 Martin Böttcher
Martin Böttcher Rebecca Baur
Rebecca Baur Andrej Stoll
Andrej Stoll Andreas Mackensen
Andreas Mackensen Dimitrios Mougiakakos
Dimitrios Mougiakakos
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 05 November 2020
Sec. Cancer Immunity and Immunotherapy
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.594782
This article is part of the Research Topic New Insights into the Complexity of Tumor Immunology in B-cell Malignancies: Disease Biology and Signaling View all 14 articles
Lymphomas represent a diverse group of malignancies that emerge from lymphocytes. Despite improvements in diagnosis and treatment of lymphomas of B-cell origin, relapsed and refractory disease represents an unmet clinical need. Therefore, it is of utmost importance to better understand the lymphomas’ intrinsic features as well as the interactions with their cellular microenvironment for developing novel therapeutic strategies. In fact, the role of immune-based approaches is steadily increasing and involves amongst others the use of monoclonal antibodies against tumor antigens, inhibitors of immunological checkpoints, and even genetically modified T-cells. Metabolic reprogramming and immune escape both represent well established cancer hallmarks. Tumor metabolism as introduced by Otto Warburg in the early 20th century promotes survival, proliferation, and therapeutic resistance. Simultaneously, malignant cells employ a plethora of mechanisms to evade immune surveillance. Increasing evidence suggests that metabolic reprogramming does not only confer cell intrinsic growth and survival advantages to tumor cells but also impacts local as well as systemic anti-tumor immunity. Tumor and immune cells compete over nutrients such as carbohydrates or amino acids that are critical for the immune cell function. Moreover, skewed metabolic pathways in malignant cells can result in abundant production and release of bioactive metabolites such as lactic acid, kynurenine or reactive oxygen species (ROS) that affect immune cell fitness and function. This “metabolic re-modeling” of the tumor microenvironment shifts anti-tumor immune reactivity toward tolerance. Here, we will review molecular events leading to metabolic alterations in B-cell lymphomas and their impact on anti-tumor immunity.
Metabolic reprogramming is a well-established hallmark of cancer (1). In fact, emerging evidence suggests that metabolic reprogramming does not only confer bioenergetic advantages but also impacts immune surveillance, thus being closely interconnected with immune escape, another hallmark of cancer (1). In this mini-review on B-cell-derived lymphomas, we will focus on metabolic alterations, the underlying molecular mechanisms, and their impact on anti-tumor immunity.
In general, cells meet their energetic demands in form of adenosine triphosphate (ATP) by different degrees of either glycolysis or mitochondrial oxidative phosphorylation (OxPhos). Although both metabolic axes happen simultaneously, there is a distinct fine-tuning of the balance between glycolysis and OxPhos. Non-proliferating, quiescent or differentiated cells primarily rely on OxPhos in the presence of oxygen and only switch to glycolysis under hypoxia. Highly proliferative cells obtain most of their ATP by aerobic glycolysis despite the availability of oxygen, a mechanism introduced by Otto Warburg in the 1920s and referred to as the “Warburg effect” (2).
Although aerobic glycolysis is less efficient, energy generation is faster and provides intermediates such as nucleotides, amino acids, and carbons for fatty acid (FA)/lipid synthesis; all crucial components for dividing cells. Consequently, aerobic glycolysis is predominantly found in activated immune cells such as effector T-cells (3) but also in most dividing malignant cells (4).
OxPhos provides an enhanced metabolic flexibility as it can be fueled by different sources, primarily glucose, glutamine, and FAs. Cell types that preferentially utilize OxPhos are slow-dividing, long-lived immune cells such as regulatory and memory T-cells (5) or leukemia stem cells (6).
Apart from the energy provision, metabolic processes impact a plethora of cellular functions by amongst others interfering with translation, epigenetics, and post-translational modifications as reviewed by Patel et al. (7). As such, metabolism represents a central regulator of cell fate and function.
Owed to its vital importance metabolism is tightly regulated by both intrinsic and extrinsic mechanisms. Malignant cells display aberrations in those regulatory circuits leading to metabolic profiles that favor survival, growth, and immune escape. In the follow, we will discuss the most prominent metabolic alterations described in B-cell-derived Non-Hodgkin lymphomas (B-NHLs) including chronic lymphocytic leukemia (CLL).
As the most common form of B-cell lymphomas, aggressive DLBCL accounts for about 35% of all newly diagnosed B-NHLs (8). The DLBCL displays a pronounced heterogeneity in terms of genetic background and outcome. The cell of origin (COO) algorithm categorizes DLBCLs based on the gene expression profiles (GEPs) into “germinal center B-cell like” (GCB) types resembling normal germinal center B-cells and “activated B-cell like” (ABC) types with GEPs reminiscent of in vitro activated B-cells (9–11). An additional classification framework known as consensus cluster classification (CCC) revealed three separate clusters with distinct metabolic fingerprints: OxPhos-DLBCL, B-cell receptor (BCR)-DLBCL, and host response (HR)-DLBCL (12). OxPhos-DLBCLs display a prominent mitochondrial component, with elevated OxPhos, an overall increased mitochondrial contribution to the total energy turnover, and a greater incorporation of carbons derived from FAs and glucose into the tricarboxylic acid (TCA) cycle. In contrast, non-OxPhos-DLBCLs are metabolically rewired toward aerobic glycolysis (13). Immunohistochemical studies in DLBCL revealed expression of transporters of lactate (i.e., MCT1 and TOMM20) that can fuel the TCA cycle of malignant cells in a process better known as the “reverse Warburg effect” (14). Interestingly, OxPhos-DLBCL exhibited marked susceptibility toward inhibition of mitochondrial FA oxidation (FAO) and of PPARγ that regulates FA uptake and storage (13). BCR-DLBCLs were susceptible to pharmacological SYK inhibition (15), which in turn leads to a downregulation of glycolytic components (such as GLUT1 and hexokinase 2) (16).
As a central hub for the integration of metabolic processes, mammalian target of rapamycin (mTOR) controls nutrient/amino acid sensing, glycolysis, OxPhos, and consequently proliferation and survival. It serves as the core component of two multi-protein complexes (mTORC1 and mTORC2) that regulate different cell processes [reviewed in (17)]. Non-GCB DLBCLs depict increased mTOR-activity, which is linked to inferior survival (18). However, an in vitro study conducted on different DLBCL cell lines demonstrated therapeutic efficacy of mTOR inhibitors independent of COO. Overall, no clear link between COO- or CCC-based classifications and mTOR activity could be established yet.
Furthermore, DLBCLs (over-)express indoleamine-2,3-dioxygenase (IDO), which catalyzes breakdown of the essential amino acid L-tryptophan into the catabolite L-kynurenine (19). The latter one could promote expression of the pro-oncogenic Bcl-6 in DLCBL (20). In fact, both enhanced IDO expression (21) as well as elevated serum L-kynurenine levels (22) were linked to reduced response rates and inferior 3-yr overall survival (OS).
