 Franklin Ning
Franklin Ning Christopher B. Cole
Christopher B. Cole Christina M. Annunziata
Christina M. Annunziata
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 15 January 2021
Sec. Cancer Molecular Targets and Therapeutics
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.604084
This article is part of the Research Topic Targeting the Microenvironment Niche in Solid Tumors View all 13 articles
Ovarian cancer is the leading cause of death among gynecological neoplasms, with an estimated 14,000 deaths in 2019. First-line treatment options center around a taxane and platinum-based chemotherapy regimen. However, many patients often have recurrence due to late stage diagnoses and acquired chemo-resistance. Recent approvals for bevacizumab and poly (ADP-ribose) polymerase inhibitors have improved treatment options but effective treatments are still limited in the recurrent setting. Immunotherapy has seen significant success in hematological and solid malignancies. However, effectiveness has been limited in ovarian cancer. This may be due to a highly immunosuppressive tumor microenvironment and a lack of tumor-specific antigens. Certain immune cell subsets, such as regulatory T cells and tumor-associated macrophages, have been implicated in ovarian cancer. Consequently, therapies augmenting the immune response, such as immune checkpoint inhibitors and dendritic cell vaccines, may be unable to properly enact their effector functions. A better understanding of the various interactions among immune cell subsets in the peritoneal microenvironment is necessary to develop efficacious therapies. This review will discuss various cell subsets in the ovarian tumor microenvironment, current immunotherapy modalities to target or augment these immune subsets, and treatment challenges.
Ovarian cancer presents a unique tumor microenvironment (TME) with its predilection to metastasize in the peritoneal cavity and generate malignant ascites. The cancer spreads by direct shedding into the ascites and movement throughout the peritoneal cavity. Common sites of tumor deposits are on the mesenteric and serosal surfaces of the abdominal organs. The immune microenvironment in this location is characterized by interactions among the tumor cells, myeloid and lymphoid immune cells, as well as fibroblasts and adipocytes in the peritoneum that promote tumor growth. Growth factors, such as fibroblast growth factor and vascular endothelial growth factor (VEGF), promote angiogenesis and direct fibroblast differentiation towards cancer-associated fibroblasts that promote metastases (1). Adipocytes in the omentum can also provide energy for tumor growth and metastases (2). Little is known about how these cells interact with immune cells and if they promote immunosuppression. Additional information is therefore needed about cellular interactions and trafficking in the peritoneal TME in order to better develop immunotherapies for ovarian cancer.
Advances in immunotherapy, such as immune checkpoint inhibitors and chimeric antigen-receptor T cells (CAR-T), have demonstrated efficacy in various cancers. However, performance in ovarian cancer patients has remained poor. Multiple studies have demonstrated that the highly immunosuppressive TME and low mutational burden of ovarian cancer is a barrier to effective treatment (3). Inhibitiory cells in the TME, such as regulatory T and B cells, myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages (TAMs) inevitably contribute to tumor growth through a milieu of inhibitory effects (4–7). In this review, we discuss key subsets of adaptive and innate immunity that play a role in the ovarian TME and current efforts to target or augment these populations (Figure 1).
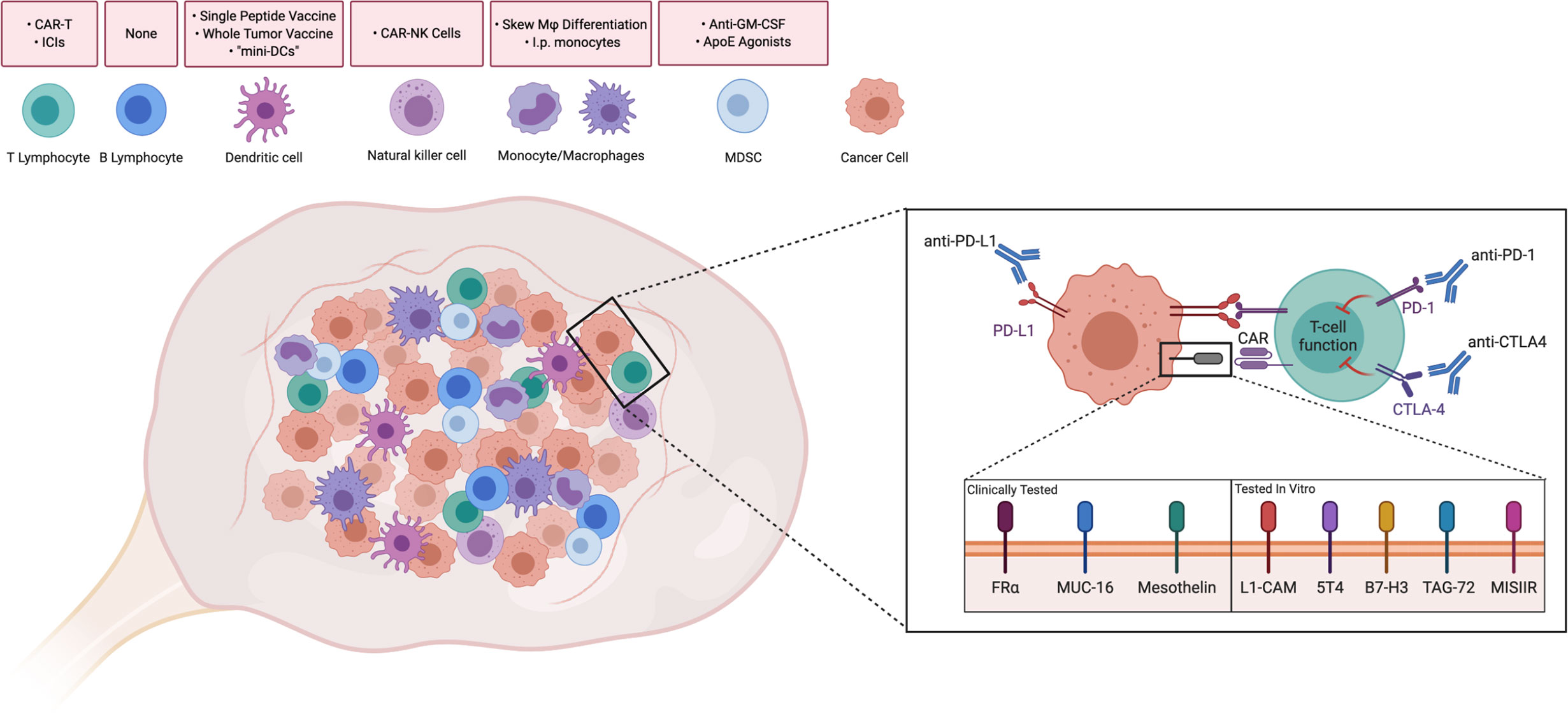
Figure 1 Driving Immune Responses in the Ovarian Tumor Microenvironment. Immune cells are present intratumorally and in the ovarian tumor microenvironment. Strategies discussed throughout the paper have been summarized above the corresponding cell type. Attempts to improve T cell functionality in the ovarian TME include Immune checkpoint inhibitors, such as anti-PD-1, anti-PD-L1, and anti-CTLA4, which have been used in clinical trials to reduce inhibitory signaling. Similarly, T cells with chimeric antigen receptors (CAR-Ts) against folate receptor-alpha (FRα), MUC-16, and mesothelin have been tested in clinical trials in order to recognize tumor-associated antigens. CAR-Ts have been tested in vitro against novel antigens. CAR-T: Chimeric antigen receptor T-cell. ICI, Immune checkpoint inhibitor; CAR-NK, Chimeric antigen receptor-Natural Killer cell; Mφ, Macrophage; I.p., Intraperitoneal; GM-CSF, Granulocyte-monocyte colony stimulating factor; ApoE, ApolipoproteinE; MDSC, Myeloid-derived suppressor cell. Created with Biorender.com.
T cells play a significant role in anti-tumor processes by recognizing tumor neoantigens and facilitating and directly inducing apoptosis of tumor cells. CD3+ tumor-infiltrating T lymphocytes (TILs) were shown to be correlated with improved clinical outcome in ovarian cancer (8). Of 186 tumor samples, 102 samples were identified to have CD3+ cells within the tumor and 72 did not have any. Between these two groups, the 5-year overall survival (OS) for patients with TILs was 38% while those without TILs was 4.5%, suggesting a beneficial effect of TILs in women treated with standard chemotherapy. Interestingly, the absence of TILs correlated with increased levels of VEGF.
TILs, CD3+, can be further divided into CD4+ and CD8+ cells. In brief, CD4+ T cells, also known as helper T cells, recognize MHC class II and shape the adaptive immune response while CD8+ T, also known as cytotoxic T cells, recognize MHC class I and mediate direct killing Studies looking at patient survival have shown increased CD8+ T cells within the tumor predict better prognoses (9). An increase in intra-tumoral CD4+ T cells have also shown to be correlated with increased survival (10).
To boost the tumor-specific T cell response, adoptive cell therapy (ACT) has been used to increase the number of T cells that can recognize a tumor-associated antigen (TAA). ACT requires apheresis of a patient’s T cells and expanding them ex vivo to suitable levels after stimulation with lysed tumor cells. Recent advances in autologous therapy now include genetically modifying the T-cell receptor (TCR) or generating chimeric antigen receptor T-cells (CAR-T) to engineer a stronger, more precise, immune response to pre-determined tumor neoantigens (11).
Briefly, CAR-T cells are T cells that have been transfected to express a transmembrane protein with 1) a single chain fraction variable, also known as the antigen-recognizing domain, and 2) a TCR zeta chain, allowing for intracellular signaling. Since then, new generations have modified the CAR for improved immune responses. Second generation CAR-Ts added in either CD28 or 4-1BB as a costimulatory gene, third generation CAR-Ts allowed for two downstream signaling domains and the possibility of using OX40, and fourth generation CAR-Ts further improved effector functions by giving the receptor the ability to induce cytokines, such as IL-12 (12).
ACT and CAR-T have shown great promise in hematological tumors. However, they have demonstrated poor efficacy in solid tumors (13, 14). Part of this has been attributed to a lack of tumor specific antigens, a highly immunosuppressive TME, and a lack of persistence in the tumor. Some CAR-Ts that have made it to Phase 1 clinical trials for ovarian cancer include those targeting folate receptor, MUC-16, and mesothelin (Figure 1). Other examples in pre-clinical testing are also discussed.
Folate receptor-α (FRα) is one of a number of high affinity receptors that facilitates the uptake of folate into the cell (15). While it is rarely found in normal tissues, its overexpression has been identified in multiple malignancies, including ovarian cancer (16). When overexpressed in ovarian cancer, FRα has been correlated with a poor response to chemotherapy (17–20). Interestingly, correlation with survival has been inconclusive. Studies on FR overexpression have ranged from negatively prognostic to having no impact on survival to even an improvement in survival (19–22). Additional studies will be needed to determine the prognostic value of FRα.
In the first CAR-T treatment against FR, 14 patients with recurrent epithelial FR+ ovarian cancer were enrolled. Cohort 1 was given interleukin-2 (IL-2) and generic T cells transfected with a first generation anti-FR CAR while Cohort 2 was given T cells that were endogenously specific to FR (23). Cohort 1 was given 3 × 109 T cells, with possible dose escalation to 1 × 1010 and 3 to 5 × 1010 cells, with a dose of 720,000 IU/kg IL-2 each cycle. Cohort 2 was given 2 × 109 to 4 × 109 T cells. Most common Grade 1-2 drug-related side effects in Cohort 1 were fatigue and nausea compared to erythema at the site of injection for Cohort 2. Cohort 1 also experienced some Grade 3–4 side-effects, such as hypotension and dyspnea, that were attributed to the addition of IL-2. Unfortunately, no reduction in tumor burden or improvement in survival was seen in either cohort. A noted issue was the lack of persistence of these improved autologous T cells in circulation.
As a means of increasing persistence, investigators have added the co-stimulatory molecule CD137, or 4-1BB, to the CAR in order to improve cytokine secretion and the antitumor response in vivo (24). Intravenous (i.v.) injection of these CAR-Ts into NSG mice, inoculated subcutaneously with FR+ SKOV3, showed improved antitumor effects and reduced tumor volume. These T cells also showed improved persistence in circulating blood. Upon waiting 30 days for metastases in their model, only mice treated with the CAR-T cells were devoid of malignant ascites.
Although a thorough discussion is outside the scope of this review, a number of monoclonal antibodies (mAbs) have also been designed to target FR expression in ovarian cancer. In brief, farletuzumab was evaluated in a Phase 3 clinical trial, but did not reach the primary endpoint of PFS. Mirvetuximab soravtansine, an anti-FR coupled to tubulin-targeting agent DM4, was shown to be well-tolerated when given as a monotherapy or with bevacizumab (25–27).
Cancer antigen 125 (CA-125) is cleaved from the cell surface and is commonly used as a circulating serum marker for ovarian cancer relapse. MUC-16 is the remnant of the protein that is retained on the cancer cell membrane after cleavage. It is known that there is some overlap in MUC-16 expression with derivates of the fetal coelomic epithelia, such as the uterus, fallopian tubes, and the trachea (28).
