
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 19 October 2020
Sec. Cancer Metabolism
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.593017
This article is part of the Research TopicMetabolism Meets Function: The Multifaced Role of Metabolism in CancerView all 24 articles
 Zhaodong Ji1,2†
Zhaodong Ji1,2† Yan Shen3†Xu Feng1,2†Yue Kong1,2
Yan Shen3†Xu Feng1,2†Yue Kong1,2 Yang Shao1,2Jiao Meng1,2
Yang Shao1,2Jiao Meng1,2 Xiaofei Zhang4*Gong Yang1,2,5*
Xiaofei Zhang4*Gong Yang1,2,5*Ovarian cancer is one of the most malignant gynecological cancers around the world. In spite of multiple treatment options, the five-year survival rate is still very low. Several metabolism alterations are described as a hallmark in cancers, but alterations of lipid metabolism in ovarian cancer have been paid less attention. To explore new markers/targets for accurate diagnosis, prognosis, and therapeutic treatments based on metabolic enzyme inhibitors, here, we reviewed available literature and summarized several key metabolic enzymes in lipid metabolism of ovarian cancer. In this review, the rate limiting enzymes associated with fatty acid synthesis (FASN, ACC, ACLY, SCD), the lipid degradation related enzymes (MAGL, CPT, 5-LO, COX2), and the receptors related to lipid uptake (FABP4, CD36, LDLR), which promote the development of ovarian cancer, were analyzed and evaluated. We also focused on the review of application of current metabolic enzyme inhibitors for the treatment of ovarian cancer through which the potential therapeutic agents may be developed for ovarian cancer therapy.
Ovarian cancer, as one malignant gynecological cancer, is the eighth leading cause in cancer-related death around world (1). According to the latest statistical cohort from the Surveillance, Epidemiology and End Results (SEER) in 2017, there was an annual incidence of 11.6 cases/100,000 women per year, with an estimated 224,940 women living with this disease in the world (2). Because of hidden symptoms and lack of effective diagnostic methods, about 70% of patients are diagnosed in advanced stage when they receive treatment for the first time (3), which underlines the status of ovarian cancer as a serious public health concern for women. Based on the various research and epidemiological investigations, the pathogenesis of ovarian cancer mainly include viral infection, endocrine disorders, genetics, and environmental pollution (4–7). Ovarian cancer is characterized by widespread and rapid metastasis in the peritoneal cavity, which facilitates metastatic dissemination and poor disease progression. Malignant ascites constitute a unique tumor microenvironment providing a physical structure for the accumulation of many components. A large number of cancer-promoting components such as cytokines, proteins, and metabolites in ascites are reported to promote cancer invasion and resistance to chemotherapy through surface-specific receptors on tumor cells (8–10). Meanwhile, the malignant progress of ovarian cancer also brings a series of changes in its own metabolism including glycometabolism, lipid metabolism, and amino acid metabolism, which may further strengthen the malignancy of the disease (11–14).
Lipids, as important nutrients for the body, are a class of water-insoluble substances including triacylglycerol, glycerol phosphates, sterols, and sphingolipids. In addition to providing a large amount of energy, lipids are also widely distributed in cellular organelles and used as biologically vital active molecules in a variety of signaling pathways to participate in process of inflammation, immunity, cell proliferation, and differentiation (15, 16). Four major routes demonstrate how lipids are routed and used in the cell: uptake, lipogenesis, storage, and degradation. The lipogenesis refers to the fatty acid synthesis pathway and the mevalonate pathway, the latter mainly leading to cholesterol and isoprenoid synthesis. The important raw material for the de novo synthesis of fatty acids is acetyl-CoA, which comes from two approaches: one is citric acid from the tricarboxylic acid cycle. Citrate is transported across the inner mitochondrial membrane by the transport protein CIC (citrate carrier) and then catalyzed by ATP-citrate lyase (ACLY) to produce acetyl-CoA and oxaloacetate. The other is that cells uptake acetic acid directly from the outside and catalyze the production of acetyl CoA through acetyl CoA synthetase (17, 18). Deregulation of lipid metabolism including the increasing de novo synthesis and degradation of fatty acid often occurs in a variety of cancer diseases, which could provide cancer cell a strong support for proliferation, invasion and metastasis. A large number of studies have found that in multiple cancers, the expression and activity of various enzymes involved in the synthesis and catabolic pathways of fatty acids (phospholipids and cholesterol) are significantly up-regulated. In addition, other lipid-metabolizing enzymes such as lipoxygenase (LOX) and cyclooxygenase (COX) gradually become cancer research hotspots in recent years. Oncogenes highly expressed in cancer cells can activate the PI3K/AKT/mTOR signaling pathway to allow the related proteins such as ErbB2 and HIF-1 to promote the expression of lipid synthetases (19–21).
Lipid droplets (LDs) occurring in specialized cytoplasm are considered to be special lipid storage organelles because they can synthesize and store triglycerides. LDs are composed of a core of neutral lipids, surrounded by phospholipids and cholesterol, and specific proteins are embedded or associated with their surroundings. More and more evidence shows that LDs are not only passive reservoirs of lipids, but are actually dynamic organelles that play a central role in lipid and energy metabolism (22).
Since fatty acids are essential for cancer malignant progression, the availability of rate limiting enzymes in lipid metabolism could be therapeutic targets. Lipid metabolism could be regulated by suppressing fatty acid synthesis, accelerating fatty acid degradation via oxidation, diverting fatty acid to storage, retarding fatty acid release from storage, and blocking fatty acids intake (23). Limiting lipid metabolism through these mechanisms could be accomplished in alone or in a combinatorial manner, which could pave the way for the therapy of ovarian cancer (Table 1). This article summarizes the effects of lipid metabolism disorders in ovarian cancer from two aspects: exogenous lipid metabolism and endogenous lipid metabolism.
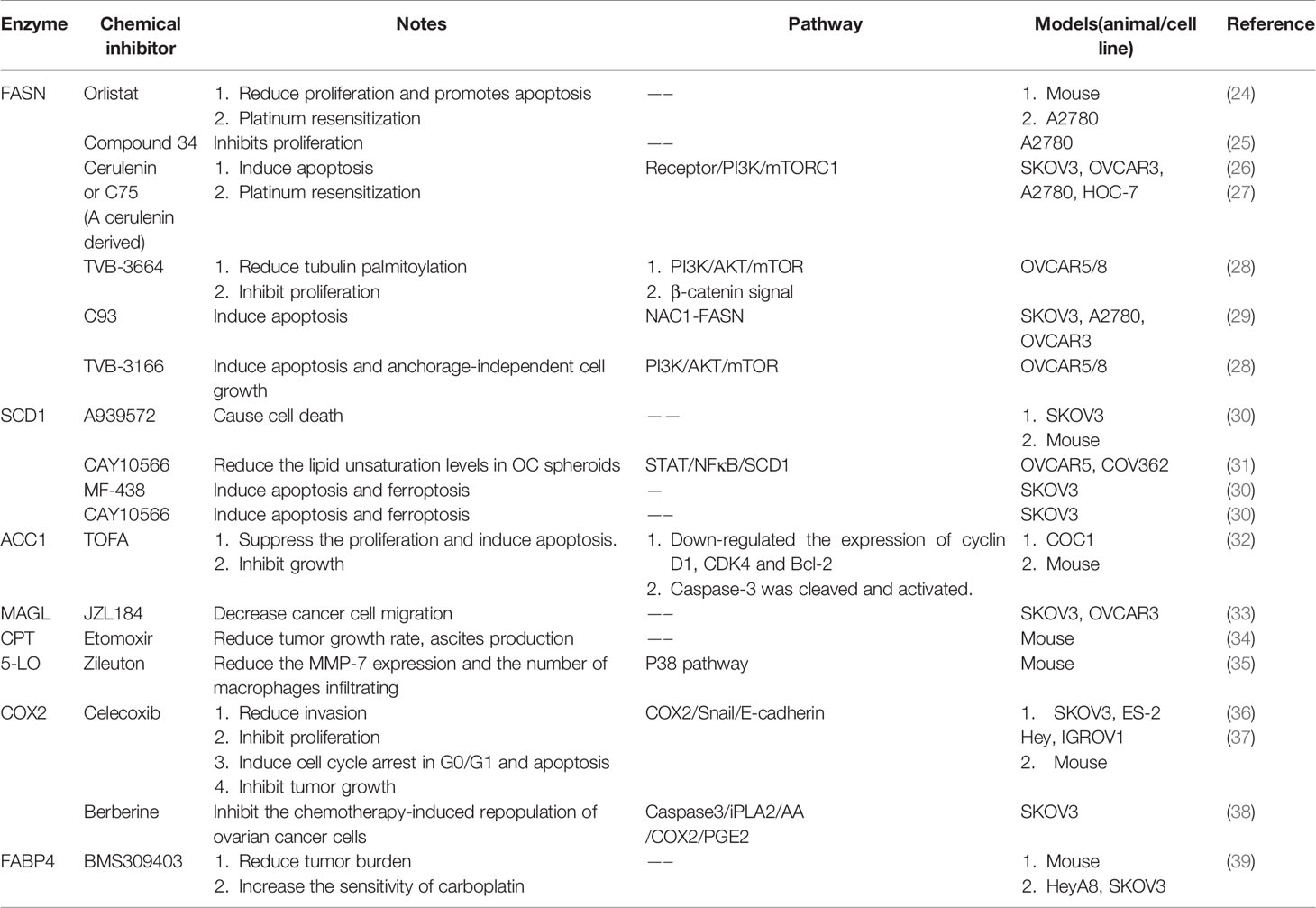
Table 1 Chemical Inhibitors targeting enzymes of lipid metabolism in ovarian cancer.
