 Gaoyang Zhu
Gaoyang Zhu Chaoyun Pan2
Chaoyun Pan2 Bo Li
Bo Li Yang Xu
Yang Xu Xuemei Fu
Xuemei Fu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 06 November 2020
Sec. Molecular and Cellular Oncology
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.595187
This article is part of the Research Topic Mutant p53 in Cancer Progression and Personalized Therapeutic Treatments View all 14 articles
TP53 is the most frequently mutated tumor suppressor gene in human cancer. The majority of mutations of p53 are missense mutations, leading to the expression of the full length p53 mutant proteins. Mutant p53 (Mutp53) proteins not only lose wild-type p53-dependent tumor suppressive functions, but also frequently acquire oncogenic gain-of-functions (GOF) that promote tumorigenesis. In this review, we summarize the recent advances in our understanding of the oncogenic GOF of mutp53 and the potential therapies targeting mutp53 in human cancers. In particular, we discuss the promising drugs that are currently under clinical trials as well as the emerging therapeutic strategies, including CRISPR/Cas9 based genome edition of mutant TP53 allele, small peptide mediated restoration of wild-type p53 function, and immunotherapies that directly eliminate mutp53 expressing tumor cells.
Tumor suppressor p53 is the principal cellular responder to various stress signals such as oncogene activation, DNA damage, hypoxia, reactive oxygen species (ROS), etc. Upon activation, p53 induces numerous cellular responses including cell cycle arrest to restore genetic integrity, or apoptosis, senescence, or ferroptosis to eliminate unrecoverable cells. Therefore, p53 is considered the “Guardian of the genome” to prevent accumulation of oncogenic mutations that lead to malignant tumor (1, 2).
Mutations in TP53 are found in over half of human cancers, thus is known as the most commonly mutated gene in human cancers (3, 4). Different from many other tumor suppressor genes which generally undergo deletion or truncation in cancer cells, mutations in TP53 allele are predominantly missense mutations which give rise to a single amino acid substitution in the full-length mutant protein (5). p53 protein is composed of three functional domains including a transactivation and proline rich domain, a central DNA-binding domain (DBD), and an oligomerization domain (6). While mutations can occur spontaneously throughout the p53 gene, the majority of p53 missense mutations are located in the central DBD region of the p53 gene, which binds to its consensus DNA binding sites to regulate its target gene expression. These missense mutations are divided into two categories: DNA contact mutations such as R248Q and R273H that directly occur at the amino acids mediating p53-DNA interaction, and conformational mutations that indirectly disrupt p53-DNA interaction by inducing local (R249S and G245S) or global (R175H and R282W) conformational changes due to the reduced thermostability caused by these mutations (5). Nearly one third of all p53 mutations occur at these six “hotspot” mutational residues (3).
Mutations in the p53 gene can appear at either the initial-stage or the late-stage during tumorigenesis depending on the origin of cancer types, and strongly facilitate the onset or progression of cancers (7). Functionally, p53 mutants (mutp53) not only lead to the loss of wild-type p53 functions, but can also result in a dominant negative effect by forming hetero-tetramers with the remaining wild-type p53 expressed from the other wild-type allele. p53 mutations are usually followed by the loss of heterozygosity (LOH) at the remaining wild-type TP53 allele, leading to the complete loss of wild-type p53 in late-stage tumors, and further confer these cancer cells a selective advantage during cancer development (8, 9). Most p53 missense mutants acquire oncogenic gain-of-function (GOF) activities. For example, conformational changes of mutp53 enable them to interact with many transcription factors such as p63, p73, NF-Y, Sp1, ETS1/2, NF-κB, ATM, and SMADs, altering the transcription, cell cycle, apoptosis and metabolism of cancer cells. This changes lead to increased genetic instability, cellular proliferation, metastasis and chemo-/radio-resistance (10). In addition, the new transcriptional targets acquired by mutp53 is another well established GOF mechanism for mutp53 to promote cancer progression (5). Therefore, to compete for survival in a nutrient-deprived and hypoxic environment, the human tumor cells are under stringent selection for the loss of wild-type p53 function and acquirement of p53 mutants that possess GOF to promote the survival of tumor cells.
In this review, we focus on the gain-of-functions of mutp53 in malignant tumor progression and the current strategies targeting mutp53 for personalized therapeutic treatments, aiming to provide insights into targeted treatment of human cancers with p53 mutation.
As the “Guardian of the genome”, the fundamental goal of WT p53 is to maintain genetic stability by preventing the passage of genetic mutations to daughter cells (1). While p53 null cells still retain certain levels of checkpoint and DNA repair capacities, cells harboring p53 mutant proteins showed a dramatic higher level of genomic instability such as interchromosomal translocations and aneuploidy, indicating the oncogenic GOF activity of p53 mutants (8, 11, 12). These variations largely contribute to genetic diversity that expedites malignant tumor development. Mechanistically, the common p53 mutants can disrupt the earliest stage of DNA double-stranded break (DSB) damage responses by interacting with the nuclease Mre11 to suppress the recruitment of Mre11/Rad50/NBS1 (MRN) complex to the site of DNA DSB damage, leading to inactivation of ATM, the key DNA DSB damage sensor, and the resultant G2/M checkpoint impairment (8). Mutp53 can also induce genomic abnormality by inactivating DNA replication process. For example, some mutp53 proteins activate cyclin A to promote the formation of DNA replication origin and the intra-S phase checkpoint kinase CHK1 to stabilize the replication forks, facilitating the duplication of aberrant genomic DNAs (13).
Accumulating evidence has revealed that mutp53 promotes the limitless replicative potential and insensitivity to anti-growth signals during the malignant transformation of a normal cell, which are two of the key “hallmarks of cancer” (14, 15). Mutp53 was reported to physically interact with the major cell cycle regulator nuclear transcription factor Y (NF-Y), and recruits either acetyltransferase p300 or the main effector of Hippo pathway, YAP, to activate NF-Y target genes including cyclin A, cyclin B, cdk1 and cdc25C (16, 17). Mutant p53 and YAP have also been found to form another trimeric transcriptional complex with TEAD to induce the expression of circular RNA circPVT1, which activates proliferative genes such as aurka and mki67 (18). Mutp53 also regulates the expression of MicroRNA miR-27a, which promotes a sustained EGF-induced ERK1/2 activation, thereby facilitating cellular proliferation and tumorigenesis (19). In addition, p53 mutants also target key chromatin regulators including methyltransferases MLL1 and MLL2 and acetyltransferase MOZ, leading to genome-wide increases of active histone modifications H3K4me3 and H3K9ac to enhance proliferation (20). In addition, the TP53 R249S mutant, frequently detected in HBV positive human hepatocellular carcinoma (HCC), has a unique GOF in regulating proliferation and survival of HCC cells by promoting c-Myc-dependent rDNA transcription essential for ribosomal biogenesis (21).
