 Luis M. Valor
Luis M. Valor Irati Hervás-Corpión
Irati Hervás-Corpión
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 02 December 2020
Sec. Neuro-Oncology and Neurosurgical Oncology
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.602378
This article is part of the Research Topic Neural Stem Cells of the Subventricular Zone: from Neurogenesis to Glioblastoma Origin View all 14 articles
Glioma stem cells (GSCs) are crucial in the formation, perpetuation and recurrence of glioblastomas (GBs) due to their self-renewal and proliferation properties. Although GSCs share cellular and molecular characteristics with neural stem cells (NSCs), GSCs show unique transcriptional and epigenetic features that may explain their relevant role in GB and may constitute druggable targets for novel therapeutic approaches. In this review, we will summarize the most important findings in GSCs concerning epigenetic-dependent mechanisms.
GB is the most common and aggressive primary brain cancer in adults. Despite the combined clinical therapy of surgical resection, radiotherapy and chemotherapy with the first-line agent temozolomide (TMZ), the prognosis is still unfavorable, with a median overall survival of 15 months and a high risk of recurrence (>90%) (1). This ability to resist chemo- and radiotherapy can be explained by the presence of a subpopulation of cells within the perivascular and hypoxic niches of the tumor known as GSCs or brain tumor-initiating cells. The subventricular zone (SVZ) is a neurogenic niche containing NSCs and progenitor cells and is suspected to be the origin of different brain tumor types due to the generation of GSCs (2–4). GSCs share functional characteristics with NSCs, including the capacity for self-renewal and long-term proliferation required to maintain and propagate the tumor, respectively (5). In addition, GSCs exhibit other properties of cancer cells, such as angiogenesis, invasion and immunosuppression, that promote disease progression and complicate treatment (6). Cells positive for stemness markers (e.g., CD133) have the ability to form tumors in vivo and oncospheres in vitro (reminiscent of neurosphere-derived NSCs) (6). In fact, understanding the hallmarks of GSCs can offer novel therapeutic strategies targeted at these cells to achieve an effective treatment for this disease.
The nucleosome is the structural unit of chromatin and is composed of 147 bp of DNA wrapped around an octamer of histones (H2A, H2B, H3, and H4). The chromatin organization and its degree of compaction are modulated by DNA and histone covalent modifications, ATP-dependent chromatin remodeling and certain non-coding RNAs (ncRNAs). Epigenetic mechanisms contribute to the cellular hierarchy of tumoral tissue in GB (7) and are crucial to understanding tumorigenesis and response to treatment in gliomas. For example, promoter hypermethylation of the O-6-methylguanine-DNA methyltransferase (MGMT) gene can predict good outcomes in TMZ treatment (8, 9). Additionally, mutations in arginine 132 of the tricarboxylic acid cycle component IDH1 (or in arginine 172 of IDH2), which are associated with longer survival, induce the overproduction of the 2-hydroxybutyrate metabolite that inhibits the α-ketoglutarate-dependent activity of epigenetic enzymes such as JumonjiC histone demethylases and TET hydroxymethylases, affecting both histone and DNA methylation (10).
The gene expression profiles of GSCs resemble those of normal NSCs (11, 12), but differential gene expression patterns between both types of cells can identify a transcriptional signature that is correlated with patient survival (13); however, copy number variations only explain a small portion of such gene expression alterations and other mechanisms (e.g., epigenetics) should be more relevant. For instance, changes in the patterns of DNA methylation, H3K27me3 and H3K4me3 are important in neural lineage differentiation (14–16), and a comparison of the genome-wide distribution of these and other epigenetic marks revealed important differences between GSCs and normal NSCs, affecting genes involved in neural differentiation and cancer processes (17, 18). These glioma-specific patterns of epigenetic marks can be found in DNA elements that are important for gene regulation:
- Bivalent promoters are considered a feature of embryonic stem cells (ESCs) due to their high prevalence in these cells (16, 19) and are characterized by the coexistence of epigenetic marks associated with active and repressed genes (generally H3K4me3 and H3K27me3). Genes under the control of such promoters are poised, i.e., maintained in silent state but ready to be activated under appropriate external or developmental stimuli (20). Genome-wide analyses identified a high diversity of bivalent regions within GSCs, which were shown to have significantly distinct patterns compared to NSCs and ESCs (17, 21). Loss of bivalency in GSCs affected a very low number of promoters but associated with the potential activation of proto-oncogenes and genes related to transcription, and the potential repression of genes linked to cell adhesion and ion channels (17). Moreover, consistent bivalent genes across several GSCs were members of the Wnt pathway and HOX family as well as potassium channels and solute carriers that can be associated with overall survival (21).
- Enhancers often regulate cell-specific gene expression and are defined by the simultaneous occupancy of H3K27ac and H3K4me1. Although enhancer patterns are relatively conserved between GSCs and NSCs, unique GSC patterns are mainly linked to genes with functions in DNA damage response, p53 signaling and angiogenesis; prominent examples are HOX cluster genes, which acquire enhancer histone modifications in GSCs and become highly expressed despite promoter methylation (22). In contrast, NSC-specific enhancers are more associated with stem cell differentiation, apoptosis and epigenetic regulation (22).
Overall, GSCs are characterized by an impairment of differentiation due to a permanent epigenetic block that maintains the self-renewal capacity of these cells (18, 23). Nonetheless, GSCs can rapidly adapt to diverse microenvironments by modulating their transcriptomes and DNA methylomes (24), indicating that such alterations are at least partially reversible, contrary to genetic variations. Reversibility of epigenetic marks was demonstrated in reprogramming experiments of glioma cells: with the appropriate combination of transcription factors they can be reversed into an early embryonic state that was accompanied by a widespread resetting of cancer-associated DNA methylation (23). Still, this resetting was not sufficient to abolish the malignant behavior of these cancer cells, indicating that we need to decipher how epigenetic-related activities work in GSCs to explaining their malignancy. In the following sections we review the experimental evidences found in GSCs about the role of epigenetics in malignancy and potential treatments.
