 Mario Capasso1,2*
Mario Capasso1,2* Annalaura Montella2
Annalaura Montella2 Matilde Tirelli2,3
Matilde Tirelli2,3 Teresa Maiorino1,2
Teresa Maiorino1,2 Sueva Cantalupo1,2
Sueva Cantalupo1,2 Achille Iolascon1,2
Achille Iolascon1,2- 1Dipartimento di Medicina Molecolare e Biotecnologie Mediche, Università degli Studi di Napoli Federico II, Naples, Italy
- 2CEINGE Biotecnologie Avanzate, Naples, Italy
- 3European School of Molecular Medicine, Università Degli Studi di Milano, Milan, Italy
Progresses over the past years have extensively improved our capacity to use genome-scale analyses—including high-density genotyping and exome and genome sequencing—to identify the genetic basis of pediatric tumors. In particular, exome sequencing has contributed to the evidence that about 10% of children and adolescents with tumors have germline genetic variants associated with cancer predisposition. In this review, we provide an overview of genetic variations predisposing to solid pediatric tumors (medulloblastoma, ependymoma, astrocytoma, neuroblastoma, retinoblastoma, Wilms tumor, osteosarcoma, rhabdomyosarcoma, and Ewing sarcoma) and outline the biological processes affected by the involved mutated genes. A careful description of the genetic basis underlying a large number of syndromes associated with an increased risk of pediatric cancer is also reported. We place particular emphasis on the emerging view that interactions between germline and somatic alterations are a key determinant of cancer development. We propose future research directions, which focus on the biological function of pediatric risk alleles and on the potential links between the germline genome and somatic changes. Finally, the importance of developing new molecular diagnostic tests including all the identified risk germline mutations and of considering the genetic predisposition in screening tests and novel therapies is emphasized.
Introduction
Genomic sequencing studies have highlighted that pediatric cancers typically have few somatic mutations but a higher prevalence of germline alterations in cancer predisposition genes (1). The contribution of germline variants in pediatric tumors has been estimated between 8 and 12% (2, 3). Genetic variants are generally classified on the basis of their clinical effect: pathogenic variant means any sequence change that, differing from the consensus wild-type sequence, directly contributes to the development of the disease; likely pathogenic variants, instead, are genetic changes with a high likelihood of being disease-causing, but additional evidence is expected to confirm their clinical significance. Variant classification can arise from different methodologies and algorithms, which can assign different weights to collected data. However, studies cited in the present review generally refer to the American College of Medical Genetics and Genomics (ACMG) guidelines for variants interpretation (4). In this process, multiple categories of data (such as frequency in affected and unaffected populations, computational prediction tools, functional studies, and gene- or disease-specific information) are taken into account and combined to determine a variant pathogenicity classification.
It is also important to note that genetic variants can be detected through different genomic approaches and the type of identified alteration depends on the nature of the assay used. Large-scale genomic analyses such as whole-exome sequencing (WES) or whole-genome sequencing (WGS) can identify uncommon, moderate penetrant variants. Since WES investigates only the coding regions of the genome, it has proved very useful in detecting most of the causative variants of Mendelian diseases (5, 6). Furthermore, it has recently been used also to identify rare and uncommon causative mutations of complex diseases (7). On the other hand, WGS can capture nearly all known genetic variations, including those falling in regulatory elements, with much more uniform coverage of the genome, but it does not allow to detect mosaic variants with low clonality or variations causing DNA repetitions (8). Common, low-penetrance genetic variants, instead, are mostly identified by genome association study (GWAS), which assesses genotype–phenotype associations through testing of variants across genomes of many individuals, based on data obtained using numerous technologies, mostly WGS or genome-wide single-nucleotide polymorphism (SNP) arrays. Consequently, GWAS limitations are linked to the technology on which it is based: e.g., SNP array-based GWAS rely on pre-existing genetic variant reference panels (9). Finally, besides SNP array, copy-number variations (CNV) can be identified also through CGH array. Anyway, array methods cannot be used to detect single base pair changes, indels, balanced chromosome rearrangements, and low-percent mosaicism (10).
Recently, in addition to germline pathogenic and/or likely pathogenic variants in known cancer-predisposing genes, it has been estimated that a high percentage (61%) of children, adolescent and young adult patients with solid tumors carry germline pathogenic and likely pathogenic variants in new candidate genes, including PRKN, SMACAL1, SMAD7, and TMPRSS3 (3). The detection of cancer predisposition can lead to clinical benefits for patients, both for the molecular diagnosis and for the presence of specific biological features, as well as to eventually refine therapeutic choices. We provide an overview of the most significant knowledge of germline predisposition for the main pediatric solid tumors, which are central nervous system tumors (medulloblastoma, ependymoma and astrocytoma), neuroblastoma, retinoblastoma, Wilms tumor, osteosarcoma, rhabdomyosarcoma, and Ewing sarcoma, altogether accounting for 34.8% of all childhood cancers (Figure 1). Each tumor description is organized into two subsections: “familial cancer” and “sporadic tumor.” Familial cancer means a form of cancer that has higher incidence in families than in the general population due to rare, high-penetrance genetic variants. In this group, we also included rare genetic syndromes that are not usually considered as cancer syndromes but that predispose to the development of solid pediatric tumors. The second group, sporadic tumor, is referred to cancers which do not run in families and are intended as multifactorial diseases whose onset can be attributed to the combined effect of environmental and genetic factors. In sporadic cancers, genetic factors can be categorized into two types: uncommon, moderate-penetrance genetic variants, which for the studies considered in this review show a frequency lower than 1–0.001% in the general population and are not so rare as those associated with familial cancer, and common, low-penetrance genetic variants.
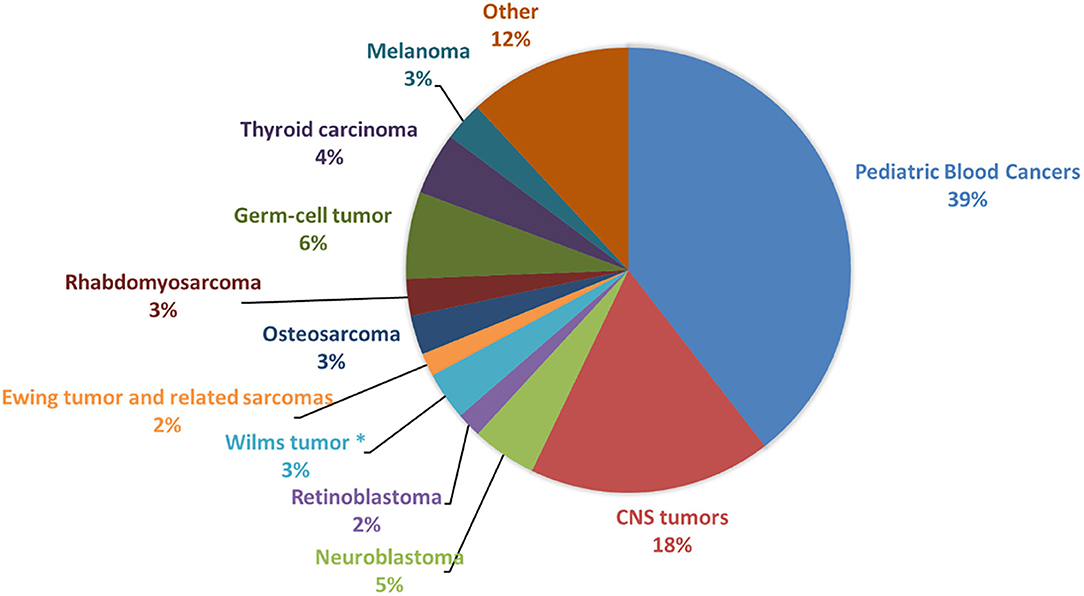
Figure 1. Frequency of pediatric cancers in patients younger than 19 years. The figure shows the prevalence of the main pediatric cancer types among patients younger than 19 years of age, calculated from Centers for Disease Control and Prevention (CDC) data (United States Cancer Statistics Data, https://wonder.cdc.gov/cancer.html) and based on incidence in United States for the years 1999–2016. CNS, Central Nervous System. *This frequency is related to Wilms tumor and other non-epithelial renal tumors.
The knowledge of genetic mutations responsible for syndromic disorders associated with the risk of developing pediatric cancer has greatly increased over the past years (11). Indeed, several tumor predisposing syndromes are the underlying cause of at least 8.5% of cancers in pediatric patients (12). Thus, the role of general practitioners and pediatricians in recognizing the major cancer genetic-associated syndromes, in making appropriate referrals for genetic counseling and testing when indicated, is crucial for a specific monitoring and management of the patient.
Most cancer susceptibility genes are involved in fundamental biological pathways such as cell-cycle control, chromatin remodeling, or DNA repair. Therefore, alterations in these genes compromise the normal control of cell growth and lead to a substantial increase in the risk of developing cancer. Another element of great interest discussed here is the presence of cooperation between germline and somatic alterations, which can represent an early tool for evaluating the clinical outcome and for the stratification of patients in risk subgroups. We also discuss evidence that points to a need for more collaborative investigations in identifying driver events in pediatric cancers.
Central Nervous System Tumors
Central nervous system (CNS) tumors represent the most frequent types of cancer in children aged 0–14 years, with a mortality rate of 0.72 per 100,000 population (13). The three most frequent tumors are medulloblastoma (MB), ependymoma (EP), and astrocytoma (AS) (Figure 2).
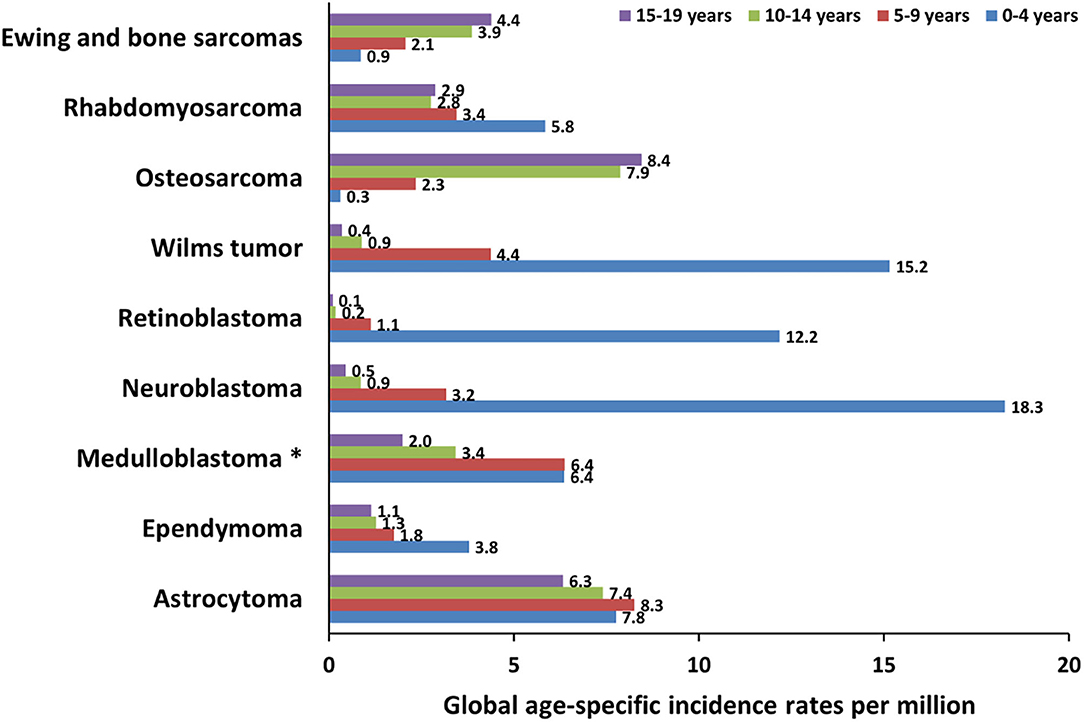
Figure 2. Global incidence of pediatric cancers in patients younger than 19 years. The graph shows the global age-specific incidence rates (ASR) per million for individual age groups (0–4 years, 5–9 years, 10–14 years, and 15–19 years) of pediatric cancer types discussed in this review. ASR reported next to the bars are calculated from International Incidence of Childhood Cancer (IICC, https://iicc.iarc.fr/) data. *These ASR include also less frequent embryonal central nervous system tumors.
Medulloblastoma
MB is an embryonal tumor of cerebellum (14) that affects children under the age of 14, with an average onset of about 6–8 years (Figure 2) and with a 5-year overall survival for standard-risk patients of 70–85% (14). It is classified into four genetic and molecular groups: the first two groups, WNT-activated (MBWNT) and Sonic Hedgehog activated (MBSHH), are named for the signaling pathways that play prominent roles in the pathogenesis of those subgroups, while, since less is known about the biology of the remaining two subgroups, they are numerically designated as “Group 3” and “Group 4” (14). Damaging germline mutations in known cancer-predisposing genes play an important role in two main subgroups, MBWNT and MBSHH, in which genetic testing is highly recommended (15). MBWNT is characterized at somatic level by activating mutations in exon 3 of β-catenin (CTNNB1) and monosomy of chromosome 6, while MBSHH by amplification of GLI2 and MYCN, as well as loss of 17p (16).
Familial Medulloblastoma
To date, only germline mutations in ELP1 have been found in two independent families with MBSHH (17). Although inherited or familial MB is extremely rare, there are few rare inherited syndromes that are associated with increased risk of developing this tumor (Table 2). Germline mutations of PTCH1 and SUFU, by causing activation of the SHH signaling pathway, predispose to MBSHH in Gorlin syndrome, an autosomal dominant disease caused by mutations in PTCH1 (67, 124). In Turcot syndrome, a rare disorder characterized by the association of colonic polyposis and primary brain tumors, germline mutations of APC predispose to the development of MBWNT (114). In MBWNT, activation of the WNT pathway is due to somatic mutations of CTNNB1 in most of tumors but it is also observed in patients with only germline mutations of APC, stressing the importance of genetic predisposition in high-risk patients (15, 114). Germline mutations in BRCA2 and PALB2, associated or not associated with Fanconi anemia, have been found in MBSHH (58, 125) and are often observed in association with somatic homologous recombination repair defects (15). The role of germline mutations in TP53 in MB is still widely debated today. TP53 germline mutations affect MB prognosis differently according to the different subgroups: germline mutations in MBSHH are associated with poor prognosis, while both germline and somatic mutations in MBWNT are associated with better prognosis. This may be due to a different origin of the MB itself (14). Patients with germline TP53 mutations can have tumors characterized by catastrophic DNA chromothripsis and are often associated with Li–Fraumeni syndrome (LFS), a cancer predisposition disorder caused by germline mutations of the tumor-suppressor p53 (71). Other MB-associated syndromes are Bloom's syndrome (31), ataxia telangiectasia (18), and Greig's cephalopolysyndactyly syndrome (14, 40, 45, 85, 122) (Table 2).
Sporadic Medulloblastoma
The association between MB and genetic syndromes explains most of the genetic predisposition to MB. However, sporadic forms are known in literature and are partially explained through uncommon, moderate penetrant mutations identified by whole-exome sequencing (WES) or whole-genome sequencing (WGS), or common, low-penetrance genetic variants identified by genome wide association study (GWAS) (Table 1 and Table 3).
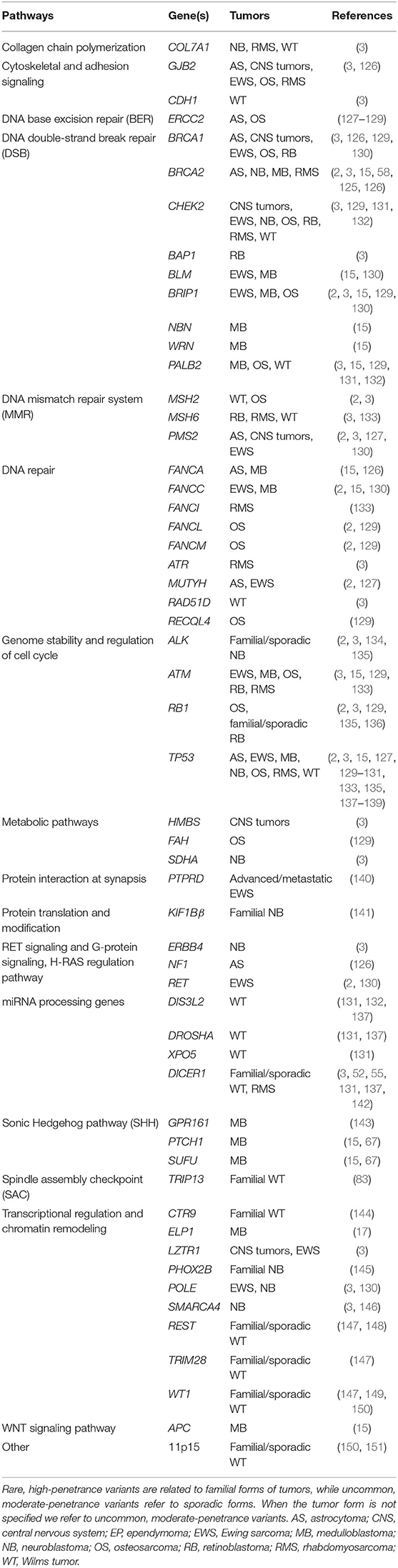
Table 1. Rare, high-penetrance, and uncommon, moderate-penetrance variants in genes predisposing to pediatric tumors and main biological pathways.