Cellular metabolism of glucose, lipid, and nucleotide are the fundamental basis for cell survival, which undergo dramatic changes during malignant transformation. Emerging evidence shows that mutp53 proteins contribute to various aspects of these processes (22, 23). Rapidly dividing tumor cells rely mainly on aerobic glycolysis to meet their high energy and biosynthetic demand, a phenomenon known as Warburg effect (24). Mutp53 has been shown to activate the small GTPase RhoA and its downstream effector ROCK, to promote GLUT1 translocation to the plasma membrane and thus enhance glucose uptake and glycolytic rate (25). Under conditions of energy stress, mutp53 preferentially binds to the AMPKα subunit and directly inhibits the metabolic functions of AMPK signaling, leading to increased aerobic glycolysis as well as lipid production (26). Notably, the roles of mutp53 in promoting lipid metabolism are partly mediated by the mevalonate pathway, which is responsible for de novo cholesterol synthesis and generation of many important nonsterol isoprenoid derivatives. Mutp53 is recruited to the promoters of several mevalonate pathway genes to induce their expression through its interaction with the master transcription factor SREBP1/SREBP2 (27). In addition, the mevalonate pathway-DNAJA1 axis as well as the STAT3-mevalonate pathway axis are both found to prevent mutp53 from being degraded by CHIP ubiquitin ligase, forming a positive-feedback loop to ensure rapid lipid synthesis (28, 29). Mutp53 proteins were also reported to promote nucleotide synthesis through its interaction with ETS2 to activate numerous nucleotide metabolism genes (RRM2, dCK, TK1, GMPS, IMPDH1, PAICS) involved in both the de novo and the salvage pathways required for nucleotide synthesis, leading to elevated nucleotide pools and the subsequent enhancement of GTP dependent protein (GTPase) activity (30, 31). Collectively, these findings highlighted the metabolic reprogramming roles of mutp53 in cancer cells.
Metastasis is another “hallmark of cancer” and contributes to over 90% of cancer-associated deaths (15, 32). Epithelial-to-mesenchymal transition (EMT) is the first and the most essential step of metastasis that allows the cells to change their morphology to gain enhanced migration and invasion capacity. Mutp53 were reported to promote the expression of several key EMT-related transcription factors including ZEB1, SLUG, and TWIST1 through transcriptional, post-translational and epigenetic modifications, possibly in a cell type dependent manner (33–35). In endometrial cancer tissues, mutp53 represses the expression of miR-130b, which negatively regulates ZEB1 (33). In non-small-cell lung cancers (NSCLCs), mutp53 inactivates MDM2 mediated SLUG degradation and result in high SLUG and low E-cadherin expression (34). While in prostate cells, mutp53 induces the reduction of H3K27me3 repression mark on TWIST1 promoter (35). Besides, several lines of evidence suggest that p63 is an effector of mutant p53 mediated metastasis. Mutp53 forms a ternary complex with p63 and phosphorylated Smad2 in the presence of TGF-β signaling, which repressed the activation of p63 downstream metastasis suppressor genes Cyclin G2 and Sharp-1 (36). Mutp53 also inhibits p63 mediated inactivation of Rab-coupling protein (RCP), resulting in enhanced α5β1-integrin and EGFR trafficking to the plasma membrane and the constitutive activation of EGFR/integrin signaling and its downstream pro-metastatic Akt signaling (37, 38). Besides, the activation of RCP by mutp53 also enhances HSP90α secretion, which increases cell motility through interaction with extracellular matrix (ECM) (39). Metabolism reprogramming is also involved in mutp53 induced metastasis. Using a p53R172H/+ mice model (R175H in human), Xiong. et al. found that the interaction between mutp53 and ETS2 also induces Pla2g16 expression, which encodes a phospholipase that catalyzes phosphatidic acid into lysophosphatidic acid and free fatty acid, and both of which have been implicated in promoting migration and metastasis (40–42). Besides, the common polymorphism Pro72Arg at mutp53 enhances migration and metastasis of tumors through its ability to bind and regulate PGC-1α target genes, which is a key regulator in mitochondrial biogenesis and oxidative phosphorylation (43, 44).
Chemotherapy and radiotherapy are currently the most widely used therapies for metastatic cancers. However, tumor cells always develop ways to evolve radio- and chemo-resistance capacity to survive these therapies, and mutation in the p53 gene is one of the crucial attempts (45). In this context, mutp53 proteins regulate the expression of several chemo- and radio-resistant genes. MDR1 (multi-drug resistance 1) encodes an energy-dependent efflux pump that mediates the resistance of tumor cells to various hydrophobic cytotoxic drugs (46). Mutp53 proteins strongly upregulate MDR1 expression through ETS1 mediated promoter binding, while the restoration of WT p53 could abolish MDR1 activity by reducing its phosphorylation (47–49). Mutp53 activates the expression of NRF2, which is known to confer both chemo- and radio-resistance (50), including chemo-resistance of cisplatin, apigenin, and radio-resistance of tumor cells (51–53). In triple-negative breast cancer cells, the cooperation between mutp53 and NRF2 was reported to activate proteasome gene transcription, resulting in resistance to proteasome inhibitor carfilzomib (54). Therefore, targeting NRF2 pathway has the potential to increase the curcumin compound induced cell death of mutp53-carrying cancer cells (55). In addition, in cells with WT p53, DNA damage caused by radiotherapy and most chemotherapeutic agents would lead to p53 accumulation and apoptosis. Whereas certain mutp53 has been reported to inhibit caspase-9 and p63/73-dependent induction of Bax and Noxa, contributing to the anti-apoptotic effects of mutp53 and the insensitivity of mutp53 harboring cells to radio- and chemo-therapies (56–58).
Emerging evidence suggests that the radio- and chemo-resistance capacity are primarily achieved by cancer stem cells (CSC) (59–61). Mutp53 proteins play vital roles in CSC formation and maintenance (62). High prevalence of p53 mutations is reported in poorly differentiated carcinomas and contributes to a stem cell-like transcriptome (63, 64). WT p53 has been reported to repress the expression of several CSC markers including CD44, c-KIT, NANOG and OCT4, while mutations in p53 would lead to loss of repression on these CSC markers, subsequent CSC transformation and the resultant enhanced radio- and chemo-resistance (65).
It is now accepted that tumor progression and response to therapeutic treatments are not simply dependent on cell autonomous characteristics. The tumor microenvironment consisting mainly of ECM, stromal cells, immune cells, and blood vessels plays a key role in the tumorigenesis and chemoresistance capacity (15). Mutp53 can modulate tumor microenvironment by inducing the secretion of pro-inflammatory cytokines and angiogenesis (66, 67). The p53R248W and D281G mutants can activate the activity of matrix metalloproteinases (MMPs) by repressing the transcription of TIMP3 (68). Consistently, colorectal carcinomas expressing p53R273H,V216M show significant upregulation of MMP9 expression (69). The increased MMP activity results in the degradation of ECM surrounding the tumor cells, leading to enhanced metastasis and invasiveness (68, 70). Mutp53 has also been reported to form complex with HIF1 to upregulate ECM components Viia1 collagen and laminin-γ2 to promote tumor progression (71). In addition, the crosstalk between Mutp53 and the master inflammatory regulator NF-κB pathway has been largely implicated in modulating tumor development and migration (69, 72–74), through the upregulation of a cancer-related gene signature including CXC-chemokines, interleukins (ILs) and ECM-related genes (73–75). Finally, mutp53 was reported to positively regulate the expression of pro-angiogenic factors including IL-8, GRO-α, and VEGF to promote tumor neo-angiogenesis, which is another “hallmark of cancer” (15, 76, 77).
The reliance of tumors on mutp53 makes it an ideal target for cancer therapy. Therapeutic strategies targeting mutp53 can be divided into three categories, restoring the WT conformation and transcriptional activity of mutp53, targeting mutp53 for degradation, and inducing synthetic lethality (78, 79). To achieve these therapeutic goals, small molecular compounds, synthetic small peptides, CRISPR/Cas9 mediated genome editing, small interference RNAs (RNAi) as well as immunotherapies have been explored (Figure 1).
