 Anaïs Jiménez-Reinoso
Anaïs Jiménez-Reinoso Daniel Nehme-Álvarez
Daniel Nehme-Álvarez Carmen Domínguez-Alonso
Carmen Domínguez-Alonso Luis Álvarez-Vallina
Luis Álvarez-Vallina
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol., 16 February 2021
Sec. Cancer Immunity and Immunotherapy
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.593848
Immunotherapy has emerged as an effective and life-changing approach for several types of cancers, both liquid and solid tumors. In combination with traditional treatments such as radiotherapy and/or chemotherapy, immune checkpoints inhibitors have improved prognosis and overall survival of patients with advanced melanoma and many other cancers. Among adoptive cell therapies (ACT), while chimeric antigen receptor T cell therapies have demonstrated remarkable efficacy in some hematologic malignancies, such as B cell leukemias, their success in solid tumors remains scarce due to the characteristics of the tumor microenvironment. On the other hand, ACT using tumor-infiltrating lymphocytes (TILs) is arguably the most effective treatment for metastatic melanoma patients, but even if their isolation has been achieved in epithelial tumors, their success beyond melanoma remains limited. Here, we review several aspects impacting TIL- and gene-modified “synthetic” TIL-based therapies and discuss future challenges that must be addressed with these approaches.
The spectrum of cancer treatments has been increasing in recent years with the incorporation of different immunotherapy strategies that take advantage of the effectiveness and potential of the immune system to fight cancer cells. Cancer immunotherapy has been named 2013 “Breakthrough of the year” by the prestigious Science journal (1). Different immunotherapy approaches are currently under development aiming to improve outcomes for cancer patients, such as immune checkpoint inhibitors [CTLA4 and PD-1/PD-L1 axis (2–5)], monospecific (6) and bispecific (7) monoclonal antibodies, immune-stimulatory agents such as BCG (8), cancer vaccines (9), and the adoptive transfer of tumor-reactive immune cells (10).
Adoptive cell therapy (ACT) is a personalized strategy that involves infusion of ex vivo-expanded endogenous (pre-existing) tumor-reactive T cell repertoires, such as tumor-infiltrating lymphocytes (TILs) (11), and endogenous T cell therapy (ETC) (12), or the generation of artificial tumor-reactive T cells (13), such as engineered T cells expressing transgenic T cell receptors (TCR) or chimeric antigen receptors (CAR) (14). While TILs are tumor-specific lymphocytes directly isolated from tumor resections, ETC are tumor-reactive T cells isolated from the peripheral blood of patients (15). Engineered TCR- and CAR-T cells are leukapheresis blood-derived T cells genetically modified ex vivo in order to specifically recognize a tumor-associated antigen (TAA) via mRNA electroporation (16), lentiviral (17) or retroviral (18) transduction, transposon mediated modification (19) or via CRISPR/Cas9 gene editing (20).
TILs are T cells isolated from tumor fragments, ex vivo-expanded and reinfused back into pre-conditioned patients under a non-myeloablative lymphodepletion chemotherapy with high doses of interleukin-2 (IL-2) (21). TILs have shown impressive results in patients with metastatic melanoma (MM), where objective response rates of 40%–50% including complete tumor regression in 10%–20% of treated patients have consistently been reported by several independent centers (22–27). Although TILs can also be obtained from epithelial cancers (11, 28) such as breast (29, 30), ovarian (31), renal (32), gastrointestinal (33), pancreatic (34), cervical (35) or prostate (36) tumors, the reported response rates have been very modest (37).
In CAR-T-based ACT the major histocompatibility complex (MHC)-restricted peptide presentation is bypassed, but only a user-defined cell surface TAA can be recognized by the CAR. However, the use of CAR-T cells in solid tumors has been limited by organ toxicities related to activation of T cell effector functions through the CAR, since most TAAs are also found in normal tissues, raising the risk of on-target off-tumor toxicities (38). In contrast, TILs products are highly polyclonal; thus TIL-based ACT benefits from a multitarget T cell attack directed against multiple different and largely unknown antigens (39). Due to the complexity of identifying the antigen repertoire present in every tumor for which TIL detection has been reported it has been difficult to assess the specific antigens that are detected by TILs, but cloning studies have divided them into non-self and self-antigens, which can be further divided into another 3 major groups: cancer germline antigens (developmental proteins that are re-expressed in some cancerous but not adult/normal tissues), differentiation antigens (which can also be present in normal tissues but in a limited distribution) and foreign antigens (that arise from viral proteins in viral-associated cancers) (39). Nevertheless, several studies demonstrated that the effectiveness of TIL-based ACT in MM is based on the specific recognition of neoantigens (nonsynonymous somatic mutations) (40, 41).
Although TIL therapy is not yet approved by the FDA, several clinical trials are being performed (mainly for MM) (42), and current efforts for its approval are focused on optimizing the manufacturing process (43), and its application to other cancer types. In contrast to MM, the effectiveness of TIL-based ACT relies on the fact that the number of TILs isolated from other tumor types is lower and difficult to expand, as well as on the mutational burden and the characteristics of the tumor microenvironment (44). The “standard TIL” isolation protocol (“selected TILs”), is based on an initial pre-Rapid Expansion Protocol (preREP) stage which comprise the resection of fresh tumor specimens into small segments, their fragmentation and culture under high-dose IL-2 (6,000 IU/ml) conditions for 3–5 weeks (45). The outcome of this first expansion phase is rather variable, since the number of TILs present in the original tumor does not always correlates with the efficiency of the preREP process (46). Following this preREP step, individual TIL micro-cultures are assayed with IFN-γ ELISA/ELISPOT for the ability to recognize autologous tumor cells or HLA-A matched allogeneic melanoma cell lines (43). TIL micro-cultures displaying tumor reactivity against HLA-matched or autologous tumor cells are selected and expanded further in the REP stage under an allogeneic feeder co-culture with healthy donor irradiated (40 Gy) peripheral blood mononuclear cells (PBMC) in a 1:200 ratio with 30 ng/ml anti-CD3 (clone OKT3) and 6,000 IU/ml IL-2. After this REP stage, expanded TILs are transferred into culture bags, prepared and reinfused back into the patient. In addition to the manufacturing obstacles derived from TIL therapy, —it is an extremely personalized therapy which required specialized personal to manage pre- and REP stages as well as highly controlled conditions that guarantee their clinical use—, TIL expansion is a time-consuming protocol that in some cases is not viable due to the rapid clinical deterioration of some melanoma patients from which those TIL were initially isolated. Due to the limitations of the selected TIL method, a modified TIL production protocol was developed and tested in clinical trials both at the Surgery Branch, NCI and the Sheba Medical Center, Israel (47–49). With this modified method, named the “young TIL” protocol, all TIL micro-cultures generated from individual fragments are pooled together as one single bulk TIL culture, eliminating the tumor-reactivity assay (50).
