 Bárbara Paranhos Coelho†
Bárbara Paranhos Coelho† Camila Felix de Lima Fernandes†
Camila Felix de Lima Fernandes† Jacqueline Marcia Boccacino
Jacqueline Marcia Boccacino Maria Clara da Silva Souza
Maria Clara da Silva Souza Maria Isabel Melo-Escobar
Maria Isabel Melo-Escobar Rodrigo Nunes Alves
Rodrigo Nunes Alves Mariana Brandão PradoRebeca Piatniczka Iglesia
Mariana Brandão PradoRebeca Piatniczka Iglesia Giovanni CangianoGiulia La Rocca Mazzaro
Giovanni CangianoGiulia La Rocca Mazzaro Marilene Hohmuth Lopes*
Marilene Hohmuth Lopes*- Laboratory of Neurobiology and Stem Cells, Institute of Biomedical Sciences, Department of Cell and Developmental Biology, University of São Paulo, São Paulo, Brazil
Tumor cells can employ epithelial-mesenchymal transition (EMT) or autophagy in reaction to microenvironmental stress. Importantly, EMT and autophagy negatively regulate each other, are able to interconvert, and both have been shown to contribute to drug-resistance in glioblastoma (GBM). EMT has been considered one of the mechanisms that confer invasive properties to GBM cells. Autophagy, on the other hand, may show dual roles as either a GBM-promoter or GBM-suppressor, depending on microenvironmental cues. The Wingless (WNT) signaling pathway regulates a plethora of developmental and biological processes such as cellular proliferation, adhesion and motility. As such, GBM demonstrates deregulation of WNT signaling in favor of tumor initiation, proliferation and invasion. In EMT, WNT signaling promotes induction and stabilization of different EMT activators. WNT activity also represses autophagy, while nutrient deprivation induces β-catenin degradation via autophagic machinery. Due to the importance of the WNT pathway to GBM, and the role of WNT signaling in EMT and autophagy, in this review we highlight the effects of the WNT signaling in the regulation of both processes in GBM, and discuss how the crosstalk between EMT and autophagy may ultimately affect tumor biology.
Introduction
Autophagy and epithelial-mesenchymal transition (EMT) are cellular processes that present an intricate correlation, being associated with the development and progression of different tumors. Particularly, autophagy is an essential process for cell survival in healthy tissues and in several types of cancer. Autophagy allows tumor cells to obtain energy from their own cellular structures, consequently turning cell components into possible energy and nutrient reserves, preserving metabolic rates (1–3). It directly contributes to resistance to stress in the tumor microenvironment that is associated with hypoxia, nutritional deficit, acidosis or genotoxicity (3–6). Additionally, autophagy is able to interconvert with EMT, that, in turn, is a cellular process in which the epithelial cells lose their cell-cell junction proteins, apical-basal polarity, and interaction with the basement membrane to acquire mesenchymal cell characteristics. This leads to functional changes and increased migratory capacity with invasive properties (7, 8). The crosstalk between autophagy and EMT, as well as all different molecular pathways involved in these processes, is of great interest in physiological and pathological contexts, especially in cancer.
In tumor cells, EMT is activated in metastasis and invasion, which allows cells to detach from the basement membrane and connect with other cells, extravasating from its initial location and invading adjacent tissues (6, 9). However, in some types of cancer, such as glioblastoma (GBM), EMT appears to be favored by downregulation of autophagic processes (10), and a better comprehension of this entangled regulation might help to clarify GBM biology.
GBM is the most common and malignant glioma, classified as a grade IV brain tumor derived from glial cells (11). Besides high mitotic activity, microvascular proliferation, necrosis, cellular polymorphism, and substantial infiltrative capacity, this tumor is highly heterogeneous and is mainly characterized by resistance to treatment (12, 13). The standard treatment for newly diagnosed tumors is surgical resection with radiation, followed by chemotherapy with temozolomide (TMZ) (13). Along with radiotherapy and TMZ, bevacizumab, a monoclonal antibody against vascular endothelial growth factor (VEGF), and lomustine, an alkylating agent, are current options for recurrent GBM (14). However, a high recurrence rate and failure to respond to therapy lead patients to an average survival time of 15 months (15, 16). A subpopulation of GBM cells with stem-like features termed glioblastoma stem cells (GSCs) is characterized by self-renewal and differentiation into distinct neural cell types, which greatly contributes to the intratumor heterogeneity of GBM (17). GSCs are associated with GBM maintenance, progression and resistance to therapy, contributing to the highly aggressive phenotype (18, 19).
Several signaling pathways upregulated in GBM are involved in cell survival, growth and invasiveness that sustain tumor development and confer resistance to therapy and to harsh microenvironments (20). Among these pathways, current literature suggests that aberrant activation of the Wingless (WNT) signaling contributes to GBM pathology through different cell processes, such as proliferation (21, 22), motility (23–25), cell fate specification (26), and maintenance of stemness properties (27, 28). Interestingly, WNT signaling pathway emerged as a pivotal player to mediate the crosstalk between autophagy and EMT, regulating molecules and connecting to other important signal transduction cascades, such as the mTOR signaling pathway.
Mammalian target of rapamycin (mTOR) signaling pathway is also upregulated in GBM (29). mTOR can establish two distinct multiprotein complexes: mTORC1, that promotes its effects downstream of PI3K/AKT signaling, regulating a diverse variety of cellular processes such as glucose, lipid and nucleotide metabolism, protein biosynthesis and degradation (30); and mTORC2, responsible for phosphorylation of AKT on Ser473, which stimulates its maximal activation (31). In GBM, the PTEN/PI3K/AKT/mTOR pathway is one of the most deregulated, contributing to tumor development and progression (32). Furthermore, mTOR is highly associated with WNT signaling (33), and both WNT pathway and mTOR complexes are involved with autophagy and EMT in GBM, as will be discussed throughout this review.
Thus, we analyze the current knowledge on the role of WNT signaling in autophagy and EMT in the context of GBM, discussing the existent crosstalk between these processes. Notably, we consider how autophagy, EMT and WNT signaling may be interconnected to promote GBM development, progression and therapy resistance.
WNT Signaling Pathway and Its Role in Glioblastoma
WNTs are secreted glycoproteins rich in cysteine and composed of 300–400 amino acids (34). WNT signaling is classified as the canonical or β-catenin-dependent pathway, and the non-canonical or β-catenin-independent pathway.
In the canonical WNT signaling (Figure 1), WNT proteins (WNT1, WNT3A, WNT7A) bind to the membrane receptor complex of Frizzled (FZD) and low-density lipoprotein receptor-related protein 5/6 (LRP5/6), recruiting Dishevelled (DVL). This ultimately leads to stabilization and translocation of the transcription factor β-catenin to the nucleus, where it activates WNT target genes, such as AXIN2, C-MYC and CCND1 (cyclin D1), through binding with the complementary transcription factors T-cell factor (TCF) and the lymphoid enhancer factor (LEF) (Figure 1) (34). In the lack of WNT binding, β-catenin is phosphorylated by casein kinase 1 (CK1) and glycogen synthase kinase 3 (GSK3), which leads to the rapid degradation of β-catenin by the proteasome through a destruction complex with involvement of axin-1 and adenomatous polyposis coli (APC) (34). The existence of a low concentration of β-catenin in the nucleus induces the formation of the transcriptional co-repressor Groucho-TCF/LEF complex, which downregulates the expression of WNT target genes (35).
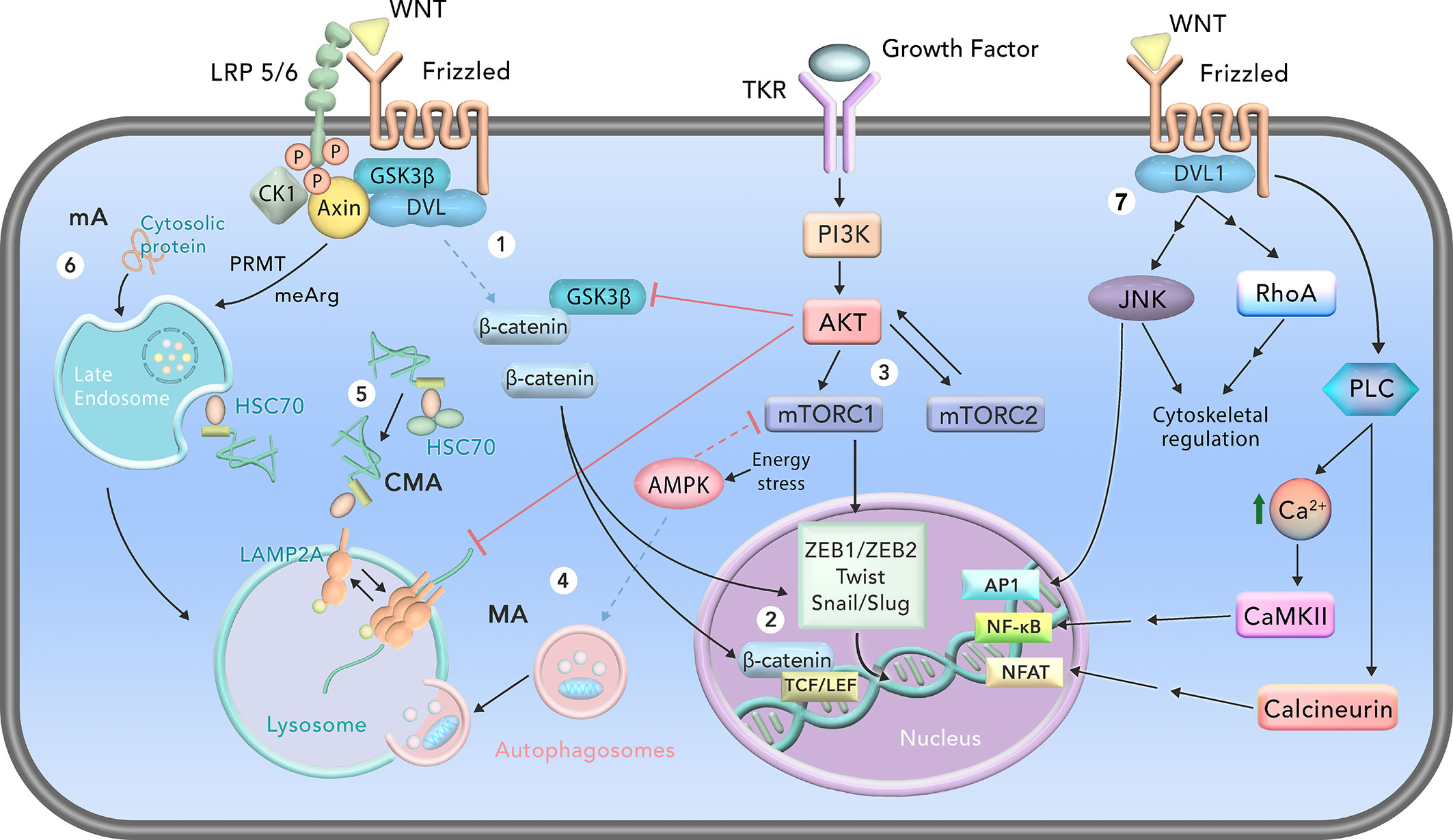
Figure 1 Schematic representation of cell signaling pathways involved in autophagy and epithelial-mesenchymal-transition (EMT). 1) In the canonical WNT signaling, β-catenin stabilizes and translocates to the nucleus after binding of WNT to Frizzled and low-density lipoprotein receptor-related protein 5/6 (LRP5/6) receptors. GSK3 is sequestered together with proteins of the destruction complex of β-catenin (DVL, axin, CK1). 2) In the nucleus, β-catenin binds to TCF/LEF transcriptional factors, activating its target genes. β-catenin also stimulates transcription of important EMT transcriptional factors such as ZEB1/ZEB2, Twist and Snail/Slug. 3) EMT is also induced by the mammalian Target of Rapamycin Complex 1 (mTORC1) signaling, through activation of the transcriptional factors ZEB1/ZEB2, Twist and Snail/Slug. mTORC1 and mTORC2 are activated by tyrosine kinase receptor (TKR) signaling through PI3K/AKT. mTORC2 also stimulates AKT. 4) Depending on the energy status of the cell, activation of AMP Kinase (AMPK) inhibits mTORC1 and induces macroautophagy (MA). 5) Chaperone-mediated autophagy (CMA) degrades cytosolic proteins that possess the KFERQ or KFERQ-like motif through recognition and binding of the heat shock-cognate chaperone of 71 kDa (HSC70) and a cochaperone complex. HSC70 targets the substrate protein to the lysosomal membrane, where it binds monomeric lysosome-associated membrane protein type 2A (LAMP2A) and induces its multimerization and stabilization. The substrate protein is unfolded and translocated into the lysosome for degradation through multimerized LAMP2A. 6) In mammals, microautophagy (mA) occurs in the late endosome, in a process called endosomal microautophagy. It can degrade cytosolic proteins, and some are recognized by HSC70. Some proteins, such as GSK3, are targeted for mA through arginine methylation (meArg) by protein arginine methyltransferases (PRMTs). After entering the late endosome, this organelle fuses with lysosomes to complete the mA cycle. 7) In the noncanonical WNT signaling, WNTs bind to Frizzled receptors and DVL1 is recruited to the membrane. c-Jun N-terminal kinases (JNKs), RhoA and phosphoinositide phospholipase C (PLC) can be activated by noncanonical WNT. JNK and RhoA further regulate the cytoskeleton, and JNK induces gene transcription through activator protein 1 (AP1). PLC increases cytosolic Ca2+ levels, leading to activation of Ca2+/calmodulin-dependent protein kinase II (CAM-KII) and calcineurin, which activate nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and nuclear factor of activated T cells (NFAT), respectively. Solid black lines represent activation, while solid red lines represent inhibition. Double black lines represent indirect activation. Dashed lines indicate protein interactions when the WNT/β-catenin and mTOR signaling are not activated.
The non-canonical pathway is further divided into two processes: the planar cell polarity (PCP) and the WNT/Ca2+ cascade pathways (Figure 1). In the first, activation of the PCP pathway leads to determination of the polarity of cells, in addition to affecting cellular shape and migration. In the PCP pathway, WNT ligands (WNT4, WNT5A and WNT11) bind to the FZD receptor and to one of two co-receptors: receptor-like tyrosine kinase (RYK) or receptor tyrosine kinase-like orphan receptor (ROR). This binding induces DVL, which activates DVL-associated activator of morphogenesis 1 (DAAM1), profilin, and the protein Rho- and RAS-related C3 botulinum toxin substrate 1 (RAC1). DAAM1 then activates the RAS homolog family member A (RhoA), which regulates the cytoskeleton through the Rho-associated kinase (ROCK). RAC1 induces c-Jun terminal kinases (JNKs), which directly or indirectly activate cytoskeletal alterations (20, 35). Secondly, the WNT/Ca2+ pathway is important during embryogenesis, in the formation of the embryonal dorsal axis, gastrulation and tissue morphogenesis. WNT ligands bind to the FZD receptor, which promotes activation of DVL1 and a G-protein. This complex results in the intracellular release of Ca2+, which activates the Ca2+/calmodulin-dependent protein kinase II (CAM-KII) and calcineurin, along with two transcription factors: nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and nuclear factor of activated T cells (NFAT). Additionally, certain WNT ligands of the non-canonical pathways can inhibit the canonical WNT pathway (20).