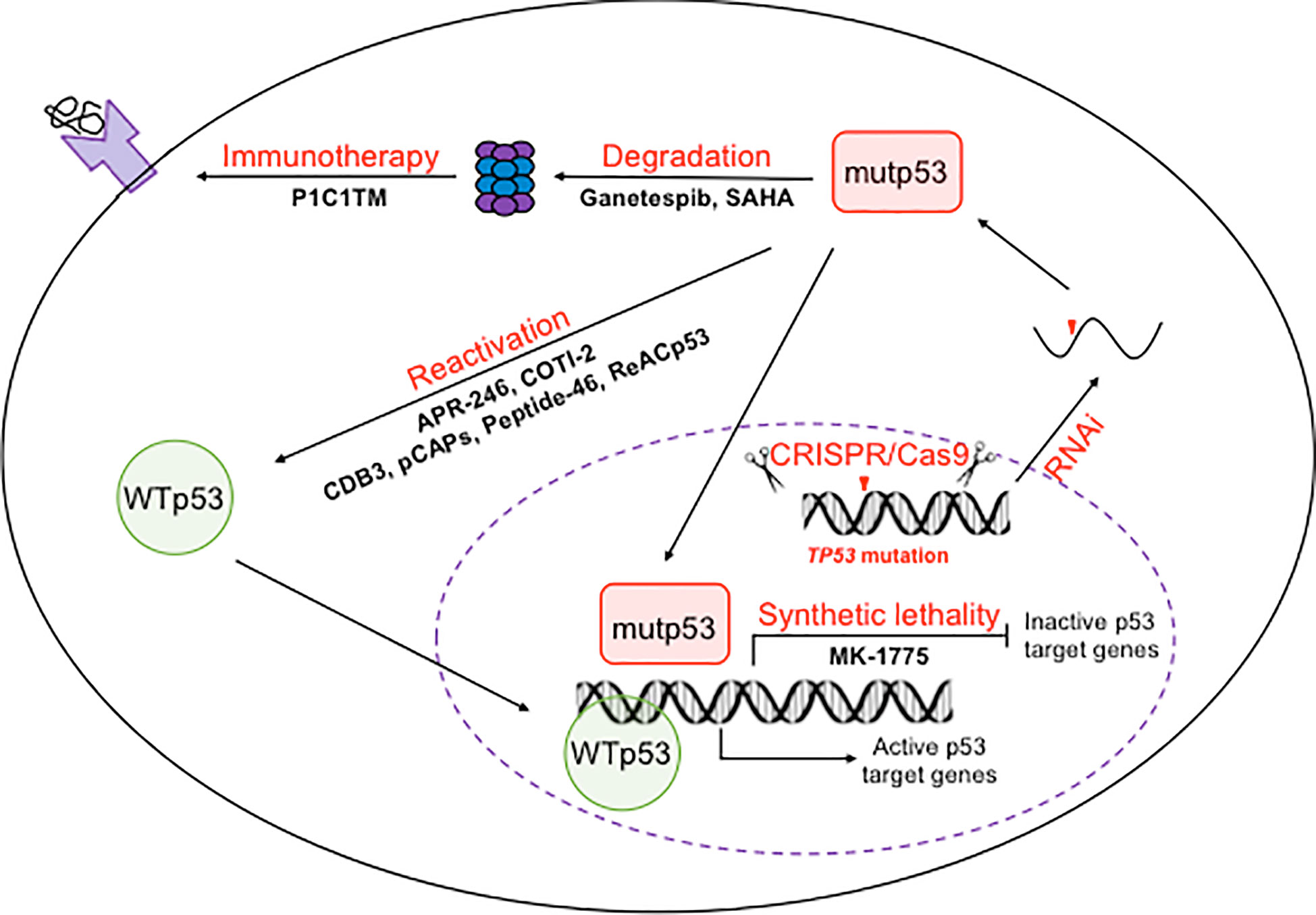
Figure 1 Therapeutic strategies to target p53 mutants. On the DNA level, mutations in TP53 allele could be reversed back to wild-type ones using CRISPR/Cas9 mediated genome editing. One the mRNA levels, mutp53 mRNA could be silenced by RNAi. On the protein level, mutp53 could be reactivated or trageted for degradation by both small molecule compounds and small peptides. The inability of mutp53 to activate its downstream target genes provides an opportunity for synthetic lethality based therapy. The mutant peptides produced by degradation of mutp53 makes immunotherapies possible.
Pharmaceutical targeting of mutp53 is more challenging than targeting oncogenic kinases, which can be easily inactivated by small molecule inhibitors (80). While intensively pursued, no mutp53 targeting regiment has yet been clinically approved. Several promising small molecule drugs under clinical trials are reviewed below (ClinicalTrials.gov). Some other drugs that have achieved promising results in preclinical studies have been extensively reviewed elsewhere (79, 80).
APR-246 is a methylated analogue of PRIMA-1, which was identified as a low-molecular-weight compound that restores wildtype function of mutp53 (81, 82). While methylene quinuclidinone (MQ) is the common bioactive decomposition product of both APR-246 and PRIMA-1, the bioactivity of APR-246 is much higher, possibly due to its higher lipophilicity and cell permeability (80, 82). The covalent binding of MQ to p53 core domain, primarily via cysteines 124 and 277, enhances the thermostability of mutp53 and contributes to the refolding of mutp53 to WT conformation, thus enables the re-induction of p53 target genes such as CDKN1A (83–85). Numerous preclinical studies using rodent models have revealed the tumor suppressive effect of APR-246 on mutp53-expressing tumor cells of various origins (86–90). Furthermore, the phase I study has verified that APR-246 is safe at predicted therapeutic plasma levels with a favorable pharmacokinetic profile, and most importantly, can induce considerable p53-dependent biological effects in cancer patients with p53 mutations (91, 92). Therefore, APR-246 is considered as a promising first-in-class mutp53-targeting drug. Now, several phase II clinical trials of APR-246 are ongoing, including a systemic carboplatin combination chemotherapy with APR-246 in patients with platinum sensitive recurrent high grade serous ovarian cancer with mutated p53 (NCT02098343), a combination of APR-246 with azacytidine in p53 mutant myeloid neoplasms (NCT03072043) and a combination of APR-246 with 5-FU and cisplatin in oesophageal cancer (NCT02999893).
COTI-2 is a novel thiosemicarbazone derivative that is active against multiple human cancers from different origins (93). The anti-tumor activity of COTI-2 is at least partially achieved by promoting the refolding and therefore the DNA binding capacity of mutp53, leading to the reactivation of wildtype p53 target genes including CDKN1A, PUMA, and NOXA. Besides, MAPK and mTOR pathways are also involved in COTI-2 induced apoptosis or senescence (80, 94, 95). COTI-2 is effective at nanomolar concentrations in vitro, and is proved to be safe and well-tolerated in xenograft mouse models (93). Following studies revealed that COTI-2 was synergistic in combinations with cytotoxic chemotherapeutics without exerting significant toxicities in vivo. In addition, tumor cells resistant to chemotherapeutic agents exhibit no or little cross-resistance to COTI-2, highlighted the potential of COTI-2 in salvage treatment after current first- and second-line treatment failures (96). Based on these observations, a phase I trial of COTI-2 as monotherapy or combination therapy in gynecological tumors and head and neck squamous cell carcinoma (HNSCC) with confirmed p53 mutations is currently being performed (NCT02433626).
In contrast to the relative low levels of wildtype p53 in unstressed physiological conditions, mutp53 is in most cases highly expressed in tumor cell, which is achieved by its cooperation with HSP90 chaperone machinery that inhibit the activity of its primary E3 ubiquitin ligase MDM2 and CHIP (97, 98). This hyperstabilization of mutp53 largely contributes to its dominant-negative and oncogenic GOF activities, and is the foundation of anti-tumor therapies aimed to induce mutp53 degradation. Ganetespib is a highly efficient HSP90 inhibitor (99), which is 50-fold more potent than the first-generation HSP90 inhibitor 17AAG in degrading mutp53 and killing mutp53 cancer cells (100). In vivo studies suggested that ganetespib extended the survival of tumor-bearing R172H (corresponding to R175H in human) and R248Q Trp53 knock-in mice, while have no effect on their corresponding Trp53-/- littermates (100). Meanwhile, in Trp53R248Q/- mice bearing T-lymphomas, ganetespib synergizes with cyclophosphamide to suppress tumor growth and extend survival (101). However, it is worth noticing that mutp53 is not the only target of HSP90, instead, HSP90 regulates the activation and stability of a diverse array of oncogenic proteins including HER2, mutant EGFR, and mutant BRAF (99). Even though phase II clinical trials in metastatic breast cancer, malignant peripheral nerve sheath tumors and advanced non-small cell lung cancer all reported that the effect of ganetespib alone or in combination with other anti-tumor drugs did not meet the criteria for overall response rate, subgroups of these patients showed positive responses, which might be attributed to their specific genetic background (102–104). Therefore, more extensive clinical trials with ganetespib are needed.
