
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 15 January 2021
Sec. Hematologic Malignancies
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.587809
This article is part of the Research Topic New Insights into the Complexity of Tumor Immunology in B-cell Malignancies: Disease Biology and Signaling View all 14 articles
 Rosario Munguía-Fuentes1‡Raúl Antonio Maqueda-Alfaro2‡Rommel Chacón-Salinas3Leopoldo Flores-Romo2†Juan Carlos Yam-Puc4*
Rosario Munguía-Fuentes1‡Raúl Antonio Maqueda-Alfaro2‡Rommel Chacón-Salinas3Leopoldo Flores-Romo2†Juan Carlos Yam-Puc4*Gaining knowledge of the neoplastic side of the three main cells—B cells, Follicular Helper T (Tfh) cells, and follicular dendritic cells (FDCs) —involved in the germinal center (GC) reaction can shed light toward further understanding the microuniverse that is the GC, opening the possibility of better treatments. This paper gives a review of the more complex underlying mechanisms involved in the malignant transformations that take place in the GC. Whilst our understanding of the biology of the GC-related B cell lymphomas has increased—this is not reviewed in detail here—the dark side involving neoplasms of Tfh cells and FDCs are poorly studied, in great part, due to their low incidence. The aggressive behavior of Tfh lymphomas and the metastatic potential of FDCs sarcomas make them clinically relevant, merit further attention and are the main focus of this review. Tfh cells and FDCs malignancies can often be misdiagnosed. The better understanding of these entities linked to their molecular and genetic characterization will lead to prediction of high-risk patients, better diagnosis, prognosis, and treatments based on molecular profiles.
The germinal center (GC), a specialized microstructure with a high rate of cell division, is the site where antigen-driven somatic hypermutation (SHM) occurs (1, 2), a process that ultimately will produce high-affinity antibodies during adaptive immune responses (3). Over weeks, memory B cells and high-affinity antibody producing plasma cells will generate from GCs, which are necessary to