 Marco V. Haselager
Marco V. Haselager Arnon P. Kater
Arnon P. Kater Eric Eldering
Eric Eldering
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 08 October 2020
Sec. Hematologic Malignancies
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.592205
This article is part of the Research Topic New Insights into the Complexity of Tumor Immunology in B-cell Malignancies: Disease Biology and Signaling View all 14 articles
Chronic lymphocytic leukemia (CLL) cells cycle between lymphoid tissue sites where they actively proliferate, and the peripheral blood (PB) where they become quiescent. Strong evidence exists for a crucial role of B cell receptor (BCR) triggering, either by (self-)antigen or by receptor auto-engagement in the lymph node (LN) to drive CLL proliferation and provide adhesion. The clinical success of Bruton’s tyrosine kinase (BTK) inhibitors is widely accepted to be based on blockade of the BCR signal. Additional signals in the LN that support CLL survival derive from surrounding cells, such as CD40L-presenting T helper cells, myeloid and stromal cells. It is not quite clear if and to what extent these non-BCR signals contribute to proliferation in situ. In vitro BCR triggering, in contrast, leads to low-level activation and does not result in cell division. Various combinations of non-BCR signals delivered via co-stimulatory receptors, Toll-like receptors (TLRs), and/or soluble cytokines are applied, leading to comparatively modest and short-lived CLL proliferation in vitro. Thus, an unresolved gap exists between the condition in the patient as we now understand it and applicable knowledge that can be harnessed in the laboratory for future therapeutic applications. Even in this era of targeted drugs, CLL remains largely incurable with frequent relapses and emergence of resistance. Therefore, we require better insight into all aspects of CLL growth and potential rewiring of signaling pathways. We aim here to provide an overview of in vivo versus in vitro signals involved in CLL proliferation, point out areas of missing knowledge and suggest future directions for research.
CLL is the most frequent hematologic cancer and is characterized by the clonal expansion of CD5+CD19+ malignant B cells (1). CLL patients can be distinguished into 2 categories with distinct clinical outcome, based on the presence or absence of somatic hypermutation in the immunoglobulin heavy chain variable region (IGHV) genes of the clonotypic B cell receptor (BCR). Patients with low IGHV mutation levels (<2% change from the germline sequence), referred to as unmutated (um-CLL), experience a significantly more aggressive disease than those with mutations, referred to as mutated (m-CLL). IGHV mutation status remains one of the most robust prognostic markers in CLL, yet it does not entirely reflect the heterogeneity of the disease (2).
In addition, CLL is a prime example of a B cell malignancy that is crucially dependent on signals from the microenvironment. CLL cells cycle between lymphoid tissue sites and peripheral blood (PB). CLL cells accumulating in the PB become quiescent, whereas active CLL cells at lymphoid tissue sites are provided with signals from surrounding cells, such as CD40L-presenting T helper cells, myeloid, and stromal cells (3). Since CLL cells are strictly microenvironment-dependent, the crosstalk with the surrounding microenvironment in promoting CLL survival and proliferation has been a focus of intense research.
In order not to perish by neglect, CLL cells need to return to the proliferation sites in lymphoid tissues. This notion is supported by BCR kinase inhibitors that have entered the clinic, foremost the Bruton's tyrosine kinase (BTK) inhibitor ibrutinib, which was found to induce lymphocytosis in patients due to the release of activated CLL cells from lymphoid tissue sites into the PB, preventing migration back into the lymphoid tissue sites and thereby halting disease progression. However, stopping ibrutinib treatment reverses remission and some patients may relapse even on ibrutinib treatment, highlighting the need for greater understanding of the mechanisms that promote CLL proliferation (4).
Strong evidence exists for a crucial role of BCR signaling to drive CLL disease progression, especially the success of inhibitors targeting BCR-associated kinases (5). However, in vitro BCR triggering only leads to low-level activation without induction of proliferation, suggesting that additional factors that play a role in vivo are missing (6). Several other receptors are known to mediate interactions between CLL cells and the microenvironment, such as CD40 or Toll-like receptor (TLRs), in combination with cytokine receptors which have been shown to induce proliferation upon in vitro stimulation (7, 8). It is not quite clear if and to what extent these non-BCR signals contribute to proliferation in vivo. Perhaps a combination of stimuli is what may really drive CLL proliferation in vivo, or makes CLL develop into more aggressive disease.
In this review, we aim to provide an overview of in vivo versus in vitro signals involved in CLL proliferation. With focus on BCR, CD40 and TLR signaling, we will attempt to separately describe in vivo and in vitro data and, in each case, discuss how these receptor-mediated signaling modes may drive CLL. By integrating multiple facets of the CLL microenvironment, we aim to bridge the gap between in vivo and vitro studies, point out areas of missing knowledge and suggest future directions for research.
The conceptual framework of CLL biology has changed over the past decades. The traditional view was that CLL is a disease deriving from an inherent defect in apoptosis, in which slowly proliferating CLL cells accumulate due to diminished cell death. In this view, CLL cells continue to accumulate until they reached a level that is detrimental to the patient (9). However, in vivo labeling studies using deuterated water have changed this view by documenting the dynamic cellular kinetics of CLL cells. These studies showed that CLL is a dynamic condition, comprising of CLL cells that multiply and die at variable rates (9). Proliferation rates in patients with stable white blood cell count (WBC) indicated that CLL cells are continually dying and replenishing. Therefore, fast clonal birth is not necessarily associated with high WBC increase, suggesting that WBC does not reflect underlying cellular dynamics but rather the net effect of clonal turnover between cell birth and death rates (10). These studies consistently estimated that between 0.07–1.75% of CLL cells circulating in the PB are added to the population each day (9–13). Importantly, patients with birth rates >0.35% were much more likely to exhibit active or progressive disease than patients with lower birth rates (10). Also in patients with recently diagnosed disease, high CLL birth rate was a strong predictor of the need for earlier initial treatment, reinforcing the concept that enhanced cell proliferation is an important driver in the biology of disease progression (13, 14).
Interestingly, both birth rates and death rates of CLL cells were lower than those of healthy B cells, suggesting that CLL cells divide slower and have a lower turnover than their normal counterpart (11, 12). In addition, telomere lengths of CLL cells were shorter than those of healthy B cells in age-matched healthy donors (HDs), showing that CLL cells completed more rounds of proliferation than healthy B cells (15). These observations indicate that CLL progression appears to be the consequence of an imbalance of decreased cell turnover combined with excess proliferation, resulting in a longer replicative history of CLL cells (11).
The observed in vivo proliferation rates of CLL cells promote the acquisition of genetic mutations (16). Combined with the fact that CLL cells are less susceptible to apoptosis, CLL cells are able to obtain a more extensive replicative history, suggesting that disease progression is not a result of accumulation but rather of stochastic generation of subclones. Over time, more pathological subclones could be selected which may further affect CLL birth and death rates (9). Importantly, the accumulation of genetic changes may eventually result in subclones that may prevail of microenvironmental control at later stages (17). The insight that CLL is a dynamic disease with both substantial proliferation and death rates is important, since this allows novel clonal variants to expand more quickly to a substantial level (10). Clonal evolution with outgrowth of novel variants harboring genetic alterations has been well described in CLL and has significant impact on clinical outcome (18). However, in vivo labeling studies have not managed to link such genetic aberrations to increased proliferation rates (13), suggesting that they are a consequence rather than the cause of increased proliferation in CLL.
In aforementioned in vivo labeling studies, the fraction of labeled CLL cells in many patients continued to increase for many weeks after the end of the labeling phase, implying they had to have remained in a separate compartment for some time prior to being released into the PB (10). Subsequent in vivo labeling studies identified the LN as the anatomical site harboring the largest fraction of newly born CLL cells, with birth rates as high as 3.3% of CLL cells circulating in the PB per day. In contrast, the BM did not seem to be a major proliferation site (12). Gene expression profiling and Ki-67 staining support that active proliferation occurs in the LN from which newborn cells enter the PB (14, 19). Finally, immunohistochemical studies have demonstrated the presence of proliferating CLL cells within specific structures in the LN, resembling proliferation centers, otherwise known as pseudofollicles (20, 21).
Extensive immunophenotyping and intraclonal analyses suggest a spectrum of circulating CLL cells with at one end the proliferative fraction, enriched in recently divided cells that have recently emigrated from the LNs (CXCR4low/CD5high), and at the other end the resting fraction, enriched in older, less vital cells that need to either immigrate back to the LN or die (CXCR4high/CD5low) (Figure 1) (14, 22). Moreover, gene expression analysis indicated higher levels of pro-proliferation and anti-apoptotic genes in the proliferative CXCR4low/CD5high fraction (22).
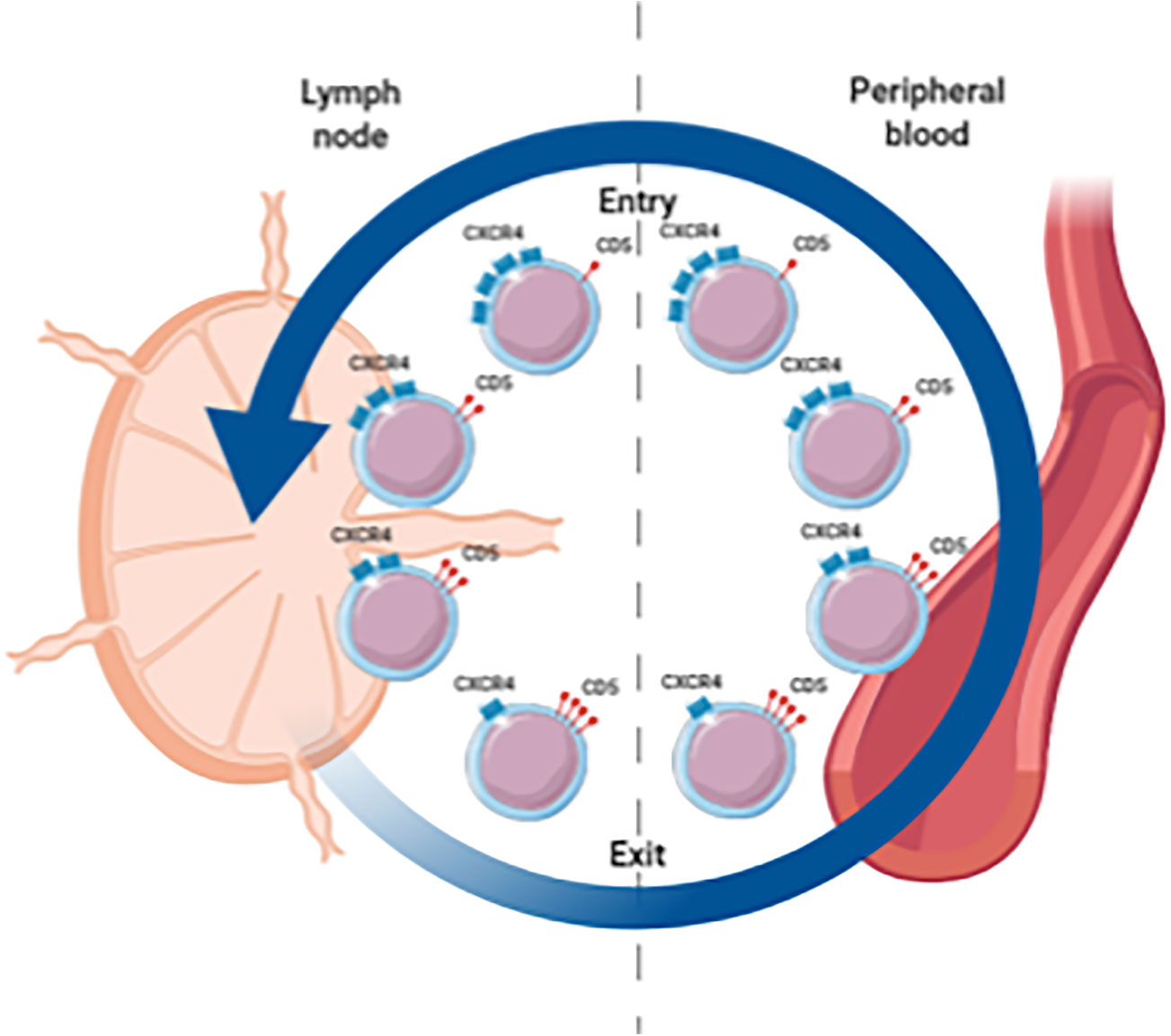
Figure 1 Spectrum of circulating CLL cells illustrating the proliferative fraction enriched in LN emigrants versus the resting fraction enriched in LN immigrants. CXCR4high/CD5low cells are in a resting state. When these cells are activated in the LN, CD5 is upregulated and CXCR4 is internalized. These CXCR4low/CD5high cells are released from the LN and migrate into the PB. Eventually these cells become more quiescent leading to downregulation of CD5 and renewed surface expression of CXCR4 which increases the likelihood of a return to the LN.