Uncommon, Moderate-Penetrance Variants
In a study on 1,022 MB patients, novel partial or total APC deletions were found (15). These mutations were not associated with any familial syndrome and predisposed to MBWNT. In the same study, 1% of patients (classified as MBSHH) had TP53 mutations but only 5/11 patients showed family history of cancer, emphasizing the role of TP53 germline mutations in predisposing to sporadic MB. Notably, germline missense, frameshift, or non-sense mutations in the DNA-binding domain of TP53 were found to be associated with a series of events at the somatic level such as rearrangements, chromothripsis, and loss of heterozygosity in MBSHH patients, whereas germline mutations in SUFU and PTCH1 co-occurred with somatic loss of heterozygosity (15) (Table 4). These results further provide evidence that novel associations between germline variants and specific somatic events, beyond those reported by Knudson in 1971, can play a role in carcinogenesis. Indeed, recent body of literature supports the hypothesis that specific germline variants determine which somatic events and mutations are generated and selected in cancer cells during tumorigenesis (179).
MB can also arise in patients with germline mutations in other known cancer genes such as ATM, FANCA, FANCC, NBN, WRN, BLM, and BRIP1 and in candidate genes like CHEK2, CREBBP, RAD51, ERCC2, and ERCC4. All of these genes are involved in cell-cycle regulation and DNA repair (15). Frameshift, protein-truncating, and missense mutations occurring in GPR161, a gene never previously associated with MB, were found in 6 MBSHH cases (143) that, at the somatic level, showed loss of heterozygosity with retention of the mutated allele, confirming its role as driver gene in MBSHH. GPR161 functions are essential for embryonic development and for the proliferation of granular cells (143). Germline mutations in ELP1 have been very recently found to predispose to MBSHH and to be associated with two consecutive somatic events: loss of the 9q arm, with consequent loss of the wild-type copy of PTCH1 and ELP1, and a second independent mutation event in PTCH1 (17) (Table 4). This study, importantly, showed that 40% of MBSHH patients carry disease-predisposing mutations and that genetic predisposition to proteome instability may be a determinant in the pathogenesis of pediatric brain cancers (17) (Table 1).
Common, Low-Penetrance Variants
To date, there are no relevant GWAS conducted to identify common variants associated with MB. Only one study has been performed in a small sample including 244 MB cases and 247 control subjects from Sweden and Denmark, but no locus reached the significance threshold (154). The most significant locus was 18p11.23 including PTPRM (154). A different approach that starts from the most frequently mutated genes in MB such as CCND2, CTNNB1, DDX3X, GLI2, SMARCA4, MYC, MYCN, PTCH1, TP53, and KMT2D was proposed to identify MB-associated common variants (162). Eight variants, located in CCND2, PTCH1, and GLI2, associated with the risk of developing MB (162) (Table 3). However, these findings need further validation in independent cohorts of cases and controls.
Microsatellites are tandem repeats of 1–6 base pairs, and their variability is associated with numerous tumors, including MB. In a recent work, starting from WES and WGS data, the authors developed an algorithm able to identify a signature of 43 microsatellites that distinguished with high-sensitivity and specificity MB subjects from controls in two independent sets of MB cases and controls (180). Interestingly, in silico analyses revealed that genes harboring these microsatellite loci had cellular functions important for tumorigenesis (180).
Other Brain Tumors
EP originates from the walls of the ventricular system (79), arises between 0 and 4 years (Figure 2) (79), and has a 5-year overall survival of about 60% (181). EP is diagnosed in ~33–53% of patients with type 2 neurofibromatosis, with high occurrence of truncating mutations in NF2 (97). EP has recently been associated with Kabuki syndrome, with mutations in KMT2D (70) and rarely occurs in Turcot and MEN1 syndromes with mutations in MSH2 and MEN1, respectively (79) (Table 2). To date, large studies on common variants and sporadic forms are lacking (Table 1). AS is classified into several forms including pilocytic, anaplastic, diffuse, and glioblastoma (182). Pilocytic AS is the most common form in children and young adults, with an average age at onset between 0 and 9 years (13) (Figure 2) and a 5-year survival of 94.1% (13). Regarding the genetic predisposition, one large study reported germline splicing mutations in the tumor-suppressor genes MUTYH and ERCC2 and point mutations in TP53 and PMS2 (127) (Table 1). Pathogenic mutations in NF1, BRCA2, FANCA, and GJB2 have been also identified in a recent study involving 280 patients with different forms of AS (126).
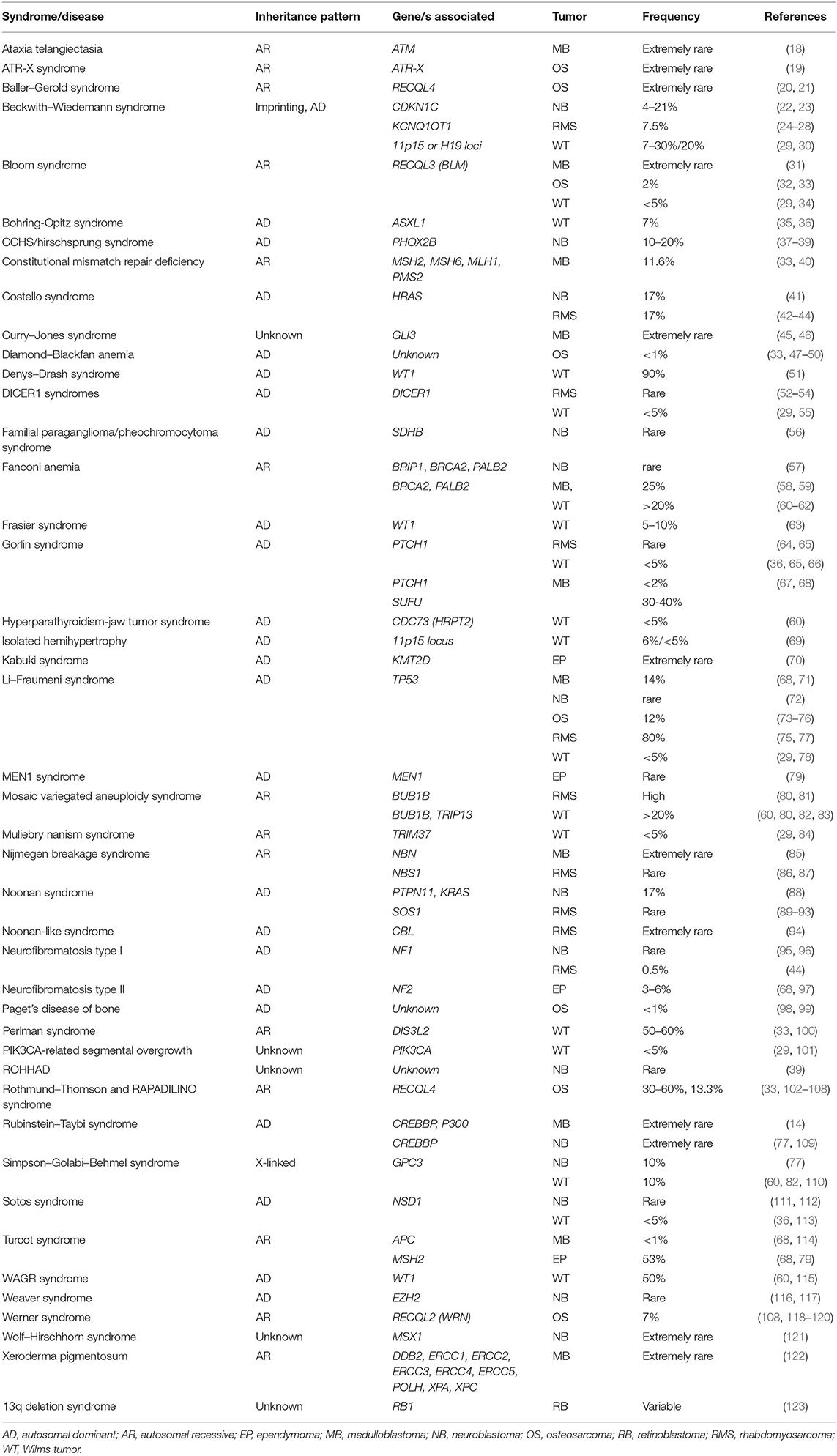
Table 2. Syndromes associated with pediatric tumors. Frequencies reported refer to the occurrence rate of pediatric cancers in patients with genetic syndromes.
Neuroblastoma
Neuroblastoma (NB) originates from neural crest cells and affects the nervous sympathetic system (183). NB exhibits unique features, such as early age of onset, high frequency of metastatic disease at diagnosis in patients over 1 year of age (Figure 2), and the tendency for spontaneous regression of tumors in infants. In high-risk cases, the survival rate is only 50% (183). NB tumors, as well as other pediatric cancers, present few recurrent somatic mutations but frequent chromosomic aberrations such MYCN amplification, 17q gain, 1p deletion, and 11q deletion (184).
Familial Neuroblastoma
Familial NB represents 1–2% of cases, with PHOX2B and ALK as major susceptibility genes (184) (Table 1). The first identified familial gene is PHOX2B (37, 145), already associated with congenital central hypoventilation syndrome (CCHS) (185) and encoding a transcription factor driving neural crest differentiation (186). NB-exclusive mutations are mainly missense and frameshift (187). PHOX2B germline mutations account for ~10% of familial NB (188), but this gene is also mutated in 2% of sporadic cases (189). Subsequently, the major susceptibility gene was identified in ALK. Its gain-of-function mutations, which account for 75% of familial cases (134, 188), are mainly located in the kinase domain of the encoded tyrosine kinase receptor and show incomplete penetrance (190). ALK somatic mutations are also reported in 10–12% of primary sporadic NB tumors (134, 191). Additional NB-predisposing genes have not yet been discovered. Mutations in KIF1Bβ (141) and GALNT14 (192) and in 16p12–13, 4p16, and 1p loci (193–195) (Table 1) have been reported in related patients, but further validations are needed.
Children suffering from specific cancer predisposition syndromes such as LFS and others (Table 2) show an increased NB risk (22, 38, 39, 41, 56, 57, 72, 77, 88, 95, 111, 116, 121). Thus, protocols for NB surveillance need to be established.
Sporadic Neuroblastoma
Only a small subset of sporadic NB cases has an identifiable somatic oncogenic point mutation (196, 197), suggesting that predisposing genetic factors found in GWAS studies could cooperate to increase disease occurrence (198, 199).
Uncommon, Moderate-Penetrance Variants
Recent studies focused on uncommon germline variants, which presumably have a larger effect on predisposition compared to common ones. In different studies, pathogenic and likely pathogenic variants were identified in predisposition genes such as ALK, CHEK2, BRCA2, SMARCA4, and TP53 (Table 1) but also in candidate genes like AXIN2, PALB2, BARD1, PINK1, APC, BRCA1, SDHB, and LZTR1 (2, 135, 146, 196, 197, 200) Specifically, TP53 variants are strongly associated with NB susceptibility (201). All the mentioned genes are involved in DNA repair and maintenance of genomic integrity (Table 1).
Common, Low-Penetrance Variants
GWAS studies identified several NB susceptibility loci (Table 3) including CASC15 (160), BARD1 (157), LMO1 (175), HACE1, and LIN28B (155) associated with high-risk NB, whereas DUSP12, HSD17B12, DDX4, and IL31RA associated with the low-risk NB group (161, 198). Functional studies of these loci have highlighted the key role of GWAS in elucidating NB carcinogenesis. A SNP in the long non-coding RNA (lcnRNA) CASC15 produces a truncated isoform, whose lower expression correlates with advanced disease (202). Loss of another lncRNA, NBAT-1, at the same locus, contributes to aggressive NB by increasing proliferation and impairing differentiation of neuronal precursors (203). Diverse functional studies have elucidated the role of BARD1 and its variants in NB development (204). Variants in the BARD1 promoter decrease the expression of the tumor-suppressor form which protects NB cells from DNA damage (205, 206), whereas variants in introns increase the expression of an oncogenic isoform, BARD1β, which stabilizes the Aurora kinases (207, 208). LMO1 decreased expression, caused by a variant in a super-enhancer which disrupts GATA binding (209), reduces NB cell proliferation. Finally, the activation of LIN28B, due to genetic variants, can enhance MYCN levels via let-7 microRNA suppression (155, 210, 211). The genetic landscape of sporadic NB has been amplified with the discovery of additional susceptibility genes including RSRC1/MLF1 and CPZ (159), SPAG16 (177), NEFL (156), and CDKN1B (170).
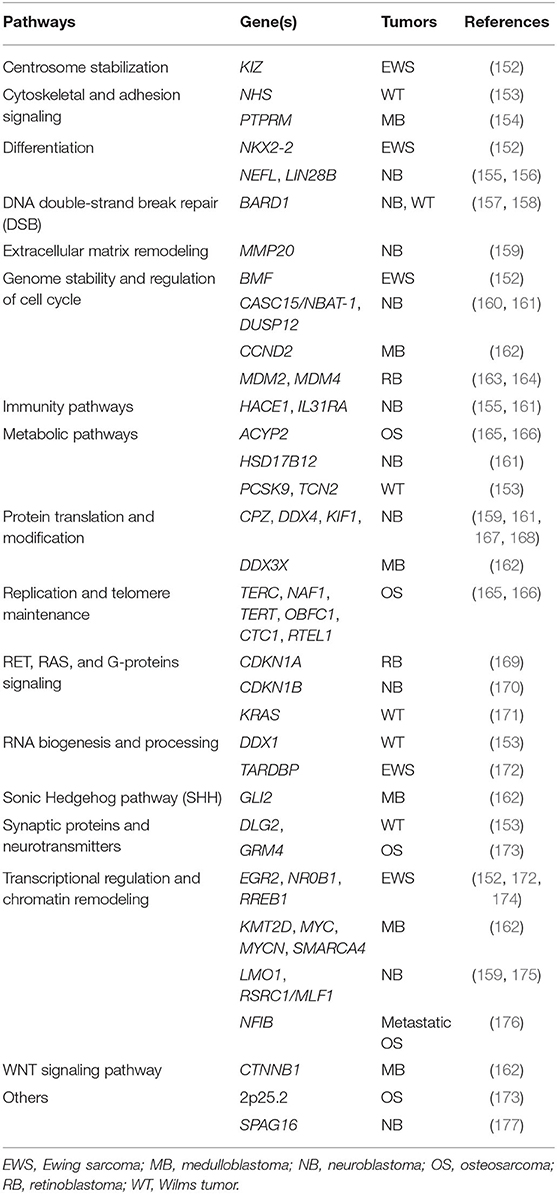
Table 3. Common, low-penetrance variants in genes predisposing to pediatric tumors and main biological pathways.
Reanalyses of GWAS data have discovered novel mechanisms and genetic factors that promote NB development (Table 3). Two studies clearly demonstrate a cooperation between predisposing variants and somatic aberrations in NB initiation (Table 4). Indeed, SNPs in MMP20 (167) and KIF15 (168) increase NB susceptibility in the presence of 11q deletion and MYCN amplification, respectively, whereas another study shows that specific mtDNA haplogroups can influence the risk of NB (212). We have provided evidence that SNPs in PARP1 and IL6 might be predictive biomarkers of response to chemotherapy and prognosis (213, 214). Finally, our recent works found that NB shares risk loci with other complex diseases and tumors. Indeed, SNPs in 2q35, 3q25.32, and 4p16.2 are cross-associated with congenital heart disease (CHD) and NB (215), while 1p13.2 showed cross-association with NB and melanoma (216). Very recently, a cross-match investigation between germline alterations in pediatric patients with different solid tumors and CHD-related genes has identified that NB is among the tumors with the highest enrichment of germline pathogenic and likely pathogenic variants in these genes (3).
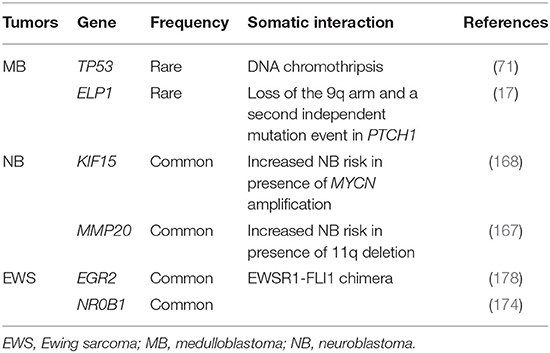
Table 4. Germline–somatic interactions identified in genes predisposing to pediatric tumors.
Constitutional Chromosomal Abnormalities
Highly associated with NB are hemizygous deletion in 1q21.1, disruption in NBPF23 (217), and microdeletion in 16p11.2, containing SEZ6L2 and PRRT2 (218). Deletion including SLFN11, duplication of SOX4, and partial deletion of PARK2 have been identified in three different patients, respectively (219).
Retinoblastoma
Retinoblastoma (RB) is a pediatric malignancy of the neural retina, commonly initiated by biallelic inactivation of RB1 (220) and affecting one (unilateral) or both eyes (bilateral). The median age at diagnosis is 12 months in bilateral tumors and 24 months in unilateral ones (220) (Figure 2). Patient survival is >95% in high-income countries but <30% globally (220). The first studies on RB unveiled the importance of genetics in cancer; indeed, the “two-hit hypothesis” formulated by Knudson (221) on RB1 has been paradigmatic for the understanding of tumor-suppressor genes and the study of familial cancers.