The second most common type of B-NHL is the indolent follicular lymphoma (FL) (23). In FL SYK is, similar to DLBCL, highly activated and regulates mTOR (24). In addition, recurrent somatic mutations of RRAGC that encodes for the Ras-related GTP-binding protein C are the leading cause for mTORC1 activation in FL (25) and render FL cells more susceptible toward mTOR-induced cytotoxicity (26). Beyond that, metabolism of FLs remains largely unexplored. Notably, transformation into DLBCL is associated with an enhanced expression of the glycolytic machinery, which is in line with the increased glucose uptake as revealed by 18F-FDG PET/CT in transformed lymphomas (27, 28).
MCL represents about 5-10% of B-NHLs. Despite being classified as indolent, it has a rather aggressive disease course. MCL cell lines display constitutive mTOR activation (29). A dysregulation of the upstream PI3K/AKT pathway has been implicated as a driver of mTOR in MCL. This notion is further corroborated by the observation that the phosphatase and tensin homologue (PTEN), which acts as an intrinsic PI3K/AKT inhibitor, can be reduced or undetectable in MCL (30). Inhibiting mTOR was effective in targeting MCL metabolism (31) and is approved for the relapsed/refractory (r/r) situation based on positive data from clinical studies (32). The Bruton tyrosine kinase (BTK) inhibitor ibrutinib abolishes BCR signaling and has emerged as a potent therapeutic option for r/r MCL. BTK-blockade markedly affected the (ibrutinib-responsive) MCLs’ metabolic activity including glycolysis and the TCA cycle (33). Interestingly, Zhang et al. reported that ibrutinib-resistant MCLs depict a metatobolic rewiring toward glutaminolysis-fueled OxPhos (34). These drug-resistant cells were readily targeted by a OxPhos inhibitor, showing promising efficacy in patient-derived preclinical models.
CLL as the most common adult leukemia of the Western world is a heterogeneous disease characterized by accumulating monoclonal B-lymphocytes (35). Circulating CLL cells are mainly quiescent and proliferation predominantly occurs in lymph nodes (LNs) and the bone marrow (BM). Nevertheless, circulating CLL cells possess a marked metabolic activity that differs from healthy B-lymphocytes. As they traffic between hypoxic (i.e., LN and BM) and normoxic compartments (i.e., peripheral blood), CLL cells were found to constitutively express hypoxia-inducible factor (HIF-1α), which gets further upregulated within LNs thus promoting aerobic glycolysis (36, 37). Hypoxia-induced upregulation of glycolytic genes is further supported by adenosine signaling, which is triggered by the CLL cells’ ectonucleotidases CD39 and CD73 (37).
CLL cells per se contain more mitochondria than conventional B-lymphocytes. Endogenous nitric oxide (NO) levels correlate positively with mitochondrial mass (38). In fact, NO can drive mitochondrial biogenesis, as NO supplementation increases mitochondrial mass in B-NHL-derived cell lines, whereas NO inhibition antagonizes this process (39). Correspondingly, Jitschin et al. demonstrated enhanced mitochondrial OxPhos, respiration, and respiratory capacity (40). The thereby amplified electron turnover via the mitochondrial electron transport chain yielded increased levels of ROS within the CLL cells but also systemically. Oxidative stress led then to a compensatory upregulation of heme-oxygenase-1 in CLL cells, a key cellular antioxidant, which also functions as a positive switch for the key activator of mitochondrial transcription factor A. Mitochondrial biogenesis, increased respiration, and oxidative stress appear to form a positive self-reinforcing feedback loop. As previously shown for solid tumors, pyruvate can act as a scavenger of mitochondrial superoxide in CLL cells. Increased oxidative stress under hypoxia led to enhanced pyruvate uptake while normoxic conditions led to a pyruvate release (36).
In fact, CLL cells from patients with higher disease stages and those with molecular features associated with a poor prognosis, like unmutated IGHV (U-CLL) and ZAP-70 positivity, showed higher mitochondrial respiration (41, 42). Both aforementioned genetic risk factors foster BCR signaling. Consistently, BCR-targeting reduces the metabolic activity (41). In general, CLL cells and conventional B-cells did not differ in terms of their basal glycolytic rate. However, CLL cells showed an elevated glycolytic capacity and glycolytic (together with respiratory) reserve. Furthermore, patients with U-CLL had higher lactic acid serum concentrations and displayed an elevated glycolytic capacity as compared to their mutated counterparts (M-CLL) (43). This suggests that CLL cells are better equipped to adapt to fluctuations of bioenergetic resources. In fact, microenvironmental stimuli further support the CLL cells’ metabolic flexibility as contact to LN- or BM-resident stromal cells elicits a glycolytic switch in a Notch/Myc-dependent manner (44). Transferring these findings into the clinics, patients with higher glycolytic capacity and reserve have a worse OS and a shorter time-to-treatment (43). Furthermore, CLL samples with higher glycolytic flexibility showed an increased resistance to novel drugs affecting the mitochondria, such as venetoclax and navitoclax (43).
Another metabolically important aspect is the role of free fatty acids (FFAs). Lipoprotein lipase (LPL) is the major enzyme catalyzing hydrolysis of triglycerides into FFAs and is mainly present in adipose tissue, playing a key role in lipid metabolism. CLL cells carry LPL on their cell membrane, while LPL gene expression is elevated in U-CLL cells (45, 46). LPL facilitates lipoprotein uptake, which enables CLL cells (unlike normal B-cells) to store and metabolize FFAs (46). Intracellular FFAs can then be used to promote the already more active OxPhos in CLL cells (47). Moreover, FFAs may themselves drive mitochondrial biogenesis through activation of PPARγ (48). Upregulation of LPL in CLL cells is at least partly mediated by STAT3, since STAT3 binds to the LPL promotor and STAT3 knockout downregulated LPL protein levels (46). In addition to LPL, CD36 (a cell surface FA translocase) density is higher on CLL cells as compared to non-malignant B-cells. CD36 expression (and the consecutive FA uptake) were driven by STAT3 and inhibition of FA uptake reduced CLL cell viability (49). Again, BTK-inhibition reduced LPL levels and FFA metabolism both in vitro and in patients at least partly through interfering with STAT3 (50). Interestingly, CLL ibrutinib-resistant cases could be targeted by an FAO-inhibitor highlighting the importance of FAO metabolism and OxPhos (51). Emphasizing the relevance of FFA metabolism in CLL, the OS of patients with high LPL levels was worse than for those with low ones (45).
Taken together, metabolic adaptations and flexibility occur in multiple facets in DLBCL, FL, MCL, and CLL (Figure 1). They confer enhanced survival, proliferation, and therapeutic resistance but at the same time, we can therapeutically exploit them.