Chekmosova et al. demonstrated that second generation CAR-Ts against MUC-16 were able to lyse MUC-16+ cells in vitro (29). These CAR-T cells were subsequently injected into SCID mice, inoculated i.p. or i.v. with MUC-16+ OVCAR3, and showed significant survival improvement when compared to untreated mice or those given anti-CD19 CAR-Ts. Later improvement to MUC-16 specific CAR-T cells included the ability to secrete IL-12 and an “elimination” gene to improve immune signaling and minimalize off-target effects (30). This “elimination” gene is a truncated portion of epidermal growth factor receptor (EGFR) that does not signal, but retains the ability to be bound by cetuximab and induce antibody-dependent cellular cytotoxicity or complement-mediated cytotoxicity against cells expressing this chimeric receptor. Mice inoculated i.p. with SKOV3 and treated with this novel CAR-T showed enhanced survival compared to CAR-Ts without the IL-12 domain. Interestingly, i.p. administration was found to be more effective than an i.v. route. A phase 1 clinical trial was proposed to test its efficacy in recurrent platinum-resistant ovarian cancer (31).
Similar to FR, mAbs have also been developed against CA-125. One of the leading antibodies, oregovomab, enhanced anti-cancer activity when given with carboplatin and paclitaxel, although oregovomab did not show clinical efficacy when treating patients with ovarian cancer as a monotherapy (32, 33). The combination significantly improved PFS to 41.8 months, when given front-line, compared to 12.2 months for patients given standard carboplatin-paclitaxel (34). Interestingly, patients who had better survival outcomes had lower levels of HLA-DR-CD14+ MDSCs and a lower neutrophil-and-monocyte to lymphocyte ratio (35).
Mesothelin is a surface antigen found overexpressed on malignant mesothelioma as well as pancreatic, ovarian, and lung cancers (36). However, it is also found expressed at low levels on other mesothelial surfaces, such as the pleura, pericardium, and peritoneum.
In ovarian cancer, a mesothelin-specific CAR-T was developed using mRNA transfection in order to minimalize off-target toxicity due to CAR-T persistence (37, 38). Five patients with ovarian cancer were inoculated i.v. with second-generation CARs against mesothelin in a phase 1 clinical trial (39). Up to 1 to 3*108/m2 CAR-T cells were given with or without lymphodepletion by 1.5 g/m2 cyclophosphamide. Only one patient demonstrated a sizable reduction in tumor burden, but did not qualify as a partial reduction per formal criteria for objective response. This may have been due to a lack of persistence of CAR-Ts by day 28 in most patients and the single-chain variable fraction being murine-based. There were also no significant changes in cytokine levels during the first month post-infusion. The most common low-grade adverse events (AEs) included fatigue, nausea, and emesis, while development of Grade 3 ascites was the most common high-grade AE. A similar Phase 1 trial is evaluating MCY-M11, a mRNA-based anti-mesothelin therapy, when given intraperitoneally (i.p.) for platinum-resistant OC patients, with or without cyclophosphamide (NCT03608618). Another clinical trial evaluating anti-mesothelin CAR-T cells, but generated using a lentiviral delivery system, is ongoing (NCT03054298).
Recently, Hassan et al. published the first in-human clinical trial evaluating anetumab, an anti-mesothelin mAb in a Phase 1 clinical trial (40). Sixty-four patients were enrolled with ovarian cancer, and of those, they noted 1 CR, 4 PRs, and 29 patients with SD. Interestingly, all patients who responded had high mesothelin expression, defined as ≥60% by immunohistochemistry staining. A Phase 1b clinical trial is currently ongoing (NCT02751918).
Other surface antigens have been identified in pre-clinical studies as possible neoantigens with anti-tumor activity and high specificity for ovarian cancer. Hong et al. showed that L1-CAM was overexpressed in a wide panel of ovarian samples (41). Administration of second-generation anti-L1-CAM CAR-Ts, to NSG mice, inoculated i.p. with SK-OV3, improved median survival time to 104.5 days when compared to mock (50 days) or anti-CD19 CAR-T cells (56.5 days). Similar tissue expression, in vitro, and in vivo studies can be seen with 5T4, B7-H3, TAG-72, and MISIIR (42–45). Interestingly, Du et al. demonstrated the addition of 4-1BB to the anti-B7-H3 CAR mediated lower expression of PD-1 in transfected CD8+ T cells, possibly enabling them to better enact effector functions. In this pre-clinical study, i.p. administration of CAR-T cells was shown to have improved survival benefit when compared to an i.v. route.
Other mechanisms of promoting immune cell activity are immune checkpoint inhibitors that target surface proteins such as cytotoxic T-lymphocyte-associated protein 4 (CTLA4), programmed cell death protein 1 (PD-1), and its ligand (PD-L1; Figure 1). These proteins normally function to prevent autoimmunity, but their upregulation on a tumor prevents an appropriate immune response. CTLA-4 competes with B7 (CD80) in binding to CD28 on T cells, inducing a suppressive phenotype rather than activating. PD-1 and PD-L1 belong to the CD28 and B7 family of receptors on T cells, respectively, and have been shown to induce a suppressive phenotype in peripheral tissues.
Drugs targeting CTLA-4, such as ipilimumab, have shown great success in other cancers. In unresectable Grade 3 and 4 melanoma, ipilimumab improved 1-year survival from 25% to 46% when used in combination with a gp-100 peptide vaccine, granting it FDA approval in March 2011. In a phase 1 clinical trial with nine Stage IV ovarian cancer patients, three instances of stable disease (SD) were observed (46). Multiple toxicities occurred, including two cases of Grade 3 inflammation in the GI tract, one case of Sweet’s syndrome, and multiple dermatological reactions. A phase 2 clinical trial was later done in patients with recurrent platinum-sensitive ovarian cancer (NCT01611558). Patients received 10 mg/kg of ipilimumab once every 3 weeks for four doses then once every 12 weeks. The overall response rate (ORR) was low at 10.3%. 18 patients experienced drug-related severe AEs, the most common being small intestinal obstruction, diarrhea, pneumonitis, and adrenal insufficiency.
Trials combining ipilimumab with other treatment regiments have also been conducted. Recently, Zamarin et al. published a Phase 2 trial comparing nivolumab to nivolumab and ipilimumab for recurrent ovarian cancer. One hundred patients were either given 3 mg/kg nivolumab every 2 weeks or 3 mg/kg nivolumab plus 1 mg/kg ipilimumab every 3 weeks (47). In the nivolumab monotherapy group, 3 complete responses (CRs), 3 partial responses (PRs), and 14 patients with SD were noted compared to the 3 CRs, 16 PRs, and 20 patients with SD in the combination group. Median progression-free survival (PFS) was 2 and 3.9 months while median OS was 21.8 and 28.1 months for the monotherapy and combination therapy, respectively. There were no treatment-related deaths with the most common grade 3 or higher AEs being asymptomatic elevation of pancreatic and liver enzymes, anemia, colitis, and diarrhea. A number of other ongoing trials are evaluating anti-CTLA-4 antibodies in combination with chemotherapy, poly (ADP-ribose) polymerase inhibitors (PARPi), and other immunotherapies (Table 1).
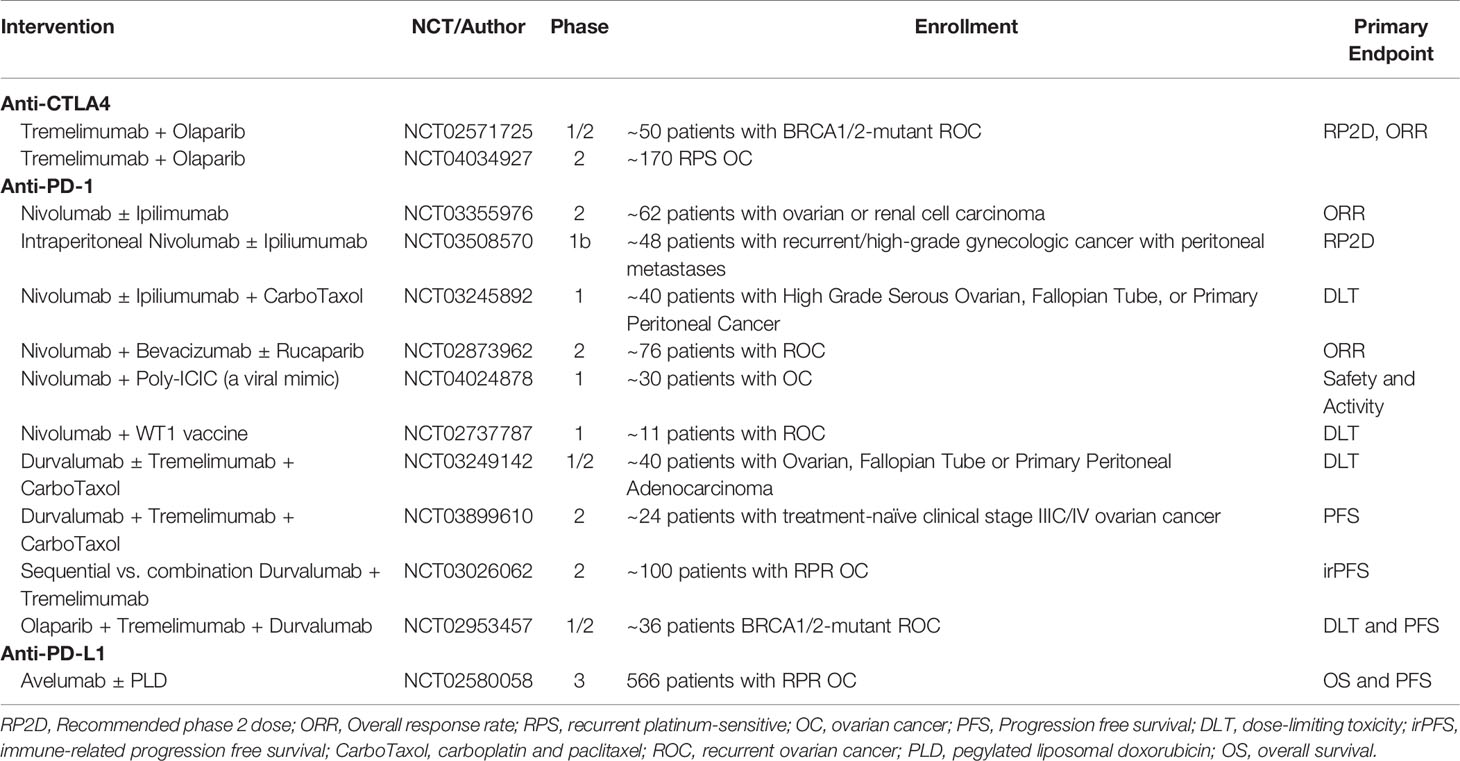
Table 1 Ongoing clinical trials of immune checkpoint inhibitors in women with ovarian cancer.
Anti-PD-1 therapy has also had some success in treating ovarian cancer. In a phase two trial, 20 patients with platinum-resistant recurrent ovarian cancer were treated with either 1 or 3 mg/kg single-dose nivolumab every 2 weeks for one year (48). In the 1 mg/kg cohort, two patients had a CR and four had SD while in the 3 mg/kg cohort, one patient had a PR while two had SD. Median OS was 20 months and PFS was 3.5 months. Low-grade AEs included mild fever, rash, arthralgia, elevated liver function tests (LFTs), and lymphocytopenia. Two patients experienced Grade 3 disorientation and gait disorder or Grade 3 fever and a deep vein thrombosis, respectively. Notably, two patients developed a PR to maintenance chemotherapy post-treatment with nivolumab (49). An open-label, randomized clinical trial is ongoing in Japan.
One Phase 2 study has been published evaluating the combination of nivolumab and bevacizumab in relapsed ovarian cancer (50). Thirty-eight women with relapsed OC were enrolled and treated with 10 mg/kg bevacizumab and 240 mg nivolumab once every 2 weeks. In platinum-sensitive patients, there were eight PRs and nine patients with SD compared to 3 PRs and 10 patients with SD in the platinum-resistant group. Median PFS was 12.1 months and 7.7 months for platinum-sensitive and -resistant patients, respectively. Grade 3 AEs reported were hypertension, myalgia, arthralgia, and elevations in LFTs and serum amylase. Two patients experienced Grade 4 increases in serum lipase levels as a result of treatment. Like anti-CTLA-4 compounds, several ongoing trials are evaluating combinations of nivolumab with chemotherapy, PARPi, or vaccines (Table 1).
Pembrolizumab, another anti-PD-1 agent, has also been evaluated as a single agent. In a phase 1b trial, 26 patients with advanced metastatic ovarian cancer received 10 mg/kg every 2 weeks for up to two years (51). ORR was documented at 11.5%, with one CR and two PRs, and seven patients experienced SD. Median PFS was 1.9 months while OS was 13.8 months. Drug-related AEs occurred in 19 patients with one patient experiencing Grade 3 increases in transaminase levels. Eight immune-related AEs occurred where the only Grade 3 AE was pancreatitis. Later, Keynote-100 looked at pembrolizumab in 396 patients with advanced recurrent ovarian cancer. Two cohorts were formed based on prior lines of treatment (52). Cohort A (285 patients) had one to three lines of prior treatment while cohort B (91 patients) had four to six. Patients received 200 mg IV every 3 weeks. ORR in cohorts A and B was 7.4% and 9.9%, respectively. Median PFS was 2.1 months for both cohorts while median OS was not reached in cohort A and 17.6 months in cohort B. The most common low-grade treatment-related AE was fatigue while the most common immune-related AE was hypothyroidism. The most common Grade 3 immune-related AEs were skin reactions and colitis. More severely, two patient deaths were attributed to treatment, one due to hyperaldosteronism and the other to Stevens-Johnson syndrome. These recurring AEs led to the need for early recognition and treatment to prevent devastating complications from immune checkpoint inhibitor therapy (53).