protect against invading microorganisms (4). However, the more potent the immune response, the greater the risk of autoreactivity or malignancy. This is particularly relevant for the GC, where B cells may have an unfavorable outcome driving to lymphomagenesis. Importantly, most of B−cell lymphomas originate from GC B cells (5–7).
To succeed during GC reactions, B cells need the help of other crucial cells, such as Follicular Helper T (Tfh) cells and follicular dendritic cells (FDCs). Here, we focus on one dark side of GCs: malignancies derived from their aforementioned three players, B-cells, Tfh cells, and FDCs (Figure 1), with greater emphasis on Tfh lymphomas and FDC sarcomas (Table 1).
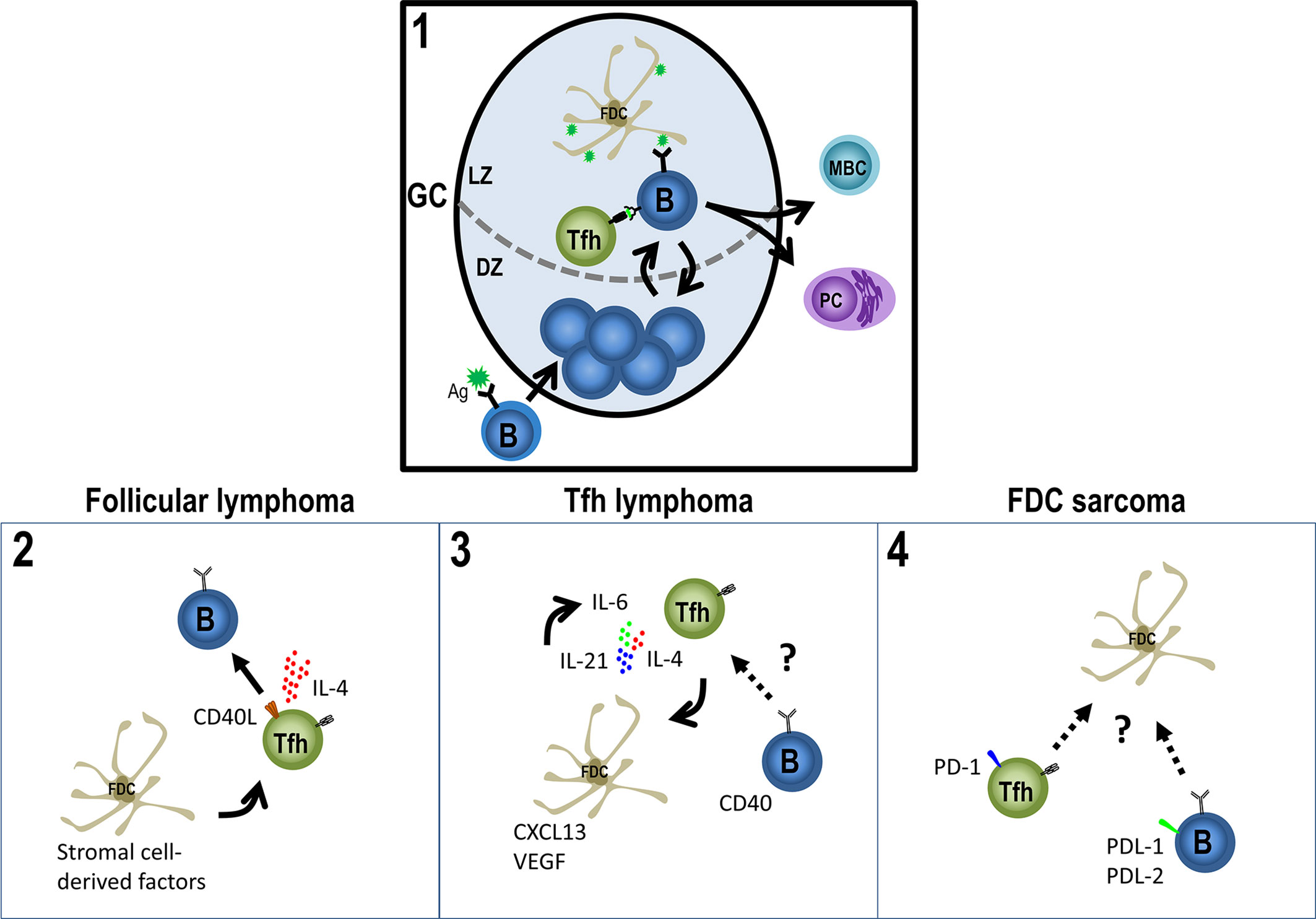
Figure 1 Germinal Center-derived malignancies’ players. The GC (1) is the site for B cell affinity maturation for the antigen through somatic hypermutation (in the DZ) and for antigen-driven selection of B cells which have improved their affinity (in the LZ). To success the reaction, the help from Tfh cells is fundamental, as well as, the presence of the antigen on iccosomes on FDCs. Eventually, GC B cells will differentiate to PCs or MBCs. However, the GC response may have a detrimental role, the development of malignancies from their three main players: B-cells, Tfh cells and FDCs. Follicular lymphoma cells (2) derive from follicles partly resembling normal GCs and depend on Tfh cells and FDCs to survive, while Tfh cells provide a high production of IL-4 and CD40L, FDCs provide a scaffold attracting FL cells and Tfh cells around them. (3) Infiltration of different immune cells and the proliferation of FDCs and HEV in AITL are probably caused by a stimulatory niche, secreting IL-21, IL-4 or/and IL-6, CXLC13 and VEGF, promoting a loop of Tfh cell generation and FDCs growth. (4) Tfh and Treg cells seem to be enriched in FDC sarcomas with high levels of PD-1 and its ligands PD-L1 and PD-L2 and the B/T cells mixed with the neoplastic population, altogether supporting the neoplastic niche and the evasion of effector immune cells. See text for further details.