Familial Retinoblastoma
Hereditary RB encompasses about 40% of all cases with most having bilateral tumors, 15% unilateral, and 5% trilateral (associated with midline brain tumor) (220). Familial RB is distinctly associated with the RB1 tumor-suppressor gene, which encodes pRB, a crucial regulator of the cell cycle. Germline mutations in RB1 are inherited in 25% of cases in an autosomal-dominant manner. A broad spectrum of inactivating RB1 germline mutations have been described, mainly nonsense and frameshifts affecting the coding region, few large deletions, and <5% silencing gene promoter (136). Penetrance and expressivity can vary within families due to partially functional RB1 alleles (222, 223) or parent-of-origin effect (224). Influence of genetic modifiers such as MDM2, MDM4 (225, 226), or MED4 (227) and polymorphisms in p53 (228), CDKN1A (169), and CDKN2A (229) could also influence RB development. Reduced MDM2 and MDM4 expression may increase the RB1 haploinsufficiency, whereas variants affecting the activity of p53 pathway effectors impact cell-cycle arrest. However, studies on larger cohorts of patients are required to confirm these findings. A small subset of hereditary RB patients is not carrier of RB1 mutations. Investigation through a clinical exome gene panel within 3 families proposed FGFR4, NQO1, ACADS, CX3CR1, GBE1, KRT85, and TYR as possible candidate genes involved in RB oncogenesis, given their association with the retinoic acid pathway (230).
RB is generally described as retinoblastoma predisposition syndrome since germline RB1 mutations lead to a high risk of second primary malignancies (231). Interestingly, RB onset is reported in 13q deletion syndrome, caused by deletion of part of the long arm of chromosome 13, where RB1 is located (123, 232) (Table 2). Patients with this syndrome show a very wide phenotypic spectrum depending on the size and the location of the deletion (123, 232, 233).
Sporadic Retinoblastoma
Sporadic RB is always unilateral. Biallelic loss of RB1 is found in 98% of cases, whereas 2% show MYCN amplification (234, 235). A significant proportion of sporadic RB exhibits somatic mosaicism for RB1 mutations (236, 237).
Uncommon, Moderate-Penetrance and Common, Low-Penetrance Variants
Susceptibility variants have been investigated mostly in patients with hereditary RB. However, given the role of the p53 pathway in RB development, polymorphisms in genes such as MDM2 (163), MDM4 (164), and CDKN1A (169) could also influence the development of the sporadic form (Table 3). Uncommon variants conferring RB risk may be present in asymptomatic individuals. Indeed, high-throughput analysis revealed that several low-frequency RB1 variants are present in the human population, including rare alleles disrupting splicing (238).
Constitutional Chromosomal Abnormalities
Mosaic and non-mosaic chromosomal deletions of 13q14 region are causative of RB (123, 239). Additionally, duplication of 1q21.1, containing the oncogene BCL9, has been reported in a patient with bilateral RB (240).
Wilms Tumor
Wilms tumor (WT), also known as nephroblastoma, is the most common renal malignancy of childhood, with a median age at diagnosis between 2 and 3 years (241) (Figure 2). It is considered an embryonal tumor as it arises from the aberrant kidney development, due to genetic anomalies in genes essential for fetal nephrogenesis (29). WT treatment is successful with a 5-year overall survival of about 90% and 75% for localized and metastatic disease, respectively (82). It is estimated that about 10% of WT cases are caused by genetic predisposition factors, mainly represented by germline pathogenic variants or epigenetic alterations occurring early during embryogenesis (147, 242). The number of known susceptibility loci has significantly increased over the past years, even if our knowledge is still incomplete and further predisposition factors remain to be discovered. The landscape of somatic genetic alterations in WT is quite broad, with classical genetic changes involving WT1, the IGF2 locus, the WNT pathway, MYCN and TP53 but also driver mutations in several additional cancer genes including epigenetic remodelers, miRNA processing genes and transcription factors essential for nephrogenesis (29).
Familial Wilms Tumor
Several congenital malformation and cancer predisposition syndromes are associated with the risk of developing WT (Table 2). Some of the most known and characterized syndromes are associated with constitutional alterations in WT1 at 11p13 (60). WT1 was the first gene identified in WT and encodes a zinc-finger transcription factor, essential for renal and gonadal development (243). A syndrome frequently associated with high risk of developing WT (around 50%) is the Wilms tumor–aniridia syndrome (WAGR), caused by microdeletions of 11p13 including WT1 and PAX6 (115, 244). The second WT1-related disorder is Denys–Drash syndrome (DDS), due to missense variants in WT1 exons 8 or 9, which affect critical residues in the zinc finger domains (51). The risk of WT in children with DDS is about 90% (241). Another syndrome, phenotypically similar to DDS but with a lower risk of WT development, is Frasier syndrome (FS), caused by splicing variants that result in an imbalance of WT1 isoforms (63). The second major WT locus, identified at 11p15 (245), is also characterized by multiple germline epigenetic and genetic changes causing the overgrowth disorder Beckwith–Wiedemann syndrome (BWS). High WT risk is specifically associated with uniparental paternal disomy at 11p15 and to isolated H19 hyper-methylation that results in biallelic expression of IGF2 and over-activation of the IGF signaling pathway (30, 246). Table 2 reports other constitutional genetic mutations underlying both congenital syndromes and WT predisposition (34, 35, 61, 66, 69, 78, 80, 84, 100, 101, 110, 113).
WT is primarily a non-familial condition, with only about 2% of affected individuals belonging to familial pedigrees (29) (Table 1). A small proportion of familial cases are due to germline WT1 variants (149, 150) and mutations in the H19 region of 11p15 (151). Two further predisposition loci at 17q21 (FWT1) and 19q13 (FWT2) were identified by genetic linkage studies, but the causative genes still remain not fully characterized (247). Another cause of familial WT is the presence of inactivating mutations in the DICER1 miRNA processing gene, also causative of cancer susceptibility in DICER1 syndrome (55). Other recognized familial WT predisposition genes are CTR9 and REST (144, 148, 248). CTR9 encodes a key component of the PAF1 complex, implicated in maintenance of stem cell pluripotency (144), while REST encodes the RE1-silencing transcription repressor, well-known for its role in repressing neural development and differentiation (249). Rare biallelic TRIP13 mutations have been found in a WES study on familial WT pedigrees (83). TRIP13 encodes a member of the spindle assembly checkpoint complex, whose inactivation leads to chromosome segregation dysfunction and aneuploidy (83). Pathogenic inactivating mutations of TRIM28 have been found in about 8% of familial WT in a sequencing study on 890 patients (147). These mutations have been found to show a strong parent-of-origin effect and a robust association with the epithelial subtype of WT (147, 250, 251). The same study reports constitutional mutations in FBXW7, NYNRIN, and CDC73 as contributors to a small number of familial cases, and pathogenic mutations in TRIM28, FBXW7, and KDM3B as de novo events in children with sporadic tumors (147).
It is important to note that, to date, germline pathogenic variants have been identified only in a small proportion of familial WT cases and so that the underlying causative genetic events remain still obscure for the majority of individuals.
Sporadic Wilms Tumor
Many genetic causes of familial and syndromic WT also contribute to sporadic cases, e.g., constitutional WT1 mutations and germline 11p15 anomalies (150, 151). It is currently estimated that in sporadic cases the number of predisposition genes is more than 20 (147). Next-generation sequencing (NGS) and GWAS approaches have allowed researchers to discover an ever-growing number of uncommon (Table 1) and common (Table 3) genetic variants associated with WT susceptibility.
Uncommon, Moderate-Penetrance Variants
Two recent WGS and WES studies have identified new pathogenic germline variants in CHEK2 and PALB2 in children with sporadic WT (131, 132). Both PALB2 and CHEK2 are involved in DNA repair pathways and are associated with breast cancer predisposition (62, 252). Germline mutations in REST and TRIM28, in addition to their role of familial WT predisposition genes, are also responsible for uncommon sporadic cases (148, 251). Additional pathogenic and likely pathogenic variants were identified in predisposition genes such as TP53, DIS3L2, and MLLT1, but also in candidate genes like EP300, HDAC4, HACE1, ARID1A, NF1, MYCN, and GLI3 (131, 132, 137), that need to be validated in independent cohorts. Finally, exome and transcriptome sequencing studies have revealed constitutional mutations in the miRNA processing genes DROSHA, DGCR8, DICER1, and XPO5 (131, 137), some of which associated with the blastemal subtype of WT (137).
Common, Low-Penetrance Variants
The first WT related GWAS study was performed by Turnbull et al. (153), using a dataset of 757 affected and 1.879 controls from North America and subsequently validated in two independent replication series from UK and US populations. They identified two significant SNPs at 2p24 (rs807624 and rs3755132), in the promoter of DDX1, and one SNP at 11q14 (rs790356) located near DLG2. They also identified candidate predisposition loci at 5q14, 22q12, and Xp22, located near the genes PCSK9, TCN2, and NHS, which need further validation (153). More recently, the group of Fu and colleagues performed two candidate gene studies on Southern Chinese populations and found a significant association between WT risk and BARD1 (158) and KRAS (171) polymorphisms, respectively. However, both associations need to be validated in larger cohorts.
Constitutional Chromosomal Abnormalities
Few chromosomal aberrations and copy-number variations (CNVs) are known to be WT predisposing genetic factors. In addition to karyotypic abnormalities affecting 11p13 and 11p15 (60), a very small number of WT patients with gain of entire chromosomes have been reported, specifically with trisomy 18 and trisomy 13 (60). Rare chromosomal aberrations have been identified at 2q (60, 253, 254) and 7q (255, 256) regions, with terminal deletions and balanced and unbalanced translocations. A constitutional de novo balanced translocation was also identified in a child with bilateral WT, affecting the tumor-suppressor gene HACE1, also reported as NB susceptibility gene. HACE1 controls growth and apoptosis and is often somatically mutated in WT (257). Moreover, gain of MYCN (2p24), which is predominantly a somatic event, has been reported as a rare germline aberration (258). Finally, in 2020, a germline duplication of SUZ12 has been detected in a WT patient carrying other germline pathogenic variants in new candidate cancer predisposition genes (3).
Osteosarcoma
Osteosarcoma (OS) is the most common primary bone cancer. This tumor has a bimodal distribution with a high peak during adolescence and a smaller peak in elderly individuals (259) (Figure 2). Survival rates for children and young adults with non-metastatic disease have remained at 60–70%; however, outcome is reduced in patients with metastases (259). Unlike other childhood sarcomas, which are characterized by specific chromosome rearrangements and low mutation rate, complex genomic rearrangements are involved in OS. Indeed, OS exhibits extensive intra-tumoral heterogeneity and has a higher mutation rate (259).
Familial Osteosarcoma
OS is a sentinel cancer in many heritable cancer predisposition syndromes, including autosomal dominant cancer predisposition syndromes such as LFS (73–75) and Diamond–Blackfan anemia (47–50) (Table 2). Furthermore, recessive cancer syndromes associated with OS are Rothmund–Thomson syndrome (102–105), Baller–Gerold syndrome (20, 21), RAPADILINO syndrome (106, 107), Werner syndrome (118–120), Bloom syndrome (32), and ATR-X syndrome (19). OS has also been seen to arise in Paget's disease of bone (98, 99).
Sporadic Osteosarcoma
Targeted gene sequencing and WGS and WES studies have identified uncommon variants in tumor-suppressor and cancer predisposition genes (Table 1), while candidate gene, pathway studies, and GWAS have discovered common variants in genes involved in several key pathways for OS development (259) (Table 3).
Uncommon, Moderate-Penetrance Variants
In 2015, a sequencing study on 765 germline DNA samples showed the presence of uncommon TP53 germline variants that could contribute to OS development; 3.8% of these variants were associated with LFS, and 5.7% were uncommon exonic variants of uncertain clinical significance (138). Another sequencing study on 1120 cases found 7/39 OS patients carrying pathogenic and likely pathogenic variants in TP53, RB1, APC, MSH2, and PALB2 (2). In 2016, a targeted exon sequencing on 1162 patients with sarcoma found that >50% of all patients carried pathogenic variants in TP53, BRCA2, ATM, ATR, and in ERCC2 (128). Among 11% of patients with OS, one patient showed a probable pathogenic variant in ERCC2. In the same work, an excess of functionally pathogenic variants in ERCC2 was found to enhance cell sensitivity to cisplatin, commonly used in the treatment of OS (128). Recently, a sequencing study of 1244 OS patients showed that 28% of patients carried pathogenic and likely pathogenic variants in OS susceptibility genes, identifying new candidates (CDKN2A, MEN1, VHL, POT1, and ATRX) that require further confirmation in independent cohorts (129).
Common, Low-Penetrance Variants
In 2013, the first GWAS study on 941 cases and 3291 controls of European ancestry, identified two risk loci, one at 6p21.3 (rs1906953) mapping in intron 7 of GRM4, and the other at 2p25.2 (rs7591996) in an intergenic region (173). Subsequently, a GWAS study on OS metastasis at diagnosis identified rs7034162 at 9p24.1 (in NFIB) associated with metastasis (176). Functional investigations showed that reduced NFIB expression, due to the risk allele of the rs7034162 SNP, promoted an increase of OS cell migration, proliferation, and colony formation (176). In 2016, a case–control study identified that, for SNPs in genes associated with inter-individual variation in leukocyte telomere length (LTL) (ACYP2, TERC, NAF1, TERT, OBFC1, CTC1, and RTEL1), the allele associated with longer LTL increased OS risk, mainly rs9420907 in OBFC1 (165). These findings were confirmed in 537 OS cases belonging to California Cancer Registry (166).
Constitutional Chromosomal Abnormalities
Next to the heterogeneous somatic CNV scenario present in OS, in a study conducted on 54 patients with childhood tumor, two large germinal CNVs were identified in 2 OS patients: dup4q13.33 of 476 kb containing STATH, CSN1S2B, CABS1, CSN1S1, CSN2, HTN3, HTN1, CSN1S2A, C4orf40, ODAM, FDCSP, and CSN3; and dup18q21.33 of 600 kb containing RNF152, CDH20, and PIGN (240). In 2020, a duplication of DDX10 in an OS patient with a germline variant in GJB2 has been reported (3).
Rhabdomyosarcoma
Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma in childhood and represents a high-grade neoplasm of skeletal myoblast-like cells. Currently, 5-year overall survival of pediatric RMS exceeds 70% (260). The two major histological subtypes are embryonal (ERMS, 67%) and alveolar (ARMS, 32%) (261). ARMS is uniformly distributed among the different age groups (Figure 2) and has a worse prognosis; ERMS has a bimodal distribution (the first peak in early childhood and the second one in early adolescence) and has a better outcome (260, 262) (Figure 2). At somatic level, ARMS is often associated with fusion of FOXO and PAX3 or PAX7, while ERMS does not show such translocations, but it is characterized by loss of heterozygosity at 11p15.5 as well as mutations in TP53, NRAS, KRAS, HRAS, PIK3CA, CTNNB1, and FGFR4 (263). Since a small but substantial fraction of ARMS patients do not harbor one of these translocations, and tumors from those patients are biologically and clinically similar to ERMS, the disease classification has been further refined dividing RMS into “fusion-positive” RMS (FPRMS) and “fusion-negative” RMS (FNRMS) subtypes.
Familial Rhabdomyosarcoma
Although RMS is primarily sporadic (264, 265), it arises in several syndromes. Cancer predisposition syndromes appear to be more frequent in patients with ERMS than in those with ARMS (260). Among syndromes commonly associated with RMS and reported in Table 2 (24–27, 42, 43, 52–54, 64, 75, 80, 81, 86, 87, 89–92, 94, 96), a high RMS risk is associated with RASopathies-like type I neurofibromatosis (NF1) (deletions in NF1), Costello syndrome (HRAS mutations), and Noonan syndrome (germline variants activating RAS-MAPK pathway), highlighting the tight dependence of RMS on the RAS pathway, which results to be activated in 40% of sporadic ERMS (263, 266, 267). In particular, up to 25% of children affected by Costello syndrome shows high RMS risk (43, 268). In addition, children who have a first-degree relative with cancer, particularly if the cancer occurred at a young age (<30 years), show an increase in RMS risk, especially of ERMS (269).
Sporadic Rhabdomyosarcoma
Unlike OS and Ewing sarcoma, GWAS studies for RMS have not been published (260) and few studies identified uncommon germline variants associated with tumor susceptibility (2, 52, 133, 139, 142, 270) (Table 1).
Many studies have found the presence of DICER1 germline mutations in sporadic RMS patients for whom DICER syndrome has been ruled out (52, 142). WES and WGS on 1,120 patients with pediatric cancers identified germline pathogenic variants in 3/43 RMS patients in TP53 and BRCA2 (2). In a cohort of 66 patients with sarcoma, one patient with ARMS showed a protein-truncating variant (in ERCC4) co-occurring with predicted pathogenic mutations (in ATM, FANCI, and MSH6), suggesting a possible collective impact of these genetic variants on DNA repair and genomic instability, therefore conferring susceptibility to tumorigenesis (133).
Ewing Sarcoma
Ewing sarcoma (EWS) is the second most frequent primary skeletal tumor that mainly affects bone and can also arise in soft tissue. It occurs in children, adolescents, and young adult (Figure 2). It is highly aggressive, with a survival of 70–80% for patients with standard-risk and localized disease and 30% for those with metastasis at diagnosis (20–25% of those resistant to intensive therapy) (271). EWS is characterized by low somatic mutation rate (272–274), mainly including fusions between EWSR1 and members of the ETS gene family, usually EWSR1-FLI1, that play a key role in its pathogenesis. The chimeric protein EWSR1-FLI1 leads to the production of an oncogenic transcription factor that binds GGAA motifs (174, 271, 275, 276).