In lipid metabolism of ovarian cancer cells, many metabolic enzymes are abnormally expressed, which can cause lipid metabolism disorders by participating in processes that affect lipid synthesis or degradation, thereby to provide raw materials and energy for cancer development. At present, the combination of inhibitors of rate limiting metabolic enzymes with first-line chemotherapy agents has become a new strategy for treatment of ovarian cancer.
ATP-citrate lyase (ACLY), the upstream enzyme in fatty acid biosynthesis, functions physiologically to catalyze the six-carbon citric acid from the tricarboxylic acid cycle, either from glucose by glycolysis or glutamine, to oxaloacetate and acetyl on the cytosolic side, which provides raw materials for the synthesis of fat acid and cholesterol (Figure 1). Therefore, it is considered as a bridge connecting glycometabolism and lipid metabolism (40). The AKT-mediated phosphorylation of ACLY could promote histone acetylation in cancer cells and immune cells to response to the oncogenic and cytokine-induced signaling, while ACLY is transcriptionally regulated by SREBP1 (sterol regulatory element binding transcription protein-1) (41, 42). In addition, other substances such as insulin, glucagon, and TGF-β can promote the phosphorylation of ACLY.
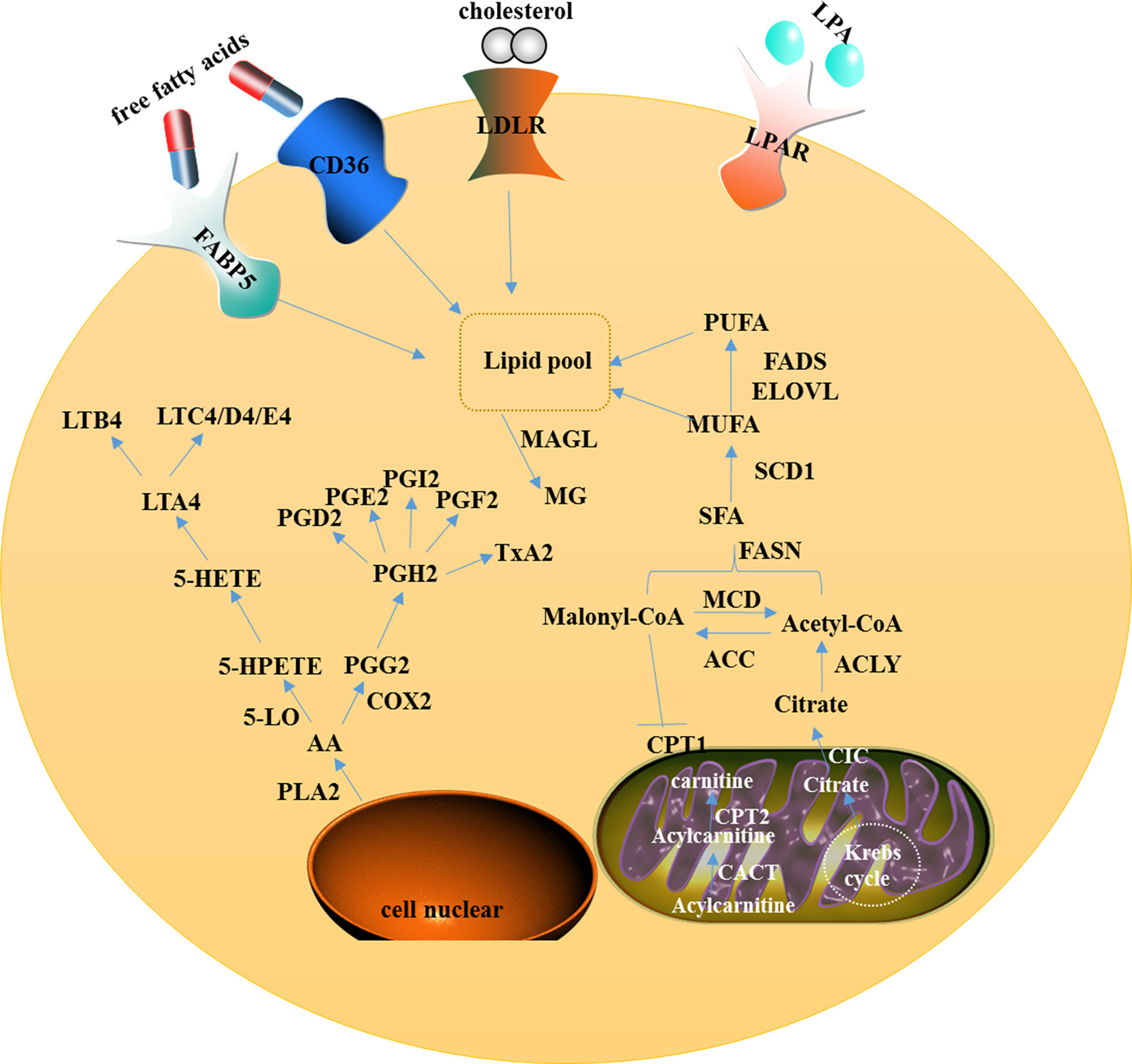
Figure 1 A model showing intracellular lipid metabolism and AA metabolism in ovarian cancer. In the cytoplasm of the cell, fatty acid metabolism includes uptake, de novo lipogenesis, and degradation. AA can be metabolized via two major pathways, namely the lipoxygenase pathway and the cyclooxygenase pathway. In mitochondria, CIC promotes the efflux of citrate from the mitochondria to the cytosol and CACT catalyzes acylcarnitine to translocate through the inner mitochondrial membrane.
Wang et al. found that ACLY expression was higher in malignant tissues than that in normal ovarian tissues. Immunohistochemical analysis showed that the increased expression level of phosphorylated ACLY in ovarian cancer tissues was related to cancer grade, FIGO stage, and poor prognosis. Mechanismlly, by knockdown of ACLY expression could inhibit the proliferation of ovarian cancer A2780 cells and cause G1 phase arrest (43, 44), suggesting that ACLY promoted cancer cell proliferation through the regulation of cell cycle.
The ubiquitin-proteasome controls protein degradation and regulatory functions. Ubiquitin-specific proteases (USPs) are the largest family of deubiquitin, which can catalyze the removal of ubiquitin from different target proteins to regulate cell function. Studies have reported that ubiquitin-specific peptidase 13 (USP13) was the main regulator of ovarian cancer metabolism (45). ACLY can be one of deubiquitinase target proteins of USP13, removing K48-related ubiquitination on ACLY to improve the stability of ACLY. The in vitro experiments found that the inhibition of USP13 expression could significantly inhibit the progression of ovarian cancers and enhance the sensitivity of cancer cells to treatment with PI3K/AKT inhibitors. Therefore, the researchers proposed that ACLY may play an important role in the USP13-mediated deubiquitination to promote cancer development (45, 46).