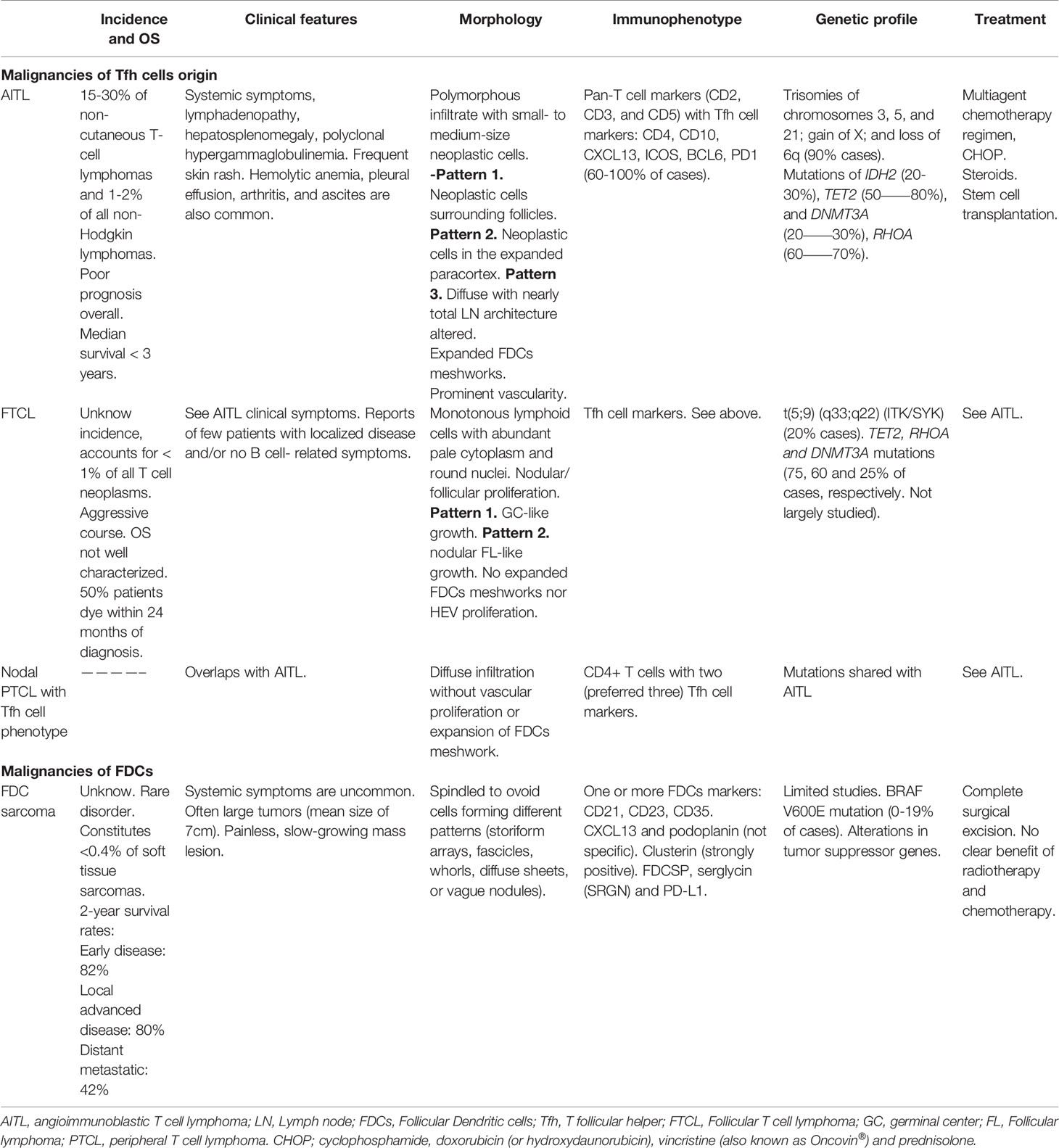
Table 1 Lymphomas of Follicular Helper T (Tfh) cells and sarcomas of follicular dendritic cells (FDCs).
GCs arise from proliferating B cells in the follicles of peripheral lymphoid tissues during T cell-dependent antibody responses. Naïve B cells encountering their antigen migrate to the T-B border, where they become fully activated during interaction with cognate CD4+ T cells (3, 4, 8–12). The engagement of CD40 by CD40L (CD154) represents the major component of the T cell help. Activated B cells can then either differentiate rapidly into antibody-secreting plasma cells in specialized extra-follicular niches or mature their affinity for the antigen into GC reactions, a microstructure of B cells, in a high-rate of cell division, Tfh cells and a network of FDCs (3, 13, 14). There, B cells begin to proliferate rapidly giving rise to the distinctive structure of the GC: a dark zone (DZ) of centroblast proliferating B cells and a light zone (LZ) with higher frequencies of smaller, non-dividing centrocytes. GC DZ B cells undergo SHM, and those cells that improved the affinity for the antigen are selected in the LZ to eventually differentiate into memory B cells or plasma cells (3, 4, 15, 16).
The GC response, beneficial for the host during immune responses against invading pathogens, may have a detrimental role, the development of malignancies. B cells inside GC reactions are mutating at much higher rates than in any other site in the body (17), these mutations might turn B cells into a dark side, B cell lymphomas. Except the relatively rare lymphoblastic and mantle-cell lymphoma subtypes, B cell non-Hodgkin lymphomas (B−NHLs)—including diffuse large B cell lymphoma (DLBCL), follicular lymphoma (FL) and Burkitt lymphoma (BL)—are derived from GC B cells. This can be demonstrated by the presence of SHM in the immunoglobulin genes, together with histological, immunophenotypic, and gene expression characteristics (5, 18–23).