Moreover, certain extracellular antagonists can control WNT signaling, by extracellularly interacting with WNT ligands or receptors, preventing the proper binding of WNT proteins and impairing the maturation of receptors (36). For instance, Dickkopf (DKK1, DKK2, DKK3, DKK4, and DKKL1) is a family of secreted glycoproteins that modulate WNT signaling mainly by binding to LRP5/6, preventing the triggering of signaling (37). These features of DKK proteins are being explored due to its role in different types of cancers (38, 39). Additionally, other antagonists such as WNT inhibitor factor 1 (WIF1), frizzled-related protein family (FRP) – also referred as secreted frizzled-related proteins (sFRP) – and Cerberus work in similar ways (40).
Growing evidence has established a critical role of WNT signaling pivotal players during the onset of GBM. Several of the aforementioned molecules can be associated with the aberrant activation of WNT pathway in GBM, and a particular interest has been attributed to GSCs. WNT5A is epigenetically activated in GSCs due to absence of the H3K27me3 repressive mark (41). Another important molecule is LEF1, and its downregulation inhibits the capacity of self-renewal and expression of stemness markers, such as CD133 and Nestin (42), which negatively impacts proliferation, migration and in vitro invasion of GBM cells, playing a role in the EMT process (43). Additionally, the RYK receptor is upregulated in GSCs of patient samples, activating WNT pathway, promoting stemness and improving cell motility (44). Moreover, one of the molecules responsible for translocating and stabilizing β-catenin in GBM is forkhead box protein M1 (FOXM1), which may induce the canonical pathway independently from ligand binding (45). This factor can also be linked to the SOX2 promoter, a classic marker of the stem cell phenotype, known for promoting clonogenic growth in GBM (46).
Using a whole-genome approach, Foltz et al. (47) found that DKK1, sFRP1 and WIF1 were epigenetically silenced in GBM cells (47). The FRP genes are frequently found hypermethylated and inhibited during tumor development. Demethylation of the FRP gene promoter in human glioma cell lines led to an increase in phosphorylated β-catenin in the cytosol, attenuating tumorigenesis (48). Expression of FRPs promotes apoptosis through a possible activation of the DNA damage machinery through FAS-p53, activating the non-canonical WNT/Ca2+ pathway and the release of reactive oxygen species (ROS) (49). FRP4 treatment, in conjunction with TMZ, inhibited the canonical WNT pathway and was associated with a decrease in the expression of mesenchymal markers such as N-cadherin, Twist and Snail, along with a greater expression of epithelial markers, such as E-cadherin, showing the role of the inhibitor in reversing EMT (49). In addition, FRP4 chemically sensitizes GSCs, which decreases stemness properties that contribute to therapeutic resistance (49).
WIF1 has a negative influence on the ability of tumor cells to invade and migrate in vitro and in vivo (24). This suppressor phenotype is due to the downregulation of the canonical and non-canonical WNT pathways, with the inhibition of β-catenin-independent pathway being mediated by the sequestration of WNT5A, a ligand overexpressed in GBM. This inhibition results in decreased phosphorylation of p38-MAPK, reduction of intracellular Ca2+ concentration, and reduction in the expression of the metastasis-associated lung adenocarcinoma transcript 1 (MALAT1), a long non-coding RNA and a key invasion regulator (24). Moreover, recovering the expression of DKK1 in GBM cells results in inhibition of the WNT pathway, leading to growth suppression and decreased colony formation (47).
WNT signaling is also fundamental in the crosstalk between microglia and GBM (50). WNT3A derived from GBM induces upregulation of stress-inducible protein 1 (STI1/HOP), interleukin 10 (IL-10), and arginase-1 (ARG-1) in tumor-associated microglia. Together, these molecules collaborate with tumor immune evasion and increase in cell proliferation and migration (51–53). Another report showed that WNT3A increases the level of expression of programmed death-ligand 1 (PD-L1) on the surface of GBM cells, which is an important protein that inhibits the activation, expansion and effector functions of cells with antitumor activity (TCD8+) (54), corroborating the importance of WNT3A in immune evasion (55). Furthermore, WNT3A also acts on the WNT/β-catenin/FRA1 axis, a cascade that plays a crucial role in the aggressiveness of glioma and in EMT activation (56).
Due to GBM heterogeneity, EMT is usually incomplete, with associated transient phenotypic states, which results in a combination of epithelial and mesenchymal characteristics (9). It is noteworthy that the infiltrative peripheral region of GBM contains cells with mesenchymal characteristics, with higher WNT/β-catenin pathway activation (5), conferring a specific type of EMT-like process in GBM. As will be further discussed in this review, EMT and autophagy are influenced by WNT signaling, and the crosstalk between these pathways plays an important role in GBM pathology.
Autophagy in the Context of Glioblastoma and WNT Signaling
Autophagy is a greatly conserved process in eukaryotes, and it has an important role in homeostasis and cell components renewal in the face of adverse conditions. Autophagy degrades defectively folded proteins and dysfunctional organelles, such as mitochondria and peroxisomes, in addition to being essential for cell survival under microenvironmental stress (57).
Mammalian autophagy is classified in three types: microautophagy (mA), chaperone-mediated autophagy (CMA), and macroautophagy (MA) (57, 58). All three types can coexist within a cell and, despite all having the same overall purpose of cargo recycling or degradation of cargo in the lysosome, each one has its own delivery mechanisms and regulation. Remarkably, the modulation of different autophagy types has been associated to GBM biology and will be discussed as follows.
Microautophagy in Glioblastoma
In mammals, mA ensues through different mechanisms that share some similarities. It can occur directly in lysosomes, in which they extend and wrap themselves around a portion of the cytoplasm, or they form arm- or flap-like protrusions that surround parts of the cytoplasm (59, 60). However, mammalian mA is mostly described as endosomal microautophagy (eMI), occurring in late endosomes/multivesicular bodies (MVBs) (61), which then fuse with lysosomes for breakdown of their cargo (Figure 1). This process is attributed to bulk degradation of proteins and other cytosolic components engulfed by late endosomes and can also selectively degrade specific proteins with the assistance of chaperones such as heat shock cognate 71 kDa (HSC70) (61). Interestingly, Sato et al. (62) demonstrated that rapamycin induces eMI, independently of MA, while lacking effect upon CMA (62). This activation is triggered by the nuclear transcription factor EB (TFEB) which increases the expression of several genes related to mA (62).
Recent studies have shown a connection between the WNT pathway and mA. WNT activation leads to the sequestration of GSK3 from the cytosol into late endosomes/MVBs through mA (63). WNT signaling also stabilizes other proteins and GSK3 substrates besides β-catenin. Moreover, WNT activation is highest in the G2/M phase of the cell cycle and the stabilization of GSK3 substrates is thought to decrease the rate of GSK3-dependent protein degradation to prepare for cell division. This shifts the molecules from proteasome degradation (which consumes a large amount of ATP) to microautophagic degradation, which depends on hydrolases inside lysosomes and does not require ATP consumption (64). Despite the research gathered about WNT and mA, there is still an extensive gap in the knowledge of the intricate regulatory mechanisms and the physiological function attributed to mA/eMI in mammals.
eMI is suggested to control protein quality (65) by limiting the intracellular levels of key proteins, working as a regulator of specific cellular processes (66, 67). Other than its physiological roles, mA may be involved with cancer growth. Albrecht et al. (64) showed, for example, that arginine methylation (meArg), promoted by protein arginine methyltransferases (PRMTs), is important for mA, being necessary for the WNT-induced GSK3 sequestration and subsequent microautophagic degradation (64). GSK3 and the tumor suppressor SMAD4 are methylated and targeted to late endosomes/MVBs upon WNT signaling (Figure 1), suggesting that inhibition of PRMT1 might oppose cancer progression. Additionally, axin has also been shown as a PRMT1 substrate. As mentioned above, axin acts as a scaffold protein and plays a major role in the destruction complex of β-catenin (64). Upon WNT activation, axin is recruited to the plasma membrane, where it binds to the WNT receptor complex. Arginine methylation in axin has been shown to increase its binding to GSK3, and the WNT coreceptors and GSK3 are then translocated together with axin to MVBs (64).
Thus, meArg has been associated with cancer progression and PRMTs have been considered as novel drug targets (68). In GBM, PRMT2 was shown to be overexpressed and is correlated with a poor prognosis (69). PRMT5 is also highly expressed in GBM, promotes self-renewal of GBM neurospheres (70), and resistance to mTOR inhibition in GBM cells lines and short-term patient cultures (71). mA, however, has not been investigated in GBM. Due to the recent discoveries on the requirement of meArg and mA to WNT signaling activation, this autophagic process might be involved with GBM biology and merits further investigation.
Chaperone-Mediated Autophagy in Glioblastoma
CMA contributes to the control of cellular quality and maintenance of the proteome (72, 73). Exposure of the KFERQ-like domain of proteins targets them to CMA and selective eIM. In CMA, exposure of this motif leads to its recognition by the chaperone HSC70, and consequent binding of this chaperone to the substrate protein, followed by targeting of this complex directly to the lysosomal membrane (74). This mechanism differs from mA and MA, in which the cargo is sequestered into vesicles before being directed to the lysosome (75). Interestingly, selective blockage of CMA induces activation of MA; however, MA cannot compensate for CMA functions under specific stress conditions, in which CMA plays an essential role (76).
Upon binding of HSC70 to the substrate protein, a chaperone-cochaperone complex is formed and assists HSC70 to redirect client proteins captured in the cytosol to the lysosome membrane (Figure 1) (77). On the lysosome membrane, the chaperone complex and its client proteins bind to the lysosome-associated membrane protein type 2A (LAMP2A), inducing LAMP2A multimerization (78). After unfolding, the substrate is then translocated into the lysosome, where the protein is rapidly degraded by hydrolases (77, 78). It is clear that CMA activity is limited by the levels of LAMP2A on the lysosomal membrane, which makes LAMP2A the rate-limiting component for CMA (79, 80).
In addition to LAMP2A levels, further regulation of CMA occurs at the lysosomal membrane. Glial fibrillary acidic protein (GFAP) and elongation factor 1α (EF1α) regulate the stability of multimeric LAMP2A (81). Lysosomal GFAP, when phosphorylated by AKT1 activated by mTORC2, remains bound to EF1α and it is unable to stabilize multimeric LAMP2A (81). Ultimately, this leads to CMA inhibition regulated by mTORC2. When CMA is necessary, the GTPase RAC1 recruits and stabilizes the phosphatase pleckstrin homology domain and leucine-rich repeat protein phosphatase 1 (PHLPP1) at the lysosomal membrane, and this phosphatase dephosphorylates AKT1. This increases non-phosphorylated GFAP and favors the formation of the LAMP2A multimeric complex (73). Thus, it is possible to hypothesize that CMA inhibition might be regulated by mTORC2 and by WNT signaling, since RAC1 is one of the effectors of the PCP pathway. Moreover, signaling of the retinoic acid receptor α (RARα), associated to differentiation of non-tumor and tumor cells, inhibits transcription of genes required for CMA, highlighting an additional pathway of regulation for this autophagic process in health and, particularly, in cancers such as GBM, in which GSCs are a prominent factor (82).
CMA activity is markedly increased in cancer cells. Noteworthy, an anti- or pro-cancer function of CMA appears to depend, at least in some extent, on the status of cell transformation (83) and stage of the specific tumor (84, 85). This facet supports the importance of a context-dependent therapy investigation, requiring specialized research and a more translational approach.
There are very few studies that specifically explore the function of CMA in GBM, but the aggressiveness and high heterogeneity of this tumor raise important questions about the extent to which CMA may be contributing to these features. Maititi et al. (86) reported that the treatment of different GBM cell lines with curcumin and solid lipid curcumin particles downregulated LAMP2A levels, consequently decreasing CMA (86). Alternatively, regarding CMA activation, TMZ-resistant and TMZ-sensitive cells respond differently, according to hypoxia-inducible factor 1α (HIF-1α) expression and their basal levels of cytoplasmic ROS. CMA was shown to be the main process through which TMZ inhibits activity of HIF-1α in sensitive cells, promoting apoptosis and increasing responsiveness to the drug (87). Downregulation of LAMP2A blocks HIF-1α degradation through CMA and is sufficient to induce a TMZ-resistant phenotype in GBM sensitive cells, reducing the expression of several apoptosis-related genes upon treatment (87). Further research showed CMA is activated following cytoplasmic release of mitochondrial ROS, since concomitant increase in LAMP2A and HSC70 is observed with induction of oxidative stress. In accordance, inhibition of mitochondrial ROS downregulates LAMP2A and HSC70 (88). TMZ-sensitive cells present low basal levels of ROS, which in turn are increased with TMZ treatment, consequently activating CMA and pro-apoptotic genes. In contrast, TMZ-resistant cells have high basal levels of ROS and fail to activate CMA upon TMZ treatment (88). Remarkably, knockout of LAMP2A leads to CMA blockage, which is sufficient to trigger the switch of sensitive to resistant responsiveness (88). In T cells, production of mitochondrial ROS by engagement of the T cell receptor induces nuclear translocation of NFAT, which binds to the LAMP2 promoter and activates CMA (89). As discussed in section 2, the non-canonical β-catenin-independent WNT/Ca2+ cascade activates NFAT (90). Although non-canonical WNT pathways are usually related to embryonic patterning and morphogenesis, their aberrant regulation increases invasiveness in GBM (20). It is interesting to interrogate whether non-canonical WNT signaling through NFAT could be involved with activation of CMA in GBM cells, since ROS can be involved with the activation of both NFAT and CMA.
Another compelling mechanism is the ability of GBM cells to induce CMA in pericytes that surround the tumor, suppressing antitumor response and facilitating tumor progression. Specifically, interaction between GBM cells and pericytes of the peritumoral area increases ROS and subsequently LAMP2A levels, inducing nanotube formation and downregulation of the secretion of vesicles containing cytotoxic molecules (91). This results in promotion of direct interaction between these cells and abrogates inflammatory response (91). Supporting this, high levels of granulocyte-macrophage colony stimulating factor (GMCSF), molecule that promotes immunosuppression, were found in GBM cells after interaction with pericytes (91).