Along with other limitations that TIL-based ACT implies, such as their reduced proliferative capacity and in vivo persistence after the reinfusion to the patient, TILs are in essence highly differentiated effector cells (39), which differentiate from a T effector phenotype to late-stage effector memory cells (51–53). Current efforts also implies selection of tumor-reactive TILs with the co-stimulatory marker 4-1BB/CD137 (34, 54–59) and alternatively, with PD-1/CD279 (60–62), although these approaches remain to be evaluated under clinical trials (63).
The success of TIL-based ACT in MM is based mainly in the high mutational burden and neoantigen emergence rates and in the sustained antitumor reactivity exhibited by this type of cancer (42). However, these characteristic are usually absent in most epithelial tumors such as those mentioned above. In contrast to MM, other solid tumors from which TIL isolation and production have been achieved lack a high mutational load or neoantigen burden and exhibit a scarce antitumor reactivity, which combined to the heterogeneous CD4+ or CD8+ lymphocyte or innate-like and myeloid infiltrates, and the wide variety of metastases types, are currently hindering the applicability of TIL-based ACT (64). In this context, genetically-modified TILs, named “synthetic TILs”, could emerge as an effective ACT beyond MM.
Given the therapeutic potential of TIL-based ACT, almost in parallel with de development of TIL protocols, the genetic modification of TILs has been explored in order to improve their tumor-homing ability after ex vivo expansion and reinfusion into patients (Table 1). In early 90’, feasibility of TIL gene transfer after transduction with the retroviral vector N2 encoding the bacterial gene for neomycin-resistance (NeoR) was analyzed, concluding that those NeoR-TILs could be used for studying TIL trafficking and survival in vivo with no growth detriment or cytokine mRNA pattern alterations (65). Based on these results, the same group studied the safety of reinfusion of genetically modified NeoR-TILs in five MM patients. No adverse effects were reported, PCR analysis detected NeoR-TILs in the circulation after three weeks and in the tumor deposits after 64 days (66, 80), and long-term viable NeoR-TILs after cell infusion were observed (67).
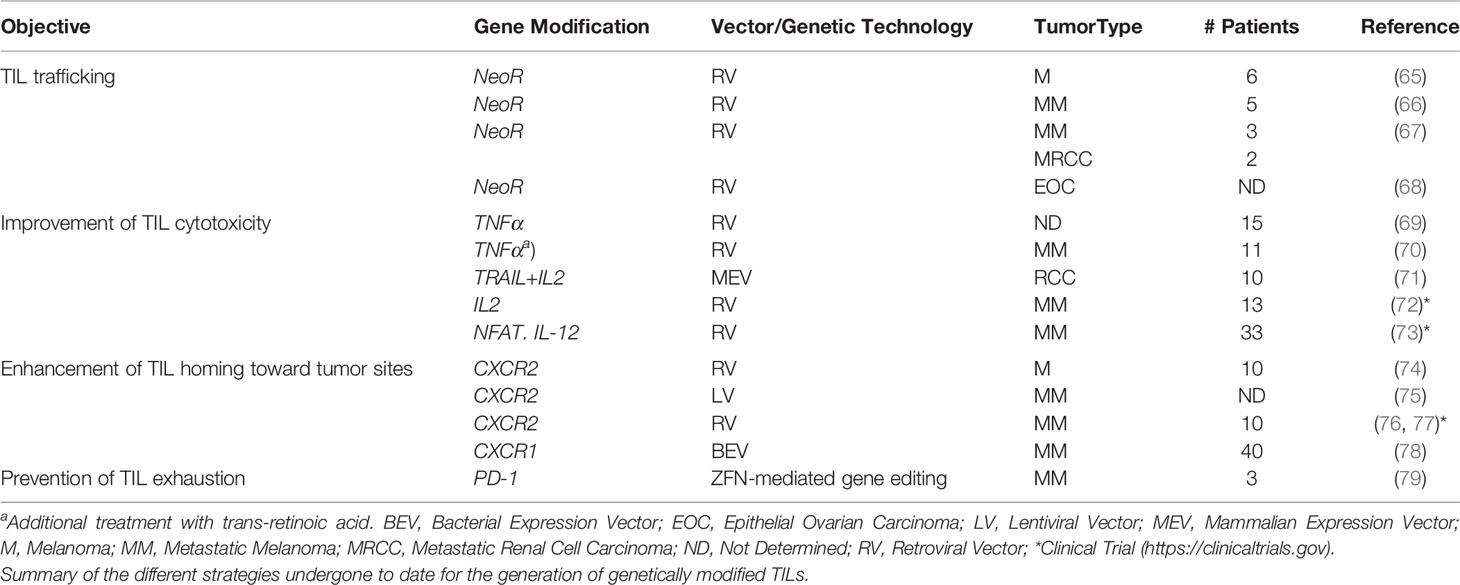
Table 1 Synthetic tumor-infiltrating lymphocytes (TILs) in cancer therapy.