Thus, LN tissues are the preferred site for CLL cell proliferation, possibly due to accessory cells within the microenvironment that promote proliferation, propagated through diverse receptors such as the BCR, CD40, and TLRs (19, 23, 24). Characterization of BCR, CD40, and TLR signaling in primary CLL cells of the proliferative fraction may pinpoint the importance of each of these individual modes of stimulation and is of interest for understanding the process by which CLL cells residing in these proliferative niches are contributing to disease progression. Aside from BCR, CD40, and TLR stimulation, various other in vitro stimulations and culture conditions have been applied in the context of CLL proliferation, including factors like BAFF and APRIL, as well as co-culture with stromal cells, follicular dendritic cells or nurse-like cells (1). These and other candidates are certainly of interest, yet the interaction with feeder cells combined with their secretion of cytokines makes the identification of essential factors difficult (6). In addition, our previous efforts have not managed to show a direct role for BAFF or APRIL in human CLL proliferation (25, 26). Therefore, we take a restricted approach and in the first part of the review we will provide an overview of the in vivo evidence of BCR, CD40, and TLR signaling in CLL proliferation, and in the second part of the review we will cover the in vitro data that support the role of BCR, CD40, and TLR signaling in the proliferation of CLL.
Signaling through the BCR pathway is a key functional step of all normal and malignant B cells and is also a critical component in CLL (27). BCR signaling activity is elevated in CLL cells compared to healthy B cells, and deregulated BCR signaling is considered a driving mechanism leading to CLL development, progression, and relapse (28). BCR triggering leads to the activation of downstream signaling pathways, including the MAPK/ERK, PI3K/AKT/mTOR, and NF-κB pathways, which play a role in CLL survival and proliferation (Figure 2) (5, 29–31).
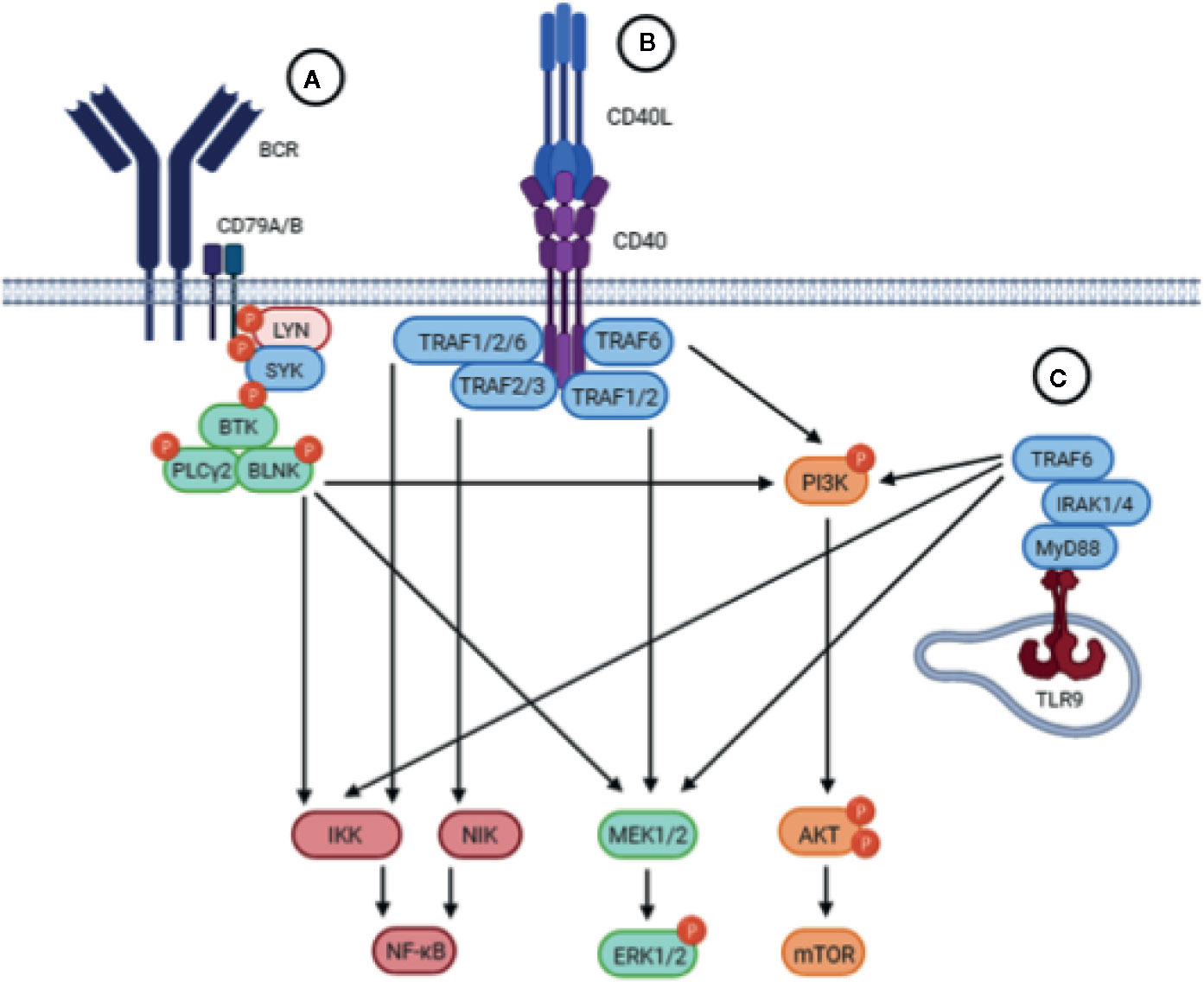
Figure 2 Schematic representation of the BCR, CD40 and TLR signaling pathways. Upstream triggering of BCR, CD40, or TLR signaling lead to activation of similar downstream pathways, including the NF-kB, MEK/ERK, and PI3K/AKT/mTOR pathways. (A) The BCR is composed of an antigen-specific surface membrane immunoglobulin paired with the signal transduction component consisting of the CD79A/B heterodimer. Engagement of the BCR by antigen results in aggregation of BCR components that leads to the phosphorylation of ITAMs in the cytoplasmic tails of CD79A/B which triggers the recruitment and activation of the proximal tyrosine kinases LYN and SYK. Subsequently, BTK is activated which will activate PLCγ2 and BLNK, resulting in activation of downstream signaling pathways. (B) Upon binding of CD40L, the CD40 receptor on CLL cells trimerizes leading to the recruitment of TRAFs to the cytoplasmic domain of CD40. TRAFs may then cooperate in order to activate different signaling pathways downstream. (C) Upon TLR9 activation in the endosomal compartment of CLL cells, the TIR domain of TLR9 engages the TIR domain of the adaptor protein MyD88. MyD88 contains an IRAK1 domain which activates TRAF6, leading to the activation of downstream pathways.
Critical evidence of the involvement of BCR stimulation in driving CLL is the expression of BCR-associated genes in LN CLL cells (19). In addition, LN CLL cells had higher pSYK levels compared to CLL cells from PB or BM, supporting BCR-dependent activation of CLL cells in vivo and suggesting that the LN is the crucial site for proliferation (19). Mouse models exhibiting spontaneous CLL development also show an important role for BCR signaling in the onset and development of CLL. These studies showed that overexpression of BTK leads to accelerated CLL onset (32). The IgH.ETµ mouse model shows that BTK expression is a prerequisite for CLL development (32) whereas the TCL1 mouse model showed that mice with ablation of BTK significantly delayed CLL development but still developed leukemia at rates similar to wild type TCL1 mice treated with ibrutinib (33–35).
In patients, CLL cells have elevated BTK expression (36) and pBTK levels compared to healthy B cells (28), as well as lower expression of surface IgM (sIgM) (37) which is additional evidence for BCR stimulation in vivo, as sIgM expression in CLL cells is downregulated after antigen stimulation which is reversed during circulation in the PB (38). This observation suggests endocytosis of sIgM in vivo which can only be due to interaction of the BCR with a ligand able to bind (27, 39). In CLL cells isolated from the PB, subgroups of cells could be distinguished with increasing sIgM and CXCR4 expression, likely regulating the ability to migrate to the LN and engage antigen in situ (38).
However, this raises questions in the context of the two CLL subsets stratified by IGHV gene mutations. Specifically, as um-CLL cells have polyreactive BCRs that may respond to a wide spectrum of epitopes whereas m-CLL cells have undergone somatic hypermutation and thus express BCRs specific to a certain epitope, and may therefore be less responsive to external signals (5, 40). Indeed, in vivo labeling studies measured higher CLL growth rates in the LNs of um-CLL patients compared to m-CLL patients (14). Consistent is the finding that um-CLL cells had much shorter telomeres than m-CLL cells, implying a more extensive replicative history (15).
Furthermore, LN CLL cells showed increased BCR signaling in um-CLL compared to m-CLL (19). This is consistent with findings in PB CLL cells, where even though BCR signaling was significantly lower, a preferential expression of BCR-regulated genes was found in um-CLL as compared to m-CLL, which most likely reflects BCR activation in the LN as cells carry a temporary imprint of their prior stimulation (19, 41). However, even in m-CLL patients, BCR signaling in LN CLL cells was significantly higher compared to PB, indicating that BCR signaling is also involved in this subset of CLL (4). ZAP-70 expression, which is one of the most prominent genes distinguishing um-CLL from m-CLL, may reinforce BCR responsiveness (42). ZAP-70 is structurally similar to the BCR-associated kinase SYK (5) and its expression was associated with greater BCR signaling capacity, implicating a role for the BCR in CLL proliferation (43). Therefore, this suggests that the ability to respond to antigen stimulation coupled to signal reinforcement may underlie the differences in disease activity between the two prognostic subsets (29, 39, 44–46).
BCR signaling can be broadly divided into two main types: one that is mediated by antigen and another that is independent of antigen, referred to as tonic BCR signalling (1, 47, 48). Antigen-dependent and antigen-independent BCR stimulation are two fundamentally different mechanisms of BCR signaling which exist in B cell lymphomas. However, it is not quite clear to what extent tonic or antigen-dependent BCR signaling play a role in driving CLL. The absence of mutations in BCR signaling components leading to antigen-independent pathway activation in CLL favors a dominant role for antigen-dependent BCR signalling (5). The fact that CLL proliferation only takes place in lymphoid tissues may suggest that relevant antigens are localized to discrete anatomic compartments, but more likely this indicates the lack of additional signals outside of these compartments that trigger CLL proliferation, such as T cell-derived signals in the LN (16).
Strong molecular evidence for antigen-dependent BCR signaling in CLL is the presence of stereotyped BCRs, which support the idea of a common selecting antigenic epitope (49, 50). A study using antigen-specific TCL1 mice showed that neither acute nor chronic exposure to specific antigen influenced disease progression. Rather, CLL clones preferentially selected light chains paired with the antigen-specific heavy chains that conferred CLL cells the ability to interact with a broad range of autoantigens (51). These results suggest that pathogens may drive CLL by selecting and expanding pathogen-specific B cells that cross-react with one or more self-antigens, indicating that the BCR may in fact shape CLL progression in vivo. The specific antigens recognized by these stereotyped CLL BCRs are not well described, especially in the case of m-CLL. Whereas m-CLL clones exhibit more restricted autoreactivity (9, 40), the majority of um-CLL clones express low-affinity BCRs that are polyreactive recognizing self-antigens such as DNA, insulin and the cytoskeletal proteins myosin and vimentin, as well as foreign antigens such as bacterial DNA and lipopolysaccharides in addition to a spectrum of molecular motifs exposed on apoptotic cells (40, 52–57). One study showed and identified specificity to an autoantigenic target in one-fourth of CLL cases independent of IGHV mutation status (58). All identified BCR targets were cytoplasmic proteins. The translocation of cytoplasmic antigens to surface membrane blebs and apoptotic bodies would enable binding to the surface BCR of CLL cells (52). Importantly, the same study showed that BCRs belonging to the same stereotyped subset target identical antigens, but surprisingly, BCRs from individual CLL patients were specific for different epitopes of the same antigens. Finally, binding of the autoantigen to the respective CLL cells induced specific activation and proliferation, suggesting that autoreactivity of CLL cells via the BCR may be a general mechanism for driving CLL. Other stereotypic subsets of m-CLL have been described showing specificities to the cytomegalovirus phosphoprotein pUL32, the Fc-tail of IgG, as well as specificity to β-(1,6)-glucan, an abundant component in yeast and filamentous fungi (50, 59–61). However, it should be noted that there have been no recent reports of new subsets exhibiting specificity to exogenous antigens, illustrating that perhaps this represents a unique attribute of only a few CLL subsets. A study with TCL1 mice expressing transgenic BCRs with different antigen specificities showed that chronic interactions with low-affinity can induce CLL in vivo, whereas interactions with high-affinity antigens cannot (62). Additionally, the authors showed that low-affinity BCRs are positively selected, whereas high-affinity BCRs are not. Consequently, m-CLL clones remain more stable overall and expand at a slower rate, likely due to high-affinity binding to restricted sets of antigenic epitopes that either occur infrequently because they are on foreign antigens or because they induce anergy due to high-affinity binding (5).