The Polycomb repressive complexes, essential for normal developmental processes, have been the most studied epigenetic modulators in GSCs. The most relevant findings are summarized in Figure 1A. Polycomb repressive complex 2 (PRC2) is necessary for neurogenesis at the SVZ (25, 26) and regulates the trimethylation of H3K27 thanks to the catalytic activity of Enhancer of Zeste Homolog 2 (EZH2), which transfers a methyl group from S-adenosyl methionine, in cooperation with Suppressor of Zeste 12 (SUZ12) and Embryonic Ectoderm Development (EED). Overexpression of EZH2 has proto-oncogenic implications in several cancers, including glioma, in which elevated EZH2 expression has been associated with high-grade disease and poor overall survival (27, 28). Moreover, EZH2 activity is required for GSC maintenance by targeting MYC expression (29). Even in cells derived from diffuse intrinsic pontine gliomas (DIPG), a brain pediatric cancer that can also affect young adults, in which the actions of EZH2 are inhibited by the H3K27M mutation, residual EZH2 activity is still retained at strong PRC2 targets to drive GSC proliferation (30). Therefore, it is not surprising that selective EZH2 inhibition can constitute a promising therapeutic approach, as treated GSCs can reduce the levels of EZH2 and H3K27me3, cell proliferation and migration, the number and diameter of oncospheres, and the growth of intracranial xenotransplanted cells in mice, reverse epithelial-mesenchymal transition (EMT), potentiate the effects of TMZ and downregulate stem cell markers while increasing the expression of differentiation markers (29, 31–33).
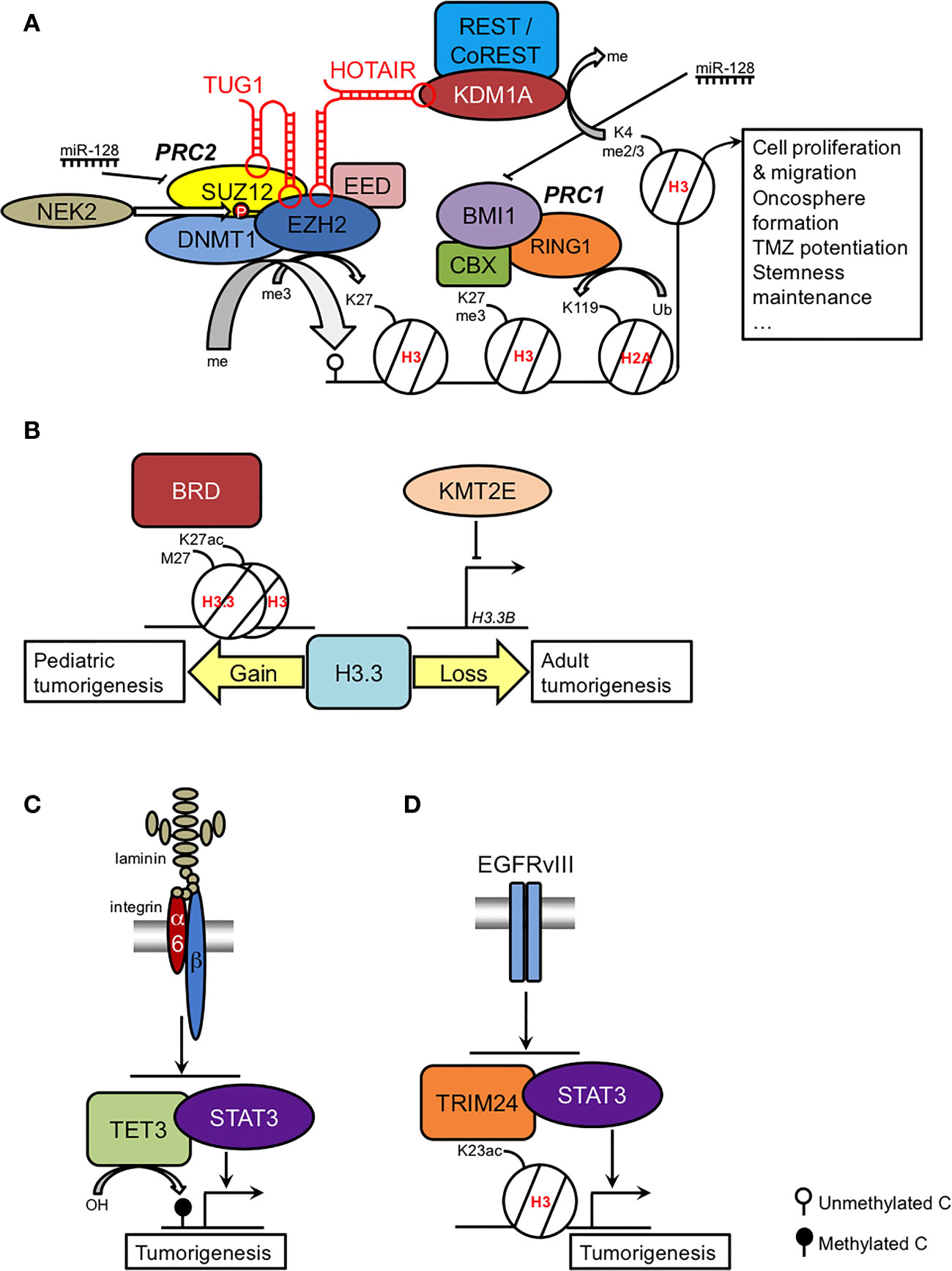
Figure 1 Summary of the epigenetic regulation of the GSC phenotype. (A) Coordinated actions and regulation of Polycomb complexes; (B) role of H3.3 in pediatric and adult GSCs as a result of gain and loss-of-function, respectively; (C) TET3-STAT regulation by the laminin-integrin signaling pathway; (D) H3K23ac-TRIM24-STAT regulation by the EGFRvIII signaling pathway. See main text for further details.
PRC2 activity is important for other epigenetic modifications. First, trimethylation of H3K27 is a prerequisite for histone H2A monoubiquitylation by Polycomb repressive complex 1 (PRC1) (34). Within this complex, the ring finger protein BMI1 is also a glioma stemness marker, and interference of its activity affects GSC malignancy in vitro and in xenotransplanted mice and enhances radiosensitivity (35–37). Second, EZH2 can recruit DNA methyltransferases (38), which explains the hypermethylation of PRC2 targets in primary GB (39, 40).