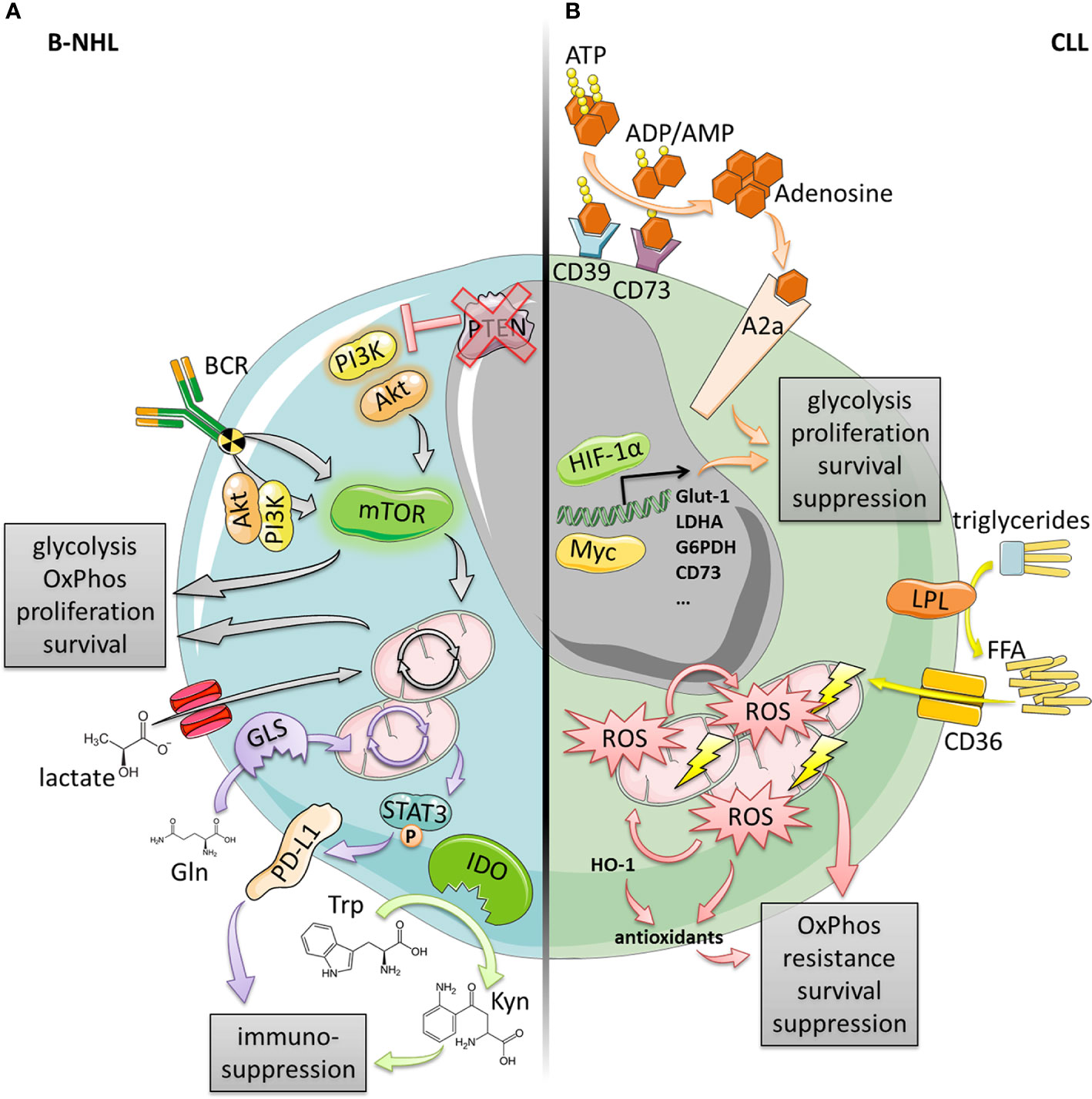
Figure 1 Metabolic alterations in B-cell lymphomas and CLL. (A) B-NHL (DLBCL, MCL, FL) often exhibit an elevated mTOR signaling activity enabling increased glycolysis, OxPhos, proliferation and survival. This can be driven by the BCR in a PI3K/AKT-dependent or independent manner, or by genetic events such as the loss of PTEN expression resulting in constitutively active PI3K/AKT. Increased OxPhos was also found to be fueled by elevated lactate shuttling into the TCA cycle due to increased lactate importer expression. Additionally, both increased expression of IDO as well as glutaminolysis-driven PD‑L1 induction provide enhanced immune-suppression. (B) CLL cells display a high mitochondrial biomass and high levels of OxPhos generating large amounts of energy and ROS that in turn drive mitochondrial biogenesis and generation of antioxidants (at least partly by HO-1). This vicious cycle confers enhanced oxidative stress resistance, survival and suppression. OxPhos and mitochondrial biogenesis could also be driven by increased activity of LPL and CD36 consuming triglycerides and importing free fatty acids (FFA). In contrast, microenvironmental trigger (e.g., hypoxia or LN-/BM-stroma) can induce transcription factors, such as Myc or HIF-1α leading to a glycolytic switch (enabled by high metabolic flexibility), and an increase of the adenosinergic axis culminating in enhanced survival, proliferation, and suppression.
Tumors including B-cell malignancies have developed a variety of mechanisms to evade the anti-tumor immunity. Amongst others, four major “metabolic strategies” have been identified (Figure 2): 1) competition over nutrients, 2) production of bioactive metabolites, 3) induction/promotion of regulatory, tolerogenic immune cells, and 4) metabolic control of immune checkpoints (ICPs).
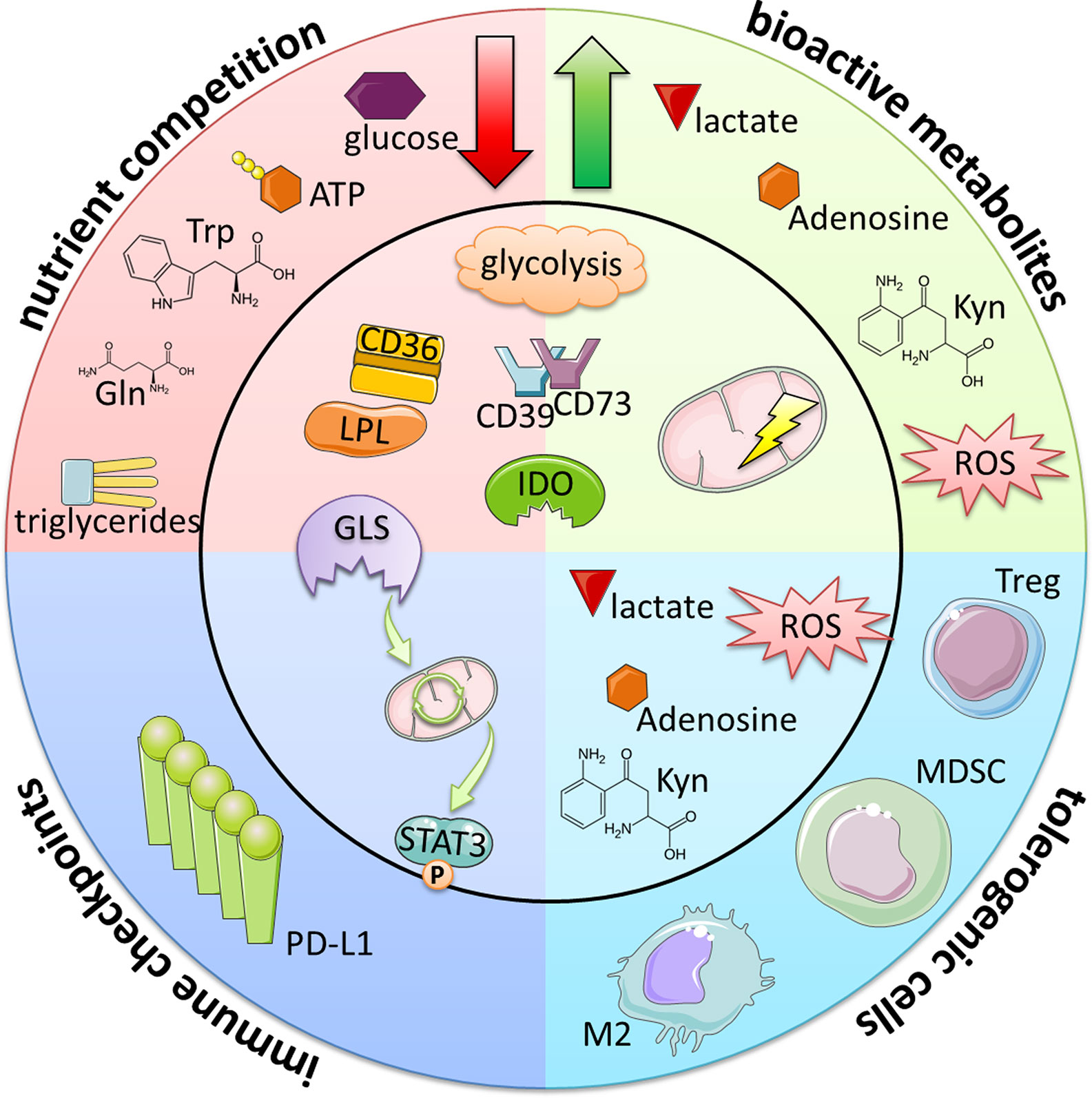
Figure 2 Immunometabolic evasion mechanisms of B-cell lymphomas and CLL. Metabolic alterations lead to the reduction of essential immune cell nutrients and at the same time to the generation of bioactive metabolites. In addition to their direct immuno-suppressive nature, these byproducts also increase the frequencies of tolerogenic, suppressive immune cells (such as Tregs, myeloid-derived suppressor cells [MDSCs] and M2-like macrophages) either indirectly via the creation of favorable conditions or directly inducing their differentiation. Furthermore, certain metabolic programs can promote the expression of immune checkpoints such as PD-L1. Taken together, these events ultimately lead to the overall suppression of immune attack and increase the tumor cells’ survival and persistence.
