 Enrico Martin
Enrico Martin Ibtissam Acem
Ibtissam Acem Dirk J. Grünhagen
Dirk J. Grünhagen Judith V. M. G. Bovée
Judith V. M. G. Bovée Cornelis Verhoef
Cornelis Verhoef
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Oncol., 22 December 2020
Sec. Cancer Genetics
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.594069
Background: Malignant peripheral nerve sheath tumors (MPNSTs) are aggressive soft tissue sarcomas with dismal prognosis. Pathological and genetic markers may predict more aggressive behavior in MPNSTs but have uncommonly been investigated, and few are used in daily practice. This study reviews the prognostic value of immunohistochemical markers and genetic alterations in MPNST.
Methods: A systematic search was performed in PubMed and Embase databases according to the PRISMA guidelines. Search terms related to ‘MPNST’ and ‘prognostic’ were used. Studies investigating the association of immunohistochemical markers or genetic alterations with prognosis were included. Qualitative synthesis was performed on all studies. A distinction was made between univariable and multivariable associations.
Results: Forty-six studies were included after full-text screening. Sixty-seven different immunohistochemical markers were investigated. Absence of S100 and H3K27me3 and high Ki67 and p53 staining was most commonly independently associated with worse survival and disease-free survival. Several genetic alterations were investigated as well with varying association to survival. TP53, CDK4, RASSF1A alterations were independently associated with worse survival, as well as changes in chromosomal length in Xp, 10q, and 16p.
Conclusions: MPNSTs harbor complex and heterogeneous biology. Immunohistochemical markers and genetic alterations have variable prognostic value. Absence of S100 and H3K27me3 and increased Ki67 can be of prognostic value. Alterations in TP53 or increase in p53 staining may distinguish MPNSTs with worse outcomes. Genetic alterations and staining of other cell cycle regulatory and Ras pathway proteins may also help stratifying patients with worse outcomes. A combination of markers can increase the prognostic value.
Malignant peripheral nerve sheath tumors (MPNSTs) are rare and aggressive soft tissue sarcomas (STS) that carry a dismal prognosis (1–3). Neurofibromatosis type 1 (NF1) patients have an increased risk of developing these tumors and encompass approximately 25–50% of MPNST patients (1–5). The NF1 gene is commonly affected in MPNSTs which causes loss of the neurofibromin protein which inhibits the Ras enzyme (6). Activation of the Ras pathway leads to upregulation of the mitogen-activated protein (MAPK) and phosphoinositide 3-kinase (PI3K) pathways (7). Besides the common knockdown of NF1, alterations in several genes including TP53, SUZ12, EED, PTEN, and CDKN2A as well as upregulation of several tyrosine kinases contribute to the formation of MPNST (8–12). MPNSTs are known for harboring complex genomic alterations, but despite our increasing understanding of underlying biology, prognosis has not ameliorated the past decades and median survival stagnates at 5–6 years (2, 3).
Staging of MPNSTs is important to increase accuracy of outcome prediction, but it may also facilitate treatment stratification. However, the clinical American Joint Committee of Cancer (AJCC) STS staging system is less applicable in MPNST (4, 5, 13). The histologic Fédération Nationale des Centres de Lutte Contre le Cancer (FNCLCC) grading system used in STS is of prognostic value since low grade MPNST (FNCLCC grade 1) has improved survival (2). However, only 10% of MPNSTs are grade 1, and the FNCLCC grading can likely only distinguish prognosis between grades 1 and 3 (2, 5). Moreover, the histological distinction between low-grade MPNST and benign neurofibroma with atypia is difficult as objective criteria are lacking, causing interobserver variability. In the context of NF1, the diagnosis of progression to MPNST is even more challenging. Recently, a consensus view has been published defining “atypical neurofibromatous neoplasm of uncertain biologic potential (ANNUBP)” as an intermediate lesion in NF1 patients (14). While driver mutations are increasingly being studied, the transition of neurofibromas to MPNSTs is not yet fully understood. Clinical parameters as predictors of outcome have been studied more commonly, but independent predictors are found inconsistently (3). Although radiation-induced MPNSTs have repeatedly been associated with worse survival, the influence of NF1 disease on survival has been subject of debate (3, 13, 15). Better classification systems for MPNSTs are therefore urgently needed.
Currently, surgery remains the only proven treatment to improve survival (1–3). Chemotherapy has limited effect in localized disease, and its use is controversial. Some studies suggest a minor benefit in high-grade, large, and deep MPNST (16–18). Moreover, 10–20% of patients present with metastatic or unresectable disease and up to 50% of patients will develop metastases over time (1–5, 13, 19). Targeted therapies are warranted, but so far none have been proven effective (20). Immunohistochemical and genetic markers may predict more aggressive behavior in MPNSTs, but their association with oncological outcome has uncommonly been investigated and few are yet used in daily practice for prognostication. For this reason this systematic review set out to summarize current knowledge on the prognostic value of immunohistochemical and genetic markers. Such markers may enhance prognostication and aid in elucidating driver mutations of malignancy.
A systematic search was performed in Embase and PubMed databases according to the Preferred Reporting Items for Systematic Reviews and Meta-analysis (PRISMA) guidelines, in order to identify all potentially relevant articles as of March 2020. The string was built with the help of a professional librarian using search terms related to ‘MPNST’ and ‘prognostic’. The exact search syntaxes for PubMed and Embase are shown in Supplementary Table 1. Studies were included that evaluated the association of immunohistochemical markers and genetic alterations to oncological outcomes in MPNST patients. Exclusion criteria included lack of full text or studies without specific analyses fitting our inclusion criteria. The initial review was conducted by two independent authors (EM. and IA). Disagreements were solved through discussion in which one additional author was involved (CV).
Data extracted from studies included: study period, total number of patients, mean age and range, percentage NF1 patients, markers and genetic alterations investigated for prognostic value, and analyses used to identify prognosticators. For all markers and genetic alterations investigated additional information was extracted: number of patients with survival data, population with ‘positive’ test, oncological outcome analyzed, and whether its prognostic value was corrected for common clinical prognostic factors. Whenever the marker was independently associated with outcome, the hazard ratio was noted. Common factors for which could have been adjusted in multivariable models included: age, presence of NF1, tumor size, tumor site, metastasis at diagnosis, tumor depth, tumor grade, and surgical margin (3). All results of the predictive value of markers were presented or re-calculated to represent the marker cut-off as a negative predictor of survival. Qualitative synthesis was performed for all studies, summarizing results based on type of analysis. Immunohistochemical markers were further stratified into markers of differentiation, receptors and their ligands, Ras pathway, cell cycle regulation, p53 pathway, vascularization, and others. For each immunohistochemical marker cumulative incidence of univariable and multivariable association to survival (disease-specific or overall) or disease-free survival (recurrence, metastasis, or both) were calculated.
After removal of duplicates, a total of 1,882 articles were identified in PubMed and Embase databases (Figure 1). Title and abstract screening resulted in 55 potentially relevant articles, of which 46 were selected for qualitative synthesis after full-text screening. Mean age differed between 11 and 50 years old (range of all patients 1–94). Prevalence of NF1 patients in study populations ranged from 0 to 100% (mean: 48.0%). Immunohistochemical markers were studied exclusively in 36 studies, genetic alterations in seven studies, and both in three studies (Table 1). A total of 67 different immunohistochemical markers and numerous genetic alterations were evaluated (Table 2, Figure 2).