The first committed step of fatty acid synthesis is mediated by acetyl- ACC, which in mammals is encoded by two subtype enzymes ACC1 (Acetyl-CoA Carboxylase Alpha) and ACC2(Acetyl-CoA Carboxylase Beta) (47). ACC1 is generally expressed in lipogenic tissues, and the ACC1-generated malonyl-CoA is utilized for the synthesis of fatty acids in cytosol. In contrast, ACC2 is highly expressed in heart and muscle and to a lesser expressed in liver. Unlike ACC1 promoting fatty acid synthesis, ACC2 is anchored at outer membrane of mitochondria in subcellular where localized malonyl-CoA production blocks carnitine palmitoyltransferase-1 (CPT1) function to prevent fatty acids from entering the mitochondria to undergo fatty acid oxidation (48). ACC is a biotin-dependent multi-domain enzyme, which has biotin carboxylase (BC) and carboxyl transferase (CT) activities in most eukaryotes. In regard to these two enzyme activities, BC catalyzes the ATP-dependent carboxylation of biotin with bicarbonate as a CO2 donor, and CT promotes the transfer of carboxyl groups from biotin to acetyl CoA. In recent years, ACC activity is tightly regulated by reverse phosphorylation and gene expression. The phosphorylation of ACC by adenosine monophosphate-activated protein kinase (AMPK) has been identified (49). Notably, ACC1 has been shown to be elevated in a number of cancers, including liver cancer, lung cancer, breast cancer, and pancreatic cancer. Inhibitors targeting ACC1 were shown to reduce cell proliferation through inhibiting fatty acid synthesis (50, 51).
TOFA, an allosteric inhibitor of ACC1, was reported to suppress the proliferation of ovarian cancer via arresting the cells in G0/G1 cell cycle phase and inducing apoptosis (32). Meanwhile, TOFA could inhibit growth of ovarian cancer xenograft in mice. One study based on a randomized multicentre phase 3 trial (MITO2) found that carboplatin/PLD might be more effective than carboplatin/paclitaxel to ovarian cancer patients in the presence of pACC overexpression, suggesting that ACC might be a new biomarker for personalizing the choice of chemotherapy regimen in ovarian cancer (52).
FASN is a key enzyme for endogenous fatty acid synthesis. This cytosolic enzyme catalyzes the synthesis of 16-carbon palmitic acid by malonyl-CoA and acetyl-CoA under the action of reducing coenzyme II (53) (Figure 1). In normal condition, the physiological function of FASN is to convert excess carbohydrates into fatty acid, which will be further esterified into triacylglycerols, and finally stored or supplied for energy through β oxidation. As a downstream effector, FASN could be activated by the PI3K/AKT/mTOR signaling pathway and the transcription factors such as SREBP-1, ZBTB7A, and p53 (54, 55). FASN is highly expressed in ovarian cancer tissues and is associated with poor prognosis and survival rate (56). Because the majority of cancers rely on the FASN-mediated de novo fatty acid synthesis pathway, FASN could be an attractive therapeutic target, and inhibition of FASN has shown antitumor effects in ovarian cancer (25, 57).
In tumor cell lines, FASN overexpression was found to cause chemotherapy resistance induced by culture in drug-containing media. This means that FASN may be involved in chemoresistance of cancer cells. O Bauerschlag et al. treated HEY cells with cerulenin, an inhibitor of FASN, and found that cerulenin markedly decreased FASN expression and cell viability, and induced apoptosis. Unlike combined administrations, sequential cerulenin, and cisplatin treatment profoundly reduced cisplatin’s half maximal inhibitory concentration in a cisplatin-resistant cell line, suggesting that cerulenin had reinduce platinum sensitivity (26). Papaevangelou et al. conducted a metabolite analysis and histopathology of ovarian cancer xenograft mice treated with the combination of the anti-obesity drugs orlistat and cisplatin, and found that orlistat reduced cancers by inhibiting FASN. At the same time, cisplatin reduced the β-oxidation of fatty acids, and combined therapy delayed the cisplatin-resistant ovarian cancer cell growth and induced apoptosis. The combination therapy of the two drugs also reduced glycometabolism, biosynthesis of nucleotides and glutathione, and β-oxidation of fatty acids (24).
Overexpression of FASN was also reported to be associated with tumor cell proliferation, metastasis, poor prognosis, and high risk of recurrence in breast cancer, prostate cancer and gastric cancer (58–60). The FASN inhibitor TVB-3166 can destroy the lipid structure on membrane of cancer cell, inhibit lipid biosynthesis, and promote cancer cell apoptosis through the PI3K-AKT-mTOR and β-catenin signaling pathways in ovarian cancer. At the same time, this inhibitor can also block the expression of the oncogene c-Myc (28). Some studies have pointed out that FASN inhibitors could also induce the cell cycle arrest at S/G2/M and apoptosis of cancer cells, but only caused cell cycle deceleration without apoptosis for normal cells (61). Therefore, FASN is proposed as a metabolic marker for ovarian cancer proliferation.
Recently, some scholars have found that the abnormal activation of FASN can blunt the anti-tumor immunity of host (62). The clinical data showed that, in the advanced stage of ovarian cancer, the abnormally increased expression of FASN was positively correlated with the state of immunosuppression. The immunosuppression was manifested in the lower number and dysfunction of infiltrating T cells. Mechanistic studies have found that FASN activation in ovarian cancer cells can induce the resulting lipid accumulation at high concentrations in the tumor microenvironment. High expression of FASN in ovarian cancer cells also caused defects in the ability of dendritic cells to present antigens and prime T cells in ascites. To further explore FASN inhibition effect in anti-tumor immune response in vivo, the use of FASN inhibitors could partially restore the immune-stimulating activity of Tumor-Infiltrating DCs (TIDCs) and evoke protective anti-tumor immune responses.
Stearoyl COA desaturase (SCD) is an endoplasmic reticulum enzyme that promotes a balance of saturated fatty acids (SFA) and mono-unsaturated fatty acids (MUFA) in cell lipids. Specifically speaking, SCD catalyzes the synthesis of MUFA SFA, principally stearic acid (18:0) and palmitic acid (16:0), to their D9-monounsaturated counterparts, oleic acid (18:1) and palmitoleic acid (16:1; ref. 8) (63). These MUFAs are major components of cell membrane phospholipids and cholesterol esters. Two SCD isoforms SCD1 and SCD5 have been identified in human, whereas other four desaturases (SCD1-SCD4) share the same enzymatic function exist in mouse (64). Among of five isoforms, SCD1 is expressed ubiquitously among tissue with a 33-amino acid sequence at the N terminus that leads to the rapid degradation of this enzyme via an ubiquitin-dependent proteasome (65). It has been identified that the promoter of SCD1 contains several binding sites with the peroxisome proliferator-activated receptor (PPAR), NF-1, AP-2, and SREBP. The enzyme activity of SCD1 is either promoted by insulin, glucose, and fructose or inhibited by unsaturated fatty acids, ethanol, TNFα, IL-11, thyroid hormones, and some steroid hormones (66). Previous studies have shown that SCD1 was overexpressed in many malignant cancers to regulate cell proliferation, cell cycle, apoptosis, metastasis, and to modulate lipid metabolism through reducing fatty acid oxidation to foster lipogenesis (67).
Roongta et al. found that the expression of SCD1 was up-regulated in ovarian cancer tissues and stem cells (68). Inhibition of SCD1 expression can induce cancer cell death. Conversely, overexpression of SCD1 or exogenous addition of palmitoleic acid can protect cells from death. Ferroptosis is an iron-dependent oxidative damage causing cell death that greatly inhibits the growth of ovarian cancer cells (69). Overexpression of SCD1 protects cells from ferroptosis through the increase of monounsaturated fatty acids, whereas inhibition of SCD1 significantly enhances the anticancer effect of ferroptosis-inducers on ovarian cancer cell lines and xenograft mouse tumors (30).
Scattering microscopy was used to observe an increase in unsaturated fatty acid level in ovarian cancer stem cells, and a significant increase in the mRNA level of SCD1 was detected by qRT-PCR. However, when SCD1 inhibitors were used to treat the primary ovarian cancer stem cells, the stemness markers were down-regulated. In addition, the treatment of ovarian cancer stem cells with SCD1 inhibitors retarded the tumor growth of cells when injected into athymic mice. Further study demonstrated that NF-κB may directly regulate the transcription of SCD1 (31).
Cancer cells usually stimulate the degradation of fatty acids to provide energy for proliferation, and this degradation process can be achieved through mitochondrial β-oxidation. Within mitochondria, fatty acids continuously undergo cyclical series of reactions to produce acetyl-CoAs that were fed into the Krebs cycle and supply energy to tissues in demand when glycogen store is out of service (70).