However, not only MM-derived TILs have been explored for gene modification. Transduced CD4+ and CD8+ TILs with the G1Na retroviral vector (encoding for NeoR) have been used to study the in vivo trafficking of ovarian-derived TILs (68).
Other approaches have explored the genetic engineering of TILs with secreted proteins such as tumor necrosis factor (TNF). Hwu et al. (69) demonstrated that TILs could be retrovirally transduced with TNF-α, and although the secreted levels in vitro were lower than expected, these levels could be increased by replacing the transmembrane region of TNF with the IFN-γ signal peptide or after treatment with retinoic acid (70).
TNF-related apoptosis-inducing ligand (TRAIL) and IL-2 have also being transfected into TILs isolated from renal cell carcinoma (RCC) resulting in improved cytotoxicity activity (71). A clinical trial explored whether retrovirally transduced IL-2 secreting MM-derived TILs could enhance their in vivo survival after adoptive transfer. IL2-secreting synthetic TILs improved their in vitro survival in the absence of added IL-2, but the in vivo survival or clinical were not enhanced (72). The potential of IL-12 as a putative enhancer of antitumor activity has been studied in TILs isolated from MM patients. Based on previous studies showing tumor cell toxicity associated with constitutive IL-12 secretion (81), Zhang et al. (73) developed a system in which a single-chain human IL-12 driven by a nuclear factor of activated T cells (NFAT) inducible promoter was selectively secreted at the tumor site after the TCR engagement. However, although objective responses were observed, the clinical toxicities likely associated with IL-12 secreted by synthetic NFAT.IL-12-TILs makes it imperative to improve the approach before undertaking further studies.
Once proved that TILs can be efficiently engineered using different strategies, recent studies have been focusing on improving TIL migration toward tumor sites after re-infusion (Figure 1). With this aim, several groups have explored the generation of synthetic TILs expressing chemokine receptors for different chemokines secreted by tumor cells, such as CXCR2, which is the receptor for several chemokines such as CXCL1 and CXCL8. Initial studies demonstrated that recombinant as well as tumor cell line-derived CXCL1 induced chemoattraction in vitro and IFN-γ secretion of CXCR2-engineered T cells (74). In vivo studies in two xenograft tumor models have also shown that melanoma antigen-specific CXCR2-engineered T cells improved tumor migration and antitumor activity in mice bearing MC38/gp100 tumors or CXCL1-expressing tumors (82). These findings have been validated in NOG mice bearing subcutaneous human melanoma xenografts, in which increased tumor homing and infiltration by CXCR2-engineered T cells was observed (75). In addition, a clinical trial with CXCR2-engineered in MM patients is currently ongoing (76). The methodology developed for this clinical trial has been described by Forget et al. (77) and includes retroviral transduction and TIL expansion (83). Another chemokine studied as a possible target for synthetic TIL generation is CXCR1, which in contrast to CXCR2, is expressed at low levels in MM-derived TILs. Sapoznik et al. (78) demonstrated that CXCR1-engineered TILs migrated in vitro more efficiently toward melanoma or recombinant CXCL8 without altering effector function of migrating TILs.
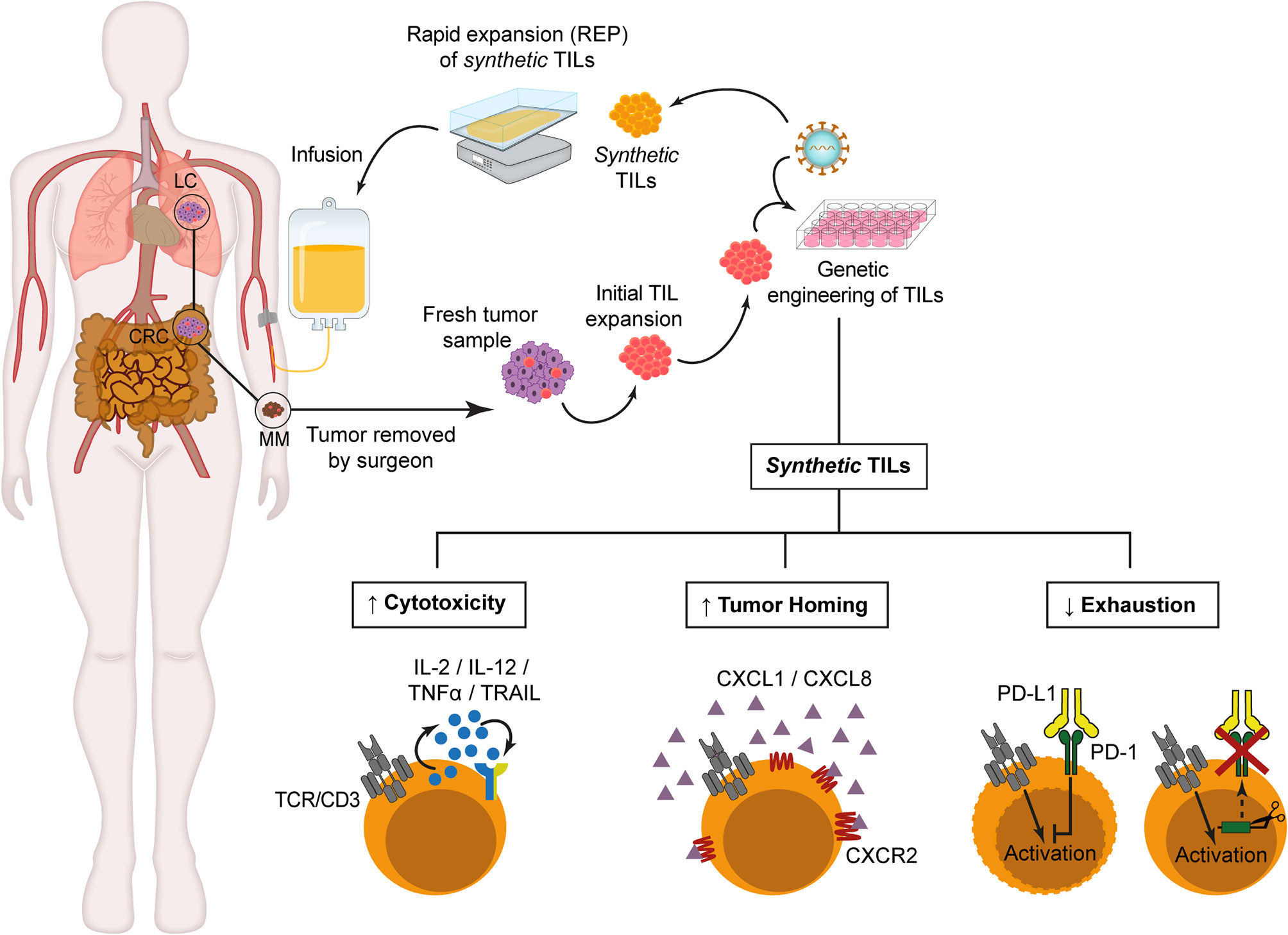
Figure 1 Synthetic TILs manufacturing. After initial TILs isolation from fresh tumor fragments, retroviral or lentiviral vectors are used to genetically modified TILs in order to improve cytotoxicity, enhance tumor homing or reduce T cell exhaustion. Then, synthetic TILs are ex vivo expanded for 14 days in the presence of allogeneic irradiated peripheral blood mononuclear cells, soluble anti-CD3 antibodies and IL-2. Prior to synthetic TIL infusion, the patients receive lymphodepleting chemotherapy to ensure TIL persistence and expansion. CRC, Colorectal Cancer. LC, Lung Cancer. MM, Metastatic Melanoma..