Familial Ewing Sarcoma
To date, no susceptibility genes to familial forms of EWS have been reported, and only case reports about siblings and cousins affected by this tumor have been documented (277, 278). On the basis of these isolated clinical cases, the presence of other cancer types among familial members of EWS patients (279, 280) suggests an important contribution of genetic susceptibility factors in this tumor. Nowadays, EWS is not considered part of predisposition syndromes because of its rare occurrence among these (281).
Sporadic Ewing Sarcoma
WES, WGS, and GWAS studies have led to the identification of uncommon (Table 1) and common (Table 3) germline variants associated with the risk of developing EWS. Despite the rarity and the paucity of information about familial cases, most of the known genetic scenario on this tumor concerns the sporadic form.
Uncommon, Moderate-Penetrance Variants
Two WGS and WES studies on EWS revealed an over-representation of uncommon pathogenic and likely pathogenic variants in DNA repair and cancer-predisposing syndrome genes (2, 130). Studies on small cohorts of patients identified other uncommon germline variants in BRCA2 (146) and in PTPRD (140).
Common, Low-Penetrance Variants
In 2012, the first GWAS on EWS found 3 susceptibility loci at 1p36.22, 10q21, and 15q15, identifying a strong association of EWS risk with rs9430161 (25 kb upstream of TARDBP) and rs224278 (5 kb upstream of EGR2), and a modest association with rs4924410 (at 15q15) (172). The second GWAS detected a tagging variant strongly associated with EWS at 15q.15.1 (rs2412476 near BMF) and new risk loci at 6p25.1, 20p11.22, and 20p11.23 (152). Expression quantitative locus (eQTL) analyses identified candidate genes at 6p25.1 (RREB1) and 20p11.23 (KIZ) (152). Independent studies showed that a different number of germline GGAA repeats in polymorphic enhancer-like GGAA microsatellites impacts the binding between these regulatory elements and EWS cancer driver mutations (EWSR1-FLI1), affecting downstream genes expression (174, 178, 282).
These studies further suggest that cooperation between regulatory germline variants and somatic mutations can drive oncogenesis and create a major source of inter-tumor heterogeneity, determining clinical outcome and drug response through modulation of a druggable key downstream player.
Constitutional Chromosomal Abnormalities
Only one study reports the presence of germline CNV associated with EWS, describing a 14-year-old male with EWS carrying an intragenic deletion in PTPRD (283). Notably, germline and somatic variants in PTPRD have been already identified in a limited number of EWS patients (140).
Conclusions
For a long time, the prevalence of childhood cancer attributed to genetic predisposition was generally considered very low. However, to date, WGS, WES, and GWAS studies performed on pediatric cancers have made it possible to highlight a strong contribution of germline variants to tumorigenesis, helping us to better understand the etiology underlying pediatric tumors. Indeed, an important body of work allows us to highlight that the prevalence of hereditable risk variants in pediatric solid cancers ranges between 6% and 18% (Figure 3). These variants generally affect the functions of genes belonging to biological processes linked to tumorigenesis, such as cell-cycle control, apoptosis, DNA repair, and transcriptional regulatory programs. The enrichment of genetic alterations in these pathways is often due to a bias because, since germline variant analysis is a highly challenging task in general, the vast majority of studies are based on a “candidate-gene” approach, which means they focus on specific subsets of genes already known to play a key role in cancer predisposition and tumorigenesis. For this reason, it may be useful exploiting a genome-wide scale approach, e.g., exome-wide association studies, to investigate the presence of genetic alterations predisposing to cancer also in genes involved in pathways others than the ones above mentioned. This approach may contribute in a meaningful way to the current knowledge of the mechanisms underlying solid pediatric tumors onset.
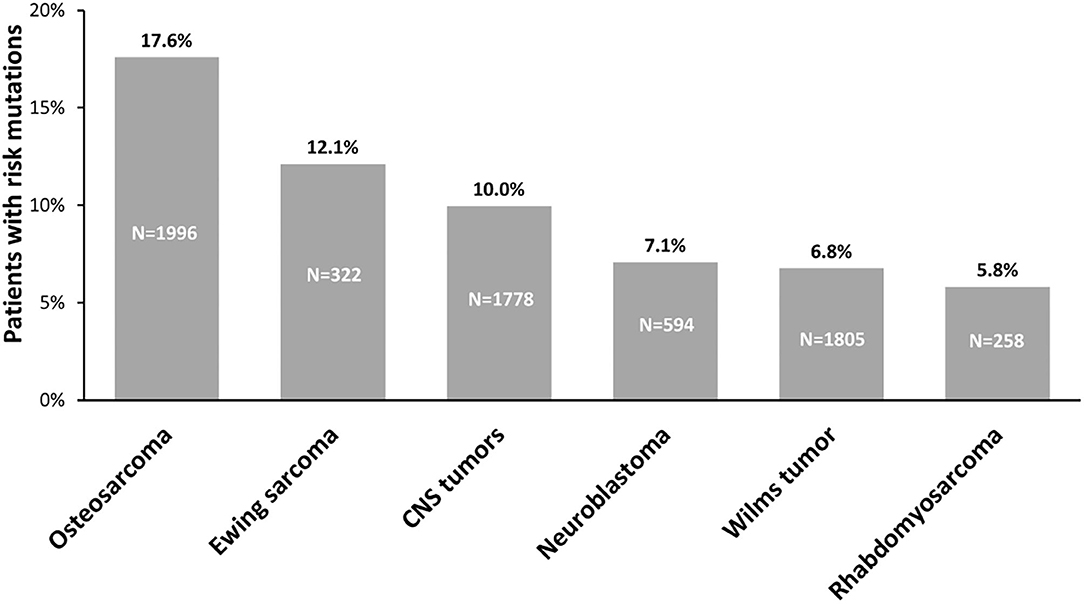
Figure 3. Prevalence of germline predisposition in pediatric tumors. The percentage of germline predisposition due to uncommon, moderate-penetrance variants, reported above the bars, has been calculated evaluating the number of patients carrying pathogenic and likely pathogenic variants on the total number of patients from the cohorts analyzed for each tumor: CNS tumors: (3, 15, 17); neuroblastoma: (2, 3, 135, 146, 196, 197, 200); Wilms tumor: (3, 131, 132, 137, 148, 150, 251); osteosarcoma: (2, 3, 129, 138); rhabdomyosarcoma: (2, 3, 52, 139); Ewing sarcoma: (2, 3, 130, 146). N, number of patients analyzed in cohorts; CNS, central nervous system.
A very recent study reports a high number of germline variants in new candidate susceptibility genes, highlighting that some of them carry druggable alterations (3). It should be emphasized that the presence of germline variants in target therapeutic genes could improve current approaches of personalized therapy, making them more efficient and less toxic to patients. Furthermore, a more in-depth investigation of the germline component underlying tumor development should also be performed on pediatric solid tumors for which there is not yet a broad knowledge of germline landscape (e.g., thyroid carcinoma, melanoma) (284–289).
Our literature review reveals that the presence of specific germline mutations is often associated with increased frequency of somatically acquired cancer-specific abnormalities (such as aberrations, rearrangements). The interplay between somatic and germline mutations may be at the basis of high interindividual tumor heterogeneity (290). For example, the cooperation between regulatory germline variants and somatic mutations underlines the importance of regulatory regions to stratify patients into risk groups to predict the clinical outcome and therapeutic approaches (290). In NB, inherited deleterious variants in genes that code for proteins involved in chromosomal segregation, centrosome segregation, DNA repair, and spindle apparatus machinery are thought to be the cause of chromosome instability at somatic levels (199). A similar germline–somatic interaction has been proposed for MB; indeed, germline TP53 mutations are often found in combination with tumors characterized by catastrophic DNA chromothripsis. Determining if germline risk alleles predispose to genomic instability in pediatric cancers is an important research objective for biologists and geneticists. Another interesting research field is related to the impact of risk alleles on genomic regions that regulate mutated cancer driver genes. The mechanisms underlying this type of interaction between germline–somatic variation have been elegantly elucidated in the EWS (174, 178, 282), and it is reasonable to think that it is common to other pediatric tumors as well. No relevant study has investigated the possible interplay between germline variations and epigenetic somatic events. For instance, there is an urgent need to find possible associations between germline risk alleles and DNA methylation of tumor. Studies integrating information on germline, somatic, and epigenomic variations using gene expression data as the intermediate phenotype may unravel the biological mechanisms underlying oncogenic interactions and cooperation of these different types of genomic variations.
The low number of recurrent somatic mutations in some pediatric cancers, compared to adult ones (135), does not explain the clinical heterogeneity and the resulting need for personalized therapies in tumors. Confirming a germline contribution to the clinical heterogeneity, some studies have highlighted that specific pathogenic variants are much more common in specific tumor histotypes (137, 147) and these associations could be used for the management and stratification of patients. Thereby, implementing screening tests with the introduction of germline detection would bring clinical benefits. In addition, screening for germline and somatic components of the tumor could lead to the identification of new prognostic markers to monitor cancer and predict clinical outcome. Finally, the use of these information in screening tests is important in the context of genetic counseling, to monitor and supervise family members of patients.
It is also important to note that many genetic syndromes such as Beckwith–Wiedemann, Costello, Fanconi anemia, Gorlin, Noonan syndrome, Li-Fraumeni, and others (Table 2) are both characterized by genetic and/or allelic heterogeneity and associated with the risk to develop different types of pediatric cancers. Therefore, NGS-based cancer gene panel tests should be performed in children with a genetic syndrome to ensure the patient a more precise diagnosis and to be able to assess the risk of developing a cancer disease. A clinical management that includes a cancer genetic test not only is useful to indicate a modification of the surveillance that also integrates periodic and cancer specific diagnostic tests, but over time it will increase our knowledge of genetic risk variants and thus will give a clearer picture of cancer risk in children affected by genetic syndrome. This surely can have a positive impact on improving patient care and survival.
Author Contributions
MC and AI contributed to the design, reviewing, and editing of this manuscript. AM, SC, TM, and MT contributed to the design, writing, and editing of this manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Associazione Italiana per la Ricerca sul Cancro (Grant no. 19255 to MC and Grant no. 20757 to AI), Fondazione Italiana per la Lotta al Neuroblastoma (to MC), Associazione Oncologia Pediatrica e Neuroblastoma (to MC), and Regione Campania SATIN grant 2018-2020 (to MC).
Conflict of Interest
All authors were employed by the company CEINGE Biotecnologie Avanzate.
References
1. Sweet-Cordero EA, Biegel JA. The genomic landscape of pediatric cancers: implications for diagnosis and treatment. Science. (2019) 363:1170–5. doi: 10.1126/science.aaw3535
2. Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. (2015) 373:2336–46. doi: 10.1056/NEJMoa1508054
3. Akhavanfard S, Padmanabhan R, Yehia L, Cheng F, Eng C. Comprehensive germline genomic profiles of children, adolescents and young adults with solid tumors. Nat Commun. (2020) 11:2206. doi: 10.1038/s41467-020-16067-1
4. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
5. Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, et al. Exome sequencing as a tool for mendelian disease gene discovery. Nat Rev Genet. (2011) 12:745–55. doi: 10.1038/nrg3031
6. Warr A, Robert C, Hume D, Archibald A, Deeb N, Watson M. Exome sequencing: current and future perspectives. G3. (2015) 5:1543–50. doi: 10.1534/g3.115.018564
7. Povysil G, Petrovski S, Hostyk J, Aggarwal V, Allen AS, Goldstein DB. Rare-variant collapsing analyses for complex traits: guidelines and applications. Nat Rev Genet. (2019) 20:747–59. doi: 10.1038/s41576-019-0177-4
8. Yin R, Kwoh CK, Zheng J. Whole genome sequencing analysis. In: Ranganathan S, Gribskov M, Nakai K, Schönbach C, editor. Encyclopedia of Bioinformatics and Computational Biology. Academic Press (2019). p. 176–83. doi: 10.1016/B978-0-12-809633-8.20095-2
9. Tam V, Patel N, Turcotte M, Bosse Y, Pare G, Meyre D. Benefits and limitations of genome-wide association studies. Nat Rev Genet. (2019) 20:467–84. doi: 10.1038/s41576-019-0127-1
10. Boone PM, Stankiewicz P. Array comparative genomic hybridization. In: Maloy S, Hughes K, editors. Brenner's Encyclopedia of Genetics, 2nd ed. Academic Press (2013). p. 193–7. doi: 10.1016/B978-0-12-374984-0.00300-4
11. Postema FAM, Hopman SMJ, Hennekam RC, Merks JHM. Consequences of diagnosing a tumor predisposition syndrome in children with cancer: a literature review. Pediatr Blood Cancer. (2018) 65. doi: 10.1002/pbc.26718
12. Brodeur GM, Nichols KE, Plon SE, Schiffman JD, Malkin D. Pediatric cancer predisposition and surveillance: an overview, and a tribute to alfred G. Knudson Jr. Clin Cancer Res. (2017) 23:e1–e5. doi: 10.1158/1078-0432.CCR-17-0702
13. Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2011-2015. Neuro Oncol. (2018) 20(Suppl. 4):iv1–86. doi: 10.1093/neuonc/noy131
14. Northcott PA, Robinson GW, Kratz CP, Mabbott DJ, Pomeroy SL, Clifford SC, et al. Medulloblastoma. Nat Rev Dis Primers. (2019) 5:11. doi: 10.1038/s41572-019-0063-6
15. Waszak SM, Northcott PA, Buchhalter I, Robinson GW, Sutter C, Groebner S, et al. Spectrum and prevalence of genetic predisposition in medulloblastoma: a retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol. (2018) 19:785–98. doi: 10.1016/S1470-2045(18)30242-0
16. Skowron P, Ramaswamy V, Taylor MD. Genetic and molecular alterations across medulloblastoma subgroups. J Mol Med. (2015) 93:1075–84. doi: 10.1007/s00109-015-1333-8
17. Waszak SM, Robinson GW, Gudenas BL, Smith KS, Forget A, Kojic M, et al. Germline elongator mutations in sonic hedgehog medulloblastoma. Nature. (2020) 580:396–401. doi: 10.1038/s41586-020-2164-5
18. Hart RM, Kimler BF, Evans RG, Park CH. Radiotherapeutic management of medulloblastoma in a pediatric patient with ataxia telangiectasia. Int J Radiat Oncol Biol Phys. (1987) 13:1237–40. doi: 10.1016/0360-3016(87)90200-8
19. Masliah-Planchon J, Levy D, Heron D, Giuliano F, Badens C, Freneaux P, et al. Does ATRX germline variation predispose to osteosarcoma? Three additional cases of osteosarcoma in two ATR-X syndrome patients. Eur J Hum Genet. (2018) 26:1217–21. doi: 10.1038/s41431-018-0147-x
20. Van Maldergem L, Piard J, Larizza L, Wang LL. Baller-Gerold syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews®. Seattle, WA: University of Washington, Seattle (2020).
21. Preis S, Majewski F, Korholz D, Gobel U. Osteosarcoma in a 16-year-old boy with Baller-Gerold syndrome. Clin Dysmorphol. (1995) 4:161–8. doi: 10.1097/00019605-199504000-00009
22. Chitayat D, Friedman JM, Dimmick JE. Neuroblastoma in a child with wiedemann-beckwith syndrome. Am J Med Genet. (1990) 35:433–6. doi: 10.1002/ajmg.1320350322
23. Maas SM, Vansenne F, Kadouch DJ, Ibrahim A, Bliek J, Hopman S, et al. Phenotype, cancer risk, and surveillance in Beckwith-Wiedemann syndrome depending on molecular genetic subgroups. Am J Med Genet A. (2016) 170:2248–60. doi: 10.1002/ajmg.a.37801
24. Smith AC, Squire JA, Thorner P, Zielenska M, Shuman C, Grant R, et al. Association of alveolar rhabdomyosarcoma with the Beckwith-Wiedemann syndrome. Pediatr Dev Pathol. (2001) 4:550–8. doi: 10.1007/s10024001-0110-6
25. Weksberg R, Nishikawa J, Caluseriu O, Fei YL, Shuman C, Wei C, et al. Tumor development in the Beckwith-Wiedemann syndrome is associated with a variety of constitutional molecular 11p15 alterations including imprinting defects of KCNQ1OT1. Hum Mol Genet. (2001) 10:2989–3000. doi: 10.1093/hmg/10.26.2989
26. Kuroiwa M, Sakamoto J, Shimada A, Suzuki N, Hirato J, Park MJ, et al. Manifestation of alveolar rhabdomyosarcoma as primary cutaneous lesions in a neonate with Beckwith-Wiedemann syndrome. J Pediatr Surg. (2009) 44:e31–35. doi: 10.1016/j.jpedsurg.2008.12.010
27. Mondi V, Piersigilli F, Salvatori G, Auriti C. The skin as an early expression of malignancies in the neonatal age: a review of the literature and a case series. Biomed Res Int. (2015) 2015:809406. doi: 10.1155/2015/809406
28. Pappas JG. The clinical course of an overgrowth syndrome, from diagnosis in infancy through adulthood: the case of Beckwith-Wiedemann syndrome. Curr Probl Pediatr Adolesc Health Care. (2015) 45:112–7. doi: 10.1016/j.cppeds.2015.03.001
29. Treger TD, Chowdhury T, Pritchard-Jones K, Behjati S. The genetic changes of wilms tumour. Nat Rev Nephrol. (2019) 15:240–51. doi: 10.1038/s41581-019-0112-0
30. Cooper WN, Luharia A, Evans GA, Raza H, Haire AC, Grundy R, et al. Molecular subtypes and phenotypic expression of beckwith-wiedemann syndrome. Eur J Hum Genet. (2005) 13:1025–32. doi: 10.1038/sj.ejhg.5201463
31. Petrella R, Hirschhorn K, German J. Triple autosomal trisomy in a pregnancy at risk for bloom's syndrome. Am J Med Genet. (1991) 40:316–8. doi: 10.1002/ajmg.1320400314
32. German J. Bloom's syndrome. XX. the first 100 cancers. Cancer Genet Cytogenet. (1997) 93:100–6. doi: 10.1016/S0165-4608(96)00336-6
33. Scollon S, Anglin AK, Thomas M, Turner JT, Wolfe Schneider K. A comprehensive review of pediatric tumors and associated cancer predisposition syndromes. J Genet Couns. (2017) 26:387–434. doi: 10.1007/s10897-017-0077-8
34. Cairney AE, Andrews M, Greenberg M, Smith D, Weksberg R. Wilms tumor in three patients with bloom syndrome. J Pediatr. (1987) 111:414–6. doi: 10.1016/s0022-3476(87)80469-9
35. Russell B, Johnston JJ, Biesecker LG, Kramer N, Pickart A, Rhead W, et al. Clinical management of patients with ASXL1 mutations and bohring-opitz syndrome, emphasizing the need for wilms tumor surveillance. Am J Med Genet A. (2015) 167A:2122–31. doi: 10.1002/ajmg.a.37131
36. PDQ Pediatric Treatment Editorial Board. Wilms tumor and other childhood kidney tumors treatment (PDQ®): patient version. In: PDQ Cancer Information Summaries. Bethesda, MD: National Cancer Institute (US) (2002).
