 Alana Serrano Campelo de Souza1,2
Alana Serrano Campelo de Souza1,2 Letícia Boslooper Gonçalves1,2
Letícia Boslooper Gonçalves1,2 Ana Paula Lepique3
Ana Paula Lepique3 Patrícia Savio de Araujo-Souza1*
Patrícia Savio de Araujo-Souza1*- 1Laboratório de Imunogenética e Histocompatibilidade (LIGH), Departamento de Genética, Setor de Ciências Biológicas, Universidade Federal do Paraná (UFPR), Curitiba, Brazil
- 2Programa de Pós-graduação em Genética, Departamento de Genética, Universidade Federal do Paraná (UFPR), Curitiba, Brazil
- 3Laboratório de Imunomodulação, Departamento de Imunologia, Instituto de Ciências Biomédicas, Universidade de São Paulo, São Paulo, Brazil
The tumor microenvironment (TME) is complex, and its composition and dynamics determine tumor fate. From tumor cells themselves, with their capacity for unlimited replication, migration, and invasion, to fibroblasts, endothelial cells, and immune cells, which can have pro and/or anti-tumor potential, interaction among these elements determines tumor progression. The understanding of molecular pathways involved in immune escape has permitted the development of cancer immunotherapies. Targeting molecules or biological processes that inhibit antitumor immune responses has allowed a significant improvement in cancer patient’s prognosis. Autophagy is a cellular process required to eliminate dysfunctional proteins and organelles, maintaining cellular homeostasis. Usually a process associated with protection against cancer, autophagy associated to cancer cells has been reported in response to hypoxia, nutrient deficiency, and oxidative stress, conditions frequently observed in the TME. Recent studies have shown a paradoxical association between autophagy and tumor immune responses. Tumor cell autophagy increases the expression of inhibitory molecules, such as PD-1 and CTLA-4, which block antitumor cytotoxic responses. Moreover, it can also directly affect antitumor immune responses by, for example, degrading NK cell-derived granzyme B and protecting tumor cells. Interestingly, the activation of autophagy on dendritic cells has the opposite effects, enhancing antigen presentation, triggering CD8+ T cells cytotoxic activity, and reducing tumor growth. Therefore, this review will focus on the most recent aspects of autophagy and tumor immune environment. We describe the dual role of autophagy in modulating tumor immune responses and discuss some aspects that must be considered to improve cancer treatment.
Introduction
According to the Cancer Immune Edition hypothesis, tumor and immune cell interactions go through three phases: elimination, equilibrium, and evasion. During cancer development, the immune system recognizes molecular changes in transformed cells and eliminates most or all of them, avoiding tumor progression. Genetic alterations that cause cell transformation generate neoantigens for immune recognition, leading to T lymphocyte activation, which can prevent tumor outgrowth, through cytotoxic activity and interferon-gamma (IFN-γ) signaling (1, 2). At the same time, less immunogenic mutations or mutations that lead to loss of the antigen recognized by the immune system allow tumor cells to escape from elimination mechanisms. As genetic alterations accumulate, generating oncogenes and preventing the expression of tumor-suppressor genes, transformed cells gain proliferative advantages, and again escape immunosurveillance, leading to tumor progression (3, 4).
The interplay between tumor and other cells composing the tumor microenvironment (TME) is determinant for tumor growth, maintenance, metastasis, and response to therapy. TME is composed of stromal cells (fibroblasts, pericytes, mesenchymal and endothelial cells), extracellular matrix (ECM), and immune cells, such as natural killer (NK) cells, tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and T and B lymphocytes. During cancer progression, tumor cells display genetic and phenotypic diversity, changing cellular metabolism and, consequently, the TME (5, 6).
Generally, TME displays low levels of oxygen and nutrients, and high production of reactive oxygen species (ROS), crucial factors for autophagy activation. Autophagy is a natural cellular survival process, usually activated to maintain cellular homeostasis (7, 8). Despite that, recent studies have suggested that autophagy is also important for cancer development and progression, neurodegenerative and infectious diseases, once it can affect immune cells and modulate immune responses (9–11).
In this review, we will present the major mechanisms by which the immune system interferes in the TME, and how autophagy can influence it. Then we will focus on the modifications of cancer immune responses in TME influenced by autophagy and how it can affect cancer therapy.
Cancer Immune Response and Tumor Microenvironment
Besides the TME elements already mentioned, soluble molecules as cytokines, metabolites, and inflammation mediators also contribute to the interaction among the cellular elements. These biochemical signals orchestrate cell death, proliferation, survival, and other cells recruitment. Leukocyte activity is essential in cancer progression (reviewed in 12). Since Rudolph Virchow has described the presence of lymphoreticular infiltrate in human tumors, it was discovered that leukocytes play an essential role in tumor progression, either by eliminating tumor cells or by facilitating progression and growth (13).
Immune responses can inhibit tumor growth and even eliminate tumor cells completely. However, chronic inflammation is considered a risk factor for many types of cancers (reviewed in 12). An interesting example of this dual role is HIV infection, AIDS (Acquired Immune Deficiency Syndrome), and cancer risk. In the early ‘90s, patients with AIDS were at high risk of Kaposi sarcoma and non-Hodgkin lymphoma development, in part due to cellular and molecular mechanisms, but in large part due to immunodeficiency. Highly active antiretroviral therapy (HAART) decreased AIDS-related cancer (14) by increasing T lymphocyte levels and consequently immune responses. Still, HAART treated HIV infected patients display chronic inflammation and early aging, with increased plasma levels of interleukin-(IL)6 and C reactive protein, which have a role in carcinogenesis. Indeed, HAART treated patients display increased risk to develop AIDS-unrelated cancer, such as cervical, lung, anal cancer, and Hodgkin lymphoma (14, 15).
The immune responses mediated by NK cells, CD8+ T lymphocytes, and CD4+ T helper (Th) 1 and Th17 lymphocytes are considered cytotoxic responses. These cells can control tumor growth through either directly killing tumor cells, as NK and CD8+ T cells, or indirectly, in the case of CD4+ T cells, which secrete cytokines capable of activating other effector leukocytes (16–18).
NK cells and CD8+ T lymphocytes are bona fide cytotoxic cells. NK cells are innate lymphoid cells that recognize target cells through activating and inhibitory receptors. The signaling triggered by these sets of receptors determines the cytotoxic activity. Among the inhibitory receptors, there are the killer immunoglobulin-like inhibitory receptors (KIRs), which recognize human leukocyte antigen (HLA) class I molecules and CD94/NKG2A, which specifically binds to the non-classical HLA-E molecule. The last one causes NK inhibition to ensure that normal cells cannot be lysed. However, transformed cells that downregulate the HLA-I surface molecules are not able to inhibit NK cells. The stimulatory receptors bind to stress-inducible molecules in the target cell surface, as sialic acid, Fcγ, adhesion molecules, and others, to trigger cytotoxic activity. Displaying a different strategy for target recognition, CD8+ T lymphocytes activation depends on TCR (T cell receptor) binding to specific antigens presented by classical HLA-I molecules in target cells (19). In spite of the different development and recognition receptors, both NK and CD8+ T cells display similar cytotoxic mechanisms, leading to the activation of cell death pathways in cancer cells (20).
Th1 and Th17 cells can either assist in CD8+ T lymphocytes and dendritic cells (DCs) activation, through CD40L signaling, and cytokine secretion as IL-2, or activate other effector cells, such as macrophages, neutrophils, NK cells through IFN-γ and tumor necrosis factor-alpha (TNF-α). Moreover, TNF-α, through its receptor, can trigger cell death, and IFN-γ and cytokines secreted by Th17 cells, through activation of stromal cells, can stimulate ROS production and neutrophils, enhancing the cytotoxic effects on cancer cells (21).
These antitumor responses are counteracted by tolerogenic responses, enabling tumor growth. There are several known immune escape mechanisms. Chemokines secreted by cells in the TME favors the recruitment of MDSCs and regulatory T cells (Treg), well-characterized suppressors of effector T lymphocytes function. Moreover, it is well known that cancer cells display reduction in antigen presentation potential, decreasing tumor cell recognition by CD8 T lymphocytes. One classic example, from a virus associated cancer is the HPV E7 oncoprotein, which binds to interferon regulatory factor 1 (IRF1) in the IFN type I (IFN-I) signaling pathway, and recruits histone deacetylase (HDAC) to the promoter sequences responsive to IRF1, repressing genes that otherwise would be transcribed in response to the virus (22). IFN-I are important activators of innate responses, as well as antigen-presenting activity, therefore playing a role in T lymphocyte activation and phenotype (23). More recently, it has become clear that human oncogenes also play a role in immune escape mechanisms (24). Stabilization of β-catenin, in the Wnt pathway, for example, reduces the expression of CCL4, a chemokine that attracts DCs, impairing tumor antigen presentation (25).
Oncogenes also drive the reprogramming of tumor cell metabolism, the so-called Warburg effect. Tumor cells display different metabolic strategies to maintain energy production and catabolism at a rate to allow continuous cell proliferation. Some cells use glycolysis almost exclusively, while others also required amino acids and fatty acids as well, and keep the Krebs cycle and oxidative phosphorylation active. In either case, tumor cells usually increase the glucose uptake and secrete lactate in higher concentrations than other cells in the body (26). Both the decrease in glucose and the increase in lactate concentration have consequences for immune responses. Activated T lymphocytes and M1 macrophages display a metabolic profile similar to tumor cells, therefore, dependent on glucose. Low glucose concentration inhibits T lymphocyte proliferation and macrophage function. Additionally, lactate is a regulatory molecule, modulating the phenotype of DCs, inducing suppressor phenotype on macrophages, and inhibiting T lymphocytes (27).