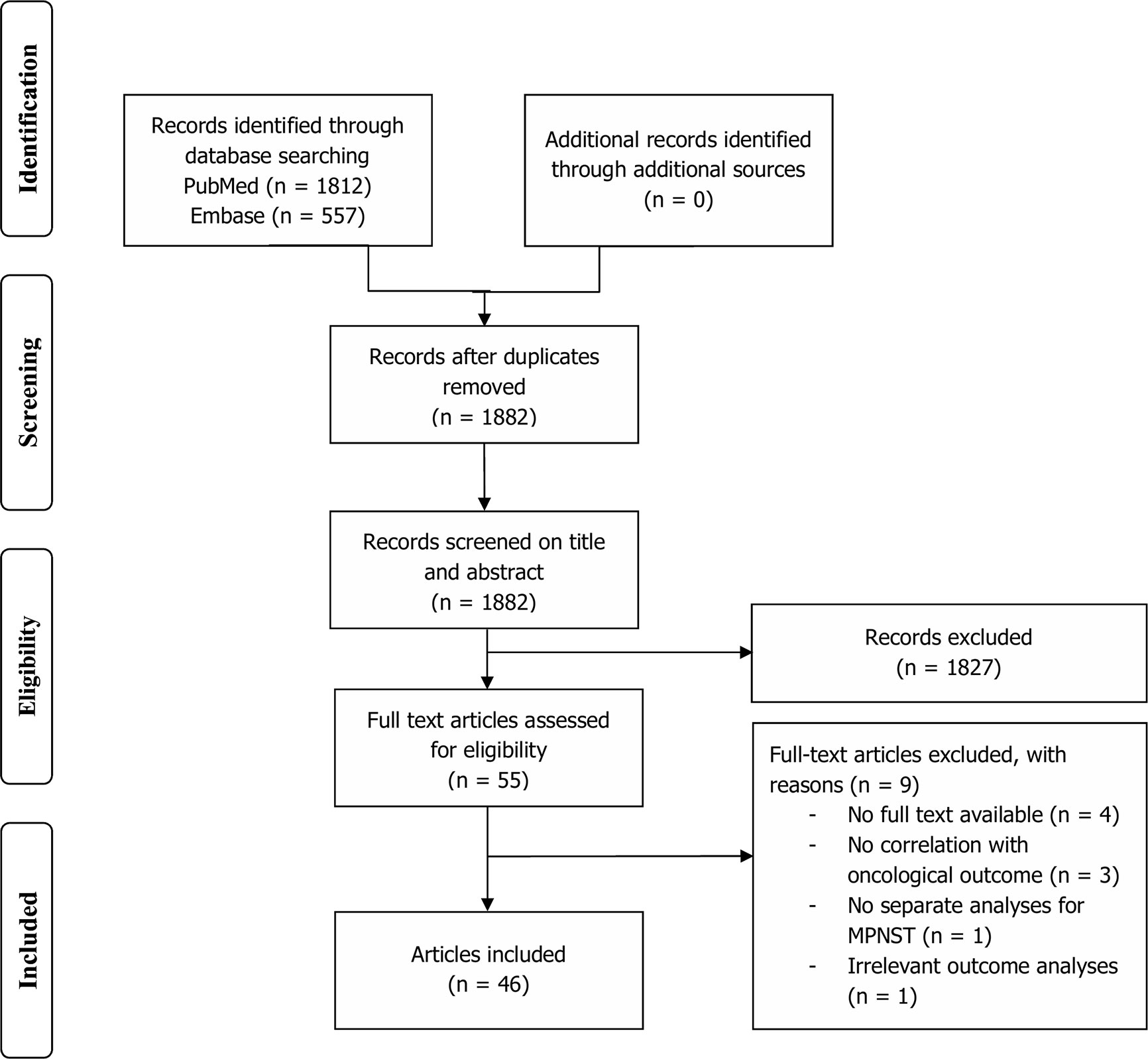
Figure 1 Flowchart depicting study selection.
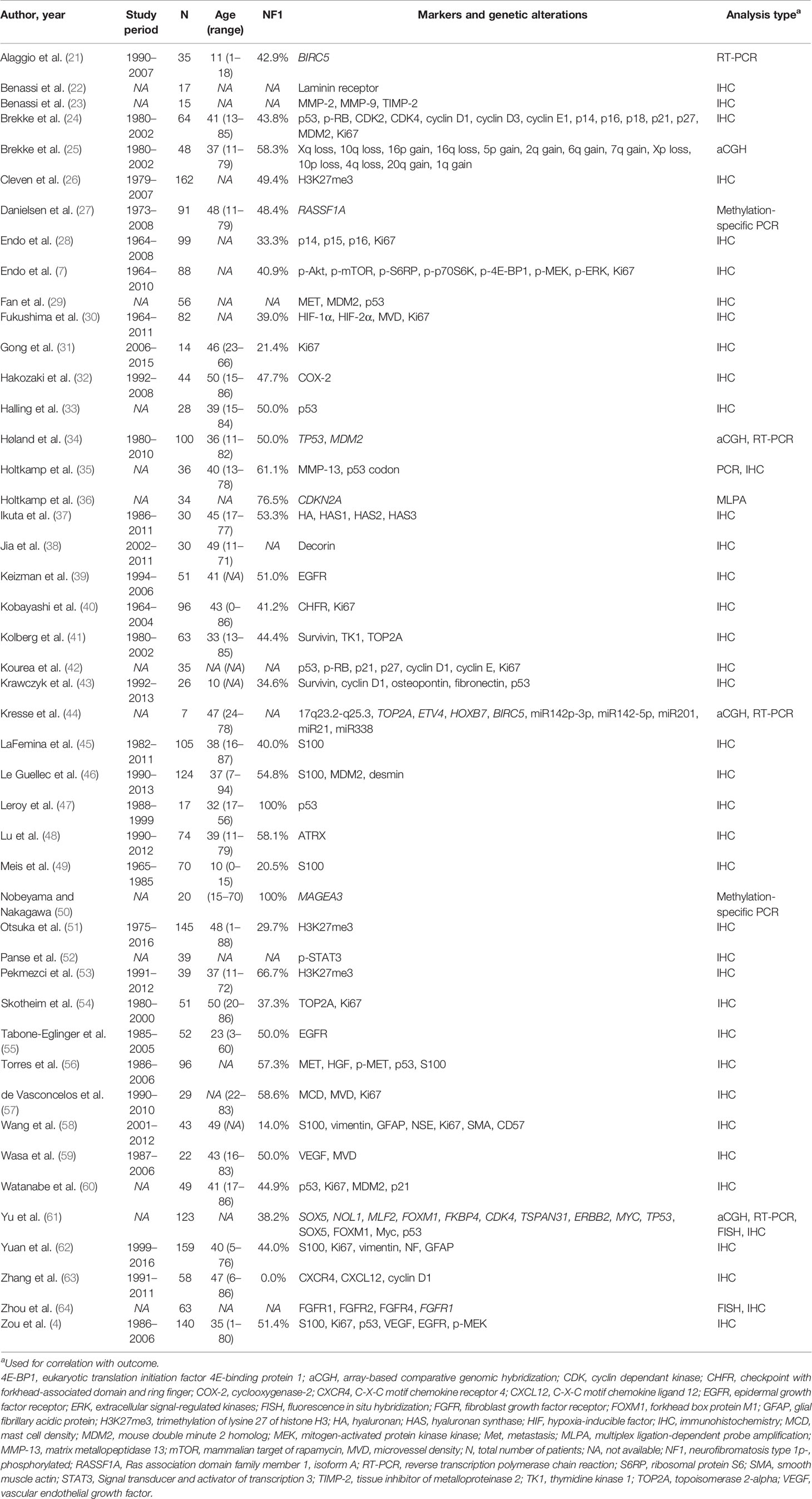
Table 1 Study characteristics of included studies.