37. Trochet D, Bourdeaut F, Janoueix-Lerosey I, Deville A, de Pontual L, Schleiermacher G, et al. Germline mutations of the paired-like homeobox 2B (PHOX2B) gene in neuroblastoma. Am J Hum Genet. (2004) 74:761–4. doi: 10.1086/383253
38. Rohrer T, Trachsel D, Engelcke G, Hammer J. Congenital central hypoventilation syndrome associated with hirschsprung's disease and neuroblastoma: case of multiple neurocristopathies. Pediatr Pulmonol. (2002) 33:71–6. doi: 10.1002/ppul.10031
39. Barr EK, Applebaum MA. Genetic predisposition to neuroblastoma. Children. (2018) 5:119. doi: 10.3390/children5090119
40. Taeubner J, Wimmer K, Muleris M, Lascols O, Colas C, Fauth C, et al. Diagnostic challenges in a child with early onset desmoplastic medulloblastoma and homozygous variants in MSH2 and MSH6. Eur J Hum Genet. (2018) 26:440–4. doi: 10.1038/s41431-017-0071-5
41. Gripp KW, Lin AE. Costello syndrome: a Ras/mitogen activated protein kinase pathway syndrome (rasopathy) resulting from HRAS germline mutations. Genet Med. (2012) 14:285–92. doi: 10.1038/gim.0b013e31822dd91f
42. Kratz CP, Franke L, Peters H, Kohlschmidt N, Kazmierczak B, Finckh U, et al. Cancer spectrum and frequency among children with Noonan, Costello, and cardio-facio-cutaneous syndromes. Br J Cancer. (2015) 112:1392–7. doi: 10.1038/bjc.2015.75
43. Sanchez-Montenegro C, Vilanova-Sanchez A, Barrena-Delfa S, Tenorio J, Santos-Simarro F, Garcia-Minaur S, et al. Costello syndrome and umbilical ligament rhabdomyosarcoma in two pediatric patients: case reports and review of the literature. Case Rep Genet. (2017) 2017:1587610. doi: 10.1155/2017/1587610
44. Burgu B, Wilcox DT. Chapter 51 - Rhabdomyosarcoma. In: Gearhart JP, Rink RC, Mouriquand PDE, editors. Pediatric Urology, 2nd ed. W.B. Saunders (2010). p. 684–693. doi: 10.1016/B978-1-4160-3204-5.00051-7
45. Twigg SRF, Hufnagel RB, Miller KA, Zhou Y, McGowan SJ, Taylor J, et al. A recurrent mosaic mutation in SMO, encoding the hedgehog signal transducer smoothened, is the major cause of curry-jones syndrome. Am J Hum Genet. (2016) 98:1256–65. doi: 10.1016/j.ajhg.2016.04.007
46. Porath B, Farooki S, Gener M, Amudhavalli SM, Grote L, Cooley LD, et al. Occurrence and characterization of medulloblastoma in a patient with Curry-Jones syndrome. Clin Genet. (2020) 97:670–1. doi: 10.1111/cge.13681
47. Lee RS, Higgs D, Haddo O, Pringle J, Briggs TW. Osteosarcoma associated with diamond-blackfan anaemia: a case of a child receiving growth hormone therapy. Sarcoma. (2004) 8:47–9. doi: 10.1080/13577140410001679266
48. Narla A, Ebert BL. Ribosomopathies: human disorders of ribosome dysfunction. Blood. (2010) 115:3196–205. doi: 10.1182/blood-2009-10-178129
49. Vlachos A, Rosenberg PS, Atsidaftos E, Alter BP, Lipton JM. Incidence of neoplasia in diamond blackfan anemia: a report from the diamond blackfan anemia registry. Blood. (2012) 119:3815–9. doi: 10.1182/blood-2011-08-375972
50. Vlachos A, Rosenberg PS, Atsidaftos E, Kang J, Onel K, Sharaf RN, et al. Increased risk of colon cancer and osteogenic sarcoma in diamond-blackfan anemia. Blood. (2018) 132:2205–8. doi: 10.1182/blood-2018-05-848937
51. Pelletier J, Bruening W, Kashtan CE, Mauer SM, Manivel JC, Striegel JE, et al. Germline mutations in the wilms' tumor suppressor gene are associated with abnormal urogenital development in denys-drash syndrome. Cell. (1991) 67:437–47. doi: 10.1016/0092-8674(91)90194-4
52. Doros L, Yang J, Dehner L, Rossi CT, Skiver K, Jarzembowski JA, et al. DICER1 mutations in embryonal rhabdomyosarcomas from children with and without familial PPB-tumor predisposition syndrome. Pediatr Blood Cancer. (2012) 59:558–60. doi: 10.1002/pbc.24020
53. de Kock L, Geoffrion D, Rivera B, Wagener R, Sabbaghian N, Bens S, et al. Multiple DICER1-related tumors in a child with a large interstitial 14q32 deletion. Genes Chromosomes Cancer. (2018) 57:223–30. doi: 10.1002/gcc.22523
54. McCluggage WG, Apellaniz-Ruiz M, Chong AL, Hanley KZ, Velazquez Vega JE, McVeigh TP, et al. Embryonal rhabdomyosarcoma of the ovary and fallopian tube: rare neoplasms associated with germline and somatic DICER1 mutations. Am J Surg Pathol. (2020) 44:738–47. doi: 10.1097/PAS.0000000000001442
55. Palculict TB, Ruteshouser EC, Fan Y, Wang W, Strong L, Huff V. Identification of germline DICER1 mutations and loss of heterozygosity in familial Wilms tumour. J Med Genet. (2016) 53:385–8. doi: 10.1136/jmedgenet-2015-103311
56. Schimke RN, Collins DL, Stolle CA. Paraganglioma, neuroblastoma, and a SDHB mutation: resolution of a 30-year-old mystery. Am J Med Genet A. (2010) 152A:1531–5. doi: 10.1002/ajmg.a.33384
57. Nalepa G, Clapp DW. Fanconi anaemia and cancer: an intricate relationship. Nat Rev Cancer. (2018) 18:168–85. doi: 10.1038/nrc.2017.116
58. Miele E, Mastronuzzi A, Po A, Carai A, Alfano V, Serra A, et al. Characterization of medulloblastoma in fanconi Anemia: a novel mutation in the BRCA2 gene and SHH molecular subgroup. Biomark Res. (2015) 3:13. doi: 10.1186/s40364-015-0038-z
59. Villani A, Malkin D, Tabori U. Syndromes predisposing to pediatric central nervous system tumors: lessons learned and new promises. Curr Neurol Neurosci Rep. (2012) 12:153–64. doi: 10.1007/s11910-011-0244-5
60. Scott RH, Stiller CA, Walker L, Rahman N. Syndromes and constitutional chromosomal abnormalities associated with wilms tumour. J Med Genet. (2006) 43:705–15. doi: 10.1136/jmg.2006.041723
61. Reid S, Renwick A, Seal S, Baskcomb L, Barfoot R, Jayatilake H, et al. Biallelic BRCA2 mutations are associated with multiple malignancies in childhood including familial wilms tumour. J Med Genet. (2005) 42:147–51. doi: 10.1136/jmg.2004.022673
62. Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet. (2007) 39:162–4. doi: 10.1038/ng1947
63. Barbaux S, Niaudet P, Gubler MC, Grunfeld JP, Jaubert F, Kuttenn F, et al. Donor splice-site mutations in WT1 are responsible for frasier syndrome. Nat Genet. (1997) 17:467–70. doi: 10.1038/ng1297-467
64. Hettmer S, Teot LA, Kozakewich H, Werger AM, Davies KJ, Fletcher CD, et al. Myogenic tumors in nevoid Basal cell carcinoma syndrome. J Pediatr Hematol Oncol. (2015) 37:147–9. doi: 10.1097/MPH.0000000000000115
65. Cajaiba MM, Bale AE, Alvarez-Franco M, McNamara J, Reyes-Mugica M. Rhabdomyosarcoma, Wilms tumor, and deletion of the patched gene in Gorlin syndrome. Nat Clin Pract Oncol. (2006) 3:575–80. doi: 10.1038/ncponc0608
66. Isidor B, Bourdeaut F, Lafon D, Plessis G, Lacaze E, Kannengiesser C, et al. Wilms' tumor in patients with 9q22.3 microdeletion syndrome suggests a role for PTCH1 in nephroblastomas. Eur J Hum Genet. (2013) 21:784–87. doi: 10.1038/ejhg.2012.252
67. Guerrini-Rousseau L, Dufour C, Varlet P, Masliah-Planchon J, Bourdeaut F, Guillaud-Bataille M, et al. Germline SUFU mutation carriers and medulloblastoma: clinical characteristics, cancer risk, and prognosis. Neuro Oncol. (2018) 20:1122–32. doi: 10.1093/neuonc/nox228
68. Johansson G, Andersson U, Melin B. Recent developments in brain tumor predisposing syndromes. Acta Oncol. (2016) 55:401–11. doi: 10.3109/0284186X.2015.1107190
69. Shuman C, Smith AC, Steele L, Ray PN, Clericuzio C, Zackai E, et al. Constitutional UPD for chromosome 11p15 in individuals with isolated hemihyperplasia is associated with high tumor risk and occurs following assisted reproductive technologies. Am J Med Genet A. (2006) 140:1497–503. doi: 10.1002/ajmg.a.31323
70. Roma D, Palma P, Capolino R, Figa-Talamanca L, Diomedi-Camassei F, Lepri FR, et al. Spinal ependymoma in a patient with kabuki syndrome: a case report. BMC Med Genet. (2015) 16:80. doi: 10.1186/s12881-015-0228-4
71. Rausch T, Jones DT, Zapatka M, Stutz AM, Zichner T, Weischenfeldt J, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. (2012) 148:59–71. doi: 10.1016/j.cell.2011.12.013
72. Birch JM, Alston RD, McNally RJ, Evans DG, Kelsey AM, Harris M, et al. Relative frequency and morphology of cancers in carriers of germline TP53 mutations. Oncogene. (2001) 20:4621–8. doi: 10.1038/sj.onc.1204621
73. Malkin D, Li FP, Strong LC, Fraumeni JFJr, Nelson CE, Kim DH, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. (1990) 250:1233–8. doi: 10.1126/science.1978757
74. Ognjanovic S, Olivier M, Bergemann TL, Hainaut P. Sarcomas in TP53 germline mutation carriers: a review of the IARC TP53 database. Cancer. (2012) 118:1387–96. doi: 10.1002/cncr.26390
76. Hameed M, Mandelker D. Tumor syndromes predisposing to osteosarcoma. Adv Anat Pathol. (2018) 25:217–22. doi: 10.1097/PAP.0000000000000190
77. Ripperger T, Bielack SS, Borkhardt A, Brecht IB, Burkhardt B, Calaminus G, et al. Childhood cancer predisposition syndromes-A concise review and recommendations by the cancer predisposition working group of the society for pediatric oncology and hematology. Am J Med Genet A. (2017) 173:1017–37. doi: 10.1002/ajmg.a.38142
78. Hartley AL, Birch JM, Tricker K, Wallace SA, Kelsey AM, Harris M, et al. Wilms' tumor in the li-fraumeni cancer family syndrome. Cancer Genet Cytogenet. (1993) 67:133–5. doi: 10.1016/0165-4608(93)90166-j
79. Vitanza NA, Partap S. Pediatric ependymoma. J Child Neurol. (2016) 31:1354–66. doi: 10.1177/0883073815610428
80. Hanks S, Coleman K, Reid S, Plaja A, Firth H, Fitzpatrick D, et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat Genet. (2004) 36:1159–61. doi: 10.1038/ng1449
81. Nishitani-Isa M, Hiraumi Y, Nishida Y, Usami I, Maihara T. Rhabdomyosarcoma with premature chromatid separation-mosaic variegated aneuploidy syndrome: reduced-intensity chemotherapy. Pediatr Int. (2019) 61:613–6. doi: 10.1111/ped.13849
82. Scott RH, Walker L, Olsen OE, Levitt G, Kenney I, Maher E, et al. Surveillance for wilms tumour in at-risk children: pragmatic recommendations for best practice. Arch Dis Child. (2006) 91:995–9. doi: 10.1136/adc.2006.101295
83. Yost S, de Wolf B, Hanks S, Zachariou A, Marcozzi C, Clarke M, et al. Biallelic TRIP13 mutations predispose to Wilms tumor and chromosome missegregation. Nat Genet. (2017) 49:1148–51. doi: 10.1038/ng.3883
84. Karlberg N, Karlberg S, Karikoski R, Mikkola S, Lipsanen-Nyman M, Jalanko H. High frequency of tumours in mulibrey nanism. J Pathol. (2009) 218:163–71. doi: 10.1002/path.2538
85. Distel L, Neubauer S, Varon R, Holter W, Grabenbauer G. Fatal toxicity following radio- and chemotherapy of medulloblastoma in a child with unrecognized Nijmegen breakage syndrome. Med Pediatr Oncol. (2003) 41:44–8. doi: 10.1002/mpo.10275
86. Tekin M, Dogu F, Tacyildiz N, Akar E, Ikinciogullari A, Ogur G, et al. 657del5 mutation in the NBS1 gene is associated with Nijmegen breakage syndrome in a Turkish family. Clin Genet. (2002) 62:84–8. doi: 10.1034/j.1399-0004.2002.620112.x
87. Meyer S, Kingston H, Taylor AM, Byrd PJ, Last JI, Brennan BM, et al. Rhabdomyosarcoma in Nijmegen breakage syndrome: strong association with perianal primary site. Cancer Genet Cytogenet. (2004) 154:169–74. doi: 10.1016/j.cancergencyto.2004.02.022
88. Cotton JL, Williams RG. Noonan syndrome and neuroblastoma. Arch Pediatr Adolesc Med. (1995) 149:1280–1. doi: 10.1001/archpedi.1995.02170240098019
89. Denayer E, Devriendt K, de Ravel T, Van Buggenhout G, Smeets E, Francois I, et al. Tumor spectrum in children with Noonan syndrome and SOS1 or RAF1 mutations. Genes Chromosomes Cancer. (2010) 49:242–52. doi: 10.1002/gcc.20735
90. Hastings R, Newbury-Ecob R, Ng A, Taylor R. A further patient with Noonan syndrome due to a SOS1 mutation and rhabdomyosarcoma. Genes Chromosomes Cancer. (2010) 49:967–8. doi: 10.1002/gcc.20800
91. Jongmans MC, Hoogerbrugge PM, Hilkens L, Flucke U, van der Burgt I, Noordam K, et al. Noonan syndrome, the SOS1 gene and embryonal rhabdomyosarcoma. Genes Chromosomes Cancer. (2010) 49:635–41. doi: 10.1002/gcc.20773
92. Salem B, Hofherr S, Turner J, Doros L, Smpokou P. Childhood rhabdomyosarcoma in association with a rasopathy clinical phenotype and mosaic germline SOS1 duplication. J Pediatr Hematol Oncol. (2016) 38:e278–82. doi: 10.1097/MPH.0000000000000566
93. Garren B, Stephan M, Hogue JS. NRAS associated RASopathy and embryonal rhabdomyosarcoma. Am J Med Genet A. (2020) 182:195–200. doi: 10.1002/ajmg.a.61395
94. Ji J, Navid F, Hiemenz MC, Kaneko M, Zhou S, Saitta SC, et al. Embryonal rhabdomyosarcoma in a patient with a germline CBL pathogenic variant. Cancer Genet. (2019) 231–2:62–6. doi: 10.1016/j.cancergen.2018.12.006
95. Origone P, Defferrari R, Mazzocco K, Lo Cunsolo C, De Bernardi B, Tonini GP. Homozygous inactivation of NF1 gene in a patient with familial NF1 and disseminated neuroblastoma. Am J Med Genet A. (2003) 118A:309–13. doi: 10.1002/ajmg.a.10167
96. Crucis A, Richer W, Brugieres L, Bergeron C, Marie-Cardine A, Stephan JL, et al. Rhabdomyosarcomas in children with neurofibromatosis type I: a national historical cohort. Pediatr Blood Cancer. (2015) 62:1733–8. doi: 10.1002/pbc.25556
97. Coy S, Rashid R, Stemmer-Rachamimov A, Santagata S. An update on the CNS manifestations of neurofibromatosis type 2. Acta Neuropathol. (2020) 139:643–65. doi: 10.1007/s00401-019-02029-5
98. Hansen MF, Seton M, Merchant A. Osteosarcoma in Paget's disease of bone. J Bone Miner Res. (2006) 21(Suppl. 2):P58–63. doi: 10.1359/jbmr.06s211
99. Scotto di Carlo F, Pazzaglia L, Esposito T, Gianfrancesco F. The loss of profilin 1 causes early onset paget's disease of bone. J Bone Miner Res. (2020) 35:1387–98. doi: 10.1002/jbmr.3964
100. Astuti D, Morris MR, Cooper WN, Staals RH, Wake NC, Fews GA, et al. Germline mutations in DIS3L2 cause the perlman syndrome of overgrowth and wilms tumor susceptibility. Nat Genet. (2012) 44:277–84. doi: 10.1038/ng.1071
101. Gripp KW, Baker L, Kandula V, Conard K, Scavina M, Napoli JA, et al. Nephroblastomatosis or Wilms tumor in a fourth patient with a somatic PIK3CA mutation. Am J Med Genet A. (2016) 170:2559–69. doi: 10.1002/ajmg.a.37758
102. Wang LL, Levy ML, Lewis RA, Chintagumpala MM, Lev D, Rogers M, et al. Clinical manifestations in a cohort of 41 Rothmund-Thomson syndrome patients. Am J Med Genet. (2001) 102:11–7. doi: 10.1002/1096-8628(20010722)102:1<11::AID-AJMG1413>3.0.CO;2-A
103. Kellermayer R. The versatile RECQL4. Genet Med. (2006) 8:213–6. doi: 10.1097/01.gim.0000214457.58378.1a
104. Zils K, Klingebiel T, Behnisch W, Mueller HL, Schlegel PG, Fruehwald M, et al. Osteosarcoma in patients with rothmund-thomson syndrome. Pediatr Hematol Oncol. (2015) 32:32–40. doi: 10.3109/08880018.2014.987939
105. Maciaszek JL, Oak N, Chen W, Hamilton KV, McGee RB, Nuccio R, et al. Enrichment of heterozygous germline RECQL4 loss-of-function variants in pediatric osteosarcoma. Cold Spring Harb Mol Case Stud. (2019) 5:a004218. doi: 10.1101/mcs.a004218