One of the critical challenges that must be solved for clinical approval of TIL-based ACT concerns the durability of TIL responses (Figure 1). PD-1 ligands, PD-L1/L2 are expressed in several tumor types, and the interaction with its receptor triggers effector T cell function inhibition and T cell exhaustion, developing a suppressive microenvironment which prevents antitumor activity. Rosenberg’s group had previously described that isolated tumor-reactive TILs from MM patients expressed PD-1 (61, 84), so they analyzed whether a permanent inhibition of PD-1 in engineered-TILs through zinc finger endonucleases (ZFN) gene-editing technique could improve the effectiveness of TIL after infusion. Beane et al. (79) demonstrate a significant decrease in the number of tumors after treatment with ZFN-PD-1 KO TILs as well as an improved in vitro effector function through TNF-α, GM-CSF and IFN-γ secretion after co-culture with tumor cells, with a predominant effector memory-like phenotype in engineered TILs, and no detectable proliferative defects or tumor formation in NSG mice (79). Nevertheless, safety and efficiency for clinical treatment remains to be tested.
TIL-based ACT in combination with high-dose IL-2 have been shown to be an effective clinical strategy in MM patients, and to a lesser extent, in other tumors. Although many issues remain to be addressed, especially regarding the relatively long generation process and the requirement for GMP facilities and trained personnel, the early-treatment costs are considerably lower than those of anti-CTLA4 mAb in MM (85). Given than TILs are naturally infiltrating cells, they can also serve as biomarker to predict the clinical efficacy of immunotherapies enhancing antitumor adaptive responses (86) Interestingly, different approaches have recently demonstrated that during the ex vivo process necessary for TIL generation, these tumor-specific T cells can be efficiently genetically modified in order to enhance their cytotoxicity, tumor homing or to reduce T cell exhaustion. The resulting cellular product, called synthetic TILs in this review, is at the very beginning of their evolution and could eventually transform the current immunotherapy landscape. By using different genetic engineering strategies and/or gene editing systems, we can speculate that it will be possible to generate personalized synthetic TIL-based ACTs addressing the particular tumor characteristics, with the aim of counteracting the specific tumor evasion mechanisms that are operative in a given patient, or redirecting other immune cells against the tumor. Furthermore, the identification of the TIL mutanome, the specific mutated neoantigens recognize by TILs (86), will provide rationale to develop “à la carte” neoantigen-specific synthetic TILs, or combinations thereof that could be significantly more effective than populations of potentially tumor-reactive TILs obtained by conventional enrichment protocols.
AJ-R and LA-V contributed to the conception of the work and wrote the manuscript. AJ-R and CD-A conceptualized and performed the figure and DN-A developed the table. All authors contributed to the article and approved the submitted version.
AJ-R was supported by CRIS Cancer Foundation. CD-A was supported by the Spanish Ministry of Economy and Competitiveness (PRE2018-083445). LA-V was supported by grants from the Spanish Ministry of Economy and Competitiveness (SAF2017-89437-P, RTC-2017-5944-1), the CRIS Cancer Foundation (FCRIS-IFI-2018), and the Spanish Association Against Cancer (AECC, 19084).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ACT, adoptive cell therapy; CAR, chimeric antigen receptor; ETC, endogenous T cell therapy; IL-2, inteleukin-2; mAb, monoclonal antibody; MHC, major histocompatibility complex; MM, metastatic melanoma; NeoR, bacterial gene for neomycin-resistance; NFAT, nuclear factor of activated T cells; PBMC, peripheral blood mononuclear cells; preREP, pre-Rapid Expansion Protocol; RCC, renal cell carcinoma; REP, rapid expansion protocol; TAA, tumor-associated antigen; TIL, tumor-infiltrating lymphocyte; TCR, T cell receptor; TNF, tumor necrosis factor; TRAIL, TNF-related apoptosis-inducing ligand; ZFN, zinc finger endonucleases.