The most studied example of nutrient competition is the increased glucose consumption by malignant cells caused by elevated expression levels of glucose transporters and enzymes of the glycolytic machinery {as seen in BCR-DLBCL [e.g., GAPDH expression (52) and lactate secretion (13)], transformed FL [e.g., GAPDH and aldolase A (27, 28)], MCL [e.g., glycolytic flux (33)], and CLL in the LN-/BM-niche [e.g., glycolytic flux and key glycolytic enzymes (44)]}. This is detrimental for T- and NK-cells as their proliferation, activation, and differentiation is highly dependent on glucose as a fuel for both aerobic glycolysis and OxPhos [reviewed in (53)]. Similarly, tumoricidal (M1) macrophages depend on glucose to fully mount their effector cytokine response [reviewed in (54)]. Additionally, increased activity of lipid/FFA-consumptive enzymes (i.e., LPL and CD36 as seen in CLL) as well as of glutaminase (as seen in DLBCL) contribute to a nutrient-poor environment. Apart from the depletion of basic bioenergetic substrates, increased expression of the ectonucleotidases CD39/CD73 (as seen in CLL) reduces extracellular ATP (exATP) by enzymatic conversion to adenosine. exATP was shown to be particularly important for long-lived T-cell immunity through purinergic signaling promoting mitochondrial fitness (55). Expression of L-tryptophan-depleting IDO (as seen in DLBCL) has been linked to inferior progression free survival and OS in B-cell malignancies (21). In T-cells L-tryptophan shortage leads to cell cycle arrest (56) at least partly due to a stress response caused by uncharged transfer-RNAs (57), and to reduced proliferation caused by mTOR inhibition (58). Furthermore, tryptophan is essential for clonal expansion and effector T-cell differentiation via metabolic reprogramming through mTOR (59).
Simultaneously, the different rewired metabolic activities lead to accumulation of various bioactive (and potentially immunomodulatory) metabolites within the tumor microenvironment (TME). This includes lactate (as a result of enhanced glycolytic activity) which leads to the inhibition of T- and NK-cells via blunted lactate export acidifying the cytoplasm (60) as well as reduced NFAT levels (61) resulting in diminished cytokine production and effector function. Similarly, the IDO catabolite L-kynurenine inhibits immune-cell function (62), e.g., by induction of T-cell exhaustion (63, 64) and deregulation of NK-cell activating receptors (65). In fact, increased L-kynurenine serum concentration is associated with inferior OS in DLBCL (22). Accumulating extracellular adenosine converted from exATP by CD39/CD73 (as seen in CLL) can blunt immune responses by activating adenosine receptors that signal via cyclic AMP and protein kinase A. T-cells respond with reduced proliferation (66, 67), NFκB activity (68), and cytokine production (67, 69, 70) as well as increased exhaustion (71). NK cells are similarly affected by adenosine (67, 72). Abundant ROS (as seen in CLL, DLBCL, and FL) regularly leads to oxidative stress in malignancies. Again, T- and NK-cells are particularly sensitive toward ROS-induced cytotoxicity (73, 74), e.g., through impairment of T-cell receptor signaling (75, 76) leading to reduced cytokine production (77). This is reflected by the negative prognostic impact of oxidative stress in DLBCL (78) and FL (79).
However, those bioactive metabolites are not only capable of direct immune cell suppression, but can also favor preferential survival and/or induction of tolerogenic cell types. Actually, regulatory T-cells (Tregs), myeloid-derived suppressor cells (MDSCs), and pro-tumorigenic M2-like macrophages accumulate in B-cell-derived malignancies. As a prime example, kynurenine directly promotes reprogramming toward Tregs by inducing their master transcription factor FOXP3 (80). At the same time, Tregs are more resistant than conventional (potentially tumor-directed) T-cells toward detrimental effects caused by abundant lactate (81) or ROS (82) thereby enjoying a survival benefit.
As stated above, a novel, fourth mechanistic axis has been established interconnecting metabolic activity and expression of ICPs such as PD-L1 that prevent mounting of an effective anti-tumor immunity. PD-L1 is found in B-NHL and can have a negative prognostic impact (e.g., for DLBCL) (83, 84). Consequently, ICP blockade is currently undergoing clinical evaluation. Recent studies have demonstrated that glucose uptake and glutaminolysis are required for a stable PD-L1 expression. In DLBCL, glutaminolysis contributes to STAT3 induction, which positively regulates PD-L1 (85). Glucose serves as a substrate for posttranslational protein glycosylation, while N-glycosylation of PD-L1 maintains its stability and interaction with its cognate receptor (86).
Overall, metabolic reprogramming is closely linked to immunoevasion. However, many of the here described phenomena are extrapolated from basic studies or translational research within different disease contexts. Thus, immunometabolic research in B-cell malignancies needs to be further extended to build a sound basis for novel treatment strategies.
