SL, et al. An MTCH2 pathway repressing mitochondria metabolism regulates haematopoietic stem cell fate. Nat Commun (2015) 6:7901. doi: 10.1038/ncomms8901
66. Khatib-Massalha E, Bhattacharya S, Massalha H, Biram A, Golan K, Kollet O, et al. Lactate released by inflammatory bone marrow neutrophils induces their mobilization via endothelial GPR81 signaling. Nat Commun (2020) 11:3547. doi: 10.1038/s41467-020-17402-2
67. Boise LH, Shanmugam M. Stromal Support of Metabolic Function through Mitochondrial Transfer in Multiple Myeloma. Cancer Res (2019) 79:2102–3. doi: 10.1158/0008-5472.CAN-19-0500
68. Marlein CR, Piddock RE, Mistry JJ, Zaitseva L, Hellmich C, Horton RH, et al. CD38-Driven Mitochondrial Trafficking Promotes Bioenergetic Plasticity in Multiple Myeloma. Cancer Res (2019) 79:2285–97. doi: 10.1158/0008-5472.CAN-18-0773
69. Burt R, Dey A, Aref S, Aguiar M, Akarca A, Bailey K, et al. Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress. Blood (2019) 134:1415–29. doi: 10.1182/blood.2019001398
70. Kolba MD, Dudka W, Zaręba-Kozioł M, Kominek A, Ronchi P, Turos L, et al. Tunneling nanotube-mediated intercellular vesicle and protein transfer in the stroma-provided imatinib resistance in chronic myeloid leukemia cells. Cell Death Dis (2019) 10:817. doi: 10.1038/s41419-019-2045-8
71. Rodriguez AM, Nakhle J, Griessinger E, Vignais ML. Intercellular mitochondria trafficking highlighting the dual role of mesenchymal stem cells as both sensors and rescuers of tissue injury. Cell Cycle (2018) 17:712–21. doi: 10.1080/15384101.2018.1445906
72. Griessinger E, Moschoi R, Biondani G, Peyron JF. Mitochondrial Transfer in the Leukemia Microenvironment. Trends Cancer (2017) 3:828–39. doi: 10.1016/j.trecan.2017.10.003
73. Marlein CR, Zaitseva L, Piddock RE, Robinson SD, Edwards DR, Shafat MS, et al. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood (2017) 130:1649–60. doi: 10.1182/blood-2017-03-772939
74. Moschoi R, Imbert V, Nebout M, Chiche J, Mary D, Prebet T, et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood (2016) 128:253–64. doi: 10.1182/blood-2015-07-655860
75. Rasche L, Kortüm KM, Raab MS, Weinhold N. The Impact of Tumor Heterogeneity on Diagnostics and Novel Therapeutic Strategies in Multiple Myeloma. Int J Mol Sci (2019) 20:1248. doi: 10.3390/ijms20051248
76. Araf S, Wang J, Korfi K, Pangault C, Kotsiou E, Rio-Machin A, et al. Genomic profiling reveals spatial intra-tumor heterogeneity in follicular lymphoma. Leukemia (2018) 32:1261–5. doi: 10.1038/s41375-018-0043-y
77. Rasche L, Chavan SS, Stephens OW, Patel PH, Tytarenko R, Ashby C, et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat Commun (2017) 8:268. doi: 10.1038/s41467-017-00296-y
78. Mithraprabhu S, Khong T, Ramachandran M, Chow A, Klarica D, Mai L, et al. Circulating tumour DNA analysis demonstrates spatial mutational heterogeneity that coincides with disease relapse in myeloma. Leukemia (2017) 31:1695–705. doi: 10.1038/leu.2016.366
79. Opinto G, Vegliante MC, Negri A, Skrypets T, Loseto G, Pileri SA, et al. The Tumor Microenvironment of DLBCL in the Computational Era. Front Oncol (2020) 10:351. doi: 10.3389/fonc.2020.00351
80. Romano A, Parrinello NL, Chiarenza A, Motta G, Tibullo D, Giallongo C, et al. Immune off-target effects of Brentuximab Vedotin in relapsed/refractory Hodgkin Lymphoma. Br J Haematol (2019) 185:468–79. doi: 10.1111/bjh.15801