GSC characteristics display regional variations depending on the tumor niche. Whereas the regions defined by the disruption of the blood-brain barrier in angiogenesis foci were characterized by a high expression of proneural genes, an enrichment of EZH2/SUZ12/H3K27me3 targets and GSCs primarily positive to the proneural markers SOX2 and OLIG2, the hypoxic necrotic regions contained high expression of mesenchymal genes, a strong association with H2A119ub, an enrichment of BMI1 targets and GSCs primarily positive to the mesenchymal markers CD44 and YKL40 (41). Selective inhibition of either EZH2 or BMI1 was highly effective against the survival of proneural and mesenchymal GSCs, respectively. Thus, the combined strategy to abolish the activity of both PRCs can target different tumor compartments, increasing the efficacy of the therapy (41).
Research on GSCs is starting to disentangle EZH2-dependent oncogenic mechanisms. In certain GSCs, astroglial differentiation mediated by the bone morphogenic protein (BMP) and ciliary neurotrophic factor (CNTF) signaling pathways is impaired due to the silencing of the BMP receptor subtype gene BMPR1B by hypermethylation of its promoter, mediated by the EZH2-dependent recruitment of DNMT1 (42). Whereas incubation with BMP2 or CNTF can induce an increase in the differentiation markers GFAP or β-III tubulin in cultured NSCs and GSCs, in GSCs with impaired expression of BMPR1B, these trophic factors enhance proliferation (42). These pleiotropic actions are reminiscent of the role of the BMP signaling pathway in embryonic NSCs to promote either NSC proliferation or neuronal differentiation, depending on the expression of the BMP receptor subunit (43). To add more complexity to the EZH2 involvement in gliomagenesis, EZH2 can methylate non-histone proteins such as oncogenic STAT3. This association leads to enhanced activation of STAT3 to positively regulate GSC self-renewal and survival (44).
How PRC2 activity is deregulated in GSCs has been intensively explored. For instance, EZH2-dependent resistance of GSCs to radiotherapy can be explained by the transcriptional upregulation of EZH2 induced by maternal embryonic leucine zipper kinase (MELK) and activation of EZH2 through phosphorylation by NIMA-related kinase 2 (NEK2) (33, 45). Moreover, it has been proposed that dysfunction of miR-128 is an early event of gliomagenesis that increases the levels of both SUZ12 and BMI1, augmenting the histone modifications they regulate: H3K27me3 and H2AK119ub. These observations suggested a coordinated regulation of PRC1 and PRC2 activities. Therefore, restoring miR-128 expression diminishes proliferation and confers radiosensitivity (46). Additionally, EZH2 activity can be regulated by the lncRNA HOX transcript antisense RNA (HOTAIR), which is associated with poor survival in diverse cancers (47). In CD133+ cells, HOTAIR recruits both EZH2 and the lysine demethylase KDM1A/LSD1 to repress the tumor suppressor gene PDCD4 (48). In addition, another lncRNA, taurine upregulated gene 1 (TUG1), also binds to EZH2 and SUZ12 to repress neuronal differentiation genes such as BDNF, NGF, and NTF3 (49).
The histone H3 variant H3.3 can play a determinant role in pediatric GB. H3.3 is an independent replication variant that replaces the canonical histones H3.1 and H3.2 during brain development, becoming predominant in adulthood (50). H3.3 is encoded by two genes: H3F3A (H3.3A) and H3F3B (H3.3B). Mutations in H3F3A are present in approximately one-third of pediatric gliomas, affecting either lysine 27 (H3K27M) or glycine 34 (H3G34R/V), although the former mutation can also be found to a much lesser extent in the HIST1H3B (H3C2) gene (51–53). H3K27M is a relevant driver mutation in the pathogenesis of DIPG and is sufficient to immortalize NSCs from human embryo pons (54). In DIPG-derived cell lines, H3K27M specifically increases the acetylation of H3K27 and creates heterotopic H3K27M/H3K27ac nucleosomes that can be targeted by inhibitors of bromodomain (BRD) proteins to modulate the expression of the cell cycle arrest gene CDKN1A, the neuronal mature markers TUBB3 and MAP2, and the Zn finger protein GLI2 (55), a relevant downstream target of the Sonic Hedgehog pathway that is implicated in the etiology of DIPG (56) (Figure 1B).
In adult GB, dominant negative mutations in histone H3 are extremely rare. Instead, downregulation of the H3F3B gene has been reported to lead to a deficit of H3.3 function in GSCs as a result of the action of the lysine methyltransferase KMT2E (myeloid/lymphoid leukemia MLL5), maintaining the self-renewal capacity of GSCs and interfering with their differentiation (57) (Figure 1B). These findings suggest that H3.3 impairment in adult GB may produce similar chromatin rearrangements as the H3.3 mutation in pediatric GB, given the similar DNA methylation patterns in both types of tumors (57).
In addition to PRCs and H3.3, other epigenetic-related factors have been implicated in the GSC phenotype and are listed in Table 1.