Besides tumor cell-intrinsic metabolism, other cells in the TME also display metabolic pathways that lead to tolerance. DCs, the essential population for naive T lymphocyte activation, can acquire tolerogenic phenotype due to signals from tumor cells, but also from binding, via CD80 or CD86, to cytotoxic T lymphocyte-associated protein 4 (CTLA-4) expressed by Treg. The signal triggered by this interaction promotes indoleamine-2,3-deoxygenase (IDO) expression in DCs. This enzyme, which physiological function is the protection of immune-privileged tissues, catabolizes the reaction that converts the essential amino acid tryptophan in kynurenine, which through the aryl hydrocarbon receptor, promotes regulatory phenotype in T lymphocytes (28). Therefore, this mechanism works as an amplification of the regulatory cycle in the TME. Furthermore, tumor cells can also overexpress IDO, as observed in oral squamous cell carcinoma from smoker patients (29). In general, IDO expression depends on IFN-γ stimulation, which in cancer, characterizes it as a negative feedback mechanism for effector immune responses.
Programmed cell death-ligand 1 (PD-L1) is also an inhibitory molecule expressed upon IFN-γ stimulation, both in antigen-presenting cells (APCs) and cancer cells (30). The receptor for PD-L1, programmed cell death-1 (PD-1), is upregulated upon T lymphocytes activation. Several transcription factors, such as NFAT, AP-1, FoxO1, and NFκB, mediate the PDCD1 expression. Moreover, chromatin changes are also important to control PD-1 expression and are observed in exhausted CD8+ T cells (31). PD-1 contains immunoreceptor tyrosine-based inhibitory (ITIM) domains and can inhibit TCR signaling, rendering T cells inactive. PD-1 expressing stem-like CD8+ memory T cells can be found in lymphoid follicles in the tumor (32). These cells, when activated, differentiate in exhausted cells. There are two PD-1 ligands: CD274 (PD-L1), which has a basal expression in several cell types, and programmed cell death ligand-2 (PD-L2), which expression is usually limited to DCs and macrophages. A variety of cancers display constitutive PD-L1 expression, which can be triggered by genetic and epigenetic alterations in its promoter region, cytokine stimulation, such as IFN-γ and IL-6, growth factors, hypoxia, among others (28). The PD-1/PD-L1 signaling, which induces T cell exhaustion, is an important effect resulting from the chronicity of antigen presentation in cancer. Whenever antigens are chronically presented, negative feedback mechanisms are activated to protect the organism. This protective response is usurped by cancer to create an immune-privileged situation, and immune evasion (33).
As mentioned before, CTLA-4 binds to the co-stimulatory molecules CD80 and CD86. It competes with the activating receptor CD28, which also binds to these molecules, but with lower affinity. CD28 signaling is essential for T lymphocyte activation since it triggers the PI3K/Akt pathway and causes stabilization of the antigen activation signal. Not only CTLA-4 competes for biding to co-stimulatory molecules, but also, through a process called trans-endocytosis, this biding removes CD80 and CD86 from the APC surface, eliminating the possibility of CD28 activation, and consequently preventing T cell activity (34).
Many of these mechanisms can happen simultaneously in the TME, in a dynamic process that varies through time. To add to this complex situation, other factors should also be considered. Some tumors are very immunogenic, and tumor antigen-specific T lymphocytes can be found in the TME, where it can also be observed evasion mechanisms and chronic antigen signaling that can, eventually, inhibit anti-tumor responses. When immune responses persist, it results in chronic inflammation leading to cancer progression. Other tumors are less immunogenic and recruit mainly myeloid cells, which display a tolerogenic phenotype, helping cancer cells meet their metabolic demands, and promoting angiogenesis. M2 macrophages, for example, display arginase activity, causing conversion of arginine to ornithine, which is a substrate to the synthesis of polyamines, necessary for catabolism and cell proliferation (35).
Autophagy Modulates Tumor Immune Environment
Autophagy
Autophagy is a survival cellular process in which organelles and other cytoplasmic components are directed to the lysosomes for degradation (7, 36). This mechanism is highly conserved in eukaryotic cells and its activation occurs in face of starvation, hypoxia, and/or oxidative stress conditions (8). Up to now, three classes of autophagy are known: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). In macroautophagy, an isolation membrane enclosures a portion of the cytoplasm, molecules, and organelles, forming a double-membrane vesicle associated with light chain protein-3 (LC3), called an autophagosome. LC3 is processed and cleaved, generating LC3-I, which receives carboxyl glycine radical and turns into LC3-II. LC3-II acts as a receptor in autophagosome membrane binding to p62, through the LC3-interacting region. P62 is a multidomain protein, involved in the cell death and survival process, which delivers ubiquitin radicals to LC3-II. This induces autophagosome-lysosome fusion to form an autolysosome, autophagy is then activated and p62 is degraded (37, 38). In microautophagy, an invagination of the lysosome membrane engulfs cytoplasmic compounds, in a similar process to endosome formation, producing a multivesicular body (39). Conversely, CMA is a type of autophagy used to degrade specific soluble proteins. A cytosolic substrate is recognized by the chaperone protein heat shock cognate 70 (Hsc 70), which binds to lysosomal-associated membrane protein-(LAMP) 2A in the lysosome membrane to transport this substrate into the lysosome lumen (40).
Macroautophagy is the main type of autophagy, therefore it will be referred to just as autophagy. In response to hypoxia and oxidative stress, hypoxia-inducible factors (HIF) 1 and 2 bind to hypoxia response elements (HREs), leading to the transcription of several genes that are involved in angiogenesis, metastasis, cell survival, immune escape, and autophagy pathways. Activation of HIF-1 subunit-1 leads to an increase in BCL2 interacting protein 3 (BNIP3) and BCL2 interacting protein 3 like (BNIP3L) expression levels. These factors are responsible for breaking the connection between Beclin1 (BECN1) and B-cell lymphoma 2 (Bcl-2), an inhibitory complex that prevents autophagy (41). Another way to induce autophagy in a hostile cellular environment is through the activation of autophagy-related genes (ATG) (42) and 5’-adenosine monophosphate-activated protein kinase (AMPK) (43). AMPK is a nutrient availability sensor and can regulate oxidative and glycolytic metabolism. It can also activate the autophagic recycling of cellular components to balance cellular energy supply. In autophagy activation, ATG and AMPK, independently of BNIP3 and BNIP3L, downregulate the mammalian target of rapamycin (mTOR), which drives autophagosome formation (43).
Despite being a natural process to maintain cellular homeostasis, autophagy activity has been described to contribute to the progression of many human diseases, such as some neurodegenerative disorders, infectious diseases, and cancer. In 1999, it was described that mono-allelic deletions and decrease in expression of Beclin1, on MCF7 human breast carcinoma cells, contributed to tumorigenesis in nude mice, indicating that autophagy could inhibit tumor growth (9). It is reasonable to assume that autophagy could stop the transformation process by eliminating oncogenic, aggregated, or erroneously folded proteins (44). Nevertheless, tumor cells and components of TME can induce autophagy to survive hostile conditions and suppress immune responses, helping tumor growth and proliferation (reviewed in 45). Thus, its role in cancer development remains unclear, and the aspects of how autophagy can modulate immune components of TME will be reviewed in the next topics.
Autophagy and Antitumor Immune Response
The Dual Role of Autophagy in Antigen Presentation
Studies have shown the influence of autophagy in antigen presentation (46–49), as well as in the anti-tumor adaptive immune response activation (50). As shown in Figure 1, adaptive anti-cancer immune responses are triggered by endogenous tumor-associated antigens (TAA) presented to T lymphocytes via the major histocompatibility complex (MHC) context by DCs (reviewed in (51, 53). Normally, MHC-I presents intracellular antigens, such as the ones derived from self-proteins and viral proteins, to CD8+ T lymphocytes while extracellular antigens are generally presented to CD4+ T lymphocytes by MHC-II (reviewed in 54). Effective tumor antigen presentation and the consequent effector T lymphocyte responses and access to the TME are essential for clinical responses to immunotherapy (55) and are associated with positive clinical outcomes (56). Reduction in MHC expression and expression of non-classical molecules is frequently observed in different types of cancers, leading to compromised antigen presentation and/or immune evasion (reviewed in 52), which can influence tumor progression and resistance to immunotherapy (57).