A thought-provoking aspect of CMA contribution to GBM is that chaperones and cochaperones that function in CMA play important roles in GBM biology (92). STI1/HOP, a known component of the CMA translocation complex (77), is upregulated in GBM cells. High levels of both STI1/HOP and its partner cellular prion protein (PrPC) are correlated with increased proliferation of GBM cells (93). Moreover, interaction between STI1/HOP-PrPC was found to sustain proliferation of GBM stem-like cells and tumor growth in vivo, contributing to its aggressiveness (94). Curiously, a mutant form of PrPC interacts with LAMP2A and HSC70, raising the question whether the non-mutated form of PrPC can also interact with CMA-related proteins (95). Interestingly, WNT3A increases STI1/HOP expression in GBM cells (53), suggesting that WNT signaling could regulate CMA, or that GBM cells take advantage of increased STI1/HOP expression to sustain tumor growth and modulate GSCs. Furthermore, since mA favors WNT signaling through GSK3 degradation, these data demonstrate how WNT signaling appears closely involved with autophagic processes, both by depending on them to be induced and by participating in their indirect activation.
In addition to STI1/HOP, other proteins of the translocation complex are proposed to be related to GBM biology, such as BAG1. BAG1 confers proliferation advantage in GBM cell lines in serum starvation conditions, a CMA-inducing environment, through interaction with BCL-2 (96). Although the mechanisms described for the function of these proteins in the biology of GBM have not yet been directly associated with CMA, it may be an interesting strategy to investigate the contribution of CMA in this context, considering the intricate relationship of its components with the aggressiveness of GBM. Thus, evidence gathered here demonstrate that, despite knowing the main processes and molecules involved in CMA, novel intermediate players that finely tune the mechanism - especially in the context of disorders such as cancer - need to be unraveled to actively consider CMA as a viable, well controlled and safe therapeutic approach.
Macroautophagy in Glioblastoma
MA involves the formation of an isolated vesicle, termed autophagosome, for transportation of the materials to the lysosome (Figure 1) (97). It consists of the following critical steps: initiation, nucleation, maturation and degradation of the cargo, with consequent release of the degradation yields to the cytosol (98). In the initiation step, there is the formation of a kinase complex comprised of Unc-51-like kinase 1 and 2 (ULK1/2), family 200-kDa interacting protein (FIP200), the mammalian ATG13 (mATG13); and ATG101 (99). mTORC1 inhibition under starvation activates ULK1/2, triggers phosphorylation of mATG13 and ULK1/2, and induces ULK-dependent phosphorylation of FIP200 (100). Moreover, studies demonstrate AMP-activated protein kinase (AMPK) binds to the ULK1-mTORC1 complex and induces ULK1-mediated autophagy through inhibition of mTORC1 by phosphorylation of Raptor (Figure 1) (101, 102).
In the stage of nucleation, the ULK1 complex targets another protein complex, which includes a class III phosphatidylinositol-3 kinase (VPS34), beclin 1, ATG14L, VPS15, and the autophagy beclin 1 regulator 1 (AMBRA-1) (103, 104). Subsequent to phagophore nucleation, in the maturation phase, there is the formation of two protein conjugations. During one of them, the processing of microtubule associated protein 1 light chain 3 (LC3) by ATG4 occurs to yield LC3-I, which in turn is bound to the target lipid phosphatidylethanolamine to generate processed LC3-II (105). LC3-II is integrated in the phagophore where it interacts with adaptor molecules, such as p62/SQSTM1, to select cargo for degradation and to label the phagophore membrane as autophagic (106). Promptly after autophagosome completion, most of the ATG proteins detach from the vesicle (107) triggering its fusion with the lysosome to form the autolysosome (108). In mammalian cells, autophagosome-lysosome fusion is regulated by LAMP2 and the small GTPase RAB7 (109, 110).
The role of MA in cancer and in GBM has been extensively covered in the literature (for more information, refer to (98, 111). Here we will focus on the involvement of the WNT pathway and MA in GBM.
In GBM cell lines, WNT5A/β-catenin signaling was demonstrated to modulate MA (112). The macroautophagic flux was increased upon impairment of WNT/β-catenin signaling via the inhibition of the downstream effector TCF (113). Similarly, extracellular DKK1 triggers MA by disrupting the communication of WNT ligands to their cell surface receptors, thus inhibiting WNT cascades (113).
Furthermore, WNT signaling proteins are also targets of MA. The WNT/β-catenin target genes AXIN2 and CCND1 become downregulated upon starvation-induced MA or mTOR blockage in GBM, although C-MYC expression shows no alteration (114). The same autophagy-inducer mechanisms were shown to decrease the expression of the DVL2 protein (115). This was also observed by Colella et al. (114), where MA led to a decrease in the expression of DVL2 and nuclear β-catenin in GBM cells (114). The study also showed that β-catenin was located in nearby plasma membrane regions and engaged with N-cadherin to form structures similar to epithelial-like cell-cell junctions (114).
The protein Von Hippel-Lindau (VHL) was demonstrated to ubiquitylate DVL2, which leads to its macroautophagic degradation (115). Noteworthy, data from the literature demonstrate that VHL also promotes the ubiquitination of the HIF-1α and 2α (HIF-2α) in GBM (116–119). Interestingly, in renal cell carcinoma, HIF-2α was shown to be degraded via both proteasome system and macroautophagic pathway in a VHL-dependent manner. Inhibition of MA led to HIF-2α degradation by the proteasome, whereas suppression of the proteasome caused HIF-2α degradation by MA (120). Furthermore, HIF-1α was demonstrated to induce MA in GBM as a mechanism of resistance to antiangiogenic therapy (121). Evidence indicates an interaction between the WNT/β-catenin pathway and HIF proteins in both physiological and pathological conditions. For instance, the crosstalk between WNT signaling and HIF-1α was demonstrated to be involved in maintenance of GSCs stemness, since their interaction transcriptionally regulates the neuronal differentiation of these cells (122).
In summary, the aforementioned studies point out to the importance of the WNT signaling and MA in GBM biology. As discussed, there is an important crosstalk between WNT/β-catenin signaling, MA and hypoxic pathways that warrants further investigation. The relationship between these pathways indicates the importance of metabolic reprogramming in GBM, in a context where nutrient availability may be scarce, and MA may be downregulated through induction of WNT/β-catenin and PI3K/AKT/mTOR signaling.
Epithelial-Mesenchymal Transition in Glioblastoma
Epithelial cells can undergo EMT, in which cells lose their apical-basolateral polarity as well as cell-cell adhesion structures in order to obtain an enhanced migratory phenotype (123). Upon transition, the cells become able to degrade the basal membrane using metalloproteases and thus migrate from their original site assisted by cytoskeleton rearrangements (124). As described in the name of the process, the transitioning cell shows increased expression of mesenchymal markers as well as loss of epithelial marker and adhesion molecules (125). Initial onset of EMT is dependent on many proteins that participate in the motility cycle, such as Rho GTPases, RAC1, and CDC42 (126), transcription factors from the SNAI family, such as Snail1 and Slug, as well as homeodomain transcription factors such as ZEB1 and ZEB2 (Figure 1) (5, 125). Additionally, many of the above-mentioned proteins - such as Snail1, Slug, ZEB1, ZEB2 and Twist - are able to regulate the switch of expression from E-cadherin to N-cadherin, an important hallmark of EMT (127, 128). This biological phenomenon has a well-known role in wound healing and development (129, 130), in addition to cancer invasion (131). Moreover, EMT is divided into 3 distinct subtypes: type 1 EMT is related to morphogenesis and is modulated by signaling pathways such as WNT/β-catenin, which in turn regulates Snail1 expression (132); type 2 EMT is related to wound healing and fibrosis and can be regulated by Slug, epidermal growth factor (EGF) and transforming growth factor β (TGF-β) pathways (130); and type 3 EMT occurs in cancer cells, playing an important role on tumor invasion and metastasis (133), and can be regulated by cytokines secreted in the tumoral niche such as TGF-β and tumor necrosis factor α (TNFα) (5).
As seen during neurodevelopment, mainly during migration of the neural crest, neuroepithelial cells may undergo transition to a more mesenchymal phenotype and, therefore, increased migratory capacity (134, 135). GBM can co-opt this process to assist cell invasion and migration. Importantly, neural tissues lack E-cadherin expression, although this protein is expressed in GSCs and in a rare subgroup of extremely aggressive GBM cells (5, 136). As GBM cells do not intrinsically express E-cadherin, the switch to N-cadherin, that is classic in EMT, is not entirely observed in this tumor (137). Moreover, distinct motility behaviors observed in GBM were associated to N-cadherin distribution in the cell instead of its expression levels (138). Therefore, GBM cells undergo a process similar to EMT, called EMT-like or glia-to-mesenchymal transition (GMT). Such process can be induced through radiation and be modulated by TGF-β-dependent and TGF-β-independent pathways through mitogen-activated protein kinases (ERK1/2) and GSK3, which in turn modulate Snail expression and localization (139). Likewise, neuronal cells also seem to undergo an EMT-like process, relying on Scratch1 and Scratch2 proteins, members of the Snail superfamily, to repress E-cadherin expression (140).
Additionally, the G protein-coupled receptor LGR5 triggers EMT in GSCs through the WNT/β-catenin cascade (141). Moreover, WNT-C59, an antagonist of the pathway, was able to impair EMT in GSCs (141). The transcription-related protein FRA1 was shown to act downstream of WNT3A signaling, promoting EMT in GBM cells and therefore contributing to the aggressive behavior of this tumor (56). Furthermore, the micro-RNA miR-504 suppresses the expression of the FZD7 gene, which causes the WNT/β-catenin pathway to be downregulated and affects EMT (142). Importantly, the transcription factors LEF1 and HOXA13 have both been shown to promote EMT in GBM via WNT signaling (143). Furthermore, the WNT/β-catenin cascade was shown to be activated in GBM cells of the mesenchymal subtype, leading to induction of expression of ZEB1, Twist1 and Slug, along with increased migratory capability of these cells (5). These recent findings support the importance of the WNT signaling for induction of EMT in GBM, pointing out to the WNT pathway as a promising therapeutic target for reducing the aggressiveness of this tumor.
Crosstalk Between Autophagy, Epithelial-Mesenchymal Transition, and WNT Signaling in Glioblastoma
When supporting cancer cells, autophagy can provide nutrients and energy for both EMT and the consequent metastasis, since it can sustain cells for survival in stressful environmental conditions (7). On the other hand, autophagy can also inhibit EMT by modulating the expression of specific proteins. As such, MA can downregulate mTOR signaling, leading to β-catenin and N-cadherin degradation and E-cadherin expression (7, 144). Therefore, it is believed that autophagy is oncosuppressive at initial stages of metastasis, by destabilizing or degrading crucial inducers of EMT, consequently inhibiting this process. If the cell is unable to prevent malignant transformation and metastasis ensues, EMT is activated and autophagy is required for tumor cell survival in environmental stress (5). Additionally, further dimensions should be added to this hypothesis, by considering the role that important pathways, such as the WNT signaling, and the different autophagic processes (mA, CMA, and MA), may have in the balance of oncosuppressive or metastatic function, especially in a highly infiltrative tumor as GBM. In this session, we discuss the evidences for the crosstalk of WNT signaling-autophagy-EMT observed in different aspects of GBM biology (Figure 2).
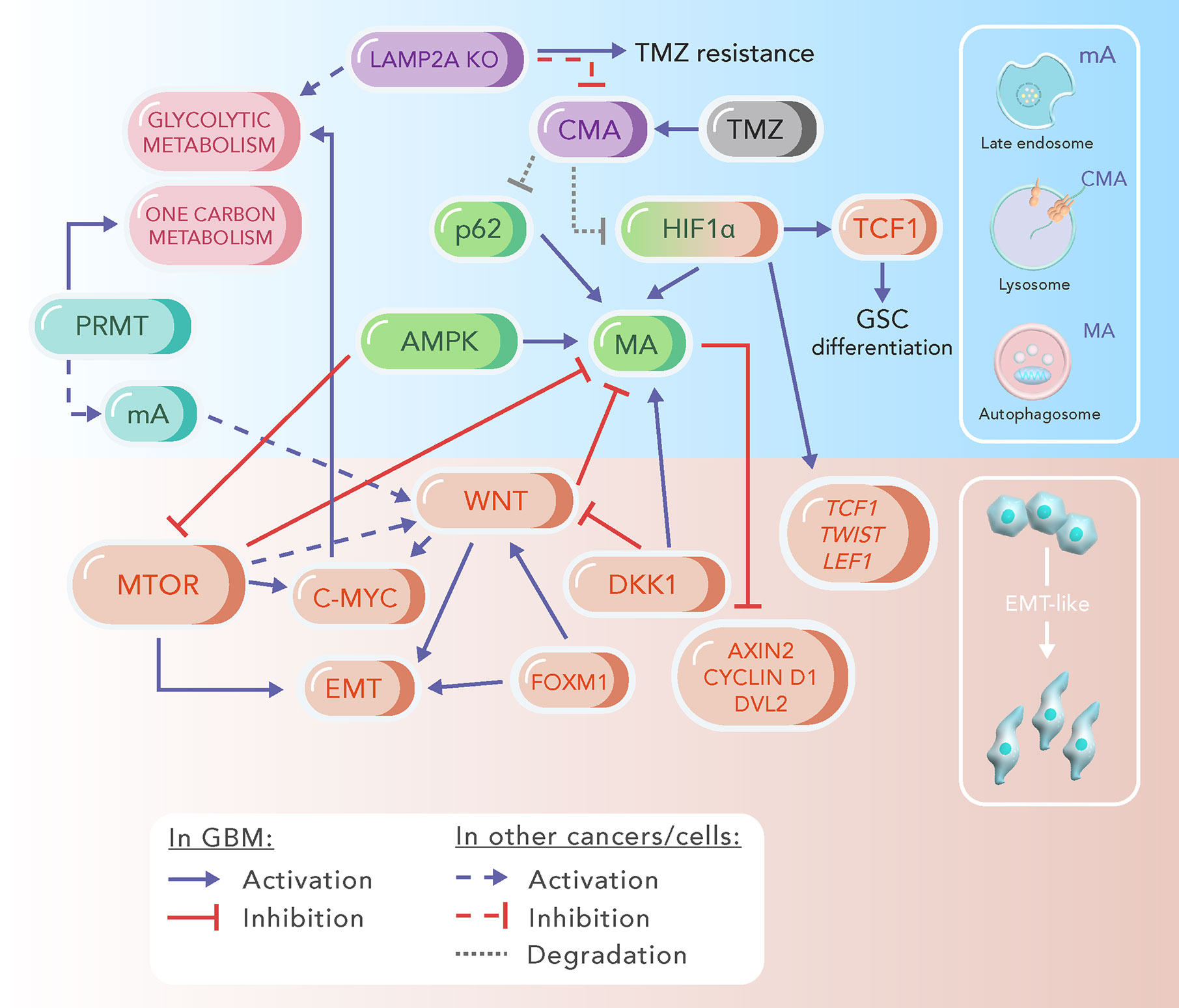
Figure 2 Schematic representation of signaling pathways involved with WNT/β-catenin, autophagy and epithelial-to-mesenchymal transition (EMT) in glioblastoma (GBM). The major players of the highlighted processes are grouped by a colour code, which differentiates microautophagy (mA, light blue), chaperone-mediated autophagy (CMA, purple), macroautophagy (MA, green), EMT (orange) and some metabolic processes (pink). AMPK, AMP Kinase; DKK, Dickkopf; DVL2, dishevelled 2; FOXM1, forkhead box protein M1; HIF1a, hypoxia-inducible factor 1a; LAMP2A KO, knockout of LAMP2A protein; LEF1, lymphoid enhancer factor 1; mTOR, mammalian target of rapamycin; PRMT, protein arginine methyltransferase; TCF1, T-cell factor 1; TMZ, temozolomide.