1. Couzin-Frankel J. Breakthrough of the year 2013. Cancer Immunother Sci (2013) 342(6165):1432–3. doi: 10.1126/science.342.6165.1432
2. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi: 10.1056/NEJMoa1003466
3. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med (2015) 373(17):1627–39. doi: 10.1056/NEJMoa1507643
4. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med (2015) 372(21):2018–28. doi: 10.1056/NEJMoa1501824
5. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature (2014) 515(7528):563–7. doi: 10.1038/nature14011
6. Maloney DG, Grillo-Lopez AJ, Bodkin DJ, White CA, Liles TM, Royston I, et al. IDEC-C2B8: results of a phase I multiple-dose trial in patients with relapsed non-Hodgkin’s lymphoma. J Clin Oncol (1997) 15(10):3266–74. doi: 10.1200/JCO.1997.15.10.3266
7. Przepiorka D, Ko CW, Deisseroth A, Yancey CL, Candau-Chacon R, Chiu HJ, et al. FDA Approval: Blinatumomab. Clin Cancer Res (2015) 21(18):4035–9. doi: 10.1158/1078-0432.CCR-15-0612
8. Aldousari S, Kassouf W. Update on the management of non-muscle invasive bladder cancer. Can Urol Assoc J (2010) 4(1):56–64. doi: 10.5489/cuaj.777
9. Guo C, Manjili MH, Subjeck JR, Sarkar D, Fisher PB, Wang XY. Therapeutic cancer vaccines: past, present, and future. Adv Cancer Res (2013) 119:421–75. doi: 10.1016/B978-0-12-407190-2.00007-1
10. Yang JC, Rosenberg SA. Adoptive T-Cell Therapy for Cancer. Adv Immunol (2016) 130:279–94. doi: 10.1016/bs.ai.2015.12.006
11. Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science (1986) 233(4770):1318–21. doi: 10.1126/science.3489291
12. Yee C. Adoptive T cell therapy: points to consider. Curr Opin Immunol (2018) 51:197–203. doi: 10.1016/j.coi.2018.04.007
13. Crowther MD, Svane IM. Met O. T-Cell Gene Therapy in Cancer Immunotherapy: Why It Is No Longer Just CARs on The Road. Cells (2020) 9(7):1588. doi: 10.3390/cells9071588
14. Met O, Jensen KM, Chamberlain CA, Donia M, Svane IM. Principles of adoptive T cell therapy in cancer. Semin Immunopathol (2019) 41(1):49–58. doi: 10.1007/s00281-018-0703-z
15. Oelke M, Maus MV, Didiano D, June CH, Mackensen A, Schneck JP. Ex vivo induction and expansion of antigen-specific cytotoxic T cells by HLA-Ig-coated artificial antigen-presenting cells. Nat Med (2003) 9(5):619–24. doi: 10.1038/nm869
16. Zhao Y, Zheng Z, Cohen CJ, Gattinoni L, Palmer DC, Restifo NP, et al. High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol Ther (2006) 13(1):151–9. doi: 10.1016/j.ymthe.2005.07.688
17. Tsuji T, Yasukawa M, Matsuzaki J, Ohkuri T, Chamoto K, Wakita D, et al. Generation of tumor-specific, HLA class I-restricted human Th1 and Tc1 cells by cell engineering with tumor peptide-specific T-cell receptor genes. Blood (2005) 106(2):470–6. doi: 10.1182/blood-2004-09-3663
18. Baumeister SH, Murad J, Werner L, Daley H, Trebeden-Negre H, Gicobi JK, et al. Phase I Trial of Autologous CAR T Cells Targeting NKG2D Ligands in Patients with AML/MDS and Multiple Myeloma. Cancer Immunol Res (2019) 7(1):100–12. doi: 10.1158/2326-6066.CIR-18-0307
19. Peng PD, Cohen CJ, Yang S, Hsu C, Jones S, Zhao Y, et al. Efficient nonviral Sleeping Beauty transposon-based TCR gene transfer to peripheral blood lymphocytes confers antigen-specific antitumor reactivity. Gene Ther (2009) 16(8):1042–9. doi: 10.1038/gt.2009.54
20. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature (2017) 543(7643):113–7. doi: 10.1038/nature21405
21. Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science (2002) 298(5594):850–4. doi: 10.1126/science.1076514
22. Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med (1988) 319(25):1676–80. doi: 10.1056/NEJM198812223192527
23. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res (2011) 17(13):4550–7. doi: 10.1158/1078-0432.CCR-11-0116
24. Pilon-Thomas S, Kuhn L, Ellwanger S, Janssen W, Royster E, Marzban S, et al. Efficacy of adoptive cell transfer of tumor-infiltrating lymphocytes after lymphopenia induction for metastatic melanoma. J Immunother (2012) 35(8):615–20. doi: 10.1097/CJI.0b013e31826e8f5f
25. Radvanyi LG, Bernatchez C, Zhang M, Fox PS, Miller P, Chacon J, et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin Cancer Res (2012) 18(24):6758–70. doi: 10.1158/1078-0432.CCR-12-1177
26. Besser MJ, Shapira-Frommer R, Itzhaki O, Treves AJ, Zippel DB, Levy D, et al. Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin Cancer Res (2013) 19(17):4792–800. doi: 10.1158/1078-0432.CCR-13-0380
27. Gorabi AM, Hajighasemi S, Sathyapalan T, Sahebkar A. Cell transfer-based immunotherapies in cancer: A review. IUBMB Life (2020) 72(4):790–800. doi: 10.1002/iub.2180
28. Feldman SA, Assadipour Y, Kriley I, Goff SL, Rosenberg SA. Adoptive Cell Therapy–Tumor-Infiltrating Lymphocytes, T-Cell Receptors, and Chimeric Antigen Receptors. Semin Oncol (2015) 42(4):626–39. doi: 10.1053/j.seminoncol.2015.05.005