Histone deacetylase (HDAC) inhibitors are another group of compounds that are widely reported to reduce the levels of mutp53. SAHA (suberoylanilide hydroxamic acid) is a FDA-approved HDAC inhibitor for the treatment of T cell lymphomas (105). Recent studies found that SAHA exhibits preferential cytotoxicity for mutp53, rather than WT and null p53 cancer cells in certain kinds of human cancers, and also strongly sensitizes mutp53 harboring cancer cells to chemotherapies (100, 106, 107). Mechanistically, SAHA could destabilize mutp53 through inhibition of the HDAC6-HSP90 chaperone axis, and at the same time, inhibit the transcription of mutp53 through HDAC8 (106–109).
p53 is mainly responsible for the G1/S cell cycle arrest, while in mutp53 harboring cancer cells, the abrogation of this checkpoint results in direct S phase entry even in the presence of DNA damage, making the cells more dependent on G2/M checkpoint to maintain genomic stability (110). In this context, further inactivation of G2/S checkpoint will lead to unscheduled mitotic entry of cells with extensive DNA damage, resulting in mitotic catastrophe (111, 112). This synthetic lethality provides an ideal opportunity for therapeutic targeting of mutp53 harboring cancer cells. Wee-1 is a tyrosine kinase that involved in DNA damage induced G2/M cell cycle arrest by inhibiting CDK1 activity (113). Its specific inhibitor MK-1775, therefore, was reported to show amplified anti-tumor activity specifically in p53 mutant cancer cells. MK-1775 significantly elevated the efficacy of cisplatin, vorinostat (HADC inhibitor), or alisertib (aurora kinase A inhibitor) in HNSCC cells expressing high-risk mutp53 both in vitro and in vivo, while tumor cells bearing wildtype p53 displayed minimal response to MK-1775 (114–117). Consistently, MK-1775 was also reported to sensitize p53 mutant colon cancer cells to the DNA damage associated drug irinotecan (118). Currently, a randomized phase II study evaluating MK-1775 in combination with paclitaxel and carboplatin in adult patients with platinum sensitive p53 mutant ovarian cancer is ongoing (NCT01357161).
CRISPR/Cas9-based genome editing appears to be a straight-forward therapeutic strategies for tumor cells expressing p53 mutants. By directly replacing the TP53 414delC frameshift mutation locus with a functional copy, Batir et al. successfully restored the wild-type TP53 genotype and phenotype in prostate cancer cells (119). CRISPR/Cas9 has also also employed in a p53 genetic sensor system which specifically and efficiently killed p53-deficient cancer cells (120). However, the high risk of genome instabiliy induced by CRISPR/Cas9 should be rigorously considered (121, 122). Small interference RNAs could specifically eliminate mutant p53 mRNA without affecting the wild-type one, However, the specificity and in vivo efficacy of such RNAi remains to be elucidated.
The goal of small peptide based therapies is to restore wild-type p53 function, either by restabilization of mutp53 or inhibition of the aggregation of mutp53. The denaturation of mutp53 at physiological temperature largely contributes to the inabilily of mutp53 to activate downstream tumor suppressive genes. Therefore, several mutp53 reactivating peptides, such as CDB3, peptide-46 and pCAPs, have been identified to restore wildtype p53 activities to cancer cells (123, 124). On the other hand, a large portion of p53 mutants have been reported to form protein aggregates, which contributes to the GOF properties that promote tumor growth. In this context, ReACp53, a cell-penetrating peptide inhibitor of mutp53’s aggregation, which resembles the transactivation inhibitory domain of p63, showed promising anti-cancer effect in both ovarain and prostate cancer models in vivo (125–127).
While the accumulated mutp53 escapes from MDM2-mediated degradation, it can still be degraded in a MDM2-independent and proteasome-dependent pathway, generating peptides that are eventually presentated on tumor cell surface by class I molecules of the major histocompatibility comples (MHC). Therefore, mutp53 and the p53-derived mutant peptide-MHCs could serve as potental therapeutic targets for immunotherapies (128, 129). Even though peptides containing mutp53 sequences are rare due to MHC-binding restrictions, an engineered T cell receptor-like (TCRL) antibody P1C1TM, which is specific for a wild-type p53125-134 peptide presented by the HLA-A24:02 (HLA-24) MHC allele, was reported to be able to discriminate between mutant and wild-type p53-expressing HLA-A24+ cells based on antigen expression levels. This elegant interaction between intracellular mutp53 and targetable cell surface peptide-MHC complex enables efficient antibody dependent cellular cytotoxicity of mutp53 expressing cells both in vitro and in vivo (129). In the future, it is worthwhile to identify new cell surface peptides specifically derived from mutp53.
The addiction of cancer cells to mutp53 makes it an attractive target for cancer therapy. By elucidating the mechanisms of GOFs of mutp53, numerous strategies have been explored to specifically target mutp53. One highly pursued strategy is to develop small molecule compound and small peptide to restore the conformation and transcriptional activity of wild-type p53 to the mutp53. This strategy is challenging due to the relative undruggable nature of mutp53 with various thermostability or conformational structures. Therefore, high-resolution structural and functional analysis of the full length WT and mutp53 will be required to design more effective small molecule compounds and small peptides to target mutp53. However, it is noteworthy that our group recently found that hepatocellular carcinomas (HCCs) often retain the wild-type p53 to suppress oxidative phosphorylation and increase glycolysis, thereby promoting HCC progression (130). In this context, strategies aiming to restore WT p53 activities of mutp53 might instead promote tumorigenesis under certain circumstances, therefore requires rigorous validation before clinical trials. Synthetic lethality, gene editing, siRNA silencing, and immunotherapy are promising strategies to target mutp53 to treat mutp53-expressing tumors, however, these approaches all have intrinsic problems that must be optimized before clinical applications. In this context, future effort should be devoted to improve the specificity, efficacy, and safety of these promising strategies to target mutp53-expressing human cancers.