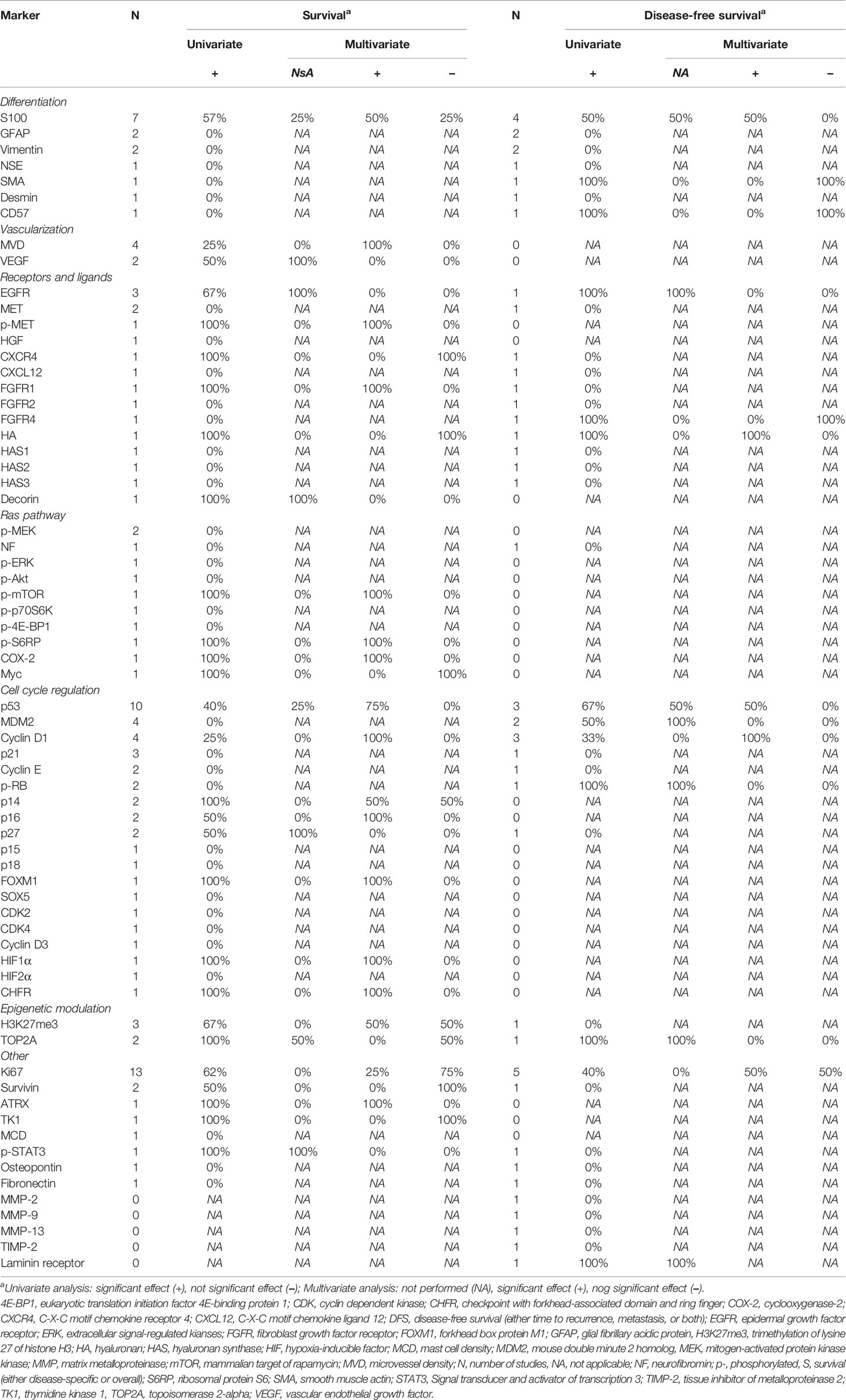
Table 2 Prognostic value of immunohistochemical markers.
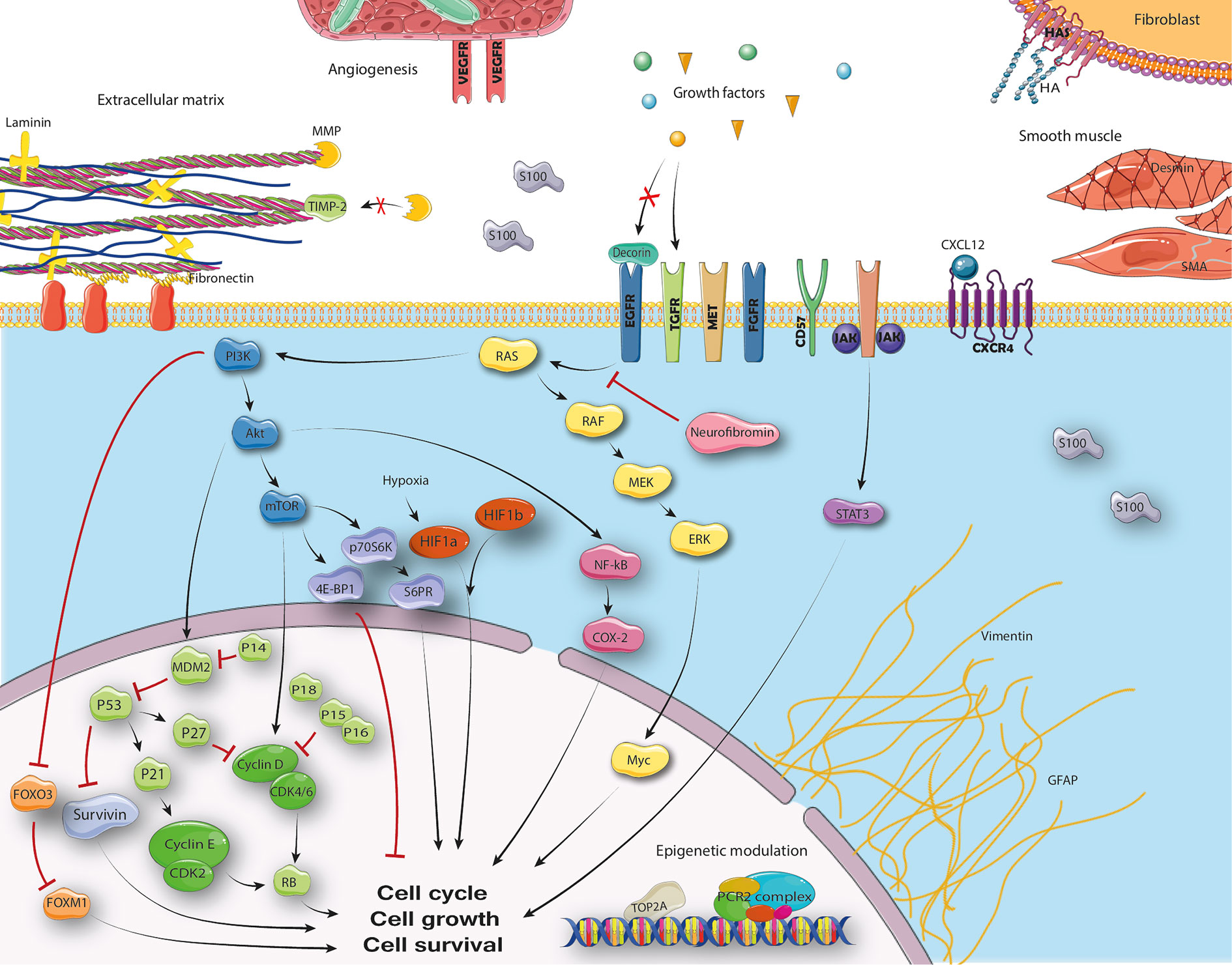
Figure 2 Cellular pathways in MPNST.