29. Aaltomaa S, Lipponen P, Eskelinen M, Kosma VM, Marin S, Alhava E, et al. Lymphocyte infiltrates as a prognostic variable in female breast cancer. Eur J Cancer (1992) 28A(4-5):859–64. doi: 10.1016/0959-8049(92)90134-N
30. Lee HJ, Kim YA, Sim CK, Heo SH, Song IH, Park HS, et al. Expansion of tumor-infiltrating lymphocytes and their potential for application as adoptive cell transfer therapy in human breast cancer. Oncotarget (2017) 8(69):113345–59. doi: 10.18632/oncotarget.23007
31. Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med (2003) 348(3):203–13. doi: 10.1056/NEJMoa020177
32. Andersen R, Westergaard MCW, Kjeldsen JW, Muller A, Pedersen NW, Hadrup SR, et al. T-cell Responses in the Microenvironment of Primary Renal Cell Carcinoma-Implications for Adoptive Cell Therapy. Cancer Immunol Res (2018) 6(2):222–35. doi: 10.1158/2326-6066.CIR-17-0467
33. Turcotte S, Gros A, Hogan K, Tran E, Hinrichs CS, Wunderlich JR, et al. Phenotype and function of T cells infiltrating visceral metastases from gastrointestinal cancers and melanoma: implications for adoptive cell transfer therapy. J Immunol (2013) 191(5):2217–25. doi: 10.4049/jimmunol.1300538
34. Sakellariou-Thompson D, Forget MA, Creasy C, Bernard V, Zhao L, Kim YU, et al. 4-1BB Agonist Focuses CD8(+) Tumor-Infiltrating T-Cell Growth into a Distinct Repertoire Capable of Tumor Recognition in Pancreatic Cancer. Clin Cancer Res (2017) 23(23):7263–75. doi: 10.1158/1078-0432.CCR-17-0831
35. Hilders CG, Ras L, van Eendenburg JD, Nooyen Y, Fleuren GJ. Isolation and characterization of tumor-infiltrating lymphocytes from cervical carcinoma. Int J Cancer (1994) 57(6):805–13. doi: 10.1002/ijc.2910570608
36. Yunger S, Bar El A, Zeltzer LA, Fridman E, Raviv G, Laufer M, et al. Tumor-infiltrating lymphocytes from human prostate tumors reveal anti-tumor reactivity and potential for adoptive cell therapy. Oncoimmunology (2019) 8(12):e1672494. doi: 10.1080/2162402X.2019.1672494
37. Garber K. Pursuit of tumor-infiltrating lymphocyte immunotherapy speeds up. Nat Biotechnol (2019) 37(9):969–71. doi: 10.1038/d41587-019-00023-6
38. Alonso-Camino V, Harwood SL, Álvarez-Méndez A, Álvarez-Vallina L. Efficacy and toxi city management of CAR-T-cell immunotherapy: a matter of responsiveness control or tumour-specificity? Biochem Soc Trans (2016) 44(2):406–11. doi: 10.1042/BST20150286
39. Robbins PF. Tumor-Infiltrating Lymphocyte Therapy and Neoantigens. Cancer J (2017) 23(2):138–43. doi: 10.1097/PPO.0000000000000267
40. Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med (2013) 19(6):747–52. doi: 10.1038/nm.3161
41. Prickett TD, Crystal JS, Cohen CJ, Pasetto A, Parkhurst MR, Gartner JJ, et al. Durable Complete Response from Metastatic Melanoma after Transfer of Autologous T Cells Recognizing 10 Mutated Tumor Antigens. Cancer Immunol Res (2016) 4(8):669–78. doi: 10.1158/2326-6066.CIR-15-0215
42. Rohaan MW, van den Berg JH, Kvistborg P, Haanen J. Adoptive transfer of tumor-infiltrating lymphocytes in melanoma: a viable treatment option. J Immunother Cancer (2018) 6(1):102. doi: 10.1186/s40425-018-0391-1
43. Hopewell EL, Cox C, Pilon-Thomas S, Kelley LL. Tumor-infiltrating lymphocytes: Streamlining a complex manufacturing process. Cytotherapy (2019) 21(3):307–14. doi: 10.1016/j.jcyt.2018.11.004
44. Ikeda H. T-cell adoptive immunotherapy using tumor-infiltrating T cells and genetically engineered TCR-T cells. Int Immunol (2016) 28(7):349–53. doi: 10.1093/intimm/dxw022
45. Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother (2003) 26(4):332–42. doi: 10.1097/00002371-200307000-00005
46. Qin Y, Petaccia de Macedo M, Reuben A, Forget MA, Haymaker C, Bernatchez C, et al. Parallel profiling of immune infiltrate subsets in uveal melanoma versus cutaneous melanoma unveils similarities and differences: A pilot study. Oncoimmunology (2017) 6(6):e1321187. doi: 10.1080/2162402X.2017.1321187
47. Tran KQ, Zhou J, Durflinger KH, Langhan MM, Shelton TE, Wunderlich JR, et al. Minimally cultured tumor-infiltrating lymphocytes display optimal characteristics for adoptive cell therapy. J Immunother (2008) 31(8):742–51. doi: 10.1097/CJI.0b013e31818403d5
48. Besser MJ, Shapira-Frommer R, Treves AJ, Zippel D, Itzhaki O, Schallmach E, et al. Minimally cultured or selected autologous tumor-infiltrating lymphocytes after a lympho-depleting chemotherapy regimen in metastatic melanoma patients. J Immunother (2009) 32(4):415–23. doi: 10.1097/CJI.0b013e31819c8bda
49. Donia M, Junker N, Ellebaek E, Andersen MH, Straten PT, Svane IM. Characterization and comparison of ‘standard’ and ‘young’ tumour-infiltrating lymphocytes for adoptive cell therapy at a Danish translational research institution. Scand J Immunol (2012) 75(2):157–67. doi: 10.1111/j.1365-3083.2011.02640.x
50. Dudley ME, Gross CA, Langhan MM, Garcia MR, Sherry RM, Yang JC, et al. CD8+ enriched “young” tumor infiltrating lymphocytes can mediate regression of metastatic melanoma. Clin Cancer Res (2010) 16(24):6122–31. doi: 10.1158/1078-0432.CCR-10-1297