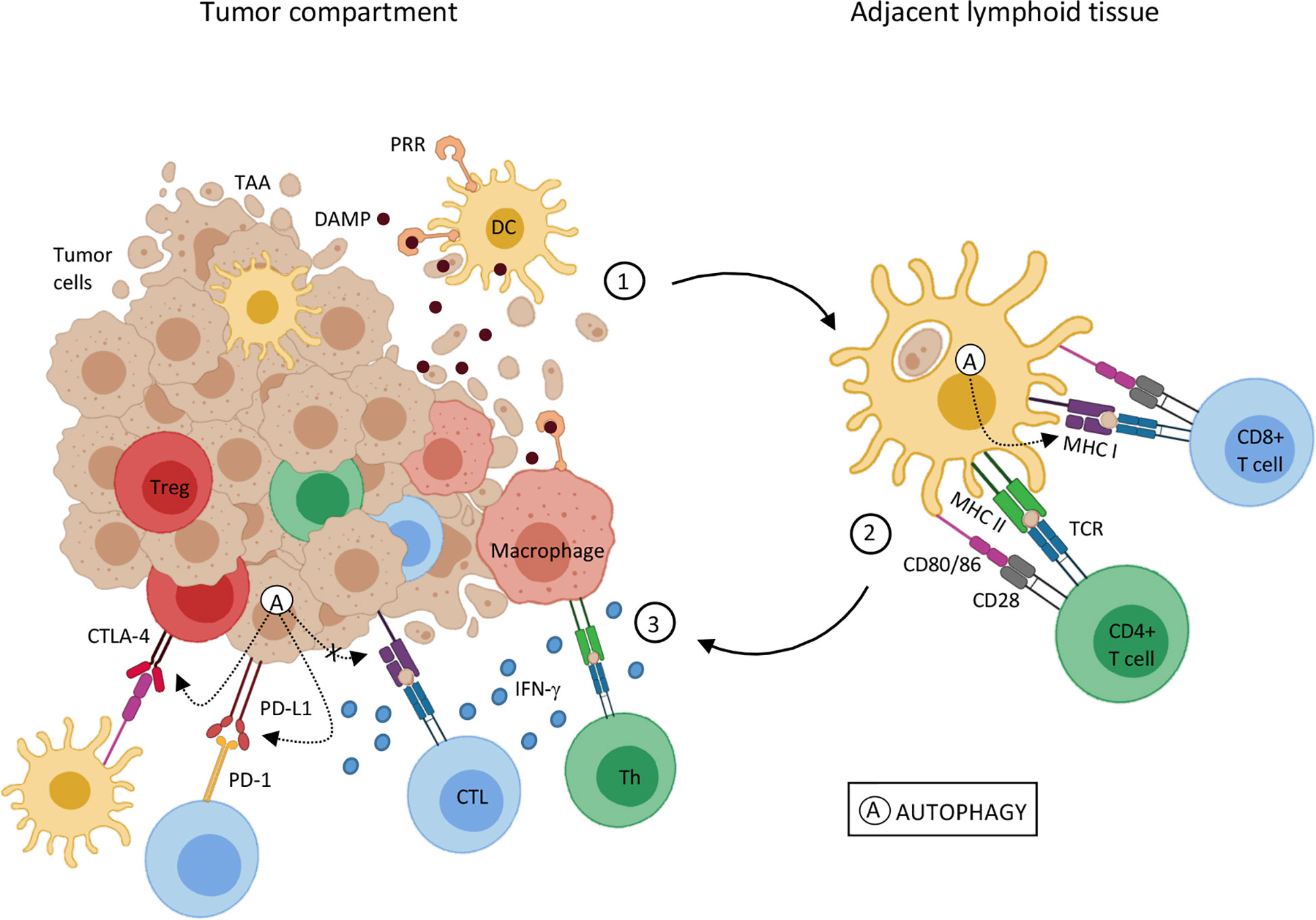
Figure 1 Autophagy influence on tumor-associated antigen presentation 1. Genetic alterations that cause cell transformation can also generate tumor-associated antigens (TAA) for immune recognition. Tumor cell death is an antigen source for antigen-presenting cells, such as dendritic cells (DC) and macrophages. DC activation by DAMPs and antigen processing leads to an upregulation of costimulatory molecules and MHC on DC surface, the cardinal signals for T lymphocyte activation, and migration to adjacent lymphoid tissue. 2. Mature DCs present TAA through MHC-I and MHC-II to naïve CD8+ T cells and CD4+ T cells, respectively. Antigen recognition results in T lymphocyte differentiation in effector cells (CTL e Th) and migration to the tumor site. 3. In the tumor microenvironment, upon TAA recognition through TCR interaction with MHC-I and MHC II, CTL and Th lymphocytes, respectively, trigger cytotoxic mechanisms, as interferon-gamma (IFN-γ) mediated ones. Despite that, inhibitory molecules, such as PD-1 and CTLA-4 in T cells, and PD-L1 in TME can interfere in T cell activation and function. IFN-γ stimulation can result in PD-L1 expression in both APCs and tumor cells, inhibiting T cell function. CTLA-4 expressed by regulatory T lymphocytes (Treg), through binding to co-stimulatory molecules, CD80/86, induces tolerogenic phenotype on DCs, amplifying the regulatory mechanisms in the TME. Autophagy (A) can either help or disturb the antigen presentation and T cell activation pathway. Autophagic activity on DCs seems to increase MHC-I expression, thus enhancing antigen presentation. On the other hand, autophagy activation on tumor cells may promote a reduction in MHC-I and an increase in PD-1 and CTLA-4 expression, leading to tumor progression. Sources (11, 28, 34, 47, 49, 51–56).
Effective Antitumor Immune Responses
Effective antitumor responses are dependent on potent antigen presentation and leukocyte infiltrates enriched with effector CD8+ T cells (49). Autophagy activation in DCs may improve antigen presentation and stimulate cytotoxic responses mediated by CD8+ T lymphocytes (47, 49). For example, nano-activators conjugated to antigens were used to stimulate DCs, triggering anti-tumor T cell responses in mice. It has been shown that nano-activators treated DCs, through autophagy-dependent mechanisms, could increase antigen presentation and cross-presentation to T lymphocytes, increasing effector CD8+ T tumor infiltrating lymphocytes (TILs) (49). Additionally, experimental data has shown that semi-synthetic vitamin E derivative alpha-tocopheryloxyacetic acid (α-TEA) could modulate autophagy in tumor cells from both Lewis lung carcinoma (LLC) and murine mammary tumor, improving antigen cross-presentation by DCs and triggering tumor antigen-specific CD8+ T lymphocytes responses. Treatment with α-TEA resulted in LC3-II increase, both in vitro and in vivo, indicating autophagic activity. These authors also found that α-TEA-generated autophagosome-enriched fraction (α-TAGS) was a competent tumor antigen carrier, which stimulated antigen cross-presentation mediated by DCs to CD8+ T cells and stimulated CD8+ T cell proliferation in an autophagy-dependent fashion. Overall, these findings demonstrated a new mechanism of immune activation by α-TEA, which stimulated tumor cell autophagy and antigen cross-presentation to CD8+ T cells (47).
Autophagy can also create new epitopes arising from stress-induced post-translational modifications, which could increase immune recognition (46). It has been shown that citrullination – the conversion of arginine residues to citrulline - can take place in cells during autophagy induced by stress and, in inflammatory conditions, it can result in MHC-II presentation of citrullinated epitopes to CD4+ T cells (46). Although autophagy modulation has not been directly investigated, the combination of citrullinated peptide based vaccine with TLR ligand adjuvant promoted a Th1 anti-tumor response in melanoma and ovarian cancer mouse models. CD4+ T TILs were associated with tumor regression. Interestingly, they also observed a Th1 response to the citrullinated peptides in ovarian cancer patients (58). Collectively, these findings indicated that autophagy is associated with efficient antigen presentation in different types of cancer. As it increases antigen availability and enhances T cell activation, it favors cytotoxic responses and clearly can act to inhibit cancer progression.
Immunosuppression: Autophagy Disrupts Antigen Presentation
Autophagy may also play a negative role in antigen presentation, facilitating tumor evasion from CD8+ T cells in both pancreatic ductal adenocarcinoma (PDAC) and melanoma (11, 59). PDAC displays low levels of MHC-I surface molecules. In these tumor cells, knockdown of the autophagy cargo receptor gene, NBR1, increased MHC-I surface expression, confirming the implication of NBR1-mediated autophagy-lysosomal pathway in the process. The authors showed that NBR1 targeted MHC-I for lysosomal degradation. Mouse PDAC cells expressing an autophagy inhibitor, restored MHC-I membrane expression, improved antigen presentation, and CD8+ TILs, leading to a reduction in tumor growth. These findings indicated that high levels of MHC-I at PDAC cell surface after autophagy inhibition were required to increase CD8+ T cell infiltration and to kill the tumor cells (11).
The activation of autophagy pathways has also been described in macrophages and DCs infiltrating B16F10 mouse melanoma. These tumors normally express T cell immunoglobulin and mucin domain protein-4 (TIM-4). Autophagy initiates when TIM-4 binds to AMPK-α1. This activation promotes the degradation of TAA through the lysosomal pathway, which led to a decrease in antigen presentation and, consequently, in specific anti-tumor CD8+ T cells. TIM-4 blockade with a monoclonal specific antibody resulted in autophagy inhibition and improvement in antigen cross-presentation and IFN-γ production (59). Moreover, chloroquine (CQ), a known autophagy inhibitor, combined with low concentrations of 5-fluorouracil (5-FU), increased DCs maturation and activation in HCT-116 colorectal cancer cells, enhancing CD8+ T lymphocyte stimulation (60). Together, these results suggested that autophagy can impair antigen presentation by interfering with different key steps in this process.
TME Induces Autophagy
Not only autophagy modulates TME components, but the opposite is also true. As shown in Figure 2, cytokines and metabolic conditions may also promote or inhibit autophagy and influence the tumor immune response. IFN-γ can induce autophagy in gastric cells, thus inhibiting carcinogenesis (61). Gastric cancer is usually associated with chronic inflammation. Transgenic mice that overexpress IFN-γ in the gastric mucosa (H+/K+-ATPase-IFN-γ and H+/K+-ATPase-IL-1β; IFN-γ) displayed protection against gastric dysplasia in comparison to controls. The authors observed that gastric cells displayed lower proliferation rates and T cell apoptosis dependent on IFN-γ expression. Furthermore, transgenes resulted in increased levels of LC3-II and Beclin-1 mRNA and protein, in the stomach, indicating autophagy activation. Additionally, transgenic animals showed higher apoptotic T cells, concurrently with inhibition of IL-6, IL-1β; and TNF-α production, and presented less chronic inflammation (61).