106. Siitonen HA, Kopra O, Kaariainen H, Haravuori H, Winter RM, Saamanen AM, et al. Molecular defect of RAPADILINO syndrome expands the phenotype spectrum of RECQL diseases. Hum Mol Genet. (2003) 12:2837–44. doi: 10.1093/hmg/ddg306
107. Siitonen HA, Sotkasiira J, Biervliet M, Benmansour A, Capri Y, Cormier-Daire V, et al. The mutation spectrum in RECQL4 diseases. Eur J Hum Genet. (2009) 17:151–8. doi: 10.1038/ejhg.2008.154
108. Calvert GT, Randall RL, Jones KB, Cannon-Albright L, Lessnick S, Schiffman JD. At-risk populations for osteosarcoma: the syndromes and beyond. Sarcoma. (2012) 2012:152382. doi: 10.1155/2012/152382
109. de Kort E, Conneman N, Diderich K. A case of Rubinstein-Taybi syndrome and congenital neuroblastoma. Am J Med Genet A. (2014) 164A:1332–3. doi: 10.1002/ajmg.a.36399
110. Cottereau E, Mortemousque I, Moizard MP, Burglen L, Lacombe D, Gilbert-Dussardier B, et al. Phenotypic spectrum of simpson-golabi-behmel syndrome in a series of 42 cases with a mutation in GPC3 and review of the literature. Am J Med Genet C Semin Med Genet. (2013) 163C:92–105. doi: 10.1002/ajmg.c.31360
111. Berdasco M, Ropero S, Setien F, Fraga MF, Lapunzina P, Losson R, et al. Epigenetic inactivation of the sotos overgrowth syndrome gene histone methyltransferase NSD1 in human neuroblastoma and glioma. Proc Natl Acad Sci USA. (2009) 106:21830–5. doi: 10.1073/pnas.0906831106
112. Nance MA, Neglia JP, Talwar D, Berry SA. Neuroblastoma in a patient with Sotos' syndrome. J Med Genet. (1990) 27:130–2. doi: 10.1136/jmg.27.2.130
113. Fagali C, Kok F, Nicola P, Kim C, Bertola D, Albano L, et al. MLPA analysis in 30 sotos syndrome patients revealed one total NSD1 deletion and two partial deletions not previously reported. Eur J Med Genet. (2009) 52:333–6. doi: 10.1016/j.ejmg.2009.07.001
114. Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, et al. The whole-genome landscape of medulloblastoma subtypes. Nature. (2017) 547:311–7. doi: 10.1038/nature22973
115. Breslow NE, Norris R, Norkool PA, Kang T, Beckwith JB, Perlman EJ, et al. Characteristics and outcomes of children with the wilms tumor-aniridia syndrome: a report from the national wilms tumor study group. J Clin Oncol. (2003) 21:4579–85. doi: 10.1200/JCO.2003.06.096
116. Tatton-Brown K, Murray A, Hanks S, Douglas J, Armstrong R, Banka S, et al. Weaver syndrome and EZH2 mutations: clarifying the clinical phenotype. Am J Med Genet A. (2013) 161A:2972–80. doi: 10.1002/ajmg.a.36229
117. Coulter D, Powell CM, Gold S. Weaver syndrome and neuroblastoma. J Pediatr Hematol Oncol. (2008) 30:758–60. doi: 10.1097/MPH.0b013e3181758974
118. Goto M, Miller RW, Ishikawa Y, Sugano H. Excess of rare cancers in Werner syndrome. (adult progeria). Cancer Epidemiol Biomarkers Prev. (1996) 5:239–46.
119. Murata K, Hatamochi A, Shinkai H, Ishikawa Y, Kawaguchi N, Goto M. A case of Werner's syndrome associated with osteosarcoma. J Dermatol. (1999) 26:682–6. doi: 10.1111/j.1346-8138.1999.tb02072.x
120. Ishikawa Y, Miller RW, Machinami R, Sugano H, Goto M. Atypical osteosarcomas in Werner Syndrome. (adult progeria). JPN J Cancer Res. (2000) 91:1345–9. doi: 10.1111/j.1349-7006.2000.tb00924.x
121. Ozcan A, Acer H, Ciraci S, Gumus H, Karakukcu M, Patiroglu T, et al. Neuroblastoma in a child with wolf-hirschhorn syndrome. J Pediatr Hematol Oncol. (2017) 39:e224–6. doi: 10.1097/MPH.0000000000000768
122. Bianchi C, Giammusso V, Berti N, Vassallo A. [Medulloblastoma in a patient with xeroderma pigmentosum]. Pathologica. (1979) 71:697–701.
123. Baud O, Cormier-Daire V, Lyonnet S, Desjardins L, Turleau C, Doz F. Dysmorphic phenotype and neurological impairment in 22 retinoblastoma patients with constitutional cytogenetic 13q deletion. Clin Genet. (1999) 55:478–82. doi: 10.1034/j.1399-0004.1999.550614.x
124. Wu F, Zhang Y, Sun B, McMahon AP, Wang Y. Hedgehog signaling: from basic biology to cancer therapy. Cell Chem Biol. (2017) 24:252–80. doi: 10.1016/j.chembiol.2017.02.010
125. Xu J, Margol AS, Shukla A, Ren X, Finlay JL, Krieger MD, et al. Disseminated medulloblastoma in a child with germline BRCA2 6174delT mutation and without fanconi anemia. Front Oncol. (2015) 5:191. doi: 10.3389/fonc.2015.00191
126. Muskens IS, de Smith AJ, Zhang C, Hansen HM, Morimoto L, Metayer C. Germline cancer predisposition variants and pediatric glioma: a population-based study in California. Neuro Oncol. (2020) 22:864–74. doi: 10.1093/neuonc/noaa014
127. Kline CN, Joseph NM, Grenert JP, van Ziffle J, Talevich E, Onodera C, et al. Targeted next-generation sequencing of pediatric neuro-oncology patients improves diagnosis, identifies pathogenic germline mutations, and directs targeted therapy. Neuro Oncol. (2017) 19:699–709. doi: 10.1093/neuonc/now254
128. Ballinger ML, Goode DL, Ray-Coquard I, James PA, Mitchell G, Niedermayr E, et al. Monogenic and polygenic determinants of sarcoma risk: an international genetic study. Lancet Oncol. (2016) 17:1261–71. doi: 10.1016/S1470-2045(16)30147-4
129. Mirabello L, Zhu B, Koster R, Karlins E, Dean M, Yeager M, et al. Frequency of pathogenic germline variants in cancer-susceptibility genes in patients with osteosarcoma. JAMA Oncol. (2020) 6:724–34. doi: 10.1001/jamaoncol.2020.0197
130. Brohl AS, Patidar R, Turner CE, Wen X, Song YK, Wei JS, et al. Frequent inactivating germline mutations in DNA repair genes in patients with Ewing sarcoma. Genet Med. (2017) 19:955–8. doi: 10.1038/gim.2016.206
131. Gadd S, Huff V, Walz AL, Ooms A, Armstrong AE, Gerhard DS, et al. A children's oncology group and TARGET initiative exploring the genetic landscape of Wilms tumor. Nat Genet. (2017) 49:1487–94. doi: 10.1038/ng.3940
132. Ciceri S, Gamba B, Corbetta P, Mondini P, Terenziani M, Catania S, et al. Genetic and epigenetic analyses guided by high resolution whole-genome SNP array reveals a possible role of CHEK2 in Wilms tumour susceptibility. Oncotarget. (2018) 9:34079–89. doi: 10.18632/oncotarget.26123
133. Chan SH, Lim WK, Ishak NDB, Li ST, Goh WL, Tan GS, et al. Germline mutations in cancer predisposition genes are frequent in sporadic sarcomas. Sci Rep. (2017) 7:10660. doi: 10.1038/s41598-017-10333-x
134. Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. (2008) 455:930–5. doi: 10.1038/nature07261
135. Grobner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature. (2018) 555:321–7. doi: 10.1038/nature25480
136. Dommering CJ, Mol BM, Moll AC, Burton M, Cloos J, Dorsman JC, et al. RB1 mutation spectrum in a comprehensive nationwide cohort of retinoblastoma patients. J Med Genet. (2014) 51:366–74. doi: 10.1136/jmedgenet-2014-102264
137. Wegert J, Ishaque N, Vardapour R, Georg C, Gu Z, Bieg M, et al. Mutations in the SIX1/2 pathway and the DROSHA/DGCR8 miRNA microprocessor complex underlie high-risk blastemal type Wilms tumors. Cancer Cell. (2015) 27:298–311. doi: 10.1016/j.ccell.2015.01.002
138. Mirabello L, Yeager M, Mai PL, Gastier-Foster JM, Gorlick R, Khanna C, et al. Germline TP53 variants and susceptibility to osteosarcoma. J Natl Cancer Inst. (2015) 107:djv101. doi: 10.1093/jnci/djv101
139. Diller L, Sexsmith E, Gottlieb A, Li FP, Malkin D. Germline p53 mutations are frequently detected in young children with rhabdomyosarcoma. J Clin Invest. (1995) 95:1606–11. doi: 10.1172/JCI117834
140. Jiang Y, Janku F, Subbiah V, Angelo LS, Naing A, Anderson PM, et al. Germline PTPRD mutations in Ewing sarcoma: biologic and clinical implications. Oncotarget. (2013) 4:884–9. doi: 10.18632/oncotarget.1021
141. Yeh IT, Lenci RE, Qin Y, Buddavarapu K, Ligon AH, Leteurtre E. A germline mutation of the KIF1B beta gene on 1p36 in a family with neural and nonneural tumors. Hum Genet. (2008) 124:279–85. doi: 10.1007/s00439-008-0553-1
142. de Kock L, Yoon JY, Apellaniz-Ruiz M, Pelletier D, McCluggage WG, Stewart CJR, et al. Significantly greater prevalence of DICER1 alterations in uterine embryonal rhabdomyosarcoma compared to adenosarcoma. Mod Pathol. (2020) 33:1207–19. doi: 10.1038/s41379-019-0436-0
143. Begemann M, Waszak SM, Robinson GW, Jager N, Sharma T, Knopp C, et al. Germline GPR161 mutations predispose to pediatric medulloblastoma. J Clin Oncol. (2020) 38:43–50. doi: 10.1200/JCO.19.00577
144. Hanks S, Perdeaux ER, Seal S, Ruark E, Mahamdallie SS, Murray A, et al. Germline mutations in the PAF1 complex gene CTR9 predispose to Wilms tumour. Nat Commun. (2014) 5:4398. doi: 10.1038/ncomms5398
145. Perri P, Bachetti T, Longo L, Matera I, Seri M, Tonini GP, et al. PHOX2B mutations and genetic predisposition to neuroblastoma. Oncogene. (2005) 24:3050–3. doi: 10.1038/sj.onc.1208532
146. Parsons DW, Roy A, Yang Y, Wang T, Scollon S, Bergstrom K, et al. Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol. (2016) 2:616–24. doi: 10.1001/jamaoncol.2015.5699
147. Mahamdallie S, Yost S, Poyastro-Pearson E, Holt E, Zachariou A, Seal S, et al. Identification of new wilms tumour predisposition genes: an exome sequencing study. Lancet Child Adolesc Health. (2019) 3:322–31. doi: 10.1016/S2352-4642(19)30018-5
148. Mahamdallie SS, Hanks S, Karlin KL, Zachariou A, Perdeaux ER, Ruark E, et al. Mutations in the transcriptional repressor REST predispose to Wilms tumor. Nat Genet. (2015) 47:1471–4. doi: 10.1038/ng.3440
149. Little SE, Hanks SP, King-Underwood L, Jones C, Rapley EA, Rahman N, et al. Frequency and heritability of WT1 mutations in nonsyndromic Wilms' tumor patients: a UK children's cancer study group study. J Clin Oncol. (2004) 22:4140–6. doi: 10.1200/JCO.2004.02.136
150. Segers H, Kersseboom R, Alders M, Pieters R, Wagner A, van den Heuvel-Eibrink MM. Frequency of WT1 and 11p15 constitutional aberrations and phenotypic correlation in childhood Wilms tumour patients. Eur J Cancer. (2012) 48:3249–56. doi: 10.1016/j.ejca.2012.06.008
151. Scott RH, Douglas J, Baskcomb L, Huxter N, Barker K, Hanks S, et al. Constitutional 11p15 abnormalities, including heritable imprinting center mutations, cause nonsyndromic Wilms tumor. Nat Genet. (2008) 40:1329–34. doi: 10.1038/ng.243
152. Machiela MJ, Grunewald TGP, Surdez D, Reynaud S, Mirabeau O, Karlins E, et al. Genome-wide association study identifies multiple new loci associated with Ewing sarcoma susceptibility. Nat Commun. (2018) 9:3184. doi: 10.1038/s41467-018-05537-2
153. Turnbull C, Perdeaux ER, Pernet D, Naranjo A, Renwick A, Seal S, et al. A genome-wide association study identifies susceptibility loci for Wilms tumor. Nat Genet. (2012) 44:681–4. doi: 10.1038/ng.2251
154. Dahlin AM, Wibom C, Andersson U, Bybjerg-Grauholm J, Deltour I, Hougaard DM, et al. A genome-wide association study on medulloblastoma. J Neurooncol. (2020) 147:309–15. doi: 10.1007/s11060-020-03424-9
155. Diskin SJ, Capasso M, Schnepp RW, Cole KA, Attiyeh EF, Hou C, et al. Common variation at 6q16 within HACE1 and LIN28B influences susceptibility to neuroblastoma. Nat Genet. (2012) 44:1126–30. doi: 10.1038/ng.2387
156. Capasso M, Diskin S, Cimmino F, Acierno G, Totaro F, Petrosino G, et al. Common genetic variants in NEFL influence gene expression and neuroblastoma risk. Cancer Res. (2014) 74:6913–24. doi: 10.1158/0008-5472.CAN-14-0431
157. Capasso M, Devoto M, Hou C, Asgharzadeh S, Glessner JT, Attiyeh EF, et al. Common variations in BARD1 influence susceptibility to high-risk neuroblastoma. Nat Genet. (2009) 41:718–23. doi: 10.1038/ng.374
158. Fu W, Zhu J, Xiong SW, Jia W, Zhao Z, Zhu SB, et al. BARD1 gene polymorphisms confer nephroblastoma susceptibility. EBioMedicine. (2017) 16:101–5. doi: 10.1016/j.ebiom.2017.01.038
159. McDaniel LD, Conkrite KL, Chang X, Capasso M, Vaksman Z, Oldridge DA, et al. Common variants upstream of MLF1 at 3q25 and within CPZ at 4p16 associated with neuroblastoma. PLoS Genet. (2017) 13:e1006787. doi: 10.1371/journal.pgen.1006787
160. Maris JM, Mosse YP, Bradfield JP, Hou C, Monni S, Scott RH, et al. Chromosome 6p22 locus associated with clinically aggressive neuroblastoma. N Engl J Med. (2008) 358:2585–93. doi: 10.1056/NEJMoa0708698
161. Nguyen le B, Diskin SJ, Capasso M, Wang K, Diamond MA, Glessner J, et al. Phenotype restricted genome-wide association study using a gene-centric approach identifies three low-risk neuroblastoma susceptibility loci. PLoS Genet. (2011) 7:e1002026. doi: 10.1371/journal.pgen.1002026
162. Dahlin AM, Hollegaard MV, Wibom C, Andersson U, Hougaard DM, Deltour I, et al. CCND2, CTNNB1, DDX3X, GLI2, SMARCA4, MYC, MYCN, PTCH1, TP53, and MLL2 gene variants and risk of childhood medulloblastoma. J Neurooncol. (2015) 125:75–8. doi: 10.1007/s11060-015-1891-1
163. Jiao Y, Jiang Z, Wu Y, Chen X, Xiao X, Yu H. A functional polymorphism (rs937283) in the MDM2 promoter region is associated with poor prognosis of retinoblastoma in chinese han population. Sci Rep. (2016) 6:31240. doi: 10.1038/srep31240
164. Yu F, Jiang Z, Song A. Association of rs11801299 and rs1380576 polymorphisms at MDM4 with risk, clinicopathological features and prognosis in patients with retinoblastoma. Cancer Epidemiol. (2019) 58:153–9. doi: 10.1016/j.canep.2018.12.010
165. Walsh KM, Whitehead TP, de Smith AJ, Smirnov IV, Park M, Endicott AA, et al. Common genetic variants associated with telomere length confer risk for neuroblastoma and other childhood cancers. Carcinogenesis. (2016) 37:576–82. doi: 10.1093/carcin/bgw037
166. Zhang C, Hansen HM, Semmes EC, Gonzalez-Maya J, Morimoto L, Wei Q, et al. Common genetic variation and risk of osteosarcoma in a multi-ethnic pediatric and adolescent population. Bone. (2020) 130:115070. doi: 10.1016/j.bone.2019.115070
167. Chang X, Zhao Y, Hou C, Glessner J, McDaniel L, Diamond MA, et al. Common variants in MMP20 at 11q22.2 predispose to 11q deletion and neuroblastoma risk. Nat Commun. (2017) 8:569. doi: 10.1038/s41467-017-00408-8
168. Hungate EA, Applebaum MA, Skol AD, Vaksman Z, Diamond M, McDaniel L, et al. Evaluation of genetic predisposition for MYCN-amplified neuroblastoma. J Natl Cancer Inst. (2017) 109:djx093. doi: 10.1093/jnci/djx093
169. Carvalho IN, Reis AH, Cabello PH, Vargas FR. Polymorphisms of CDKN1A gene and risk of retinoblastoma. Carcinogenesis. (2013) 34:2774–7. doi: 10.1093/carcin/bgt308
170. Capasso M, McDaniel LD, Cimmino F, Cirino A, Formicola D, Russell MR, et al. The functional variant rs34330 of CDKN1B is associated with risk of neuroblastoma. J Cell Mol Med. (2017) 21:3224–30. doi: 10.1111/jcmm.13226
171. Fu W, Zhuo Z, Hua RX, Fu K, Jia W, Zhu J, et al. Association of KRAS and NRAS gene polymorphisms with Wilms tumor risk: a four-center case-control study. Aging (Albany NY). (2019) 11:1551–63. doi: 10.18632/aging.101855
172. Postel-Vinay S, Veron AS, Tirode F, Pierron G, Reynaud S, Kovar H, et al. Common variants near TARDBP and EGR2 are associated with susceptibility to Ewing sarcoma. Nat Genet. (2012) 44:323–7. doi: 10.1038/ng.1085