81. Agostinelli C, Gallamini A, Stracqualursi L, Agati P, Tripodo C, Fuligni F, et al. The combined role of biomarkers and interim PET scan in prediction of treatment outcome in classical Hodgkin’s lymphoma: a retrospective, European, multicentre cohort study. Lancet Haematol (2016) 3:e467–79. doi: 10.1016/S2352-3026(16)30108-9
82. Romano A, Parrinello NL, Vetro C, Tibullo D, Giallongo C, La Cava P, et al. The prognostic value of the myeloid-mediated immunosuppression marker Arginase-1 in classic Hodgkin lymphoma. Oncotarget (2016) 7:67333–46. doi: 10.18632/oncotarget.12024
83. Romano A, Parrinello NL, Vetro C, Forte S, Chiarenza A, Figuera A, et al. Circulating myeloid-derived suppressor cells correlate with clinical outcome in Hodgkin Lymphoma patients treated up-front with a risk-adapted strategy. Br J Haematol (2015) 168:689–700. doi: 10.1111/bjh.13198
84. Vetro C, Romano A, Ancora F, Coppolino F, Brundo MV, Raccuia SA, et al. Clinical Impact of the Immunome in Lymphoid Malignancies: The Role of Myeloid-Derived Suppressor Cells. Front Oncol (2015) 5:104. doi: 10.3389/fonc.2015.00104
85. Romano A, Vetro C, Caocci G, Greco M, Parrinello NL, Di Raimondo F, et al. Immunological deregulation in classic hodgkin lymphoma. Mediterr J Hematol Infect Dis (2014) 6:e2014039. doi: 10.4084/mjhid.2014.039
86. Tiacci E, Döring C, Brune V, van Noesel CJ, Klapper W, Mechtersheimer G, et al. Analyzing primary Hodgkin and Reed-Sternberg cells to capture the molecular and cellular pathogenesis of classical Hodgkin lymphoma. Blood (2012) 120:4609–20. doi: 10.1182/blood-2012-05-428896
87. Birkenmeier K, Dröse S, Wittig I, Winkelmann R, Käfer V, Döring C, et al. Hodgkin and Reed-Sternberg cells of classical Hodgkin lymphoma are highly dependent on oxidative phosphorylation. Int J Cancer (2016) 138:2231–46. doi: 10.1002/ijc.29934
88. Birkenmeier K, Moll K, Newrzela S, Hartmann S, Dröse S, Hansmann ML. Basal autophagy is pivotal for Hodgkin and Reed-Sternberg cells’ survival and growth revealing a new strategy for Hodgkin lymphoma treatment. Oncotarget (2016) 7:46579–88. doi: 10.18632/oncotarget.10300
89. Yadav N, Kumar S, Marlowe T, Chaudhary AK, Kumar R, Wang J, et al. Oxidative phosphorylation-dependent regulation of cancer cell apoptosis in response to anticancer agents. Cell Death Dis (2015) 6:e1969. doi: 10.1038/cddis.2015.305
90. Romano A, Parrinello NL, Vetro C, Chiarenza A, Cerchione C, Ippolito M, et al. Prognostic meaning of neutrophil to lymphocyte ratio (NLR) and lymphocyte to monocyte ration (LMR) in newly diagnosed Hodgkin lymphoma patients treated upfront with a PET-2 based strategy. Ann Hematol (2018) 97:1009–18. doi: 10.1007/s00277-018-3276-y
91. Gallamini A, Tarella C, Viviani S, Rossi A, Patti C, Mulé A, et al. Early Chemotherapy Intensification With Escalated BEACOPP in Patients With Advanced-Stage Hodgkin Lymphoma With a Positive Interim Positron Emission Tomography/Computed Tomography Scan After Two ABVD Cycles: Long-Term Results of the GITIL/FIL HD 0607 Trial. J Clin Oncol (2018) 36:454–62. doi: 10.1200/JCO.2017.75.2543
92. Gallamini A, Di Raimondo F, La Nasa G, Romano A, Borra A, Greco M. Standard therapies versus novel therapies in Hodgkin lymphoma. Immunol Lett (2013) 155:56–9. doi: 10.1016/j.imlet.2013.09.011
93. Aggarwal V, Tuli HS, Varol A, Thakral F, Yerer MB, Sak K, et al. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules (2019) 9(11):735. doi: 10.3390/biom9110735