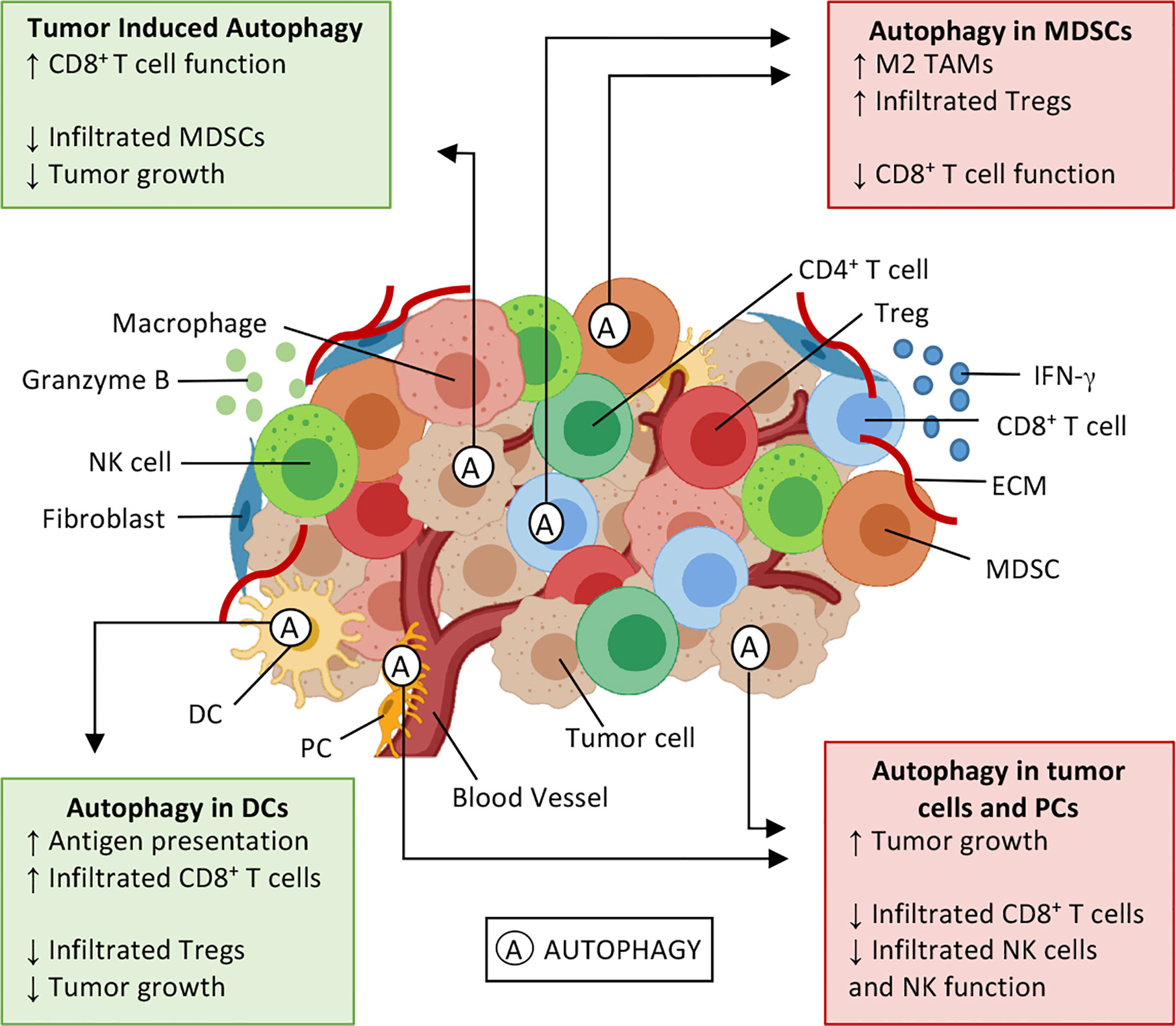
Figure 2 Effects of autophagy on the tumor microenvironment. Tumor microenvironment (TME) is composed by cytokines (e.g. IFN-γ), extracellular matrix (ECM), and several cell types: tumor cells, fibroblasts, and immune cells, such as natural killer (NK) cells, CD8+ T and T helper (Th) lymphocytes, T regulatory (Treg) cells, myeloid-derived suppressor cells (MDSC), dendritic cells (DCs), macrophages, and pericytes (PC). Autophagy (A) is a cellular survival mechanism, which is activated in stressful conditions, can be activated in TME. Autophagy can either enhance antitumor immune responses (green boxes) or induce an immunosuppressive environment (red boxes), thus playing a dual role in cancer development and progression. Autophagy activation in DCs enhances antigen presentation and results in an enrichment of CD8+ T tumor-infiltrating lymphocytes, also decreasing infiltrated Treg and tumor cell proliferation. Similar outcomes can be seen when IFN-γ induces autophagy on tumor cells, and elevated extracellular potassium induces autophagy on CD8+ T cells. In these situations, it is possible to observe a decrease in infiltrated MDSCs and T lymphocytes expressing PD-1, thus inhibiting tumor growth. Contrarily, the autophagy activation in myeloid cells and tumor cells has the opposite effect, favoring an immunosuppressive profile of TME, leading to tumor cell proliferation, through M2 macrophage polarization, enhancing Treg infiltration and inhibitory molecules (PD-1 and CTLA-4) expression. Sources (5, 6, 11, 47, 49, 61–67).
Besides the cellular components and soluble mediators, TME also display metabolic disorders such as aerobic glycolysis (68), oxygen deprivation, and higher levels of extracellular potassium (62), which are stressful conditions that can also influence cellular autophagy. Activation of autophagy in T lymphocytes exposed to elevated potassium concentrations reduced acetylation of the promoter and enhancer regions of T cell effector- and exhaustion-markers Ifng and Pdcd1. Potassium exposed T lymphocytes adoptively transferred to B16 melanoma bearing mice resulted in T cells persistence in the TME, driving tumor regression, and improving animal survival (62). Furthermore, metabolic conditions of TME, such as elevated extracellular potassium levels, were responsible for autophagy activation on T-cells and boosted antitumor responses (62).
Despite that, tumor metabolism can play tricks on antitumor immune responses, such as the ones with elevated glycolytic metabolism, that seems to inhibit autophagy pathways and fuel MDSCs development, leading to an immunosuppressive environment (68). MDSCs and Treg cells suppress T lymphocyte effector functions in the TME and are enriched in tumors with elevated glycolytic metabolism, such as triple-negative breast cancer (TNBC). These tumors secrete high levels of G-CSF, which stimulates MDSCs, and lower expression levels of LC3 mRNA. Inhibition of the glycolytic enzyme lactate dehydrogenase A (LDHA) in 4T1 and Py8119 TNBC cells using target-directed short-hairpin(sh), restored tumor autophagy, and reduced MDSC infiltration in tumors growing in BALB/C and C57/BL6 mice, respectively. The mechanism was dependent on AMPK-ULK1 signaling, which was impaired by glycolysis. At the same time, glycolysis and AMPK-ULK1 inhibition increased G-CSF expression. Consequently decreasing IFN-γ+ and TNF-α+ effector CD8+ T TILs and in tumor-draining lymph nodes, resulting in an immunosuppressive environment. Consequently, autophagy boosted antitumor response mediated by effector CD8+ T cells (68). These results described above indicate that autophagy may be an important mechanism in tumor immune responses. Sometimes, directly improving the cytotoxic activity of T cells, and others, modulating the immunosuppressive components of TME, such as infiltrated MDSCs and T-cell exhaustion-markers, leading to tumor elimination and better survival rates.
Autophagy and Immune Evasion Mechanisms
Despite the evidence that autophagy can improve antitumor immune responses, it can also inhibit both innate and adaptive responses, leading to cancer immune evasion.
Autophagy in Myeloid Cells Induces an Immunosuppressive TME
It is known that MDSCs and M2 macrophages are components of the immunosuppressive compartment of TME. The autophagic activity in MDSCs has been described by different research groups (63, 64) and has been associated with antigen presentation and cytotoxic T cell function impairment, as well as to M2 macrophage polarization and Treg recruitment (Figure 2) (48, 63).
The importance of a non-canonical autophagy pathway, the LC3-associated phagocytosis (LAP), in TME was demonstrated using Cre-lox based ablation of several genes in myeloid cells of immunocompetent mice (C57BL/6). The ablation of Becn1, Vps34, Atg5, Atg7, or Atg16l1 impacted on both conventional autophagy and LAP pathway. While myeloid cells deficient in Fip200, Ulk1, or Atg14 lacked only the canonical autophagy pathway, and the absence of Rubicon or Nox2 affected only the LAP pathway (63). In the LLC mouse model, the absence of LAP (Rubcn-/-) in myeloid cells increased the co-stimulatory molecule CD86 and reduced CD206, a mannose receptor associated with M2-like phenotype. In this model, the reduction on M2-like TAMs improved CD8+ and CD4+ T cell IFN-γ production, increased IFN-I, and IL-1β, although no quantitative alteration in the frequency of TILs was observed (63), suggesting modulation of T cell activity rather than proliferation or recruitment.
Similarly, an increase in autophagy activity was observed in MDSCs from melanoma patients and melanoma experimental model. Functional autophagy was measured by the expression of LC3, LAMP-1, and SQSTM1/p62, and colocalization analysis with p62 and LC3 in MDSCs isolated from melanoma patients (stages III and IV) and clinically healthy controls’ peripheral blood. MDSCs from melanoma patients, and also from mice with melanoma, showed higher levels of functional autophagy. In LysMcreAtg5fl/fl mice, in which myeloid cells lacked Atg5 expression, there were less Treg TILs, tumor cell proliferation rate reduction, and a significant increase in both MHC-II expression and IFN-γ production (64). Again, an indication that autophagy activity in TME myeloid cells plays a role in these cells’ immunosuppressive functions.
Autophagy Disrupts the Cytotoxic Activity of TME
TME is a hostile environment where autophagy can lead to degradation of cytotoxic molecules (granzyme B and IFN-γ), expression of T cell exhaustion markers, and quantitative changes in TILs.