Seven mesenchymal and neuronal differentiation markers were evaluated (Table 2), most commonly S100 (4, 45, 46, 56, 58, 62). In univariable analysis complete absence of S100 was found negatively associated with survival in four/six studies. Two studies showed the absence of S100 to be an independent predictor of worse survival with HR 4.5 (95% CI: 2.0–12.1) and HR 6.6 (95% CI: 1.8–23.8) (4, 62). All seven markers were also evaluated for association with disease-free survival (DFS). Negative S100 staining was associated with worse DFS in two/four studies, of which one study showed an independent association (HR 4.2, 95% CI: 1.5–12.3) (62). Negative smooth muscle actin (SMA) and CD57 staining were also found associated with worse DFS in univariable analysis in one study, but not in multivariable analysis (58).
Microvascular density (MVD) and vascular epithelial growth factor (VEGF) staining were evaluated as vascularization markers (Table 2) (4, 28, 30, 57, 59). High MVD was associated with worse survival in one/four studies. This association was also significant in multivariable analyses (HR 7.3, 95% CI: 1.4–38.5) (57). High VEGF staining was associated with worse survival in one/two studies, but this was not studied in a multivariable model (59). No markers were studied for association with DFS.
Immunohistochemical expression of nine different receptors or their ligands were evaluated, most commonly the epidermal growth factor receptor (EGFR, Table 2) (4, 29, 37–39, 55, 56, 63, 64). Increased EGFR staining was associated with worse survival in univariable analysis in two/three studies, but this was not evaluated in a multivariable model (4, 39, 54). Increased phosphorylated MET (p-MET), C-X-C motif chemokine receptor 4 (CXCR4), and low fibroblast growth factor receptor 1 (FGFR1) staining were also associated with worse survival in univariable analysis, but only p-MET (HR 1.04, 95% CI: 1.0–1.1) and FGFR1 (HR 2.8, 95% CI: 1.2–6.7) were independently associated with survival (37, 56, 63, 64). Increased EGFR and FGFR4 were associated with worse DFS, but only in univariable analyses (39, 64). On a genetic level, no amplification of FGFR1 on fluorescence in situ hybridization (FISH) was associated with worse survival and DFS in univariable analysis (Supplementary Table 2) (64). Copy number alterations in ERBB2 were not associated with survival (61).
Twelve extracellular matrix markers were studied, of which none was evaluated more than once (Table 2) (22, 23, 35, 37, 38, 43). Only increased hyaluronan (HA) and decorin staining were associated with decreased survival, but none in a multivariable model (36, 37). Increased HA and laminin receptor were associated with worse DFS, but only HA was associated with worse DFS in a multivariable model (HR 5.7, 95% CI: 1.2–26.4) (22, 37).
Ten different Ras pathway proteins were stained, but only phosphorylated MAPK kinase (MEK) was evaluated more than once (Table 2) (4, 7, 32, 61, 62). Increased phosphorylated mammalian target of rapamycin (p-mTOR), phosphorylated ribosomal protein S6 (p-S6RP), cyclooxygenase-2 (COX-2), and Myc staining were associated with worse survival univariable analysis (7, 32, 61). Only increased p-mTOR (HR 2.6, 95% CI: 1.3–5.5), p-S6RP (HR 2.5, 95% CI: 1.3–5.5), and COX-2 (HR 3.0, 95% CI: 1.1–10.2) staining were independently associated with worse survival (7, 32). No Ras pathway associated immunohistochemical marker was found associated with DFS. On a genetic level, copy number alterations of MYC were not associated with survival (61). Methylation of RASSF1A gene was associated independently with worse survival in one study (HR 5.2, 95% CI: 1.4–19.4, Supplementary Table 2) (27). This association was however only found in the NF1 subpopulation.
Sixteen immunohistochemical markers of cell cycle regulation were evaluated, most commonly p53 (Table 2) (4, 24, 26, 28, 29, 33, 40, 42, 43, 46, 47, 51, 53, 56, 60, 61, 63). Low p14, p16, checkpoint with forkhead-associated domain and ring finger (CHFR), and increase in p53, p14, cyclin D1, p27, and forkhead box protein M1 (FOXM1) staining were associated with worse survival in univariable analysis (4, 26, 28, 40, 42, 43, 51, 56, 61). Positive p53 staining was independently associated with survival in three/four studies (HR 1.8, 95% CI: 1.0–3.3, HR 2.3, 95% CI: 1.2–4.5, and HR 6.4, 95% CI: 1.5–29.0) (4, 24, 56). Increased staining of cyclin D1 (HR 15.9, 95% CI: 2.0–125.0), HIF1α (HR 8.3, 95% CI: 2.8–28.9), FOXM1 (HR 1.9, 95% CI: 1.1–3.3), and decreased staining of p16 (HR 2.2, 95% CI: 1.5–3.2) and p14 (HR 2.7, 95% CI: 1.8–4.2) were also independently associated with worse survival in one study each (28, 30, 43, 61). Positive staining of p53, MDM2, cyclin D1, and p-RB were associated with worse DFS in univariable analysis (29, 42, 43). Only cyclin D1 (HR 11.1, 95% CI: 2.8–47.6) and p53 (HR 3.2, 95% CI: 1.0–10.4) were independently associated with worse DFS in one study (43). On a genetic level, mutation, homozygous loss, or loss of heterogeneity of TP53 was associated with worse survival in two/three studies (Supplementary Table 2) (34, 35, 61). The copy number gain of MDM2 and CDK4 as well as amplification on FISH of CDK4 was associated with worse survival (34, 61). Gain (HR 4.2, 95% CI: 1.4–12.4) or amplification (HR 2.0, 95% CI: 1.0–4.0) of CDK4 was independently associated with worse survival (59). The combination of either MDM2 gain or TP53 aberration made a high risk group (16%) for worse survival with a HR 3.4 (95% CI: 1.4–8.3) (34). In the same study, a gene expression profile was made and a score of ≥0.12 was present in 66.7% of the population which was associated with worse survival as well (HR 4.0, 95% CI: 1.3–12.1). Another study on DNA copy number changes found a significant association with worse survival for gain at 17q23.2–25.3, but not in several related genes or micro-RNAs in this region (44). The association was not evaluated in a multivariable model. A gain in FOXM1 was worse survival in another study (61). Only the polymorphism of p53Pro72 was associated with worse DFS in one study (35). This association was not evaluated in a multivariable model.
Two epigenetic modulating proteins were investigated as immunohistochemical markers (Table 2) (26, 41, 51, 53, 54). Loss of trimethylation of lysine 27 of histone H3 (H3K27me3) and increased topoisomerase 2-alpha (TOP2A) staining were both associated with decreased survival (26, 41, 51, 54). Only H3K27me3 was independently associated with worse survival (HR 2.6, 95% CI: 1.2–5.7) in one out of two studies (26, 51). Increased TOP2A staining was also associated with worse DFS in one study (54). High copy number changes of TOP2A was not associated with worse survival (Supplementary Table 2) (44).