GZ, CP, JB, BL, CL, YX, and XF drafted the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (Nos. 91959204, 81930084, 815300045, 81871197, 81703092), the leading talents of Guangdong Province Program (No. 00201516), Shenzhen “Sanming” Project of Medicine (SZSM201602102), and the Development and Reform Commission of Shenzhen Municipality (S2016004730009).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
2. Levine AJ. p53: 800 million years of evolution and 40 years of discovery. Nat Rev Cancer (2020) 20:471–80. doi: 10.1038/s41568-020-0262-1
3. Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science (1991) 253:49–53. doi: 10.1126/science.1905840
4. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature (2013) 502:333–9. doi: 10.1038/nature12634
5. Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer (2009) 9:701–13. doi: 10.1038/nrc2693
6. Raj N, Attardi LD. The Transactivation Domains of the p53 Protein. Cold Spring Harb Perspect Med (2017) 7:a026047. doi: 10.1101/cshperspect.a026047
7. Levine AJ, Jenkins NA, Copeland NG. The Roles of Initiating Truncal Mutations in Human Cancers: The Order of Mutations and Tumor Cell Type Matters. Cancer Cell (2019) 35:10–5. doi: 10.1016/j.ccell.2018.11.009
8. Song H, Hollstein M, Xu Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat Cell Biol (2007) 9:573–80. doi: 10.1038/ncb1571
9. Amelio I, Melino G. Context is everything: extrinsic signalling and gain-of-function p53 mutants. Cell Death Discovery (2020) 6:16. doi: 10.1038/s41420-020-0251-x
10. D’Orazi G, Cirone M. Mutant p53 and Cellular Stress Pathways: A Criminal Alliance That Promotes Cancer Progression. Cancers (Basel) (2019) 11:614. doi: 10.3390/cancers11050614
11. Liu DP, Song H, Xu Y. A common gain of function of p53 cancer mutants in inducing genetic instability. Oncogene (2010) 29:949–56. doi: 10.1038/onc.2009.376
12. Hanel W, Moll UM. Links between mutant p53 and genomic instability. J Cell Biochem (2012) 113:433–9. doi: 10.1002/jcb.23400
13. Singh S, Vaughan CA, Frum RA, Grossman SR, Deb S, Palit Deb S. Mutant p53 establishes targetable tumor dependency by promoting unscheduled replication. J Clin Invest (2017) 127:1839–55. doi: 10.1172/JCI87724
14. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell (2000) 100:57–70. doi: 10.1016/S0092-8674(00)81683-9
15. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
16. Di Agostino S, Strano S, Emiliozzi V, Zerbini V, Mottolese M, Sacchi A, et al. Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell (2006) 10:191–202. doi: 10.1016/j.ccr.2006.08.013
17. Di Agostino S, Sorrentino G, Ingallina E, Valenti F, Ferraiuolo M, Bicciato S, et al. YAP enhances the pro-proliferative transcriptional activity of mutant p53 proteins. EMBO Rep (2016) 17:188–201. doi: 10.15252/embr.201540488
18. Verduci L, Ferraiuolo M, Sacconi A, Ganci F, Vitale J, Colombo T, et al. The oncogenic role of circPVT1 in head and neck squamous cell carcinoma is mediated through the mutant p53/YAP/TEAD transcription-competent complex. Genome Biol (2017) 18:237. doi: 10.1186/s13059-017-1368-y
19. Wang W, Cheng B, Miao L, Mei Y, Wu M. Mutant p53-R273H gains new function in sustained activation of EGFR signaling via suppressing miR-27a expression. Cell Death Dis (2013) 4:e574. doi: 10.1038/cddis.2013.97
20. Zhu J, Sammons MA, Donahue G, Dou Z, Vedadi M, Getlik M, et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature (2015) 525:206–11. doi: 10.1038/nature15251
21. Liao P, Zeng SX, Zhou X, Chen T, Zhou F, Cao B, et al. Mutant p53 Gains Its Function via c-Myc Activation upon CDK4 Phosphorylation at Serine 249 and Consequent PIN1 Binding. Mol Cell (2017) 68:1134–1146 e1136. doi: 10.1016/j.molcel.2017.11.006
22. Eriksson M, Ambroise G, Ouchida AT, Lima Queiroz A, Smith D, Gimenez-Cassina A, et al. Effect of Mutant p53 Proteins on Glycolysis and Mitochondrial Metabolism. Mol Cell Biol (2017) 37:e00328–17. doi: 10.1128/MCB.00328-17
23. Liu J, Zhang C, Hu W, Feng Z. Tumor suppressor p53 and metabolism. J Mol Cell Biol (2019) 11:284–92. doi: 10.1093/jmcb/mjy070
24. Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer (2011) 11:325–37. doi: 10.1038/nrc3038
25. Zhang C, Liu J, Liang Y, Wu R, Zhao Y, Hong X, et al. Tumour-associated mutant p53 drives the Warburg effect. Nat Commun (2013) 4:2935. doi: 10.1038/ncomms3935
26. Zhou G, Wang J, Zhao M, Xie TX, Tanaka N, Sano D, et al. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol Cell (2014) 54:960–74. doi: 10.1016/j.molcel.2014.04.024
27. Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell (2012) 148:244–58. doi: 10.1016/j.cell.2011.12.017
28. Parrales A, Ranjan A, Iyer SV, Padhye S, Weir SJ, Roy A, et al. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat Cell Biol (2016) 18:1233–43. doi: 10.1038/ncb3427
29. Romeo MA, Gilardini Montani MS, Benedetti R, Santarelli R, D’Orazi G, Cirone M. STAT3 and mutp53 Engage a Positive Feedback Loop Involving HSP90 and the Mevalonate Pathway. Front Oncol (2020) 10:1102. doi: 10.3389/fonc.2020.01102
30. Kollareddy M, Dimitrova E, Vallabhaneni KC, Chan A, Le T, Chauhan KM, et al. Regulation of nucleotide metabolism by mutant p53 contributes to its gain-of-function activities. Nat Commun (2015) 6:7389. doi: 10.1038/ncomms8389
31. Schmidt V, Nagar R, Martinez LA. Control of Nucleotide Metabolism Enables Mutant p53’s Oncogenic Gain-of-Function Activity. Int J Mol Sci (2017) 18:2759. doi: 10.3390/ijms18122759
32. Gupta GP, Massague J. Cancer metastasis: building a framework. Cell (2006) 127:679–95. doi: 10.1016/j.cell.2006.11.001
33. Dong P, Karaayvaz M, Jia N, Kaneuchi M, Hamada J, Watari H, et al. Mutant p53 gain-of-function induces epithelial–mesenchymal transition through modulation of the miR-130b–ZEB1 axis. Oncogene (2012) 32:3286–95. doi: 10.1038/onc.2012.334
34. Wang S-P, Wang W-L, Chang Y-L, Wu C-T, Chao Y-C, Kao S-H, et al. p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of Slug. Nat Cell Biol (2009) 11:694–704. doi: 10.1038/ncb1875
35. Kogan-Sakin I, Tabach Y, Buganim Y, Molchadsky A, Solomon H, Madar S, et al. Mutant p53(R175H) upregulates Twist1 expression and promotes epithelial-mesenchymal transition in immortalized prostate cells. Cell Death Differ (2011) 18:271–81. doi: 10.1038/cdd.2010.94
36. Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell (2009) 137:87–98. doi: 10.1016/j.cell.2009.01.039
37. Selivanova G, Ivaska J. Integrins and mutant p53 on the road to metastasis. Cell (2009) 139:1220–2. doi: 10.1016/j.cell.2009.12.016
38. Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, et al. Mutant p53 drives invasion by promoting integrin recycling. Cell (2009) 139:1327–41. doi: 10.1016/j.cell.2009.11.026
39. Zhang S, Wang C, Ma B, Xu M, Xu S, Liu J, et al. Mutant p53 Drives Cancer Metastasis via RCP-Mediated Hsp90alpha Secretion. Cell Rep (2020) 32:107879. doi: 10.1016/j.celrep.2020.107879
40. Xiong S, Tu H, Kollareddy M, Pant V, Li Q, Zhang Y, et al. Pla2g16 phospholipase mediates gain-of-function activities of mutant p53. Proc Natl Acad Sci U S A (2014) 111:11145–50. doi: 10.1073/pnas.1404139111
41. Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer (2003) 3:582–91. doi: 10.1038/nrc1143
42. Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell (2010) 140:49–61. doi: 10.1016/j.cell.2009.11.027