NK cells release granules containing perforins and granzymes as part of their effector mechanism. In MCF7 breast cancer cells, granzyme B suffered lysosome degradation, after hypoxia-induced autophagy, which inhibited NK cell-mediated tumor cell lysis (65). Likewise, BECN1 inhibition increased functional NK cell tumor infiltration in a melanoma mouse model. The higher frequency in infiltrating NK cells was correlated with an increase in CCL5 secretion, which is an important chemokine for NK cell proliferation and activation. The enhancement of NK cell function, after inhibition of tumor-autophagy, caused tumor regression and predicted improved patient survival (66), suggesting that tumor cell-autophagy can be a resistance mechanism to NK cell activity.
The impact of autophagy in cytotoxic immune responses also influences CD8+ and CD4+ T cells, although there is no consensus about this topic in the literature. Atg5 deficient mice displayed a reduction of CD8+ TILs in mammary and colorectal cancer models. Despite that, among the CD8+ T lymphocytes recruited to the tumors, around 80% exhibited memory phenotype and were positive for IFN-γ and TNF-α, leading to tumor rejection (69). Moreover, in the non-small cell lung cancer (NSCLC) mouse model, Atg5 tissue-specific depletion promoted tumor initiation through a mechanism dependent on Treg recruitment to the TME (48). These findings indicated that autophagy may have a positive effect on cytotoxic immune responses, through the reduction in Treg frequency in the TME and increase in IFN-γ and TNF-α release.
On the other hand, the interaction with human glioblastoma (GB) cells induced Lamp2a mRNA and protein expression in mouse brain pericytes (PC). Engraftment of PC from Lamp2a knockout mice (PC-KO) into GB grown in mice led to central-memory (CD44+, CD62+) CD4+ and CD8+ T TILs and better CD4+/CD8+ ratio of 2:1 than in tumors engrafted with wild type (WT) PC. Moreover, only 2% of TILs expressed PD-1 and CTLA-4 and there was a lower GB cell proliferation rate than in GB/PC-WT control mice. Compared to GB/PC-KO, GB/PC-WT mice displayed increased IL-2 levels, tumor proliferation, FoxP3+PD-1+CTLA-4+ TILs, higher Tgfβ, and Il10 mRNA expression levels, and a CD4+/CD8+ ratio of 4:1. Collectively, these results demonstrated that CMA activity in PC favored an immunosuppressive environment in response to GB cells (64).
Inhibitory molecules expression is associated with CD8+ T cell exhaustion phenotype, and also resistance to targeted therapy and autophagy inhibitors. PD-L1+ A375 melanoma cells showed resistance to BRAF inhibitor Vemurafenib and CQ, but not to nitrobenzoxadiazole (NBD). Both CQ and NBD inhibit autophagy, however, NBD acts at multiple levels, targeting not only the late stages of autophagy but also different apoptotic pathways (70). These findings indicate that autophagy may act as an immunosuppressive mechanism, affecting cytotoxic cells—NK and T lymphocytes—functionally and quantitatively. Additionally, autophagy modulates immune checkpoints expression, leading to T cell exhaustion phenotype and resistance to treatment.
Autophagy and Cancer Therapy
Autophagy can be a preventive mechanism to malignant transformation in healthy cells, but in transformed cells this mechanism can contribute to cancer progression. Autophagy’s main roles in tumorigenesis and cancer therapy were reviewed elsewhere (71). In this section, we will focus on the interplay between autophagy and immune responses in cancer therapies in different experimental models.
Autophagy and Conventional Therapies
Traditionally, cancer therapy is based on direct toxicity to tumor cells. But the importance of immune responses to the efficacy of traditional therapies can be observed in mouse models, in which T lymphocytes are necessary for tumor growth reduction by chemotherapeutic agents (72). Recently, it has become clear that several chemotherapeutic agents, such as doxorubicin and mitoxantrone (MTX), and even radiotherapy, may induce tumor cell immunogenic cell death (ICD) (72–74). ICD is characterized by the release of damage-associated molecular patterns (DAMPs) and consequent elicitation of immune responses. The pre-apoptotic surface exposure of calreticulin (CRT) is considered the “eat me” (immunogenicity) signal for DCs, influencing antigen presentation to T lymphocytes (73). Likewise, adenosine triphosphate (ATP) secreted or leaked to the extracellular milieu is considered a “find me” signal. The chromatin-binding protein high mobility group B1 (HMGB1) is another DAMP exposed after chemotherapy by post-apoptotic release. It induces TLR4-MyD88 signaling on DCs facilitating antigen processing and presentation (74, 75).
The ATP lysosomal secretion depends on autophagy, as autophagy-deficient CT26 colon carcinoma mouse cells (due to the lack or decrease in Atg5 or Atg7 expression) released lower amounts of ATP in response to MTX chemotherapy. The lower ATP release resulted in DCs recruitment impairment and consequent lack of T cell priming to elicit an anti-tumor immune response (76). Extracellular ATP binds to surface purinergic receptors (such as P2YR2 receptors) on immature DC precursors (77), promoting DC maturation and recruitment to the tumor site in lung cancer mouse model. In line with that, autophagy induction (by fasting or caloric restriction) resulted in ATP release and improved chemotherapy “efficacy” by decreasing TME infiltration by Treg (78).
CD39, an ectonucleotidase, also influences the extracellular ATP concentration by converting extracellular ATP into adenosine. CD39 overexpression was observed in Atg5 deficient tumors, leading to the attraction of Treg expressing adenosinergic receptors to the TME (48). Indeed, an enhanced number of initial tumor foci and increased Treg infiltration were observed in autophagy-deficient tumors in the KRasG12D-driven lung cancer mouse model (48). In contrast, similar T cell infiltration and function in autophagy-deficient (due to inhibition of autophagy essential genes Atg7 and Atg12) or competent tumors were observed in the B16 melanoma mouse model, even after Doxorubicin treatment (10). These data suggest that autophagy-dependent immune modulation may be specific to the clinical context and time (10). However, the complexity of autophagy role in carcinogenesis can be seen in the KRasG12D-driven lung cancer mouse model, because mice with autophagy-deficient-tumors presented a prolonged survival and reduced malignant progression of adenomas to adenocarcinomas, in a TP53 dependent manner. Although the autophagy modulation needs to be investigated in each case, these data indicated that tumor-specific loss of Atg5 favored Treg TILs. Thus, autophagy-deficient tumors (or with CD39 overexpression) treated with ICD inducer chemotherapy agents did not recruit effector cells and it possibly contributed to chemotherapy (CT) resistance (48).
Indeed, there is evidence that autophagy may play a role in CT resistance in pancreatic cancer patients. A clinical trial to investigate if autophagy inhibition by HCQ (Hydroxychloroquine sulfate) improved overall survival of metastatic pancreatic cancer patients treated with gemcitabine hydrochloride and nab-paclitaxel was performed (79). Autophagy inhibition did not improve 1-year patients’ overall survival, but there was an improvement in the overall response rate in HCQ treated patients, indicating its role in the locally advanced setting, supporting the need for more research and biomarkers to drive this therapeutic option. Likewise, the treatment of bladder cancer cells (J82 and T24) with enzalutamide, an anti-androgen receptor drug, resulted in cytoplasmatic autophagosomes accumulation, increased expression of autophagy-related genes (AMPK, ATG5, LC3B, ULK1, and LC3-II) and had no effect on apoptosis and proliferation rates. However, the treatment with a combination of enzalutamide and autophagy inhibitors (CQ, 3-methyladenine, and bafilomycin A1) impaired tumor growth, indicating that the combined treatment may be a potential strategy to avoid enzalutamide-resistant bladder cancer (80). A similar result was exhibited in docetaxel resistant prostate cancer cell lines (PC3-DR and VCaP-DR): these cells present enhanced autophagy activity through the overexpression of Forkhead box protein M1 (FOXM1). Thus, the knockout of ATG7, Beclin-1, or treatment with CQ restored the antitumor effect of docetaxel, demonstrating that either autophagy or FOXM1 may be potential targets for combined therapies with docetaxel to treat metastatic prostate cancer patients (81). On the other hand, new therapies may be used to activate autophagy, improving the efficacy of conventional treatment, such as CT. Oxaliplatin-induced ICD was not sufficient to completely eliminate both breast and colorectal tumors (82, 83). Oxaliplatin treatment together with a nanoparticle in CT26 tumor-bearing mice led to autophagy activation on tumor cells, improving antigen presentation, and consequently tumor cell death (83).
The role of autophagy in other cells beyond the tumor ones was investigated to elucidate the hypothesis that autophagy competence in the immune system would contribute to the reduction of tumor growth by ICD-inducing CT. However, the growth of autophagy competent tumors was the same in wild type and partially autophagy-deficient (Becn1± or Atg4b-/-) mice, although the MTX toxicity was higher on Atg4b-/- mice (84). These results pointed to an autophagy role in tumor cells influencing anticancer immune responses induced by CT. Nonetheless, a similar T cell profile was observed in the B16 melanoma mouse model, both in tumor-specific autophagy inhibition treated with Doxorubicin (10) and in systemic inhibition of autophagy with CQ and quinacrine, suggesting that host autophagy competence did not influence the efficacy of ICD-inducing CT in this model (10).
Response to radiotherapy may also be influenced by autophagy. Radiation therapy induced MHC-I expression in NSCLC cell lines A549 and H1975 in parallel with an increase in the LC3-II/LC3-I ratio, while p62 detection decreased (85). Treatment of both cell lines with autophagy inhibitor CQ after radiation resulted in decreased MHC-I expression, indicating that radiation-induced MHC-I expression and CD8+ T cell infiltration were dependent on autophagy (85). It was also demonstrated that imiquimod (TLR7 agonist) activated oxidative stress, inducing autophagy, and sensibilization of melanoma cells to γ-ionizing radiation (86). Mouse treatment with 3-MA, an autophagy inhibitor, after radiation restored the B16F10 or B16F1 tumor growth. The combined imiquimod and radiation therapy increased IFN-γ and TNF-α secreting CD8+ T lymphocytes and decreased Treg and MDSCs in the TME (86), indicating the influence of autophagy in the regulation of therapy-induced immune responses.