The microenvironment surrounding GBM presents hypoxic niches, which contribute to scarcity of nutrients and to dependence on glycolytic metabolism (145). Hypoxia, and consequent HIF-1α signaling activation, is a known inducer of MA, and also a known EMT promoter in different types of cancers (146–150). In GBM, an intricate relationship of those pathways with WNT signaling can be postulated, since strong correlation in the expression of HIF-1α and β-catenin was demonstrated in tumor cells (151). In addition, HIF-1α promotes the expression of the β-catenin transcriptional partners TCF1 and LEF1, regulating β-catenin nuclear retention (152, 153). This contributes to the activation of target genes associated with GBM tumor invasion and proliferation, such as Snail/Slug and cyclin D1 (154). Remarkably, MA and CMA appear to have opposed roles during GBM responsiveness to hypoxia. As covered in Chaperone-Mediated Autophagy in Glioblastoma, CMA decreases HIF-1α activity in GBM cells. On the other hand, hypoxia induction increases MA. Furthermore, the response to curcumin treatment is also opposed: while the treatment induces MA, concomitantly decreasing the expression of β-catenin targets, it also decreases CMA. However, this increased MA by curcumin treatment could be a feedback response of the tumor cell to the CMA inhibition.
Alternatively, HIF-1α/TCF1 activation can also result in neuronal differentiation in GBM cells subjected to hypoxia, and this pathway is primarily activated in the subpopulation of GSCs, being responsible for a pro-differentiation phenotype (122, 155). Interestingly, HIF-1α/TCF4 activation in normoxia triggers transcriptional inhibition of the same regions once activated in hypoxia (122). The differences between the outcomes for tumor biology might rely on the highly heterogenous cell populations in GBM tumors, and the initial spatial-molecular context of these cells. Despite that, different reports advocate hypoxia-mediated pathways working in tumor supporting mechanisms, contributing to chemotherapy resistance and invasion of healthy tissue.
Besides contributing to the distinct above-mentioned mechanisms, HIF-1α is able to bind the hypoxia-response element of TWIST proximal promoter and mediate its expression (156). Twist and Snail are targets of β-catenin signaling (156, 157), and are known EMT transcription factors (158), whose levels can be controlled by MA (159). In GBM particularly, a recent report describes the associated increase in MA markers with decreased levels of Twist and Snail in tumor cells treated with a combination of TMZ and curcumin (160). Moreover, Snail and Twist are found increased in GBM, together with other EMT-related transcripts (161). Thus, regulation of the levels of these transcription factors by autophagy represents one of the feedback correlations contributing to the crosstalk between autophagy and EMT, ultimately dictated by WNT upstream signaling.
An additional factor to be considered when exploring the implications of WNT pathway, EMT and autophagy in GBM is FOXM1. FOXM1 is upregulated in GBM (162) and plays a key role in the EMT-like process, directly controlling the expression of the proteins matrix metallopeptidase 2 (MMP2) (163) and A disintegrin and metalloproteinase 17 (ADAM17) (164). WNT signaling works in the stabilization of FOXM1 (165), and FOXM1 is able to directly translocate β-catenin to the nucleus of GBM cells, independently of extracellular ligands (45). Recently, it was shown that FOXM1 binds to the promoter region and controls the overexpression of the ubiquitin-conjugating enzyme E2C (UBE2C), protecting GBM cells from autophagic cell death, probably through PI3K-AKT-mTOR activation (166).
According with this overall increase in WNT activation in GBM, protein expression of the DVL family is also enhanced in this tumor (167–169). Interestingly, autophagic decrease in DVL2 levels impairs β-catenin nuclear localization, concomitant with an epithelial-like phenotype (114). Additionally, the dishevelled-associated antagonist of β-catenin 2 (DACT2) is a DVL-interacting protein that decreases the level of activated β-catenin resulting in suppression of WNT/β-catenin target genes (170). In GBM, decreased levels of DACT2 were proportionally associated with shorter patient survival, greater tumor aggressiveness and in vivo growth (171), suggesting that an apparent decrease in the levels of this protein favors tumor maintenance. The control of the levels of DACT2 in GBM has not yet been directly associated with autophagy, but the expression of other proteins of the same family were found strongly associated with autophagic processes in cancer cells (172, 173). Those evidences support the fundamental role of the intricate relationship between the EMT-like process, WNT signaling and autophagy for the progression of GBM tumors.
In the context of GBM, angiogenesis provides nutrients for the tumor to grow and favors metastasis (174). Thus, antiangiogenic therapy has been employed for GBM, especially with bevacizumab as adjuvant therapy (175). Although bevacizumab was shown to increase progression-free survival by 6 months, its effects on overall survival are controversial (175). Moreover, bevacizumab treatment generated hypoxia in the tumor microenvironment in vivo, and this hypoxia induced by the drug altered the metabolism of GBM cells in vitro (176). Interestingly, endostatin is another antiangiogenic drug being considered for GBM therapy. Endostatin is a terminal fragment of collagen XVIII-α 1, binds to α5β1 integrin on endothelial cells and induces autophagy through VPS34, beclin 1 and LC3-II activation (177, 178). Moreover, endostatin was shown to antagonize WNT signaling and to target β-catenin for degradation (179). Endostatin also decreased EMT markers on basal cell carcinoma (180). Additionally, a negative correlation between β-catenin and beclin 1 was reported (181), which could explain the decrease of EMT markers by endostatin, through inhibition of WNT signaling and induction of autophagy. Other reports in the literature confirm a role for WNT signaling in inhibiting autophagy in GBM (182).
Therefore, other therapeutic approaches against GBM are aiming to induce autophagy through mTOR inhibition. The first line of mTOR inhibitors, however, were able to block only mTOCR1 activity (183). As seen throughout this review, mTORC2 is involved with GBM invasion and could inhibit CMA. Moreover, during osteoblast differentiation, WNT3A/LRP5 induces mTORC2/AKT signaling downstream of RAC1 (184). To circumvent this ineffectiveness of initial mTOR inhibitors, novel ATP-competitive mTOR kinase inhibitors (TORKIs) were developed to target both mTOR complexes and are being investigated against GBM (5). Torin 1, for example, induced nutrient deprivation and inhibition of mTOR complexes, resulting in β-catenin re-localization and inhibition of migration (114). Moreover, other molecules that induce autophagy in GBM, such as arsenic trioxide, sodium selenite, and cannabinoids, combined with traditional therapy, were able to increase drug-induced cell death (185–187).
In contrast, autophagy inhibition was also proposed as a therapeutic approach against GBM. TMZ was shown to induce autophagy in GBM cells instead of apoptosis. The use of an inhibitor of late stages of autophagy restored TMZ-induced cell death (188). In accordance with this observation, chloroquine and hydroxychloroquine (HCQ) are being considered promising adjuvants in GBM therapy and are currently under phase III clinical trials (5, 189). Up until recently, these drugs were thought to improve median survival on GBM patients through autophagy inhibition. However, the maximum tolerated dose of HCQ was ineffective for autophagy inhibition, demonstrating the drug exerts its antitumoral effects through unknown mechanisms (190). Ultimately, the question of whether activation of autophagy would be beneficial or detrimental as a possible treatment for GBM remains unanswered. Knowledge on the crosstalk of autophagy with other cellular and molecular mechanisms can enlighten the discussion of how induction or suppression of this process should be safely applied in GBM treatment.
The comprehension of the metabolic features of GBM, highly associated with autophagic process, may contribute to circumvent the challenges faced in anti-tumor therapy. In fact, metabolic reprogramming is one of the hallmarks of cancer (191) and, in GBM, it fuels tumor survival, proliferation and invasion (192). Specially for this type of cancer, the upregulated WNT and AKT/mTOR signaling pathways are directly or indirectly involved with regulation of a diverse range of metabolic pathways (192, 193), intricately regulating autophagy processes and EMT.
As discussed in Macroautophagy in Glioblastoma, an overall downregulation of mTOR signaling exerted by MA was noted, leading to β-catenin degradation and decrease in WNT signaling (7, 144). On the other hand, mA is important for canonical WNT signaling activation. Evidence suggests methionine is required for the WNT-induced mA through the universal methyl donor S-adenosylmethionine (SAM), which would assist the internalization of GSK3 into late endosomes/MVBs (194). SAM is a known hub metabolite generated through one-carbon metabolism, which involves both folate and methionine cycles (195). Recently, WNT signaling has been emerging as a key regulator of cellular endocytic capacity and studies show endocytosis of extracellular proteins could provide cancer cells with recycle of nutrients required to grow, leading to adaptation and survival in a hostile microenvironment (64). Albrecht et al. (194) demonstrated methionine and SAM are necessary for this WNT-induced endolysosomal activity and extracellular protein degradation (194). In that sense, one-carbon metabolism has been shown to contribute to the de novo synthesis of purines and pyrimidines, in addition to nucleotide salvage in several tumors (193). In GBM, the scavenging of hypoxanthine is being considered as a reason for the resistance to anti-folate therapy (193). Furthermore, nearly half of GBM tumors have deleted 5-methylthioadenosine phosphorylase (MTAP), a key enzyme in the methionine salvage pathway (196). This enhances the dependence of the tumor cells for PRMT5 and studies have demonstrated PRMT5 inhibitors show antitumor effects against GBM (197–200). As such, PRMT5 inhibitors are currently being tested in clinical trials in GBM and several solid tumors (NCT02783300).
As one type of autophagic process, CMA has great impact on glucose metabolism (201). Studies with CMA-incompetent mice showed increased glycolysis in hepatocytes, due to decreased degradation of the glycolytic enzymes pyruvate kinase (PK), enolase 1 (eno1) and aldolase A (aldoA) (202). The glycolytic enzymes hexokinase 2 (HK2) and the M2 isoform of pyruvate kinase (PKM2) were also shown to be substrates of CMA degradation, in ovarian and non-small cell lung cancers, respectively (203, 204). GBM significantly boosts glycolysis for energy production through transcriptional and allosteric upregulation of key glycolytic enzymes such as HK2, enolase 2 (eno2) and phosphofructokinase (PFK), while decreasing the entering of pyruvate to the TCA cycle through inhibition of PDH (for more details on GBM metabolism, refer to 193). Interestingly, WNT3A/LRP5 signaling was shown to increase glycolysis during osteoblast differentiation through induction of the glycolytic enzymes GLUT1, HK2, PFK, PDK1 and others (184). Additionally, mTORC2 was shown to influence glycolytic metabolism in GBM through c-MYC activation (205). It would be interesting to evaluate whether canonical WNT signaling could be involved with glycolytic metabolism and CMA regulation in GBM cells, since the canonical WNT pathway is also upregulated in this tumor. Thus, in contrast to the possibility of CMA and MA hindering GBM metabolism and invasiveness, activation of mA may be a strategy employed by this tumor to survive in the surrounding microenvironment.
Given the importance of WNT signaling in GBM biology, the use of WNT pathway inhibitors are being evaluated for GBM therapy [reviewed in (206)]. LGK974 is a promising small-molecule inhibitor that interferes with the palmitoylation of WNTs, an indispensable step for their secretion and consequent binding to receptors. LGK97 was recently shown to have a synergistic effect with TMZ in vitro, reducing the clonogenic potential, with decreased expression of CD133, Nestin and SOX2. Importantly, these effects were shown to be independent of O6-alkylguanine DNA alkyltransferase (MGMT) promoter methylation status (207). Some WNT signaling inhibitors are currently being tested in clinical trials, such as Enzastaurin and alisertib. Enzastaurin inhibits phosphorylation of GSK3 and AKT, through protein kinase Cβ inhibition. However, a phase II trial of Enzastaurin in combination with bevacizumab had response and progression free survival similar to bevacizumab monotherapy, although the drug was well-tolerated (208). Alisertib is an Aurora A kinase (AURKA) inhibitor that decreased WNT signaling in GBM in vivo and in vitro. AURKA interferes with GSK3/axin/β-catenin through interaction with axin. In patient-derived orthotopic models of GBM resistant to bevacizumab, alisertib prolonged survival, induced mitotic arrest and decreased histone H3 phosphorylation (209). In a recent phase I clinical trial for GBM patients, alisertib was safe and well tolerated (210). Hence, although WNT signaling inhibitors show promising therapeutic rationale and results against GBM, the intricate crosstalk between EMT-autophagy-WNT needs further evaluation to gain a clearer understanding of how these processes and signaling pathways may be used against GBM.
Concluding Remarks
GBM is a highly heterogeneous and invasive solid tumor and, although there has been certain progress in the search for new therapeutic strategies, treatment of primary and recurrent GBM still remains a great challenge. Several of the signaling and metabolic abnormalities of GBMs are well described, especially those involving WNT, mTOR and EMT (Figure 2). However, the contributions of autophagic processes to GBM biology are largely unknown and debated, with clinical trials aiming at apparent opposing objectives. Some focus on autophagy inhibition, while others target mTOR inhibition to stimulate autophagy (211).
Moreover, as a highly heterogeneous tumor, GBM cells in different tumor layers may present distinct autophagic and EMT phenotypes that confer them with the molecular tools necessary to survive, proliferate, and evade therapy. As such, single-cell omics technologies are greatly contributing to the understanding of GBM heterogeneity and identification of subpopulations in the tumor bulk. However, it is crucial to first understand the molecular control and role of autophagy and EMT in GBM, to add to the identification of subpopulations of cells that would be the best target to overcome this disease.