Thirteen other immunohistochemical markers were studied, most commonly the proliferation marker Ki67 (4, 7, 24, 30, 31, 40–43, 48, 52, 54, 57, 58, 60–62). On average a cut-off at 20.9% (range: 5–30%) for high Ki67 staining was used and it was significantly associated with worse survival in 8/12 studies, of which two studies showed an independent association (HR 2.4, 95% CI: 1.1–4.9 and HR 10.2, 95% CI: 3.6–32.1) (28, 30). Increased survivin, thymidine kinase 1 (TK1), phosphorylated signal transducer and activator of transcription 3 (p-STAT3), and hypoxia-induced factor 1-alpha (HIF1α) and decreased ATRX staining were associated with worse survival (30, 41, 48). Both decreased ATRX (HR 5.3, 95% CI: 1.4–20.4) and positive HIF1α staining (HR 8.3, 95% CI: 2.8–28.9) were independently associated with worse survival (30, 48). One study showed that when there was high survivin and high TK1 staining or low survivin and high TOP2A staining a high risk group of patients could be stratified with HR 4.6 (95% CI: 1.5–14.4) (41). Increased staining of Ki67 and laminin receptor were associated with worse DFS (22, 31, 62). Only high Ki67 staining was shown to have an independent association with worse DFS in one/two studies (HR 3.8, 95% CI: 1.7–8.5) (62). Four studies investigated several other genetic alterations, including two on BIRC5, the gene encoding surviving (21, 25, 44, 50). One out of two studies showed that an increase in BIRC5 mRNA was associated with worse survival in univariable analysis (21). Gain at 17q23.2–25.3 was associated with worse survival in univariable analysis in another study (44). One study investigated the effect of chromosomal gains and losses and showed an independent effect on worse survival for Xq loss (HR 3.6, 95% CI: 1.6–8.3), 10q loss (HR 3.2, 95% CI: 1.4–7.7), and 16p gain (HR 2.5, 95% CI: 1.0–6.2) (25). Together a high risk group (63% of population) was obtained for either gain or loss which resulted in a HR 11.0 (95% CI: 3.5–35.0) after correction for several clinical characteristics. A gain in SOX5 and NOL1 were associated with worse survival in one study, but only in univariable analyses (61). Finally, methylation of MAGEA3 was also associated with worse survival in univariable analysis (50).
he underlying biology of MPNSTs remains complex as is highlighted by the diverse findings of studies included in this review. Many markers and genetic alterations have been proposed to be of prognostic value, yet outcomes are infrequently repeated. Alterations in TP53 or its resulting increased p53 staining were commonly found associated with survival and DFS as were several other proteins and genes involved in cell cycle regulation. Epigenetic modulatory proteins, especially loss of H3K27me3, and more general markers as absence of S100 and increased Ki67 were commonly found to be of prognostic value too.
The predictive value of clinical parameters including patient and tumor characteristics has been studied more commonly than immunohistochemical or genetic biomarkers in MPNST. Increasing age, large tumor size, metastatic disease at diagnosis, and tumors not amenable to complete resection are the most commonly found predictors of worse survival in MPNST (2, 3, 5, 13, 45, 65). This emphasizes the importance of early diagnosis of MPNST in order to completely resect tumors, along with finding new systemic therapies to improve the prognosis of irresectable and metastatic disease. Non-extremity tumor sites have also been shown to have a negative impact on survival; however, this may be more true for those arising in retroperitoneal or pelvic sites (1, 3, 5, 13, 66). Tumor depth used to be incorporated for prognostication in the AJCC staging system for STS, but has varyingly been shown to be of prognostic value in MPNST (2, 3, 5, 13, 45, 65). The importance of NF1 disease has also been subject of debate. A meta-analysis in 2012 showed no difference in survival for patients in papers published after 2000 (15). However, recent large cohorts did find an independent association with worse survival for NF1 patients (3, 13, 67, 68). Altogether, clinical parameters seem to be able to predict some part of a patient’s course of disease. The addition of tumor biology to clinical parameters may further increase our ability to stratify subgroups of patients based on prognosis. TP53 is one of the few recurrently mutated genes found in MPNST. TP53 mutations and high p53 staining were independently associated with survival or DFS in five different studies (4, 24, 34, 43, 56). This may indicate that aberrations in this gene may indeed be of clinical importance. Other genes involved in cell cycle regulation such as CDKN2A and downstream proteins are commonly altered and may not only contribute to tumorigenesis but also be of clinical significance, supporting a belief that dysregulations in this cellular pathway are of overall importance. Loss of polycomb regressive complex 2 (PRC2) complex has recently been shown to be common in MPNSTs due to mutations in EED and SUZ12 (9, 69). This results in loss of H3K27me3 which can reliably distinguish high-grade MPNSTs from their benign counterparts by immunohistochemistry (26, 70). MPNSTs without loss of H3K27me3 staining may also be associated with less aggressive behavior as many low-grade MPNSTs are known to retain this expression (14, 26). Preclinical research on targeted therapies has most frequently shown promising results targeting proteins in the Ras pathway, especially when combined with other target drugs, but unfortunately no clinical trial has proven benefit to date (20). Activated proteins in the Ras pathway, including p-mTOR, p-4E-BP1, p-S6RP, COX-2, and Myc as well as methylation of RASSF1A may however predict worse survival (6, 7, 27, 32). Targeting vascular pathways in MPNSTs may be beneficial, but unfortunately few studies have focused on this. Studies included in this review also showed that increased vascularity, as evidenced by increased microvascular density as well as increased expression of VEGF, may be associated with more aggressive biological behavior (57, 59). It seems that many other targets may be of prognostic value as well emphasizing the need for further research into MPNST tumor biology. Survivin markers may for instance stratify a subgroup of patients and survivin has been shown a viable target in a xenograft mouse model (71). Seeing as MPNSTs are heterogenic and markers such as p53 are not be MPNST specific, combined scores of different markers and genetic alterations may be of most clinical importance. Four studies in this review highlight this phenomenon demonstrating increased prognostic value when markers are combined (25, 28, 34, 41).
Unfortunately, due to the large heterogeneity of published studies meta-analyses were not presumed feasible. All studies included in this review were retrospective of nature inherently harboring bias. None of the markers and genetic alterations found in these studies were prospectively validated. Moreover, many did not evaluate the prognostic value of their markers in a multivariable model nor on their discriminative ability. Studies that evaluated the prognostic value of markers in a multivariable model were nonetheless not always capable to correct for all common clinical variables. MPNSTs are rare sarcomas, which in combination with their complex biology, make it difficult to obtain enough cases to create valuable models. But as shown in this review, several markers and genetic alterations may already be of clinical importance as they have shown an independent association with survival in addition to clinical parameters. Future research should therefore be encouraged to replicate these results using larger datasets obtained by large-scale international collaborations. Important immunohistochemical staining may include Ki67, S100, p53, and H3K27me3 in all patients, and possibly further staining of proteins associated with cell cycle regulation. In turn individual prediction models for MPNST patients specifically may arise taking their significant heterogeneity into account. Such models may better elucidate patient selection for (neo)adjuvant treatment and targeted therapies, which should then be validated in a prospective database. But as MPNSTs remain rare entities one may also turn to exploratory analyses using machine learning techniques on large STS genetic databases to identify attractive genes as biomarkers or prognostic markers in subtypes of STS (72).
MPNSTs harbor complex and heterogenic biology and currently lack adequate staging systems. Immunohistochemical markers and genetic alterations are varyingly of prognostic value. Absence of S100 and H3K27me3 and increased Ki67 staining were commonly found to be of independent prognostic value alongside of clinical parameters. Alterations in TP53 or its consequential increase in p53 staining seems to distinguish a subgroup of MPNSTs with worse outcomes. Immunohistochemical staining and associated genetic alterations of proteins involved in cell cycle regulation and the Ras pathway may also help stratifying patients with worse outcomes. Other markers will likely need further evaluation for validation. A combination of markers may increase the prognostic value.
All datasets presented in this study are included in the article/Supplementary Material.