Anergy is one of the mechanisms of the immune system to silence autoreactive B cells upon recognition of self-antigens. A state of BCR desensitization is induced by chronic binding of antigen in vivo, resulting in unresponsiveness when cells are stimulated with antigen in vitro. The fact that m-CLL is associated with a favorable disease course and bind antigen more specifically with higher affinity than um-CLL, suggests that exposure to antigen in vivo may lead to anergy of CLL cells (63). In mouse models, anergic B cells showed features of low levels of sIgM as the result of constant BCR internalization, increased basal intracellular calcium concentrations and constitutive activation of ERK1/2. This biochemical program is reversible and lasts as long as B cells are exposed to the antigen (64). Notably, repetitive BCR stimulation in healthy B cells resulted in anergy and CD5 expression, which is a feature of CLL (65). In addition, mouse models have shown that CD5 is necessary to maintain anergy in B cells as knocking out CD5 in these mice resulted in a loss of self-tolerance (66). In vitro studies analyzing responses to BCR triggering have identified a subset of CLL patients exhibiting indolent disease with CLL cells containing anergic features (67), including a lack of BCR signaling capacity and constitutive activation of ERK1/2 (64). Additionally, the BCR-associated kinase LYN initiates a negative feedback loop via recruitment of the phosphatase SHP-1 which inhibits BCR signaling and is overexpressed in CLL, further illustrating an anergic phenotype (68). Several studies have shown that the anergic phenotype of CLL cells, including sIgM expression and signaling capacity, reverses spontaneously after culture in vitro or following capping and endocytosis (39). This shows that downstream signaling pathways appear to be intact and that anergy can thus be attributed to surface immunoglobulins (sIgs), and this also provides direct evidence for engagement of putative antigen in vivo (39, 64).
The lack of antigen reactivity in m-CLL may indicate a role for antigen-independent BCR signaling, which is supported by the observation of phosphorylated LYN, SYK and canonical NF-κB in unstimulated CLL cells (5). Ligand-independent signaling is frequent in other malignancies where proliferation is subverted by the acquisition of genetic mutations in signaling components mimicking physiological stimulation of receptors (69). Such mutations in BCR signaling components are absent in CLL (5).
A special type of antigen-independent BCR activation has been described in CLL, which involves the binding of auto-epitopes existing on adjacent or in the same sIgs. Reactivity with sIg auto-epitopes could occur on a continuous basis. This also suggests a mutual BCR recognition on CLL cells which was confirmed by the binding of secreted CLL-derived BCRs to the surface of cell lines expressing either CLL or HD BCRs, but not to cells that did not express a BCR (70). Likewise, serum immunoglobulins from HD plasma were not able to bind HD B cells whereas serum immunoglobulins from CLL plasma were (71). It was shown that the binding was mediated by a conserved epitope in the second framework region of VH domains of the CLL BCR, as point mutations inside this motif abolished autonomous signaling. This motif was located outside of the antigen-recognition site, indicating that the induced signaling is not mediated by external antigens. Consistently, VH domain point mutations of antigen-specific BCRs in m-CLL were still able to signal upon recognition of antigen, indicating that this type of tonic signaling is antigen-independent but does not rule out the involvement of extrinsic antigens in the pathogenesis of CLL (70). Consistent with these observations are imaging data showing that HD BCRs exist predominantly as monomers and dimers in the plasma membranes of resting B cells and form oligomeric clusters upon stimulation with antigen. In contrast, CLL BCRs form dimers and oligomers in the absence of a stimulus, reflecting an antigen-independent tonic activity of the BCR (72). A single amino acid exchange reverted the organization to monomers and thus prevented auto-aggregation of CLL BCRs (72).
As tonic signaling would essentially result in constitutive BCR signaling, this would thus lead to a tolerogenic signal that should result in anergy (5). Consistently, a lack of external BCR stimulation does not lead to spontaneous CLL proliferation in vitro (5). Therefore it can be hypothesized that binding of extrinsic cognate antigens is essential to overcome anergy of CLL cells and is thus required for CLL proliferation (50, 73). A study using the TCL1 transgenic mouse model showed that this unique autonomous signaling capacity is a prerequisite for CLL development. Moreover, the capacity of CLL cells to respond to antigen inversely correlated with time to leukemia development, suggesting that signals induced by external antigens contribute to the aggressiveness of the disease (62).
In summary, both antigen-dependent and antigen-independent BCR signaling have been described in CLL, and CLL cells can receive both continuous and intermittent BCR signals that may facilitate proliferation (5). Yet, ligand-dependent and tonic BCR signaling may not be mutually exclusive. CLL clones could originate as antigen-dependent, but evolve to become more autonomous if the critical BCR regions are mutated. Substantiating this possibility would require comparison of BCR sequences in a cohort containing early MBL and later CLL stages of the same patient, which to our knowledge has not been performed. Alternatively, these separate mechanisms could reflect different routes for clonal expansion after initial transformation (69).
The BCR signaling pathway has emerged as an important therapeutic target for B cell malignancies, including CLL (74). Several BCR-targeted agents, including inhibitors of BTK (ibrutinib), PI3Kδ (idelalisib), and SYK (fostamatinib) have demonstrated clinical efficacy, which led to FDA approval of idelalisib and ibrutinib (28, 75–78). Especially the introduction of ibrutinib has dramatically changed the management of CLL, allowing for treatment without chemotherapy (74). Ibrutinib inhibits the activation of BTK, which plays a role in proliferation, survival, migration and tissue adhesion of CLL cells (4, 79, 80). Treatment of patients with ibrutinib leads to lymphocytosis due to efflux of CLL cells from the proliferative LN compartment into the PB (28, 81), thereby depriving CLL cells from microenvironmental signals and halting disease progression (17).
The impact of ibrutinib treatment on in vivo CLL kinetics of CLL cells showed that no newly divided CLL cells entered the PB upon ibrutinib treatment. The average pretreatment birth rate decreased upon ibrutinib treatment whereas death rates increased (81). In addition, ibrutinib treatment resulted in a reduction of the proliferative CXCR4low/CD5high fraction (82). Even though ibrutinib targets a key pathogenic pillar of CLL by depriving cells from antigen and interactions with the lymphoid microenvironment, it is not sufficient to eradicate disease, as stopping treatment reverses remission (17). The fact that the BCR components BTK and PLCG2 are specifically mutated in ibrutinib-resistant CLL underlines that therapeutic success depends critically on inhibition of this pathway (83).
Importantly, it was shown that maximum inhibition of BCR signaling in vivo was already achieved after one administration of the drug whereas maximum inhibition of downstream NF-κB signaling required repeated dosing (4). This indicates that aside from direct effects, continued treatment of ibrutinib leads to changes in the microenvironment that have indirect effects on CLL cells, highlighting the role of accessory cells mediating signaling via alternative receptors such as CD40 and TLRs. Next, we will discuss the existing evidence for in vivo CD40 and TLR signaling and their roles in CLL proliferation.
CD40 expressed on CLL cells can be stimulated by its physiological ligand CD154 (CD40L) expressed on, for instance, activated CD4 T cells and follicular T helper cells (84, 85). Interaction of CD40 and CD40L stimulates the proliferation and differentiation of healthy B cells (85). CD40 stimulation on CLL cells activates downstream signaling pathways including MAPK/ERK, PI3K/AKT/mTOR, and NF-κB (86), thus largely overlapping with downstream BCR signaling pathways (Figure 2). As a result, CLL cells are activated and provided with strong survival signals rendering them highly resistant to a wide variety of therapeutics (7, 85). In addition, CD40 stimulation propels both CLL cells and healthy B cells in a proliferative state (7, 87). Interestingly, a few studies have reported the expression of CD40L on CLL cells as well, suggesting a mechanism in which activated CLL cells may directly stimulate CD40 on resting bystander CLL cells in a paracrine manner (88, 89).
The earliest observation for a role of CD40 signaling in CLL was the infiltration of CD4 T cells that express CD40L in CLL pseudofollicles that co-localize with Ki-67+ CLL cells in these proliferation centers (23, 90). This is suggested to be a mechanism to regulate CLL proliferation, which was supported by in vitro stimulation of PB CLL cells with CD40L, inducing expression of CCL22, which serves as an attractant for CD4 T cells which in turn express CD40L (23). Moreover, co-culture of CLL cells with activated autologous T cells results in proliferation of CLL cells (7, 91). Importantly, interference with CD40 signaling collapses LN germinal centers necessary for B cell development, differentiation and somatic hypermutation (92).
Interestingly, in lymphocytes isolated from PB of CLL patients, a fraction of proliferating T helper cells were observed in the presence, but not in the absence of CLL cells. Moreover, these CLL-associated T helper cells induce HLA class II-dependent activation and proliferation of CLL cells in vitro, suggesting that CLL is a disease driven by immune responses via a process in which T helper cells engage CLL cells in response to antigen presented on the CLL cells’ own HLA class II molecules (93). It would be worthwhile if these intriguing findings can be confirmed by other studies.
Even though direct evidence for T cell-dependent CLL proliferation in patients is lacking, several mouse models have provided more insight. CLL cells xenografted in NOD-SCID mice require activated autologous T cells in order for the CLL cells to proliferate. Moreover, depletion of CD4 T cells inhibited proliferation whereas depletion of CD8 T cells did not (94). Also, LMP1/CD40 mice express a chimeric protein containing part of the Epstein-Barr viral Latent Membrane Protein 1, mimicking constitutively active CD40 triggering (95). These mice showed an increase of B cells in secondary lymphoid organs with an activated phenotype, increased proliferation and prolonged survival. In addition, they showed significantly impaired T cell-dependent immune responses, thus resembling CLL in many aspects (96). Moreover, LMP1/CD40 cells proliferated spontaneously in vitro in a CD40-dependent manner (95).
Combined, these observations indicate that CD40 may in fact be an important mediator in CLL proliferation, which is currently less widely recognized compared to the contribution of BCR signaling.
Recurrent mutations in CLL include MYD88, a gene which encodes a downstream component of TLR signaling (69). TLRs are part of the innate immune system and respond to specific molecular patterns found in various microorganisms, including bacteria (43). These receptors are expressed in CLL cells and biologically active, suggesting an additional route of stimulation besides BCR signaling (69). TLR signaling leads to the activation of several downstream signaling pathways, including MAPK/ERK, PI3K/AKT/mTOR, and NF-κB (97), resulting in the activation and proliferation of CLL cells (43) (Figure 2).
The most evident observations for a role of TLR signaling in CLL are increased expression of TLR9 in CLL cells compared to healthy B cells (98, 99), TLR pathway activation in LN CLL cells as shown by gene array studies (19, 24), as well as in situ proximity ligation assay experiments that showed the interaction of pIκBα with TLR9 and MYD88 in LN CLL cells (24). CLL BCR specificity for DNA or antigens physically linked to DNA further suggest a role for TLR signaling in driving CLL (8). Moreover, apoptosis-associated antigens bound by sIgs can also be recognized by TLRs after entrance to the endosomal compartment via sIgs (52). Interestingly, the observation that um-CLL cells are more responsive to BCR activation than m-CLL cells is mirrored by TLR activation in vivo and in vitro (24). A possible explanation is that the antigen-specific BCRs in m-CLL make them less likely to internalize antigen-linked TLR ligands compared to um-CLL, whose BCRs are polyreactive and bind with low affinity to a wide variety of antigens (52, 54, 100). Yet, this does not hold for CpG stimulation in vitro, as these are internalized in a BCR-independent fashion. See below in the section in vitro TLR signaling for additional observations that TLR and BCR signaling may cooperate to promote CLL proliferation in um-CLL.