As discussed in the present review, the WNT/β-catenin pathway plays a major role on EMT induction and invasiveness in GBM, while contributing to inhibition of MA (Figure 2). Interestingly, mA and one carbon metabolism are facilitators of β-catenin activation, and one carbon metabolism has been viewed as a possible therapeutic target in GBM (193). CMA was linked to acquired TMZ resistance in GBM cells, with very few reports about CMA in this tumor. Importantly, CMA assists the metabolic regulation of glycolytic enzymes and lipolysis and can also inhibit MA via p62 and HIF-1α degradation (Figure 2). Thus, there is an increasing demand for investigating mA, CMA and MA separately from each other in cancer and GBM (Figure 3). This could unveil the intricate relationship between these different autophagic processes, and how they influence cancer metabolism, aggressiveness, and invasion. Although these processes have overlapping signaling control, they can be activated or inhibited separately, which could confer tumor cells with a vast array of possible responses to survive and thrive even in harsh microenvironments.
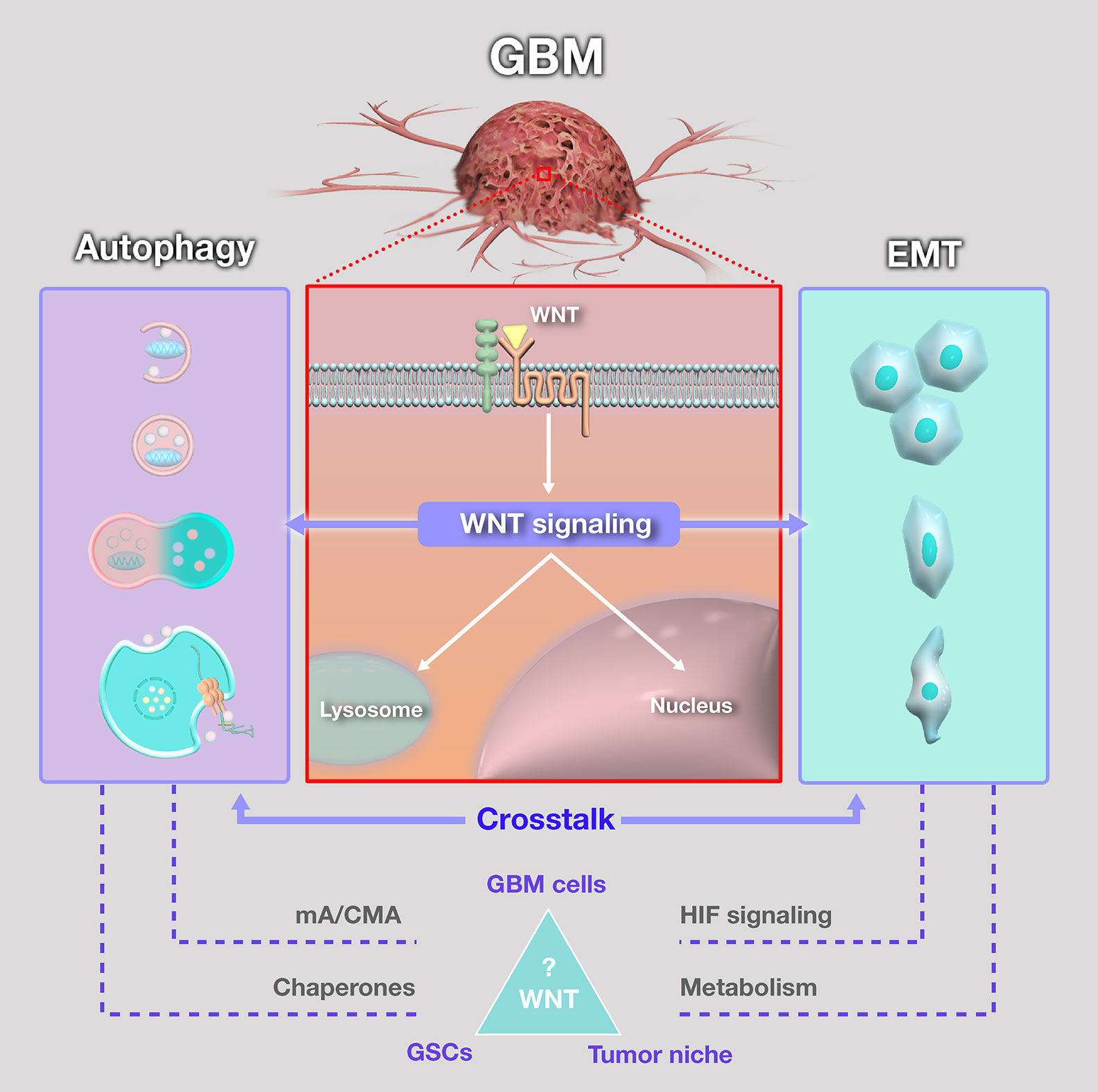
Figure 3 Schematic summary of the crosstalk between WNT signaling, autophagy and epithelial-mesenchymal transition (EMT) in glioblastoma (GBM). Despite the extensively discussed relevance, the importante of WNT pathways in the crosstalk between autophagy and EMT in GBM is still surrounded by open questions. Among them, the role of microautophagy (mA) and chaperone-mediated autophagy (CMA) is largely unknown. Moreover, function of chaperones in this context might represent a target for crosstalk regulation in GBM, in glioblastoma stem-like cells (GSCs), tumor bulk and tumor niche, and warrant further investigation. Accordingly, the role of HIF signaling and metabolic reprogramming on this crosstalk should also be a subject for future research efforts.
Author Contributions
Review conceptualization: BPC and MHL. Writing: BPC, CFLF, RPI, RNA, MIME, MBP, MCSS, JMB, GLRM, and GC. Review and editing: BPC, CFLF, RNA, MIME, MBP, MCSS, JMB, and MHL. Post-revision writing: BPC, CFLF, JMB, MCSS, GC, and GLRM. Post-revision editing: BPC, CFLF, and MHL. Figure conceptualization: BPC and MHL. Figure design: MIME. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, Processes numbers: BPC: 2019/14952-0; CFLF: 2019/14741-9; RPI: 2019/12710-9; MBP: 2017/26158-0; MIME: 2019/11097-1; RNA: 2020/04687-4; MCSS: 2019/06971-4; JMB: 2020/07450-5; GLRM: 2019/14552-1; MHL: 2018/15557-4;) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Process number: JMB: 101796/2020-0).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AMPK, AMP Kinase; DKK, Dickkopf; DVL2, dishevelled 2; FOXM1, forkhead box protein M1; HIF1α, hypoxia-inducible factor 1α; LAMP2A KO, knockout of LAMP2A protein; LEF1, lymphoid enhancer factor 1; mTOR, mammalian target of rapamycin; PRMT, protein arginine methyltransferase; TCF1, T-cell factor 1; TMZ, temozolomide.
References
1. Liu EY, Ryan KM. Autophagy and cancer–issues we need to digest. J Cell Sci (2012) 125(Pt 10):2349–58. doi: 10.1242/jcs.093708
2. Kimmelman AC, White E. Autophagy and Tumor Metabolism. Cell Metab (2017) 25(5):1037–43. doi: 10.1016/j.cmet.2017.04.004
3. Nazio F, Bordi M, Cianfanelli V. Autophagy and cancer stem cells: molecular mechanisms and therapeutic applications. Cell Death Differ (2019) 26(4):690–702. doi: 10.1038/s41418-019-0292-y
4. Kaza N, Kohli L, Roth KA. Autophagy in brain tumors: a new target for therapeutic intervention. Brain Pathol (Zurich Switzerland) (2012) 22(1):89–98. doi: 10.1111/j.1750-3639.2011.00544.x
5. Colella B, Faienza F, Di Bartolomeo S. EMT regulation by autophagy: a new perspective in glioblastoma biology. Cancers (2019) 11(3):312. doi: 10.3390/cancers11030312
6. Rojas-Sanchez G, Cotzomi-Ortega I, Pazos-Salazar NG, Reyes-Leyva J, Maycotte P. Autophagy and Its Relationship to Epithelial to Mesenchymal Transition: When Autophagy Inhibition for Cancer Therapy Turns Counterproductive. Biology (Basel) (2019) 8(4). doi: 10.3390/biology8040071
7. Chen H-T, Liu H, Mao M-J, Tan Y, Mo X-Q, Meng X-J, et al. Crosstalk between autophagy and epithelial-mesenchymal transition and its application in cancer therapy. Mol Cancer (2019) 18(1):1–19. doi: 10.1186/s12943-019-1030-2
8. Dongre A, Weinberg RA. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol (2019) 20(2):69–84. doi: 10.1038/s41580-018-0080-4
9. Yang J, Antin P, Berx G, Blanpain C, Brabletz T, Bronner M, et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat Rev Mol Cell Biol (2020) 21(6):341–52. doi: 10.1038/s41580-020-0237-9
10. Catalano M, D’Alessandro G, Lepore F, Corazzari M, Caldarola S, Valacca C, et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol Oncol (2015) 9(8):1612–25. doi: 10.1016/j.molonc.2015.04.016
11. Wirsching HG, Galanis E, Weller M. Glioblastoma. Handb Clin Neurol (2016) 134:381–97. doi: 10.1016/b978-0-12-802997-8.00023-2
12. Chen Z, Hambardzumyan D. Immune microenvironment in glioblastoma subtypes. Front Immunol (2018) 9:1004. doi: 10.3389/fimmu.2018.01004
13. Noch EK, Ramakrishna R, Magge R. Challenges in the Treatment of Glioblastoma: Multisystem Mechanisms of Therapeutic Resistance. World Neurosurg (2018) 116:505–17. doi: 10.1016/j.wneu.2018.04.022
14. Weller M, Le Rhun E. How we treat glioblastoma. Curr Oncol Rep (2019) 4(Suppl 2):e000520. doi: 10.1136/esmoopen-2019-000520
15. Campos B, Olsen LR, Urup T, Poulsen H. A comprehensive profile of recurrent glioblastoma. Oncogene (2016) 35(45):5819–25. doi: 10.1038/onc.2016.85
16. Batash R, Asna N, Schaffer P, Francis N, Schaffer M. Glioblastoma multiforme, diagnosis and treatment; recent literature review. Curr Med Chem (2017) 24(27):3002–9. doi: 10.2174/0929867324666170516123206
17. Sattiraju A, Sai KKS, Mintz A. Glioblastoma Stem Cells and Their Microenvironment. Adv Exp Med Biol (2017) 1041:119–40. doi: 10.1007/978-3-319-69194-7_7
18. Hatiboglu MA, Wei J, Wu AS, Heimberger AB. Immune therapeutic targeting of glioma cancer stem cells. Targeted Oncol (2010) 5(3):217–27. doi: 10.1007/s11523-010-0151-8
19. Liebelt BD, Shingu T, Zhou X, Ren J, Shin SA, Hu J. Glioma Stem Cells: Signaling, Microenvironment, and Therapy. Stem Cells Int (2016) 2016:7849890. doi: 10.1155/2016/7849890
20. Tompa M, Kalovits F. Contribution of the Wnt Pathway to Defining Biology of Glioblastoma. Neuromolecular Med (2018) 20(4):437–51. doi: 10.1007/s12017-018-8514-x
21. Liu X, Wang L, Zhao S, Ji X, Luo Y, Ling F. β-Catenin overexpression in malignant glioma and its role in proliferation and apoptosis in glioblastma cells. Med Oncol (Northwood London England) (2011) 28(2):608–14. doi: 10.1007/s12032-010-9476-5
22. Yu CY, Liang GB, Du P, Liu YH. Lgr4 promotes glioma cell proliferation through activation of Wnt signaling. Asian Pacific J Cancer Prevent: APJCP (2013) 14(8):4907–11. doi: 10.7314/apjcp.2013.14.8.4907
23. Kamino M, Kishida M, Kibe T, Ikoma K, Iijima M, Hirano H, et al. Wnt-5a signaling is correlated with infiltrative activity in human glioma by inducing cellular migration and MMP-2. Cancer Sci (2011) 102(3):540–8. doi: 10.1111/j.1349-7006.2010.01815.x
24. Vassallo I, Zinn P, Lai M, Rajakannu P, Hamou MF, Hegi ME. WIF1 re-expression in glioblastoma inhibits migration through attenuation of non-canonical WNT signaling by downregulating the lncRNA MALAT1. Oncogene (2016) 35(1):12–21. doi: 10.1038/onc.2015.61
25. Wang M, Li Q, Yu S, Zhang Z, Qiu P, Zhang Y, et al. Coronin 3 Promotes the Development of Oncogenic Properties in Glioma Through the Wnt/β-Catenin Signaling Pathway. OncoTarg Ther (2020) 13:6661–73. doi: 10.2147/ott.s257001
26. Rajakulendran N, Rowland KJ, Selvadurai HJ, Ahmadi M, Park NI, Naumenko S, et al. Wnt and Notch signaling govern self-renewal and differentiation in a subset of human glioblastoma stem cells. Genes Dev (2019) 33(9-10):498–510. doi: 10.1101/gad.321968.118
27. Huang M, Zhang D. Wnt-mediated endothelial transformation into mesenchymal stem cell-like cells induces chemoresistance in glioblastoma. Sci Transl Med (2020) 12(532):eaay7522. doi: 10.1126/scitranslmed.aay7522
28. Manoranjan B, Chokshi C, Venugopal C, Subapanditha M, Savage N, Tatari N, et al. A CD133-AKT-Wnt signaling axis drives glioblastoma brain tumor-initiating cells. Oncogene (2020) 39(7):1590–9. doi: 10.1038/s41388-019-1086-x
29. Mecca C, Giambanco I, Donato R, Arcuri C. Targeting mTOR in Glioblastoma: Rationale and Preclinical/Clinical Evidence. Dis Markers (2018) 2018:9230479. doi: 10.1155/2018/9230479
30. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell (2012) 149(2):274–93. doi: 10.1016/j.cell.2012.03.017
31. Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell (2006) 22(2):159–68. doi: 10.1016/j.molcel.2006.03.029
32. Li X, Wu C, Chen N, Gu H, Yen A, Cao L, et al. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget (2016) 7(22):33440–50. doi: 10.18632/oncotarget.7961
33. Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol (2014) 15(3):155–62. doi: 10.1038/nrm3757
34. Nusse R, Clevers H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell (2017) 169(6):985–99. doi: 10.1016/j.cell.2017.05.016
35. Lee Y, Lee JK, Ahn SH, Lee J, Nam DH. WNT signaling in glioblastoma and therapeutic opportunities. Lab Investigation; J Tech Methods Pathol (2016) 96(2):137–50. doi: 10.1038/labinvest.2015.140
36. Cruciat C-M, Niehrs C. Secreted and transmembrane wnt inhibitors and activators. Cold Spring Harbor Perspect Biol (2013) 5(3):a015081. doi: 10.1101/cshperspect.a015081
37. Niehrs C. Function and biological roles of the Dickkopf family of Wnt modulators. Oncogene (2006) 25(57):7469–81. doi: 10.1038/sj.onc.1210054
38. Katase N, Nagano K, Fujita S. DKK3 expression and function in head and neck squamous cell carcinoma and other cancers. J Oral Biosci (2020) 62(1):9–15. doi: 10.1016/j.job.2020.01.008
39. Li J, Gao Y, Yue W. The Clinical Diagnostic and Prognostic Value of Dickkopf-1 in Cancer. Cancer Manag Res (2020) 12:4253–60. doi: 10.2147/cmar.s254596
40. Esteve P, Bovolenta P. The advantages and disadvantages of sfrp1 and sfrp2 expression in pathological events. Tohoku J Exp Med (2010) 221(1):11–7. doi: 10.1620/tjem.221.11
41. Hu B, Wang Q, Wang YA, Hua S, Sauvé CG, Ong D, et al. Epigenetic Activation of WNT5A Drives Glioblastoma Stem Cell Differentiation and Invasive Growth. Cell (2016) 167(5):1281–95.e18. doi: 10.1016/j.cell.2016.10.039
42. Gao X, Mi Y, Ma Y, Jin W. LEF1 regulates glioblastoma cell proliferation, migration, invasion, and cancer stem-like cell self-renewal. Tumour Biol: J Int Soc Oncodevelopmental Biol Med (2014) 35(11):11505–11. doi: 10.1007/s13277-014-2466-z
43. Santiago L, Daniels G, Wang D, Deng FM, Lee P. Wnt signaling pathway protein LEF1 in cancer, as a biomarker for prognosis and a target for treatment. Am J Cancer Res (2017) 7(6):1389–406.