The concept of immunometabolic regulation has emerged as an important research field. Interplay between cells does not only occur via signaling molecules and receptor-ligand interactions but also through metabolic communication. Tumor cells have adapted their metabolic regulatory circuits (Figure 1), which improves their survivability and resistance toward anti-tumor immunity and/or (immune-based) therapies (Figure 2). However, metabolic reprogramming can cause novel metabolic dependencies and/or vulnerabilities rendering malignant cells more susceptible toward interferences within their metabolic framework as already described for DLBCL (13, 15), FL (24, 26), MCL (31, 32) and CLL (41, 43, 50, 51).
Targeting key (dysregulated) metabolic molecules would represent one very obvious strategy for re-establishing a (for immune cells) more favorable environment without lack of nutrients or presence of detrimental catabolites. However, it needs to be taken into consideration that bioenergetic processes of malignant cells and of activated immune cells are very similar. As such, mTOR as a central hub for nutrient sensing and bioenergetic regulation in various types of B-NHLs would represent a bona fide target. Activated T-cells are also strongly dependent on mTOR-regulated uptake of glucose and amino acids [reviewed in (87)]. Thus, targeting mTOR would inevitably affect the T-cell’s metabolic competence (and consequently anti-tumor function) as seen in preclinical models (88). Here, focusing on pathways that are not directly associated with the cell’s bioenergetics and self-evidently do not overlap between malignant and immune cells is more promising. Blockade of IDO (that is not expressed in T- and/or NK-cells) for example is currently investigated in a number of malignancies (89). Notably, reducing ROS production (by histamine application) coupled with IL-2-triggered T- and NK-cell activation has led to solid clinical effects in patients with acute myeloid leukemia (90) and comparable observations were reported when combining bicarbonate that neutralizes an acidic milieu with ICP blockade in preclinical melanoma models (91).
Nowadays, adoptive transfer of genetically engineered chimeric antigen receptor (CAR) T-cells has heralded a new era in the immunotherapy of cancer in particular of B-NHLs (92). The efficacy of CAR T-cell treatment and their adequate anti-tumor effect rely on sustained metabolic activity and energy supply as well as in vivo persistence. In analogy to the intrinsic anti-tumor immune responses, the TME can represent a metabolic barrier for CAR T-cells as convincingly shown for IDO and anti-CD19 CAR T-cells in a preclinical lymphoma model (93). Manipulating the metabolic equipment of CAR T-cells itself to empower their function in the TME [reviewed in (94)] poses a promising approach for optimizing CAR T-cell therapy in the foreseeable future. Strategies to do so, include additional genetic manipulation that, e.g., has led to the design of ROS-resistant CAR T-cells, the expansion of CAR T-cells in presence of cytokines that promote metabolic fitness such as IL-21 (95), and their combined use with agents such as adenosine receptor antagonist that target tumor metabolism-triggered detrimental effects (96).
In summary, the multifaceted metabolic alterations in B-NHL and CLL have been the subject of intense research. However, more research is needed to better understand the complex immunometabolic interactions in order to help us to further improve the efficacy of emerging immunotherapies such as CAR T-cells or immune cell engaging antibodies.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
RB, AM, and DM were funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) - Project-ID 324392634 - TRR 221.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Hanahan D, Weinberg Robert A. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
2. Warburg O. On the Origin of Cancer Cells. Science (1956) 123(3191):309–14. doi: 10.1126/science.123.3191.309
3. Pearce EL, Poffenberger MC, Chang C-H, Jones RG. Fueling Immunity: Insights into Metabolism and Lymphocyte Function. Science (2013) 342(6155):1242454. doi: 10.1126/science.1242454
4. Cascone T, McKenzie JA, Mbofung RM, Punt S, Wang Z, Xu C, et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab (2018) 27(5):977–87.e4. doi: 10.1016/j.cmet.2018.02.024
5. Geltink RIK, Kyle RL, Pearce EL. Unraveling the Complex Interplay Between T Cell Metabolism and Function. Annu Rev Immunol (2018) 36(1):461–88. doi: 10.1146/annurev-immunol-042617-053019
6. Vetrie D, Helgason GV, Copland M. The leukaemia stem cell: similarities, differences and clinical prospects in CML and AML. Nat Rev Cancer (2020) 20(3):158–73. doi: 10.1038/s41568-019-0230-9
7. Patel CH, Leone RD, Horton MR, Powell JD. Targeting metabolism to regulate immune responses in autoimmunity and cancer. Nat Rev Drug Discov (2019) 18(9):669–88. doi: 10.1038/s41573-019-0032-5
8. The Non-Hodgkin’s Lymphoma Classification P. A Clinical Evaluation of the International Lymphoma Study Group Classification of Non-Hodgkin’s Lymphoma. Blood (1997) 89(11):3909–18. doi: 10.1182/blood.V89.11.3909
9. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature (2000) 403(6769):503–11. doi: 10.1038/35000501
10. Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. New Engl J Med (2002) 346(25):1937–47. doi: 10.1056/NEJMoa012914
11. Wright G, Tan B, Rosenwald A, Hurt EH, Wiestner A, Staudt LM. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc Natl Acad Sci U S A (2003) 100(17):9991–6. doi: 10.1073/pnas.1732008100
12. Monti S, Savage KJ, Kutok JL, Feuerhake F, Kurtin P, Mihm M, et al. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood (2005) 105(5):1851–61. doi: 10.1182/blood-2004-07-2947
13. Caro P, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB, et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell (2012) 22(4):547–60. doi: 10.1016/j.ccr.2012.08.014
14. Gooptu M, Whitaker-Menezes D, Sprandio J, Domingo-Vidal M, Lin Z, Uppal G, et al. Mitochondrial and glycolytic metabolic compartmentalization in diffuse large B-cell lymphoma. Semin Oncol (2017) 44(3):204–17. doi: 10.1053/j.seminoncol.2017.10.002