94. Rabilloud T, Heller M, Gasnier F, Luche S, Rey C, Aebersold R, et al. Proteomics analysis of cellular response to oxidative stress. Evidence for in vivo overoxidation of peroxiredoxins at their active site. J Biol Chem (2002) 277:19396–401. doi: 10.1074/jbc.M106585200
95. Bur H, Haapasaari KM, Turpeenniemi-Hujanen T, Kuittinen O, Auvinen P, Marin K, et al. Oxidative stress markers and mitochondrial antioxidant enzyme expression are increased in aggressive Hodgkin lymphomas. Histopathology (2014) 65:319–27. doi: 10.1111/his.12389
96. Vial G, Detaille D, Guigas B. Role of Mitochondria in the Mechanism(s) of Action of Metformin. Front Endocrinol (2019) 10:294. doi: 10.3389/fendo.2019.00294
97. Chukkapalli V, Gordon LI, Venugopal P, Borgia JA, Karmali R. Metabolic changes associated with metformin potentiates Bcl-2 inhibitor, Venetoclax, and CDK9 inhibitor, BAY1143572 and reduces viability of lymphoma cells. Oncotarget (2018) 9:21166–81. doi: 10.18632/oncotarget.24989
98. Casasnovas RO, Bouabdallah R, Brice P, Lazarovici J, Ghesquieres H, Stamatoullas A, et al. PET-adapted treatment for newly diagnosed advanced Hodgkin lymphoma (AHL2011): a randomised, multicentre, non-inferiority, phase 3 study. Lancet Oncol (2019) 20:202–15. doi: 10.1016/S1470-2045(18)30784-8
99. Cunha Júnior AD, Pericole FV, Carvalheira JBC. Metformin and blood cancers. Clinics (Sao Paulo) (2018) 73:e412s. doi: 10.6061/clinics/2018/e412s
100. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature (2000) 403:503–11. doi: 10.1038/35000501
101. Jaffe ES, Harris NL, Stein H, Isaacson PG. Classification of lymphoid neoplasms: the microscope as a tool for disease discovery. Blood (2008) 112:4384–99. doi: 10.1182/blood-2008-07-077982
102. Monti S, Savage KJ, Kutok JL, Feuerhake F, Kurtin P, Mihm M, et al. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood (2005) 105:1851–61. doi: 10.1182/blood-2004-07-2947
103. Liu X, Wang L, Jiang W, Lu W, Yang J, Yang W. B cell lymphoma with different metabolic characteristics show distinct sensitivities to metabolic inhibitors. J Cancer (2018) 9:1582–91. doi: 10.7150/jca.24331
104. Chen L, Monti S, Juszczynski P, Ouyang J, Chapuy B, Neuberg D, et al. SYK inhibition modulates distinct PI3K/AKT- dependent survival pathways and cholesterol biosynthesis in diffuse large B cell lymphomas. Cancer Cell (2013) 23:826–38. doi: 10.1016/j.ccr.2013.05.002
105. Chen L, Monti S, Juszczynski P, Daley J, Chen W, Witzig TE, et al. SYK-dependent tonic B-cell receptor signaling is a rational treatment target in diffuse large B-cell lymphoma. Blood (2008) 111:2230–7. doi: 10.1182/blood-2007-07-100115
106. Doughty CA, Bleiman BF, Wagner DJ, Dufort FJ, Mataraza JM, Roberts MF, et al. Antigen receptor-mediated changes in glucose metabolism in B lymphocytes: role of phosphatidylinositol 3-kinase signaling in the glycolytic control of growth. Blood (2006) 107:4458–65. doi: 10.1182/blood-2005-12-4788
107. Broecker-Preuss M, Becher-Boveleth N, Bockisch A, Dührsen U, Müller S. Regulation of glucose uptake in lymphoma cell lines by c-MYC- and PI3K-dependent signaling pathways and impact of glycolytic pathways on cell viability. J Transl Med (2017) 15:158. doi: 10.1186/s12967-017-1258-9
108. Bhalla K, Jaber S, Nahid MN, Underwood K, Beheshti A, Landon A, et al. Role of hypoxia in Diffuse Large B-cell Lymphoma: Metabolic repression and selective translation of HK2 facilitates development of DLBCL. Sci Rep (2018) 8:744. doi: 10.1038/s41598-018-25251-9