The absence of TIR8, a negative regulator of TLR signaling, was shown to accelerate disease progression in TCL1 mice (101). Moreover, epidemiological studies found an increased risk for the development of CLL following episodes of sinusitis, pharyngitis, influenza, cellulitis, and herpes zoster, where risk increased with increasing severity or frequency of infection (102–104). These studies suggest that infectious agents can promote disease onset and progression of CLL, which may be related to TLR activation (102).
It is not yet clear to what extent these non-BCR signals contribute to proliferation in situ, but it is apparent that both BCR, CD40, and TLR activation all show marked similarities in the downstream signaling pathways involved, including the MAPK/ERK, PI3K/AKT/mTOR, and NF-κB pathways (43) (Figure 2). Perhaps a combination of stimuli is what may really drive CLL proliferation in vivo or helps develop it into more aggressive disease.
Despite CLL being a proliferative disease with significant cell turnover, primary CLL cells rapidly undergo apoptosis in the absence of microenvironmental survival signals (105). What further illustrates that essential in vivo factors are missing in in vitro systems, is that co-culture with stromal cells or the addition of soluble factors can significantly extend CLL survival, yet only for a limited amount of time and thus far, no system permits the long-term expansion of CLL cells in vitro (7, 19, 94, 106, 107). In vitro studies typically analyze CLL cells isolated from PB and consequently, the contribution of the host microenvironment to the proliferation of CLL cells in vivo remains ill-defined (19). The difficulties of mimicking a physiologic microenvironment supporting CLL proliferation hinder in vitro studies and as a result, a large variety of culture systems have been developed in order to investigate CLL proliferation (105). This raises difficulties in comparing data achieved with these highly variable approaches and hinders systematic characterization of culture systems or back-to-back comparisons in terms of CLL proliferation (105, 106). In the next section, we will elaborate on CLL proliferation in the light of in vitro data and we will specifically discuss models utilizing BCR, CD40 or TLR stimulation in combination with costimulatory cytokine signals.
Despite the consensus regarding the role of BCR signaling in the biology of CLL, the response of CLL cells to BCR stimulation in vitro is notoriously heterogeneous among patient samples (43). In vitro BCR stimulation of CLL cells is performed using anti-IgM, either soluble or immobilized. In vitro responses to BCR stimulation differ between um-CLL and m-CLL as demonstrated by several groups using multiple assays, including global protein tyrosine phosphorylation, gene expression profiling, cellular metabolic activity, apoptotic response and proliferative activity (29–31). Several studies have reported that response to BCR stimulation was correlated with sIgM levels (31). Some studies found higher sIgM levels in um-CLL (108), whereas others found little to no differences between um-CLL and m-CLL (39, 46), claiming a role for additional factors contributing to BCR responsiveness (39). Many studies showed a correlation of CD38 and ZAP-70 expression with BCR responsiveness, however, CD38 does not influence BCR signaling in vitro and ZAP-70 is not required for response. Therefore, these correlations most likely do not result from functional interactions, but are a result of um-CLL expressing higher levels of CD38 and ZAP-70 (39). Some discrepancies found in literature concerning in vitro responses to BCR stimulation can, at least partially, be attributed to the lack of a standardized protocol to trigger the CLL BCR in vitro (109). For example, it has been shown that immobilized anti-IgM provides a more potent in vitro CLL stimulus than soluble anti-IgM (109, 110).
Another possibility is that the variability in BCR responses stems from variable degrees of sIg clustering, which may be associated with natural genomic heterogeneity in BCRs and/or response to antigen (71). Indeed, induction of stable BCR clustering on healthy B cells modulated BCR responsiveness. In fact, by titrating the amount of anti-IgM crosslinking, healthy B cells could be induced to recapitulate the diversity in signaling observed in CLL cells, confirming that BCR clustering can modulate BCR responsiveness and thereby phenocopy the signaling dysfunction in CLL (71). As for the heterogeneity of BCR responsiveness, CLL cells could be divided into 3 subgroups of SHP-1low/pPLCG2high to SHP-1high/pPLCG2low expression, where each subset displayed unique deviations in their BCR signaling responses (71). As increasing levels of SHP-1 and decreasing levels of pPLCG2 correlated with weakened BCR responsiveness, this suggests that phenotypic variability within isogenic populations of cells may result from heterogeneous levels of signaling regulators (71).
Whereas BCR stimulation of healthy B cells significantly induces their proliferation, in vitro BCR stimulation of CLL cells does not lead to proliferation (107), which is another reminder that the in vitro CLL systems are missing a crucial aspect that is active in patients. In vitro engagement of the BCR in CLL promotes G1 cell cycle progression as shown by increased levels of cyclin D2 and CDK4, but does not induce cell division, associated with constitutively high levels of the cell cycle inhibitor p27 (30). Accordingly, CLL cells within proliferation centers of the LN showed high expression of cyclin D2 and downregulation of p27 (111). Therefore, CLL proliferation may depend on costimulatory signals such as those delivered through CD40 or TLRs, possibly in combination with cytokines (6, 7, 16). The interleukins are a family of cytokines that serve as key regulatory elements within the immune system and a number of specific interleukins have been identified as being associated with the proliferation of CLL cells in vitro (43). Importantly, IL-4 promotes the expression and function of surface IgM in CLL cells, thereby enhancing in vitro BCR responsiveness (112). However, proliferation of CLL cells has been mostly described in an antigen-independent context using combined stimulations with CD40L/IL-21, CD40L/CpG, or CpG/IL-15 (107), which we will describe in the next sections.
In vitro CD40 stimulation of CLL cells can be performed using either soluble agonists or via co-culture with CD40L-presenting cells. Importantly, soluble agonists like anti-CD40 antibodies or soluble CD40L are inferior to co-culture with CD40L-presenting cell lines which are able to support proliferation more efficiently (85, 113, 114). Again, as various soluble CD40 agonists and CD40L co-culture systems are used, this hinders direct comparison of in vitro studies on CD40-mediated CLL proliferation (85).
Similar to BCR stimulation, crosslinking of CD40 provides only weak proliferative responses in CLL whereas healthy B cells proliferate well (87). In contrast to single BCR or CD40 stimulation, co-culture of CLL cells with activated CD4 T cells promoted CLL proliferation to the same extent of that in healthy B cells, which respond equally well to single CD40 stimulation or co-culture with T cells (87, 93). Although a monolayer of CD40L-presenting fibroblasts induced a highly similar gene profile as induced by co-culture with T cells (7), it did not induce CLL proliferation like activated T cells (106), highlighting the role of additional T cell-derived signals in the proliferation of CLL.
CD40 triggering upregulates the IL-21 receptor, making CLL cells more receptive for the T cell cytokine IL-21, which has been implicated in CLL proliferation (115). IL-21 significantly increased CD40-mediated proliferation, and CLL proliferation by activated T cells was shown to be IL-21-dependent as well (7, 116). Importantly, in vitro T cell activation induced IL-21 mRNA production, specifically in follicular T helper cells, which have been shown to be present and produce IL-21 in LNs in vivo (7, 117). Another T cell cytokine implicated in CD40-mediated CLL proliferation is IL-4. In vitro stimulation with soluble CD40L caused a slight increase of CD40 expression on CLL cells but stimulation with IL-4 resulted in a significant increase of CD40 expression (118). Combined stimulation of CD40 and IL-4 however results in only modest proliferation of CLL cells (116), but it primes cells for proliferation via IL-21, as combined stimulation of CD40L, IL-4 and IL-21 results in increased CLL proliferation (117).
Interestingly, in vitro BCR stimulation of CLL cells also resulted in increased expression of CD40, suggesting potential crosstalk between BCR and CD40 signaling (31, 93). In fact, whereas BCR stimulation rapidly activates pSYK in both healthy B cells and CLL cells, stimulation with CD40L also activates pSYK in CLL cells but has no effect on pSYK in healthy B cells (119). Likewise, inhibition of SYK hampers CD40-mediated proliferation of CLL cells but not in healthy B cells. In addition to SYK, BTK is also activated upon CD40 stimulation, suggesting that CLL cells exploit CD40 stimulation by increasing BCR pathway activity (119). A possible explanation may be that CD40 acts as a gatekeeper for BCR signaling by inhibiting negative feedback components like LYN and SHP-1, so that CD40-dependent activation of the BCR pathway is required to overcome negative feedback signals in anergic CLL cells (119) (Figure 3).
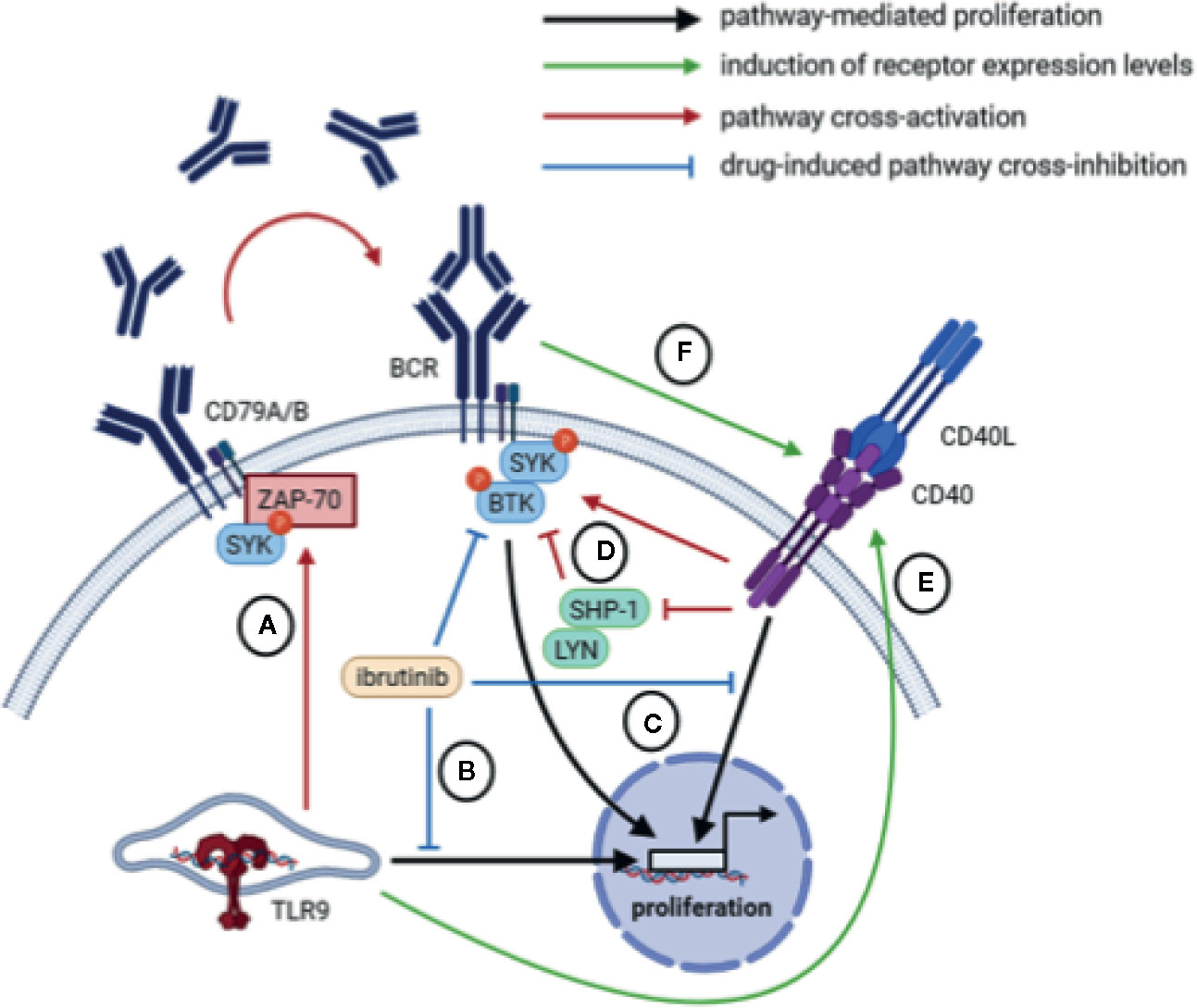
Figure 3 Overview of potential crosstalk mechanisms between BCR, CD40, and TLR signaling. (A) TLR signaling in um-CLL may mount an autocrine feedback loop mediated by ZAP-70 and SYK involving the production and secretion of IgM which may subsequently trigger BCR signaling in an autocrine/paracrine manner. (B) Inhibition of BCR signaling via BTK inhibits CpG-mediated proliferation. (C) Inhibition of the BCR-associated kinases BTK or SYK inhibits CD40-mediated proliferation. (D) Upon CD40 stimulation, the BCR-associated kinases BTK and SYK are activated, possibly via inhibition of negative feedback components LYN and SHP-1. (E) TLR9 stimulation via CpG increases expression of the CD40 receptor. (F) Activation of BCR signaling increases expression of the CD40 receptor.