173. Savage SA, Mirabello L, Wang Z, Gastier-Foster JM, Gorlick R, Khanna C, et al. Genome-wide association study identifies two susceptibility loci for osteosarcoma. Nat Genet. (2013) 45:799–803. doi: 10.1038/ng.2645
174. Johnson KM, Mahler NR, Saund RS, Theisen ER, Taslim C, Callender NW, et al. Role for the EWS domain of EWS/FLI in binding GGAA-microsatellites required for Ewing sarcoma anchorage independent growth. Proc Natl Acad Sci USA. (2017) 114:9870–5. doi: 10.1073/pnas.1701872114
175. Wang K, Diskin SJ, Zhang H, Attiyeh EF, Winter C, Hou C, et al. Integrative genomics identifies LMO1 as a neuroblastoma oncogene. Nature. (2011) 469:216–20. doi: 10.1038/nature09609
176. Mirabello L, Koster R, Moriarity BS, Spector LG, Meltzer PS, Gary J, et al. A genome-wide scan identifies variants in NFIB associated with metastasis in patients with osteosarcoma. Cancer Discov. (2015) 5:920–31. doi: 10.1158/2159-8290.CD-15-0125
177. Gamazon ER, Pinto N, Konkashbaev A, Im HK, Diskin SJ, London WB, et al. Trans-population analysis of genetic mechanisms of ethnic disparities in neuroblastoma survival. J Natl Cancer Inst. (2013) 105:302–9. doi: 10.1093/jnci/djs503
178. Grunewald TG, Bernard V, Gilardi-Hebenstreit P, Raynal V, Surdez D, Aynaud MM, et al. Chimeric EWSR1-FLI1 regulates the Ewing sarcoma susceptibility gene EGR2 via a GGAA microsatellite. Nat Genet. (2015) 47:1073–8. doi: 10.1038/ng.3363
179. Ramroop JR, Gerber MM, Toland AE. Germline variants impact somatic events during tumorigenesis. Trends Genet. (2019) 35:515–26. doi: 10.1016/j.tig.2019.04.005
180. Rivero-Hinojosa S, Kinney N, Garner HR, Rood BR. Germline microsatellite genotypes differentiate children with medulloblastoma. Neuro Oncol. (2020) 22:152–62. doi: 10.1093/neuonc/noz179
181. McGuire CS, Sainani KL, Fisher PG. Both location and age predict survival in ependymoma: a SEER study. Pediatr Blood Cancer. (2009) 52:65–9. doi: 10.1002/pbc.21806
182. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. (2016) 131:803–20. doi: 10.1007/s00401-016-1545-1
183. Matthay KK, Maris JM, Schleiermacher G, Nakagawara A, Mackall CL, Diller L, et al. Neuroblastoma. Nat Rev Dis Primers. (2016) 2:16078. doi: 10.1038/nrdp.2016.78
184. Capasso M, Diskin SJ. Genetics and genomics of neuroblastoma. Cancer Treat Res. (2010) 155:65–84. doi: 10.1007/978-1-4419-6033-7_4
185. Amiel J, Laudier B, Attie-Bitach T, Trang H, de Pontual L, Gener B, et al. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat Genet. (2003) 33:459–61. doi: 10.1038/ng1130
186. Pattyn A, Morin X, Cremer H, Goridis C, Brunet JF. The homeobox gene Phox2b is essential for the development of autonomic neural crest derivatives. Nature. (1999) 399:366–70. doi: 10.1038/20700
187. Bachetti T, Ceccherini I. Causative and common PHOX2B variants define a broad phenotypic spectrum. Clin Genet. (2020) 97:103–13. doi: 10.1111/cge.13633
188. Ritenour LE, Randall MP, Bosse KR, Diskin SJ. Genetic susceptibility to neuroblastoma: current knowledge and future directions. Cell Tissue Res. (2018) 372:287–307. doi: 10.1007/s00441-018-2820-3
189. van Limpt V, Schramm A, van Lakeman A, Sluis P, Chan A, van Noesel M, et al. The Phox2B homeobox gene is mutated in sporadic neuroblastomas. Oncogene. (2004) 23:9280–8. doi: 10.1038/sj.onc.1208157
190. Bresler SC, Weiser DA, Huwe PJ, Park JH, Krytska K, Ryles H, et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell. (2014) 26:682–94. doi: 10.1016/j.ccell.2014.09.019
191. Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. (2008) 455:967–70. doi: 10.1038/nature07398
192. De Mariano M, Gallesio R, Chierici M, Furlanello C, Conte M, Garaventa A, et al. Identification of GALNT14 as a novel neuroblastoma predisposition gene. Oncotarget. (2015) 6:26335–46. doi: 10.18632/oncotarget.4501
193. Lo Cunsolo C, Iolascon A, Cavazzana A, Cusano R, Strigini P, Mazzocco K, et al. Neuroblastoma in two siblings supports the role of 1p36 deletion in tumor development. Cancer Genet Cytogenet. (1999) 109:126–30. doi: 10.1016/s0165-4608(98)00154-x
194. Maris JM, Weiss MJ, Mosse Y, Hii G, Guo C, White PS, et al. Evidence for a hereditary neuroblastoma predisposition locus at chromosome 16p12-13. Cancer Res. (2002) 62:6651–8.
195. Perri P, Longo L, Cusano R, McConville CM, Rees SA, Devoto M, et al. Weak linkage at 4p16 to predisposition for human neuroblastoma. Oncogene. (2002) 21:8356–60. doi: 10.1038/sj.onc.1206009
196. Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. (2013) 45:279–84. doi: 10.1038/ng.2529
197. Lasorsa VA, Formicola D, Pignataro P, Cimmino F, Calabrese FM, Mora J, et al. Exome and deep sequencing of clinically aggressive neuroblastoma reveal somatic mutations that affect key pathways involved in cancer progression. Oncotarget. (2016) 7:21840–52. doi: 10.18632/oncotarget.8187
198. Capasso M, Diskin SJ, Totaro F, Longo L, De Mariano M, Russo R, et al. Replication of GWAS-identified neuroblastoma risk loci strengthens the role of BARD1 and affirms the cumulative effect of genetic variations on disease susceptibility. Carcinogenesis. (2013) 34:605–11. doi: 10.1093/carcin/bgs380
199. Tonini GP, Capasso M. Genetic predisposition and chromosome instability in neuroblastoma. Cancer Metastasis Rev. (2020) 39:275–85. doi: 10.1007/s10555-020-09843-4
200. Mody RJ, Wu YM, Lonigro RJ, Cao X, Roychowdhury S, Vats P, et al. Integrative clinical sequencing in the management of refractory or relapsed cancer in youth. JAMA. (2015) 314:913–25. doi: 10.1001/jama.2015.10080
201. Diskin SJ, Capasso M, Diamond M, Oldridge DA, Conkrite K, Bosse KR, et al. Rare variants in TP53 and susceptibility to neuroblastoma. J Natl Cancer Inst. (2014) 106:dju047. doi: 10.1093/jnci/dju047
202. Russell MR, Penikis A, Oldridge DA, Alvarez-Dominguez JR, McDaniel L, Diamond M, et al. CASC15-S is a tumor suppressor lncRNA at the 6p22 neuroblastoma susceptibility locus. Cancer Res. (2015) 75:3155–66. doi: 10.1158/0008-5472.CAN-14-3613
203. Pandey GK, Mitra S, Subhash S, Hertwig F, Kanduri M, Mishra K, et al. The risk-associated long noncoding RNA NBAT-1 controls neuroblastoma progression by regulating cell proliferation and neuronal differentiation. Cancer Cell. (2014) 26:722–37. doi: 10.1016/j.ccell.2014.09.014
204. Cimmino F, Formicola D, Capasso M. Dualistic role of BARD1 in cancer. Genes. (2017) 8:375. doi: 10.3390/genes8120375
205. Cimmino F, Avitabile M, Diskin SJ, Vaksman Z, Pignataro P, Formicola D, et al. Fine mapping of 2q35 high-risk neuroblastoma locus reveals independent functional risk variants and suggests full-length BARD1 as tumor-suppressor. Int J Cancer. (2018) 143:2828–37. doi: 10.1002/ijc.31822
206. Cimmino F, Avitabile M, Lasorsa VA, Pezone L, Cardinale A, Montella A, et al. Functional characterization of full-length BARD1 strengthens its role as a tumor suppressor in neuroblastoma. J Cancer. (2020) 11:1495–504. doi: 10.7150/jca.36164
207. Ryser S, Dizin E, Jefford CE, Delaval B, Gagos S, Christodoulidou A, et al. Distinct roles of BARD1 isoforms in mitosis: full-length BARD1 mediates Aurora B degradation, cancer-associated BARD1beta scaffolds aurora B and BRCA2. Cancer Res. (2009) 69:1125–34. doi: 10.1158/0008-5472.CAN-08-2134
208. Bosse KR, Diskin SJ, Cole KA, Wood AC, Schnepp RW, Norris G, et al. Common variation at BARD1 results in the expression of an oncogenic isoform that influences neuroblastoma susceptibility and oncogenicity. Cancer Res. (2012) 72:2068–78. doi: 10.1158/0008-5472.CAN-11-3703
209. Oldridge DA, Wood AC, Weichert-Leahey N, Crimmins I, Sussman R, Winter C, et al. Genetic predisposition to neuroblastoma mediated by a LMO1 super-enhancer polymorphism. Nature. (2015) 528:418–21. doi: 10.1038/nature15540
210. Molenaar JJ, Domingo-Fernandez R, Ebus ME, Lindner S, Koster J, Drabek K, et al. LIN28B induces neuroblastoma and enhances MYCN levels via let-7 suppression. Nat Genet. (2012) 44:1199–206. doi: 10.1038/ng.2436
211. Powers JT, Tsanov KM, Pearson DS, Roels F, Spina CS, Ebright R, et al. Multiple mechanisms disrupt the let-7 microRNA family in neuroblastoma. Nature. (2016) 535:246–51. doi: 10.1038/nature18632
212. Chang X, Bakay M, Liu Y, Glessner J, Rathi KS, Hou C, et al. Mitochondrial DNA haplogroups and susceptibility to neuroblastoma. J Natl Cancer Inst. (2020) djaa024. doi: 10.1093/jnci/djaa024
213. Totaro F, Cimmino F, Pignataro P, Acierno G, De Mariano M, Longo L, et al. Impact of interleukin-6−174 G>C gene promoter polymorphism on neuroblastoma. PLoS ONE. (2013) 8:e76810. doi: 10.1371/journal.pone.0076810
214. Avitabile M, Lasorsa VA, Cantalupo S, Cardinale A, Cimmino F, Montella A, et al. Association of PARP1 polymorphisms with response to chemotherapy in patients with high-risk neuroblastoma. J Cell Mol Med. (2020) 24:4072–81. doi: 10.1111/jcmm.15058
215. Testori A, Lasorsa VA, Cimmino F, Cantalupo S, Cardinale A, Avitabile M, et al. Exploring shared susceptibility between two neural crest cells originating conditions: neuroblastoma and congenital heart disease. Genes. (2019) 10:663. doi: 10.3390/genes10090663
216. Avitabile M, Succoio M, Testori A, Cardinale A, Vaksman Z, Lasorsa VA, et al. Neural crest-derived tumor neuroblastoma and melanoma share 1p13.2 as susceptibility locus that shows a long-range interaction with the SLC16A1 gene. Carcinogenesis. (2019) 41:284–95. doi: 10.1093/carcin/bgz153
217. Diskin SJ, Hou C, Glessner JT, Attiyeh EF, Laudenslager M, Bosse K, et al. Copy number variation at 1q21.1 associated with neuroblastoma. Nature. (2009) 459:987–91. doi: 10.1038/nature08035
218. Egolf LE, Vaksman Z, Lopez G, Rokita JL, Modi A, Basta PV, et al. Germline 16p11.2 microdeletion predisposes to neuroblastoma. Am J Hum Genet. (2019) 105:658–68. doi: 10.1016/j.ajhg.2019.07.020
219. Gambale A, Russo R, Andolfo I, Quaglietta L, De Rosa G, Contestabile V, et al. Germline mutations and new copy number variants among 40 pediatric cancer patients suspected for genetic predisposition. Clin Genet. (2019) 96:359–65. doi: 10.1111/cge.13600
220. Dimaras H, Corson TW, Cobrinik D, White A, Zhao J, Munier FL, et al. Retinoblastoma. Nat Rev Dis Primers. (2015) 1:15021. doi: 10.1038/nrdp.2015.21
221. Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. (1971) 68:820–3. doi: 10.1073/pnas.68.4.820
222. Lohmann DR, Gallie BL. Retinoblastoma: revisiting the model prototype of inherited cancer. Am J Med Genet C Semin Med Genet. (2004) 129C:23–28. doi: 10.1002/ajmg.c.30024
223. Taylor M, Dehainault C, Desjardins L, Doz F, Levy C, Sastre X, et al. Genotype-phenotype correlations in hereditary familial retinoblastoma. Hum Mutat. (2007) 28:284–93. doi: 10.1002/humu.20443
224. Imperatore V, Pinto AM, Gelli E, Trevisson E, Morbidoni V, Frullanti E, et al. Parent-of-origin effect of hypomorphic pathogenic variants and somatic mosaicism impact on phenotypic expression of retinoblastoma. Eur J Hum Genet. (2018) 26:1026–37. doi: 10.1038/s41431-017-0054-6
225. Castera L, Sabbagh A, Dehainault C, Michaux D, Mansuet-Lupo A, Patillon B, et al. MDM2 as a modifier gene in retinoblastoma. J Natl Cancer Inst. (2010) 102:1805–8. doi: 10.1093/jnci/djq416
226. de Oliveira Reis AH, de Carvalho IN, de Sousa Damasceno PB, Ferman SE, Lucena E, Lopez-Camelo JS, et al. Influence of MDM2 and MDM4 on development and survival in hereditary retinoblastoma. Pediatr Blood Cancer. (2012) 59:39–43. doi: 10.1002/pbc.24014
227. Dehainault C, Garancher A, Castera L, Cassoux N, Aerts I, Doz F, et al. The survival gene MED4 explains low penetrance retinoblastoma in patients with large RB1 deletion. Hum Mol Genet. (2014) 23:5243–50. doi: 10.1093/hmg/ddu245
228. Epistolato MC, Disciglio V, Livide G, Berchialla P, Mencarelli MA, Marozza A, et al. p53 Arg72Pro and MDM2 309 SNPs in hereditary retinoblastoma. J Hum Genet. (2011) 56:685–6. doi: 10.1038/jhg.2011.82
229. Indovina P, Acquaviva A, De Falco G, Rizzo V, Onnis A, Luzzi A, et al. Downregulation and aberrant promoter methylation of p16INK4A: a possible novel heritable susceptibility marker to retinoblastoma. J Cell Physiol. (2010) 223:143–50. doi: 10.1002/jcp.22019
230. Akdeniz D, Tuncer SB, Kebudi R, Celik B, Kuru G, Kilic S, et al. Investigation of new candidate genes in retinoblastoma using the TruSight one “clinical exome” gene panel. Mol Genet Genomic Med. (2019) 7:e785. doi: 10.1002/mgg3.785
231. Aerts I, Lumbroso-Le Rouic L, Gauthier-Villars M, Brisse H, Doz F, Desjardins L. Retinoblastoma. Orphanet J Rare Dis. (2006) 1:31. doi: 10.1186/1750-1172-1-31
232. Bojinova RI, Schorderet DF, Addor MC, Gaide AC, Thonney F, Pescia G, et al. Further delineation of the facial 13q14 deletion syndrome in 13 retinoblastoma patients. Ophthalmic Genet. (2001) 22:11–8. doi: 10.1076/opge.22.1.11.2235
233. Quelin C, Bendavid C, Dubourg C, de la Rochebrochard C, Lucas J, Henry C, et al. Twelve new patients with 13q deletion syndrome: genotype-phenotype analyses in progress. Eur J Med Genet. (2009) 52:41–46. doi: 10.1016/j.ejmg.2008.10.002
234. Dimaras H, Kimani K, Dimba EA, Gronsdahl P, White A, Chan HS, et al. Retinoblastoma. Lancet. (2012) 379:1436–46. doi: 10.1016/S0140-6736(11)61137-9
235. Rushlow DE, Mol BM, Kennett JY, Yee S, Pajovic S, Theriault BL, et al. Characterisation of retinoblastomas without RB1 mutations: genomic, gene expression, and clinical studies. Lancet Oncol. (2013) 14:327–34. doi: 10.1016/S1470-2045(13)70045-7
236. Chen Z, Moran K, Richards-Yutz J, Toorens E, Gerhart D, Ganguly T, et al. Enhanced sensitivity for detection of low-level germline mosaic RB1 mutations in sporadic retinoblastoma cases using deep semiconductor sequencing. Hum Mutat. (2014) 35:384–91. doi: 10.1002/humu.22488
237. Amitrano S, Marozza A, Somma S, Imperatore V, Hadjistilianou T, De Francesco S, et al. Next generation sequencing in sporadic retinoblastoma patients reveals somatic mosaicism. Eur J Hum Genet. (2015) 23:1523–30. doi: 10.1038/ejhg.2015.6
238. Cygan KJ, Soemedi R, Rhine CL, Profeta A, Murphy EL, Murray MF, et al. Defective splicing of the RB1 transcript is the dominant cause of retinoblastomas. Hum Genet. (2017) 136:1303–12. doi: 10.1007/s00439-017-1833-4
239. Kivela T, Tuppurainen K, Riikonen P, Vapalahti M. Retinoblastoma associated with chromosomal 13q14 deletion mosaicism. Ophthalmology. (2003) 110:1983–8. doi: 10.1016/S0161-6420(03)00484-6
240. Krepischi AC, Capelli LP, Silva AG, de Araujo ES, Pearson PL, Heck B, et al. Large germline copy number variations as predisposing factor in childhood neoplasms. Future Oncol. (2014) 10:1627–33. doi: 10.2217/fon.14.41
241. PDQ Pediatric Treatment Editorial Board. Wilms tumor and other childhood kidney tumors treatment (PDQ®): health professional version. In: PDQ Cancer Information Summaries [Internet]. Bethesda, MD: National Cancer Institute (US) (2002).