Recently, a single-arm phase I/II trial evaluated niraparib in combination with pembrolizumab in patients with recurrent platinum-resistant OC (54). 62 patients with OC were enrolled between Phase I and II. ORR was 18% with 3 CRs and 8 confirmed PRs. Another 28 patients were noted to have SD. Median PFS was 3.4 months. Most common low-grade AEs were fatigue, nausea, anemia, and constipation while high-grade AEs were noted to be anemia and thrombocytopenia. Extensive ongoing work is exploring combination therapies with pembrolizumab.
Durvalumab, another anti-PD-1 compound, has been tested in combination with PARPi in both Phase I and II trials. In a Phase 1 trial, seven patients with ovarian cancer, along with one endometrial and triple negative breast cancer, were treated with olaparib and cediranib, a VEGFR1-3 inhibitor (55). ORR was 44% with four partial responses and three patients with SD. Most common low-grade AEs were fatigue, nausea, and increased LFTs. Five patients experienced grade 3 hematological AEs, three with lymphopenia and two with anemia). In a subsequent Phase 2, 35 OC patients were enrolled in a single-center study (56). ORR was noted to be 14% with five patients achieving a partial response while 20 patients had SD. Overall, median PFS was noted to be 3.9 months. Like the Phase 1, hematologic toxicity, mainly anemia, was the most common high-grade AE, affecting eleven patients. The study also found significant increases in VEGFR3 were correlated with worse PFS. Of note, when evaluating durvalumab and olaparib in PARPi-naïve patients with platinum-sensitive and mutated BRCA in a Phase II trial, a 63% RR was noted, with six patients achieving a CR and fourteen achieving PRs (57). The most common grade three or higher AEs were anemia, increased lipase and amylase, and neutropenia.
Avelumab, an anti-PD-L1 therapy, though not directly inhibiting the suppression of T cells, plays a role in preventing the inactivation of T cells. In a phase 1b trial, 125 ovarian cancer patients with Stage III or IV disease received 10 mg/kg avelumab every 2 weeks (58). Confirmed objective response was seen in only twelve patients with one CR and eleven PRs. Median OS was 11.2 months. The most common treatment-related AEs were fatigue, diarrhea, and nausea while the most common high-grade was an increase in lipase-levels. Low grade immune-related AEs mainly were hypothyroidism while three patients separately experienced high-grade colitis, type 2 diabetes, or myositis. Ongoing clinical trials are evaluating avelumab with chemotherapy and PARPi (Table 1).
Discovered in 1995, Tregs were originally shown to be involved in immune homeostasis, preventing an over-activation of the immune system towards self (59). They have been characterized to express CD4, CD25, and, most notably, FoxP3 (4). Tregs enact their function by suppressing activation of immune cells, inducing cell death of effector cells, and secreting anti-inflammatory cytokines, such as TGF-beta and IL-10 (60). Since their discovery, Tregs have been shown to be involved in a number of disease processes, including cancer.
In ovarian cancer, Tregs have been shown to be an indicator of poor prognosis. Curiel et al. showed that in 104 patients, increased numbers of intratumoral Tregs predicted poor survival (61). Absolute Treg counts may overlook certain cellular interactions, because in a study of 117 patients, intraepithelial Treg counts did not correlate with a significant difference in survival (9). Instead, increased ratios of CD8+ T cells to CD4+CD25+FoxP3+ T cells were shown to significantly associate with improved survival. Later studies also showed that a high CD8+/Treg ratio as well as CD4+/Treg ratio were associated with better survival outcomes (62).
Treg function can also be influenced by the ovarian TME. TAMs and tumor cells have been shown to increase levels of CCL22, which aid in Treg recruitment to the ovarian TME (61). In a set of 75 ovarian cancer patients, CCL22 levels were shown to be elevated in the peritoneal fluid, possibly contributing to Treg recruitment and cancer progression (63). Tregs have also been shown to be highly activated when found intratumorally in ovarian cancer. CD45RA-FoxP3hi effector Tregs expressed significantly higher levels of 4-1BB, ICOS, OX40, and CTLA4 compared to CD45RA-FoxP3lo effector T cells (64). Tregs expressing high PD-1 and 4-1BB were subsequently both more responsive to stimulation by anti-CD3/anti-CD28 and were able to better suppress T cells in vitro.
One method of reducing Treg effector functions is to directly deplete Tregs. Low-dose cyclophosphamide has been tested for use in conjunction with cancer vaccines due to its ability to deplete FoxP3 Tregs (65). However, Tregs treated with low-dose cyclophosphamide were unable to suppress the proliferation of CD4+ and CD8+ T cells in vitro. When tested in ovarian cancer, a combination therapy of low-dose cyclophosphamide with a p53-SLP vaccine did not directly suppress either Treg counts or functionality (66). However, overall T cell counts were higher and persisted longer in the combination group when compared to vaccination alone. Similarly, a phase 1/2 clinical trial found a single i.v. dose of cyclophosphamide had no effect on circulating Tregs (67).
In ovarian cancer, there is evidence infiltrating B cells can be either good or bad prognostic indicators. Milne et al. found that intraepithelial CD20+ B cells, when present in patients optimally debulked from high-grade serous OC, positively correlated with disease-specific survival (68). Patients who had residual disease or another histological subtype, however, did not demonstrate any significant survival benefit with infiltrating CD20+ B cells. In a follow-up study, tumor-infiltrating CD20+ cells were found to have responded to antigen, having undergone class switching, somatic hypermutation, and clonal expansion (69). These CD20+ B cells, when found co-localized with CD8+ T cells in tumor, also correlated with improved patient survival compared to tumor-infiltrating T cells alone. In an independent study, Santoiemma et al. also found tumor-infiltrating CD20+ B cells to positively correlate with OS (70). Later, these CD20+ B cells, in addition to CD138+ plasma cells and CD4+ TILs, were found to co-localize with CD8+ T cells in tertiary lymphoid structures and indicate better prognosis (71). Interestingly, Kroeger et al. also found tumor-infiltrating plasma cells, in high-grade serous ovarian cancer, to express IgG and CXCR3, the latter being normally expressed under immuno-stimulatory environments (72).
Contrary to this finding, Lundgren et al. found CD138+ plasma cells to positively correlate with tumor grade and negatively with OS (73). Furthermore, they found CD20+ B cells only correlated with tumor grade and had no significant correlation with survival. Other studies have also shown infiltrating B cells to be detrimental. Yang et al. showed that high levels of CD19+ B cells in the omentum correlated with poor survival (74). Ultimately, additional phenotype characterization of tumor-infiltrating B cells needs to be completed to determine their impact in the TME and patient outcomes.
Recently, a subset of B cells known as B regulatory cells has been found in ovarian cancer patients (75). More specifically, IL-10+ B cells were increased in ascites compared to peripheral blood. These B cells were inversely correlated with the number of CD8+ T cells in ascites and positively correlated with FoxP3+CD4+ T cells. Ex vivo studies showed these B cells were capable of suppressing IFN gamma secretion by T cells even under stimulation by anti-CD28. Finally, accumulation of these cells in the ascites correlated with more late-stage and aggressive disease. In models of spontaneous ovarian cancer, increased CD25+ pre-B-like cells were found intratumorally (76). These pre-B-like cells were shown in breast cancer models to develop into tBregs (CD19+ CD25HiCD69Hi) and promote metastases. There are no modalities currently available that specifically target Bregs. Additional research is necessary to determine which markers best define this subset and how they function in ovarian cancer.
Despite recent advances in the use of checkpoint inhibitors and augmenting the T cell response, attempts at augmenting adaptive immunity have been unsuccessful in treating ovarian cancer patients. Recent efforts have shifted to include exploiting the innate immune response (77). Myeloid cells of the innate immune system, such as monocytes, classical macrophages, natural killer cells, and dendritic cells, all play key roles in promoting an effective adaptive response; the lack of accounting for these interactions may be where current immunotherapies fall short. Alternatively, cell types such as TAMs and MDSCs induce a highly immuno-suppressive TME and may be targets of future therapeutic strategies (6, 7).
As the main mediator of responses between the innate and adaptive immune response, conventional human DCs (cDCs, CD141+, or CD1c+), are critical for the adaptive response by up-taking antigen and skewing helper T cell differentiation (78). However, cancer cells subvert proper antigen presentation by down-regulating MHC, reducing TAAs on their surface, and can be suppressed by numerous cytokines (79). DCs can also induce T cell suppression themselves through PD-1 and CD277 in the ovarian cancer TME (80, 81). Human plasmacytoid dendritic cells (pDCs, CD303+) increase immunosuppression in the ovarian cancer TME through upregulation of Tregs (82, 83). Furthermore, tumor-associated pDCs have been correlated with poor prognosis and early relapse for ovarian cancer patients, possibly due to their influence on CD4+ T cells to produce increased IL-10 (84).
Multiple clinical trials have looked at the benefit of utilizing DCs, obtained through leukapheresis or derived from monocytes, that are pulsed with specific antigens as immunotherapy. In one trial, autologous DCs were pulsed with Her2/neu, human telomerase reverse transcriptase, and pan-DR epitope with or without a single-dose of cyclophosphamide. Five of eleven patients had no evidence of disease at the time of publication with only one patient dying of disease within 36 months after the initial vaccination (67). No grade 3 or 4 treatment-related AEs were reported. Two studies introduced IL-2 in conjunction with DC treatment. Rahma et al. evaluated the optimal mechanism of peptide delivery to DCs, with 6 OC patients receiving DCs pulsed ex vivo with wild-type p53 peptide 264-272 (85). PFS and median OS was found to be 8.7 and 29.6 months, respectively, comparable to those patients receiving subcutaneous injections of solely peptide. Notably, all Grade 3 or 4 AEs occurred during cycles of IL-2 administration—the most common being fatigue, lymphopenia, and elevated liver enzymes. IL-2 administration also led to increases in Tregs. In another study by Baek et al., 10 patients with minimal residual disease were treated with DCs pulsed with keyhole limpet haemocyanin (KLH) and IL-2 (86). KLH has previously been used as a surrogate marker for DC vaccination. Three patients underwent complete remission with the most common AEs being flu-like symptoms, attributed to IL-2 administration. Contrastingly, this study found treatment decreased CD4+CD25+ T cells, albeit they did not characterize FoxP3 expression. A recent Phase I/II study enrolled three ovarian cancer patients and treated them with DCs pulsed with Wilms’ tumor protein 1 (87). One patient reached SD by RECIST criteria while the other two had progressive disease. No Grade 3 or greater AEs were reported.
Alternatively, DCs could be pulsed with whole tumor lysate as a means of eliciting a response to a variety of neoantigens as opposed to a single one. In a phase I study with six ovarian cancer patients, autologous DCs were pulsed with autologous tumor lysate and (KLH) (84). No grade 2 or higher AEs reported. Most common Grade 1 AEs included pain, fatigue, nausea, and abdominal pain. In another clinical study, 25 immunotherapy-naïve OC patients were treated with either a) intranodal injections of DCs pulsed with oxidized whole tumor lysate, b) whole-tumor lysate pulsed DCs with bevacizumab or c) the prior combination with cyclophosphamide (88). No toxicities greater than Grade 2 were reported due to the treatment. The most common Grade 1 AEs overall were pain, fatigue, nausea and abdominal pain. There were two PRs and thirteen patients had SD. Of note, patients without intratumoral T cells reactive for autologous tumor had poorer outcomes, again suggesting the success of DC vaccinations relies on the ability to generate a specific T cell response. Interestingly, the addition of cyclophosphamide improved both immune response, as measured by IFN-gamma release, and ultimately patient survival.
But despite some success of these DC vaccines, limitations include the ability to generate a consistent immuno-stimulatory effect and the difficulty of vaccine production (89). In an attempt to generate a more efficient T-cell response in a preclinical setting, Mirandola et al. treated DCs infected with a recombinant adeno-associated virus (rAAV) containing cancer/testis antigen mSP17 with a p38 MAPK inhibitor (90). A p38 inhibitor was used due to previous studies showing its use improving monocyte-derived DC survival and decreasing Treg production (91, 92). Murine DCs were infected with rAAV-mSP17 and treated with a p38 MAPK inhibitor. Survival analyses showed that when C57BL/6 mice, injected i.p. with 1 × 106 ID8 cells, were treated with DCs plus p38 inhibitor, 95% of mice survived up to 300 days, as compared to those receiving solely DCs surviving up to 98 days. Furthermore, the addition of p38 inhibitor increased lymphocyte trafficking to the tumor. When using human DCs, the addition of p38 inhibitor significantly decreased PD-L1 expression, reversing one contributor to the highly immunosuppressive TME. In a separate attempt to alternatively produce DCs for use in OC patients, Cheng et al. developed “mini DCs” through the use of cell membrane coating nanotechnology, i.e., fusing cell membranes onto synthetic polymer cores (93). To traffic tumor antigens to the membrane prior to fusion, ID8 murine cells were lysed and pulsed onto bone marrow-derived DCs. Additionally, IL-2 was loaded into the nanoparticle prior to emulsion. Mice inoculated with ID8 cells subcutaneously showed significant growth reduction when treated with the mini DCs compared to normal DCs and empty nanoparticles. Increased CD8+ T cell infiltration and decreased Tregs were also observed intratumorally in the mini DC treated mice. When evaluating the effect of mini DCs on metastases, mice injected i.p. with ID8 cells and treated with mini DCs had significantly fewer nodules on the peritoneal wall when compared to vehicle or those treated with normal DCs. No changes in body weight or liver and kidney functions were observed in mice treated with mini DCs, indicating good biocompatibility.