Monoacylglycerol lipase, a member of the serine hydrolase superfamily, mainly functions as a key enzyme to catalyze the decomposition of monoacylglycerol into free fatty acids and glycerol (Figure 1). Furthermore, MAGL controls several physiological processes including pain and nociperception through hydrolysis of the endocannabinoid 2-arachidonoylglycerol (2-AG). MAGL was highly expressed in ovarian and breast cancer tissues, and identified to contribute to tumorigenesis and metastasis through up-regulation of free fatty acids (33). MAGL also promotes epithelial-mesenchymal transition (EMT) and may serve as a gene expression signature for cancer stem cells (71, 72).
Other studies also found that the multiple inhibitors of MAGL could inhibit the proliferation of ovarian cancer cells (73, 74). The knockdown of MAGL expression inhibited the proliferation, migration and invasion of ovarian cancer cells (33).
When cancer cells lack glucose, energy is generated through the increased β-oxidation. CPT is a key enzyme that catalyzes the conversion of long-chain fatty acids into acylcarnitine, which can be inhibited by malonyl-CoA (Figure 1). Two subtypes of CPT (CPT1 and CPT2) differently catalyze the decomposition of long-chain fatty acids and β-oxidation. CPT1 resides at the outer membrane of mitochondria and transports long-chain fatty acids into mitochondria for β-oxidation. CPT2 is located on the mitochondrial inner membrane and catalyzes the production of acyl-CoA from acyl-carnitine-derived acyl groups and free coenzymes to shuttle across the inner mitochondrial membrane CACT (carnitine acylcarnitine translocase), which helps acylcarnitine to translocate through the inner mitochondrial membrane and to be converted back to acyl-CoA for β-oxidation and energy substrate generation. Increasing studies have reported that β-oxidation abnormality can be induced through the high expression of CPT1 to promote cancer progression (75).
Three different CPT1 isozymes are identified. CPT1A is widely distributed in multiple tissues with stronger enzyme activity. CPT1B is mainly expressed in skeletal muscle cells and cardiac muscle cells, while CPT1C is mainly found in testis and central nervous tissues. With the improvements of metabolic studies, it has revealed that CPT1 may promote cancer cell proliferation and survival (76).
Shao et al. found that CPT1A was highly expressed in ovarian cancer cell lines and primary ovarian serous carcinomas. Analysis of database revealed that overexpression of CPT1A was associated with poor survival in ovarian cancer patients. Knockdown of CPT1A expression reduced the cellular level of ATP and induced the cell cycle arrest at G0/G1 in ovarian cancer cells, indicating that the CPT1A-mediated β-oxidation controlled the proliferation through regulating cell cycle process. Knockdown of CPT1A stimulated the phosphorylation of the transcription factor FOXO through the AMPK/p38/JNK signaling pathway and up-regulated P21 to arrest cell cycle (77).
Roy et al. found that overexpression of CPT1A can increase the β-oxidation of fatty acids and ATP levels to promote cancer cell proliferation. In contrast, Etomoxir, a specific inhibitor of CPT1A, can inhibit the proliferation of ovarian cancer cells (34).
Arachidonic acid (AA) is located in the phospholipid bilayer of the cell membrane and the precursor of main signal molecules. The metabolism of AA is closely associated with the development of cancer cells (78, 79). As a member of the arachidonic acid lipoxygenase family, 5-LO is composed of 674 amino acids and a monomeric enzyme containing iron ions. 5-LO can be transcriptionally regulated by t Egr, Sp1, nuclear factor-κB (NF-κB), and GATA (80).
5-LO is activated by 5-LO activating protein (ALOXAP) to catalyze AA which is released from the phospholipid bilayer by phospholipase A2. AA is transformed to 5-hydroxyeicosatetraenoic acid which can be metabolized by glutathione peroxidase into 5-hydroxyeicosatetranoic acid (5-HETE), which is further converted into either 5-oxo-eicosatetraenoic acid or LTA4. LTA4 is further converted into LTB4, LTC4, LTD4, or LTE4 depending on the different catalytic enzymes (81) (Figure 1). By immunohistochemistry, researchers found that the expression of 5-LO was high in epithelial ovarian cancer tissues and was associated with poor prognosis (35).
Z Wen et al. found that the high expression of 5-LO was strongly correlated with the density of TAMs in hypoxic areas of human ovarian tumor tissues. Leukotrienes (LTs) from 5-LO metabolites promoted migration and invasion of macrophages, which was mediated by up-regulation of matrix metalloproteinase-7 (MMP7) expression (35). Zileuton, a selective and specific 5-LO inhibitor, can reduce the expression of MMP-7 and the number of infiltrating macrophages in xenograft tumor tissues.
As another rate-limiting enzyme in AA metabolism, cyclooxygenase mainly catalyzes AA to produce prostaglandins (PGs) (Figure 1). Cyclooxygenase includes two isozymes, COX-1 and COX-2. COX1 maintains the homeostasis, while COX-2 can be induced by various stimulants, including cytokines, mitogens, hormones, and hypoxia. Growing evidence proves that COX-2 is highly expressed in cancers such as skin cancer, liver cancer, and breast cancer. Some studies reported that COX-2 and its derivative prostaglandin E2 (PGE2) were highly expressed in ovarian cancer cells and might promote cancer cell proliferation and metastasis (36, 82).
Angiogenesis is the physiological basis of solid cancer growth and metastasis. The high expression of COX-2 and its metabolite PGE2 promote angiogenesis through up-regulating of the angiogenic factors such as vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF). COX-2 can also promote the metastasis and invasion of ovarian cancer through induction of matrix metalloproteinases (MMPs) in extracellular matrix and the decomposition of collagen matrix which may be involved in activation of the PI3K/AKT signaling pathway (83). Inhibition of COX-2 with its specific inhibitor NS-398 can increase the expression of E-cadherin and inhibit the expression of slug, vimentin, MMP2, and MMP9, thereby to suppress invasion and metastasis of ovarian cancer cells under estrogen treatment (84). Moreover, overexpression of COX-2 in ovarian cancer cells can directly up-regulate Bcl-2 expression through the increased synthesis of PGs. Celecoxib, a selective COX-2 inhibitor, can decrease cell growth, increase the cleaved caspase-3 activity and induce cell cycle G1 phase arrest in a dose-dependent manner in ovarian cancer cells (37).
The interactions between ovarian cancer cells and human peritoneal adipocytes in ascites are believed to be important for tumor progression. Co-culture of human primary omental adipocytes with ovarian cancer cells could transfer lipids directly from adipocytes to ovarian cancer cells, indicating that adipocytes may serve as an energy source for cancer cells (57).
The family of FABPs is a type of intracellular lipid chaperones that coordinate cellular lipid responses through binding to and redistributing intracellular fatty acids, so FABPs are also called lipid chaperone proteins (85). The function of FABP4 is to promote the uptake of long-chain fatty acids and to participate in lipid transport and metabolic regulation. Overexpression of FABP4 is reported in various types of tumors such as ovarian cancer. As a key mediator in adipocytes and cancer progression, FABP4 can be a worthy predictor of residual disease in ovarian cancer. Recent studies have found that miR-409-3p can target the 3’UTR region of FABP4 and regulate the expression of FABP4 (86).
Nieman et cal. compared primary ovarian cancers with corresponding omental metastatic tissues by immunohistochemical staining, and found that FABP4 was increased in ovarian cancer cells at the adipocyte-cancer interface, but was not detected in ovarian cancer cells and benign tissues adjacent to ovarian cancers far from the adipocyte-cancer interface (87). Co-culture of adipocytes with ovarian cancer cells showed that the adipocytes significantly promoted the metastasis of the ovarian cancer cells, whereas treatment of the co-cultured cells with FABP4 inhibitor, lipid accumulation and adipocyte-mediated invasion of the cancer cells were greatly reduced. In the latest research, knockdown of FABP4 in ovarian cancer cells resulted in the increasing level of 5-hydroxymethylcytosine, the downregulated expression of genes was associated with metastasis and the number of clone formation. BMS309403, a small molecule inhibitor of FABP4, was used and the results showed that it not only significantly reduced tumor burden in a syngeneic orthotopic mouse model but also increased the sensitivity of cancer cells towards carboplatin (39).