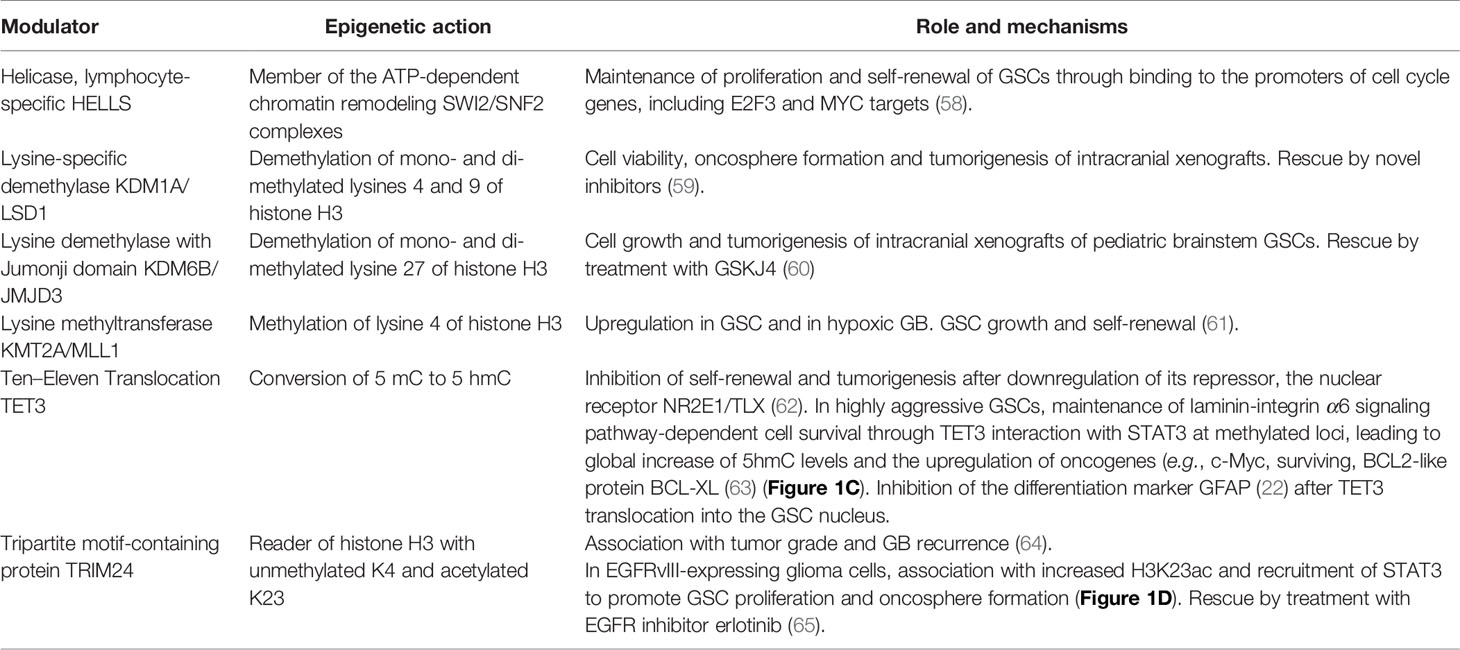
Table 1 List of other epigenetic-related factors in GSC studies.
Considering that altered gene expression levels have been reported for histone deacetylases (HDACs) in GB (66, 67), most therapeutic approaches have been focused on histone deacetylase inhibitors (HDACis) due to their recognized antiproliferative effects in multiple cancer models and their benefits and tolerability in the amelioration of several neurological conditions in vivo at the preclinical stage; in addition, some of these compounds have been approved as therapeutic agents in other types of cancers. Histone acetylation is regulated by the opposing enzymatic activities of lysine acetyltransferases and HDACs: whereas the former enzymes transfer the acetyl group from an acetyl-CoA molecule to the lysines of the protruding histone tails (an activity that is associated with active genes), HDACs catalyze this removal, which is associated with gene repression. Inhibition of HDACs can induce cell cycle arrest, apoptosis and cellular differentiation and can interfere with cancer angiogenesis (68). One interesting target of HDACis is the phosphatase DUSP1, an inhibitor of the JNK, ERK1/2 and p38 MAPK pathways that is associated with GSC differentiation and good prognosis (69).
Among the tested HDACis in clinical trials, vorinostat/SAHA, romidepsin/FK228/FR901228 and panobinostat/LBH-589 demonstrated very limited efficacy as therapeutic agents in single therapies in both newly diagnosed and recurrent GB. However, the most promising effect of HDACis is as sensitizers to current therapeutic approaches such as radiotherapy and TMZ therapy [see (70) for a review on this topic]. Some considerations should be kept in mind to understand the potential benefits and limitations of HDACi-based treatment in vivo. First, acetylation increase by HDACis is not exclusive of histones (71); second, antineoplastic actions of HDACis can be achieved independently of (or in addition to) HDAC inhibition (72); third, the solubility of HDACis in water is usually poor, resulting in inefficient transport through the blood-brain barrier with oral administration (73); last, chemoresistance has been reported in long-term treatments (74). In any case, the prospects of using HDACis are still promising, and research on GSCs can help in elucidating the underlying anticancer mechanisms of HDAC inhibition and in proposing novel formulations to improve drug delivery (e.g., loading these hydrophobic compounds into nanomicelles) (75). Efforts are being mainly focused on valproic acid (VPA), with proven antitumoral effects (72, 76, 77). Often administered as an anticonvulsant agent to treat epilepsy in brain tumors (78), retrospective clinical studies reported that treatment with this compound increased the overall survival of GB patients (79) although this effect was not found in other reports and still remains controversial (80, 81). VPA is capable of inducing a predifferentiation state in GSCs (74) and can be combined with other antineoplastic compounds for synergistic effects, as reported for the antimitotic paclitaxel (82). However, VPA failed to sensitize GSCs to TMZ (74), although another study reported sensitization to both TMZ and nimustine (ACNU), especially in MGMT-expressing cells (83). VPA is able to modify the DNA methylomes of GSCs (74), leading to the activation of the Wnt/β-catenin pathway which was related with growth inhibition, reduced migration and EMT impairment (84). This is in conflict with the suppression of the Wnt/β-catenin pathway by SAHA, which partially rescues the downregulation of histocompatibility complex class I and antigen-processing machinery genes, as a plausible strategy to potentiate the activation of cytotoxic T cells in vivo (85). Side effects have also been reported as VPA can exacerbate the unfolded protein response program, leading to protein homeostasis dysregulation and proteostasis stress in GSCs (86).
Research on the epigenetics of GSCs has the potential to elucidate the self-renewal and perpetuation mechanisms of these cells through the identification of the epigenetic program that governs aberrant gene activation and repression in cancer. Less known epigenetic modifications should be further explored, as they can provide further insights into tumorigenesis, as in the case of 5’-formylcytosine (5fC) and 5′-carboxylcytosine (5caC) (22). In addition, a systematic and detailed description of direct target genes of epigenetic activities is required to understand the complex mechanisms of epigenetic dysregulation in gliomas. As we have seen through this review, multiple epigenetic activities can be involved in glioma malignancy in a complex manner; therefore, the simultaneous modulation of various epigenetic activities may be highly effective, as demonstrated by the dual inhibition of HDACs and KDM1A/LSD1 (87, 88).
All authors contributed to the article and approved the submitted version.