44. Adamo A, Fiore D, De Martino F, Roscigno G, Affinito A, Donnarumma E, et al. RYK promotes the stemness of glioblastoma cells via the WNT/β-catenin pathway. Oncotarget (2017) 8(8):13476. doi: 10.18632/oncotarget.14564
45. Zhang N, Wei P, Gong A, Chiu WT, Lee HT, Colman H, et al. FoxM1 promotes β-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell (2011) 20(4):427–42. doi: 10.1016/j.ccr.2011.08.016
46. Lee Y, Kim KH, Kim DG, Cho HJ, Kim Y, Rheey J, et al. FoxM1 Promotes Stemness and Radio-Resistance of Glioblastoma by Regulating the Master Stem Cell Regulator Sox2. PloS One (2015) 10(10):e0137703. doi: 10.1371/journal.pone.0137703
47. Foltz G, Yoon JG, Lee H, Ma L, Tian Q, Hood L, et al. Epigenetic regulation of wnt pathway antagonists in human glioblastoma multiforme. Genes Cancer (2010) 1(1):81–90. doi: 10.1177/1947601909356103
48. Schiefer L, Visweswaran M, Perumal V, Arfuso F, Groth D, Newsholme P, et al. Epigenetic regulation of the secreted frizzled-related protein family in human glioblastoma multiforme. Cancer Gene Ther (2014) 21(7):297–303. doi: 10.1038/cgt.2014.30
49. Bhuvanalakshmi G, Gamit N, Patil M, Arfuso F, Sethi G, Dharmarajan A, et al. Stemness, pluripotentiality, and Wnt antagonism: sFRP4, a Wnt antagonist mediates pluripotency and stemness in glioblastoma. Cancers (2019) 11(1):25. doi: 10.3390/cancers11010025
50. Roos A, Ding Z, Loftus JC, Tran NL. Molecular and Microenvironmental Determinants of Glioma Stem-Like Cell Survival and Invasion. Front Oncol (2017) 7:120:120. doi: 10.3389/fonc.2017.00120
51. Fonseca AC, Romão L, Amaral RF, Assad Kahn S, Lobo D, Martins S, et al. Microglial stress inducible protein 1 promotes proliferation and migration in human glioblastoma cells. Neuroscience (2012) 200:130–41. doi: 10.1016/j.neuroscience.2011.10.025
52. Zhang I, Alizadeh D, Liang J, Zhang L, Gao H, Song Y, et al. Characterization of Arginase Expression in Glioma-Associated Microglia and Macrophages. PloS One (2016) 11(12):e0165118. doi: 10.1371/journal.pone.0165118
53. Matias D, Dubois LG, Pontes B, Rosário L, Ferrer VP, Balça-Silva J, et al. GBM-Derived Wnt3a Induces M2-Like Phenotype in Microglial Cells Through Wnt/β-Catenin Signaling. Mol Neurobiol (2019) 56(2):1517–30. doi: 10.1007/s12035-018-1150-5
54. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell (2017) 168(4):707–23. doi: 10.1016/j.cell.2017.01.017
55. Du L, Lee JH, Jiang H, Wang C, Wang S, Zheng Z, et al. β-Catenin induces transcriptional expression of PD-L1 to promote glioblastoma immune evasion. J Exp Med (2020) 217(11):e20191115. doi: 10.1084/jem.20191115
56. Zhang L, Liu H, Mu X, Cui J, Peng Z. Dysregulation of Fra1 expression by Wnt/β-catenin signalling promotes glioma aggressiveness through epithelial-mesenchymal transition. Biosci Rep (2017) 37(2):BSR20160643. doi: 10.1042/bsr20160643
57. Lefebvre C, Legouis R, Culetto E. ESCRT and autophagies: Endosomal functions and beyond. Semin Cell Dev Biol (2018) 74:21–8. doi: 10.1016/j.semcdb.2017.08.014
58. Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signaling (2014) 20(3):460–73. doi: 10.1089/ars.2013.5371
59. Drews U. Cholinesterase in embryonic development. Prog Histochem Cytochem (1975) 7(3):1–52. doi: 10.1016/S0079-6336(75)80008-8
60. Sakai M, Ogawa K. Energy-dependent lysosomal wrapping mechanism (LWM) during autophagolysosome formation. Histochemistry (1982) 76(4):479–88. doi: 10.1007/bf00489903
61. Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, Follenzi A, et al. Microautophagy of cytosolic proteins by late endosomes. Dev Cell (2011) 20(1):131–9. doi: 10.1016/j.devcel.2010.12.003
62. Sato M, Seki T, Konno A, Hirai H, Kurauchi Y, Hisatsune A, et al. Rapamycin activates mammalian microautophagy. J Pharmacol Sci (2019) 140(2):201–4. doi: 10.1016/j.jphs.2019.05.007
63. Tejeda-Muñoz N, Albrecht LV, Bui MH, De Robertis EM. Wnt canonical pathway activates macropinocytosis and lysosomal degradation of extracellular proteins. Proc Natl Acad Sci U States America (2019) 116(21):10402–11. doi: 10.1073/pnas.1903506116
64. Albrecht LV, Ploper D, Tejeda-Muñoz N, De Robertis EM. Arginine methylation is required for canonical Wnt signaling and endolysosomal trafficking. Proc Natl Acad Sci (2018) 115(23):E5317–E25. doi: 10.1073/pnas.1804091115
65. Cannizzo ES, Clement CC, Morozova K, Valdor R, Kaushik S, Almeida LN, et al. Age-related oxidative stress compromises endosomal proteostasis. Cell Rep (2012) 2(1):136–49. doi: 10.1016/j.celrep.2012.06.005
66. Uytterhoeven V, Lauwers E, Maes I, Miskiewicz K, Melo MN, Swerts J, et al. Hsc70-4 Deforms Membranes to Promote Synaptic Protein Turnover by Endosomal Microautophagy. Neuron (2015) 88(4):735–48. doi: 10.1016/j.neuron.2015.10.012
67. Huber LA, Teis D. Lysosomal signaling in control of degradation pathways. Curr Opin Cell Biol (2016) 39:8–14. doi: 10.1016/j.ceb.2016.01.006
68. Jarrold J, Davies CC. PRMTs and Arginine Methylation: Cancer’s Best-Kept Secret? Trends Mol Med (2019) 25(11):993–1009. doi: 10.1016/j.molmed.2019.05.007
69. Dong F, Li Q, Yang C, Huo D, Wang X, Ai C, et al. PRMT2 links histone H3R8 asymmetric dimethylation to oncogenic activation and tumorigenesis of glioblastoma. Nat Commun (2018) 9(1):1–14. doi: 10.1038/s41467-018-06968-7
70. Banasavadi-Siddegowda Y, Russell L, Frair E, Karkhanis V, Relation T, Yoo J, et al. PRMT5–PTEN molecular pathway regulates senescence and self-renewal of primary glioblastoma neurosphere cells. Oncogene (2017) 36(2):263–74. doi: 10.1038/onc.2016.199
71. Holmes B, Benavides-Serrato A, Saunders JT, Landon KA, Schreck AJ, Nishimura RN, et al. The protein arginine methyltransferase PRMT5 confers therapeutic resistance to mTOR inhibition in glioblastoma. J Neurooncol (2019) 145(1):11–22. doi: 10.1007/s11060-019-03274-0
72. Zhang C, Cuervo AM. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med (2008) 14(9):959–65. doi: 10.1038/nm.1851
73. Arias E, Cuervo AM. Chaperone-mediated autophagy in protein quality control. Curr Opin Cell Biol (2011) 23(2):184–9. doi: 10.1016/j.ceb.2010.10.009
74. Chiang H-L, Plant C, Dice J. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science (4928) 1989) 246:382–5. doi: 10.1126/science.2799391
75. Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol (2018) 19(6):365–81. doi: 10.1038/s41580-018-0001-6
76. Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM. Consequences of the selective blockage of chaperone-mediated autophagy. Proc Natl Acad Sci U States America (2006) 103(15):5805–10. doi: 10.1073/pnas.0507436103
77. Agarraberes FA, Dice JF. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J Cell Sci (2001) 114(13):2491–9.
78. Bandyopadhyay U, Kaushik S, Varticovski L, Cuervo AM. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol (2008) 28(18):5747–63. doi: 10.1128/MCB.02070-07
79. Cuervo A, Dice J. Unique properties of lamp2a compared to other lamp2 isoforms. J Cell Sci (2000) 113(24):4441–50.
80. Cuervo AM, Dice J. Regulation of lamp2a levels in the lysosomal membrane. Traffic (2000) 1(7):570–83. doi: 10.1034/j.1600-0854.2000.010707.x
81. Bandyopadhyay U, Sridhar S, Kaushik S, Kiffin R, Cuervo AM. Identification of regulators of chaperone-mediated autophagy. Mol Cell (2010) 39(4):535–47. doi: 10.1016/j.molcel.2010.08.004
82. Anguiano J, Garner TP, Mahalingam M, Das BC, Gavathiotis E, Cuervo AM. Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat Chem Biol (2013) 9(6):374. doi: 10.1038/nchembio.1230
83. Gomes LR, Menck CFM, Cuervo AM. Chaperone-mediated autophagy prevents cellular transformation by regulating MYC proteasomal degradation. Autophagy (2017) 13(5):928–40. doi: 10.1080/15548627.2017.1293767
84. Kon M, Kiffin R, Koga H, Chapochnick J, Macian F, Varticovski L, et al. Chaperone-mediated autophagy is required for tumor growth. Sci Trans Med (2011) 3(109):109ra17. doi: 10.1126/scitranslmed.3003182
85. Peng JQ, Han SM, Chen ZH, Yang J, Pei YQ, Bao C, et al. Chaperone-mediated autophagy regulates apoptosis and the proliferation of colon carcinoma cells. Biochem Biophys Res Commun (2020) 522(2):348–54. doi: 10.1016/j.bbrc.2019.11.081
86. Maiti P, Scott J, Sengupta D, Al-Gharaibeh A, Dunbar GL. Curcumin and Solid Lipid Curcumin Particles Induce Autophagy, but Inhibit Mitophagy and the PI3K-Akt/mTOR Pathway in Cultured Glioblastoma Cells. Int J Mol Sci (2019) 20(2):399. doi: 10.3390/ijms20020399
87. Lo Dico A, Martelli C, Diceglie C, Lucignani G, Ottobrini L. Hypoxia-Inducible Factor-1α Activity as a Switch for Glioblastoma Responsiveness to Temozolomide. Front Oncol (2018) 8:249:249. doi: 10.3389/fonc.2018.00249
88. Lo Dico A, Salvatore D. Intracellular Redox-Balance Involvement in Temozolomide Resistance-Related Molecular Mechanisms in Glioblastoma. Cells (2019) 8(11):1315. doi: 10.3390/cells8111315
89. Valdor R, Mocholi E, Botbol Y, Guerrero-Ros I, Chandra D, Koga H, et al. Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat Immunol (2014) 15(11):1046–54. doi: 10.1038/ni.3003
90. Komiya Y, Habas R. Wnt signal transduction pathways. Organogenesis (2008) 4(2):68–75. doi: 10.4161/org.4.2.5851
91. Valdor R, García-Bernal D, Riquelme D, Martinez CM. Glioblastoma ablates pericytes antitumor immune function through aberrant up-regulation of chaperone-mediated autophagy. Proc Natl Acad Sci U S A (2019) 116(41):20655–65. doi: 10.1073/pnas.1903542116
92. Iglesia RP, Fernandes CFL. Heat Shock Proteins in Glioblastoma Biology: Where Do We Stand? Int J Mol Sci (2019) 20(22):5794. doi: 10.3390/ijms20225794
93. Lopes MH, Santos TG, Rodrigues BR, Queiroz-Hazarbassanov N, Cunha IW, Wasilewska-Sampaio AP, et al. Disruption of prion protein-HOP engagement impairs glioblastoma growth and cognitive decline and improves overall survival. Oncogene (2015) 34(25):3305–14. doi: 10.1038/onc.2014.261
94. Iglesia RP, Prado MB, Cruz L, Martins VR, Santos TG, Lopes MH. Engagement of cellular prion protein with the co-chaperone Hsp70/90 organizing protein regulates the proliferation of glioblastoma stem-like cells. Stem Cell Res Ther (2017) 8(1):76. doi: 10.1186/s13287-017-0518-1
95. Wang H, Tian C, Sun J, Chen LN, Lv Y, Yang XD, et al. Overexpression of PLK3 Mediates the Degradation of Abnormal Prion Proteins Dependent on Chaperone-Mediated Autophagy. Mol Neurobiol (2017) 54(6):4401–13. doi: 10.1007/s12035-016-9985-0
96. Roth W, Grimmel C, Rieger L, Strik H, Takayama S, Krajewski S, et al. Bag-1 and Bcl-2 gene transfer in malignant glioma: modulation of cell cycle regulation and apoptosis. Brain Pathol (Zurich Switzerland) (2000) 10(2):223–34. doi: 10.1111/j.1750-3639.2000.tb00256.x
97. Chen Y, Klionsky DJ. The regulation of autophagy–unanswered questions. J Cell Sci (2011) 124(2):161–70. doi: 10.1242/jcs.064576
98. Onorati AV, Dyczynski M, Ojha R, Amaravadi RK. Targeting autophagy in cancer. Cancer (2018) 124(16):3307–18. doi: 10.1002/cncr.31335
99. Hosokawa N, Sasaki T, Iemura S, Natsume T, Hara T, Mizushima N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy (2009) 5(7):973–9. doi: 10.4161/auto.5.7.9296
100. Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell (2009) 20(7):1992–2003. doi: 10.1091/mbc.e08-12-1249
101. Lee JW, Park S, Takahashi Y, Wang HG. The association of AMPK with ULK1 regulates autophagy. PloS One (2010) 5(11):e15394. doi: 10.1371/journal.pone.0015394
102. Zhao M, Klionsky DJ. AMPK-dependent phosphorylation of ULK1 induces autophagy. Cell Metab (2011) 13(2):119–20. doi: 10.1016/j.cmet.2011.01.009
103. He C, Song H, Yorimitsu T, Monastyrska I, Yen WL, Legakis JE, et al. Recruitment of Atg9 to the preautophagosomal structure by Atg11 is essential for selective autophagy in budding yeast. J Cell Biol (2006) 175(6):925–35. doi: 10.1083/jcb.200606084
104. Zhou C, Ma K, Gao R, Mu C, Chen L, Liu Q, et al. Regulation of mATG9 trafficking by Src- and ULK1-mediated phosphorylation in basal and starvation-induced autophagy. Cell Res (2017) 27(2):184–201. doi: 10.1038/cr.2016.146
105. Tanida I, Ueno T, Kominami E. LC3 and Autophagy. Methods Mol Biol (Clifton NJ) (2008) 445:77–88. doi: 10.1007/978-1-59745-157-4_4
106. Shvets E, Fass E, Scherz-Shouval R, Elazar Z. The N-terminus and Phe52 residue of LC3 recruit p62/SQSTM1 into autophagosomes. J Cell Sci (2008) 121(Pt 16):2685–95. doi: 10.1242/jcs.026005
107. Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Topics Microbiol Immunol (2009) 335:1–32. doi: 10.1007/978-3-642-00302-8_1
108. Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol (2010) 221(1):3–12. doi: 10.1002/path.2697
109. Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lüllmann-Rauch R, et al. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature (2000) 406(6798):902–6. doi: 10.1038/35022595
110. Jäger S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, et al. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci (2004) 117(Pt 20):4837–48. doi: 10.1242/jcs.01370
111. Yang K, Niu L, Bai Y, Le W. Glioblastoma: Targeting the autophagy in tumorigenesis. Brain Res Bull (2019) 153:334–40. doi: 10.1016/j.brainresbull.2019.09.012
112. Wang T, Chen Z, Zhang W. Regulation of autophagy inhibition and inflammatory response in glioma by Wnt signaling pathway. Oncol Lett (2017) 14(6):7197–200. doi: 10.3892/ol.2017.7103
113. Nàger M, Sallán MC, Visa A, Pushparaj C, Santacana M, Macià A, et al. Inhibition of WNT-CTNNB1 signaling upregulates SQSTM1 and sensitizes glioblastoma cells to autophagy blockers. Autophagy (2018) 14(4):619–36. doi: 10.1080/15548627.2017.1423439
114. Colella B, Faienza F, Carinci M, D’Alessandro G, Catalano M, Santoro A, et al. Autophagy induction impairs Wnt/β-catenin signalling through β-catenin relocalisation in glioblastoma cells. Cell Signal (2019) 53:357–64. doi: 10.1016/j.cellsig.2018.10.017
115. Gao C, Cao W, Bao L, Zuo W, Xie G, Cai T, et al. Autophagy negatively regulates Wnt signalling by promoting Dishevelled degradation. Nat Cell Biol (2010) 12(8):781–90. doi: 10.1038/ncb2082
116. Ziello JE, Jovin IS, Huang Y. Hypoxia-Inducible Factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J Biol Med (2007) 80(2):51–60.