Taken together, these studies suggest that targeting FABP4 in ovarian cancer may inhibit the ability to adapt lipid-rich cancer microenvironment and to reduce tumor aggressiveness.
CD36 is a transmembrane glycoprotein, which is one of the most abundantly expressed members in the class B scavenger receptor family. CD36 not only uptakes of free fatty acids and cholesterol, and the transfer of intracellular signals, but also pertains to the cancer-associated antigen presentation, inflammation, and angiogenesis (88). Studies have found that CD36 is highly expressed in ovarian cancer tissues and also metastatic tissues, which shows that CD36 may participate in the metastasis and proliferation of ovarian cancer.
Ladanyi et al. found that co-culture of ovarian cancer cells with human primary adipocytes (HPAs) increased the expression of CD36 in ovarian cancer cells. However, the inhibition of CD36 caused a decrease in fatty acid intake of cancer cells and reduced the accumulation of cholesterol and lipid droplets and the intracellular reactive oxygen species (ROS) in cancer cells. Knockdown of CD36 can also diminish adipocyte-mediated invasion and migration of cancer cells. Intraperitoneal injection of CD36-deficient cells significantly reduced the number of metastatic nodules in the abdominal of xenograft mouse tumor model (89). Thus, CD36 inhibition can effectively reduce fat acid uptake from microenvironment in ovarian cancer cells to suppress adipocyte-mediated tumor progression.
LDLR is a trans-membrane protein that mediates the uptake of cellular cholesterol (90). Reports about LDLR mainly focus on the mechanism of LDLR-mediated chemo-resistance in ovarian cancer cells.
LDLR expression was reported to be correlated with the poor prognosis in patients with epithelial ovarian cancer (EOCs) treated with platinum-based drugs according to the cDNA chip database. Knockdown of LDLR can increase the sensitivity of cells to platinum, whereas overexpression of LDLR can promote chemotherapy resistance. The LDLR/LPC/FAM83B/FGFRs axis is involved in the LDLR-mediated resistance to platinum based chemotherapy. Zheng et al. determined that both SREBP2 and LDLR expression levels were increased in ovarian cancer cisplatin-resistant cell lines. Bioinformatics analysis predicts that SREBP2 may mediate ovarian cancer resistance through binding to LDLR (91).
Lysophosphatidic acid (LPA) is a kind of growth factor-like lipid signal molecule, and is secreted from platelets, nerve cells, and endothelial cells by endocrine and paracrine. LPA exerts its biological function through binding to the heterotrimeric transmembrane G protein coupled receptor (including Gα q, Gα 12/13, Gα i/o, and Gα s) on cell surface. At least six members of the receptor family are identified, named as LPA1-6 (92). These LPA receptors can be divided into two subfamilies, of which LPA1-3 are the member of vascular endothelial gene (edg) family and LPA4-6 belong to the family of non-vascular endothelial factors. LPA1 is widely distributed in heart, brain and kidney; LPA2 is distributed in testis, pancreas, and prostate; LPA3 is distributed in testis and prostate. All LPAs bind to cell surface receptors and are quickly degraded into inactive monoacylglycerol (MAG) and phosphatidic acid by phospholipase (93).
Ovarian cancer cells can uptake the lysophosphatidic acid through membrane receptors to promote proliferation (94). Studies have found that compared with normal ovarian tissues, LPA2 and LPA3 receptors were highly expressed in ovarian cancer tissues, while LPA1 receptor expression was still low. Inhibition of LPA2 or LPA3 receptor expression led to decreased cancer cell migration and invasiveness. Treatment of cells with LPA1 and LPA3 receptor-specific antagonist VPC32183 reduced the uptake of LPA and caused apoptosis through inhibition of the phosphorylation of ERK1/2. LPA and its receptors can regulate the promoter activity of cyclin D1 through the downstream signaling pathway of LTA receptor, which thereby promoting cell proliferation (95).
LPA and its receptors are also involved in cancer metastasis-related signaling pathways. Xu et al. found that the thyroid receptor interference protein 6 (TRIP6) can affect the LPA-induced cancer cell migration through directly binding to LPA2 receptor. The specific manifestation is that overexpression of TRIP6 enhanced the LPA-induced cell migration, while in contrast, inhibition of TRIP6 expression suppressed the LPA-induced cell migration, suggesting that TRIP6 may mediate the LPA2-induced cancer cell migration (96). Park et al. found that LPA could activate the downstream Gα 12/13/RhoA signaling pathway through LPA 1/2 receptor to induce the phosphorylation of ERM proteins (Ezrin/Radixin/Moesin), which promotes the metastasis of ovarian cancer cell line OVC-3 (97).
The combination of paclitaxel and cisplatin is a first-line chemotherapeutic strategy for ovarian cancer treatment. The researchers pretreated ovarian cancer cells with LPA followed by paclitaxel and found that LPA reduced mitochondrial ROS production while the LPA receptor agent VPC32183 increased the content of mitochondrial ROS. Further ROS could cause mitochondrial membrane damage and cancer cell apoptosis (98).
Lipid metabolism of ovarian cancer is a complex process, including lipid uptake, lipid synthesis or storage, and fatty acid degradation by oxidation. So far, the researchers mainly clarified that the enzymes related to fatty acid synthesis (FASN, ACC, ACLY, SCD) and lipid degradation related enzymes (MAGL, CPT, 5-LO, COX2), and receptors related to lipid uptake (FABP4, CD36, LDLR) play important roles in promoting cancer development (Figure 1). However, the study of lipid metabolomics for ovarian cancer markers is still in the primary stage.
In this review, we systematically summarized the process metabolism of fatty acid and the rate-limiting enzymes in this framework. Meanwhile, a number of promising agents targeting the lipid metabolism axis are being developed and applied in clinical treatment, which can provide new strategies for clinical treatment of ovarian cancer.
GY and XZ were responsible for the revision of the manuscripts. ZJ completed the writing. YShe and XF were involved in the design of the manuscripts. JM, YSha, and YK completed the documentation and figure drawing. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.593017/full#supplementary-material
Supplementary Table 1 | Abbreviation List. AA, arachidonic acid; ACC, acetyl-CoA carboxylase; ACLY, ATP citrate lyase; CACT, carnitine-acylcarnitine translocase; CIC, citrate carrier protein; COX2, cyclooxygenase-2; CPT1, carnitine palmitoyl transferase 1; CPT2, carnitine palmitoyl transferase 2; DGAT, diacylglycerol acyltransferase; ELOVL, elongation of very long-chain fatty acids diacylglycerol acyltransferase; FADS, fatty acid desaturase; FSAN, fatty acid synthase; FABP4, fatty acid binding protein 4; LDLR, low density lipoprotein receptor; LTA4, lipoxin A4; LTB4, leukotriene B4; LTC4, leukotriene C4; LTD4, leukotriene D4; LTE4, leukotriene E4; LPAR, lysophosphatidic acid receptor; MAGL, monoacylglycerol lipase; MCD, malonyl-CoA decarboxylase; MG, monoglyceride; MUFA, monounsturated fatty acids; PGD2, prostaglandin D2; PGE2, prostaglandin E2; PGF2, prostaglandin F2; PGH2, prostaglandin H2; PGI2, prostaglandin I2; PLA2, phospholipase A2; PUFA, polyunsturated fatty acids; SCD1, stearoyl COA desaturase1; SFA, saturated fatty acids; TxA2, thromboxane A2; 5-HETE, 5-hydroxyeicosatetraenoic acid; 5-HPETE, 5-hydroperoxy-eicosatetraenoic acid; 5-LO, 5-Lipoxygenase.
1. Torre LA, Trabert B, DeSantis CE, Miler KD, Goli S, Runowicz CD, et al. Ovarian cancer statistics, 2018. CA Cancer J Clin (2018) 68(4):284–96. doi: 10.3322/caac.21456
2. Eisenhauer EA. Real-world evidence in the treatment of ovarian cancer. Ann Oncol (2017) 28:viii61–5. doi: 10.1093/annonc/mdx443
3. Liu J, Matulonis UA. New strategies in ovarian cancer: translating the molecular complexity of ovarian cancer into treatment advances. Clin Cancer Res (2014) 20(20):5150–6. doi: 10.1158/1078-0432.CCR-14-1312
4. Zhang P-P, Zhou L, Cao J-S. Possible Epithelial Ovarian Cancer Association with HPV18 or HPV33 Infection. Asian Pac J Cancer Prev (2016) 17(6):2959–64.