Study conceptualization: EM, CV. Study design: EM, IA, DG, JB, CV. Data acquisition: EM, IA. Drafting manuscript: EM, IA. Study supervision: DG, JB, CV. Manuscript reviewing: DG, JB, CV. Approval of final manuscript: EM, IA, DG, JB, CV. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.594069/full#supplementary-material
1. Stucky C-CH, Johnson KN, Gray RJ, Pockaj BA, Ocal IT, Rose PS, et al. Malignant peripheral nerve sheath tumors (MPNST): the Mayo Clinic experience. Ann Surg Oncol (2012) 19:878–85. doi: 10.1245/s10434-011-1978-7
2. Valentin T, Le Cesne A, Ray-Coquard I, Italiano A, Decanter G, Bompas E, et al. Management and prognosis of malignant peripheral nerve sheath tumors: The experience of the French Sarcoma Group (GSF-GETO). Eur J Cancer (2016) 56:77–84. doi: 10.1016/j.ejca.2015.12.015
3. Martin E, Coert JH, Flucke UE, Slooff W-BM, Ho VKY, van der Graaf WT, et al. A nationwide cohort study on treatment and survival in patients with malignant peripheral nerve sheath tumours. Eur J Cancer (2019) 124:77–87. doi: 10.1016/j.ejca.2019.10.014
4. Zou C, Smith KD, Liu J, Lahat G, Myers S, Wang W-L, et al. Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg (2009) 249:1014–22. doi: 10.1097/SLA.0b013e3181a77e9a
5. Anghileri M, Miceli R, Fiore M, Mariani L, Ferrari A, Mussi C, et al. Malignant peripheral nerve sheath tumors: prognostic factors and survival in a series of patients treated at a single institution. Cancer (2006) 107:1065–74. doi: 10.1002/cncr.22098
6. Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature (1992) 356:713–5. doi: 10.1038/356713a0
7. Endo M, Yamamoto H, Setsu N, Kohashi K, Takahashi Y, Ishii T, et al. Prognostic significance of AKT/mTOR and MAPK pathways and antitumor effect of mTOR inhibitor in NF1-related and sporadic malignant peripheral nerve sheath tumors. Clin Cancer Res (2013) 19:450–61. doi: 10.1158/1078-0432.CCR-12-1067
8. Beert E, Brems H, Daniels B, De Wever I, Van Calenbergh F, Schoenaers J, et al. Atypical neurofibromas in neurofibromatosis type 1 are premalignant tumors. Genes Chromosomes Cancer (2011) 50:1021–32. doi: 10.1002/gcc.20921
9. De Raedt T, Beert E, Pasmant E, Luscan A, Brems H, Ortonne N, et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature (2014) 514:247–51. doi: 10.1038/nature13561
10. Legius E, Dierick H, Wu R, Hall BK, Marynen P, Cassiman JJ, et al. TP53 mutations are frequent in malignant NF1 tumors. Genes Chromosomes Cancer (1994) 10:250–5. doi: 10.1002/gcc.2870100405
11. Masliah-Planchon J, Pasmant E, Luscan A, Laurendeau I, Ortonne N, Hivelin M, et al. MicroRNAome profiling in benign and malignant neurofibromatosis type 1-associated nerve sheath tumors: evidences of PTEN pathway alterations in early NF1 tumorigenesis. BMC Genomics (2013) 14:473. doi: 10.1186/1471-2164-14-473
12. Kluwe L, Friedrich RE, Mautner VF. Allelic loss of the NF1 gene in NF1-associated plexiform neurofibromas. Cancer Genet Cytogenet (1999) 113:65–9. doi: 10.1016/S0165-4608(99)00006-0
13. Miao R, Wang H, Jacobson A, Lietz AP, Choy E, Raskin KA, et al. Radiation-induced and neurofibromatosis-associated malignant peripheral nerve sheath tumors (MPNST) have worse outcomes than sporadic MPNST. Radiother Oncol (2019) 137:61–70. doi: 10.1016/j.radonc.2019.03.015
14. Miettinen MM, Antonescu CR, Fletcher CDM, Kim A, Lazar AJ, Quezado MM, et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum Pathol (2017) 67:1–10. doi: 10.1016/j.humpath.2017.05.010
15. Kolberg M, Holand M, Agesen TH, Brekke HR, Liestol K, Hall KS, et al. Survival meta-analyses for >1800 malignant peripheral nerve sheath tumor patients with and without neurofibromatosis type 1. Neuro Oncol (2013) 15:135–47. doi: 10.1093/neuonc/nos287
16. Gronchi A, Ferrari S, Quagliuolo V, Broto JM, Pousa AL, Grignani G, et al. Histotype-tailored neoadjuvant chemotherapy versus standard chemotherapy in patients with high-risk soft-tissue sarcomas (ISG-STS 1001): an international, open-label, randomised, controlled, phase 3, multicentre trial. Lancet Oncol (2017) 18:812–22. doi: 10.1016/S1470-2045(17)30334-0
17. Higham CS, Steinberg SM, Dombi E, Perry A, Helman LJ, Schuetze SM, et al. SARC006: Phase II Trial of Chemotherapy in Sporadic and Neurofibromatosis Type 1 Associated Chemotherapy-Naive Malignant Peripheral Nerve Sheath Tumors. Sarcoma (2017) 2017:8685638. doi: 10.1155/2017/8685638
18. Carli M, Ferrari A, Mattke A, Zanetti I, Casanova M, Bisogno G, et al. Pediatric malignant peripheral nerve sheath tumor: the Italian and German soft tissue sarcoma cooperative group. J Clin Oncol (2005) 23:8422–30. doi: 10.1200/JCO.2005.01.4886
19. Zehou O, Bularca S, Bastuji-Garin S, Ortonne N, Valeyrie-Allanore L, Wolkenstein P, et al. Neurofibromatosis 1 phenotype associated to malignant peripheral nerve sheath tumours: A case-control study. J Eur Acad Dermatol Venereol (2013) 27:1044–7. doi: 10.1111/j.1468-3083.2012.04485.x
20. Martin E, Lamba N, Flucke UE, Verhoef C, Coert JH, Versleijen-Jonkers YMH, et al. Non-cytotoxic systemic treatment in malignant peripheral nerve sheath tumors (MPNST): A systematic review from bench to bedside. Crit Rev Oncol Hematol (2019) 138:223–32. doi: 10.1016/j.critrevonc.2019.04.007
21. Alaggio R, Turrini R, Boldrin D, Merlo A, Gambini C, Ferrari A, et al. Survivin expression and prognostic significance in pediatric malignant peripheral nerve sheath tumors (MPNST). PloS One (2013) 8:e80456. doi: 10.1371/journal.pone.0080456
22. Benassi MS, Ragazzini P, Gamberi G, Sollazzo MR, Molendini L, Ferrari C, et al. Adhesion molecules in high-grade soft tissue sarcomas: Correlation to clinical outcome. Eur J Cancer (1998) 34:496–502. doi: 10.1016/S0959-8049(97)10097-1
23. Benassi MS, Gamberi G, Magagnoli G, Molendini L, Ragazzini P, Merli M, et al. Metalloproteinase expression and prognosis in soft tissue sarcomas. Ann Oncol (2001) 12:75–80. doi: 10.1023/A:1008318614461
24. Brekke HR, Kolberg M, Skotheim RI, Hall KS, Bjerkehagen B, Risberg B, et al. Identification of p53 as a strong predictor of survival for patients with malignant peripheral nerve sheath tumors. Neuro Oncol (2009) 11:514–28. doi: 10.1215/15228517-2008-127