15. Chen L, Monti S, Juszczynski P, Daley J, Chen W, Witzig TE, et al. SYK-dependent tonic B-cell receptor signaling is a rational treatment target in diffuse large B-cell lymphoma. Blood (2008) 111(4):2230–7. doi: 10.1182/blood-2007-07-100115
16. Sheng D, Li T, Wang W-G, Li M-J, Jiang K-L, Gao A-H, et al. Diffuse large B-cell lymphoma with low 18F-fluorodeoxyglucose avidity features silent B-cell receptor signaling. Leukemia Lymphoma (2020) 61(6):1364–71. doi: 10.1080/10428194.2020.1713317
17. Jhanwar-Uniyal M, Wainwright JV, Mohan AL, Tobias ME, Murali R, Gandhi CD, et al. Diverse signaling mechanisms of mTOR complexes: mTORC1 and mTORC2 in forming a formidable relationship. Adv Biol Regul (2019) 72:51–62. doi: 10.1016/j.jbior.2019.03.003
18. Sebestyén A, Sticz TB, Márk A, Hajdu M, Timár B, Nemes K, et al. Activity and complexes of mTOR in diffuse large B-cell lymphomas–a tissue microarray study. Modern Pathol (2012) 25(12):1623–8. doi: 10.1038/modpathol.2012.141
19. Munn DH, Mellor AL. IDO and tolerance to tumors. Trends Mol Med (2004) 10(1):15–8. doi: 10.1016/j.molmed.2003.11.003
20. Sun D, Stopka-Farooqui U, Barry S, Aksoy E, Parsonage G, Vossenkämper A, et al. Aryl Hydrocarbon Receptor Interacting Protein Maintains Germinal Center B Cells through Suppression of BCL6 Degradation. Cell Rep (2019) 27(5):1461–71.e4. doi: 10.1016/j.celrep.2019.04.014
21. Ninomiya S, Hara T, Tsurumi H, Hoshi M, Kanemura N, Goto N, et al. Indoleamine 2,3-dioxygenase in tumor tissue indicates prognosis in patients with diffuse large B-cell lymphoma treated with R-CHOP. Ann Hematol (2011) 90(4):409–16. doi: 10.1007/s00277-010-1093-z
22. Yoshikawa T, Hara T, Tsurumi H, Goto N, Hoshi M, Kitagawa J, et al. Serum concentration of L-kynurenine predicts the clinical outcome of patients with diffuse large B-cell lymphoma treated with R-CHOP. Eur J Haematol (2010) 84(4):304–9. doi: 10.1111/j.1600-0609.2009.01393.x
23. Pasqualucci L, Khiabanian H, Fangazio M, Vasishtha M, Messina M, Holmes AB, et al. Genetics of follicular lymphoma transformation. Cell Rep (2014) 6(1):130–40. doi: 10.1016/j.celrep.2013.12.027
24. Leseux L, Hamdi SM, Al Saati T, Capilla F, Recher C, Laurent G, et al. Syk-dependent mTOR activation in follicular lymphoma cells. Blood (2006) 108(13):4156–62. doi: 10.1182/blood-2006-05-026203
25. Okosun J, Wolfson RL, Wang J, Araf S, Wilkins L, Castellano BM, et al. Recurrent mTORC1-activating RRAGC mutations in follicular lymphoma. Nat Genet (2016) 48(2):183–8. doi: 10.1038/ng.3473
26. Ortega-Molina A, Deleyto-Seldas N, Carreras J, Sanz A, Lebrero-Fernández C, Menéndez C, et al. Oncogenic Rag GTPase signaling enhances B cell activation and drives follicular lymphoma sensitive to pharmacological inhibition of mTOR. Nat Metab (2019) 1(8):775–89. doi: 10.1038/s42255-019-0098-8
27. Ludvigsen M, Madsen C, Kamper P, Hamilton-Dutoit SJ, Bendix K, d’Amore F, et al. Histologically transformed follicular lymphoma exhibits protein profiles different from both non-transformed follicular and de novo diffuse large B-cell lymphoma. Blood Cancer J (2015) 5(3):e293. doi: 10.1038/bcj.2015.18
28. Monrad I, Madsen C, Lauridsen KL, Honoré B, Plesner TL, Hamilton-Dutoit S, et al. Glycolytic biomarkers predict transformation in patients with follicular lymphoma. PloS One (2020) 15(5):e0233449. doi: 10.1371/journal.pone.0233449
29. Witzig TE, Reeder CB, LaPlant BR, Gupta M, Johnston PB, Micallef IN, et al. A phase II trial of the oral mTOR inhibitor everolimus in relapsed aggressive lymphoma. Leukemia (2011) 25(2):341–7. doi: 10.1038/leu.2010.226
30. Rudelius M, Pittaluga S, Nishizuka S, Pham TH, Fend F, Jaffe ES, et al. Constitutive activation of Akt contributes to the pathogenesis and survival of mantle cell lymphoma. Blood (2006) 108(5):1668–76. doi: 10.1182/blood-2006-04-015586
31. Sekihara K, Saitoh K, Han L, Ciurea S, Yamamoto S, Kikkawa M, et al. Targeting mantle cell lymphoma metabolism and survival through simultaneous blockade of mTOR and nuclear transporter exportin-1. Oncotarget (2017) 8(21):34552–64. doi: 10.18632/oncotarget.16602
32. Hess G, Herbrecht R, Romaguera J, Verhoef G, Crump M, Gisselbrecht C, et al. Phase III study to evaluate temsirolimus compared with investigator’s choice therapy for the treatment of relapsed or refractory mantle cell lymphoma. J Clin Oncol (2009) 27(23):3822–9. doi: 10.1200/JCO.2008.20.7977
33. Lee SC, Shestov AA, Guo L, Zhang Q, Roman JC, Liu X, et al. Metabolic Detection of Bruton’s Tyrosine Kinase Inhibition in Mantle Cell Lymphoma Cells. Mol Cancer Res MCR (2019) 17(6):1365–77. doi: 10.1158/1541-7786.MCR-18-0256
34. Zhang L, Yao Y, Zhang S, Liu Y, Guo H, Ahmed M, et al. Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma. Sci Trans Med (2019) 11(491):eaau1167. doi: 10.1126/scitranslmed.aau1167
35. Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. New Engl J Med (2005) 352(8):804–15. doi: 10.1056/NEJMra041720
36. Koczula KM, Ludwig C, Hayden R, Cronin L, Pratt G, Parry H, et al. Metabolic plasticity in CLL: adaptation to the hypoxic niche. Leukemia (2016) 30(1):65–73. doi: 10.1038/leu.2015.187
37. Serra S, Vaisitti T, Audrito V, Bologna C, Buonincontri R, Chen S-S, et al. Adenosine signaling mediates hypoxic responses in the chronic lymphocytic leukemia microenvironment. Blood Adv (2016) 1(1):47–61. doi: 10.1182/bloodadvances.2016000984
38. Carew JS, Nawrocki ST, Xu RH, Dunner K, McConkey DJ, Wierda WG, et al. Increased mitochondrial biogenesis in primary leukemia cells: the role of endogenous nitric oxide and impact on sensitivity to fludarabine. Leukemia (2004) 18(12):1934–40. doi: 10.1038/sj.leu.2403545
39. Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, et al. Mitochondrial Biogenesis in Mammals: The Role of Endogenous Nitric Oxide. Science (2003) 299(5608):896–9. doi: 10.1126/science.1079368
40. Jitschin R, Hofmann AD, Bruns H, Gießl A, Bricks J, Berger J, et al. Mitochondrial metabolism contributes to oxidative stress and reveals therapeutic targets in chronic lymphocytic leukemia. Blood (2014) 123(17):2663–72. doi: 10.1182/blood-2013-10-532200
41. Vangapandu HV, Havranek O, Ayres ML, Kaipparettu BA, Balakrishnan K, Wierda WG, et al. B-cell Receptor Signaling Regulates Metabolism in Chronic Lymphocytic Leukemia. Mol Cancer Res (2017) 15(12):1692–703. doi: 10.1158/1541-7786.MCR-17-0026