CD40 stimulation in combination with IL-21 or IL-4 is sufficient to induce CLL proliferation in vitro whereas for BCR stimulation this is not the case (6, 7). However, CLL cells treated with ibrutinib in vivo did not proliferate upon CD40/IL-21 stimulation in vitro, an effect which can be recapitulated by in vitro inhibition of either BTK, PI3K or SYK, indicating the requirement of BCR kinases in CD40-mediated proliferation (107). CLL proliferation is significantly increased further when BCR stimulation is added to the combined stimulation of CD40 and cytokines whereas for healthy B cells this is not the case, showing that these separate signaling nodes may potentially cooperate to drive CLL proliferation in vivo (6).
In vitro activation of TLR signaling is carried out using CpG, resulting in endocytosis and subsequent binding of TLR9 in the endosomal compartment, thereby mimicking the interaction of CLL cells with bacteria (120). In vitro stimulation with CpG activates p-p65 and decreases IRAK1 levels, recapitulating the effects of TLR activation as observed in LN CLL cells in vivo (24). In vitro stimulation of CLL cells with CpG resulted in quite variable reports of proliferation (99, 107, 121, 122). Similar to in vitro BCR activation, um-CLL cells can more often be induced to proliferate upon CpG stimulation (97, 123). Importantly, response is not correlated with TLR9 mRNA or protein expression (97). The in vitro proliferative response to CpG was found to be highly predictive of progression-free survival, time to treatment and overall survival in m-CLL, whereas prognosis of um-CLL was equally worse, with or without proliferative response to CpG in vitro (124). Although we still do not fully understand the mechanistic basis of TLR signaling in CLL proliferation, these findings do support the relevance of TLR signaling in driving CLL.
In vitro CpG stimulation was shown to upregulate CD122, which is a shared subunit of the receptor for IL-2 and IL-15 (125). Consistently, addition of the T cell cytokine IL-2 significantly enhances CpG-mediated proliferation of CLL cells (7). IL-15, produced by monocytes, synergistically promotes CpG-mediated CLL proliferation independent of CLL mutation status, thus reversing the difference as usually seen between um-CLL and m-CLL (8, 107). Moreover, in vitro proliferation mediated by CpG/IL-15 could not be linked to prior treatment or in vivo growth rates. As for genetic abnormalities, only TRI-12 was associated with a significantly greater propensity for proliferation in response to CpG/IL-15 (8). Examples of robust in vitro proliferation of clones with slow in vivo growth rates support the notion that the availability of stimulatory signals within the in vivo microenvironment may be as relevant as a cell’s intrinsic potential for proliferation (8). Notably, the abundant presence of IL-15-expressing cells in LNs of CLL patients makes this mechanism clinically relevant (125). Similar to CD40 stimulation, CpG induces upregulation of the IL-21 receptor (126). Whereas healthy B cells proliferated much more than CLL cells following single CpG stimulation, addition of IL-21 rescued enhanced the proliferative activity of CLL cells to the same level of healthy B cells (123).
Links between TLR and BCR signaling have also been described, as for example in vitro stimulation of CLL cells with CpG induced phosphorylation of CD79A, LYN and SYK which appears to rely on the expression of ZAP-70 (127). The strong association between CpG-mediated CLL proliferation and IGHV mutation status may suggest that TLR stimulation is modulated by BCR signaling (97, 123). For example, anergic cells that are usually m-CLL and lack expression of ZAP-70, show reduced proliferative responses to CpG in vitro (124). Studies have shown that engagement of TLR signaling in CLL is able to mount an autocrine feedback loop involving the production and secretion of IgM leading to activation of the cell’s own BCR, which reinforces the concept of tonic BCR signaling in the absence of antigen (107, 127) (Figure 3).
Similar to in vitro BCR activation, stimulation with CpG also caused upregulation of CD40 on CLL cells, showing links of TLR stimulation with CD40 signaling as well (99, 122, 128, 129). Activation of TLR9 via CpG significantly increased CLL proliferation when combined with CD40 stimulation, similar to CD40/IL-21 stimulation (6, 7, 128, 130). Importantly, combined TLR/CD40 stimulation overcomes the hyporesponsiveness to CpG as often seen in m-CLL (130). On the contrary, CLL cells that do not proliferate in vitro in a T-cell dependent manner, can be triggered to proliferate upon addition of CpG/IL-2 (93).
Finally, inhibition of BCR signaling via treatment with ibrutinib or via inhibition of SYK significantly inhibits both CD40- and CpG-mediated CLL proliferation in vitro, showing the role of BCR-associated kinases (36, 107, 131). CD40 and CpG-induced proliferation do however differ in their involvement of the BCR complex. CD40 involves the recruitment of BTK independent of upstream BCR components whereas CpG indirectly triggers the BCR via IgM secretion (107). Therefore, BCR-targeted agents effectively target the aforementioned TLR-BCR feedback loop (107).
Concerning the potential cooperation of signaling pathways, it is important to note that a divergence exists between crosstalk of separate pathways and active rewiring of distinct signaling pathways. For example, in diffuse large B cell lymphoma it was found that the GC and ABC subsets depended on the BCR subunits CD79A/B, but engaged divergent downstream signaling pathways (132). In patients with MYD88 mutations, a new complex consisting of TLR9 and MYD88 was found, revealing interactions with the BCR subunits CD79A/B. It was shown that TLR and BCR signaling cooperate to assemble MYD88 in a signalosome which activates mTOR and NF-κB signaling, providing a mechanistic insight as to why these patients were particularly sensitive to the BCR-targeted agent ibrutinib. This same rewiring was not found in CLL samples, but the expression of ZAP-70 represents another example where single activation of TLR9 is sufficient to fully engage BCR signaling (127, 132). ZAP-70 therefore represents an important candidate for signaling pathway rewiring in CLL. It was shown that TLR-mediated BCR activation was not dependent on the kinase activity of ZAP-70, which is compatible with ZAP-70 functioning as a scaffold in a signaling complex that relays TLR9 signals to SYK, thereby integrating innate into adaptive immune responses (42, 127). However, studying rewiring of signaling pathways is difficult and usually requires techniques such as mass spectrometry or proximity ligation assays to study the interactome of proteins, as ultimately protein-protein interactions assemble and regulate signaling pathways (133). Finally, multiomic analyses allow to study changes in signaling networks under specific conditions, and a novel kinomics approach applied in CLL revealed that rewiring of signaling pathways is not strictly oncogenic but can also be influenced by therapy (134). Comparison of kinase fingerprints between treatment-naïve patients and patients who had undergone prior chemoimmunotherapy, revealed SYK as a critical kinase to be differentially active upon BCR stimulation which correlated with proliferative capacity in vitro (134).
In summary, various crosstalk mechanisms between BCR, CD40, and TLR signaling have been described in CLL based on receptor expression levels, the activation of downstream mediators as well as the use of targeted inhibitors (Figure 3).
In summary, most evidence for driving CLL proliferation in vivo is currently attributed to BCR engagement. A major discrepancy is that BCR stimulation in vitro does not induce proliferation, indicating that BCR-induced CLL proliferation in vivo likely requires additional (costimulatory) signals that are missing in vitro. We have highlighted the role of T cells in the proliferation of CLL cells in vivo, as T cell-derived signals including CD40L, IL-21 and IL-4 significantly promote in vitro proliferation of CLL cells (6). Moreover, it is important to consider that crosstalk between BCR, CD40, and TLR signaling occurs in vitro, and may thus play an important role in vivo. Consequently, we propose that combined triggering of multiple nodes of BCR, CD40, and TLR signaling in combination with costimulatory signals by cytokines orchestrate CLL proliferation, both in vitro and in vivo (Figure 3).
Modeling the CLL microenvironment is an area of intense investigation, and most studies have been performed using CLL cells isolated from PB as tissue-residing CLL cells are not easily obtained. Current experimental methods relying on the investigation of PB CLL cells lack the ability to appropriately mimic the lymphoid microenvironment due to a lack of in vitro cultures that allow the long-term expansion of CLL cells. Crucially, this indicates that essential factors or aspects are missing in current in vitro models. Despite these limitations, many in vitro observations have helped to elucidate the role of (the combination of) individual signals in the proliferation of CLL in vivo, including the development of new therapies to target CLL proliferation.
New innovative in vitro CLL models continue to be developed and most promising could be 3D cultures that may overcome some of the current limitations of in vitro studies. Current 2D culture systems do not reflect the true 3D microenvironment present in human tissues, where various types of cell-cell interactions and interactions with the extracellular matrix occur, which may be fundamental to study CLL proliferation (135). Although the use of 3D models is new in the field of CLL, a recent study reported a significant increase in proliferative response, proliferation rates and number of cell generations compared to 2D cultures, irrespective of the biological characteristics of CLL cells (6). Therefore, 3D models may contribute pathophysiological relevance to in vitro culture systems of CLL and will be valuable for future studies. Similar to 2D CLL culture systems, many types of 3D models have been developed for solid tumors, including the use of scaffolds, gels, spheroid cultures and fluidic systems (136). However, unlike solid tumors, secondary lymphoid tissues do not derive from a single stem cell progenitor and thus the advantages and limitations of each of these systems have to be evaluated in terms of accurate mimicking of the CLL microenvironment. We may safely predict that in the near future a variety of 3D CLL systems will be reported. The ultimate goal is to implement a standardized system for in vitro proliferation, that will allow novel drug testing, as well as meaningful study of various CLL clonotypes.
MH wrote the review and prepared figures. EE conceptualized and wrote the review. AK edited the review and contributed text. All authors contributed to the article and approved the submitted version.
MH was funded by the Blaauboer Fund via the AMC Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank our colleague Sanne Tonino for critical reading of the manuscript and for her helpful suggestions.