Autophagy Improves New Therapeutic Strategies, by Modulating TME Components
Recently, immunotherapy has emerged as a therapeutic option able to elicit high rates of durable anti-tumor responses in cancer patients, including the ones with previously refractory responses to CRT (87). Immunotherapy aims to activate and recruit immune cells to the TME to target transformed cells. The best results have been achieved in patients with immunogenic tumors, which express high levels of neoantigens, such as metastatic melanoma. Even so, a proportion of patients with no clinical benefit after immune checkpoint blockade therapy has been reported in several studies, and autophagy may play a role in this resistance to therapy. A set of melanoma-associated antigen (MAGE) cancer-germline antigens was identified as a predictor of CTLA-4 blockade resistance in melanoma patients (88). Interestingly, autophagy markers, including LC3B, were enriched in MAGE negative tumors, suggesting that autophagy suppression (in MAGE positive tumors) may contribute to resistance to CTLA-4 inhibitor therapy (88). Another autophagy mechanism that influences the TME was observed in head and neck squamous cell carcinoma (HNSCC). Despite the high immune cell infiltration in HNSCC, response rates to immune checkpoint blockade therapy are low. It was identified that the SOX2 oncoprotein elicited an autophagy-dependent degradation of STING, which mediates IFN-I activation, important for Th1 chemokines production and M1-like macrophage polarization (89). In mice with HNSCC, the immunosuppressive TME could be reversed by vaccination with nanosatellite SatVax, which enhanced the potency of STING agonist and delivered high-density tumor antigens, improving tumor-specific T cell infiltration. Associated with anti-PD-L1 therapy, it could prevent the CD8+ T cell exhaustion (the therapy expanded CTL effectors and reduces the CD8+ T cells exhausted) (89).
Along with that, autophagy-deficient 4T1 cells, through Atg5 or Beclin1 depletion with specific single guide(sg)RNA, generated larger tumors with reduced CD4+ and CD8+ TILs and IFNγ+ T cells in Balb/c mice, when compared to autophagy competent 4T1 cells. Moreover, the antitumor effect of the anti-PD1 antibody was limited in autophagy-deficient tumors, while a significant reduction in tumor volume and increased cytotoxic activity of TILs was observed in the control group (90). Similarly, a combination of anti-PD1 immunotherapy with anti-angiogenic endostatin, Endostar, promoted the activation of autophagy pathway PI3K/AKT/mTOR in LLC-bearing mice. Combined therapy suppressed tumor growth and modulated TME, decreasing IL-17 and TGFβ1 while reducing infiltrated MDSCs and reversing CD8+ T cell suppression (91). These results indicated that the complex relationship between autophagy and TME components may be important not only to the modulation of immune responses but also to define treatment efficacy and resistance.
In addition to immunotherapy, a new therapeutic approach using RP-182, a synthetic peptide analog to naturally occurring antimicrobial peptides, triggered a conformational switch in the mannose receptor CD206, M2 macrophage marker. It resulted in endocytosis, autophagy, and apoptosis of these cells and also a shift toward an M1 phenotype in the remaining cells. This treatment enriched M1-like macrophages in TME and increased the antitumor immune response in the pancreatic cancer animal model, as well as CT-26 and B16 models (92).
Conclusions
In conclusion, we described data suggesting that autophagic activity plays a dual role in cancer development and progression, modulating TME in many different ways, that can either help or inhibit tumor development, as shown in Figure 2. DC-autophagy and TME-induced autophagy are usually associated with better antitumor responses and improvement of antigen presentation and cytotoxic activity, inhibiting regulatory T lymphocytes and MDSCs. However, myeloid cells and tumor cells autophagy seem to have the opposite effect. It improves immunosuppressive TME, through the recruitment of MDSCs and M2-like macrophage polarization, leading to tumor progression and worst prognosis. The influence of autophagy also reaches cancer treatment. The activation of autophagy pathways modulates TME by inducing macrophage polarization (M1-like phenotype), reducing CD8+ T cell exhaustion and Tregs infiltration. Therefore, targeting autophagy could improve ICD-induced by conventional and non-conventional therapies.
Author Contributions
This review was drafted by AS, LG, APL, and PA-S, then critically revised by PA-S. Figures were elaborated by AS and PA-S. All authors contributed to the article and approved the submitted version.
Funding
We thank the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for the scholarship provided to LG (141955/2020-1) and fellowship to AL (307841/2018-9), and the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) for the scholarship provided to AS (88882.382114/2019-01). This work was partially funded by LIGH-FUNPAR Alliance.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
IFN-γ; interferon gamma; TME, tumor microenvironment; ECM, extracellular matrix; NK, natural killer; TAMs, tumor-associated macrophages; MDSC, myeloid-derived suppressor cell; ROS, reactive oxygen species; AIDS, Acquired Immune Deficiency Syndrome; HAART, highly active antiretroviral therapy; IL, interleukin; Th, T helper; KIR, killer immunoglobulin-like inhibitory receptor; HLA, human leucocyte antigen; TCR, T cell receptor; DC, dendritic cell; Treg, regulatory T cells; IRF1, interferon regulatory factor 1; HDAC, histone deacetylase; IFN-I, type I interferon; TNF-α, tumor necrosis factor alpha; CTLA-4, cytotoxic T lymphocyte associated protein 4; IDO, indoleamine-2,3-deoxygenase; PD-L1, Programmed cell death-ligand 1; APC, antigen presenting cell; PD-1, programmed cell death-1; ITIM, immunoreceptor tyrosine-based inhibitory; PD-L2, programmed cell death ligand-2; CMA, chaperone-mediated autophagy; LC3, light chain protein-3; Hsc 70, heat shock cognate 70; LAMP, lysosomal-associated membrane protein; HIF, hypoxia-inducible factor; HREs, hypoxia response elements; BNIP3, BCL2 interacting protein 3; BNIP3L, BCL2 interacting protein 3 like; Bcl-2, B-cell lymphoma 2; ATG, autophagy-related genes; AMPK, adenosine monophosphate-activated protein kinase; mTOR, mammalian target of rapamycin; TAA, tumor-associated antigens; MHC, major histocompatibility complex; TILs, tumor infiltrate lymphocytes; α-TEA, alpha-tocopheryloxyacetic acid; LLC, Lewis lung carcinoma; TLR, toll-like receptor; PDAC, pancreatic ductal adenocarcinoma; TIM-4, T cell immunoglobulin and mucin domain protein-4; CQ, chloroquine; TNBC, triple negative breast cancer; LDHA, lactate dehydrogenase A; G-CSF, granulocyte colony- stimulating factor; LAP, LC3-associated phagocytosis; NSCLC, non-small cell lung cancer; GB, glioblastoma; PC, pericytes; NBD, nitrobenzoxadiazole; MTX, mitoxantrone; ICD, immunogenic cell death; DAMPs, damage-associated molecular patterns; CRT, calreticulin; ATP, adenosine triphosphate; CT, chemotherapy; HCQ, Hydroxychloroquine sulfate; MAGE, melanoma-associated antigen; HNSCC, head and neck squamous cell carcinoma.