Natural killer cells, CD56+, have become increasingly popular as an immunotherapy due to their ability to kill without prior sensitization to antigen. Instead, they integrate activating and inhibitory receptors in order to mediate their cytotoxic effect. Receptors, such as killer cell immunoglobulin-like receptors (KIRs) and NKG2A-C that recognize MHC and NKG2D that recognize stress molecules, cooperate to sense “missing self,” “induced self,” or “altered self (94).” Similar to T cells, these cells are able to kill by perforin-granzyme and also by FAS and TRAIL-mediated mechanisms (95). In OC, NK cells have been reported to both be positively and negatively prognostic. One study evaluated the prognostic value of intra-tumoral NK cells in 82 patients with mixed histologies (96). Researchers found patients with only intra-epithelial infiltration of NK cells had an increased OS (106 months) compared to those with only intra-stromal infiltration (58 months); no difference was seen in PFS between these two groups. However, tumor infiltration of CD56+ NK cells did not correlate with prognosis. Additionally, this study evaluated the presence of ULBP2 and MICA/B on patient outcomes—both activating ligands of NKG2D thought of commonly to mark cells for elimination. Interestingly, high levels of ULBP2 on tumor samples was found to indicate a poor prognosis for cancer patients, while MICA/B did not correlate with prognosis. This may be due to high levels of ULBP2 inhibiting proper T cell functioning. Samples from 283 patients with high-grade serous carcinoma were evaluated by immunohistochemistry for NK cell infiltration (97). Median OS in patients with high levels of CD57+ NK cells (≥10 cells/mm2) was improved, compared to patients with low levels (<10 cells/mm2), 45 vs. 29 months, respectively. Interestingly, higher CD56+ NK cells:lymphocyte fraction in ascites was associated with both a better PFS and OS in 20 OC patients (98). It was noted though that by selecting for patients that had enough ascites, patients with poor prognosis were inadvertently selected. Further studies will be needed to evaluate the importance of intra-tumoral NK cells.
There are multiple clinical trials in progress evaluating the benefits of augmenting NK cell number and function. One Phase I trial is evaluating IP FATE-NK100, a donor-derived NK product compromised from terminally differentiated cells, with IL-2, as a means of promoting NK survival, and lymphodepletion by cyclophosphamide and fludarabine (CyFlu) in women with recurrent OC (NCT03213964). Another Phase I trial is evaluating i.p. NK cells, instead generated from CD34 hematopoietic stem cells in umbilical cord blood, in twelve recurrent OC patients with lymphodepletion by CyFlu (NCT03539406). In an attempt to boost the body’s own NK cells, one Phase II trial is evaluating the use of i.p. as well as subcutaneous (s.q.) IL-15Ra super-agonist, ALT-803, after first-line chemotherapy (NCT03054909). It was previously shown that in OC, ascites-derived NK cells and healthy donor NK cells improved their reactivity when stimulated with IL-15 or ALT-803 (98). One published Phase II study evaluated i.v. NK cells, treated ex vivo with IL-2, given post-lymphodepletion by CyFlu in 14 ovarian cancer patients (99). Five patients also received total body irradiation to deplete lymphocytes and allow for NK expansion. Despite four patients reaching PRs and eight having SD, one patient developed a grade 5 toxicity due to tumor lysis syndrome. Other severe AEs, such as passenger lymphocyte syndrome and neutropenia, were attributed to the irradiation.
CARs have been also added to NK cells, as well as NKT cells (CD3+CD56+), in an attempt to utilize these cells. Briefly, NKT cells carry characteristics of both NK and T cells, enabling them to enact cytotoxic killing without prior activation. Utilizing a CAR against FR, Zuo and colleagues showed NKTs with improved cytotoxicity towards FR+ PEO1 cells in vitro when compared to CAR-Ts by transfecting NKT cells with CARs carrying both the CD28 and 4-1BB co-stimulatory signaling domain, (100). However, the NKT cells performed worse than CAR-T cells in nude mice inoculated subcutaneously with PEO1 cells. NK cells, specifically NK-92, were also used as a surrogate for a third-generation anti-FR CAR (101). Similarly, these NK cells demonstrated cytotoxic effects against SKOV3 in vitro and in B-NDG mice inoculated i.p. with SKOV3. One clinical trial utilizing anti-mesothelin NK cells, obtained from peripheral blood mononuclear cells, has been proposed (NCT03692637).
Changes in synthesis methods and receptor signaling may also be beneficial to the success of CAR-NKs. Li et al. created anti-mesothelin CAR-NKs, with additional NKG2D and 2B4 domains, from induced pluripotent stem cells (iPSCs) (102). Advantages to using iPSC include better clonal manipulation of the end product and increased speed of production. In a xenograft model injected i.p. with A1847 cells, treatment with their CAR-NK, along with IL-2 and IL-15, significantly reduced tumor burden and, ultimately, improved survival. When directly compared to a CAR-T with the same receptor, but with 4-1BB and CD28 signaling motifs instead, mice treated with the CAR-NK had less weight loss and pathogenic damage in organs such as the liver and kidney. Furthermore, those treated with CAR-NK also had improved survival when directly compared to those given CAR-T cells. Another group has created a CAR-NKs against glypican-3 (GPC3), with CD28 and 4-1BB signaling motifs, also from iPSCs derived immune cells (103). In NSG mice inoculated i.p. with KOC7c, a GPC3-expressing ovarian cancer cell line, a statistically significant difference in survival was seen when compared to PBS. Klapdor et al. has also developed dual-CAR-NKs, using NK-92, against CD24 and mesothelin with CD28 and 4-1BB signaling motifs (104). CD24 was chosen as a target due to its presence on cancer stem cells and lack thereof on normal tissues; mesothelin has been previously discussed. In A2780 and HEK293T cells previously transfected with CD24, mesothelin, or both, the dual-CAR was able to target both mesothelin-positive cells and CD24-positive cells.
Monocytes can be classified into three subsets based on CD14 and CD16 expression: classical monocytes (CD14+CD16−), intermediate monocytes (CD14+CD16+), and non-classical monocytes (CD14-CD16+). Upon inflammation, monocytes traffic to the tissue and differentiate on a spectrum ranging from classically activated macrophages, or M1-like, to alternatively activated macrophages, or M2-like. A holistic review on M1 vs. M2 macrophage differentiation is more thoroughly reviewed in (105). In brief, M1-like macrophages, induced by IFN-γ and TNF-α, secrete inflammatory cytokines, such as IL-6 and IL-12, while M2-like macrophages are induced by TGF-β and IL-4/13 and secrete anti-inflammatory cytokines and recruiting Tregs (106). Cancer cells themselves are able to induce a shift towards an M2 phenotype through the secretion of signaling molecules. In OC specifically, CSCs were shown to increase levels of CCL2, COX-2 and PGE-2 as well as activate the PPARγ pathway, all of which correlated with increased polarization towards M2 macrophages (107, 108). Unsurprisingly, a high M1/M2 ratio has been correlated with improved survival in OC patients (109). A meta-analysis also indicated the presence of CD163+ TAMs was correlated with poor prognosis (110). A decreased lymphocyte-to-monocyte ratio (LMR) also indicated both poor overall and PFS in retrospective reviews (111–114).
In order to bolster immunity against cancer, the goal would be to decrease M2-like macrophages and/or increase their M1-like counterparts. Pre-clinical studies have attempted to decrease the prominence of M2 macrophages in the OC TME by interfering with the number of TAMs. Trabectedin, an inhibitor of DNA repair and transcription, was found to activate caspase-8 in monocytes through TRAIL-R1/2, leading to decreases in TAMs (115). Paclitaxel, a microtubule inhibitor currently in use to treat ovarian cancer, was recently found to shift M2 macrophages towards M1 in a TLR4-dependent fashion (116).
In OC, the addition of IFN-α/γ to monocytes was hypothesized to maintain the M1-like phenotype when used as an anti-cancer therapy. Importantly, this combination was shown to significantly reduce tumor burden and improve survival in BALB/c mice inoculated subcutaneously with OVCAR-3 (117). Mice treated with the combination of IFNs and monocytes survived to 170 days when compared to 87 and 81 days for IFNs or monocytes alone, respectively. Intra-tumoral macrophages were identified by CD31 and CD68 staining. Further immunofluorescence (IF) characterization showed that the cells expressed M1 markers IL12, CXCL10 and NOS2, but decreased M2 markers IL-10 and arginase, indicating that the IFN-treated monocytes retained differentiation towards an M1-like phenotype. Extrapolating these findings to other ovarian cancer cell lines, Johnson et al. reinforced the ability of monocytes and IFNs to kill tumor cells synergistically, although sensitivity varied between lines (118). A Phase 1 clinical trial was created to evaluate the intraperitoneal administration of autologous monocytes treated ex vivo with pegylated inteferon α-2b and interferon γ-1b (119). Preliminary results showed a well-tolerated treatment with two PRs and four patients with SD (120).
Compared to the veteran immunosuppressive Tregs (discovered in 1969), myeloid derived suppressor cells (MDSCs) are a relatively “young” subset (121). Poorly-differentiated myeloid cells with the capacity to suppress T cell activation were reported in the 1970s (122), but the term “MDSC” was not coined until 2007 (123), and many questions relating to the origin, classification, and behavior of MDSCs remain unresolved (124).
A comprehensive review of the current understanding of MDSCs have been presented previously (7, 125). Briefly, MDSCs are myeloid derived cells which develop in bone marrow, traffic through peripheral blood to the tumor, and increase during tumor development in response to chemotactic and growth factor signals released by the tumor itself such as G-CSF, GM-CSF, VEGF, and IFN-γ (126, 127). MDSCs are functionally characterized by the ability to suppress T cell activation ex vivo (122), and to suppress the ability of immune cells in the TME to mount an antitumor response in vivo by mechanisms which are incompletely characterized, but include direct suppression of cytotoxic T cells and NK cells via PDL1/2, promotion of Treg expansion by TGF-beta, CD40L, and Il-10, and promotion of M2-like/TAM development (128). MDSCs can be identified by their expression of specific markers, allowing division into monocytic and granulocytic subtypes according to consensus guidelines, although these classification schemes remain in flux (125). In mice, monocytic MDSCs are defined as CD11b+ Ly6Chigh Ly6G– while granulocytic MDSCs are CD11b+ Ly6Clow Ly6G+. In humans, monocytic MDSCs are HLA-DR-CD11b+CD33+CD14+ while granulocytic MDSCs are HLA-DR-CD11b+CD33+CD15+.
The clinical relevance of MDSCs as drivers of ovarian cancer pathogenesis has been demonstrated by correlative studies in humans associating MDSC frequency and phenotype with worse prognosis as well as by experimental manipulations in mice, in which direct ablation of MDSCs can impede tumor development. In patient series, higher MDSC frequency in tumor biopsy (129), in peripheral blood (130), or in ascites (131) correlated with decreased OS or relapse free survival, as did high MDSC-to-dendritic cell ratio in peripheral blood (132). Syngeneic mouse model studies have demonstrated that MDSCs accumulate during the course of tumor development (133) and that ablation of MDSC by clodronate liposomes led to increased survival. Depletion of MDSC by anti-Gr1 antibodies also inhibited tumor growth in mouse and depletion of MDSC led to increased mouse survival whereas adoptive transfer of MDSC from one tumor-bearing mouse to another improved tumor growth (134, 135). Similarly, depletion of MDSCs by anti-GM-CSF therapy reversed anti-VEGF therapy resistance, reducing intra-tumoral MDSCs and increasing CD8+ TILs (136).
MDSCs have the capacity to develop into immunosuppressive M2-like macrophages, but they can also differentiate into non-immunosuppressive cell types such as conventional M1 macrophages and dendritic cells. Agents such as all-trans-retinoic acid (ATRA) and epigenetic modifiers such as histone-deacetylase inhibitors (HDACi) have been demonstrated to induce differentiation of MDSC (resulting in functional depletion) in preclinical studies. In ovarian cancer, the phase II trial of histone deacetylase inhibitor entinostat in combination with checkpoint inhibitor avelumab failed to demonstrate an advantage over avelumab alone (137). Differentiation vs depletion of MDSC by the hypomethylating agent azacytidine has been reported in mouse (138). Of note, azacytidine has also been reported to sensitize platinum resistant ovarian cancer cells to platinum by mechanisms that are not entirely understood (139), which has been the inspiration for other trials. Decitabine has been pursued as a platinum sensitizer and reported as having disease activity by CA-125 reduction but not by reduction in tumor size, i.e., RECIST criteria (140). A phase 1 trial of azacytidine and valproic acid, another HDACi, in combination with carboplatin in patients with platinum-refractory solid tumors was reported in abstract form as displaying disease activity but with a very high (78%) rate of grade 3–4 toxicity (141).