42. Rosenwald A, Alizadeh AA, Widhopf G, Simon R, Davis RE, Yu X, et al. Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. J Exp Med (2001) 194(11):1639–47. doi: 10.1084/jem.194.11.1639
43. Lu J, Böttcher M, Walther T, Mougiakakos D, Zenz T, Huber W. Energy metabolism is co-determined by genetic variants in chronic lymphocytic leukemia and influences drug sensitivity. Haematologica (2019) 104(9):1830–40. doi: 10.3324/haematol.2018.203067
44. Jitschin R, Braun M, Qorraj M, Saul D, Le Blanc K, Zenz T, et al. Stromal cell–mediated glycolytic switch in CLL cells involves Notch-c-Myc signaling. Blood (2015) 125(22):3432–6. doi: 10.1182/blood-2014-10-607036
45. Heintel D, Kienle D, Shehata M, Kröber A, Kroemer E, Schwarzinger I, et al. High expression of lipoprotein lipase in poor risk B-cell chronic lymphocytic leukemia. Leukemia (2005) 19(7):1216–23. doi: 10.1038/sj.leu.2403748
46. Rozovski U, Grgurevic S, Bueso-Ramos C, Harris DM, Li P, Liu Z, et al. Aberrant LPL Expression, Driven by STAT3, Mediates Free Fatty Acid Metabolism in CLL Cells. Mol Cancer Res (2015) 13(5):944–53. doi: 10.1158/1541-7786.MCR-14-0412
47. Rodríguez-Enríquez S, Hernández-Esquivel L, Marín-Hernández A, El Hafidi M, Gallardo-Pérez JC, Hernández-Reséndiz I, et al. Mitochondrial free fatty acid β-oxidation supports oxidative phosphorylation and proliferation in cancer cells. Int J Biochem Cell Biol (2015) 65:209–21. doi: 10.1016/j.biocel.2015.06.010
48. Garcia-Roves P, Huss JM, Han DH, Hancock CR, Iglesias-Gutierrez E, Chen M, et al. Raising plasma fatty acid concentration induces increased biogenesis of mitochondria in skeletal muscle. Proc Natl Acad Sci U S A (2007) 104(25):10709–13. doi: 10.1073/pnas.0704024104
49. Rozovski U, Harris DM, Li P, Liu Z, Jain P, Ferrajoli A, et al. STAT3-activated CD36 facilitates fatty acid uptake in chronic lymphocytic leukemia cells. Oncotarget (2018) 9(30):21268–80. doi: 10.18632/oncotarget.25066
50. Rozovski U, Harris DM, Li P, Liu Z, Jain P, Ferrajoli A, et al. Ibrutinib inhibits free fatty acid metabolism in chronic lymphocytic leukemia. Leuk Lymphoma (2018) 59(11):2686–91. doi: 10.1080/10428194.2018.1439167
51. Galicia-Vazquez G, Aloyz R. Ibrutinib Resistance Is Reduced by an Inhibitor of Fatty Acid Oxidation in Primary CLL Lymphocytes. Front Oncol (2018) 8:411. doi: 10.3389/fonc.2018.00411
52. Chiche J, Reverso-Meinietti J, Mouchotte A, Rubio-Patiño C, Mhaidly R, Villa E, et al. GAPDH Expression Predicts the Response to R-CHOP, the Tumor Metabolic Status, and the Response of DLBCL Patients to Metabolic Inhibitors. Cell Metab (2019) 29(6):1243–57.e10. doi: 10.1016/j.cmet.2019.02.002
53. Kedia-Mehta N, Finlay DK. Competition for nutrients and its role in controlling immune responses. Nat Commun (2019) 10(1):2123. doi: 10.1038/s41467-019-10015-4
54. O’Neill LAJ, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med (2015) 213(1):15–23. doi: 10.1084/jem.20151570
55. Borges da Silva H, Beura LK, Wang H, Hanse EA, Gore R, Scott MC, et al. The purinergic receptor P2RX7 directs metabolic fitness of long-lived memory CD8+ T cells. Nature (2018) 559(7713):264–8. doi: 10.1038/s41586-018-0282-0
56. Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T Cell Proliferation by Macrophage Tryptophan Catabolism. J Exp Med (1999) 189(9):1363–72. doi: 10.1084/jem.189.9.1363
57. Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 Kinase in T Cells Mediates Proliferative Arrest and Anergy Induction in Response to Indoleamine 2,3-Dioxygenase. Immunity (2005) 22(5):633–42. doi: 10.1016/j.immuni.2005.03.013
58. Metz R, Rust S, DuHadaway JB, Mautino MR, Munn DH, Vahanian NN, et al. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. OncoImmunology (2012) 1(9):1460–8. doi: 10.4161/onci.21716
59. Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol (2013) 14(5):500–8. doi: 10.1038/ni.2556
60. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell–derived lactic acid on human T cells. Blood (2007) 109(9):3812–9. doi: 10.1182/blood-2006-07-035972
61. Brand A, Singer K, Gudrun E K, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab (2016) 24(5):657–71. doi: 10.1016/j.cmet.2016.08.011
62. Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-derived Catabolites Are Responsible for Inhibition of T and Natural Killer Cell Proliferation Induced by Indoleamine 2,3-Dioxygenase. J Exp Med (2002) 196(4):459–68. doi: 10.1084/jem.20020121
63. Liu Y, Liang X, Dong W, Fang Y, Lv J, Zhang T, et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8(+) T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell (2018) 33(3):480–94.e7. doi: 10.1016/j.ccell.2018.02.005
64. Rad Pour S, Morikawa H, Kiani NA, Yang M, Azimi A, Shafi G, et al. Exhaustion of CD4+ T-cells mediated by the Kynurenine Pathway in Melanoma. Sci Rep (2019) 9(1):12150. doi: 10.1038/s41598-019-48635-x
65. Chiesa MD, Carlomagno S, Frumento G, Balsamo M, Cantoni C, Conte R, et al. The tryptophan catabolite l-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood (2006) 108(13):4118–25. doi: 10.1182/blood-2006-03-006700
66. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med (2007) 204(6):1257–65. doi: 10.1084/jem.20062512
67. Bastid J, Regairaz A, Bonnefoy N, Déjou C, Giustiniani J, Laheurte C, et al. Inhibition of CD39 Enzymatic Function at the Surface of Tumor Cells Alleviates Their Immunosuppressive Activity. Cancer Immunol Res (2015) 3(3):254–65. doi: 10.1158/2326-6066.CIR-14-0018
68. Majumdar S, Aggarwal BB. Adenosine suppresses activation of nuclear factor-κB selectively induced by tumor necrosis factor in different cell types. Oncogene (2003) 22(8):1206–18. doi: 10.1038/sj.onc.1206184
69. Linnemann C, Schildberg FA, Schurich A, Diehl L, Hegenbarth SI, Endl E, et al. Adenosine regulates CD8 T-cell priming by inhibition of membrane-proximal T-cell receptor signalling. Immunology (2009) 128(1pt2):e728–e37. doi: 10.1111/j.1365-2567.2009.03075.x
70. Mastelic-Gavillet B, Navarro Rodrigo B, Décombaz L, Wang H, Ercolano G, Ahmed R, et al. Adenosine mediates functional and metabolic suppression of peripheral and tumor-infiltrating CD8+ T cells. J ImmunoTher Cancer (2019) 7(1):257. doi: 10.1186/s40425-019-0719-5