25. Brekke HR, Ribeiro FR, Kolberg M, Ågesen TH, Lind GE, Eknæs M, et al. Genomic changes in chromosomes 10, 16, and X in malignant peripheral nerve sheath tumors identify a high-risk patient group. J Clin Oncol (2010) 28:1573–82. doi: 10.1200/JCO.2009.24.8989
26. Cleven AHG, Sannaa GAA, Briaire-de Bruijn I, Ingram DR, van de Rijn M, Rubin BP, et al. Loss of H3K27 tri-methylation is a diagnostic marker for malignant peripheral nerve sheath tumors and an indicator for an inferior survival. Mod Pathol (2016) 29:582–90. doi: 10.1038/modpathol.2016.45
27. Danielsen SA, Lind GE, Kolberg M, Høland M, Bjerkehagen B, Hall KS, et al. Methylated RASSF1A in malignant peripheral nerve sheath tumors identifies neurofibromatosis type 1 patients with inferior prognosis. Neuro Oncol (2015) 17:63–9. doi: 10.1093/neuonc/nou140
28. Endo M, Kobayashi C, Setsu N, Takahashi Y, Kohashi K, Yamamoto H, et al. Prognostic significance of p14ARF, p15INK4b, and p16INK4a inactivation in malignant peripheral nerve sheath tumors. Clin Cancer Res (2011) 17:3771–82. doi: 10.1158/1078-0432.CCR-10-2393
29. Fan Q, Yang J, Wang G. Clinical and molecular prognostic predictors of malignant peripheral nerve sheath tumor. Clin Transl Oncol (2014) 16:191–9. doi: 10.1007/s12094-013-1061-x
30. Fukushima S, Endo M, Matsumoto Y, Fukushi JI, Matsunobu T, Kawaguchi KI, et al. Potential therapeutic target in malignant peripheral nerve sheath tumor. PloS One (2017) 12:e0178064. doi: 10.1371/journal.pone.0178064
31. Gong H, Zhang D, Wang D, He S, Yang X, Wei H, et al. Benign-appearing intraspinal malignant peripheral nerve sheath tumors: Treatments and outcomes of 14 consecutive patients. Turk Neurosurg (2018) 28:983–8. doi: 10.5137/1019-5149.JTN.22580-18.1
32. Hakozaki M, Tajino T, Konno S, Kikuchi S, Yamada H, Yanagisawa M, et al. Overexpression of cyclooxygenase-2 in malignant peripheral nerve sheath tumor and selective cyclooxygenase-2 inhibitor-induced apoptosis by activating caspases in human malignant peripheral nerve sheath tumor cells. PloS One (2014) 9:e88035. doi: 10.1371/journal.pone.0088035
33. Halling KC, Scheithauer BW, Halling AC, Nascimento AG, Ziesmer SC, Roche PC, et al. p53 Expression in neurofibroma and malignant peripheral nerve sheath tumor: An immunohistochemical study of sporadic and NF1-associated tumors. Am J Clin Pathol (1996) 106:282–8. doi: 10.1093/ajcp/106.3.282
34. Holand M, Kolberg M, Danielsen SA, Bjerkehagen B, Eilertsen IA, Hektoen M, et al. Inferior survival for patients with malignant peripheral nerve sheath tumors defined by aberrant TP53. Mod Pathol (2018) 31:1694–707. doi: 10.1038/s41379-018-0074-y
35. Holtkamp N, Atallah I, Okuducu A-F, Mucha J, Hartmann C, Mautner V-F, et al. MMP-13 and p53 in the progression of malignant peripheral nerve sheath tumors. Neoplasia (2007) 9:671–7. doi: 10.1593/neo.07304
36. Holtkamp N, Malzer E, Zietsch J, Okaducu AF, Mucha J, Mawrin C, et al. EGFR and erbB2 in malignant peripheral nerve sheath tumors and implications for targeted therapy. Neuro Onco (2008) 10:946–57. doi: 10.1215/15228517-2008-053
37. Ikuta K, Urakawa H, Kozawa E, Arai E, Zhuo L, Futamura N, et al. Hyaluronan expression as a significant prognostic factor in patients with malignant peripheral nerve sheath tumors. Clin Exp Metastasis (2014) 31:715–25. doi: 10.1007/s10585-014-9662-5
38. Jia X, Chen C, Chen L, Yu C, Kondo T. Decorin as a prognostic biomarker in patients with malignant peripheral nerve sheath tumors. Oncol Lett (2019) 17:3517–22. doi: 10.3892/ol.2019.9959
39. Keizman D, Issakov J, Meller I, Meimon N, Ish-Shalom M, Sher O, et al. Expression and significance of EGFR in malignant peripheral nerve sheath tumor. J Neurooncol (2009) 94:383–8. doi: 10.1007/s11060-009-9862-z
40. Kobayashi C, Oda Y, Takahira T, Izumi T, Kawaguchi K, Yamamoto H, et al. Aberrant expression of CHFR in malignant peripheral nerve sheath tumors. Mod Pathol (2006) 19:524–32. doi: 10.1038/modpathol.3800548
41. Kolberg M, Holand M, Lind GE, Agesen TH, Skotheim RI, Hall KS, et al. Protein expression of BIRC5, TK1, and TOP2A in malignant peripheral nerve sheath tumours–A prognostic test after surgical resection. Mol Oncol (2015) 9:1129–39. doi: 10.1016/j.molonc.2015.02.005
42. Kourea HP, Cordon-Cardo C, Dudas M, Leung D, Woodruff JM. Expression of p27(kip) and other cell cycle regulators in malignant peripheral nerve sheath tumors and neurofibromas: The emerging role of p27(kip) in malignant transformation of neurofibromas. Am J Pathol (1999) 155:1885–91. doi: 10.1016/S0002-9440(10)65508-3
43. Krawczyk MA, Karpinsky G, Izycka-Swieszewska E, Gabrych A, Kunc M, Fatyga A, et al. Immunohistochemical assessment of cyclin D1 and p53 is associated with survival in childhood malignant peripheral nerve sheath tumor. Cancer Biomark (2019) 24:351–61. doi: 10.3233/CBM-181572
44. Kresse SH, Skårn M, Ohnstad HO, Namløs HM, Bjerkehagen B, Myklebost O, et al. DNA copy number changes in high-grade malignant peripheral nerve sheath tumors by array CGH. Mol Cancer (2008) 7. doi: 10.1186/1476-4598-7-48
45. LaFemina J, Qin L-X, Moraco NH, Antonescu CR, Fields RC, Crago AM, et al. Oncologic outcomes of sporadic, neurofibromatosis-associated, and radiation-induced malignant peripheral nerve sheath tumors. Ann Surg Oncol (2013) 20:66–72. doi: 10.1245/s10434-012-2573-2
46. Le Guellec S, Decouvelaere A-V, Filleron T, Valo I, Charon-Barra C, Robin Y-M, et al. Malignant Peripheral Nerve Sheath Tumor Is a Challenging Diagnosis: A Systematic Pathology Review, Immunohistochemistry, and Molecular Analysis in 160 Patients From the French Sarcoma Group Database. Am J Surg Pathol (2016) 40:896–908. doi: 10.1097/PAS.0000000000000655
47. Leroy K, Dumas V, Martin-Garcia N, Falzone MC, Voisin MC, Wechsler J, et al. Malignant peripheral nerve sheath tumors associated with neurofibromatosis type 1: a clinicopathologic and molecular study of 17 patients. Arch Dermatol (2001) 137:908–13.