Blocking of MDSC migration to tumor might be achieved by a variety of manipulations, including blocking of cytokines such as VEGF, G-CSF, GM-CSF, and M-CSF, as discussed above. VEGF inhibition by bevacizumab already has an established role in treatment of ovarian cancer, with approvals in both the first-line and later-line settings. Interestingly, mouse data in bevacizumab-resistant tumors demonstrated that MDSCs recruited by GM-CSF, produced by the tumor drove immunosuppression which was reversible by blockade of GM-CSF production (136). In the clinic, early phase I/II experimental approaches are focusing on blocking the receptors on MDSCs which mediate response to the above cytokines, namely CXCR2 (NCT02370238 and NCT02499328), CCR5 (NCT01736813), and CSF1R (NCT01349036). In addition, a phase 1 combination trial of the anti-CSF1R antibody cabiralizumab plus nivolumab (NCT02526017) including ovarian cancer patients is underway.
Finally, inhibition of MDSC function has been hampered to some extent by an incomplete understanding of the complex mechanisms by which MDSCs downregulate antitumor immunity. Depletion of L-arginine in the TME via expression of arginase-1 and inducible nitric oxide synthase (NOS-1) is thought to directly inhibit T cell function and result in cell cycle arrest (142). Disruption of this process may be achieved in vitro by inhibiting upstream inflammatory cyclooxygenase-2 (COX-2), prostaglandin E2 (PGE2) or phosphodiesterase-5 (PDE-5) signaling (143). Similarly, use of PDE-5 inhibitors such as sildenafil and tadalafil, which are already FDA approved for non-cancer indications, is being tested in combination trials in a variety of solid tumors. A phase I study of the anti-VEGFR2 molecule regorafenib and sildenafil in multiple solid tumors was reported, with evidence of disease activity including two ovarian cancer patients who achieved SD for >24 weeks (144).
Ovarian cancer remains a lethal disease due to late-stage diagnoses and a lack of suitable treatment options in the recurrent setting. Despite recent advances in PARPi and anti-VEGF treatment modalities, a better understanding of the immune cell subsets and their interactions in the peritoneal TME may bring forth novel targeted therapies and additional combination therapies. Herein, we discussed key subsets of the many immune cells that play a role in the immuno-suppressive and tumor-promoting microenvironment of ovarian cancer, and recent attempts to therapeutically employ both adaptive (Table 2) and innate (Table 3) immunity. Tumor-specific lymphocytes have been found in ovarian cancer, and have been associated with better prognoses, but anti-inflammatory cytokines produced by Tregs and other cells may overwhelm their effector functions. Depleting Tregs by cyclophosphamide has not been shown to directly affect Treg counts, but may promote overall T cell count. Reducing counts of other immunosuppressive cells, mainly those of innate immunity, may be significant in future treatments. MDSCs or M2-like macrophages may be important target populations.
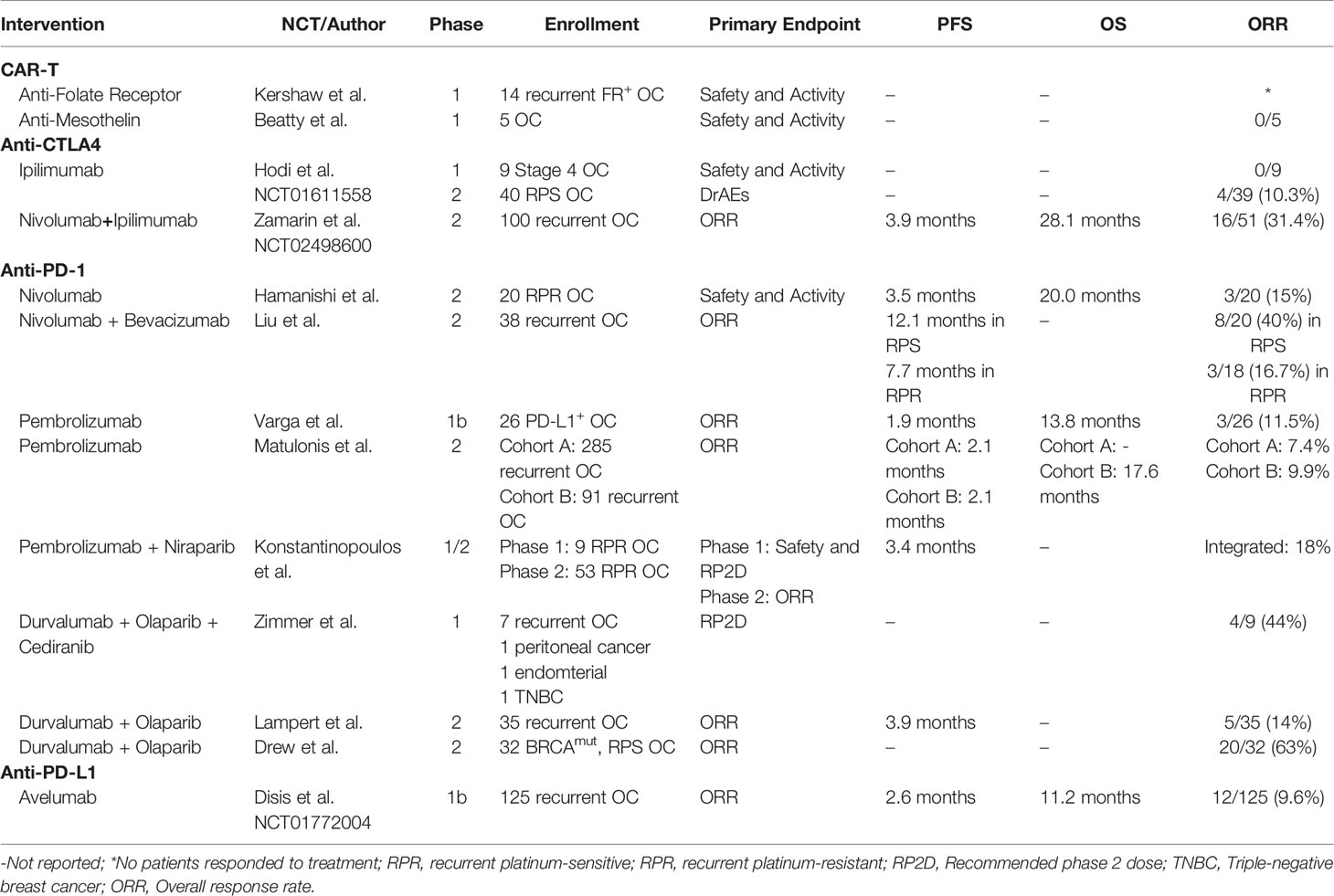
Table 2 Published clinical trials of immunotherapies modulating adaptive immunity of ovarian cancer patients.
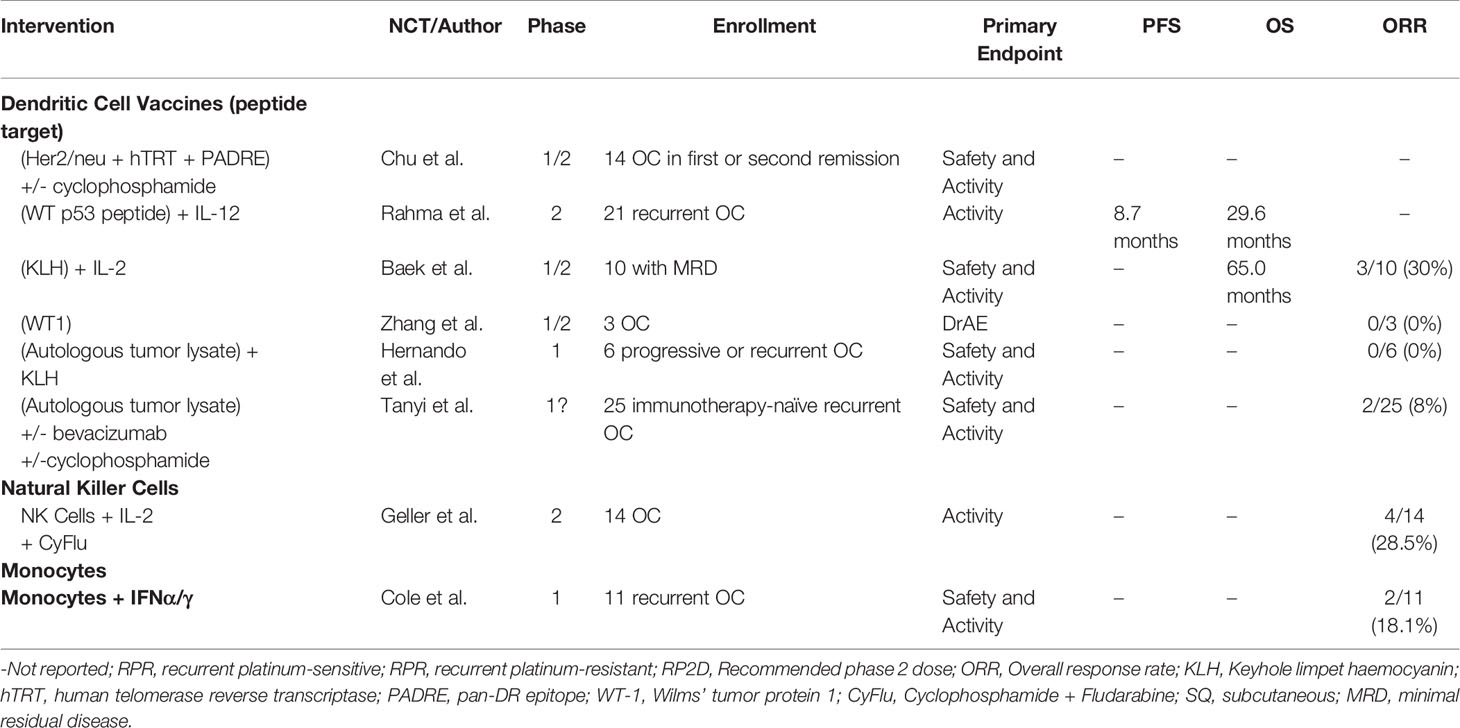
Table 3 Published clinical trials of immunotherapies modulating innate immunity in ovarian cancer patients.
A lack of tumor associated antigens and proper stimulation may also hinder a targeted immune response. A main problem seen in CAR-T therapies included a lack of persistence within circulation and poor penetrance into the peritoneal TME. Therapies such as checkpoint inhibitors and CAR-T cells may have demonstrated poor efficacy due to cellular interactions between adaptive and innate immunity that are yet to be fully characterized. Bregs are one class of cells that have yet to be fully understood. MDSCs have also been shown to play a significant role in decreasing immune function in the TME by secreting cytokines and directly inhibiting adaptive immune cells, but need further characterization.
Augmenting the innate immune response may be a mechanism by which to improve the anti-tumor response. Multiple groups have evaluated DCs as ways to augment a tumor-specific T cell response. Trials evaluating various peptides and whole tumor lysate have shown varied results though, but with minimal AEs. Future studies evaluating additional mechanisms of sustaining DC activation as well as possible bio-nanotechnology to replace synthesis of DCs will be critical in developing this option. Another possible mechanism is to use NK or NKT cells as an effector cell because they do not require additional activation. These cells have demonstrated the capacity to be transfected with CARs and clinical trials are currently underway. Similarly, efforts to utilize monocytes as effector cells are underway. Targeting of MDSCs is an exciting avenue for ovarian cancer therapy, with the multiple agents and combinations discussed above being tested, and many others with promising preclinical data. However, there are outstanding questions which need to be addressed in order for these approaches to be maximally beneficial. One invaluable is given the rapidly increasing number of available therapeutic combinations, how do we rationally choose which combinations to test in our patients? Ultimately, further understanding of the interactions amongst tumor and immune cells in the unique peritoneal microenvironment will allow us to better develop optimal targeted therapies for the treatment of ovarian cancer.
FN: conceived and designed the manuscript, performed the research, and wrote the manuscript. CC: wrote the manuscript and edited the manuscript. CA: conceived and designed the manuscript and edited the manuscript. All authors contributed to the article and approved the submitted version.