References
1. Dighe AS, Richards E, Old LJ, Schreiber RD. Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFNγ receptors. Immunity (1994) 1:447–56. doi: 10.1016/1074-7613(94)90087-6
2. Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNgamma and lympohcytes prevent primary tomour development and shape tomour immunogenicity. Nature (2001) 410:1107–11. doi: 10.1038/35074122
3. Dunn GP, Old LJ, Schreiber RD. The Three Es of Cancer Immunoediting. Annu Rev Immunol (2004) 22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803
4. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science (80- ) (2011) 331:1565–70. doi: 10.1126/science.1203486
5. Roma-Rodrigues C, Mendes R, Baptista PV, Fernandes AR. Targeting tumor microenvironment for cancer therapy. Int J Mol Sci (2019) 20:840. doi: 10.3390/ijms20040840
6. Jarosz-Biej M, Smolarczyk R, Cichoń T, Kułach N. Tumor microenvironment as a “game changer” in cancer radiotherapy. Int J Mol Sci (2019) 20:18–20. doi: 10.3390/ijms20133212
7. Deter RL, Baudhuin P, De Duve C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J Cell Biol (1967) 35:C11–6. doi: 10.1083/jcb.35.2.c11
8. Tsukada M. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett (1993) 333:169–74. doi: 10.1016/0014-5793(93)80398-E
9. Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature (1999) 402:672–6. doi: 10.1038/45257
10. Starobinets H, Ye J, Broz M, Barry K, Goldsmith J, Marsh T, et al. Antitumor adaptive immunity remains intact following inhibition of autophagy and antimalarial treatment. J Clin Invest (2016) 126:4417–29. doi: 10.1172/JCI85705
11. Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature (2020) 581:100–5. doi: 10.1038/s41586-020-2229-5
12. Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
13. Garner H, de Visser KE. Immune crosstalk in cancer progression and metastatic spread: a complex conversation. Nat Rev Immunol (2020) 20(8):483–97. doi: 10.1038/s41577-019-0271-z
14. Engels EA, Pfeiffer RM, Goedert JJ, Virgo P, Mcneel TS, Scoppa SM, et al. Trends in cancer risk among people with AIDS in the United States 1980 – 2002. AIDS (2006) 20:1645–54. doi: 10.1097/01.aids.0000238411.75324.59
15. Franzetti M, Ricci E, Bonfanti P. The Pattern of Non-AIDS-defining Cancers in the HIV Population: Epi- demiology, Risk Factors and Prognosis. A Review. Curr HIV Res (2019) 17:1–12. doi: 10.2174/1570162X17666190327153038
16. Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, et al. Identification of essential genes for cancer immunotherapy. Nature (2017) 548(7669):537–42. doi: 10.1038/nature23477
17. Zhang C, Liu Y. Targeting NK Cell Checkpoint Receptors or Molecules for Cancer Immunotherapy. Front Immunol (2020) 11:1295. doi: 10.3389/fimmu.2020.01295
18. Knochelmann HM, Dwyer CJ, Smith AS, Bowers JS, Wyatt MM, Nelson MH, et al. IL-6 fuels durable memory for Th17 cell-mediated responses to tumors. Cancer Res (2020) 80(18):3920–32. doi: 10.1158/0008-5472.can-19-3685
19. Minetto P, Guolo F, Pesce S, Greppi M, Obino V, Ferretti E, et al. Harnessing NK Cells for Cancer Treatment. Front Immunol (2019) 10:2836. doi: 10.3389/fimmu.2019.02836
20. Martinez-Lostao L, Anel A, Pardo J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin Cancer Res (2015) 21:5047–56. doi: 10.1158/1078-0432.CCR-15-0685
21. Tay RE, Richardson EK, Toh HC. Revisiting the role of CD4 + T cells in cancer immunotherapy — new insights into old paradigms. Cancer Gene Ther (2020). doi: 10.1038/s41417-020-0183-x
22. Park J, Kim E, Kwon H, Hwang E, Namkoong S, Um S. Inactivation of Interferon Regulatory Factor-1 Tumor Suppressor Protein by HPV E7 Oncoprotein. J Biol Chem (2000) 275:6764–9. doi: 10.1074/jbc.275.10.6764
23. Aricò E, Castiello L, Capone I, Gabriele L, Belardelli F. Type i interferons and cancer: An evolving story demanding novel clinical applications. Cancers (Basel) (2019) 11(12):1943. doi: 10.3390/cancers11121943
24. Bussey KA, Brinkmann MM. Strategies for immune evasion by human tumor viruses. Curr Opin Virol (2018) 32:30–9. doi: 10.1016/j.coviro.2018.08.015
25. Keerthivasan S, Aghajani K, Dose M, Molinero L, Khan W, Venkatesvaran V, et al. Wnt/β-catenin signaling in T-cells drives epigenetic imprinting of pro-inflammatory properties and promotes colitis and colon cancer. Sci Transl Med (2014) 6(225):225ra28. doi: 10.1126/scitranslmed.3007607.Wnt/
26. Kroemer G, Pouyssegur J. Tumor Cell Metabolism: Cancer’s Achilles’ Heel. Cancer Cell (2008) 13(6):472–82. doi: 10.1016/j.ccr.2008.05.005
27. Domblides C, Lartigue L, Faustin B. Control of the Antitumor Immune Response by Cancer Metabolism. Cells (2019) 8(2):104. doi: 10.3390/cells8020104
28. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol (2011) 185:3190–8. doi: 10.4049/jimmunol.0903670.AN
29. Hornyák L, Dobos N, Koncz G, Karányi Z, Páll D, Szabó Z, et al. The Role of indoleamine-2, 3- Dioxygenase in Cancer Development, Diagnostics, and Therapy. Front Immunol (2018) 9:151. doi: 10.3389/fimmu.2018.00151
30. Alspach E, Lussier DM, Schreiber RD. Interferon γ and its important roles in promoting and inhibiting spontaneous and therapeutic cancer immunity. Cold Spring Harb Perspect Biol (2019) 11(3):a028480. doi: 10.1101/cshperspect.a028480
31. Bally APR, Austin JW, Boss JM. Genetic and epigenetic regulation of PD-1 expression. J Immunol (2017) 196:2431–7. doi: 10.4049/jimmunol.1502643.Genetic
32. Jansen CS, Prokhnevska N, Master VA, Sanda MG, Carlisle JW, Bilen MA, et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature (2019) 576:465–73. doi: 10.1038/s41586-019-1836-5
33. Kurachi M. CD8+ T cell exhaustion. Semin Immunopathol (2019) 41:327–37. doi: 10.1007/s00281-019-00744-5
34. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86 : a molecular basis for the cell extrinsic function of CTLA-4. Science (80- ) (2011) 332:600–3. doi: 10.1126/science.1202947.Trans-endocytosis
35. Genard G, Lucas S, Michiels C. Reprogramming of tumor-associated macrophages with anticancer therapies: Radiotherapy versus chemo- and immunotherapies. Front Immunol (2017) 8:828. doi: 10.3389/fimmu.2017.00828
36. Duve C D, Pressman BC, Gianetto R, Wattiaux R, Appelmans F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem J (1955) 60:604–17. doi: 10.1042/bj0600604
37. Moscat J, Diaz-Meco MT. p62 at the Crossroads of Autophagy, Apoptosis, and Cancer. Cell (2009) 137(6):1001–4. doi: 10.1016/j.cell.2009.05.023
38. Glick D, Barth S, Macleod KF. Autophagy: Cellular and molecular mechanisms. J Pathol (2010) 221(1):3–12. doi: 10.1002/path.2697
39. Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, Follenzi A, et al. Microautophagy of Cytosolic Proteins by Late Endosomes. Dev Cell (2011) 20:131–9. doi: 10.1016/j.devcel.2010.12.003
40. Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science (80- ) (1996) 273:501–3. doi: 10.1126/science.273.5274.501
41. Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, et al. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol Cell Biol (2009) 29:2570–81. doi: 10.1128/mcb.00166-09
42. Egan DF, Shackelford DB, Mihaylova MM, Gelino SR, Kohnz RA, Mair W, et al. and SR. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science (80- ) (2011) 28:456–61. doi: 10.1126/science.1196371
43. Papandreou I, Lim AL, Laderoute K, Denko NC. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ (2008) 15:1572–81. doi: 10.1038/cdd.2008.84
44. Yang Y, Klionsky DJ. Autophagy and disease: unanswered questions. Cell Death Differ (2020) 27(3):858–87. doi: 10.1038/s41418-019-0480-9
45. White E, Mehnert JM, Chan CS. Autophagy, Metabolism, and Cancer. Clin Cancer Res (2015) 21:5037–46. doi: 10.1158/1078-0432.CCR-15-0490
46. Ireland JM, Unanue ER. Autophagy in antigen-presenting cells results in presentation of citrullinated peptides to CD4 T cells. J Exp Med (2011) 208:2625–32. doi: 10.1084/jem.20110640
47. Li Y, Hahn T, Garrison K, Cui ZH, Thorburn A, Thorburn J, et al. The vitamin E analogue α-TEA stimulates tumor autophagy and enhances antigen cross-presentation. Cancer Res (2012) 72:3535–45. doi: 10.1158/0008-5472.CAN-11-3103
48. Rao S, Tortola L, Perlot T, Wirnsberger G, Novatchkova M, Nitsch R, et al. A dual role for autophagy in a murine model of lung cancer. Nat Commun (2014) 5:3056. doi: 10.1038/ncomms4056
49. Wang Y, Lin YX, Wang J, Qiao SL, Liu YY, Dong WQ, et al. In Situ Manipulation of Dendritic Cells by an Autophagy-Regulative Nanoactivator Enables Effective Cancer Immunotherapy. ACS Nano (2019) 13:7568–77. doi: 10.1021/acsnano.9b00143
50. Khateri M, Abdoli A, Motevalli F, Fotouhi F, Bolhassani A, Arashkia A, et al. Evaluation of autophagy induction on HEV 239 vaccine immune response in a mouse model. IUBMB Life (2018) 70:207–14. doi: 10.1002/iub.1719
51. Munz C. Enhancing immunity through autophagy. Annu Rev Immunol (2009) 27:423–49. doi: 10.1146/annurev.immunol.021908.132537 10.1146/annurev.immunol.021908.132537.