LV is supported by the Plan Propio INiBICA (Grant L19-07IN-CO07) and by the Programa Estatal de Generación de Conocimiento y Fortalecimiento del Sistema Español de I+D+i, financed by the Instituto de Salud Carlos III and Fondo Europeo de Desarrollo Regional 2014-2020 (Grants CP15/00180 and PI16/00722). LV is the recipient of a Miguel Servet I contract (CP15/00180) financed by the Instituto de Salud Carlos III and Fondo Social Europeo 2014-2020, Programa Estatal de Promoción del Talento y su empleabilidad en I+D+i.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
BMP, Bone morphogenic protein; BRD, Brodomain; CNTF, Ciliary neurotrophic factor; DIPG, Diffuse intrinsic pontine gliomas; EED, Embryonic ectoderm development; EMT, Epithelial–mesenchymal transition; ESC, Embryonic stem cell; EZH2, Enhancer of Zeste homolog 2; GB, Glioblastoma; GSC, Glioma stem cell; HDAC, Histone deacetylase; HDACi, HDAC inhibitor; HOTAIR, HOX transcript antisense RNA; KAT, Lysine acetyltransferase; lncRNA, Long Non-coding RNA; MELK, Maternal embryonic leucine zipper kinase; MGMT, O-6-methylguanine-DNA methyltransferase; NEK2, NIMA-related kinase 2; NSC, Neural stem cell; PRC1/2, Polycomb repressive complex 1/2; SUZ12, Suppressor of Zeste 12; SVZ, subventricular zone; TMZ, Temozolomide; TUG1, Taurine upregulated gene 1; VPA, Valproic acid.
1. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol (2009) 10(5):459–66. doi: 10.1016/S1470-2045(09)70025-7
2. Lim DA, Cha S, Mayo MC, Chen MH, Keles E, VandenBerg S, et al. Relationship of glioblastoma multiforme to neural stem cell regions predicts invasive and multifocal tumor phenotype. Neuro Oncol (2007) 9(4):424–9. doi: 10.1215/15228517-2007-023
3. Gilbertson RJ, Gutmann DH. Tumorigenesis in the brain: location, location, location. Cancer Res (2007) 67(12):5579–82. doi: 10.1158/0008-5472.CAN-07-0760
4. Haskins WE, Zablotsky BL, Foret MR, Ihrie RA, Alvarez-Buylla A, Eisenman RN, et al. Molecular Characteristics in MRI-Classified Group 1 Glioblastoma Multiforme. Front Oncol (2013) 3:182. doi: 10.3389/fonc.2013.00182
5. Vescovi AL, Galli R, Reynolds BA. Brain tumour stem cells. Nat Rev Cancer (2006) 6(6):425–36. doi: 10.1038/nrc1889
6. Heddleston JM, Hitomi M, Venere M, Flavahan WA, Yang K, Kim Y, et al. Glioma stem cell maintenance: the role of the microenvironment. Curr Pharm Des (2011) 17(23):2386–401. doi: 10.2174/138161211797249260
7. Suva ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science (2013) 339(6127):1567–70. doi: 10.1126/science.1230184
8. Thon N, Kreth S, Kreth FW. Personalized treatment strategies in glioblastoma: MGMT promoter methylation status. Onco Targets Ther (2013) 6:1363–72. doi: 10.2147/OTT.S50208
9. Dahlrot RH, Dowsett J, Fosmark S, Malmstrom A, Henriksson R, Boldt H, et al. Prognostic value of O-6-methylguanine-DNA methyltransferase (MGMT) protein expression in glioblastoma excluding nontumour cells from the analysis. Neuropathol Appl Neurobiol (2018) (442):172–84. doi: 10.1111/nan.12415
10. Chang S, Yim S, Park H. The cancer driver genes IDH1/2, JARID1C/ KDM5C, and UTX/ KDM6A: crosstalk between histone demethylation and hypoxic reprogramming in cancer metabolism. Exp Mol Med (2019) 51(6):1–17. doi: 10.1038/s12276-019-0230-6
11. Stangeland B, Mughal AA, Grieg Z, Sandberg CJ, Joel M, Nygard S, et al. Combined expressional analysis, bioinformatics and targeted proteomics identify new potential therapeutic targets in glioblastoma stem cells. Oncotarget (2015) 6(28):26192–215. doi: 10.18632/oncotarget.4613
13. Engstrom PG, Tommei D, Stricker SH, Ender C, Pollard SM, Bertone P. Digital transcriptome profiling of normal and glioblastoma-derived neural stem cells identifies genes associated with patient survival. Genome Med (2012) 4(10):76. doi: 10.1186/gm377
14. Takizawa T, Nakashima K, Namihira M, Ochiai W, Uemura A, Yanagisawa M, et al. DNA methylation is a critical cell-intrinsic determinant of astrocyte differentiation in the fetal brain. Dev Cell (2001) 1(6):749–58. doi: 10.1016/S1534-5807(01)00101-0
15. Jepsen K, Solum D, Zhou T, McEvilly RJ, Kim HJ, Glass CK, et al. SMRT-mediated repression of an H3K27 demethylase in progression from neural stem cell to neuron. Nature (2007) 450(7168):415–9. doi: 10.1038/nature06270
16. Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature (2007) 448(7153):553–60. doi: 10.1038/nature06008
17. Yoo S, Bieda MC. Differences among brain tumor stem cell types and fetal neural stem cells in focal regions of histone modifications and DNA methylation, broad regions of modifications, and bivalent promoters. BMC Genomics (2014) 15:724. doi: 10.1186/1471-2164-15-724
18. Baronchelli S, Bentivegna A, Redaelli S, Riva G, Butta V, Paoletta L, et al. Delineating the cytogenomic and epigenomic landscapes of glioma stem cell lines. PLoS One (2013) 8(2):e57462. doi: 10.1371/journal.pone.0057462
19. Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell (2006) 125(2):315–26. doi: 10.1016/j.cell.2006.02.041
20. Vastenhouw NL, Zhang Y, Woods IG, Imam F, Regev A, Liu XS, et al. Chromatin signature of embryonic pluripotency is established during genome activation. Nature (2010) 464(7290):922–6. doi: 10.1038/nature08866