Intramural Research Program, National Cancer Institute (CMA, BC011775).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Nwani NG, Sima LE, Nieves-Neira W, Matei D. Targeting the microenvironment in high grade serous ovarian cancer. Cancers (2018) 10(8):1–22. doi: 10.3390/cancers10080266
2. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med (2011) 17:1498–503. doi: 10.1038/nm.2492
3. Hamanishi J, Mandai M, Konishi I. Immune checkpoint inhibition in ovarian cancer. Int Immunol (2016) 28:339–48. doi: 10.1093/intimm/dxw020
4. Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. Annual Review of Immunology Regulatory T Cells and Human Disease. Annu Rev (2020) 38:541–66. doi: 10.1146/annurev-immunol-042718
5. Rosser EC, Mauri C. Regulatory B Cells: Origin, Phenotype, and Function. Immunity (2015) 42:607–12. doi: 10.1016/j.immuni.2015.04.005
6. Noy R, Pollard JW. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity (2014) 41:49–61. doi: 10.1016/j.immuni.2014.06.010
7. Ostrand-Rosenberg S, Fenselau C. Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J Immunol (2018) 200:422–31. doi: 10.4049/jimmunol.1701019
8. Zhang L, Conejo-Garcia JR, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A, et al. Intratumoral T Cells, Recurrence, and Survival in Epithelial Ovarian Cancer. N Eng J Med (2003) 348:203–13. doi: 10.1056/NEJMoa020177
9. Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8 tumor-infiltrating lymphocytes and a high CD8 regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. PNAS (2005) 102(51):18538–43. doi: 10.1073/pnas.0509182102
10. Pinto MP, Balmaceda C, Bravo ML, Kato S, Villarroel A, Owen GI, et al. Patient inflammatory status and CD4+/CD8+ intraepithelial tumor lymphocyte infiltration are predictors of outcomes in high-grade serous ovarian cancer. Gynecol Oncol (2018) 151:10–7. doi: 10.1016/j.ygyno.2018.07.025
11. Li D, Li X, Zhou WL, Huang Y, Liang X, Jiang L, et al. Genetically engineered t cells for cancer immunotherapy. Signal Transduct Target Ther (2019) 4:1–17. doi: 10.1038/s41392-019-0070-9
12. van Schandevyl S, Kerre T. Chimeric antigen receptor T-cell therapy: design improvements and therapeutic strategies in cancer treatment. Acta Clin Belgica: Int J Clin Lab Med (2020) 75:26–32. doi: 10.1080/17843286.2018.1545373
13. Martinez M, Moon EK. CAR T cells for solid tumors: New strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front Immunol (2019) 10:128. doi: 10.3389/fimmu.2019.00128
14. Wagner J, Wickman E, DeRenzo C, Gottschalk S. CAR T Cell Therapy for Solid Tumors: Bright Future or Dark Reality? Mol Ther (2020) 28:2320–39. doi: 10.1016/j.ymthe.2020.09.015
15. Scaranti M, Cojocaru E, Banerjee S, Banerji U. Exploiting the folate receptor α in oncology. Nat Rev Clin Oncol (2020) 17:349–59. doi: 10.1038/s41571-020-0339-5
16. Lu Y, Low PS. Immunotherapy of folate receptor-expressing tumors: Review of recent advances and future prospects. J Controlled Release (Elsevier) (2003) 91(1-2):17–29. doi: 10.1016/S0168-3659(03)00215-3
17. Siu MKY, Kong DSH, Chan HY, Wong ESY, Ip PPC, Jiang LL, et al. Paradoxical Impact of Two Folate Receptors, FRα and RFC, in Ovarian Cancer: Effect on Cell Proliferation, Invasion and Clinical Outcome. PLoS One (2012) 7(11):e47201. doi: 10.1371/journal.pone.0047201
18. Chen YL, Chang MC, Huang CY, Chiang YC, Lin HW, Chen CA, et al. Serous ovarian carcinoma patients with high alpha-folate receptor had reducing survival and cytotoxic chemo-response. Mol Oncol (2012) 6:360–9. doi: 10.1016/j.molonc.2011.11.010
19. Chen J, Li L. Aberrant expression of folate metabolism enzymes and its diagnosis and survival prediction in Ovarian Carcinoma. Anal Cell Pathol (2019) 2019:1–9. doi: 10.1155/2019/1438628
20. Toffoli G, Russo A, Gallo A, Cernigoi C, Miotti S, Sorio R, et al. Expression Of Folate Binding Protein As A Prognostic Factor For Response To Platinum-Containing Chemotherapy And Survival In Human Ovarian Cancer. Wiley-Liss, Inc (1998).
21. Notaro S, Reimer D, Fiegl H, Schmid G, Wiedemair A, Rössler J, et al. Evaluation of folate receptor 1 (FOLR1) mRNA expression, its specific promoter methylation and global DNA hypomethylation in type I and type II ovarian cancers. BMC Cancer (2016) 16:1–13. doi: 10.1186/s12885-016-2637-y
22. Köbel M, Madore J, Ramus SJ, Clarke BA, Pharoah PDP, Deen S, et al. Evidence for a time-dependent association between FOLR1 expression and survival from ovarian carcinoma: Implications for clinical testing. An Ovarian Tumour Tissue Analysis consortium study. Br J Cancer (2014) 111:2297–307. doi: 10.1038/bjc.2014.567
23. Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res (2006) 12:6106–15. doi: 10.1158/1078-0432.CCR-06-1183
24. Song DG, Ye Q, Carpenito C, Poussin M, Wang LP, Ji C, et al. In vivo persistence, tumor localization, and antitumor activity of CAR-engineered T cells is enhanced by costimulatory signaling through CD137 (4-1BB). Cancer Res (2011) 71:4617–27. doi: 10.1158/0008-5472.CAN-11-0422
25. Vergote I, Armstrong D, Scambia G, Teneriello M, Sehouli J, Schweizer C, et al. A randomized, double-blind, placebo-controlled, phase 3 study to assess efficacy and safety of weekly farletuzumab in combination with carboplatin and taxane in patients with ovarian cancer in first platinum-sensitive relapse. J Clin Oncol (2016) 34:2271–8. doi: 10.1200/JCO.2015.63.2596
26. Moore KN, Martin LP, O’Malley DM, Matulonis UA, Konner JA, Perez RP, et al. Safety and activity of mirvetuximab soravtansine (IMGN853), a folate receptor alpha-targeting antibody-drug conjugate, in platinum-resistant ovarian, fallopian tube, or primary peritoneal cancer: A phase i expansion study. J Clin Oncol (2017) 35:1112–8. doi: 10.1200/JCO.2016.69.9538
27. O’Malley DM, Matulonis UA, Birrer MJ, Castro CM, Gilbert L, Vergote I, et al. Phase Ib study of mirvetuximab soravtansine, a folate receptor alpha (FRα)-targeting antibody-drug conjugate (ADC), in combination with bevacizumab in patients with platinum-resistant ovarian cancer. Gynecol Oncol (2020) 157:379–85. doi: 10.1016/j.ygyno.2020.01.037
28. Wang Y, Cheon DJ, Lu Z, Cunningham SL, Chen CM, Luo RZ, et al. MUC16 expression during embryogenesis, in adult tissues, and ovarian cancer in the mouse. Differentiation (2008) 76:1081–92. doi: 10.1111/j.1432-0436.2008.00295.x
29. Chekmasova AA, Rao TD, Nikhamin Y, Park KJ, Levine DA, Spriggs DR, et al. Successful eradication of established peritoneal ovarian tumors in SCID-Beige mice following adoptive transfer of T cells genetically targeted to the MUC16 antigen. Clin Cancer Res (2010) 16:3594–606. doi: 10.1158/1078-0432.CCR-10-0192
30. Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. OncoImmunology (2015) 4:1–11. doi: 10.4161/2162402X.2014.994446
31. Koneru M, O’Cearbhaill R, Pendharkar S, Spriggs DR, Brentjens RJ. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16ecto directed chimeric antigen receptors for recurrent ovarian cancer. J Trans Med (2015) 13:1–11. doi: 10.1186/s12967-015-0460-x
32. Berek JS, Taylor PT, Gordon A, Cunningham MJ, Finkler N, Orr J, et al. Randomized, placebo-controlled study of oregovomab for consolidation of clinical remission in patients with advanced ovarian cancer. J Clin Oncol (2004) 22:3507–16. doi: 10.1200/JCO.2004.09.016
33. Braly P, Nicodemus CF, Chu C, Collins Y, Edwards R, Alan Gordon J, et al. The Immune Adjuvant Properties of Front-line Carboplatin-Paclitaxel: A Randomized Phase 2 Study of Alternative Schedules of Intravenous Oregovomab Chemoimmunotherapy in Advanced Ovarian Cancer. J Immunother (2008) 32(1):54–65. doi: 10.1097/CJI.0b013e31818b3dad
34. Brewer M, Angioli R, Scambia G, Lorusso D, Terranova C, Panici PB, et al. Front-line chemo-immunotherapy with carboplatin-paclitaxel using oregovomab indirect immunization in advanced ovarian cancer: A randomized phase II study. Gynecol Oncol (2020) 156:523–9. doi: 10.1016/j.ygyno.2019.12.024
35. Battaglia A, Buzzonetti A, Fossati M, Scambia G, Fattorossi A, Madiyalakan MR, et al. Translational immune correlates of indirect antibody immunization in a randomized phase II study using scheduled combination therapy with carboplatin/paclitaxel plus oregovomab in ovarian cancer patients. Cancer Immunol Immunother (2020) 69:383–97. doi: 10.1007/s00262-019-02456-z
36. Argani P, Iacobuzio-Donahue C, Ryu B, Rosty C, Goggins M, Wilentz RE, et al. Mesothelin Is Overexpressed in the Vast Majority of Ductal Adenocarcinomas of the Pancreas: Identification of a New Pancreatic Cancer Marker by Serial Analysis of Gene Expression (SAGE) 1. Clin Cancer Res (2001) 7:3862–8.
37. Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. PNAS (2009) 106(9):3360–5. doi: 10.1073/pnas.0813101106
38. Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res (2014) 2:112–20. doi: 10.1158/2326-6066.CIR-13-0170
39. Haas AR, Tanyi JL, O’Hara MH, Gladney WL, Lacey SF, Torigian DA, et al. Phase I Study of Lentiviral-Transduced Chimeric Antigen Receptor-Modified T Cells Recognizing Mesothelin in Advanced Solid Cancers. Mol Ther (2019) 27:1919–29. doi: 10.1016/j.ymthe.2019.07.015
40. Hassan R, Blumenschein GR, Moore KN, Santin AD, Kindler HL, Nemunaitis JJ, et al. First-in-Human, Multicenter, Phase I Dose-Escalation and Expansion Study of Anti-Mesothelin Antibody-Drug Conjugate Anetumab Ravtansine in Advanced or Metastatic Solid Tumors. J Clin Oncol (2020) 38:1824–35. doi: 10.1200/JCO.19
41. Hong H, Brown CE, Ostberg JR, Priceman SJ, Chang WC, Weng L, et al. L1 cell adhesion molecule-specific chimeric antigen receptor-redirected Human T cells exhibit specific and efficient antitumor activity against human ovarian cancer in mice. PLoS One (2016) 11(1):e0146885. doi: 10.1371/journal.pone.0146885
42. Owens GL, Sheard VE, Kalaitsidou M, Blount D, Lad Y, Cheadle EJ, et al. Preclinical Assessment of CAR T-Cell Therapy Targeting the Tumor Antigen 5T4 in Ovarian Cancer. J Immunother (2017) 41(3):130–40. doi: 10.1097/CJI.0000000000000203
43. Murad JP, Kozlowska AK, Lee HJ, Ramamurthy M, Chang WC, Yazaki P, et al. Effective targeting of TAG72+peritoneal ovarian tumors via regional delivery of CAR-engineered T cells. Front Immunol (2018) 9:2268. doi: 10.3389/fimmu.2018.02268
44. Du H, Hirabayashi K, Ahn S, Kren NP, Montgomery SA, Wang X, et al. Antitumor Responses in the Absence of Toxicity in Solid Tumors by Targeting B7-H3 via Chimeric Antigen Receptor T Cells. Cancer Cell (2019) 35:221–37.e8. doi: 10.1016/j.ccell.2019.01.002
45. Rodriguez-Garcia A, Sharma P, Poussin M, Boesteanu AC, Minutolo NG, Gitto SB, et al. CAR T Cells Targeting MISIIR for the Treatment of Ovarian Cancer and Other Gynecologic Malignancies. Mol Ther (2020) 28:548–60. doi: 10.1016/j.ymthe.2019.11.028
46. Hodi FS, Butler M, Oble DA, Seiden MV, Haluska FG, Kruse A, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. PNAS (2008) 105(8):3005–10. doi: 10.1073/pnas.0712237105
47. Zamarin D, Burger RA, Sill MW, Powell DJ Jr, Lankes HA, Feldman MD, et al. Randomized Phase II Trial of Nivolumab Versus Nivolumab and Ipilimumab for Recurrent or Persistent Ovarian Cancer: An NRG Oncology Study. J Clin Oncol (2020) 38(16):1814–23. doi: 10.1200/JCO.19.02059
48. Hamanishi J, Mandai M, Ikeda T, Minami M, Kawaguchi A, Murayama T, et al. Safety and antitumor activity of Anti-PD-1 antibody, nivolumab, in patients with platinum-resistant ovarian cancer. J Clin Oncol (2015) 33:4015–22. doi: 10.1200/JCO.2015.62.3397
49. Inayama Y, Hamanishi J, Matsumura N, Murakami R, Abiko K, Yamaguchi K, et al. Antitumor Effect of Nivolumab on Subsequent Chemotherapy for Platinum-Resistant Ovarian Cancer. Oncol (2018) 23:1382–4. doi: 10.1634/theoncologist.2018-0167
50. Liu JF, Herold C, Gray KP, Penson RT, Horowitz N, Konstantinopoulos PA, et al. Assessment of Combined Nivolumab and Bevacizumab in Relapsed Ovarian Cancer: A Phase 2 Clinical Trial. JAMA Oncol (2019) 5:1731–8. doi: 10.1001/jamaoncol.2019.3343
51. Varga A, Piha-Paul S, Ott PA, Mehnert JM, Berton-Rigaud D, Morosky A, et al. Pembrolizumab in patients with programmed death ligand 1–positive advanced ovarian cancer: Analysis of KEYNOTE-028. Gynecol Oncol (2019) 152:243–50. doi: 10.1016/j.ygyno.2018.11.017
52. Matulonis UA, Shapira-Frommer R, Santin AD, Lisyanskaya AS, Pignata S, Vergote I, et al. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: results from the phase II KEYNOTE-100 study. Ann Oncol (2019) 30:1080–7. doi: 10.1093/annonc/mdz135
53. Gutierrez C, McEvoy C, Munshi L, Stephens RS, Detsky ME, Nates JL, et al. Critical Care Management of Toxicities Associated With Targeted Agents and Immunotherapies for Cancer. Crit Care Med (2019) 48(1):10–21. doi: 10.1097/CCM.0000000000004087
54. Konstantinopoulos PA, Waggoner S, Vidal GA, Mita M, Moroney JW, Holloway R, et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination with Pembrolizumab in Patients with Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol (2019) 5:1141–9. doi: 10.1001/jamaoncol.2019.1048
55. Zimmer AS, Nichols E, Cimino-Mathews A, Peer C, Cao L, Lee MJ, et al. A phase i study of the PD-L1 inhibitor, durvalumab, in combination with a PARP inhibitor, olaparib, and a VEGFR1-3 inhibitor, cediranib, in recurrent women’s cancers with biomarker analyses. J Immunother Cancer (2019) 7:1–8. doi: 10.1186/s40425-019-0680-3
56. Lampert EJ, Zimmer AS, Padget MR, Cimino-Mathews A, Nair JR, Liu Y, et al. Combination of PARP inhibitor olaparib, and PD-L1 inhibitor durvalumab, in recurrent ovarian cancer: a proof-of-concept phase 2 study. Clin Cancer Res (2020) 26:4268–4279. doi: 10.1158/1078-0432.ccr-20-0056. clincanres.0056.2020.