109. Nakajima K, Kawashima I, Koshiisi M, Kumagai T, Suzuki M, Suzuki J, et al. Glycolytic enzyme hexokinase II is a putative therapeutic target in B-cell malignant lymphoma. Exp Hematol (2019) 78:46–55.e3. doi: 10.1016/j.exphem.2019.09.023
110. Czuczman MS, Olejniczak S, Gowda A, Kotowski A, Binder A, Kaur H, et al. Acquirement of rituximab resistance in lymphoma cell lines is associated with both global CD20 gene and protein down-regulation regulated at the pretranscriptional and posttranscriptional levels. Clin Cancer Res (2008) 14:1561–70. doi: 10.1158/1078-0432.CCR-07-1254
111. Gu JJ, Singh A, Xue K, Mavis C, Barth M, Yanamadala V, et al. Up-regulation of hexokinase II contributes to rituximab-chemotherapy resistance and is a clinically relevant target for therapeutic development. Oncotarget (2017) 9(3):4020–33. doi: 10.18632/oncotarget.23425
112. Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol (2003) 206:2049–57. doi: 10.1242/jeb.00241
113. Chiche J, Reverso-Meinietti J, Mouchotte A, Rubio-Patiño C, Mhaidly R, Villa E, et al. GAPDH Expression Predicts the Response to R-CHOP, the Tumor Metabolic Status, and the Response of DLBCL Patients to Metabolic Inhibitors. Cell Metab (2019) 29:1243–57.e10. doi: 10.1016/j.cmet.2019.02.002
114. Yang K, Chen Z, Gao J, Shi W, Li L, Jiang S, et al. The Key Roles of GSK-3β in Regulating Mitochondrial Activity. Cell Physiol Biochem (2017) 44:1445–59. doi: 10.1159/000485580
115. Varano G, Raffel S, Sormani M, Zanardi F, Lonardi S, Zasada C, et al. The B-cell receptor controls fitness of MYC-driven lymphoma cells via GSK3β inhibition. Nature (2017) 546:302–6. doi: 10.1038/nature22353
116. Messmer BT, Messmer D, Allen SL, Kolitz JE, Kudalkar P, Cesar D, et al. In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. J Clin Invest (2005) 115:755–64. doi: 10.1172/JCI23409
117. Roy Chowdhury S, Bouchard EDJ, Saleh R, Nugent Z, Peltier C, Mejia E, et al. Mitochondrial Respiration Correlates with Prognostic Markers in Chronic Lymphocytic Leukemia and Is Normalized by Ibrutinib Treatment. Cancers (Basel) (2020) 12(3):650. doi: 10.3390/cancers12030650
118. Roy Chowdhury S, Banerji V. Targeting Mitochondrial Bioenergetics as a Therapeutic Strategy for Chronic Lymphocytic Leukemia. Oxid Med Cell Longev (2018) 2018:2426712. doi: 10.1155/2018/2426712
119. Jitschin R, Hofmann AD, Bruns H, Giessl A, Bricks J, Berger J, et al. Mitochondrial metabolism contributes to oxidative stress and reveals therapeutic targets in chronic lymphocytic leukemia. Blood (2014) 123:2663–72. doi: 10.1182/blood-2013-10-532200
120. Galicia-Vázquez G, Aloyz R. Metabolic rewiring beyond Warburg in chronic lymphocytic leukemia: How much do we actually know? Crit Rev Oncol Hematol (2019) 134:65–70. doi: 10.1016/j.critrevonc.2018.12.003
121. Vaisitti T, Gaudino F, Ouk S, Moscvin M, Vitale N, Serra S, et al. Targeting metabolism and survival in chronic lymphocytic leukemia and Richter syndrome cells by a novel NF-κB inhibitor. Haematologica (2017) 102:1878–89. doi: 10.3324/haematol.2017.173419
122. Owen OE, Kalhan SC, Hanson RW. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem (2002) 277:30409–12. doi: 10.1074/jbc.R200006200
123. Guièze R, Liu VM, Rosebrock D, Jourdain AA, Hernández-Sánchez M, Martinez Zurita A, et al. Mitochondrial Reprogramming Underlies Resistance to BCL-2 Inhibition in Lymphoid Malignancies. Cancer Cell (2019) 36:369–84.e13. doi: 10.1016/j.ccell.2019.08.005