1. Herishanu Y, Katz BZ, Lipsky A, Wiestner A. Biology of chronic lymphocytic leukemia in different microenvironments: clinical and therapeutic implications. Hematol Oncol Clin North Am (2013) 27(2):173–206. doi: 10.1016/j.hoc.2013.01.002
2. Vardi A, Agathangelidis A, Sutton LA, Ghia P, Rosenquist R, Stamatopoulos K. Immunogenetic studies of chronic lymphocytic leukemia: revelations and speculations about ontogeny and clinical evolution. Cancer Res (2014) 74(16):4211–6. doi: 10.1158/0008-5472.CAN-14-0630
3. Damle RN, Calissano C, Chiorazzi N. Chronic lymphocytic leukaemia: a disease of activated monoclonal B cells. Best Pract Res Clin Haematol (2010) 23(1):33–45. doi: 10.1016/j.beha.2010.02.001
4. Herman SE, Niemann CU, Farooqui M, Jones J, Mustafa RZ, Lipsky A, et al. Ibrutinib-induced lymphocytosis in patients with chronic lymphocytic leukemia: correlative analyses from a phase II study. Leukemia (2014) 28(11):2188–96. doi: 10.1038/leu.2014.122
5. Burger JA, Chiorazzi N. B cell receptor signaling in chronic lymphocytic leukemia. Trends Immunol (2013) 34(12):592–601. doi: 10.1016/j.it.2013.07.002
6. Schleiss C, Ilias W, Tahar O, Guler Y, Miguet L, Mayeur-Rousse C, et al. BCR-associated factors driving chronic lymphocytic leukemia cells proliferation ex vivo. Sci Rep (2019) 9(1):701. doi: 10.1038/s41598-018-36853-8
7. Pascutti MF, Jak M, Tromp JM, Derks IA, Remmerswaal EB, Thijssen R, et al. IL-21 and CD40L signals from autologous T cells can induce antigen-independent proliferation of CLL cells. Blood (2013) 122(17):3010–9. doi: 10.1182/blood-2012-11-467670
8. Mongini PK, Gupta R, Boyle E, Nieto J, Lee H, Stein J, et al. TLR-9 and IL-15 Synergy Promotes the In Vitro Clonal Expansion of Chronic Lymphocytic Leukemia B Cells. J Immunol (2015) 195(3):901–23. doi: 10.4049/jimmunol.1403189
9. Chiorazzi N. Cell proliferation and death: forgotten features of chronic lymphocytic leukemia B cells. Best Pract Res Clin Haematol (2007) 20(3):399–413. doi: 10.1016/j.beha.2007.03.007
10. Messmer BT, Messmer D, Allen SL, Kolitz JE, Kudalkar P, Cesar D, et al. In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. J Clin Invest (2005) 115(3):755–64. doi: 10.1172/JCI23409
11. Defoiche J, Debacq C, Asquith B, Zhang Y, Burny A, Bron D, et al. Reduction of B cell turnover in chronic lymphocytic leukaemia. Br J Haematol (2008) 143(2):240–7. doi: 10.1111/j.1365-2141.2008.07348.x
12. van Gent R, Kater AP, Otto SA, Jaspers A, Borghans JA, Vrisekoop N, et al. In vivo dynamics of stable chronic lymphocytic leukemia inversely correlate with somatic hypermutation levels and suggest no major leukemic turnover in bone marrow. Cancer Res (2008) 68(24):10137–44. doi: 10.1158/0008-5472.CAN-08-2325
13. Murphy EJ, Neuberg DS, Rassenti LZ, Hayes G, Redd R, Emson C, et al. Leukemia-cell proliferation and disease progression in patients with early stage chronic lymphocytic leukemia. Leukemia (2017) 31(6):1348–54. doi: 10.1038/leu.2017.34
14. Herndon TM, Chen SS, Saba NS, Valdez J, Emson C, Gatmaitan M, et al. Direct in vivo evidence for increased proliferation of CLL cells in lymph nodes compared to bone marrow and peripheral blood. Leukemia (2017) 31(6):1340–7. doi: 10.1038/leu.2017.11
15. Damle RN, Batliwalla FM, Ghiotto F, Valetto A, Albesiano E, Sison C, et al. Telomere length and telomerase activity delineate distinctive replicative features of the B-CLL subgroups defined by immunoglobulin V gene mutations. Blood (2004) 103(2):375–82. doi: 10.1182/blood-2003-04-1345
16. Chiorazzi N, Ferrarini M. Evolving view of the in-vivo kinetics of chronic lymphocytic leukemia B cells. Hematol Am Soc Hematol Educ Program (2006) 2006(1):273–8, 512. doi: 10.1182/asheducation-2006.1.273
17. Forconi F. Five years of ibrutinib in CLL. Blood (2018) 131(21):2280–1. doi: 10.1182/blood-2018-03-837864
18. Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell (2013) 152(4):714–26. doi: 10.1016/j.cell.2013.01.019
19. Herishanu Y, Perez-Galan P, Liu D, Biancotto A, Pittaluga S, Vire B, et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood (2011) 117(2):563–74. doi: 10.1182/blood-2010-05-284984
20. Stein H, Bonk A, Tolksdorf G, Lennert K, Rodt H, Gerdes J. Immunohistologic analysis of the organization of normal lymphoid tissue and non-Hodgkin’s lymphomas. J Histochem Cytochem (1980) 28(8):746–60. doi: 10.1177/28.8.7003001
21. Soma LA, Craig FE, Swerdlow SH. The proliferation center microenvironment and prognostic markers in chronic lymphocytic leukemia/small lymphocytic lymphoma. Hum Pathol (2006) 37(2):152–9. doi: 10.1016/j.humpath.2005.09.029
22. Calissano C, Damle RN, Marsilio S, Yan XJ, Yancopoulos S, Hayes G, et al. Intraclonal complexity in chronic lymphocytic leukemia: fractions enriched in recently born/divided and older/quiescent cells. Mol Med (2011) 17(11-12):1374–82. doi: 10.2119/molmed.2011.00360
23. Ghia P, Strola G, Granziero L, Geuna M, Guida G, Sallusto F, et al. Chronic lymphocytic leukemia B cells are endowed with the capacity to attract CD4+, CD40L+ T cells by producing CCL22. Eur J Immunol (2002) 32(5):1403–13. doi: 10.1002/1521-4141(200205)32:5<1403::AID-IMMU1403>3.0.CO;2-Y
24. Dadashian EL, McAuley EM, Liu D, Shaffer AL,3, Young RM, Iyer JR, et al. TLR Signaling Is Activated in Lymph Node-Resident CLL Cells and Is Only Partially Inhibited by Ibrutinib. Cancer Res (2019) 79(2):360–71. doi: 10.1158/0008-5472.CAN-18-0781
25. van Attekum M, Terpstra S, Reinen E, Kater AP, Eldering E. Macrophage-mediated chronic lymphocytic leukemia cell survival is independent of APRIL signaling. Cell Death Discovery (2016) 2:16020. doi: 10.1038/cddiscovery.2016.20
26. van Attekum MH, Kater AP, Eldering E. The APRIL paradox in normal versus malignant B cell biology. Cell Death Dis (2016) 7(6):e2276. doi: 10.1038/cddis.2016.183
27. Stevenson FK, Forconi F, Packham G. The meaning and relevance of B-cell receptor structure and function in chronic lymphocytic leukemia. Semin Hematol (2014) 51(3):158–67. doi: 10.1053/j.seminhematol.2014.05.003
28. Cheng S, Ma J, Guo A, Lu P, Leonard JP, Coleman M, et al. BTK inhibition targets in vivo CLL proliferation through its effects on B-cell receptor signaling activity. Leukemia (2014) 28(3):649–57. doi: 10.1038/leu.2013.358
29. Lanham S, Hamblin T, Oscier D, Ibbotson R, Stevenson F, Packham G. Differential signaling via surface IgM is associated with VH gene mutational status and CD38 expression in chronic lymphocytic leukemia. Blood (2003) 101(3):1087–93. doi: 10.1182/blood-2002-06-1822
30. Deglesne PA, Chevallier N, Letestu R, Baran-Marszak F, Beitar T, Salanoubat C, et al. Survival response to B-cell receptor ligation is restricted to progressive chronic lymphocytic leukemia cells irrespective of Zap70 expression. Cancer Res (2006) 66(14):7158–66. doi: 10.1158/0008-5472.CAN-06-0085
31. Guarini A, Chiaretti S, Tavolaro S, Maggio R, Peragine N, Citarella F, et al. BCR ligation induced by IgM stimulation results in gene expression and functional changes only in IgV H unmutated chronic lymphocytic leukemia (CLL) cells. Blood (2008) 112(3):782–92. doi: 10.1182/blood-2007-12-127688
32. Kil LP, de Bruijn MJ, van Nimwegen M, Corneth OB, van Hamburg JP, Dingjan GM, et al. Btk levels set the threshold for B-cell activation and negative selection of autoreactive B cells in mice. Blood (2012) 119(16):3744–56. doi: 10.1182/blood-2011-12-397919
33. Woyach JA, Bojnik E, Ruppert AS, Stefanovski MR, Goettl VM, Smucker KA, et al. Bruton’s tyrosine kinase (BTK) function is important to the development and expansion of chronic lymphocytic leukemia (CLL). Blood (2014) 123(8):1207–13. doi: 10.1182/blood-2013-07-515361
34. Holler C, Pinon JD, Denk U, Heyder C, Hofbauer S, Greil R, et al. PKCbeta is essential for the development of chronic lymphocytic leukemia in the TCL1 transgenic mouse model: validation of PKCbeta as a therapeutic target in chronic lymphocytic leukemia. Blood (2009) 113(12):2791–4. doi: 10.1182/blood-2008-06-160713
35. Sanchez-Aguilera A, Rattmann I, Drew DZ, Muller LU, Summey V, Lucas DM, et al. Involvement of RhoH GTPase in the development of B-cell chronic lymphocytic leukemia. Leukemia (2010) 24(1):97–104. doi: 10.1038/leu.2009.217
36. Herman SE, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood (2011) 117(23):6287–96. doi: 10.1182/blood-2011-01-328484
37. Oppezzo P, Dighiero G. “Role of the B-cell receptor and the microenvironment in chronic lymphocytic leukemia’’. Blood Cancer J (2013) 3:e149. doi: 10.1038/bcj.2013.45
38. Coelho V, Krysov S, Steele A, Sanchez Hidalgo M, Johnson PW, Chana PS, et al. Identification in CLL of circulating intraclonal subgroups with varying B-cell receptor expression and function. Blood (2013) 122(15):2664–72. doi: 10.1182/blood-2013-02-485425
39. Mockridge CI, Potter KN, Wheatley I, Neville LA, Packham G, Stevenson FK. Reversible anergy of sIgM-mediated signaling in the two subsets of CLL defined by VH-gene mutational status. Blood (2007) 109(10):4424–31. doi: 10.1182/blood-2006-11-056648
40. Herve M, Xu K, Ng YS, Wardemann H, Albesiano E, Messmer BT, et al. Unmutated and mutated chronic lymphocytic leukemias derive from self-reactive B cell precursors despite expressing different antibody reactivity. J Clin Invest (2005) 115(6):1636–43. doi: 10.1172/JCI24387
41. Packham G, Krysov S, Allen A, Savelyeva N, Steele AJ, Forconi F, et al. The outcome of B-cell receptor signaling in chronic lymphocytic leukemia: proliferation or anergy. Haematologica (2014) 99(7):1138–48. doi: 10.3324/haematol.2013.098384
42. Chen L, Huynh L, Apgar J, Tang L, Rassenti L, Weiss A, et al. ZAP-70 enhances IgM signaling independent of its kinase activity in chronic lymphocytic leukemia. Blood (2008) 111(5):2685–92. doi: 10.1182/blood-2006-12-062265
43. Crassini K, Shen Y, Mulligan S, Giles Best O. Modeling the chronic lymphocytic leukemia microenvironment in vitro. Leuk Lymphoma (2017) 58(2):266–79. doi: 10.1080/10428194.2016.1204654
44. Chen L, Widhopf G, Huynh L, Rassenti L, Rai KR, Weiss A, et al. Expression of ZAP-70 is associated with increased B-cell receptor signaling in chronic lymphocytic leukemia. Blood (2002) 100(13):4609–14. doi: 10.1182/blood-2002-06-1683
45. Chen L, Apgar J, Huynh L, Dicker F, Giago-McGahan T, Rassenti L, et al. ZAP-70 directly enhances IgM signaling in chronic lymphocytic leukemia. Blood (2005) 105(5):2036–41. doi: 10.1182/blood-2004-05-1715
46. Allsup DJ, Kamiguti AS, Lin K, Sherrington PD, Matrai Z, Slupsky JR, et al. B-cell receptor translocation to lipid rafts and associated signaling differ between prognostically important subgroups of chronic lymphocytic leukemia. Cancer Res (2005) 65(16):7328–37. doi: 10.1158/0008-5472.CAN-03-1563
47. Monroe JG. Ligand-independent tonic signaling in B-cell receptor function. Curr Opin Immunol (2004) 16(3):288–95. doi: 10.1016/j.coi.2004.03.010
48. Yasuda S, Zhou Y, Wang Y, Yamamura M, Wang JY. A model integrating tonic and antigen-triggered BCR signals to predict the survival of primary B cells. Sci Rep (2017) 7(1):14888. doi: 10.1038/s41598-017-13993-x