57. Drew Y, de Jonge M, Hong SH, Park YH, Wolfer A, Brown J, et al. An open-label, phase II basket study of olaparib and durvalumab (MEDIOLA): Results in germline BRCA-mutated (gBRCAm) platinum-sensitive relapsed (PSR) ovarian cancer (OC). Gynecol Oncol (2018) 149:246–7. doi: 10.1016/j.ygyno.2018.04.555
58. Disis ML, Taylor MH, Kelly K, Beck JT, Gordon M, Moore KM, et al. Efficacy and Safety of Avelumab for Patients with Recurrent or Refractory Ovarian Cancer: Phase 1b Results from the JAVELIN Solid Tumor Trial. JAMA Oncol (2019) 5:393–401. doi: 10.1001/jamaoncol.2018.6258
59. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. autoimmune diseases. mechanism of self-tolerance causes various alpha-chains (CD25). Breakdown of a single activated T cells expressing IL-2 receptor Immunologic self-tolerance maintained by. J Immunol (1995) 155(3):1151–64. http://www.jimmunol.org/content/155/3/1151.
60. Ou Y, Cannon MJ, Nakagawa M. Regulatory T Cells in Gynecologic Cancer. MOJ Immunology (2018) 6(2):34–42. doi: 10.15406/moji.2018.06.00189
61. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med (2004) 10:942–9. doi: 10.1038/nm1093
62. Knutson KL, Maurer MJ, Preston CC, Moysich KB, Goergen K, Hawthorne KM, et al. Regulatory T cells, inherited variation, and clinical outcome in epithelial ovarian cancer. Cancer Immunol Immunother (2015) 64:1495–504. doi: 10.1007/s00262-015-1753-x
63. Wertel I, Surówka J, Polak G, Barczyński B, Bednarek W, Jakubowicz-Gil J, et al. Macrophage-derived chemokine CCL22 and regulatory T cells in ovarian cancer patients. Tumor Biol (2015) 36:4811–7. doi: 10.1007/s13277-015-3133-8
64. Toker A, Nguyen LT, Stone SC, Cindy Yang SY, Katz SR, Shaw PA, et al. Regulatory T cells in ovarian cancer are characterized by a highly activated phenotype distinct from that in melanoma. Clin Cancer Res (2018) 24:5685–96. doi: 10.1158/1078-0432.CCR-18-0554
65. Lutsiak MEC, Semnani RT, de Pascalis R, Kashmiri SVS, Schlom J, Sabzevari H. Inhibition of CD4+25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood (2005) 105:2862–8. doi: 10.1182/blood-2004-06-2410
66. Vermeij R, Leffers N, Hoogeboom BN, Hamming ILE, Wolf R, Reyners AKL, et al. Potentiation of a p53-SLP vaccine by cyclophosphamide in ovarian cancer: A single-arm phase II study. Int J Cancer (2012) 131:e670–80. doi: 10.1002/ijc.27388
67. Chu CS, Boyer J, Schullery DS, Gimotty PA, Gamerman V, Bender J, et al. Phase I/II randomized trial of dendritic cell vaccination with or without cyclophosphamide for consolidation therapy of advanced ovarian cancer in first or second remission. Cancer Immunol Immunother (2012) 61:629–41. doi: 10.1007/s00262-011-1081-8
68. Milne K, Köbel M, Kalloger SE, Barnes RO, Gao D, Gilks CB, et al. Systematic analysis of immune infiltrates in high-grade serous ovarian cancer reveals CD20, FoxP3 and TIA-1 as positive prognostic factors. PLoS One (2009) 4(7):1–14. doi: 10.1371/journal.pone.0006412
69. Nielsen JS, Sahota RA, Milne K, Kost SE, Nesslinger NJ, Watson PH, et al. CD20+ tumor-infiltrating lymphocytes have an atypical CD27 - memory phenotype and together with CD8+ T cells promote favorable prognosis in ovarian cancer. Clin Cancer Res (2012) 18:3281–92. doi: 10.1158/1078-0432.CCR-12-0234
70. Santoiemma PP, Reyes C, Wang LP, McLane MW, Feldman MD, Tanyi JL, et al. Systematic evaluation of multiple immune markers reveals prognostic factors in ovarian cancer. Gynecol Oncol (2016) 143:120–7. doi: 10.1016/j.ygyno.2016.07.105
71. Kroeger DR, Milne K, Nelson BH. Tumor-infiltrating plasma cells are associated with tertiary lymphoid structures, cytolytic T-cell responses, and superior prognosis in ovarian cancer. Clin Cancer Res (2016) 22:3005–15. doi: 10.1158/1078-0432.CCR-15-2762
72. Schwartz M, Zhang Y, Rosenblatt JD. B cell regulation of the anti-tumor response and role in carcinogenesis. J Immunother Cancer (2016) 4:1–15. doi: 10.1186/s40425-016-0145-x
73. Lundgren S, Berntsson J, Nodin B, Micke P, Jirström K. Prognostic impact of tumour-associated B cells and plasma cells in epithelial ovarian cancer. J Ovarian Res (2016) 9:1–9. doi: 10.1186/s13048-016-0232-0
74. Yang C, Lee H, Jove V, Deng J, Zhang W, Liu X, et al. Prognostic Significance of B-Cells and pSTAT3 in Patients with Ovarian Cancer. PLoS One (2013) 8(1):1–6. doi: 10.1371/journal.pone.0054029
75. Wei X, Jin Y, Tian Y, Zhang H, Wu J, Lu W, et al. Regulatory B cells contribute to the impaired antitumor immunity in ovarian cancer patients. Tumor Biol (2016) 37:6581–8. doi: 10.1007/s13277-015-4538-0
76. Ragonnaud E, Moritoh K, Bodogai M, Gusev F, Garaud S, Chen C, et al. Tumor-Derived Thymic Stromal Lymphopoietin Expands Bone Marrow B-cell Precursors in Circulation to Support Metastasis. Cancer Res (2019) 79:5826–38. doi: 10.1158/0008-5472.CAN-19-1058
77. Demaria O, Cornen S, Daëron M, Morel Y, Medzhitov R, Vivier E. Harnessing innate immunity in cancer therapy. Nature (2019) 574:45–56. doi: 10.1038/s41586-019-1593-5
78. Eisenbarth SC. Dendritic cell subsets in T cell programming: location dictates function. Nat Rev Immunol (2019) 19:89–103. doi: 10.1038/s41577-018-0088-1
79. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol (2020) 20:7–24. doi: 10.1038/s41577-019-0210-z
80. Cubillos-Ruiz JR, Martinez D, Scarlett UK, Rutkowski MR, Nesbeth YC, Camposeco-Jacobs AL, et al. cd277 is a Negative Co-stimulatory Molecule Universally Expressed by Ovarian Cancer Microenvironmental Cells. Oncotarget (2010) 1(5):329–38. doi: 10.18632/oncotarget.165
81. Krempski J, Karyampudi L, Behrens MD, Erskine CL, Hartmann L, Dong H, et al. Tumor-Infiltrating Programmed Death Receptor-1 + Dendritic Cells Mediate Immune Suppression in Ovarian Cancer. J Immunol (2011) 186:6905–13. doi: 10.4049/jimmunol.1100274
82. Labidi-Galy SI, Sisirak V, Meeus P, Gobert M, Treilleux I, Bajard A, et al. Quantitative and functional alterations of plasmacytoid dendritic cells contribute to immune tolerance in ovarian cancer. Cancer Res (2011) 71:5423–34. doi: 10.1158/0008-5472.CAN-11-0367
83. Labidi-Galy SI, Treilleux I, Goddard-Leon S, Combes JD, Blay JY, Ray-Coquard I, et al. Plasmacytoid dendritic cells infiltrating ovarian cancer are associated with poor prognosis. OncoImmunology (2012) 1:380–2. doi: 10.4161/onci.18801
84. Hernando J, Park TW, Kübler K, Offergeld R, Schlebusch H, Bauknecht T. Vaccination with autologous tumour antigen-pulsed dendritic cells in advanced gynaecological malignancies: Clinical and immunological evaluation of a phase I trial. Cancer Immunol Immunother (2002) 51:45–52. doi: 10.1007/s00262-001-0255-1
85. Rahma OE, Ashtar E, Czystowska M, Szajnik ME, Wieckowski E, Bernstein S, et al. A gynecologic oncology group phase II trial of two p53 peptide vaccine approaches: Subcutaneous injection and intravenous pulsed dendritic cells in high recurrence risk ovarian cancer patients. Cancer Immunol Immunother (2012) 61:373–84. doi: 10.1007/s00262-011-1100-9
86. Baek S, Kim YM, Kim SB, Kim CS, Kwon SW, Kim YM, et al. Therapeutic DC vaccination with IL-2 as a consolidation therapy for ovarian cancer patients: A phase I/II trial. Cell Mol Immunol (2015) 12:87–95. doi: 10.1038/cmi.2014.40
87. Zhang W, Lu X, Cui P, Piao C, Xiao M, Liu X, et al. Phase I/II clinical trial of a Wilms’ tumor 1-targeted dendritic cell vaccination-based immunotherapy in patients with advanced cancer. Cancer Immunol Immunother (2019) 68:121–30. doi: 10.1007/s00262-018-2257-2
88. Tanyi JL, Bobisse S, Ophir E, Tuyaerts S, Roberti A, Genolet R, et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci Transl Med (2018) 10(436):Eaao5931. doi: 10.1126/scitranslmed.aao5931
89. Perez CR, de Palma M. Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat Commun (2019) 10:1–10. doi: 10.1038/s41467-019-13368-y
90. Mirandola L, Chiriva-Internati M, Bresalier R, Piccotti L, Grizzi F, Marincola FM. A novel method for efficient generation of antigen-specific effector T-cells using dendritic cells transduced with recombinant adeno-associated virus and p38 kinase blockade. J Trans Med (2019) 17:1–9. doi: 10.1186/s12967-019-02163-4
91. Xie J, Qian J, Yang J, Wang S, Freeman ME, Yi Q. Critical roles of Raf/MEK/ERK and PI3K/AKT signaling and inactivation of p38 MAP kinase in the differentiation and survival of monocyte-derived immature dendritic cells. Exp Hematol (2005) 33:564–72. doi: 10.1016/j.exphem.2005.03.001
92. Jarnicki AG, Conroy H, Brereton C, Donnelly G, Toomey D, Walsh K, et al. Attenuating Regulatory T Cell Induction by TLR Agonists through Inhibition of p38 MAPK Signaling in Dendritic Cells Enhances Their Efficacy as Vaccine Adjuvants and Cancer Immunotherapeutics 1. J Immunol (2008) 180(6):3797–806. doi: 10.4049/jimmunol.180.6.3797
93. Cheng S, Xu C, Jin Y, Li Y, Zhong C, Ma J, et al. Artificial Mini Dendritic Cells Boost T Cell–Based Immunotherapy for Ovarian Cancer. Adv Sci (2020) 7:1–14. doi: 10.1002/advs.201903301
94. Long EO, Rajagopalan S. Stress signals activate natural killer cells. J Exp Med (2002) 196:1399–402. doi: 10.1084/jem.20021747
95. Zamai L, Ahmad M, Bennett IM, Azzoni L, Alnemri ES, Perussia B. Natural Killer (NK) Cell-mediated Cytotoxicity: Differential Use of TRAIL and Fas Ligand by Immature and Mature Primary Human NK Cells. J Exp Med (1998) 188:2375–80. doi: 10.1084/jem.188.12.2375
96. Li K, Mandai M, Hamanishi J, Matsumura N, Suzuki A, Yagi H, et al. Clinical significance of the NKG2D ligands, MICA/B and ULBP2 in ovarian cancer: High expression of ULBP2 is an indicator of poor prognosis. Cancer Immunol Immunother (2009) 58:641–52. doi: 10.1007/s00262-008-0585-3
97. Henriksen JR, Donskov F, Waldstrøm M, Jakobsen A, Hjortkjaer M, Petersen CB, et al. Favorable prognostic impact of Natural Killer cells and T cells in high-grade serous ovarian carcinoma. Acta Oncol (2020) 59:652–9. doi: 10.1080/0284186X.2019.1711173
98. Hoogstad-Van Evert JS, Maas RJ, van der Meer J, Cany J, van der Steen S, Jansen JH, et al. Peritoneal NK cells are responsive to IL-15 and percentages are correlated with outcome in advanced ovarian cancer patients. Oncotarget (2018) 9(78):34810–20. doi: 10.18632/oncotarget.26199
99. Geller MA, Cooley S, Judson PL, Ghebre R, Carson LF, Argenta PA, et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy (2011) 13:98–107. doi: 10.3109/14653249.2010.515582