117. Yang L, Lin C, Wang L, Guo H, Wang X. Hypoxia and hypoxia-inducible factors in glioblastoma multiforme progression and therapeutic implications. Exp Cell Res (2012) 318(19):2417–26. doi: 10.1016/j.yexcr.2012.07.017
118. Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest (2013) 123(9):3664–71. doi: 10.1172/jci67230
119. DePavia A, Jonasch E, Liu X-D. Autophagy degrades hypoxia inducible factors. Mol Cell Oncol (2016) 3(2):e1104428. doi: 10.1080/23723556.2015.1104428
120. Liu XD, Yao J, Tripathi DN, Ding Z, Xu Y, Sun M, et al. Autophagy mediates HIF2α degradation and suppresses renal tumorigenesis. Oncogene (2015) 34(19):2450–60. doi: 10.1038/onc.2014.199
121. Hu YL, DeLay M, Jahangiri A, Molinaro AM, Rose SD, Carbonell WS, et al. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res (2012) 72(7):1773–83. doi: 10.1158/0008-5472.can-11-3831
122. Boso D, Rampazzo E, Zanon C, Bresolin S, Maule F, Porcù E, et al. HIF-1α/Wnt signaling-dependent control of gene transcription regulates neuronal differentiation of glioblastoma stem cells. Theranostics (2019) 9(17):4860. doi: 10.7150/thno.35882
123. Kim DH, Xing T, Yang Z, Dudek R, Lu Q, Chen YH. Epithelial Mesenchymal Transition in Embryonic Development, Tissue Repair and Cancer: A Comprehensive Overview. J Clin Med (2017) 7(1):1. doi: 10.3390/jcm7010001
124. Horejs CM. Basement membrane fragments in the context of the epithelial-to-mesenchymal transition. Eur J Cell Biol (2016) 95(11):427–40. doi: 10.1016/j.ejcb.2016.06.002
125. Iser IC, Pereira MB, Lenz G, Wink MR. The Epithelial-to-Mesenchymal Transition-Like Process in Glioblastoma: An Updated Systematic Review and In Silico Investigation. Med Res Rev (2017) 37(2):271–313. doi: 10.1002/med.21408
126. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol (2014) 15(3):178–96. doi: 10.1038/nrm3758
127. Wheelock MJ, Shintani Y, Maeda M, Fukumoto Y, Johnson KR. Cadherin switching. J Cell Sci (2008) 121(Pt 6):727–35. doi: 10.1242/jcs.000455
128. Loh CY, Chai JY, Tang TF, Wong WF. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells (2019) 8(10):1118. doi: 10.3390/cells8101118
129. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest (2009) 119(6):1420–8. doi: 10.1172/jci39104
130. Haensel D, Dai X. Epithelial-to-mesenchymal transition in cutaneous wound healing: Where we are and where we are heading. Dev Dyn (2018) 247(3):473–80. doi: 10.1002/dvdy.24561
131. Pearson GW. Control of Invasion by Epithelial-to-Mesenchymal Transition Programs during Metastasis. J Clin Med (2019) 8(5):646. doi: 10.3390/jcm8050646
132. Ferrer-Vaquer A, Viotti M, Hadjantonakis AK. Transitions between epithelial and mesenchymal states and the morphogenesis of the early mouse embryo. Cell Adhesion Migration (2010) 4(3):447–57. doi: 10.4161/cam.4.3.10771
133. Kalluri R. EMT: when epithelial cells decide to become mesenchymal-like cells. J Clin Invest (2009) 119(6):1417–9. doi: 10.1172/jci39675
134. Mayor R, Theveneau E. The neural crest. Dev (Cambridge England) (2013) 140(11):2247–51. doi: 10.1242/dev.091751
135. Campbell K. Contribution of epithelial-mesenchymal transitions to organogenesis and cancer metastasis. Curr Opin Cell Biol (2018) 55:30–5. doi: 10.1016/j.ceb.2018.06.008
136. Lewis-Tuffin LJ, Rodriguez F, Giannini C, Scheithauer B, Necela BM, Sarkaria JN, et al. Misregulated E-cadherin expression associated with an aggressive brain tumor phenotype. PloS One (2010) 5(10):e13665. doi: 10.1371/journal.pone.0013665
137. Kubelt C, Hattermann K, Sebens S, Mehdorn HM, Held-Feindt J. Epithelial-to-mesenchymal transition in paired human primary and recurrent glioblastomas. Int J Oncol (2015) 46(6):2515–25. doi: 10.3892/ijo.2015.2944
138. Perego C, Vanoni C, Massari S, Raimondi A, Pola S, Cattaneo MG, et al. Invasive behaviour of glioblastoma cell lines is associated with altered organisation of the cadherin-catenin adhesion system. J Cell Sci (2002) 115(Pt 16):3331–40.
139. Mahabir R, Tanino M, Elmansuri A, Wang L, Kimura T, Itoh T, et al. Sustained elevation of Snail promotes glial-mesenchymal transition after irradiation in malignant glioma. Neuro Oncol (2014) 16(5):671–85. doi: 10.1093/neuonc/not239
140. Itoh Y, Moriyama Y, Hasegawa T, Endo TA, Toyoda T, Gotoh Y. Scratch regulates neuronal migration onset via an epithelial-mesenchymal transition-like mechanism. Nat Neurosci (2013) 16(4):416–25. doi: 10.1038/nn.3336
141. Zhang J, Cai H, Sun L, Zhan P, Chen M, Zhang F, et al. LGR5, a novel functional glioma stem cell marker, promotes EMT by activating the Wnt/β-catenin pathway and predicts poor survival of glioma patients. J Exp Clin Cancer Res: CR (2018) 37(1):225. doi: 10.1186/s13046-018-0864-6
142. Liu Q, Guan Y, Li Z, Wang Y, Liu Y, Cui R, et al. miR-504 suppresses mesenchymal phenotype of glioblastoma by directly targeting the FZD7-mediated Wnt-β-catenin pathway. J Exp Clin Cancer Res: CR (2019) 38(1):358. doi: 10.1186/s13046-019-1370-1
143. Zuccarini M, Giuliani P, Ziberi S, Carluccio M, Iorio PD, Caciagli F, et al. The Role of Wnt Signal in Glioblastoma Development and Progression: A Possible New Pharmacological Target for the Therapy of This Tumor. Genes (2018) 9(2):105. doi: 10.3390/genes9020105
144. Guo S, Liang X, Guo M, Zhang X, Li Z. Migration inhibition of water stress proteins from Nostoc commune Vauch. via activation of autophagy in DLD-1 cells. Int J Biol Macromol (2018) 119:669–76. doi: 10.1016/j.ijbiomac.2018.07.188
145. Colwell N, Larion M, Giles AJ, Seldomridge AN, Sizdahkhani S, Gilbert MR, et al. Hypoxia in the glioblastoma microenvironment: shaping the phenotype of cancer stem-like cells. Neuro-oncology (2017) 19(7):887–96. doi: 10.1093/neuonc/now258
146. Zhu H, Wang D, Zhang L, Xie X, Wu Y, Liu Y, et al. Upregulation of autophagy by hypoxia-inducible factor-1α promotes EMT and metastatic ability of CD133+ pancreatic cancer stem-like cells during intermittent hypoxia. Oncol Rep (2014) 32(3):935–42. doi: 10.3892/or.2014.3298
147. Qin W, Li C, Zheng W, Guo Q, Zhang Y, Kang M, et al. Inhibition of autophagy promotes metastasis and glycolysis by inducing ROS in gastric cancer cells. Oncotarget (2015) 6(37):39839–54. doi: 10.18632/oncotarget.5674
148. Huang W, Ding X, Ye H, Wang J, Shao J, Huang T. Hypoxia enhances the migration and invasion of human glioblastoma U87 cells through PI3K/Akt/mTOR/HIF-1α pathway. Neuroreport (2018) 29(18):1578–85. doi: 10.1097/wnr.0000000000001156
149. Deng M, Zhang W, Yuan L, Tan J, Chen Z. HIF-1a regulates hypoxia-induced autophagy via translocation of ANKRD37 in colon cancer. Exp Cell Res (2020) 395(1):112175. doi: 10.1016/j.yexcr.2020.112175
150. Tam SY, Wu VWC, Law HKW. Hypoxia-Induced Epithelial-Mesenchymal Transition in Cancers: HIF-1α and Beyond. Front Oncol (2020) 10:486:486. doi: 10.3389/fonc.2020.00486
151. Ghosh S, Paul A, Sen E. Tumor necrosis factor α-induced hypoxia-inducible factor 1α-β-catenin axis regulates major histocompatibility complex class I gene activation through chromatin remodeling. Mol Cell Biol (2013) 33(14):2718–31. doi: 10.1128/mcb.01254-12
152. Mazumdar J, O’Brien WT, Johnson RS, LaManna JC, Chavez JC, Klein PS, et al. O2 regulates stem cells through Wnt/β-catenin signalling. Nat Cell Biol (2010) 12(10):1007–13. doi: 10.1038/ncb2102
153. Fu Y, Zheng S, Zheng Y, Huang R, An N, Liang A, et al. Glioma derived isocitrate dehydrogenase-2 mutations induced up-regulation of HIF-1α and β-catenin signaling: possible impact on glioma cell metastasis and chemo-resistance. Int J Biochem Cell Biol (2012) 44(5):770–5. doi: 10.1016/j.biocel.2012.01.017
154. Náger M, Santacana M, Bhardwaj D, Valls J, Ferrer I, Nogués P, et al. Nuclear phosphorylated Y142 β-catenin accumulates in astrocytomas and glioblastomas and regulates cell invasion. Cell Cycle (Georgetown Tex) (2015) 14(22):3644–55. doi: 10.1080/15384101.2015.1104443
155. Rampazzo E, Persano L, Pistollato F, Moro E, Frasson C, Porazzi P, et al. Wnt activation promotes neuronal differentiation of glioblastoma. Cell Death Dis (2013) 4(2):e500. doi: 10.1038/cddis.2013.32
156. Shin S, Im HJ, Kwon YJ, Ye DJ, Baek HS, Kim D, et al. Human steroid sulfatase induces Wnt/β-catenin signaling and epithelial-mesenchymal transition by upregulating Twist1 and HIF-1α in human prostate and cervical cancer cells. Oncotarget (2017) 8(37):61604–17. doi: 10.18632/oncotarget.18645
157. Ha JH, Ward JD, Radhakrishnan R, Jayaraman M, Song YS, Dhanasekaran DN. Lysophosphatidic acid stimulates epithelial to mesenchymal transition marker Slug/Snail2 in ovarian cancer cells via Gαi2, Src, and HIF1α signaling nexus. Oncotarget (2016) 7(25):37664–79. doi: 10.18632/oncotarget.9224
158. Goossens S, Vandamme N, Van Vlierberghe P, Berx G. EMT transcription factors in cancer development re-evaluated: Beyond EMT and MET. Biochim Biophys Acta Rev Cancer (2017) 1868(2):584–91. doi: 10.1016/j.bbcan.2017.06.006
159. Liu T, Zou XZ, Huang N, Ge XY, Yao MZ, Liu H, et al. Down-regulation of miR-204 attenuates endothelial-mesenchymal transition by enhancing autophagy in hypoxia-induced pulmonary hypertension. Eur J Pharmacol (2019) 863:172673. doi: 10.1016/j.ejphar.2019.172673
160. Bagherian A, Mardani R, Roudi B, Taghizadeh M, Banfshe HR, Ghaderi A, et al. Combination therapy with nanomicellar-curcumin and temozolomide for in vitro therapy of glioblastoma multiforme via Wnt signaling pathways. J Mol Neurosci (2020) 70(10):1471–83. doi: 10.1007/s12031-020-01639-z
161. Shen X, Wu S, Zhang J, Li M, Xu F, Wang A, et al. Wild−type IDH1 affects cell migration by modulating the PI3K/AKT/mTOR pathway in primary glioblastoma cells. Mol Med Rep (2020) 22(3):1949–57. doi: 10.3892/mmr.2020.11250
162. Liu M, Dai B, Kang SH, Ban K, Huang FJ, Lang FF, et al. FoxM1B is overexpressed in human glioblastomas and critically regulates the tumorigenicity of glioma cells. Cancer Res (2006) 66(7):3593–602. doi: 10.1158/0008-5472.can-05-2912
163. Dai B, Kang S-H, Gong W, Liu M, Aldape K, Sawaya R, et al. Aberrant FoxM1B expression increases matrix metalloproteinase-2 transcription and enhances the invasion of glioma cells. Oncogene (2007) 26(42):6212–9. doi: 10.1038/sj.onc.1210443
164. Zhang C, Han X, Xu X, Zhou Z, Chen X, Tang Y, et al. FoxM1 drives ADAM17/EGFR activation loop to promote mesenchymal transition in glioblastoma. Cell Death Dis (2018) 9(5):469. doi: 10.1038/s41419-018-0482-4