5. Leung PC, Choi JH. Endocrine signaling in ovarian surface epithelium and cancer. Hum Reprod Update (2007) 13(2):143–62. doi: 10.1093/humupd/dml002
6. Ilenkovan N, Gourley C. Pathogenesis, Genetics, and Genomics of Non-High Grade Serous Ovarian Cancers. Hematol Oncol Clin North Am (2018) 32(6):929–42. doi: 10.1016/j.hoc.2018.07.004
7. Hanchette C, Zhang CH, Schwartz GG. Ovarian Cancer Incidence in the U.S. and Toxic Emissions from Pulp and Paper Plants: A Geospatial Analysis. Int J Environ Res Public Health (2018) 15(8):1619. doi: 10.3390/ijerph15081619
8. Kipps E, Tan DSP, Kaye SB. Meeting the challenge of ascites in ovarian cancer: new avenues for therapy and research. Nat Rev Cancer (2013) 13(4):273–82. doi: 10.1038/nrc3432
9. Ahmed N, Stenvers KL. Getting to know ovarian cancer ascites: opportunities for targeted therapy-based translational research. Front Oncol (2013) 3:256. doi: 10.3389/fonc.2013.00256
10. Kim S, Kim B, Song YS. Ascites modulates cancer cell behavior, contributing to tumor heterogeneity in ovarian cancer. Cancer Sci (2016) 107(9):1173–8. doi: 10.1111/cas.12987
11. Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr. Cellular fatty acid metabolism and cancer. Cell Metab (2013) 18(2):153–61. doi: 10.1016/j.cmet.2013.05.017
12. Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer (2013) 13(4):227–32. doi: 10.1038/nrc3483
13. Yang Y, Cao Y, Chen L, Liu F, Qi Z, Cheng X, et al. Cryptotanshinone suppresses cell proliferation and glucose metabolism via STAT3/SIRT3 signaling pathway in ovarian cancer cells. Cancer Med (2018) 7(9):4610–8. doi: 10.1002/cam4.1691
14. Dillon EL, Volpi E, Wolfe RR. Amino Acid Metabolism and Inflammatory Burden in Ovarian Cancer Patients Undergoing Intense Oncological Therapy. Clin Nutr (2007) 26(6):736–43. doi: 10.1016/j.clnu.2007.07.004
15. Fahy E, Cotter D, Sud M, Subramaniam S. Lipid classification, structures and tools. Biochim Biophys Acta (2011) 1811(11):637–47. doi: 10.1016/j.bbalip.2011.06.009
16. Swinnen JV, Brusselmans K, Verhoeven G. Increased lipogenesis in cancer cells: new players, novel targets. Curr Opin Clin Nutr Metab Care (2006) 9(4):358–65. doi: 10.1097/01.mco.0000232894.28674.30.
17. Kouba S, Ouldamer L, Garcia C, Fontaine D, Chantome A, Vandier C, et al. Lipid metabolism and Calcium signaling in epithelial ovarian cancer. Cell Calcium (2019) 81:38–50. doi: 10.1016/j.ceca.2019.06.002
18. Schug ZT, Vande Voorde J, Gottlieb E. The metabolic fate of acetate in cancer. Nat Rev Cancer (2016) 16(11):708–17. doi: 10.1038/nrc.2016.87
19. Mylonis I, Simos G, Paraskeva E. Hypoxia-Inducible Factors and the Regulation of Lipid Metabolism. Cells (2019) 8(3):214. doi: 10.3390/cells8030214
20. Maiti P, Scott J, Sengupta D, Al-Gharaibeh A, Dunbar GL. Curcumin and Solid Lipid Curcumin Particles Induce Autophagy, but Inhibit Mitophagy and the PI3K-Akt/mTOR Pathway in Cultured Glioblastoma Cells. Int J Mol Sci (2019) 20(2). doi: 10.3390/ijms20020399
21. Chen J, Chen J, Huang J. HIF-2α upregulation mediated by hypoxia promotes NAFLD-HCC progression by activating lipid synthesis via the PI3K-AKT-mTOR pathway. Aging (Albany NY) (2019) 11(23):10839–60. doi: 10.18632/aging.102488
22. Arrese EL, Soulages JL. Insect fat body: energy, metabolism, and regulation. Annu Rev Entomol (2010) 55:207–25. doi: 10.1146/annurev-ento-112408-085356.
23. Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab (2016) 23(1):27–47. doi: 10.1016/j.cmet.2015.12.006
24. Papaevangelou E, Almeida GS, Box C, Desouza NM, Chuang YL. The effect of FASN inhibition on the growth and metabolism of a cisplatin-resistant ovarian carcinoma model. Int J Cancer (2018) 143(4):992–1002. doi: 10.1002/ijc.31392
25. Lu T, Schubert C, Cummings MD, Bignan G, Connolly PJ, Smans K, et al. Design and synthesis of a series of bioavailable fatty acid synthase (FASN) KR domain inhibitors for cancer therapy. Bioorg Med Chem Lett (2018) 28(12):2159–64. doi: 10.1016/j.bmcl.2018.05.014
26. Bauerschlag DO, Maass N, Leonhardt P, Verburg FA, Pecks U, Zeppernick F, et al. Fatty acid synthase overexpression: target for therapy and reversal of chemoresistance in ovarian cancer. J Trans Med (2015) 13(1):13535. doi: 10.1186/s12967-015-0511-3
27. Wagner R, Stübiger G, Veigel D. Multi-level suppression of receptor-PI3K-mTORC1 by fatty acid synthase inhibitors is crucial for their efficacy against ovarian cancer cells. Oncotarget (2017) 8(7):11600–13. doi: 10.18632/oncotarget.14591.
28. Ventura R, Mordec K, Waszczuk J, Wang Z, Lai J, Fridlib M, et al. Inhibition of de novo Palmitate Synthesis by Fatty Acid Synthase Induces Apoptosis in Tumor Cells by Remodeling Cell Membranes, Inhibiting Signaling Pathways, and Reprogramming Gene Expression. EBioMedicine (2015) 2(8):808–24. doi: 10.1016/j.ebiom.2015.06.020
29. Ueda SM, Yap KL, Davidson B, Tian Y, Murthy V, Wang TL, et al. Expression of Fatty Acid Synthase Depends on NAC1 and Is Associated with Recurrent Ovarian Serous Carcinomas. J Oncol (2010) 2010:285191. doi: 10.1155/2010/285191
30. Tesfay L, Paul BT, Konstorum A. Steroyl-CoA Desaturase 1 (SCD1) protects ovarian cancer cells from ferroptotic cell death. Cancer Res (2019) 79(20):5355–66. doi: 10.1158/0008-5472.CAN-19-0369
31. Li J, Condello S, Thomes-Pepin J, Ma X, Xia Y, Hurley TD, et al. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell (2017) 20(3):303–14. doi: 10.1016/j.stem.2016.11.004
32. Li S, Qiu L, Wu B, Shen H, Zhu J, Zhou L, et al. TOFA suppresses ovarian cancer cell growth in vitro and in vivo. Mol Med Rep (2013) 8(2):373–8. doi: 10.3892/mmr.2013.1505
33. Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell (2010) 140(1):49–61. doi: 10.1016/j.cell.2009.11.027
34. Sawyer BT, Qamar L, Yamamoto TM, McMellen A, Watson ZL, Richer JK, et al. Targeting fatty acid oxidation to promote anoikis and inhibit ovarian cancer progression. Mol Cancer Res (2020) 18(7):1088–98. doi: 10.1158/1541-7786.MCR-19-1057
35. Wen Z, Liu H, Li M, Li B, Gao W, Shao Q, et al. Increased metabolites of 5-lipoxygenase from hypoxic ovarian cancer cells promote tumor-associated macrophage infiltration. Oncogene (2014) 34(10):1241–52. doi: 10.1038/onc.2014.85
36. Wang Y-P, Wang Q-Y, Li C-H, Li XW. COX-2 inhibition by celecoxib in epithelial ovarian cancer attenuates E-cadherin suppression through reduced Snail nuclear translocation. Chem Biol Interact (2018) 292:24–9. doi: 10.1016/j.cbi.2018.06.020
37. Suri A, Sheng X. The effect of celecoxib on tumor growth in ovarian cancer cells and a genetically engineered mouse model of serous ovarian cancer. Oncotarget (2016) 7(26):39582–94. doi: 10.18632/oncotarget.8659