124. Romano A, Parrinello NL, Simeon V, Puglisi F, La Cava P, Bellofiore C, et al. High-density neutrophils in MGUS and multiple myeloma are dysfunctional and immune-suppressive due to increased STAT3 downstream signaling. Sci Rep (2020) 10:1983. doi: 10.1038/s41598-020-58859-x
125. Maura F, Bolli N, Angelopoulos N, Dawson KJ, Leongamornlert D, Martincorena I, et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat Commun (2019) 10:3835. doi: 10.1101/388611
126. Zhu YX, Shi CX, Bruins LA, Jedlowski P, Wang X, Kortüm KM, et al. Loss of FAM46C Promotes Cell Survival in Myeloma. Cancer Res (2017) 77:4317–27. doi: 10.1158/0008-5472.CAN-16-3011
127. Herrero AB, Quwaider D, Corchete LA, Mateos MV, García-Sanz R, Gutiérrez NC. FAM46C controls antibody production by the polyadenylation of immunoglobulin mRNAs and inhibits cell migration in multiple myeloma. J Cell Mol Med (2020) 24:4171–82. doi: 10.1111/jcmm.15078
128. Pengo N, Scolari M, Oliva L, Milan E, Mainoldi F, Raimondi A, et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol (2013) 14:298–305. doi: 10.1038/ni.2524
129. Lam TT, Zhu H, Guan Y, Holmes EC. Genomic Analysis of the Emergence, Evolution, and Spread of Human Respiratory RNA Viruses. Annu Rev Genomics Hum Genet (2016) 17:193–218. doi: 10.1146/annurev-genom-083115-022628
130. Bartolome F, Esteras N, Martin-Requero A, Boutoleau-Bretonniere C, Vercelletto M, Gabelle A, et al. Pathogenic p62/SQSTM1 mutations impair energy metabolism through limitation of mitochondrial substrates. Sci Rep (2017) 7:1666. doi: 10.1038/s41598-017-01678-4
131. Glavey SV, Manier S, Natoni A, Sacco A, Moschetta M, Reagan MR, et al. The sialyltransferase ST3GAL6 influences homing and survival in multiple myeloma. Blood (2014) 124:1765–76. doi: 10.1182/blood-2014-03-560862
132. Bolzoni M, Chiu M, Accardi F, Vescovini R, Airoldi I, Storti P, et al. Dependence on glutamine uptake and glutamine addiction characterize myeloma cells: a new attractive target. Blood (2016) 128:667–79. doi: 10.1182/blood-2016-01-690743
133. Zhan X, Yu W, Franqui-Machin R, Bates ML, Nadiminti K, Cao H, et al. Alteration of mitochondrial biogenesis promotes disease progression in multiple myeloma. Oncotarget (2017) 8:111213–24. doi: 10.18632/oncotarget.22740
134. Tibullo D, Barbagallo I, Giallongo C, Vanella L, Conticello C, Romano A, et al. Heme oxygenase-1 nuclear translocation regulates bortezomibinduced cytotoxicity and mediates genomic instability in myeloma cells. Oncotarget (2016) 7:28868–80. doi: 10.18632/oncotarget.7563
135. Camiolo G, Barbato A, Giallongo C, Vicario N, Romano A, Parrinello NL, et al. Iron regulates myeloma cell/macrophage interaction and drives resistance to bortezomib. Redox Biol (2020) 36:101611. doi: 10.1016/j.redox.2020.101611
136. Soriano GP, Besse L, Li N, Kraus M, Besse A, Meeuwenoord N, et al. Proteasome inhibitor-adapted myeloma cells are largely independent from proteasome activity and show complex proteomic changes, in particular in redox and energy metabolism. Leukemia (2016) 30:2198–207. doi: 10.1038/leu.2016.102
137. Thompson RM, Dytfeld D, Reyes L, Robinson RM, Smith B, Manevich Y, et al. Glutaminase inhibitor CB-839 synergizes with carfilzomib in resistant multiple myeloma cells. Oncotarget (2017) 8:35863–76. doi: 10.18632/oncotarget.16262