52. Reeves E, James E. Antigen processing and immune regulation in the response to tumours. Immunology (2017) 150:16–24. doi: 10.1111/imm.12675
53. Münz C. Autophagy Beyond Intracellular MHC Class II Antigen Presentation. Trends Immunol (2016) 37:755–63. doi: 10.1016/j.it.2016.08.017
54. Dendrou CA, Petersen J, Rossjohn J, Fugger L. HLA variation and disease. Nat Rev Immunol (2018) 18:325–39. doi: 10.1038/nri.2017.143
55. Tumeh PC, Hellmann MD, Hamid O, Tsai KK, Loo KL, Gubens MA, et al. Liver metastasis and treatment outcome with anti-PD-1 monoclonal antibody in patients with melanoma and NSCLC. Cancer Immunol Res (2017) 5:417–24. doi: 10.1158/2326-6066.CIR-16-0325
56. Takahashi Y, Yasui T, Minami K, Tamari K, Otani K, Seo Y, et al. Radiation enhances the efficacy of antitumor immunotherapy with an immunocomplex of interleukin-2 and its monoclonal antibody. Anticancer Res (2017) 37:6799–806. doi: 10.21873/anticanres.12140
57. Peggs KS, Segal NH, Allison JP. Targeting Immunosupportive Cancer Therapies: Accentuate the Positive, Eliminate the Negative. Cancer Cell (2007) 12:192–9. doi: 10.1016/j.ccr.2007.08.023
58. Brentville VA, Metheringham RL, Daniels I, Atabani S, Symonds P, Cook KW, et al. Combination vaccine based on citrullinated vimentin and enolase peptides induces potent CD4-mediated anti-tumor responses. J Immunother Cancer (2020) 8:6–9. doi: 10.1136/jitc-2020-000560
59. Baghdadi M, Yoneda A, Yamashina T, Nagao H, Komohara Y, Nagai S, et al. TIM-4 Glycoprotein-Mediated Degradation of Dying Tumor Cells by Autophagy Leads to Reduced Antigen Presentation and Increased Immune Tolerance. Immunity (2013) 39:1070–81. doi: 10.1016/j.immuni.2013.09.014
60. Ramirez JAZ, Romagnoli GG, Falasco BF, Gorgulho CM, Sanzochi Fogolin C, dos Santos DC, et al. Blocking drug-induced autophagy with chloroquine in HCT-116 colon cancer cells enhances DC maturation and T cell responses induced by tumor cell lysate. Int Immunopharmacol (2020) 84:106495. doi: 10.1016/j.intimp.2020.106495
61. Tu SP, Quante M, Bhagat G, Takaishi S, Cui G, Yang XD, et al. IFN- g Inhibits Gastric Carcinogenesis by Inducing Epithelial Cell Autophagy and T-cell Apoptosis. Cancer Res (2011) 71:4247–59. doi: 10.1158/0008-5472.CAN-10-4009
62. Vodnala SK, Eil R, Kishton RJ, Sukumar M, Yamamoto TN, Ha N, et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science (80- ) (2019) 363:1–12. doi: 10.1126/science.aau0135
63. Cunha LD, Yang M, Carter R, Guy C, Harris L, Crawford C, et al. LC3-associated phagocytosis in myeloid cells promotes tumor immune tolerance. Cell (2019) 175:429–41. doi: 10.1016/j.cell.2018.08.061.LC3-associated
64. Alissafi T, Hatzioannou A, Mintzas K, Barouni RM, Banos A, Sormendi S, et al. Autophagy orchestrates the regulatory program of tumor-associated myeloid-derived suppressor cells. J Clin Invest (2018) 128:3840–52. doi: 10.1172/JCI120888
65. Baginska J, Viry E, Berchem G, Poli A, Noman MZ, Van Moer K, et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci U S A (2013) 110:17450–5. doi: 10.1073/pnas.1304790110
66. Mgrditchian T, Arakelian T, Paggetti J, Noman MZ, Viry E, Moussay E, et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc Natl Acad Sci U S A (2017) 114:E9271–9. doi: 10.1073/pnas.1703921114
67. Valdor R, García-Bernal D, Riquelme D, Martinez CM, Moraleda JM, Cuervo AM, et al. Glioblastoma ablates pericytes antitumor immune function through aberrant up-regulation of chaperone-mediated autophagy. Proc Natl Acad Sci U S A (2019) 116:20655–65. doi: 10.1073/pnas.1903542116
68. Li W, Tanikawa T, Kryczek I, Xia H, Li G, Wu K, et al. Aerobic Glycolysis Controls Myeloid-Derived Suppressor Cells and Tumor Immunity via a Specific Article Aerobic Glycolysis Controls Myeloid-Derived Suppressor Cells and Tumor Immunity via a Specific CEBPB Isoform in Triple-Negative Breast Cancer. Cell Metab (2018) 28:87–103. doi: 10.1016/j.cmet.2018.04.022
69. DeVorkin L, Pavey N, Carleton G, Comber A, Ho C, Lim J, et al. Autophagy Regulation of Metabolism Is Required for CD8 + T Cell Anti-tumor Immunity. Cell Rep (2019) 27:502–513.e5. doi: 10.1016/j.celrep.2019.03.037
70. Sciarretta F, Fulci C, Palumbo C, Rotili D, Tentori L, Graziani G, et al. Effects of Glutathione Transferase-Targeting Nitrobenzoxadiazole Compounds in Relation to PD-L1 Status in Human Melanoma Cells. Chemotherapy (2019) 64:138–45. doi: 10.1159/000503339
71. Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, et al. Autophagy in malignant transformation and cancer progression. EMBO J (2015) 34:856–80. doi: 10.15252/embj.201490784
72. Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med (2005) 202:1691–701. doi: 10.1084/jem.20050915
73. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med (2007) 13:54–61. doi: 10.1038/nm1523
74. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med (2007) 13:1050–9. doi: 10.1038/nm1622
75. Saenz R, Futalan D, Leutenez L, Eekhout F, Fecteau JF, Sundelius S, et al. TLR4-dependent activation of dendritic cells by an HMGB1-derived peptide adjuvant. J Transl Med (2014) 12:1–11. doi: 10.1186/1479-5876-12-211
76. Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science (80- ) (2011) 334:1573–7. doi: 10.1126/science.1208347
77. Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity (2013) 38:729–41. doi: 10.1016/j.immuni.2013.03.003
78. Pietrocola F, Pol J, Vacchelli E, Rao S, Enot DP, Baracco EE, et al. Caloric Restriction Mimetics Enhance Anticancer Immunosurveillance. Cancer Cell (2016) 30:147–60. doi: 10.1016/j.ccell.2016.05.016
79. Karasic TB, O’Hara MH, Loaiza-Bonilla A, Reiss KA, Teitelbaum UR, Borazanci E, et al. Effect of Gemcitabine and nab-Paclitaxel with or Without Hydroxychloroquine on Patients with Advanced Pancreatic Cancer: A Phase 2 Randomized Clinical Trial. JAMA Oncol (2019) 5:993–8. doi: 10.1001/jamaoncol.2019.0684
80. Quan Y, Lei H, Wahafu W, Liu Y, Ping H, Zhang X. Inhibition of autophagy enhances the anticancer effect of enzalutamide on bladder cancer. BioMed Pharmacother (2019) 120:109490. doi: 10.1016/j.biopha.2019.109490
81. Lin JZ, Wang WW, Hu TT, Zhu GY, Li LN, Zhang CY, et al. FOXM1 contributes to docetaxel resistance in castration-resistant prostate cancer by inducing AMPK/mTOR-mediated autophagy. Cancer Lett (2020) 469:481–9. doi: 10.1016/j.canlet.2019.11.014
82. Feng B, Zhou F, Hou B, Wang D, Wang T, Fu Y, et al. Binary Cooperative Prodrug Nanoparticles Improve Immunotherapy by Synergistically Modulating Immune Tumor Microenvironment. Adv Mater (2018) 30:e1803001. doi: 10.1002/adma.201803001
83. Wang X, Li M, Ren K, Xia C, Li J, Yu Q, et al. On-Demand Autophagy Cascade Amplification Nanoparticles Precisely Enhanced Oxaliplatin-Induced Cancer Immunotherapy. Adv Mater (2020) 32:e2002160. doi: 10.1002/adma.202002160
84. Castoldi F, Vacchelli E, Zitvogel L, Maiuri MC, Pietrocola F, Kroemer G. Systemic autophagy in the therapeutic response to anthracycline-based chemotherapy. Oncoimmunology (2019) 8:1–6. doi: 10.1080/2162402X.2018.1498285
85. Zeng H, Zhang W, Gong Y, Xie C. Radiotherapy activates autophagy to increase CD8+ T cell infiltration by modulating major histocompatibility complex class-I expression in non-small cell lung cancer. J Int Med Res (2019) 47:3818–30. doi: 10.1177/0300060519855595
86. Cho JH, Lee HJ, Ko HJ, Yoon BI, Choe J, Kim KC, et al. The TLR7 agonist imiquimod induces anti-cancer effects via autophagic cell death and enhances anti-tumoral and systemic immunity during radiotherapy for melanoma. Oncotarget (2017) 8:24932–48. doi: 10.18632/oncotarget.15326
87. Robert C, Marabelle A, Herrscher H, Caramella C, Rouby P, Fizazi K, et al. Immunotherapy discontinuation — how, and when? Data from melanoma as a paradigm. Nat Rev Clin Oncol (2020) 17:707–15. doi: 10.1038/s41571-020-0399-6
88. Shukla SA, Bachireddy P, Schilling B, Galonska C, Zhan Q, Bango C, et al. Cancer-germline antigen expression discriminates clinical outcome to CTLA-4 blockade. Cell (2018) 173:624–33. doi: 10.1016/j.cell.2018.03.026
89. Tan YS, Sansanaphongpricha K, Xie Y, Christopher R, Luo X, Heath BR, et al. Mitigating SOX2-potentiated immune escape of Head and Neck Squamous Cell Carcinoma with a STING-inducing nanosatellite vaccine. Clin Cancer Res (2018) 24:4242–55. doi: 10.1158/1078-0432.CCR-17-2807
90. Li ZL, Zhang HL, Huang Y, Huang JH, Sun P, Zhou NN, et al. Autophagy deficiency promotes triple-negative breast cancer resistance to T cell-mediated cytotoxicity by blocking tenascin-C degradation. Nat Commun (2020) 11:3806. doi: 10.1038/s41467-020-17395-y
91. Wu J, Zhao X, Sun Q, Jiang Y, Zhang W, Luo J, et al. Synergic effect of PD-1 blockade and endostar on the PI3K/AKT/mTOR-mediated autophagy and angiogenesis in Lewis lung carcinoma mouse model. BioMed Pharmacother (2020) 125:109746. doi: 10.1016/j.biopha.2019.109746
Keywords: macroautophagy, tumor microenvironment, antitumor immunity, tumor immune evasion, onco immunology
Citation: de Souza ASC, Gonçalves LB, Lepique AP and de Araujo-Souza PS (2020) The Role of Autophagy in Tumor Immunology—Complex Mechanisms That May Be Explored Therapeutically. Front. Oncol. 10:603661. doi: 10.3389/fonc.2020.603661
Received: 07 September 2020; Accepted: 02 November 2020;
Published: 01 December 2020.
Edited by:
Daniel Hector Grasso, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), ArgentinaReviewed by:
Daniela Laura Papademetrio, University of Buenos Aires, ArgentinaEswari Dodagatta-Marri, University of California, San Francisco, United States
Copyright © 2020 de Souza, Gonçalves, Lepique and de Araujo-Souza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrícia Savio de Araujo-Souza, cHNhc0B1ZnByLmJy; orcid.org/0000-0002-4489-9148