38. Zhao Y, Cui L, Pan Y, Shao D, Zheng X, Zhang F, et al. Berberine inhibits the chemotherapy-induced repopulation by suppressing the arachidonic acid metabolic pathway and phosphorylation of FAK in ovarian cancer. Cell Prolif (2017) 50(6). doi: 10.1111/cpr.12393
39. Mukherjee A, Chiang C-Y, Daifotis HA, Nieman KM, Fahrmann JF, Lastra RR, et al. Adipocyte-induced FABP4 expression in ovarian cancer cells promotes metastasis and mediates carboplatin resistance. Cancer Res (2020) 80(8):1748–61. doi: 10.1158/0008-5472.CAN-19-1999
40. Granchi C. ATP citrate lyase (ACLY) inhibitors: An anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur J Med Chem (2018) 157:1276–91. doi: 10.1016/j.ejmech.2018.09.001
41. Xu H, Luo J, Ma G, Zhang X, Yao D, Li M, et al. Acyl-CoA synthetase short-chain family member 2 (ACSS2) is regulated by SREBP-1 and plays a role in fatty acid synthesis in caprine mammary epithelial cells. J Cell Physiol (2018) 233(2):1005–16. doi: 10.1002/jcp.25954
42. Covarrubias AJ, Aksoylar HI, Yu J, Snyder NW, Worth AJ, Iyer SS, et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. Elife (2016) 5:e11612. doi: 10.7554/eLife.11612
43. Wang Y, Wang Y, Shen L, Pang Y, Qiao Z, Liu P. Prognostic and therapeutic implications of increased ATP citrate lyase expression in human epithelial ovarian cancer. Oncol Rep (2012) 27(4):1156–62. doi: 10.3892/or.2012.1638
44. Wojnarowicz PM, Breznan A, Arcand SL, Filali-Mouhim A, Provencher MD, Mes-Masson A-M, et al. Construction of a chromosome 17 transcriptome in serous ovarian cancer identifies differentially expressed genes. Int J Gynecol Cancer (2008) 18(5):963–75. doi: 10.1111/j.1525-1438.2007.01134.x
45. Han C, Yang L. Amplification of USP13 drives ovarian cancer metabolism. Nat Commun (2016) 7:13525. doi: 10.1038/ncomms13525
46. Han C, Lu X, Nagrath D. Regulation of protein metabolism in cancer. Mol Cell Oncol (2018) 5(5):e1285384. doi: 10.1080/23723556.2017.1285384
47. Wakil SJ, Abu-Elheiga LA. Fatty acid metabolism: target for metabolic syndrome. J Lipid Res (2009) 50(Supplement):S138–43. doi: 10.1194/jlr.R800079-JLR200
48. McGarry JD, Leatherman GF, Foster DW. The site of inhibition of hepatic fatty acid oxidation by malonyl-CoA. J Biol Chem (1978) 253:4128–36.
49. Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiol Rev (2009) 89(3):1025–78. doi: 10.1152/physrev.00011.2008
50. Brusselmans K, De Schrijver E, Verhoeven G, Swinnen JV. RNA Interference–Mediated Silencing of the Acetyl-CoACarboxylase-a Gene Induces Growth Inhibition and Apoptosis of Prostate Cancer Cells. Cancer Res (2005) 65:6719–25. doi: 10.1158/0008-5472.CAN-05-0571
51. Chajes V, Cambot M, Moreau K, Lenoir G, Joulin V. Acetyl-CoA carboxylase alpha is essential to breast cancer cell survival. Cancer Res (2006) 66(10):5287–94. doi: 10.1158/0008-5472.CAN-05-1489
52. Perrone F, Baldassarre G, Indraccolo S. Biomarker analysis of the MITO2 phase III trial of first-line treatment in ovarian cancer_ predictive value of DNA-PK and phosphorylated ACC. Oncotarget (2016) 7(45):72654–61. doi: 10.18632/oncotarget.12056.
53. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer (2007) 7(10):763–77. doi: 10.1038/nrc2222
54. Jiang Y, Yin X, Wu L, Qin Q, Xu J. MAPK/P53-mediated FASN expression in bone tumors. Oncol Lett (2017) 13(6):4035–8. doi: 10.3892/ol.2017.6015
55. Choi WI, Jeon BN, Park H, Yoo JY, Kim YS, Koh DI, et al. Proto-oncogene FBI-1 (Pokemon) and SREBP-1 synergistically activate transcription of fatty-acid synthase gene (FASN). J Biol Chem (2008) 283(43):29341–54. doi: 10.1074/jbc.M802477200
56. Cai Y, Wang J, Zhang L, Wu D, Yu D, Tian X, et al. Expressions of fatty acid synthase and HER2 are correlated with poor prognosis of ovarian cancer. Med Oncol (2015) 32(1):391. doi: 10.1007/s12032-014-0391-z
57. Chen RR, Yung MMH, Xuan Y, Zhan S, Leung LL, Liang RR, et al. Targeting of lipid metabolism with a metabolic inhibitor cocktail eradicates peritoneal metastases in ovarian cancer cells. Commun Biol (2019) 2:281. doi: 10.1038/s42003-019-0508-1
58. Luu TH, Bard JM, Carbonnelle D, Chaillou C, Huvelin JM, Bobin-Dubigeon C, et al. Lithocholic bile acid inhibits lipogenesis and induces apoptosis in breast cancer cells. Cell Oncol (Dordr) (2018) 41(1):13–24. doi: 10.1007/s13402-017-0353-5
59. Stoykova GE, Schlaepfer IR. Lipid Metabolism and Endocrine Resistance in Prostate Cancer, and New Opportunities for Therapy. Int J Mol Sci (2019) 20(11):2626. doi: 10.3390/ijms20112626
60. Ezzeddini R, Taghikhani M, Somi MH, Samadi N, Rasaee M. Clinical importance of FASN in relation to HIF-1alpha and SREBP-1c in gastric adenocarcinoma. Life Sci (2019) 224:169–76. doi: 10.1016/j.lfs.2019.03.056
61. Veigel D, Wagner R, Stubiger G, Wuczkowski M, Filipits M, Horvat R, et al. Fatty acid synthase is a metabolic marker of cell proliferation rather than malignancy in ovarian cancer and its precursor cells. Int J Cancer (2015) 136(9):2078–90. doi: 10.1002/ijc.29261
62. Jiang L, Fang X, Wang H, Li D, Wang X. Ovarian Cancer-Intrinsic Fatty Acid Synthase Prevents Anti-tumor Immunity by Disrupting Tumor-Infiltrating Dendritic Cells. Front Immunol (2018) 9:2927. doi: 10.3389/fimmu.2018.02927
63. ALJohani AM, Syed DN, Ntambi JM. Insights into Stearoyl-CoA Desaturase-1 Regulation of Systemic Metabolism. Trends Endocrinol Metab (2017) 28(12):831–42. doi: 10.1016/j.tem.2017.10.003
64. Hodson L, Fielding BA. Stearoyl-CoA desaturase: rogue or innocent bystander? Prog Lipid Res (2013) 52(1):15–42. doi: 10.1016/j.plipres.2012.08.002
65. Liu X, Ntambi JM. Stearoyl CoA Desaturase 1: Role in Cellular Inflammation and Stress. Adv Nutr (2011) 2:15–22. doi: 10.3945/an.110.000125. MS Strable.
66. Koeberle A, Loser K, Thurmer M. Stearoyl-CoA desaturase-1 and adaptive stress signaling. Biochim Biophys Acta (2016) 1861(11):1719–26. doi: 10.1016/j.bbalip.2016.08.009
67. Igal RA. Stearoyl CoA desaturase-1: New insights into a central regulator of cancer metabolism. Biochim Biophys Acta (2016) 1861(12 Pt A):1865–80. doi: 10.1016/j.bbalip.2016.09.009
68. Roongta UV, Pabalan JG, Wang X, Ryseck RP, Fargnoli J, Henley B, et al. Cancer cell dependence on unsaturated fatty acids implicates stearoyl-CoA desaturase as a target for cancer therapy. Mol Cancer Res (2011) 9(11):1551–61. doi: 10.1158/1541-7786.MCR-11-0126
69. Stockwell BR, Angeli JFP, Bayir H, Bush AI, Conrad M, Dixon S, et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell (2017) 171(2):273–85. doi: 10.1016/j.cell.2017.09.021
70. Currie E, Schulze A, Zechner R. Cancer metabolism fatty acid oxidation in the limelight. Cell Metab (2013) 18(2):153–61. doi: 10.1016/j.cmet.2013.05.017.