49. Messmer BT, Albesiano E, Efremov DG, Ghiotto F, Allen SL, Kolitz J, et al. Multiple distinct sets of stereotyped antigen receptors indicate a role for antigen in promoting chronic lymphocytic leukemia. J Exp Med (2004) 200(4):519–25. doi: 10.1084/jem.20040544
50. Hoogeboom R, van Kessel KP, Hochstenbach F, Wormhoudt TA, Reinten RJ, Wagner K, et al. A mutated B cell chronic lymphocytic leukemia subset that recognizes and responds to fungi. J Exp Med (2013) 210(1):59–70. doi: 10.1084/jem.20121801
51. Jimenez de Oya N, De Giovanni M, Fioravanti J, Ubelhart R, Di Lucia P, Fiocchi A, et al. Pathogen-specific B-cell receptors drive chronic lymphocytic leukemia by light-chain-dependent cross-reaction with autoantigens. EMBO Mol Med (2017) 9(11):1482–90. doi: 10.15252/emmm.201707732
52. Catera R, Silverman GJ, Hatzi K, Seiler T, Didier S, Zhang L, et al. Chronic lymphocytic leukemia cells recognize conserved epitopes associated with apoptosis and oxidation. Mol Med (2008) 14(11-12):665–74. doi: 10.2119/2008-00102.Catera
53. Chu CC, Catera R, Hatzi K, Yan XJ, Zhang L, Wang XB, et al. Chronic lymphocytic leukemia antibodies with a common stereotypic rearrangement recognize nonmuscle myosin heavy chain IIA. Blood (2008) 112(13):5122–9. doi: 10.1182/blood-2008-06-162024
54. Lanemo Myhrinder A, Hellqvist E, Sidorova E, Soderberg A, Baxendale H, Dahle C, et al. A new perspective: molecular motifs on oxidized LDL, apoptotic cells, and bacteria are targets for chronic lymphocytic leukemia antibodies. Blood (2008) 111(7):3838–48. doi: 10.1182/blood-2007-11-125450
55. Binder M, Lechenne B, Ummanni R, Scharf C, Balabanov S, Trusch M, et al. Stereotypical chronic lymphocytic leukemia B-cell receptors recognize survival promoting antigens on stromal cells. PloS One (2010) 5(12):e15992. doi: 10.1371/journal.pone.0015992
56. Radic M, Marion T, Monestier M. Nucleosomes are exposed at the cell surface in apoptosis. J Immunol (2004) 172(11):6692–700. doi: 10.4049/jimmunol.172.11.6692
57. Chang MK, Binder CJ, Miller YI, Subbanagounder G, Silverman GJ, Berliner JA, et al. Apoptotic cells with oxidation-specific epitopes are immunogenic and proinflammatory. J Exp Med (2004) 200(11):1359–70. doi: 10.1084/jem.20031763
58. Zwick C, Fadle N, Regitz E, Kemele M, Stilgenbauer S, Buhler A, et al. Autoantigenic targets of B-cell receptors derived from chronic lymphocytic leukemias bind to and induce proliferation of leukemic cells. Blood (2013) 121(23):4708–17. doi: 10.1182/blood-2012-08-447904
59. Steininger C, Widhopf GF,2, Ghia EM, Morello CS, Vanura K, Sanders R, et al. Recombinant antibodies encoded by IGHV1-69 react with pUL32, a phosphoprotein of cytomegalovirus and B-cell superantigen. Blood (2012) 119(10):2293–301. doi: 10.1182/blood-2011-08-374058
60. Hoogeboom R, Wormhoudt TA, Schipperus MR, Langerak AW, Dunn-Walters DK, Guikema JE, et al. A novel chronic lymphocytic leukemia subset expressing mutated IGHV3-7-encoded rheumatoid factor B-cell receptors that are functionally proficient. Leukemia (2013) 27(3):738–40. doi: 10.1038/leu.2012.238
61. Kostareli E, Gounari M, Janus A, Murray F, Brochet X, Giudicelli V, et al. Antigen receptor stereotypy across B-cell lymphoproliferations: the case of IGHV4-59/IGKV3-20 receptors with rheumatoid factor activity. Leukemia (2012) 26(5):1127–31. doi: 10.1038/leu.2011.311
62. Iacovelli S, Hug E, Bennardo S, Duehren-von Minden M, Gobessi S, Rinaldi A, et al. Two types of BCR interactions are positively selected during leukemia development in the Emu-TCL1 transgenic mouse model of CLL. Blood (2015) 125(10):1578–88. doi: 10.1182/blood-2014-07-587790
63. Slupsky JR. Does B cell receptor signaling in chronic lymphocytic leukaemia cells differ from that in other B cell types? Scientifica (Cairo) (2014) 2014:208928. doi: 10.1155/2014/208928
64. Apollonio B, Scielzo C, Bertilaccio MT, Ten Hacken E, Scarfo L, Ranghetti P, et al. Targeting B-cell anergy in chronic lymphocytic leukemia. Blood (2013) 121(19):3879–3888, S3871-3878. doi: 10.1182/blood-2012-12-474718
65. Garcia-Munoz R, Galiacho VR, Llorente L. Immunological aspects in chronic lymphocytic leukemia (CLL) development. Ann Hematol (2012) 91(7):981–96. doi: 10.1007/s00277-012-1460-z
66. Hippen KL, Tze LE, Behrens TW. CD5 maintains tolerance in anergic B cells. J Exp Med (2000) 191(5):883–90. doi: 10.1084/jem.191.5.883
67. Muzio M, Apollonio B, Scielzo C, Frenquelli M, Vandoni I, Boussiotis V, et al. Constitutive activation of distinct BCR-signaling pathways in a subset of CLL patients: a molecular signature of anergy. Blood (2008) 112(1):188–95. doi: 10.1182/blood-2007-09-111344
68. Tibaldi E, Pagano MA, Frezzato F, Trimarco V, Facco M, Zagotto G, et al. Targeted activation of the SHP-1/PP2A signaling axis elicits apoptosis of chronic lymphocytic leukemia cells. Haematologica (2017) 102(8):1401–12. doi: 10.3324/haematol.2016.155747
69. Greaves M. Clonal expansion in B-CLL: fungal drivers or self-service? J Exp Med (2013) 210(1):1–3. doi: 10.1084/jem.20122739
70. Duhren-von Minden M, Ubelhart R, Schneider D, Wossning T, Bach MP, Buchner M, et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature (2012) 489(7415):309–12. doi: 10.1038/nature11309
71. Ziegler CGK, Kim J, Piersanti K, Oyler-Yaniv A, Argyropoulos KV, van den Brink MRM, et al. Constitutive Activation of the B Cell Receptor Underlies Dysfunctional Signaling in Chronic Lymphocytic Leukemia. Cell Rep (2019) 28(4):923–937 e923. doi: 10.1016/j.celrep.2019.06.069
72. Gomes de Castro MA, Wildhagen H, Sograte-Idrissi S, Hitzing C, Binder M, Trepel M, et al. Differential organization of tonic and chronic B cell antigen receptors in the plasma membrane. Nat Commun (2019) 10(1):820. doi: 10.1038/s41467-019-08677-1
73. Zikherman J, Parameswaran R, Weiss A. Endogenous antigen tunes the responsiveness of naive B cells but not T cells. Nature (2012) 489(7414):160–4. doi: 10.1038/nature11311
74. O’Brien SM, Byrd JC, Hillmen P, Coutre S, Brown JR, Barr PM, et al. Outcomes with ibrutinib by line of therapy and post-ibrutinib discontinuation in patients with chronic lymphocytic leukemia: Phase 3 analysis. Am J Hematol (2019) 94(5):554–62. doi: 10.1002/ajh.25436
75. Herman SE, Gordon AL, Wagner AJ, Heerema NA, Zhao W, Flynn JM, et al. Phosphatidylinositol 3-kinase-delta inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood (2010) 116(12):2078–88. doi: 10.1182/blood-2010-02-271171
76. Buchner M, Baer C, Prinz G, Dierks C, Burger M, Zenz T, et al. Spleen tyrosine kinase inhibition prevents chemokine- and integrin-mediated stromal protective effects in chronic lymphocytic leukemia. Blood (2010) 115(22):4497–506. doi: 10.1182/blood-2009-07-233692
77. Byrd JC, Brown JR, O’Brien S, Barrientos JC, Kay NE, Reddy NM, et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med (2014) 371(3):213–23. doi: 10.1056/NEJMoa1400376
78. Brown JR, Byrd JC, Coutre SE, Benson DM, Flinn IW, Wagner-Johnston ND, et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110delta, for relapsed/refractory chronic lymphocytic leukemia. Blood (2014) 123(22):3390–7. doi: 10.1182/blood-2013-11-535047
79. Ponader S, Chen SS, Buggy JJ, Balakrishnan K, Gandhi V, Wierda WG, et al. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood (2012) 119(5):1182–9. doi: 10.1182/blood-2011-10-386417
80. de Rooij MF, Kuil A, Geest CR, Eldering E, Chang BY, Buggy JJ, et al. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood (2012) 119(11):2590–4. doi: 10.1182/blood-2011-11-390989
81. Burger JA, Li KW, Keating MJ, Sivina M, Amer AM, Garg N, et al. Leukemia cell proliferation and death in chronic lymphocytic leukemia patients on therapy with the BTK inhibitor ibrutinib. JCI Insight (2017) 2(2):e89904. doi: 10.1172/jci.insight.89904
82. Morande PE, Sivina M, Uriepero A, Seija N, Berca C, Fresia P, et al. Ibrutinib therapy downregulates AID enzyme and proliferative fractions in chronic lymphocytic leukemia. Blood (2019) 133(19):2056–68. doi: 10.1182/blood-2018-09-876292
83. Mertens D, Stilgenbauer S. Ibrutinib-resistant CLL: unwanted and unwonted! Blood (2017) 129(11):1407–9. doi: 10.1182/blood-2017-01-761536
84. von Bergwelt-Baildon M, Maecker B, Schultze J, Gribben JG. CD40 activation: potential for specific immunotherapy in B-CLL. Ann Oncol (2004) 15(6):853–7. doi: 10.1093/annonc/mdh213
85. Neron S, Nadeau PJ, Darveau A, Leblanc JF. Tuning of CD40-CD154 interactions in human B-lymphocyte activation: a broad array of in vitro models for a complex in vivo situation. Arch Immunol Ther Exp (Warsz) (2011) 59(1):25–40. doi: 10.1007/s00005-010-0108-8
86. Chatzigeorgiou A, Lyberi M, Chatzilymperis G, Nezos A, Kamper E. CD40/CD40L signaling and its implication in health and disease. Biofactors (2009) 35(6):474–83. doi: 10.1002/biof.62
87. Tretter T, Schuler M, Schneller F, Brass U, Esswein M, Aman MJ, et al. Direct cellular interaction with activated CD4(+) T cells overcomes hyporesponsiveness of B-cell chronic lymphocytic leukemia in vitro. Cell Immunol (1998) 189(1):41–50. doi: 10.1006/cimm.1998.1360
88. Schattner EJ, Mascarenhas J, Reyfman I, Koshy M, Woo C, Friedman SM, et al. Chronic lymphocytic leukemia B cells can express CD40 ligand and demonstrate T-cell type costimulatory capacity. Blood (1998) 91(8):2689–97. doi: 10.1182/blood.V91.8.2689.2689_2689_2697
89. Cols M, Barra CM, He B, Puga I, Xu W, Chiu A, et al. Stromal endothelial cells establish a bidirectional crosstalk with chronic lymphocytic leukemia cells through the TNF-related factors BAFF, APRIL, and CD40L. J Immunol (2012) 188(12):6071–83. doi: 10.4049/jimmunol.1102066
90. Pizzolo G, Chilosi M, Ambrosetti A, Semenzato G, Fiore-Donati L, Perona G. Immunohistologic study of bone marrow involvement in B-chronic lymphocytic leukemia. Blood (1983) 62(6):1289–96. doi: 10.1182/blood.V62.6.1289.bloodjournal6261289
91. Patten PE, Buggins AG, Richards J, Wotherspoon A, Salisbury J, Mufti GJ, et al. CD38 expression in chronic lymphocytic leukemia is regulated by the tumor microenvironment. Blood (2008) 111(10):5173–81. doi: 10.1182/blood-2007-08-108605
92. Han S, Hathcock K, Zheng B, Kepler TB, Hodes R, Kelsoe G. Cellular interaction in germinal centers. Roles of CD40 ligand and B7-2 in established germinal centers. J Immunol (1995) 155(2):556–67.
93. Os A, Burgler S, Ribes AP, Funderud A, Wang D, Thompson KM, et al. Chronic lymphocytic leukemia cells are activated and proliferate in response to specific T helper cells. Cell Rep (2013) 4(3):566–77. doi: 10.1016/j.celrep.2013.07.011
94. Bagnara D, Kaufman MS, Calissano C, Marsilio S, Patten PE, Simone R, et al. A novel adoptive transfer model of chronic lymphocytic leukemia suggests a key role for T lymphocytes in the disease. Blood (2011) 117(20):5463–72. doi: 10.1182/blood-2010-12-324210
95. Hatzivassiliou E, Miller WE, Raab-Traub N, Kieff E, Mosialos G. A fusion of the EBV latent membrane protein-1 (LMP1) transmembrane domains to the CD40 cytoplasmic domain is similar to LMP1 in constitutive activation of epidermal growth factor receptor expression, nuclear factor-kappa B, and stress-activated protein kinase. J Immunol (1998) 160(3):1116–21.