48. Lu H-C, Eulo V, Apicelli AJ, Pekmezci M, Tao Y, Luo J, et al. Aberrant ATRX protein expression is associated with poor overall survival in NF1-MPNST. Oncotarget (2018) 9:23018–28. doi: 10.18632/oncotarget.25195
49. Meis JM, Enzinger FM, Martz KL, Neal JA. Malignant peripheral nerve sheath tumors (malignant schwannomas) in children. Am J Surg Pathol (1992) 16:694-707. doi: 10.1097/00000478-199207000-00008
50. Nobeyama Y, Nakagawa H. Aberrant demethylation and expression of MAGEB2 in a subset of malignant peripheral nerve sheath tumors from neurofibromatosis type 1. J Dermatol Sci (2016) 81:118–23. doi: 10.1016/j.jdermsci.2015.11.004
51. Otsuka H, Kohashi K, Yoshimoto M, Ishihara S, Toda Y, Yamada Y, et al. Immunohistochemical evaluation of H3K27 trimethylation in malignant peripheral nerve sheath tumors. Pathol Res Pract (2018) 214:417–25. doi: 10.1016/j.prp.2017.12.015
52. Panse G, Leung CH, Ingram DR, Wani K, Torres KE, Lin H, et al. The role of phosphorylated signal transducer and activator of transcription 3 (pSTAT3) in peripheral nerve sheath tumours. Histopathology (2017) 70:946–53. doi: 10.1111/his.13154
53. Pekmezci M, Cuevas-Ocampo AK, Perry A, Horvai AE. Significance of H3K27me3 loss in the diagnosis of malignant peripheral nerve sheath tumors. Mod Pathol (2017) 30:1710–9. doi: 10.1038/modpathol.2017.97
54. Skotheim RI, Kallioniemi A, Bjerkhagen B, Mertens F, Brekke HR, Monni O, et al. Topoisomerase-IIα is upregulated in malignant peripheral nerve sheath tumors and associated with clinical outcome. J Clin Oncol (2003) 21:4586–91. doi: 10.1200/JCO.2003.07.067
55. Tabone-Eglinger S, Bahleda R, Côté J-F, Terrier P, Vidaud D, Cayre A, et al. Frequent EGFR Positivity and Overexpression in High-Grade Areas of Human MPNSTs. Sarcoma (2008) 2008:1–7. doi: 10.1155/2008/849156
56. Torres KE, Zhu Q-S, Bill K, Lopez G, Ghadimi MP, Xie X, et al. Activated MET is a molecular prognosticator and potential therapeutic target for malignant peripheral nerve sheath tumors. Clin Cancer Res (2011) 17:3943–55. doi: 10.1158/1078-0432.CCR-11-0193
57. de Vasconcelos RAT, Guimarães Coscarelli P, Vieira TM, Noguera WS, Rapozo DCM, Acioly MA. Prognostic significance of mast cell and microvascular densities in malignant peripheral nerve sheath tumor with and without neurofibromatosis type 1. Cancer Med (2019) 8:972–81. doi: 10.1002/cam4.1977
58. Wang T, Yin H, Han S, Yang X, Wang J, Huang Q, et al. Malignant peripheral nerve sheath tumor (MPNST) in the spine: a retrospective analysis of clinical and molecular prognostic factors. J Neurooncol (2015) 122:349–55. doi: 10.1007/s11060-015-1721-5
59. Wasa J, Nishida Y, Suzuki Y, Tsukushi S, Shido Y, Hosono K, et al. Differential expression of angiogenic factors in peripheral nerve sheath tumors. Clin Exp Metastasis (2008) 25:819–25. doi: 10.1007/s10585-008-9197-8
60. Watanabe T, Oda Y, Tamiya S, Kinukawa N, Masuda K, Tsuneyoshi M. Malignant peripheral nerve sheath tumours: High Ki67 labelling index is the significant prognostic indicator. Histopathology (2001) 39:187–97. doi: 10.1046/j.1365-2559.2001.01176.x
61. Yu J, Deshmukh H, Payton JE, Dunham C, Scheithauer BW, Tihan T, et al. Array-based comparative genomic hybridization identifies CDK4 and FOXM1 alterations as independent predictors of survival in malignant peripheral nerve sheath tumor. Clin Cancer Res (2011) 17:1924–34. doi: 10.1158/1078-0432.CCR-10-1551
62. Yuan ZN, Xu LB, Zhao ZG, Xu SF, Zhang XX, Liu T, et al. Clinicopathological features and prognosis of malignant peripheral nerve sheath tumor: a retrospective study of 159 cases from 1999 to 2016. Oncotarget (2017) 8:104785–95. doi: 10.18632/oncotarget.18975
63. Zhang C, Chang F-Y, Zhou W-Y, Yang J-L. The prognostic value of C-X-C motif chemokine receptor 4 in patients with sporadic malignant peripheral nerve sheath tumors. Chin J Cancer (2017) 36:1–8. doi: 10.1186/s40880-017-0246-z
64. Zhou W, Du X, Song F, Zheng H, Chen K, Zhang W, et al. Prognostic roles for fibroblast growth factor receptor family members in malignant peripheral nerve sheath tumor. Oncotarget (2016) 7:22234–44. doi: 10.18632/oncotarget.8067
65. Watson KL, Al Sannaa GA, Kivlin CM, Ingram DR, Landers SM, Roland CL, et al. Patterns of recurrence and survival in sporadic, neurofibromatosis Type 1-associated, and radiation-associated malignant peripheral nerve sheath tumors. J Neurosurg (2017) 126:319–29. doi: 10.3171/2015.12.JNS152443
66. Martin E, Muskens IS, Coert JH, Smith TR, Broekman MLD. Treatment and survival differences across tumor sites in malignant peripheral nerve sheath tumors: a SEER database analysis. Neurooncol Pract (2018) 6:1–10. doi: 10.1093/nop/npy025
67. Martin E, Coert JH, Flucke UE, Slooff W-BM, van de Sande MAJ, van Noesel MM, et al. Neurofibromatosis-associated malignant peripheral nerve sheath tumors in children have a worse prognosis: A nationwide cohort study. Pediatr Blood Cancer (2019) 67:e28138. doi: 10.1002/pbc.28138
68. van Noesel MM, Orbach D, Brennan B, Kelsey A, Zanetti I, de Salvo GL, et al. Outcome and prognostic factors in pediatric malignant peripheral nerve sheath tumors: An analysis of the European Pediatric Soft Tissue Sarcoma Group (EpSSG) NRSTS-2005 prospective study. Pediatr Blood Cancer (2019) 66:e27833. doi: 10.1002/pbc.27833
69. Lee W, Teckie S, Wiesner T, Ran L, Prieto Granada CN, Lin M, et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet (2014) 46:1227–32. doi: 10.1038/ng.3095
70. Schaefer I-M, Fletcher CD, Hornick JL. Loss of H3K27 trimethylation distinguishes malignant peripheral nerve sheath tumors from histologic mimics. Mod Pathol (2016) 29:4–13. doi: 10.1038/modpathol.2015.134
71. Ghadimi MP, Young ED, Belousov R, Zhang Y, Lopez G, Lusby K, et al. Survivin is a viable target for the treatment of malignant peripheral nerve sheath tumors. Clin Cancer Res (2012) 18:2545–57. doi: 10.1158/1078-0432.CCR-11-2592
72. van IJzendoorn DGP, Szuhai K, Briaire-de Bruijn IH, Kostine M, Kuijjer ML, Bovee JVMG. Machine learning analysis of gene expression data reveals novel diagnostic and prognostic biomarkers and identifies therapeutic targets for soft tissue sarcomas. PloS Comput Biol (2019) 15:e1006826. doi: 10.1371/journal.pcbi.1006826
Keywords: malignant peripheral nerve sheath tumors, prognosis, molecular, clinicopathologic, markers, genes
Citation: Martin E, Acem I, Grünhagen DJ, Bovée JVMG and Verhoef C (2020) Prognostic Significance of Immunohistochemical Markers and Genetic Alterations in Malignant Peripheral Nerve Sheath Tumors: A Systematic Review. Front. Oncol. 10:594069. doi: 10.3389/fonc.2020.594069
Received: 12 August 2020; Accepted: 19 November 2020;
Published: 22 December 2020.
Edited by:
Parvin Mehdipour, Tehran University of Medical Sciences, IranReviewed by:
Jilong Yang, Tianjin Medical University Cancer Institute and Hospital, ChinaCopyright © 2020 Martin, Acem, Grünhagen, Bovée and Verhoef. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Enrico Martin, ZS5tYXJ0aW4tM0B1bWN1dHJlY2h0Lm5s
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.