71. Shi L, Feng M, Du S, Wei X, Song H, Yixin X, et al. Adenosine Generated by Regulatory T Cells Induces CD8+ T Cell Exhaustion in Gastric Cancer through A2aR Pathway. BioMed Res Int (2019) 2019:4093214. doi: 10.1155/2019/4093214
72. Young A, Ngiow SF, Gao Y, Patch A-M, Barkauskas DS, Messaoudene M, et al. A2AR Adenosine Signaling Suppresses Natural Killer Cell Maturation in the Tumor Microenvironment. Cancer Res (2018) 78(4):1003–16. doi: 10.1158/0008-5472.CAN-17-2826
73. Larbi A, Kempf J, Pawelec G. Oxidative stress modulation and T cell activation. Exp Gerontol (2007) 42(9):852–8. doi: 10.1016/j.exger.2007.05.004
74. Mellqvist UH, Hansson M, Brune M, Dahlgren C, Hermodsson S, Hellstrand K. Natural killer cell dysfunction and apoptosis induced by chronic myelogenous leukemia cells: role of reactive oxygen species and regulation by histamine. Blood (2000) 96(5):1961–8. doi: 10.1182/blood.V96.5.1961.h8001961_1961_1968
75. Otsuji M, Kimura Y, Aoe T, Okamoto Y, Saito T. Oxidative stress by tumor-derived macrophages suppresses the expression of CD3 zeta chain of T-cell receptor complex and antigen-specific T-cell responses. Proc Natl Acad Sci U S A (1996) 93(23):13119–24. doi: 10.1073/pnas.93.23.13119
76. Gringhuis SI, Papendrecht-van der Voort EA, Leow A, Nivine Levarht EW, Breedveld FC, Verweij CL. Effect of redox balance alterations on cellular localization of LAT and downstream T-cell receptor signaling pathways. Mol Cell Biol (2002) 22(2):400–11. doi: 10.1128/MCB.22.2.400-411.2002
77. Malmberg K-J, Arulampalam V, Ichihara F, Petersson M, Seki K, Andersson T, et al. Inhibition of Activated/Memory (CD45RO+) T Cells by Oxidative Stress Associated with Block of NF-κB Activation. J Immunol (2001) 167(5):2595–601. doi: 10.4049/jimmunol.167.5.2595
78. Peroja P, Pasanen AK, Haapasaari KM, Jantunen E, Soini Y, Turpeenniemi-Hujanen T, et al. Oxidative stress and redox state-regulating enzymes have prognostic relevance in diffuse large B-cell lymphoma. Exp Hematol Oncol (2012) 1(1):2. doi: 10.1186/2162-3619-1-2
79. Arakaki H, Osada Y, Takanashi S, Ito C, Aisa Y, Nakazato T. Oxidative Stress Is Associated with Poor Prognosis in Patients with Follicular Lymphoma. Blood (2016) 128(22):1787–. doi: 10.1182/blood.V128.22.1787.1787
80. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol (Baltimore Md 1950) (2010) 185(6):3190–8. doi: 10.4049/jimmunol.0903670
81. Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab (2017) 25(6):1282–93.e7. doi: 10.1016/j.cmet.2016.12.018
82. Mougiakakos D, Johansson CC, Jitschin R, Böttcher M, Kiessling R. Increased thioredoxin-1 production in human naturally occurring regulatory T cells confers enhanced tolerance to oxidative stress. Blood (2011) 117(3):857–61. doi: 10.1182/blood-2010-09-307041
83. Kiyasu J, Miyoshi H, Hirata A, Arakawa F, Ichikawa A, Niino D, et al. Expression of programmed cell death ligand 1 is associated with poor overall survival in patients with diffuse large B-cell lymphoma. Blood (2015) 126(19):2193–201. doi: 10.1182/blood-2015-02-629600
84. Xu-Monette ZY, Zhou J, Young KH. PD-1 expression and clinical PD-1 blockade in B-cell lymphomas. Blood (2018) 131(1):68–83. doi: 10.1182/blood-2017-07-740993
85. Xia X, Zhou W, Guo C, Fu Z, Zhu L, Li P, et al. Glutaminolysis Mediated by MALT1 Protease Activity Facilitates PD-L1 Expression on ABC-DLBCL Cells and Contributes to Their Immune Evasion. Front Oncol (2018) 8:632. doi: 10.3389/fonc.2018.00632
86. Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun (2016) 7:12632. doi: 10.1038/ncomms12632
87. Waickman AT, Powell JD. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunological Rev (2012) 249(1):43–58. doi: 10.1111/j.1600-065X.2012.01152.x
88. Herrero-Sánchez MC, Rodríguez-Serrano C, Almeida J, San Segundo L, Inogés S, Santos-Briz Á, et al. Targeting of PI3K/AKT/mTOR pathway to inhibit T cell activation and prevent graft-versus-host disease development. J Hematol Oncol (2016) 9(1):113. doi: 10.1186/s13045-016-0343-5
89. Moon YW, Hajjar J, Hwu P, Naing A. Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J Immunother Cancer (2015) 3:51. doi: 10.1186/s40425-015-0094-9
90. Brune M, Castaigne S, Catalano J, Gehlsen K, Ho AD, Hofmann WK, et al. Improved leukemia-free survival after postconsolidation immunotherapy with histamine dihydrochloride and interleukin-2 in acute myeloid leukemia: results of a randomized phase 3 trial. Blood (2006) 108(1):88–96. doi: 10.1182/blood-2005-10-4073
91. Pilon-Thomas S, Kodumudi KN, El-Kenawi AE, Russell S, Weber AM, Luddy K, et al. Neutralization of Tumor Acidity Improves Antitumor Responses to Immunotherapy. Cancer Res (2016) 76(6):1381–90. doi: 10.1158/0008-5472.CAN-15-1743
92. Vitale C, Strati P. CAR T-Cell Therapy for B-Cell non-Hodgkin Lymphoma and Chronic Lymphocytic Leukemia: Clinical Trials and Real-World Experiences. Front Oncol (2020) 10:849. doi: 10.3389/fonc.2020.00849
93. Ninomiya S, Narala N, Huye L, Yagyu S, Savoldo B, Dotti G, et al. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood (2015) 125(25):3905–16. doi: 10.1182/blood-2015-01-621474
94. Xu X, Gnanaprakasam JNR, Sherman J, Wang R. A Metabolism Toolbox for CAR T Therapy. Front Oncol (2019) 9(322). doi: 10.3389/fonc.2019.00322
95. Singh H, Figliola MJ, Dawson MJ, Huls H, Olivares S, Switzer K, et al. Reprogramming CD19-specific T cells with IL-21 signaling can improve adoptive immunotherapy of B-lineage malignancies. Cancer Res (2011) 71(10):3516–27. doi: 10.1158/0008-5472.CAN-10-3843
Keywords: B-cell-derived Non-Hodgkin lymphoma, chronic lymphocytic leukemia, metabolism, immune escape, immune therapeutics
Citation: Böttcher M, Baur R, Stoll A, Mackensen A and Mougiakakos D (2020) Linking Immunoevasion and Metabolic Reprogramming in B-Cell–Derived Lymphomas. Front. Oncol. 10:594782. doi: 10.3389/fonc.2020.594782
Received: 14 August 2020; Accepted: 14 October 2020;
Published: 05 November 2020.
Edited by:
Jérôme Paggetti, Luxembourg Institute of Health, LuxembourgReviewed by:
Johannes Meiser, Luxembourg Institute of Health, LuxembourgCopyright © 2020 Böttcher, Baur, Stoll, Mackensen and Mougiakakos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dimitrios Mougiakakos, RGltaXRyaW9zLk1vdWdpYWtha29zQHVrLWVybGFuZ2VuLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.