96. Homig-Holzel C, Hojer C, Rastelli J, Casola S, Strobl LJ, Muller W, et al. Constitutive CD40 signaling in B cells selectively activates the noncanonical NF-kappaB pathway and promotes lymphomagenesis. J Exp Med (2008) 205(6):1317–29. doi: 10.1084/jem.20080238
97. Longo PG, Laurenti L, Gobessi S, Petlickovski A, Pelosi M, Chiusolo P, et al. The Akt signaling pathway determines the different proliferative capacity of chronic lymphocytic leukemia B-cells from patients with progressive and stable disease. Leukemia (2007) 21(1):110–20. doi: 10.1038/sj.leu.2404417
98. Liang X, Moseman EA, Farrar MA, Bachanova V, Weisdorf DJ, Blazar BR, et al. Toll-like receptor 9 signaling by CpG-B oligodeoxynucleotides induces an apoptotic pathway in human chronic lymphocytic leukemia B cells. Blood (2010) 115(24):5041–52. doi: 10.1182/blood-2009-03-213363
99. Jahrsdorfer B, Muhlenhoff L, Blackwell SE, Wagner M, Poeck H, Hartmann E, et al. B-cell lymphomas differ in their responsiveness to CpG oligodeoxynucleotides. Clin Cancer Res (2005) 11(4):1490–9. doi: 10.1158/1078-0432.CCR-04-1890
100. Chu CC, Catera R, Zhang L, Didier S, Agagnina BM, Damle RN, et al. Many chronic lymphocytic leukemia antibodies recognize apoptotic cells with exposed nonmuscle myosin heavy chain IIA: implications for patient outcome and cell of origin. Blood (2010) 115(19):3907–15. doi: 10.1182/blood-2009-09-244251
101. Bertilaccio MT, Simonetti G, Dagklis A, Rocchi M, Rodriguez TV, Apollonio B, et al. Lack of TIR8/SIGIRR triggers progression of chronic lymphocytic leukemia in mouse models. Blood (2011) 118(3):660–9. doi: 10.1182/blood-2011-01-329870
102. Anderson LA, Landgren O, Engels EA. Common community acquired infections and subsequent risk of chronic lymphocytic leukaemia. Br J Haematol (2009) 147(4):444–9. doi: 10.1111/j.1365-2141.2009.07849.x
103. Landgren O, Gridley G, Check D, Caporaso NE, Morris Brown L. Acquired immune-related and inflammatory conditions and subsequent chronic lymphocytic leukaemia. Br J Haematol (2007) 139(5):791–8. doi: 10.1111/j.1365-2141.2007.06859.x
104. Landgren O, Rapkin JS, Caporaso NE, Mellemkjaer L, Gridley G, Goldin LR, et al. Respiratory tract infections and subsequent risk of chronic lymphocytic leukemia. Blood (2007) 109(5):2198–201. doi: 10.1182/blood-2006-08-044008
105. Hamilton E, Pearce L, Morgan L, Robinson S, Ware V, Brennan P, et al. Mimicking the tumour microenvironment: three different co-culture systems induce a similar phenotype but distinct proliferative signals in primary chronic lymphocytic leukaemia cells. Br J Haematol (2012) 158(5):589–99. doi: 10.1111/j.1365-2141.2012.09191.x
106. Asslaber D, Grossinger EM, Girbl T, Hofbauer SW, Egle A, Weiss L, et al. Mimicking the microenvironment in chronic lymphocytic leukaemia - where does the journey go? Br J Haematol (2013) 160(5):711–4. doi: 10.1111/bjh.12151
107. Slinger E, Thijssen R, Kater AP, Eldering E. Targeting antigen-independent proliferation in chronic lymphocytic leukemia through differential kinase inhibition. Leukemia (2017) 31(12):2601–7. doi: 10.1038/leu.2017.129
108. D’Avola A, Drennan S, Tracy I, Henderson I, Chiecchio L, Larrayoz M, et al. Surface IgM expression and function are associated with clinical behavior, genetic abnormalities, and DNA methylation in CLL. Blood (2016) 128(6):816–26. doi: 10.1182/blood-2016-03-707786
109. Rombout A, Lust S, Offner F, Naessens E, Verhasselt B, Philippe J. Mimicking the tumour microenvironment of chronic lymphocytic leukaemia in vitro critically depends on the type of B-cell receptor stimulation. Br J Cancer (2016) 114(6):704–12. doi: 10.1038/bjc.2016.35
110. Petlickovski A, Laurenti L, Li X, Marietti S, Chiusolo P, Sica S, et al. Sustained signaling through the B-cell receptor induces Mcl-1 and promotes survival of chronic lymphocytic leukemia B cells. Blood (2005) 105(12):4820–7. doi: 10.1182/blood-2004-07-2669
111. Igawa T, Sato Y, Takata K, Fushimi S, Tamura M, Nakamura N, et al. Cyclin D2 is overexpressed in proliferation centers of chronic lymphocytic leukemia/small lymphocytic lymphoma. Cancer Sci (2011) 102(11):2103–7. doi: 10.1111/j.1349-7006.2011.02046.x
112. Aguilar-Hernandez MM, Blunt MD, Dobson R, Yeomans A, Thirdborough S, Larrayoz M, et al. IL-4 enhances expression and function of surface IgM in CLL cells. Blood (2016) 127(24):3015–25. doi: 10.1182/blood-2015-11-682906
113. Banchereau J, de Paoli P, Valle A, Garcia E, Rousset F. Long-term human B cell lines dependent on interleukin-4 and antibody to CD40. Science (1991) 251(4989):70–2. doi: 10.1126/science.1702555
114. Garrone P, Neidhardt EM, Garcia E, Galibert L, van Kooten C, Banchereau J. Fas ligation induces apoptosis of CD40-activated human B lymphocytes. J Exp Med (1995) 182(5):1265–73. doi: 10.1084/jem.182.5.1265
115. de Totero D, Meazza R, Zupo S, Cutrona G, Matis S, Colombo M, et al. Interleukin-21 receptor (IL-21R) is up-regulated by CD40 triggering and mediates proapoptotic signals in chronic lymphocytic leukemia B cells. Blood (2006) 107(9):3708–15. doi: 10.1182/blood-2005-09-3535
116. Chapman EA, Oates M, Mohammad IS, Davies BR, Stockman PK, Zhuang J, et al. Delineating the distinct role of AKT in mediating cell survival and proliferation induced by CD154 and IL-4/IL-21 in chronic lymphocytic leukemia. Oncotarget (2017) 8(61):102948–64. doi: 10.18632/oncotarget.22292
117. Ahearne MJ, Willimott S, Pinon L, Kennedy DB, Miall F, Dyer MJ, et al. Enhancement of CD154/IL4 proliferation by the T follicular helper (Tfh) cytokine, IL21 and increased numbers of circulating cells resembling Tfh cells in chronic lymphocytic leukaemia. Br J Haematol (2013) 162(3):360–70. doi: 10.1111/bjh.12401
118. Plander M, Seegers S, Ugocsai P, Diermeier-Daucher S, Ivanyi J, Schmitz G, et al. Different proliferative and survival capacity of CLL-cells in a newly established in vitro model for pseudofollicles. Leukemia (2009) 23(11):2118–28. doi: 10.1038/leu.2009.145
119. Parente-Ribes A, Skanland SS, Burgler S, Os A, Wang D, Bogen B, et al. Spleen tyrosine kinase inhibitors reduce CD40L-induced proliferation of chronic lymphocytic leukemia cells but not normal B cells. Haematologica (2016) 101(2):e59–62. doi: 10.3324/haematol.2015.135590
120. Zent CS, Smith BJ, Ballas ZK, Wooldridge JE, Link BK, Call TG, et al. Phase I clinical trial of CpG oligonucleotide 7909 (PF-03512676) in patients with previously treated chronic lymphocytic leukemia. Leuk Lymphoma (2012) 53(2):211–7. doi: 10.3109/10428194.2011.608451
121. Jahrsdorfer B, Blackwell S, Weiner G. The effects of CpG ODN on CLL proliferation, apoptosis or phenotype could have an impact on its clinical utility. Leukemia (2007) 21(11):2354–2355; author reply 2355-2356. doi: 10.1038/sj.leu.2404870
122. Jahrsdorfer B, Hartmann G, Racila E, Jackson W, Muhlenhoff L, Meinhardt G, et al. CpG DNA increases primary malignant B cell expression of costimulatory molecules and target antigens. J Leukoc Biol (2001) 69(1):81–8.
123. Ghalamfarsa G, Jadidi-Niaragh F, Hojjat-Farsangi M, Asgarian-Omran H, Yousefi M, Tahmasebi F, et al. Differential regulation of B-cell proliferation by IL21 in different subsets of chronic lymphocytic leukemia. Cytokine (2013) 62(3):439–45. doi: 10.1016/j.cyto.2013.03.023
124. Tarnani M, Laurenti L, Longo PG, Piccirillo N, Gobessi S, Mannocci A, et al. The proliferative response to CpG-ODN stimulation predicts PFS, TTT and OS in patients with chronic lymphocytic leukemia. Leuk Res (2010) 34(9):1189–94. doi: 10.1016/j.leukres.2009.12.020
125. Gupta R, Yan XJ, Barrientos J, Kolitz JE, Allen SL, Rai K, et al. Mechanistic Insights into CpG DNA and IL-15 Synergy in Promoting B Cell Chronic Lymphocytic Leukemia Clonal Expansion. J Immunol (2018) 201(5):1570–85. doi: 10.4049/jimmunol.1800591
126. Jahrsdorfer B, Blackwell SE, Wooldridge JE, Huang J, Andreski MW, Jacobus LS, et al. B-chronic lymphocytic leukemia cells and other B cells can produce granzyme B and gain cytotoxic potential after interleukin-21-based activation. Blood (2006) 108(8):2712–9. doi: 10.1182/blood-2006-03-014001
127. Wagner M, Oelsner M, Moore A, Gotte F, Kuhn PH, Haferlach T, et al. Integration of innate into adaptive immune responses in ZAP-70-positive chronic lymphocytic leukemia. Blood (2016) 127(4):436–48. doi: 10.1182/blood-2015-05-646935
128. Decker T, Schneller F, Sparwasser T, Tretter T, Lipford GB, Wagner H, et al. Immunostimulatory CpG-oligonucleotides cause proliferation, cytokine production, and an immunogenic phenotype in chronic lymphocytic leukemia B cells. Blood (2000) 95(3):999–1006. doi: 10.1182/blood.V95.3.999.003k10_999_1006
129. Hagn M, Blackwell SE, Beyer T, Ebel V, Fabricius D, Lindner S, et al. B-CLL cells acquire APC- and CTL-like phenotypic characteristics after stimulation with CpG ODN and IL-21. Int Immunol (2014) 26(7):383–95. doi: 10.1093/intimm/dxu001
130. Tromp JM, Tonino SH, Elias JA, Jaspers A, Luijks DM, Kater AP, et al. Dichotomy in NF-kappaB signaling and chemoresistance in immunoglobulin variable heavy-chain-mutated versus unmutated CLL cells upon CD40/TLR9 triggering. Oncogene (2010) 29(36):5071–82. doi: 10.1038/onc.2010.248
131. Guo A, Lu P, Galanina N, Nabhan C, Smith SM, Coleman M, et al. Heightened BTK-dependent cell proliferation in unmutated chronic lymphocytic leukemia confers increased sensitivity to ibrutinib. Oncotarget (2016) 7(4):4598–610. doi: 10.18632/oncotarget.6727
132. Phelan JD, Young RM, Webster DE, Roulland S, Wright GW, Kasbekar M, et al. A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature (2018) 560(7718):387–91. doi: 10.1038/s41586-018-0290-0
133. Pawson T, Warner N. Oncogenic re-wiring of cellular signaling pathways. Oncogene (2007) 26(9):1268–75. doi: 10.1038/sj.onc.1210255
134. Linley A, Griffin R, Cicconi S, D’Avola A, Macewan D, Pettitt AR, et al. Kinobead profiling reveals reprogramming of B-cell receptor signaling in response to therapy within primary CLL cells. bioRxiv (2019). doi: 10.1101/841312
135. Aljitawi OS, Li D, Xiao Y, Zhang D, Ramachandran K, Stehno-Bittel L, et al. A novel three-dimensional stromal-based model for in vitro chemotherapy sensitivity testing of leukemia cells. Leuk Lymphoma (2014) 55(2):378–91. doi: 10.3109/10428194.2013.793323
Keywords: chronic lymphocytic leukemia, proliferation, micro-environment, CD40, toll-like receptor, crosstalk
Citation: Haselager MV, Kater AP and Eldering E (2020) Proliferative Signals in Chronic Lymphocytic Leukemia; What Are We Missing? Front. Oncol. 10:592205. doi: 10.3389/fonc.2020.592205
Received: 06 August 2020; Accepted: 18 September 2020;
Published: 08 October 2020.
Edited by:
Martina Seiffert, German Cancer Research Center (DKFZ), GermanyReviewed by:
Dimitar G. Efremov, International Centre for Genetic Engineering and Biotechnology, ItalyCopyright © 2020 Haselager, Kater and Eldering. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric Eldering, ZS5lbGRlcmluZ0BhbXN0ZXJkYW11bWMubmw=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.