51. Junker N, Thor Straten P, Andersen MH, Svane IM. Characterization of ex vivo expanded tumor infiltrating lymphocytes from patients with malignant melanoma for clinical application. J Skin Cancer (2011) 2011:574695. doi: 10.1155/2011/574695
52. Powell DJ Jr., Dudley ME, Robbins PF, Rosenberg SA. Transition of late-stage effector T cells to CD27+ CD28+ tumor-reactive effector memory T cells in humans after adoptive cell transfer therapy. Blood (2005) 105(1):241–50. doi: 10.1182/blood-2004-06-2482
53. Jespersen H, Lindberg MF, Donia M, Soderberg EMV, Andersen R, Keller U, et al. Clinical responses to adoptive T-cell transfer can be modeled in an autologous immune-humanized mouse model. Nat Commun (2017) 8(1):707. doi: 10.1038/s41467-017-00786-z
54. Wolfl M, Kuball J, Ho WY, Nguyen H, Manley TJ, Bleakley M, et al. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. Blood (2007) 110(1):201–10. doi: 10.1182/blood-2006-11-056168
55. Chacon JA, Wu RC, Sukhumalchandra P, Molldrem JJ, Sarnaik A, Pilon-Thomas S, et al. Co-stimulation through 4-1BB/CD137 improves the expansion and function of CD8(+) melanoma tumor-infiltrating lymphocytes for adoptive T-cell therapy. PLoS One (2013) 8(4):e60031. doi: 10.1371/journal.pone.0060031
56. Ye Q, Song DG, Poussin M, Yamamoto T, Best A, Li C, et al. CD137 accurately identifies and enriches for naturally occurring tumor-reactive T cells in tumor. Clin Cancer Res (2014) 20(1):44–55. doi: 10.1158/1078-0432.CCR-13-0945
57. Seliktar-Ofir S, Merhavi-Shoham E, Itzhaki O, Yunger S, Markel G, Schachter J, et al. Selection of Shared and Neoantigen-Reactive T Cells for Adoptive Cell Therapy Based on CD137 Separation. Front Immunol (2017) 8:1211. doi: 10.3389/fimmu.2017.01211
58. Tavera RJ, Forget MA, Kim YU, Sakellariou-Thompson D, Creasy CA, Bhatta A, et al. Utilizing T-cell Activation Signals 1, 2, and 3 for Tumor-infiltrating Lymphocytes (TIL) Expansion: The Advantage Over the Sole Use of Interleukin-2 in Cutaneous and Uveal Melanoma. J Immunother (2018) 41(9):399–405. doi: 10.1097/CJI.0000000000000230
59. Harao M, Forget MA, Roszik J, Gao H, Babiera GV, Krishnamurthy S, et al. 4-1BB-Enhanced Expansion of CD8(+) TIL from Triple-Negative Breast Cancer Unveils Mutation-Specific CD8(+) T Cells. Cancer Immunol Res (2017) 5(6):439–45. doi: 10.1158/2326-6066.CIR-16-0364
60. Inozume T, Hanada K, Wang QJ, Ahmadzadeh M, Wunderlich JR, Rosenberg SA, et al. Selection of CD8+PD-1+ lymphocytes in fresh human melanomas enriches for tumor-reactive T cells. J Immunother (2010) 33(9):956–64. doi: 10.1097/CJI.0b013e3181fad2b0
61. Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest (2014) 124(5):2246–59. doi: 10.1172/JCI73639
62. Fernandez-Poma SM, Salas-Benito D, Lozano T, Casares N, Riezu-Boj JI, Mancheno U, et al. Expansion of Tumor-Infiltrating CD8(+) T cells Expressing PD-1 Improves the Efficacy of Adoptive T-cell Therapy. Cancer Res (2017) 77(13):3672–84. doi: 10.1158/0008-5472.CAN-17-0236
63. Chrusciel E, Urban-Wojciuk Z, Arcimowicz L, Kurkowiak M, Kowalski J, Gliwinski M, et al. Adoptive Cell Therapy-Harnessing Antigen-Specific T Cells to Target Solid Tumours. Cancers (Basel) (2020) 12(3):683. doi: 10.3390/cancers12030683
64. Rohaan MW, Wilgenhof S, Haanen J. Adoptive cellular therapies: the current landscape. Virchows Arch (2019) 474(4):449–61. doi: 10.1007/s00428-018-2484-0
65. Kasid A, Morecki S, Aebersold P, Cornetta K, Culver K, Freeman S, et al. Human gene transfer: characterization of human tumor-infiltrating lymphocytes as vehicles for retroviral-mediated gene transfer in man. Proc Natl Acad Sci U S A (1990) 87(1):473–7. doi: 10.1073/pnas.87.1.473
66. Rosenberg SA, Aebersold P, Cornetta K, Kasid A, Morgan RA, Moen R, et al. Gene transfer into humans–immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction. N Engl J Med (1990) 323(9):570–8. doi: 10.1056/NEJM199008303230904
67. Merrouche Y, Negrier S, Bain C, Combaret V, Mercatello A, Coronel B, et al. Clinical application of retroviral gene transfer in oncology: results of a French study with tumor-infiltrating lymphocytes transduced with the gene of resistance to neomycin. J Clin Oncol (1995) 13(2):410–8. doi: 10.1200/JCO.1995.13.2.410
68. Nash MA, Platsoucas CD, Wong BY, Wong PM, Cottler-Fox M, Otto E, et al. Transduction of rIL-2 expanded CD4+ and CD8+ ovarian TIL-derived T cell lines with the G1Na (neor) replication-deficient retroviral vector. Hum Gene Ther (1995) 6(11):1379–89. doi: 10.1089/hum.1995.6.11-1379
69. Hwu P, Yannelli J, Kriegler M, Anderson WF, Perez C, Chiang Y, et al. Functional and molecular characterization of tumor-infiltrating lymphocytes transduced with tumor necrosis factor-alpha cDNA for the gene therapy of cancer in humans. J Immunol (1993) 150(9):4104–15.