138. Song IS, Kim HK, Lee SR, Jeong SH, Kim N, Ko KS, et al. Mitochondrial modulation decreases the bortezomib-resistance in multiple myeloma cells. Int J Cancer (2013) 133:1357–67. doi: 10.1002/ijc.28149
139. Tibullo D, Giallongo C, Romano A, Vicario N, Barbato A, Puglisi F, et al. Mitochondrial Functions, Energy Metabolism and Protein Glycosylation are Interconnected Processes Mediating Resistance to Bortezomib in Multiple Myeloma Cells. Biomolecules (2020) 10:696. doi: 10.3390/biom10050696
140. Ma R, Zhang Y, Wang W, Wu J, Yang Q, Xu W, et al. Inhibition of autophagy enhances the antitumour activity of tigecycline in multiple myeloma. J Cell Mol Med (2018) 22:5955–63. doi: 10.1111/jcmm.13865
141. Xiang Y, Fang B, Liu Y, Yan S, Cao D, Mei H, et al. SR18292 exerts potent antitumor effects in multiple myeloma via inhibition of oxidative phosphorylation. Life Sci (2020) 256:117971. doi: 10.1016/j.lfs.2020.117971
142. Aminzadeh-Gohari S, Weber DD, Vidali S, Catalano L, Kofler B, Feichtinger RG. From old to new — Repurposing drugs to target mitochondrial energy metabolism in cancer. Semin Cell Dev Biol (2020) 98:211–23. doi: 10.1016/j.semcdb.2019.05.025
143. Bajpai R, Sharma A, Achreja A, Edgar CL, Wei C, Siddiqa AA, et al. Electron transport chain activity is a predictor and target for venetoclax sensitivity in multiple myeloma. Nat Commun (2020) 11:1228. doi: 10.1038/s41467-020-15051-z
144. Giallongo C, Tibullo D, Puglisi F, Barbato A, Vicario N, Cambria D, et al. Inhibition of TLR4 Signaling Affects Mitochondrial Fitness and Overcomes Bortezomib Resistance in Myeloma Plasma Cells. Cancers (Basel) (2020) 12(8):1999. doi: 10.3390/cancers12081999
145. Giallongo C, Tibullo D, Camiolo G, Parrinello NL, Romano A, Puglisi F, et al. TLR4 signaling drives mesenchymal stromal cells commitment to promote tumor microenvironment transformation in multiple myeloma. Cell Death Dis (2019) 10:704. doi: 10.1038/s41419-019-1959-5
146. Romano A, Parrinello NL, La Cava P, Tibullo D, Giallongo C, Camiolo G, et al. PMN-MDSC and arginase are increased in myeloma and may contribute to resistance to therapy. Expert Rev Mol Diagn (2018) 18(7):675–83. doi: 10.1080/14737159.2018.1470929
147. Barbagallo I, Giallongo C, Volti GL, Distefano A, Camiolo G, Raffaele M, et al. Heme Oxygenase Inhibition Sensitizes Neuroblastoma Cells to Carfilzomib. Mol Neurobiol (2019) 56(2):1451–60. doi: 10.1007/s12035-018-1133-6
148. Chen W-L, Wang J-H, Zhao A-H, Xu X, Wang Y-H, Chen T-L, et al. A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood (2014) 124:1645–54. doi: 10.1182/blood-2014-09-602706
149. Miraki-Moud F, Ghazaly E, Ariza-McNaughton L, Hodby KA, Clear A, Anjos-Afonso F, et al. Arginine deprivation using pegylated arginine deiminase has activity against primary acute myeloid leukemia cells in vivo. Blood (2015) 125:4060–8. doi: 10.1182/blood-2014-10-608133
150. Rabinovich S, Adler L, Yizhak K, Sarver A, Silberman A, Agron S, et al. Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature (2015) 527:379–83. doi: 10.1038/nature15529
151. Waitkus MS, Diplas BH, Yan H. Biological Role and Therapeutic Potential of IDH Mutations in Cancer. Cancer Cell (2018) 34(2):186–95. doi: 10.1016/j.ccell.2018.04.011
152. Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep (2011) 12:463–9. doi: 10.1038/embor.2011.43
153. Xu W, Yang H, Liu Y, Yang Y, Wang PP, Kim SHH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell (2011) 19:17–30. doi: 10.1016/j.ccr.2010.12.014