100. Zuo S, Wen Y, Panha H, Dai G, Wang L, Ren X, et al. Modification of cytokine-induced killer cells with folate receptor alpha (FRα)-specific chimeric antigen receptors enhances their antitumor immunity toward FRα-positive ovarian cancers. Mol Immunol (2017) 85:293–304. doi: 10.1016/j.molimm.2017.03.017
101. Ao X, Yang Y, Li W, Tan Y, Guo W, Ao L, et al. Anti-αFR CAR-engineered NK-92 Cells Display Potent Cytotoxicity Against αFR-positive Ovarian Cancer. J Immunother (2019) 42(8):284–96. doi: 10.1097/CJI.0000000000000286
102. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell (2018) 23:181–92. doi: 10.1016/j.stem.2018.06.002
103. Ueda T, Kumagai A, Iriguchi S, Yasui Y, Miyasaka T, Nakagoshi K, et al. Non–clinical efficacy, safety and stable clinical cell processing of induced pluripotent stem cell-derived anti–glypican-3 chimeric antigen receptor-expressing natural killer/innate lymphoid cells. Cancer Sci (2020) 111:1478–90. doi: 10.1111/cas.14374
104. Klapdor R, Wang S, Morgan M, Dörk T, Hacker U, Hillemanns P, et al. Characterization of a novel third-generation anti-CD24-CAR against ovarian cancer. Int J Mol Sci (2019) 20(3):1–15. doi: 10.3390/ijms20030660
105. Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res (2006) 66:605–12. doi: 10.1158/0008-5472.CAN-05-4005
106. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol (2002) 23(11):549–55. doi: 10.1016/S1471-4906(02)02302-5
107. Zhang Q, Cai DJ, Li B. Ovarian cancer stem-like cells elicit the polarization of M2 macrophages. Mol Med Rep (2015) 11:4685–93. doi: 10.3892/mmr.2015.3323
108. Deng X, Zhang P, Liang T, Deng S, Chen X, Zhu L. Ovarian cancer stem cells induce the M2 polarization of macrophages through the PPARγ and NF-κB pathways. Int J Mol Med (2015) 36(8):449–54. doi: 10.3892/ijmm.2015.2230
109. Zhang M, He Y, Sun X, Li Q, Wang W, Zhao A, et al. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J Ovarian Res (2014) 7:1–16. doi: 10.1186/1757-2215-7-19
110. Yuan X, Zhang J, Li D, Mao Y, Mo F, Du W, et al. Prognostic significance of tumor-associated macrophages in ovarian cancer: A meta-analysis. Gynecol Oncol (2017) 147:181–7. doi: 10.1016/j.ygyno.2017.07.007
111. Lu C, Zhou L, Ouyang J, Yang H, Ding J. Prognostic value of lymphocyte-to-monocyte ratio in ovarian cancer: A meta-analysis. Med (U S) (2019) 98(24):1–8. doi: 10.1097/MD.0000000000015876
112. Gong J, Jiang H, Shu C, Hu MQ, Huang Y, Liu Q, et al. Prognostic value of lymphocyte-to-monocyte ratio in ovarian cancer: A meta-analysis. J Ovarian Res (2019) 12:1–7. doi: 10.1186/s13048-019-0527-z
113. Cao Y, Ni X, Wang Y, Wang L, Yuan K, Gan G, et al. Clinical and prognostic significance of combined plasma fibrinogen concentrations and the monocyte-to-lymphocyte ratio in patients with ovarian cancer. Ann Trans Med (2019) 7:242. doi: 10.21037/atm.2019.04.78
114. Cai L, Song Y, Zhao X. Prognostic significance of lymphocyte monocyte ratio in patients with ovarian cancer. Medicine (2020) 99:e19638. doi: 10.1097/md.0000000000019638
115. Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, et al. Role of Macrophage Targeting in the Antitumor Activity of Trabectedin. Cancer Cell (2013) 23:249–62. doi: 10.1016/j.ccr.2013.01.008
116. Wanderley CW, Colón DF, Luiz JPM, Oliveira FF, Viacava PR, Leite CA, et al. Paclitaxel reduces tumor growth by reprogramming tumor-associated macrophages to an M1 profile in a TLR4-dependent manner. Cancer Res (2018) 78:5891–900. doi: 10.1158/0008-5472.CAN-17-3480
117. Nakashima H, Miyake K, Clark CR, Bekisz J, Finbloom J, Husain SR, et al. Potent antitumor effects of combination therapy with IFNs and monocytes in mouse models of established human ovarian and melanoma tumors. Cancer Immunol Immunother (2012) 61:1081–92. doi: 10.1007/s00262-011-1152-x
118. Johnson CL, Green DS, Zoon KC. Human monocytes in the presence of interferons alpha2a and gamma are potent killers of serous ovarian cancer cell lines in combination with paclitaxel and carboplatin. J Interferon Cytokine Res (2015) 35:55–62. doi: 10.1089/jir.2014.0057
119. Green DS, Nunes AT, David-Ocampo V, Ekwede IB, Houston ND, Highfill SL, et al. A Phase 1 trial of autologous monocytes stimulated ex vivo with Sylatron ® (Peginterferon alfa-2b) and Actimmune ® (Interferon gamma-1b) for intra-peritoneal administration in recurrent ovarian cancer. J Trans Med (2018) 16:1–9. doi: 10.1186/s12967-018-1569-5
120. Cole CB, Annunziata CM. First-in-human phase I study of intraperitoneally administered interferon-activated autologous monocytes in platinum-resistant or refractory ovarian cancer. J Clin Oncol (2020) 38:1. doi: 10.1200/JCO.2020.38.5_suppl.1
121. Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer (2013) 13:739–52. doi: 10.1038/nrc3581
122. Bennett JA, Rao VS, Mitchell MS. Systemic bacillus Calmette-Guerin (BCG) activates natural suppressor cells (in vitro immunization/cell-mediated immunity/suppression of immunization by non-T cells). PNAS (1978) 75(10):5142–4. doi: 10.1073/pnas.75.10.5142
123. Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The terminology issue for myeloid-derived suppressor cells [1]. Cancer Res (2007) 67:425. doi: 10.1158/0008-5472.CAN-06-3037
124. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age review-article. Nat Immunol (2018) 19:108–19. doi: 10.1038/s41590-017-0022-x
125. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun (2016) 7:1–10. doi: 10.1038/ncomms12150
126. Duwe~ AK, Singhal SK. The lmmunoregulatory Role of Bone Marrow I. Suppression of the Induction of Antibody Responses to T-Dependent and T-Independent Antigens by Cells in the Bone Marrow’. Cell Immunol (1979) 43:362–71. doi: 10.1016/0008-8749(79)90180-1
127. Young M, Young M, Wright M. Stimulation of immune-suppressive bone-marrow cells by colony-stimulating factors. Exp Hematol (1990) 18:806–11.
128. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol (2016) 37:208–20. doi: 10.1016/j.it.2016.01.004
129. Cui TX, Kryczek I, Zhao L, Zhao E, Kuick R, Roh MH, et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CTBP2. Immunity (2013) 39:611–21. doi: 10.1016/j.immuni.2013.08.025
130. Okła K, Czerwonka A, Wawruszak A, Bobiński M, Bilska M, Tarkowski R, et al. Clinical relevance and immunosuppressive pattern of circulating and infiltrating subsets of myeloid-derived suppressor cells (MDSCs) in epithelial ovarian cancer. Front Immunol (2019) 10:691. doi: 10.3389/fimmu.2019.00691
131. Wu L, Deng Z, Peng Y, Han L, Liu J, Wang L, et al. Ascites-derived IL-6 and IL-10 synergistically expand CD14 + HLA-DR-/low myeloid-derived suppressor cells in ovarian cancer patients. Oncotarget (2017) 8(44):76483–856. doi: 10.18632/oncotarget.20164
132. Santegoets SJAM, de Groot AF, Dijkgraaf EM, Simões AMC, van der Noord VE, van Ham JJ, et al. The blood mMDSC to DC ratio is a sensitive and easy to assess independent predictive factor for epithelial ovarian cancer survival. OncoImmunology (2018) 7:E1465166. doi: 10.1080/2162402X.2018.1465166
133. Baert T, Vankerckhoven A, Riva M, van Hoylandt A, Thirion G, Holger G, et al. Myeloid derived suppressor cells: Key drivers of immunosuppression in ovarian cancer. Front Immunol (2019) 10:1273. doi: 10.3389/fimmu.2019.01273
134. Horikawa N, Abiko K, Matsumura N, Hamanishi J, Baba T, Yamaguchi K, et al. Expression of vascular endothelial growth factor in ovarian cancer inhibits tumor immunity through the accumulation of myeloid-derived suppressor cells. Clin Cancer Res (2017) 23:587–99. doi: 10.1158/1078-0432.CCR-16-0387
135. Rattan R, Dar S, Rasool N, Ali-Fehmi R, Giri S, Munkarah AR. Depletion of immunosuppressive myeloid-derived suppressor cells impedes ovarian cancer growth. Gynecol Oncol (2017) 145:213–4. doi: 10.1016/j.ygyno.2017.03.491
136. Horikawa N, Abiko K, Matsumura N, Baba T, Hamanishi J, Yamaguchi K, et al. Anti-VEGF therapy resistance in ovarian cancer is caused by GM-CSF-induced myeloid-derived suppressor cell recruitment. Br J Cancer (2020) 122:778–88. doi: 10.1038/s41416-019-0725-x
137. ENCORE 603. A Phase II Randomized Study of Avelumab Plus Entinostat vs Avelumab Plus Placebo in Patients With Advanced and Recurrent Epithelial Ovarian Cancer. JCO (2019) 376(15 Supplemental):5511–5511. doi: 10.1200/JCO.2019.37.15_suppl.5511
138. Mikyšková R, Indrová M, Vlková V, Bieblová J, Šímová J, Paračková Z, et al. DNA demethylating agent 5-azacytidine inhibits myeloid-derived suppressor cells induced by tumor growth and cyclophosphamide treatment. J Leukocyte Biol (2014) 95:743–53. doi: 10.1189/jlb.0813435
139. Li Y, Hu W, Shen DY, Kavanagh JJ, Fu S. Azacitidine enhances sensitivity of platinum-resistant ovarian cancer cells to carboplatin through induction of apoptosis. Am J Obstet Gynecol (2009) 200:177.e1–9. doi: 10.1016/j.ajog.2008.08.030
140. Fang F, Balch C, Schilder J, Zhang S, Shen C, Li L, et al. Abstract #3593: Clinical and pharmacodynamic activity of decitabine in vivo for patients with recurrent, platinum-resistant, epithelial ovarian cancer. Cancer Res (2009) 69:3593.
141. Bustinza-Linares E, Falchook GS, Fu S, Naing A, Hong DS, Hu W, et al. Phase I trial of sequential azacitidine and valproic acid plus carboplatin in the treatment of patients with advanced malignancies. J Clin Oncol (2010) 28:2595. doi: 10.1200/jco.2010.28.15_suppl.2595
142. Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med (2007) 13:828–35. doi: 10.1038/nm1609
143. Zeng X, Yi S. Cyclooxygenase Inhibitors in Epithelial Ovarian Cancer Treatment. Int J Gynecol Cancer (2018) 28:1085–9. doi: 10.1097/IGC.0000000000001269
Keywords: ovarian cancer, tumor microenvironment, innate immunity, adaptive immunity, cancer therapeutics
Citation: Ning F, Cole CB and Annunziata CM (2021) Driving Immune Responses in the Ovarian Tumor Microenvironment. Front. Oncol. 10:604084. doi: 10.3389/fonc.2020.604084
Received: 08 September 2020; Accepted: 30 November 2020;
Published: 15 January 2021.
Edited by:
Marco Rossi, University of Catanzaro, ItalyReviewed by:
Victor C. Kok, Asia University, TaiwanCopyright © 2021 Ning, Cole and Annunziata. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christina M. Annunziata, YW5udW56aWNAbWFpbC5uaWguZ292
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.