242. Dome JS, Huff V. Wilms tumor predisposition. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (2020).
243. Call KM, Glaser T, Ito CY, Buckler AJ, Pelletier J, Haber DA, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 wilms' tumor locus. Cell. (1990) 60:509–20. doi: 10.1016/0092-8674(90)90601-a
244. Fischbach BV, Trout KL, Lewis J, Luis CA, Sika M. WAGR syndrome: a clinical review of 54 cases. Pediatrics. (2005) 116:984–8. doi: 10.1542/peds.2004-0467
245. Karnik P, Chen P, Paris M, Yeger H, Williams BR. Loss of heterozygosity at chromosome 11p15 in wilms tumors: identification of two independent regions. Oncogene. (1998) 17:237–40. doi: 10.1038/sj.onc.1201959
246. Scott RH, Murray A, Baskcomb L, Turnbull C, Loveday C, Al-Saadi R, et al. Stratification of wilms tumor by genetic and epigenetic analysis. Oncotarget. (2012) 3:327–35. doi: 10.18632/oncotarget.468
247. Rapley EA, Barfoot R, Bonaiti-Pellie C, Chompret A, Foulkes W, Perusinghe N, et al. Evidence for susceptibility genes to familial Wilms tumour in addition to WT1, FWT1 and FWT2. Br J Cancer. (2000) 83:177–83. doi: 10.1054/bjoc.2000.1283
248. Martins AG, Pinto AT, Domingues R, Cavaco BM. Identification of a novel CTR9 germline mutation in a family with Wilms tumor. Eur J Med Genet. (2018) 61:294–9. doi: 10.1016/j.ejmg.2017.12.010
249. Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell. (2005) 121:645–57. doi: 10.1016/j.cell.2005.03.013
250. Halliday BJ, Fukuzawa R, Markie DM, Grundy RG, Ludgate JL, Black MA, et al. Germline mutations and somatic inactivation of TRIM28 in Wilms tumour. PLoS Genet. (2018) 14:e1007399. doi: 10.1371/journal.pgen.1007399
251. Diets IJ, Hoyer J, Ekici AB, Popp B, Hoogerbrugge N, van Reijmersdal SV, et al. TRIM28 haploinsufficiency predisposes to Wilms tumor. Int J Cancer. (2019) 145:941–51. doi: 10.1002/ijc.32167
252. Southey MC, Goldgar DE, Winqvist R, Pylkas K, Couch F, Tischkowitz M, et al. PALB2, CHEK2 and ATM rare variants and cancer risk: data from COGS. J Med Genet. (2016) 53:800–11.
253. Viot-Szoboszlai G, Amiel J, Doz F, Prieur M, Couturier J, Zucker JN, et al. Wilms' tumor and gonadal dysgenesis in a child with the 2q37.1 deletion syndrome. Clin Genet. (1998) 53:278–80. doi: 10.1111/j.1399-0004.1998.tb02696.x
254. Drake KM, Ruteshouser EC, Natrajan R, Harbor P, Wegert J, Gessler M, et al. Loss of heterozygosity at 2q37 in sporadic Wilms' tumor: putative role for miR-562. Clin Cancer Res. (2009) 15:5985–92. doi: 10.1158/1078-0432.CCR-09-1065
255. Rivera H, Ruiz C, Garcia-Cruz D, Rolon A, Arroyo J, Cantu JM. Constitutional mosaic t(2;7)(q33;p22) and other rearrangements in a girl with Wilms' tumor. Ann Genet. (1985) 28:52–4.
256. Wilmore HP, White GF, Howell RT, Brown KW. Germline and somatic abnormalities of chromosome 7 in Wilms' tumor. Cancer Genet Cytogenet. (1994) 77:93–8. doi: 10.1016/0165-4608(94)90221-6
257. Slade I, Stephens P, Douglas J, Barker K, Stebbings L, Abbaszadeh F, et al. Constitutional translocation breakpoint mapping by genome-wide paired-end sequencing identifies HACE1 as a putative Wilms tumour susceptibility gene. J Med Genet. (2010) 47:342–7. doi: 10.1136/jmg.2009.072983
258. Williams RD, Chagtai T, Alcaide-German M, Apps J, Wegert J, Popov S, et al. Multiple mechanisms of MYCN dysregulation in Wilms tumour. Oncotarget. (2015) 6:7232–43. doi: 10.18632/oncotarget.3377
259. Gianferante DM, Mirabello L, Savage SA. Germline and somatic genetics of osteosarcoma - connecting aetiology, biology and therapy. Nat Rev Endocrinol. (2017) 13:480–91. doi: 10.1038/nrendo.2017.16
260. Skapek SX, Ferrari A, Gupta AA, Lupo PJ, Butler E, Shipley J, et al. Rhabdomyosarcoma. Nat Rev Dis Primers. (2019) 5:1. doi: 10.1038/s41572-018-0051-2
261. Perez EA, Kassira N, Cheung MC, Koniaris LG, Neville HL, Sola JE. Rhabdomyosarcoma in children: a SEER population based study. J Surg Res. (2011) 170:e243–251. doi: 10.1016/j.jss.2011.03.001
262. Meza JL, Anderson J, Pappo AS, Meyer WH, Children's Oncology G. Analysis of prognostic factors in patients with nonmetastatic rhabdomyosarcoma treated on intergroup rhabdomyosarcoma studies III and IV: the Children's Oncology Group. J Clin Oncol. (2006) 24:3844–51. doi: 10.1200/JCO.2005.05.3801
263. Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. (2014) 4:216–31. doi: 10.1158/2159-8290.CD-13-0639
264. Dagher R, Helman L. Rhabdomyosarcoma: an overview. Oncologist. (1999) 4:34–44. doi: 10.1634/theoncologist.4-1-34
265. Dehner LP, Jarzembowski JA, Hill DA. Embryonal rhabdomyosarcoma of the uterine cervix: a report of 14 cases and a discussion of its unusual clinicopathological associations. Mod Pathol. (2012) 25:602–14. doi: 10.1038/modpathol.2011.185
266. Martinelli S, McDowell HP, Vigne SD, Kokai G, Uccini S, Tartaglia M, et al. RAS signaling dysregulation in human embryonal Rhabdomyosarcoma. Genes Chromosomes Cancer. (2009) 48:975–82. doi: 10.1002/gcc.20702
267. Paulson V, Chandler G, Rakheja D, Galindo RL, Wilson K, Amatruda JF, et al. High-resolution array CGH identifies common mechanisms that drive embryonal rhabdomyosarcoma pathogenesis. Genes Chromosomes Cancer. (2011) 50:397–408. doi: 10.1002/gcc.20864
268. Gripp KW, Scott CI Jr, Nicholson L, McDonald-McGinn DM, Ozeran JD, Jones MC, et al. Five additional Costello syndrome patients with rhabdomyosarcoma: proposal for a tumor screening protocol. Am J Med Genet. (2002) 108:80–7. doi: 10.1002/ajmg.10241
269. Lupo PJ, Danysh HE, Plon SE, Curtin K, Malkin D, Hettmer S, et al. Family history of cancer and childhood rhabdomyosarcoma: a report from the children's oncology group and the Utah population database. Cancer Med. (2015) 4:781–90. doi: 10.1002/cam4.448
270. Taeubner J, Brozou T, Qin N, Bartl J, Ginzel S, Schaper J, et al. Congenital embryonal rhabdomyosarcoma caused by heterozygous concomitant PTCH1 and PTCH2 germline mutations. Eur J Hum Genet. (2018) 26:137–42. doi: 10.1038/s41431-017-0048-4
271. Grunewald TGP, Cidre-Aranaz F, Surdez D, Tomazou EM, de Alava E, Kovar H, et al. Ewing sarcoma. Nat Rev Dis Primers. (2018) 4:5. doi: 10.1038/s41572-018-0003-x
272. Brohl AS, Solomon DA, Chang W, Wang J, Song Y, Sindiri S, et al. The genomic landscape of the Ewing sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet. (2014) 10:e1004475. doi: 10.1371/journal.pgen.1004475
273. Crompton BD, Stewart C, Taylor-Weiner A, Alexe G, Kurek KC, Calicchio ML, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. (2014) 4:1326–41. doi: 10.1158/2159-8290.CD-13-1037
274. Tirode F, Surdez D, Ma X, Parker M, Le Deley MC, Bahrami A, et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov. (2014) 4:1342–53. doi: 10.1158/2159-8290.CD-14-0622
275. Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature. (1992) 359:162–5. doi: 10.1038/359162a0
276. Guillon N, Tirode F, Boeva V, Zynovyev A, Barillot E, Delattre O. The oncogenic EWS-FLI1 protein binds in vivo GGAA microsatellite sequences with potential transcriptional activation function. PLoS ONE. (2009) 4:e4932. doi: 10.1371/journal.pone.0004932
277. Hutter RV, Francis KC, Foote FW Jr. Ewing's Sarcoma in Siblings: Report of the Second Known Occurrence. Am J Surg. (1964) 107:598–603. doi: 10.1016/0002-9610(64)90328-9
278. Joyce MJ, Harmon DC, Mankin HJ, Suit HD, Schiller AL, Truman JT. Ewing's sarcoma in female siblings. a clinical report and review of the literature. Cancer. (1984) 53:1959–62. doi: 10.1002/1097-0142(19840501)53:9<1959::AID-CNCR2820530926>3.0.CO;2-9
279. Mc CL, Dockerty MB, Ghormley RK. Ewing's sarcoma. Cancer. (1952) 5:85–99. doi: 10.1002/1097-0142(195201)5:1<85::AID-CNCR2820050111>3.0.CO;2-T
280. Ji J, Hemminki K. Familial risk for histology-specific bone cancers: an updated study in Sweden. Eur J Cancer. (2006) 42:2343–9. doi: 10.1016/j.ejca.2005.11.043
281. Rahman N. Realizing the promise of cancer predisposition genes. Nature. (2014) 505:302–8. doi: 10.1038/nature12981
282. Musa J, Cidre-Aranaz F, Aynaud MM, Orth MF, Knott MML, Mirabeau O, et al. Cooperation of cancer drivers with regulatory germline variants shapes clinical outcomes. Nat Commun. (2019) 10:4128. doi: 10.1038/s41467-019-12071-2
283. Saskin A, Seath K, Tihy F, Lemyre E, Davis J, Halal F, et al. PTPRD copy number variants and Ewing's sarcoma: Strengthening the association and therapeutic implications. Cancer Genet. (2019) 235–6:28–30. doi: 10.1016/j.cancergen.2019.03.004
284. Lu C, Zhang J, Nagahawatte P, Easton J, Lee S, Liu Z, et al. The genomic landscape of childhood and adolescent melanoma. J Invest Dermatol. (2015) 135:816–23. doi: 10.1038/jid.2014.425
285. Bucciol G, Willems L, Hauben E, Uyttebroeck A, Proesmans M, Meyts I. Thyroid carcinoma in a child with activated phosphoinositide 3-kinase delta syndrome: somatic effect of a germline mutation. J Clin Immunol. (2017) 37:422–6. doi: 10.1007/s10875-017-0407-5
286. Goldstein AM, Stidd KC, Yang XR, Fraser MC, Tucker MA. Pediatric melanoma in melanoma-prone families. Cancer. (2018) 124:3715–23. doi: 10.1002/cncr.31641
287. Paulson VA, Rudzinski ER, Hawkins DS. Thyroid cancer in the pediatric population. Genes. (2019) 10:723. doi: 10.3390/genes10090723
288. Pellegrini C, Botta F, Massi D, Martorelli C, Facchetti F, Gandini S, et al. MC1R variants in childhood and adolescent melanoma: a retrospective pooled analysis of a multicentre cohort. Lancet Child Adolesc Health. (2019) 3:332–42. doi: 10.1016/S2352-4642(19)30005-7
289. Lee YA, Im SW, Jung KC, Chung EJ, Shin CH, Kim JI, et al. Predominant DICER1 pathogenic variants in pediatric follicular thyroid carcinomas. Thyroid. (2020) 30:1120–31. doi: 10.1089/thy.2019.0233
Keywords: genetic predisposition, germline variants, cancer predisposition genes, pediatric tumors, cancer susceptibility, germline-somatic interaction, SNP, next generation sequencing
Citation: Capasso M, Montella A, Tirelli M, Maiorino T, Cantalupo S and Iolascon A (2020) Genetic Predisposition to Solid Pediatric Cancers. Front. Oncol. 10:590033. doi: 10.3389/fonc.2020.590033
Received: 31 July 2020; Accepted: 08 September 2020;
Published: 28 October 2020.
Edited by:
Angela Mastronuzzi, Bambino Gesù Children Hospital (IRCCS), ItalyReviewed by:
Luigi Boccuto, Clemson University, United StatesJun Wei, University of Texas MD Anderson Cancer Center, United States
Xiaotu Ma, St. Jude Children's Research Hospital, United States
Copyright © 2020 Capasso, Montella, Tirelli, Maiorino, Cantalupo and Iolascon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mario Capasso, bWFyaW8uY2FwYXNzb0B1bmluYS5pdA==