21. Lin B, Lee H, Yoon JG, Madan A, Wayner E, Tonning S, et al. Global analysis of H3K4me3 and H3K27me3 profiles in glioblastoma stem cells and identification of SLC17A7 as a bivalent tumor suppressor gene. Oncotarget (2015) 6(7):5369–81. doi: 10.18632/oncotarget.3030
22. Zhou D, Alver BM, Li S, Hlady RA, Thompson JJ, Schroeder MA, et al. Distinctive epigenomes characterize glioma stem cells and their response to differentiation cues. Genome Biol (2018) 19(1):43. doi: 10.1186/s13059-018-1420-6
23. Stricker SH, Feber A, Engstrom PG, Caren H, Kurian KM, Takashima Y, et al. Widespread resetting of DNA methylation in glioblastoma-initiating cells suppresses malignant cellular behavior in a lineage-dependent manner. Genes Dev (2013) 27(6):654–69. doi: 10.1101/gad.212662.112
24. Baysan M, Woolard K, Bozdag S, Riddick G, Kotliarova S, Cam MC, et al. Micro-environment causes reversible changes in DNA methylation and mRNA expression profiles in patient-derived glioma stem cells. PLoS One (2014) 9(4):e94045. doi: 10.1371/journal.pone.0094045
25. Hwang WW, Salinas RD, Siu JJ, Kelley KW, Delgado RN, Paredes MF, et al. Distinct and separable roles for EZH2 in neurogenic astroglia. Elife (2014) 3:e02439. doi: 10.7554/eLife.02439
26. Sun B, Chang E, Gerhartl A, Szele FG. Polycomb Protein Eed is Required for Neurogenesis and Cortical Injury Activation in the Subventricular Zone. Cereb Cortex (2018) 28(4):1369–82. doi: 10.1093/cercor/bhx289
27. Zhang J, Chen L, Han L, Shi Z, Pu P, Kang C. EZH2 is a negative prognostic factor and exhibits pro-oncogenic activity in glioblastoma. Cancer Lett (2015) 356(2 Pt B):929–36. doi: 10.1016/j.canlet.2014.11.003
28. Sharma V, Malgulwar PB, Purkait S, Patil V, Pathak P, Agrawal R, et al. Genome-wide ChIP-seq analysis of EZH2-mediated H3K27me3 target gene profile highlights differences between low- and high-grade astrocytic tumors. Carcinogenesis (2017) 38(2):152–61. doi: 10.1093/carcin/bgw126
29. Suva ML, Riggi N, Janiszewska M, Radovanovic I, Provero P, Stehle JC, et al. EZH2 is essential for glioblastoma cancer stem cell maintenance. Cancer Res (2009) 69(24):9211–8. doi: 10.1158/0008-5472.CAN-09-1622
30. Mohammad F, Weissmann S, Leblanc B, Pandey DP, Hojfeldt JW, Comet I, et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med (2017) 23(4):483–92. doi: 10.1038/nm.4293
31. Stazi G, Taglieri L, Nicolai A, Romanelli A, Fioravanti R, Morrone S, et al. Dissecting the role of novel EZH2 inhibitors in primary glioblastoma cell cultures: effects on proliferation, epithelial-mesenchymal transition, migration, and on the pro-inflammatory phenotype. Clin Epigenet (2019) 11(1):173. doi: 10.1186/s13148-019-0763-5
32. Yu T, Wang Y, Hu Q, Wu W, Wu Y, Wei W, et al. The EZH2 inhibitor GSK343 suppresses cancer stem-like phenotypes and reverses mesenchymal transition in glioma cells. Oncotarget (2017) 8(58):98348–59. doi: 10.18632/oncotarget.21311
33. Wang J, Cheng P, Pavlyukov MS, Yu H, Zhang Z, Kim SH, et al. Targeting NEK2 attenuates glioblastoma growth and radioresistance by destabilizing histone methyltransferase EZH2. J Clin Invest (2017) 127(8):3075–89. doi: 10.1172/JCI89092
34. Stock JK, Giadrossi S, Casanova M, Brookes E, Vidal M, Koseki H, et al. Ring1-mediated ubiquitination of H2A restrains poised RNA polymerase II at bivalent genes in mouse ES cells. Nat Cell Biol (2007) 9(12):1428–35. doi: 10.1038/ncb1663
35. Abdouh M, Facchino S, Chatoo W, Balasingam V, Ferreira J, Bernier G. BMI1 sustains human glioblastoma multiforme stem cell renewal. J Neurosci (2009) 29(28):8884–96. doi: 10.1523/JNEUROSCI.0968-09.2009
36. Facchino S, Abdouh M, Chatoo W, Bernier G. BMI1 confers radioresistance to normal and cancerous neural stem cells through recruitment of the DNA damage response machinery. J Neurosci (2010) 30(30):10096–111. doi: 10.1523/JNEUROSCI.1634-10.2010
37. Godlewski J, Nowicki MO, Bronisz A, Williams S, Otsuki A, Nuovo G, et al. Targeting of the Bmi-1 oncogene/stem cell renewal factor by microRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res (2008) 68(22):9125–30. doi: 10.1158/0008-5472.CAN-08-2629
38. Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature (2006) 439(7078):871–4. doi: 10.1038/nature04431
39. Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, et al. Epigenetic stem cell signature in cancer. Nat Genet (2007) 39(2):157–8. doi: 10.1038/ng1941
40. Martinez R, Martin-Subero JI, Rohde V, Kirsch M, Alaminos M, Fernandez AF, et al. A microarray-based DNA methylation study of glioblastoma multiforme. Epigenetics (2009) 4(4):255–64. doi: 10.4161/epi.9130
41. Jin X, Kim LJY, Wu Q, Wallace LC, Prager BC, Sanvoranart T, et al. Targeting glioma stem cells through combined BMI1 and EZH2 inhibition. Nat Med (2017) 23(11):1352–61. doi: 10.1038/nm.4415
42. Lee J, Son MJ, Woolard K, Donin NM, Li A, Cheng CH, et al. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell (2008) 13(1):69–80. doi: 10.1016/j.ccr.2007.12.005
43. Hall AK, Miller RH. Emerging roles for bone morphogenetic proteins in central nervous system glial biology. J Neurosci Res (2004) 76(1):1–8. doi: 10.1002/jnr.20019
44. Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell (2013) 23(6):839–52. doi: 10.1016/j.ccr.2013.04.008
45. Kim SH, Joshi K, Ezhilarasan R, Myers TR, Siu J, Gu C, et al. EZH2 protects glioma stem cells from radiation-induced cell death in a MELK/FOXM1-dependent manner. Stem Cell Rep (2015) 4(2):226–38. doi: 10.1016/j.stemcr.2014.12.006
46. Peruzzi P, Bronisz A, Nowicki MO, Wang Y, Ogawa D, Price R, et al. MicroRNA-128 coordinately targets Polycomb Repressor Complexes in glioma stem cells. Neuro Oncol (2013) 15(9):1212–24. doi: 10.1093/neuonc/not055
47. Wang L, He Z. Functional Roles of Long Non-Coding RNAs (LncRNAs) in Glioma Stem Cells. Med Sci Monit (2019) 25:7567–73. doi: 10.12659/MSM.916040
48. Fang K, Liu P, Dong S, Guo Y, Cui X, Zhu X, et al. Magnetofection based on superparamagnetic iron oxide nanoparticle-mediated low lncRNA HOTAIR expression decreases the proliferation and invasion of glioma stem cells. Int J Oncol (2016) 49(2):509–18. doi: 10.3892/ijo.2016.3571
49. Katsushima K, Natsume A, Ohka F, Shinjo K, Hatanaka A, Ichimura N, et al. Targeting the Notch-regulated non-coding RNA TUG1 for glioma treatment. Nat Commun (2016) 7:13616. doi: 10.1038/ncomms13616
50. Maze I, Wenderski W, Noh KM, Bagot RC, Tzavaras N, Purushothaman I, et al. Critical Role of Histone Turnover in Neuronal Transcription and Plasticity. Neuron (2015) 87(1):77–94. doi: 10.1016/j.neuron.2015.06.014
51. Lowe BR, Maxham LA, Hamey JJ, Wilkins MR, Partridge JF. Histone H3 Mutations: An Updated View of Their Role in Chromatin Deregulation and Cancer. Cancers (Basel) (2019) 11(5):660. doi: 10.3390/cancers11050660
52. Castel D, Philippe C, Calmon R, Le Dret L, Truffaux N, Boddaert N, et al. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol (2015) 130(6):815–27. doi: 10.1007/s00401-015-1478-0
53. Jones C, Baker SJ. Unique genetic and epigenetic mechanisms driving paediatric diffuse high-grade glioma. Nat Rev Cancer (2014) 14(10):651–61. doi: 10.1038/nrc3811
54. Sun Y, Xu C, Pan C, Chen X, Geng Y, Wu Y, et al. Diffuse Intrinsic Pontine Gliomas Exhibit Cell Biological and Molecular Signatures of Fetal Hindbrain-Derived Neural Progenitor Cells. Neurosci Bull (2019) 35(2):216–24. doi: 10.1007/s12264-018-00329-6
55. Piunti A, Hashizume R, Morgan MA, Bartom ET, Horbinski CM, Marshall SA, et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat Med (2017) 23(4):493–500. doi: 10.1038/nm.4296
56. Monje M, Mitra SS, Freret ME, Raveh TB, Kim J, Masek M, et al. Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc Natl Acad Sci U S A (2011) 108(11):4453–8. doi: 10.1073/pnas.1101657108
57. Gallo M, Coutinho FJ, Vanner RJ, Gayden T, Mack SC, Murison A, et al. MLL5 Orchestrates a Cancer Self-Renewal State by Repressing the Histone Variant H3.3 and Globally Reorganizing Chromatin. Cancer Cell (2015) 28(6):715–29. doi: 10.1016/j.ccell.2015.10.005
58. Zhang G, Dong Z, Prager BC, Kim LJ, Wu Q, Gimple RC, et al. Chromatin remodeler HELLS maintains glioma stem cells through E2F3 and MYC. JCI Insight (2019) 4(7):e126140. doi: 10.1172/jci.insight.126140
59. Sareddy GR, Viswanadhapalli S, Surapaneni P, Suzuki T, Brenner A, Vadlamudi RK. Novel KDM1A inhibitors induce differentiation and apoptosis of glioma stem cells via unfolded protein response pathway. Oncogene (2017) 36(17):2423–34. doi: 10.1038/onc.2016.395
60. Hashizume R, Andor N, Ihara Y, Lerner R, Gan H, Chen X, et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat Med (2014) 20(12):1394–6. doi: 10.1038/nm.3716
61. Heddleston JM, Wu Q, Rivera M, Minhas S, Lathia JD, Sloan AE, et al. Hypoxia-induced mixed-lineage leukemia 1 regulates glioma stem cell tumorigenic potential. Cell Death Differ (2012) 19(3):428–39. doi: 10.1038/cdd.2011.109
62. Qu Q, Sun G, Li W, Yang S, Ye P, Zhao C, et al. Orphan nuclear receptor TLX activates Wnt/beta-catenin signalling to stimulate neural stem cell proliferation and self-renewal. Nat Cell Biol (2010) 12(1):31–40; sup pp 1-9. doi: 10.1038/ncb2001
63. Herrmann A, Lahtz C, Song J, Aftabizadeh M, Cherryholmes GA, Xin H, et al. Integrin alpha6 signaling induces STAT3-TET3-mediated hydroxymethylation of genes critical for maintenance of glioma stem cells. Oncogene (2020) 39(10):2156–69. doi: 10.1038/s41388-019-1134-6
64. Zhang LH, Yin AA, Cheng JX, Huang HY, Li XM, Zhang YQ, et al. TRIM24 promotes glioma progression and enhances chemoresistance through activation of the PI3K/Akt signaling pathway. Oncogene (2015) 34(5):600–10. doi: 10.1038/onc.2013.593
65. Lv D, Li Y, Zhang W, Alvarez AA, Song L, Tang J, et al. TRIM24 is an oncogenic transcriptional co-activator of STAT3 in glioblastoma. Nat Commun (2017) 8(1):1454. doi: 10.1038/s41467-017-01731-w
66. Lucio-Eterovic AK, Cortez MA, Valera ET, Motta FJ, Queiroz RG, Machado HR, et al. Differential expression of 12 histone deacetylase (HDAC) genes in astrocytomas and normal brain tissue: class II and IV are hypoexpressed in glioblastomas. BMC Cancer (2008) 8:243. doi: 10.1186/1471-2407-8-243
67. Lee P, Murphy B, Miller R, Menon V, Banik NL, Giglio P, et al. Mechanisms and clinical significance of histone deacetylase inhibitors: epigenetic glioblastoma therapy. Anticancer Res (2015) 35(2):615–25.
68. Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int J Mol Sci (2017) 18(7):1414. doi: 10.3390/ijms18071414
69. Arrizabalaga O, Moreno-Cugnon L, Auzmendi-Iriarte J, Aldaz P, Ibanez de Caceres I, Garros-Regulez L, et al. High expression of MKP1/DUSP1 counteracts glioma stem cell activity and mediates HDAC inhibitor response. Oncogenesis (2017) 6(12):401. doi: 10.1038/s41389-017-0003-9
70. Williams MJ, Singleton WG, Lowis SP, Malik K, Kurian KM. Therapeutic Targeting of Histone Modifications in Adult and Pediatric High-Grade Glioma. Front Oncol (2017) 7:45. doi: 10.3389/fonc.2017.00045
71. Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science (2009) 325(5942):834–40. doi: 10.1126/science.1175371
72. Blaheta RA, Cinatl J Jr. Anti-tumor mechanisms of valproate: a novel role for an old drug. Med Res Rev (2002) 22(5):492–511. doi: 10.1002/med.10017
73. Rasmussen TA, Tolstrup M, Moller HJ, Brinkmann CR, Olesen R, Erikstrup C, et al. Activation of latent human immunodeficiency virus by the histone deacetylase inhibitor panobinostat: a pilot study to assess effects on the central nervous system. Open Forum Infect Dis (2015) 2(1):ofv037. doi: 10.1093/ofid/ofv037
74. Riva G, Butta V, Cilibrasi C, Baronchelli S, Redaelli S, Dalpra L, et al. Epigenetic targeting of glioma stem cells: Short-term and long-term treatments with valproic acid modulate DNA methylation and differentiation behavior, but not temozolomide sensitivity. Oncol Rep (2016) 35(5):2811–24. doi: 10.3892/or.2016.4665
75. Singleton WG, Collins AM, Bienemann AS, Killick-Cole CL, Haynes HR, Asby DJ, et al. Convection enhanced delivery of panobinostat (LBH589)-loaded pluronic nano-micelles prolongs survival in the F98 rat glioma model. Int J Nanomed (2017) 12:1385–99. doi: 10.2147/IJN.S125300
76. Osuka S, Takano S, Watanabe S, Ishikawa E, Yamamoto T, Matsumura A. Valproic acid inhibits angiogenesis in vitro and glioma angiogenesis in vivo in the brain. Neurol Med Chir (Tokyo) (2012) 52(4):186–93. doi: 10.2176/nmc.52.186
77. Chen Y, Tsai YH, Tseng SH. Valproic acid affected the survival and invasiveness of human glioma cells through diverse mechanisms. J Neurooncol (2012) 109(1):23–33. doi: 10.1007/s11060-012-0871-y
78. Maschio M, Aguglia U, Avanzini G, Banfi P, Buttinelli C, Capovilla G, et al. Management of epilepsy in brain tumors. Neurol Sci (2019) 40(10):2217–34. doi: 10.1007/s10072-019-04025-9
79. Ruda R, Pellerino A, Soffietti R. Does valproic acid affect tumor growth and improve survival in glioblastomas? CNS Oncol (2016) 5(2):51–3. doi: 10.2217/cns-2016-0004
80. Lu VM, Texakalidis P, McDonald KL, Mekary RA, Smith TR. The survival effect of valproic acid in glioblastoma and its current trend: a systematic review and meta-analysis. Clin Neurol Neurosurg (2018) 174:149–55. doi: 10.1016/j.clineuro.2018.09.019
81. Happold C, Gorlia T, Chinot O, Gilbert MR, Nabors LB, Wick W, et al. Does Valproic Acid or Levetiracetam Improve Survival in Glioblastoma? A Pooled Analysis of Prospective Clinical Trials in Newly Diagnosed Glioblastoma. J Clin Oncol (2016) 34(7):731–9. doi: 10.1200/JCO.2015.63.6563
82. Riva G, Baronchelli S, Paoletta L, Butta V, Biunno I, Lavitrano M, et al. In vitro anticancer drug test: A new method emerges from the model of glioma stem cells. Toxicol Rep (2014) 1:188–99. doi: 10.1016/j.toxrep.2014.05.005
83. Li Z, Xia Y, Bu X, Yang D, Yuan Y, Guo X, et al. Effects of valproic acid on the susceptibility of human glioma stem cells for TMZ and ACNU. Oncol Lett (2018) 15(6):9877–83. doi: 10.3892/ol.2018.8551
84. Riva G, Cilibrasi C, Bazzoni R, Cadamuro M, Negroni C, Butta V, et al. Valproic Acid Inhibits Proliferation and Reduces Invasiveness in Glioma Stem Cells Through Wnt/beta Catenin Signalling Activation. Genes (Basel) (2018) 9(11):522. doi: 10.3390/genes9110522
85. Yang W, Li Y, Gao R, Xiu Z, Sun T. MHC class I dysfunction of glioma stem cells escapes from CTL-mediated immune response via activation of Wnt/beta-catenin signaling pathway. Oncogene (2020) 39(5):1098–111. doi: 10.1038/s41388-019-1045-6
86. Cattaneo M, Baronchelli S, Schiffer D, Mellai M, Caldera V, Saccani GJ, et al. Down-modulation of SEL1L, an unfolded protein response and endoplasmic reticulum-associated degradation protein, sensitizes glioma stem cells to the cytotoxic effect of valproic acid. J Biol Chem (2014) 289(5):2826–38. doi: 10.1074/jbc.M113.527754
87. Singh MM, Manton CA, Bhat KP, Tsai WW, Aldape K, Barton MC, et al. Inhibition of LSD1 sensitizes glioblastoma cells to histone deacetylase inhibitors. Neuro Oncol (2011) 13(8):894–903. doi: 10.1093/neuonc/nor049
Keywords: glioblastoma, histone, DNA, methylation, acetylation, Polycomb, H3.3, HDACi
Citation: Valor LM and Hervás-Corpión I (2020) The Epigenetics of Glioma Stem Cells: A Brief Overview. Front. Oncol. 10:602378. doi: 10.3389/fonc.2020.602378
Received: 03 September 2020; Accepted: 29 October 2020;
Published: 02 December 2020.
Edited by:
Hugo Guerrero-Cazares, Mayo Clinic, United StatesReviewed by:
Takashi Shingu, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2020 Valor and Hervás-Corpión. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luis M. Valor, bHVpc20udmFsb3JAaW5pYmljYS5lcw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.