43. Tang Q, Su Z, Gu W, Rustgi AK. Mutant p53 on the Path to Metastasis. Trends Cancer (2020) 6:62–73. doi: 10.1016/j.trecan.2019.11.004
44. Basu S, Gnanapradeepan K, Barnoud T, Kung CP, Tavecchio M, Scott J, et al. Mutant p53 controls tumor metabolism and metastasis by regulating PGC-1alpha. Genes Dev (2018) 32:230–43. doi: 10.1101/gad.309062.117
45. Lu C, El-Deiry WS. Targeting p53 for enhanced radio- and chemo-sensitivity. Apoptosis (2009) 14:597–606. doi: 10.1007/s10495-009-0330-1
46. Takara K, Sakaeda T, Okumura K. An update on overcoming MDR1-mediated multidrug resistance in cancer chemotherapy. Curr Pharm Des (2006) 12:273–86. doi: 10.2174/138161206775201965
47. Chin KV, Ueda K, Pastan I, Gottesman MM. Modulation of activity of the promoter of the human MDR1 gene by Ras and p53. Science (1992) 255:459–62. doi: 10.1126/science.1346476
48. Sampath J, Sun D, Kidd VJ, Grenet J, Gandhi A, Shapiro LH, et al. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J Biol Chem (2001) 276:39359–67. doi: 10.1074/jbc.M103429200
49. Zhan M, Yu D, Liu J, Glazer RI, Hannay J, Pollock RE. Transcriptional repression of protein kinase Calpha via Sp1 by wild type p53 is involved in inhibition of multidrug resistance 1 P-glycoprotein phosphorylation. J Biol Chem (2005) 280:4825–33. doi: 10.1074/jbc.M407450200
50. Rojo de la Vega M, Chapman E, Zhang DD. NRF2 and the Hallmarks of Cancer. Cancer Cell (2018) 34:21–43. doi: 10.1016/j.ccell.2018.03.022
51. Gilardini Montani MS, Cecere N, Granato M, Romeo MA, Falcinelli L, Ciciarelli U, et al. Mutant p53, Stabilized by Its Interplay with HSP90, Activates a Positive Feed-Back Loop Between NRF2 and p62 that Induces Chemo-Resistance to Apigenin in Pancreatic Cancer Cells. Cancers (Basel) (2019) 11:703. doi: 10.3390/cancers11050703
52. Tung MC, Lin PL, Wang YC, He TY, Lee MC, Yeh SD, et al. Mutant p53 confers chemoresistance in non-small cell lung cancer by upregulating Nrf2. Oncotarget (2015) 6:41692–705. doi: 10.18632/oncotarget.6150
53. Jeong Y, Hoang NT, Lovejoy A, Stehr H, Newman AM, Gentles AJ, et al. Role of KEAP1/NRF2 and TP53 Mutations in Lung Squamous Cell Carcinoma Development and Radiation Resistance. Cancer Discovery (2017) 7:86–101. doi: 10.1158/2159-8290.CD-16-0127
54. Walerych D, Lisek K, Sommaggio R, Piazza S, Ciani Y, Dalla E, et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat Cell Biol (2016) 18:897–909. doi: 10.1038/ncb3380
55. Garufi A, Baldari S, Pettinari R, Gilardini Montani MS, D’Orazi V, Pistritto G, et al. A ruthenium(II)-curcumin compound modulates NRF2 expression balancing the cancer cell death/survival outcome according to p53 status. J Exp Clin Cancer Res (2020) 39:122. doi: 10.1186/s13046-020-01628-5
56. Irwin MS, Kondo K, Marin MC, Cheng LS, Hahn WC, Kaelin WG Jr. Chemosensitivity linked to p73 function. Cancer Cell (2003) 3:403–10. doi: 10.1016/s1535-6108(03)00078-3
57. Chee JL, Saidin S, Lane DP, Leong SM, Noll JE, Neilsen PM, et al. Wild-type and mutant p53 mediate cisplatin resistance through interaction and inhibition of active caspase-9. Cell Cycle (2013) 12:278–88. doi: 10.4161/cc.23054
58. Liu K, Ling S, Lin WC. TopBP1 mediates mutant p53 gain of function through NF-Y and p63/p73. Mol Cell Biol (2011) 31:4464–81. doi: 10.1128/MCB.05574-11
59. Schulz A, Meyer F, Dubrovska A, Borgmann K. Cancer Stem Cells and Radioresistance: DNA Repair and Beyond. Cancers (Basel) (2019) 11:862. doi: 10.3390/cancers11060862
60. Prieto-Vila M, Takahashi RU, Usuba W, Kohama I, Ochiya T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int J Mol Sci (2017) 18:2574. doi: 10.3390/ijms18122574
61. Najafi M, Farhood B, Mortezaee K. Cancer stem cells (CSCs) in cancer progression and therapy. J Cell Physiol (2019) 234:8381–95. doi: 10.1002/jcp.27740
62. Shetzer Y, Solomon H, Koifman G, Molchadsky A, Horesh S, Rotter V. The paradigm of mutant p53-expressing cancer stem cells and drug resistance. Carcinogenesis (2014) 35:1196–208. doi: 10.1093/carcin/bgu073
63. Mizuno H, Spike BT, Wahl GM, Levine AJ. Inactivation of p53 in breast cancers correlates with stem cell transcriptional signatures. Proc Natl Acad Sci U S A (2010) 107:22745–50. doi: 10.1073/pnas.1017001108
64. Fagin JA, Matsuo K, Karmakar A, Chen DL, Tang SH, Koeffler HP. High prevalence of mutations of the p53 gene in poorly differentiated human thyroid carcinomas. J Clin Invest (1993) 91:179–84. doi: 10.1172/JCI116168
65. Olivos DJ, Mayo LD. Emerging Non-Canonical Functions and Regulation by p53: p53 and Stemness. Int J Mol Sci (2016) 17:1982. doi: 10.3390/ijms17121982
66. Stein Y, Aloni-Grinstein R, Rotter V. Mutant p53-a potential player in shaping the tumor-stroma crosstalk. J Mol Cell Biol (2019) 11:600–4. doi: 10.1093/jmcb/mjz071
67. Cordani M, Pacchiana R, Butera G, D’Orazi G, Scarpa A, Donadelli M. Mutant p53 proteins alter cancer cell secretome and tumour microenvironment: Involvement in cancer invasion and metastasis. Cancer Lett (2016) 376:303–9. doi: 10.1016/j.canlet.2016.03.046
68. Loging WT, Reisman D. Inhibition of the putative tumor suppressor gene TIMP-3 by tumor-derived p53 mutants and wild type p53. Oncogene (1999) 18:7608–15. doi: 10.1038/sj.onc.1203135
69. Rahnamoun H, Lu H, Duttke SH, Benner C, Glass CK, Lauberth SM. Mutant p53 shapes the enhancer landscape of cancer cells in response to chronic immune signaling. Nat Commun (2017) 8:754. doi: 10.1038/s41467-017-01117-y
70. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell (2010) 141:52–67. doi: 10.1016/j.cell.2010.03.015
71. Amelio I, Mancini M, Petrova V, Cairns RA, Vikhreva P, Nicolai S, et al. p53 mutants cooperate with HIF-1 in transcriptional regulation of extracellular matrix components to promote tumor progression. Proc Natl Acad Sci U S A (2018) 115:E10869–78. doi: 10.1073/pnas.1808314115
72. Di Minin G, Bellazzo A, Dal Ferro M, Chiaruttini G, Nuzzo S, Bicciato S, et al. Mutant p53 reprograms TNF signaling in cancer cells through interaction with the tumor suppressor DAB2IP. Mol Cell (2014) 56:617–29. doi: 10.1016/j.molcel.2014.10.013
73. Yeudall WA, Vaughan CA, Miyazaki H, Ramamoorthy M, Choi MY, Chapman CG, et al. Gain-of-function mutant p53 upregulates CXC chemokines and enhances cell migration. Carcinogenesis (2012) 33:442–51. doi: 10.1093/carcin/bgr270
74. Solomon H, Buganim Y, Kogan-Sakin I, Pomeraniec L, Assia Y, Madar S, et al. Various p53 mutant proteins differently regulate the Ras circuit to induce a cancer-related gene signature. J Cell Sci (2012) 125:3144–52. doi: 10.1242/jcs.099663
75. Buganim Y, Solomon H, Rais Y, Kistner D, Nachmany I, Brait M, et al. p53 Regulates the Ras circuit to inhibit the expression of a cancer-related gene signature by various molecular pathways. Cancer Res (2010) 70:2274–84. doi: 10.1158/0008-5472.CAN-09-2661
76. Fontemaggi G, Dell’Orso S, Trisciuoglio D, Shay T, Melucci E, Fazi F, et al. The execution of the transcriptional axis mutant p53, E2F1 and ID4 promotes tumor neo-angiogenesis. Nat Struct Mol Biol (2009) 16:1086–93. doi: 10.1038/nsmb.1669
77. Kieser A, Weich HA, Brandner G, Marme D, Kolch W. Mutant p53 potentiates protein kinase C induction of vascular endothelial growth factor expression. Oncogene (1994) 9:963–9.