70. Treisman J, Hwu P, Yannelli JR, Shafer GE, Cowherd R, Samid D, et al. Upregulation of tumor necrosis factor-alpha production by retrovirally transduced human tumor-infiltrating lymphocytes using trans-retinoic acid. Cell Immunol (1994) 156(2):448–57. doi: 10.1006/cimm.1994.1189
71. Tian JQ, Wang ZP, Rodriguez R, Fu JS, Lu JZ, Ma BL. In vitro enhanced cytotoxicity of tumor-infiltrating lymphocytes transfected with tumor necrosis factor-related apoptosis-inducing ligand and/or interleukin-2 gene in human renal cell carcinoma. Urology (2006) 67(5):1093–8. doi: 10.1016/j.urology.2005.11.030
72. Heemskerk B, Liu K, Dudley ME, Johnson LA, Kaiser A, Downey S, et al. Adoptive cell therapy for patients with melanoma, using tumor-infiltrating lymphocytes genetically engineered to secrete interleukin-2. Hum Gene Ther (2008) 19(5):496–510. doi: 10.1089/hum.2007.0171
73. Zhang L, Morgan RA, Beane JD, Zheng Z, Dudley ME, Kassim SH, et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin Cancer Res (2015) 21(10):2278–88. doi: 10.1158/1078-0432.CCR-14-2085
74. Kershaw MH, Wang G, Westwood JA, Pachynski RK, Tiffany HL, Marincola FM, et al. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum Gene Ther (2002) 13(16):1971–80. doi: 10.1089/10430340260355374
75. Idorn M, Skadborg SK, Kellermann L, Halldorsdottir HR, Holmen Olofsson G, Met O, et al. Chemokine receptor engineering of T cells with CXCR2 improves homing towards subcutaneous human melanomas in xenograft mouse model. Oncoimmunology (2018) 7(8):e1450715. doi: 10.1080/2162402X.2018.1450715
76. Amaria RN, Haymaker CL, Bernatchez C, Forget M-A, Patel V, Hwu W-J, et al. A phase I/II study of lymphodepletion plus adoptive cell transfer (ACT) with T cells transduced with CXCR2 and NGFR followed by high dose interleukin-2 (IL-2) in patients with metastatic melanoma (MM). J Clin Oncol (2016) 34(15_suppl):TPS9594–TPS. doi: 10.1200/JCO.2016.34.15_suppl.TPS9594
77. Forget MA, Tavera RJ, Haymaker C, Ramachandran R, Malu S, Zhang M, et al. A Novel Method to Generate and Expand Clinical-Grade, Genetically Modified, Tumor-Infiltrating Lymphocytes. Front Immunol (2017) 8:908. doi: 10.3389/fimmu.2017.00908
78. Sapoznik S, Ortenberg R, Galore-Haskel G, Kozlovski S, Levy D, Avivi C, et al. CXCR1 as a novel target for directing reactive T cells toward melanoma: implications for adoptive cell transfer immunotherapy. Cancer Immunol Immunother (2012) 61(10):1833–47. doi: 10.1007/s00262-012-1245-1
79. Beane JD, Lee G, Zheng Z, Mendel M, Abate-Daga D, Bharathan M, et al. Clinical Scale Zinc Finger Nuclease-mediated Gene Editing of PD-1 in Tumor Infiltrating Lymphocytes for the Treatment of Metastatic Melanoma. Mol Ther (2015) 23(8):1380–90. doi: 10.1038/mt.2015.71
80. Aebersold P, Kasid A, Rosenberg SA. Selection of gene-marked tumor infiltrating lymphocytes from post-treatment biopsies: a case study. Hum Gene Ther (1990) 1(4):373–84. doi: 10.1089/hum.1990.1.4-373
81. Zhang L, Kerkar SP, Yu Z, Zheng Z, Yang S, Restifo NP, et al. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol Ther (2011) 19(4):751–9. doi: 10.1038/mt.2010.313
82. Peng W, Ye Y, Rabinovich BA, Liu C, Lou Y, Zhang M, et al. Transduction of tumor-specific T cells with CXCR2 chemokine receptor improves migration to tumor and antitumor immune responses. Clin Cancer Res (2010) 16(22):5458–68. doi: 10.1158/1078-0432.CCR-10-0712
83. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science (2006) 314(5796):126–9. doi: 10.1126/science.1129003
84. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood (2009) 114(8):1537–44. doi: 10.1182/blood-2008-12-195792
85. Lindenberg M, Retel V, Rohaan M, van den Berg J, Haanen J, van Harten W. Evaluating different adoption scenarios for TIL-therapy and the influence on its (early) cost-effectiveness. BMC Cancer (2020) 20(1):712. doi: 10.1186/s12885-020-07166-9
Keywords: cancer immunotherapy, adoptive cell therapy, tumor-infiltrating lymphocytes, genetically engineered TILs, synthetic TILs
Citation: Jiménez-Reinoso A, Nehme-Álvarez D, Domínguez-Alonso C and Álvarez-Vallina L (2021) Synthetic TILs: Engineered Tumor-Infiltrating Lymphocytes With Improved Therapeutic Potential. Front. Oncol. 10:593848. doi: 10.3389/fonc.2020.593848
Received: 11 August 2020; Accepted: 30 December 2020;
Published: 16 February 2021.
Edited by:
John Maher, King’s College London, United KingdomReviewed by:
Marie-Andree Forget, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2021 Jiménez-Reinoso, Nehme-Álvarez, Domínguez-Alonso and Álvarez-Vallina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luis Álvarez-Vallina, bGF2LmltYXMxMkBoMTJvLmVz
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.