71. Nomura DK, Lombardi DP, Chang JW, Niessen S, Ward A, Long J, et al. Monoacylglycerol lipase exerts dual control over endocannabinoid and fatty acid pathways to support prostate cancer. Chem Biol (2011) 18(7):846–56. doi: 10.1016/j.chembiol.2011.05.009
72. Zhu W, Zhao Y, Zhou J, Wang X, Pan Q, Zhang N, et al. Monoacylglycerol lipase promotes progression of hepatocellular carcinoma via NF-κB-mediated epithelial-mesenchymal transition. J Hematol Oncol (2016) 9(1). doi: 10.1186/s13045-016-0361-3
73. Poli G, Lapillo M, Jha V, Mouawad N, Caligiuri I, Macchia M, et al. Computationally driven discovery of phenyl(piperazin-1-yl)methanone derivatives as reversible monoacylglycerol lipase (MAGL) inhibitors. J Enzyme Inhibit Med Chem (2019) 34(1):589–96. doi: 10.1080/14756366.2019.1571271
74. Granchi C, Caligiuri I. A patent review of Monoacylglycerol Lipase (MAGL) inhibitors (2013-2017). Expert Opin Ther Pat (2017) 27(12):1341–51. doi: 10.1080/13543776.2018.1389899
75. Adeva-Andany MM, Calvo-Castro I, Fernández-Fernández C, Donapetry-García C, Pedre-Piñeiro AM. Significance ofl-carnitine for human health. IUBMB Life (2017) 69(8):578–94. doi: 10.1002/iub.1646
76. Bonnefont JP, Djouadi F, Prip-Buus C, Gobin S, Munnich A, Bastin J. Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Mol Aspects Med (2004) 25(5-6):495–520. doi: 10.1016/j.mam.2004.06.004
77. Shao H, Mohamed EM, Xu GG. Carnitine palmitoyltransferase 1A functions to repress FoxO transcription factors to allow cell cycle progression in ovarian cancer. Oncotarget (2016) 7(4):3832–46. doi: 10.18632/oncotarget.6757.
78. Mashima R, Okuyama T. The role of lipoxygenases in pathophysiology; new insights and future perspectives. Redox Biol (2015) 6:297–310. doi: 10.1016/j.redox.2015.08.006
79. Rocconi RP, Kirby TO, Seitz RS, Beck R, Straughn J, Alvarez RD, et al. Lipoxygenase pathway receptor expression in ovarian cancer. Reprod Sci (2008) 15(3):321–6. doi: 10.1177/1933719108316390
80. Moore G, Pidgeon G. Cross-Talk between Cancer Cells and the Tumour Microenvironment: The Role of the 5-Lipoxygenase Pathway. Int J Mol Sci (2017) 18(2):236. doi: 10.3390/ijms18020236
81. Das UN. Ageing: Is there a role for arachidonic acid and other bioactive lipids? A review. J Advanced Res (2018) 11:67–79. doi: 10.1016/j.jare.2018.02.004
82. Lin Y, Cui M, Xu T, Yu W, Zhang L. Silencing of cyclooxygenase-2 inhibits the growth, invasion and migration of ovarian cancer cells. Mol Med Rep (2014) 9(6):2499–504. doi: 10.3892/mmr.2014.2131
83. Gu P, Su Y, Guo S, Teng L, Xu Y, Qi J, et al. Over-Expression of COX-2 Induces Human Ovarian Cancer Cells (CAOV-3) Viability, Migration and Proliferation in Association with PI3-k/Akt Activation. Cancer Invest (2009) 26(8):822–9. doi: 10.1080/07357900801941860
84. Feng D, Zhao T, Yan K, Liang H, Liang J, Zhou Y, et al. Gonadotropins promote human ovarian cancer cell migration and invasion via a cyclooxygenase 2-dependent pathway. Oncol Rep (2017) 38(2):1091–8. doi: 10.3892/or.2017.5784
85. Lee D, Wada K, Taniguchi Y, Al-Shareef H, Masuda T, Usami Y, et al. Expression of fatty acid binding protein 4 is involved in the cell growth of oral squamous cell carcinoma. Oncol Rep (2014) 31(3):1116–20. doi: 10.3892/or.2014.2975
86. Gharpure KM, Pradeep S, Sans M, Rupaimoole R, Ivan C, Wu SY, et al. FABP4 as a key determinant of metastatic potential of ovarian cancer. Nat Commun (2018) 9(1):2923. doi: 10.1038/s41467-018-04987-y
87. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Marion Zillhardt M, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med (2011) 17(11):1498–503. doi: 10.1038/nm.2492
88. DeFilippis RA, Chang H, Dumont N, Rabban JT, Chen YY, Fontenay GV, et al. CD36 repression activates a multicellular stromal program shared by high mammographic density and tumor tissues. Cancer Discovery (2012) 2(9):826–39. doi: 10.1158/2159-8290.CD-12-0107
89. Ladanyi A, Mukherjee A, Kenny HA, Johnson A, Mitra AK, Sundaresan S, et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene (2018) 37(17):2285–301. doi: 10.1038/s41388-017-0093-z
90. Chang XL, Liu L, Wang N, Chen ZJ, Zhang C. The function of high-density lipoprotein and low-density lipoprotein in the maintenance of mouse ovarian steroid balance. Biol Reprod (2017) 97(6):862–72. doi: 10.1093/biolre/iox134
91. Zheng L, Li L, Lu Y, Jiang F, Yang XA. SREBP2 contributes to cisplatin resistance in ovarian cancer cells. Exp Biol Med (2018) 243(7):655–62. doi: 10.1177/1535370218760283
92. Tsujiuchi T, Hirane M, Dong Y, Fukushima N. Diverse effects of LPA receptors on cell motile activities of cancer cells. J Recept Signal Transd (2014) 34(3):149–53. doi: 10.3109/10799893.2013.876042
93. Wang P, Wu X, Chen W, Liu J, Wang X. The lysophosphatidic acid (LPA) receptors their expression and significance in epithelial ovarian neoplasms. Gynecol Oncol (2007) 104(3):714–20. doi: 10.1016/j.ygyno.2006.10.016
94. Umezu-Goto M, Tanyi J, Lahad J, Liu S, Yu S, Lapushin R, et al. Lysophosphatidic acid production and action: validated targets in cancer? J Cell Biochem (2004) 92(6):1115–40. doi: 10.1002/jcb.20113
95. Hu Y-L, Albanese C, Pestell RG, Jaffe RB. Dual Mechanisms for Lysophosphatidic Acid Stimulation of Human Ovarian Carcinoma Cells. J Natl Cancer Inst (2003) 95(10):733–40. doi: 10.1093/jnci/95.10.733.
96. Xu J, Lai Y-J, Lin W-C, Lin F-T. TRIP6 Enhances Lysophosphatidic Acid-induced Cell Migration by Interacting with the Lysophosphatidic Acid 2 Receptor. J Biol Chem (2004) 279(11):10459–68. doi: 10.1074/jbc.M311891200
97. Park J, Jang JH, Oh S, Kim M, Shin C, Jeong M, et al. LPA-induced migration of ovarian cancer cells requires activation of ERM proteins via LPA1 and LPA2. Cell Signal (2018) 44:138–47. doi: 10.1016/j.cellsig.2018.01.007
Keywords: ovarian cancer, lipid metabolism, potential target, fatty acid synthesis, metabolic enzyme
Citation: Ji Z, Shen Y, Feng X, Kong Y, Shao Y, Meng J, Zhang X and Yang G (2020) Deregulation of Lipid Metabolism: The Critical Factors in Ovarian Cancer. Front. Oncol. 10:593017. doi: 10.3389/fonc.2020.593017
Received: 09 August 2020; Accepted: 28 September 2020;
Published: 19 October 2020.
Edited by:
Monica Montopoli, University of Padua, ItalyReviewed by:
Mariafrancesca Scalise, University of Calabria, ItalyCopyright © 2020 Ji, Shen, Feng, Kong, Shao, Meng, Zhang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaofei Zhang, c29waGlhX3dpbGxAMTYzLmNvbQ==; Gong Yang, eWFuZ2dvbmdAZnVkYW4uZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.