165. Chen Y, Li Y, Xue J, Gong A, Yu G, Zhou A, et al. Wnt-induced deubiquitination FoxM1 ensures nucleus β-catenin transactivation. EMBO J (2016) 35(6):668–84. doi: 10.15252/embj.201592810
166. Guo L, Ding Z, Huang N, Huang Z, Zhang N, Xia Z. Forkhead Box M1 positively regulates UBE2C and protects glioma cells from autophagic death. Cell Cycle (Georgetown Tex) (2017) 16(18):1705–18. doi: 10.1080/15384101.2017.1356507
167. Pulvirenti T, Van Der Heijden M, Droms LA, Huse JT, Tabar V, Hall A. Dishevelled 2 signaling promotes self-renewal and tumorigenicity in human gliomas. Cancer Res (2011) 71(23):7280–90. doi: 10.1158/0008-5472.can-11-1531
168. Kafka A, Tomas D. Expression Levels and Localizations of DVL3 and sFRP3 in Glioblastoma. Dis Markers (2017) 2017:9253495. doi: 10.1155/2017/9253495
169. Shevchenko V, Arnotskaya N, Zaitsev S, Sharma A, Sharma HS, Bryukhovetskiy A, et al. Proteins of Wnt signaling pathway in cancer stem cells of human glioblastoma. Int Rev Neurobiol (2020) 151:185–200. doi: 10.1016/bs.irn.2020.03.006
170. Kim DH, Kim EJ, Kim DH, Park SW. Dact2 is involved in the regulation of epithelial-mesenchymal transition. Biochem Biophys Res Commun (2020) 524(1):190–7. doi: 10.1016/j.bbrc.2019.12.090
171. Tan Y, Li QM, Huang N, Cheng S, Zhao GJ, Chen H, et al. Upregulation of DACT2 suppresses proliferation and enhances apoptosis of glioma cell via inactivation of YAP signaling pathway. Cell Death Dis (2017) 8(8):e2981. doi: 10.1038/cddis.2017.385
172. Ma B, Cao W, Li W, Gao C, Qi Z, Zhao Y, et al. Dapper1 promotes autophagy by enhancing the Beclin1-Vps34-Atg14L complex formation. Cell Res (2014) 24(8):912–24. doi: 10.1038/cr.2014.84
173. Ren Y, Chen Y, Liang X, Lu Y, Pan W, Yang M. MiRNA-638 promotes autophagy and malignant phenotypes of cancer cells via directly suppressing DACT3. Cancer Lett (2017) 390:126–36. doi: 10.1016/j.canlet.2017.01.009
174. Arbab AS, Ali MM. Glioblastoma: Targeting Angiogenesis and Tyrosine Kinase Pathways. Novel Approaches Cancer Study (2020) 4(5):398.
175. Winkler F, Osswald M, Wick W. Anti-Angiogenics: Their Role in the Treatment of Glioblastoma. Oncol Res Treat (2018) 41(4):181–6. doi: 10.1159/000488258
176. Miranda-Gonçalves V, Cardoso-Carneiro D, Valbom I, Cury FP, Silva VA, Granja S, et al. Metabolic alterations underlying Bevacizumab therapy in glioblastoma cells. Oncotarget (2017) 8(61):103657–70. doi: 10.18632/oncotarget.21761
177. Wickström SA, Alitalo K, Keski-Oja J. Endostatin associates with integrin alpha5beta1 and caveolin-1, and activates Src via a tyrosyl phosphatase-dependent pathway in human endothelial cells. Cancer Res (2002) 62(19):5580–9.
178. Sudhakar A, Sugimoto H, Yang C, Lively J, Zeisberg M, Kalluri R. Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by alpha v beta 3 and alpha 5 beta 1 integrins. Proc Natl Acad Sci U States America (2003) 100(8):4766–71. doi: 10.1073/pnas.0730882100
179. Hanai J, Gloy J, Karumanchi SA, Kale S, Tang J, Hu G, et al. Endostatin is a potential inhibitor of Wnt signaling. J Cell Biol (2002) 158(3):529–39. doi: 10.1083/jcb.200203064
180. Hu P, Ma L, Wu ZQ, Zheng GY, Li JT. Effect of endostatin on proliferation, invasion and epithelial-mesenchymal transition of basal cell carcinoma cell A431. Eur Rev Med Pharmacol Sci (2019) 23(2):877–84. doi: 10.26355/eurrev_201901_16902
181. Nguyen TM, Subramanian IV, Xiao X, Ghosh G, Nguyen P, Kelekar A, et al. Endostatin induces autophagy in endothelial cells by modulating Beclin 1 and beta-catenin levels. J Cell Mol Med (2009) 13(9b):3687–98. doi: 10.1111/j.1582-4934.2009.00722.x
182. Chu C-W, Ko H-J, Chou C-H, Cheng T-S, Cheng H-W, Liang Y-H, et al. Thioridazine enhances P62-mediated autophagy and apoptosis through Wnt/β-catenin signaling pathway in glioma cells. Int J Mol Sci (2019) 20(3):473. doi: 10.3390/ijms20030473
183. Chen H, Lu Q, Chen C, Di Y, Yn L, Min W, et al. β−catenin regulates effects of miR−24 on the viability and autophagy of glioma cells. Exp Ther Med (2019) 18(2):1285–90. doi: 10.3892/etm.2019.7680
184. Esen E, Chen J, Karner CM, Okunade AL, Patterson BW, Long F. WNT-LRP5 signaling induces Warburg effect through mTORC2 activation during osteoblast differentiation. Cell Metab (2013) 17(5):745–55. doi: 10.1016/j.cmet.2013.03.017
185. Kanzawa T, Zhang L, Xiao L, Germano IM, Kondo Y, Kondo S. Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene (2005) 24(6):980–91. doi: 10.1038/sj.onc.1208095
186. Kim EH, Sohn S, Kwon HJ, Kim SU, Kim MJ, Lee SJ, et al. Sodium selenite induces superoxide-mediated mitochondrial damage and subsequent autophagic cell death in malignant glioma cells. Cancer Res (2007) 67(13):6314–24. doi: 10.1158/0008-5472.can-06-4217
187. Torres S, Lorente M, Rodríguez-Fornés F, Hernández-Tiedra S, Salazar M, García-Taboada E, et al. A combined preclinical therapy of cannabinoids and temozolomide against glioma. Mol Cancer Ther (2011) 10(1):90–103. doi: 10.1158/1535-7163.mct-10-0688
188. Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Different (2004) 11(4):448–57. doi: 10.1038/sj.cdd.4401359
189. Sotelo J, Briceño E, López-González MA. Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Ann Internal Med (2006) 144(5):337–43. doi: 10.7326/0003-4819-144-5-200603070-00008
190. Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, Brem S, et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy (2014) 10(8):1359–68. doi: 10.4161/auto.28984
191. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
192. Masui K, Cavenee WK, Mischel PS. mTORC2 and Metabolic Reprogramming in GBM: at the Interface of Genetics and Environment. Brain Pathol (Zurich Switzerland) (2015) 25(6):755–9. doi: 10.1111/bpa.12307
193. Zhou W, Wahl DR. Metabolic Abnormalities in Glioblastoma and Metabolic Strategies to Overcome Treatment Resistance. Cancers (Basel) (2019) 11(9):1231. doi: 10.3390/cancers11091231
194. Albrecht LV, Bui MH, De Robertis EM. Canonical Wnt is inhibited by targeting one-carbon metabolism through methotrexate or methionine deprivation. Proc Natl Acad Sci (2019) 116(8):2987–95. doi: 10.1073/pnas.1820161116
195. Ducker GS, Rabinowitz JD. One-Carbon Metabolism in Health and Disease. Cell Metab (2017) 25(1):27–42. doi: 10.1016/j.cmet.2016.08.009
196. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov (2012) 2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095
197. Kryukov GV, Wilson FH, Ruth JR, Paulk J, Tsherniak A, Marlow SE, et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science (2016) 351(6278):1214–8. doi: 10.1126/science.aad5214
198. Marjon K, Cameron MJ, Quang P, Clasquin MF, Mandley E, Kunii K, et al. MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis. Cell Rep (2016) 15(3):574–87. doi: 10.1016/j.celrep.2016.03.043
199. Mavrakis KJ, McDonald ER. 3rd, Schlabach MR, Billy E, Hoffman GR, deWeck A, et al. Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science (2016) 351(6278):1208–13. doi: 10.1126/science.aad5944
200. Banasavadi-Siddegowda YK, Welker AM, An M, Yang X, Zhou W, Shi G, et al. PRMT5 as a druggable target for glioblastoma therapy. Neuro-oncology (2018) 20(6):753–63. doi: 10.1093/neuonc/nox206
201. Tasset I, Cuervo AM. Role of chaperone-mediated autophagy in metabolism. FEBS J (2016) 283(13):2403–13. doi: 10.1111/febs.13677
202. Schneider JL, Suh Y, Cuervo AM. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab (2014) 20(3):417–32. doi: 10.1016/j.cmet.2014.06.009
203. Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell (2011) 42(6):719–30. doi: 10.1016/j.molcel.2011.04.025
204. Xia HG, Najafov A, Geng J, Galan-Acosta L, Han X, Guo Y, et al. Degradation of HK2 by chaperone-mediated autophagy promotes metabolic catastrophe and cell death. J Cell Biol (2015) 210(5):705–16. doi: 10.1083/jcb.201503044
205. Masui K, Tanaka K, Akhavan D, Babic I, Gini B, Matsutani T, et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab (2013) 18(5):726–39. doi: 10.1016/j.cmet.2013.09.013
206. Suwala AK, Hanaford A, Kahlert UD, Maciaczyk J. Clipping the Wings of Glioblastoma: Modulation of WNT as a Novel Therapeutic Strategy. J Neuropathol Exp Neurol (2016) 75(5):388–96. doi: 10.1093/jnen/nlw013
207. Suwala AK, Koch K, Rios DH, Aretz P, Uhlmann C, Ogorek I, et al. Inhibition of Wnt/beta-catenin signaling downregulates expression of aldehyde dehydrogenase isoform 3A1 (ALDH3A1) to reduce resistance against temozolomide in glioblastoma in vitro. Oncotarget (2018) 9(32):22703–16. doi: 10.18632/oncotarget.25210
208. Odia Y, Iwamoto FM, Moustakas A, Fraum TJ, Salgado CA, Li A, et al. A phase II trial of enzastaurin (LY317615) in combination with bevacizumab in adults with recurrent malignant gliomas. J Neuro-Oncol (2016) 127(1):127–35. doi: 10.1007/s11060-015-2020-x
209. Kurokawa C, Geekiyanage H, Allen C, Iankov I, Schroeder M, Carlson B, et al. Alisertib demonstrates significant antitumor activity in bevacizumab resistant, patient derived orthotopic models of glioblastoma. J Neuro-Oncol (2017) 131(1):41–8. doi: 10.1007/s11060-016-2285-8
210. Song A, Andrews DW, Werner-Wasik M, Kim L, Glass J, Bar-Ad V, et al. Phase I trial of alisertib with concurrent fractionated stereotactic re-irradiation for recurrent high grade gliomas. Radiother Oncol: J Eur Soc Ther Radiol Oncol (2019) 132:135–41. doi: 10.1016/j.radonc.2018.12.019
Keywords: glioblastoma, autophagy, microautophagy, chaperone-mediated autophagy, epithelial-mesenchymal transition, metabolic reprograming, WNT signaling
Citation: Coelho BP, Fernandes CFL, Boccacino JM, Souza MCS, Melo-Escobar MI, Alves RN, Prado MB, Iglesia RP, Cangiano G, Mazzaro GLR and Lopes MH (2020) Multifaceted WNT Signaling at the Crossroads Between Epithelial-Mesenchymal Transition and Autophagy in Glioblastoma. Front. Oncol. 10:597743. doi: 10.3389/fonc.2020.597743
Received: 21 August 2020; Accepted: 19 October 2020;
Published: 12 November 2020.
Edited by:
Eugenia Morselli, Pontificia Universidad Católica de Chile, ChileReviewed by:
Fabrizio Marcucci, University of Milan, ItalySabrina Di Bartolomeo, University of Molise, Italy
Copyright © 2020 Coelho, Fernandes, Boccacino, Souza, Melo-Escobar, Alves, Prado, Iglesia, Cangiano, Mazzaro and Lopes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marilene Hohmuth Lopes, bWFyaWxlbmVobEB1c3AuYnI=
†These authors have contributed equally to this work