154. Sriskanthadevan S, Jeyaraju DV, Chung TE, Prabha S, Xu W, Skrtic M, et al. AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood (2015) 125:2120–30. doi: 10.1182/blood-2014-08-594408
155. Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R, et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov (2017) 7:716–35. doi: 10.1158/2159-8290.CD-16-0441
156. Ma J, Liu B, Yu D, Zuo Y, Cai R, Yang J, et al. SIRT3 deacetylase activity confers chemoresistance in AML via regulation of mitochondrial oxidative phosphorylation. Br J Haematol (2019) 187:49–64. doi: 10.1111/bjh.16044
157. Nayak AP, Kapur A, Barroilhet L, Patankar MS. Oxidative Phosphorylation: A Target for Novel Therapeutic Strategies Against Ovarian Cancer. Cancers (Basel) (2018) 10(9):337. doi: 10.3390/cancers10090337
158. Castelli G, Pelosi E, Testa U. Emerging Therapies for Acute Myelogenus Leukemia Patients Targeting Apoptosis and Mitochondrial Metabolism. Cancers (Basel) (2019) 11(2):260. doi: 10.3390/cancers11020260
159. Green AS, Chapuis N, Maciel TT, Willems L, Lambert M, Arnoult C, et al. The LKB1/AMPK signaling pathway has tumor suppressor activity in acute myeloid leukemia through the repression of mTOR-dependent oncogenic mRNA translation. Blood (2010) 116:4262–73. doi: 10.1182/blood-2010-02-269837
160. Vitkevičienė A, Janulis V, Žučenka A, Borutinskaitė V, Kaupinis A, Valius M, et al. Oxidative phosphorylation inhibition induces anticancerous changes in therapy-resistant-acute myeloid leukemia patient cells. Mol Carcinog (2019) 58:2008–16. doi: 10.1002/mc.23092
161. Stuart SD, Schauble A, Gupta S, Kennedy AD, Keppler BR, Bingham PM, et al. A strategically designed small molecule attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process. Cancer Metab (2014) 2:4. doi: 10.1186/2049-3002-2-4
162. Pardee TS, Lee K, Luddy J, Maturo C, Rodriguez R, Isom S, et al. A phase I study of the first-in-class antimitochondrial metabolism agent, CPI-613, in patients with advanced hematologic malignancies. Clin Cancer Res (2014) 20:5255–64. doi: 10.1158/1078-0432.CCR-14-1019
163. Pardee TS, Anderson RG, Pladna KM, Isom S, Ghiraldeli LP, Miller LD, et al. A Phase I Study of CPI-613 in Combination with High-Dose Cytarabine and Mitoxantrone for Relapsed or Refractory Acute Myeloid Leukemia. Clin Cancer Res (2018) 24:2060–73. doi: 10.1158/1078-0432.CCR-17-2282
164. Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med (2018) 24:1036–46. doi: 10.1038/s41591-018-0052-4
Keywords: OX-PHOS, mitochondria, multiple myeloma, acute myeloid leukemia, chronic lymphatic leukemia, lymphoma
Citation: Barbato A, Scandura G, Puglisi F, Cambria D, La Spina E, Palumbo GA, Lazzarino G, Tibullo D, Di Raimondo F, Giallongo C and Romano A (2020) Mitochondrial Bioenergetics at the Onset of Drug Resistance in Hematological Malignancies: An Overview. Front. Oncol. 10:604143. doi: 10.3389/fonc.2020.604143
Received: 08 September 2020; Accepted: 13 November 2020;
Published: 21 December 2020.
Edited by:
Cirino Botta, Cosenza Hospital, ItalyReviewed by:
Antonio Giovanni Solimando, University of Bari Aldo Moro, ItalyGiuseppe Maurizio Campo, University of Messina, Italy
Copyright © 2020 Barbato, Scandura, Puglisi, Cambria, La Spina, Palumbo, Lazzarino, Tibullo, Di Raimondo, Giallongo and Romano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniele Tibullo, ZC50aWJ1bGxvQHVuaWN0Lml0; Alessandra Romano, c2FuZHJpbmEucm9tYW5vQGdtYWlsLmNvbQ==