78. Shaheen M, Allen C, Nickoloff JA, Hromas R. Synthetic lethality: exploiting the addiction of cancer to DNA repair. Blood (2011) 117:6074–82. doi: 10.1182/blood-2011-01-313734
79. Parrales A, Iwakuma T. Targeting Oncogenic Mutant p53 for Cancer Therapy. Front Oncol (2015) 5:288. doi: 10.3389/fonc.2015.00288
80. Bykov VJN, Eriksson SE, Bianchi J, Wiman KG. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer (2018) 18:89–102. doi: 10.1038/nrc.2017.109
81. Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med (2002) 8:282–8. doi: 10.1038/nm0302-282
82. Bykov VJ, Zache N, Stridh H, Westman J, Bergman J, Selivanova G, et al. PRIMA-1(MET) synergizes with cisplatin to induce tumor cell apoptosis. Oncogene (2005) 24:3484–91. doi: 10.1038/sj.onc.1208419
83. Zhang Q, Bykov VJN, Wiman KG, Zawacka-Pankau J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis (2018) 9:439. doi: 10.1038/s41419-018-0463-7
84. Wassman CD, Baronio R, Demir O, Wallentine BD, Chen CK, Hall LV, et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat Commun (2013) 4:1407. doi: 10.1038/ncomms2361
85. Lambert JM, Gorzov P, Veprintsev DB, Soderqvist M, Segerback D, Bergman J, et al. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell (2009) 15:376–88. doi: 10.1016/j.ccr.2009.03.003
86. Liu DS, Read M, Cullinane C, Azar WJ, Fennell CM, Montgomery KG, et al. APR-246 potently inhibits tumour growth and overcomes chemoresistance in preclinical models of oesophageal adenocarcinoma. Gut (2015) 64:1506–16. doi: 10.1136/gutjnl-2015-309770
87. Zandi R, Selivanova G, Christensen CL, Gerds TA, Willumsen BM, Poulsen HS. PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin Cancer Res (2011) 17:2830–41. doi: 10.1158/1078-0432.CCR-10-3168
88. Mohell N, Alfredsson J, Fransson A, Uustalu M, Bystrom S, Gullbo J, et al. APR-246 overcomes resistance to cisplatin and doxorubicin in ovarian cancer cells. Cell Death Dis (2015) 6:e1794. doi: 10.1038/cddis.2015.143
89. Perdrix A, Najem A, Saussez S, Awada A, Journe F, Ghanem G, et al. PRIMA-1 and PRIMA-1(Met) (APR-246): From Mutant/Wild Type p53 Reactivation to Unexpected Mechanisms Underlying Their Potent Anti-Tumor Effect in Combinatorial Therapies. Cancers (Basel) (2017) 9:172. doi: 10.3390/cancers9120172
90. Krayem M, Journe F, Wiedig M, Morandini R, Najem A, Sales F, et al. p53 Reactivation by PRIMA-1(Met) (APR-246) sensitises (V600E/K)BRAF melanoma to vemurafenib. Eur J Cancer (2016) 55:98–110. doi: 10.1016/j.ejca.2015.12.002
91. Deneberg S, Cherif H, Lazarevic V, Andersson PO, von Euler M, Juliusson G, et al. An open-label phase I dose-finding study of APR-246 in hematological malignancies. Blood Cancer J (2016) 6:e447. doi: 10.1038/bcj.2016.60
92. Lehmann S, Bykov VJ, Ali D, Andren O, Cherif H, Tidefelt U, et al. Targeting p53 in vivo: a first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J Clin Oncol (2012) 30:3633–9. doi: 10.1200/JCO.2011.40.7783
93. Salim KY, Maleki Vareki S, Danter WR, Koropatnick J. COTI-2, a novel small molecule that is active against multiple human cancer cell lines in vitro and in vivo. Oncotarget (2016) 7:41363–79. doi: 10.18632/oncotarget.9133
94. Lindemann A, Patel AA, Silver NL, Tang L, Liu Z, Wang L, et al. COTI-2, A Novel Thiosemicarbazone Derivative, Exhibits Antitumor Activity in HNSCC through p53-dependent and -independent Mechanisms. Clin Cancer Res (2019) 25:5650–62. doi: 10.1158/1078-0432.CCR-19-0096
95. Synnott NC, O’Connell D, Crown J, Duffy MJ. COTI-2 reactivates mutant p53 and inhibits growth of triple-negative breast cancer cells. Breast Cancer Res Treat (2020) 179:47–56. doi: 10.1007/s10549-019-05435-1
96. Maleki Vareki S, Salim KY, Danter WR, Koropatnick J. Novel anti-cancer drug COTI-2 synergizes with therapeutic agents and does not induce resistance or exhibit cross-resistance in human cancer cell lines. PloS One (2018) 13:e0191766. doi: 10.1371/journal.pone.0191766
97. Lukashchuk N, Vousden KH. Ubiquitination and degradation of mutant p53. Mol Cell Biol (2007) 27:8284–95. doi: 10.1128/MCB.00050-07
98. Li D, Marchenko ND, Schulz R, Fischer V, Velasco-Hernandez T, Talos F, et al. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol Cancer Res (2011) 9:577–88. doi: 10.1158/1541-7786.MCR-10-0534
99. Proia DA, Bates RC. Ganetespib and HSP90: translating preclinical hypotheses into clinical promise. Cancer Res (2014) 74:1294–300. doi: 10.1158/0008-5472.CAN-13-3263
100. Alexandrova EM, Yallowitz AR, Li D, Xu S, Schulz R, Proia DA, et al. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature (2015) 523:352–6. doi: 10.1038/nature14430
101. Alexandrova EM, Xu S, Moll UM. Ganetespib synergizes with cyclophosphamide to improve survival of mice with autochthonous tumors in a mutant p53-dependent manner. Cell Death Dis (2017) 8:e2683. doi: 10.1038/cddis.2017.108
102. Ramalingam S, Goss G, Rosell R, Schmid-Bindert G, Zaric B, Andric Z, et al. A randomized phase II study of ganetespib, a heat shock protein 90 inhibitor, in combination with docetaxel in second-line therapy of advanced non-small cell lung cancer (GALAXY-1). Ann Oncol (2015) 26:1741–8. doi: 10.1093/annonc/mdv220
103. Kim A, Lu Y, Okuno SH, Reinke D, Maertens O, Perentesis J, et al. Targeting Refractory Sarcomas and Malignant Peripheral Nerve Sheath Tumors in a Phase I/II Study of Sirolimus in Combination with Ganetespib (SARC023). Sarcoma (2020) 2020:5784876. doi: 10.1155/2020/5784876
104. Jhaveri K, Chandarlapaty S, Lake D, Gilewski T, Robson M, Goldfarb S, et al. A phase II open-label study of ganetespib, a novel heat shock protein 90 inhibitor for patients with metastatic breast cancer. Clin Breast Cancer (2014) 14:154–60. doi: 10.1016/j.clbc.2013.12.012
105. Marks PA. Discovery and development of SAHA as an anticancer agent. Oncogene (2007) 26:1351–6. doi: 10.1038/sj.onc.1210204
106. Li D, Marchenko ND, Moll UM. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ (2011) 18:1904–13. doi: 10.1038/cdd.2011.71
107. Foggetti G, Ottaggio L, Russo D, Mazzitelli C, Monti P, Degan P, et al. Autophagy induced by SAHA affects mutant P53 degradation and cancer cell survival. Biosci Rep (2019) 39:BSR20181345. doi: 10.1042/BSR20181345
108. Wang ZT, Chen ZJ, Jiang GM, Wu YM, Liu T, Yi YM, et al. Histone deacetylase inhibitors suppress mutant p53 transcription via HDAC8/YY1 signals in triple negative breast cancer cells. Cell Signal (2016) 28:506–15. doi: 10.1016/j.cellsig.2016.02.006
109. Yan W, Liu S, Xu E, Zhang J, Zhang Y, Chen X, et al. Histone deacetylase inhibitors suppress mutant p53 transcription via histone deacetylase 8. Oncogene (2013) 32:599–609. doi: 10.1038/onc.2012.81
110. Heijink AM, Blomen VA, Bisteau X, Degener F, Matsushita FY, Kaldis P, et al. A haploid genetic screen identifies the G1/S regulatory machinery as a determinant of Wee1 inhibitor sensitivity. Proc Natl Acad Sci U S A (2015) 112:15160–5. doi: 10.1073/pnas.1505283112
111. Portugal J, Mansilla S, Bataller M. Mechanisms of drug-induced mitotic catastrophe in cancer cells. Curr Pharm Des (2010) 16:69–78. doi: 10.2174/138161210789941801
112. Geenen JJJ, Schellens JHM. Molecular Pathways: Targeting the Protein Kinase Wee1 in Cancer. Clin Cancer Res (2017) 23:4540–4.
113. Beck H, Nahse-Kumpf V, Larsen MS, O’Hanlon KA, Patzke S, Holmberg C, et al. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol Cell Biol (2012) 32:4226–36. doi: 10.1128/MCB.00412-12
114. Osman AA, Monroe MM, Ortega Alves MV, Patel AA, Katsonis P, Fitzgerald AL, et al. Wee-1 kinase inhibition overcomes cisplatin resistance associated with high-risk TP53 mutations in head and neck cancer through mitotic arrest followed by senescence. Mol Cancer Ther (2015) 14:608–19. doi: 10.1158/1535-7163
115. Moser R, Xu C, Kao M, Annis J, Lerma LA, Schaupp CM, et al. Functional kinomics identifies candidate therapeutic targets in head and neck cancer. Clin Cancer Res (2014) 20:4274–88. doi: 10.1158/1078-0432.CCR-13-2858
116. Tanaka N, Patel AA, Tang L, Silver NL, Lindemann A, Takahashi H, et al. Replication Stress Leading to Apoptosis within the S-phase Contributes to Synergism between Vorinostat and AZD1775 in HNSCC Harboring High-RiskTP53Mutation. Clin Cancer Res (2017) 23:6541–54. doi: 10.1158/1078-0432.CCR-17-0947
117. Lee JW, Parameswaran J, Sandoval-Schaefer T, Eoh KJ, Yang DH, Zhu F, et al. Combined Aurora Kinase A (AURKA) and WEE1 Inhibition Demonstrates Synergistic Antitumor Effect in Squamous Cell Carcinoma of the Head and Neck. Clin Cancer Res (2019) 25:3430–42. doi: 10.1158/1078-0432.CCR-18-0440
118. Yin Y, Shen Q, Tao R, Chang W, Li R, Xie G, et al. Wee1 inhibition can suppress tumor proliferation and sensitize p53 mutant colonic cancer cells to the anticancer effect of irinotecan. Mol Med Rep (2018) 17:3344–9. doi: 10.3892/mmr.2017.8230
119. Batir MB, Sahin E, Cam FS. Evaluation of the CRISPR/Cas9 directed mutant TP53 gene repairing effect in human prostate cancer cell line PC-3. Mol Biol Rep (2019) 46:6471–84. doi: 10.1007/s11033-019-05093-y
120. Zhan H, Xie H, Zhou Q, Liu Y, Huang W. Synthesizing a Genetic Sensor Based on CRISPR-Cas9 for Specifically Killing p53-Deficient Cancer Cells. ACS Synth Biol (2018) 7:1798–807. doi: 10.1021/acssynbio.8b00202
121. Xu S, Kim J, Tang Q, Chen Q, Liu J, Xu Y, et al. CAS9 is a genome mutator by directly disrupting DNA-PK dependent DNA repair pathway. Protein Cell (2020) 11:352–65. doi: 10.1007/s13238-020-00699-6
122. Kosicki M, Tomberg K, Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol (2018) 36:765–71. doi: 10.1038/nbt.4192
123. Binayke A, Mishra S, Suman P, Das S, Chander H. Awakening the “guardian of genome”: reactivation of mutant p53. Cancer Chemother Pharmacol (2019) 83:1–15. doi: 10.1007/s00280-018-3701-x
124. Tal P, Eizenberger S, Cohen E, Goldfinger N, Pietrokovski S, Oren M, et al. Cancer therapeutic approach based on conformational stabilization of mutant p53 protein by small peptides. Oncotarget (2016) 7:11817–37. doi: 10.18632/oncotarget.7857
125. Zhang Y, Xu L, Chang Y, Li Y, Butler W, Jin E, et al. Therapeutic potential of ReACp53 targeting mutant p53 protein in CRPC. Prostate Cancer Prostatic Dis (2020) 23:160–71. doi: 10.1038/s41391-019-0172-z
126. Soragni A, Janzen DM, Johnson LM, Lindgren AG, Thai-Quynh Nguyen A, Tiourin E, et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell (2016) 29:90–103. doi: 10.1016/j.ccell.2015.12.002
127. Kehrloesser S, Osterburg C, Tuppi M, Schafer B, Vousden KH, Dotsch V. Intrinsic aggregation propensity of the p63 and p73 TI domains correlates with p53R175H interaction and suggests further significance of aggregation events in the p53 family. Cell Death Differ (2016) 23:1952–60. doi: 10.1038/cdd.2016.75
128. Sobhani N, D’Angelo A, Wang X, Young KH, Generali D, Li Y. Mutant p53 as an Antigen in Cancer Immunotherapy. Int J Mol Sci (2020) 21:4087. doi: 10.3390/ijms21114087
129. Low L, Goh A, Koh J, Lim S, Wang CI. Targeting mutant p53-expressing tumours with a T cell receptor-like antibody specific for a wild-type antigen. Nat Commun (2019) 10:5382. doi: 10.1038/s41467-019-13305-z
Keywords: mutant p53 protein, gain-of-function, targeted therapy, tumorigenesis, drug resistance
Citation: Zhu G, Pan C, Bei J-X, Li B, Liang C, Xu Y and Fu X (2020) Mutant p53 in Cancer Progression and Targeted Therapies. Front. Oncol. 10:595187. doi: 10.3389/fonc.2020.595187
Received: 15 August 2020; Accepted: 12 October 2020;
Published: 06 November 2020.
Edited by:
Jerson L. Silva, Federal University of Rio de Janeiro, BrazilReviewed by:
Gabriella D’Orazi, G. D’Annunzio University of Chieti-Pescara, ItalyCopyright © 2020 Zhu, Pan, Bei, Li, Liang, Xu and Fu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yang Xu, eWFuZ3h1QHVjc2QuZWR1; Xuemei Fu, ZnhtemoyMDA0QDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.