Our understanding of the molecular mechanisms driving GC lymphomas has increased due to next-generation sequencing (24). Gene translocations targeting MYC, BCL2 and BCL6, as well as the disruption of the epigenome, predominantly driven by somatic mutations within KMT2D, CREBBP, EZH2, and linker histones, have been well-established (25–29). The molecular and genetic characterization of these diseases will lead to prediction of high-risk patients and treatments based on molecular profiles (24).
DLBCL and FL are the two most common forms of GC NHLs. With a greater degree of genetic heterogeneity than FL, DLBCL can be divided into at least two major subtypes: GC B cell (GCB)-like and activated B cell (ABC)-like DLBCL (24, 25, 30). Whole exome sequencing has allowed the study of recurrent mutations and the characterization of new genetic DLBCL subtypes. A recent study identified five genomic clusters based on the enriched genetic feature of each group (31). MYD88 cluster [with MYD88 (L265P), PIM1, CD79B and ETV6 mutations] were strongly associated with ABC subtype. Three clusters were associated to GCB subtype (BCL2, SOCS1/SGK1, and TET2/SGK1). The BCL2 cluster showed mutations of EZH2, BCL2, CREBBP, TNFRSF14, KMT2D, and MEF2B. The SOCS1/SGK1 cluster with mutations in SOCS1, CD83, SGK1, NFKBIA, HIST1H1E, and STAT3; and the TET2/SGK1 cluster characterized by mutations including TET2, SGK1, KLHL6, ZFP36L1, BRAF, MAP2K1, and KRAS. A NOTCH2 cluster with mutations on NOTCH2, BCL10, TNFAIP3, CCND3, SPEN, TMEM30A FAS, and CD70 showed a mixture of ABC, GCB and unclassified DLBCL. This study correlated with two recent studies classifying the disease (32, 33). Importantly, patient outcome was evaluated with the worst prognosis in the MYD88 group (42% 5-year overall survival, OS). Patients within the GCB-associated clusters had better 5-year OS (> 60%) while NOTCH2 cluster had intermediate survival (53.6% 5-year OS). Patient outcome correlated with previous studies (24, 34–38).
FL is the most frequent indolent and incurable NHL. Over time FL may progress to DLBCL, with a more aggressive clinical course requiring more aggressive treatment (39, 40). Malignant cells morphologically resemble the two B cell subsets found in reactive GCs (centrocytes and centroblasts). Low-grade FL cases (grade 1–2) contain <15 centroblasts per high-power microscopic field (40 x objective, 0.159 mm2) while grade 3 contains >15 centroblasts, evaluated in 10 different follicles. Grade 3 FL is further separated in 3A with a background of centrocytes present or grade 3B with follicles composed entirely of centroblasts. A diffuse pattern of 3B grade cells is compatible with DLBCL diagnosis (41, 42). The molecular pathogenesis of FL includes the high recurrence of two mutations: chromosomal translocations that lead to the ectopic expression of BCL2 and somatic mutations in the histone methyltransferase MLL2 (also known as KMT2D) (43). Also, histone modifiers such as EZH2, CREBBP and EP300 are frequently altered in FL (44–46). While BCL2 translocation is thought to occur in B-cell precursors in the bone marrow, the translocation is found also in healthy humans (40%) (47). This supports the hypothesis that the translocation is necessary, but not sufficient for FL and probably lymphomagenesis is consequence of antigen stimulation (48, 49).
FL derives from follicles partly resembling normal GCs, with the FL cells depending on Tfh cells and FDCs. While Tfh cells provide a high production of IL-4 and CD40L as survival factors of FL cells, FDCs provide a scaffold attracting FL cells and Tfh cells around them. FDCs also contributes with a positive feedback through the overexpression of stromal cell-derived factors, supporting the abnormal production of IL-4 (50, 51).
BL is a highly aggressive NHL associated with Epstein-Barr virus (EBV), human immunodeficiency virus (HIV) or Plasmodium infection. Three clinical variants of BL are recognized: endemic BL, sporadic BL and immunodeficiency-associated BL. Whilst EBV and Plasmodium are associated to endemic and sporadic BL, HIV is associated to the immunodeficiency-related variant (52, 53). BL derives from DZ GC B cells, as indicated by its genetic profile (20, 23, 54). Although the potential pathological role of EBV in BL it is still controversial, the virus is present in all BL cases. Aberrant expression of MYC and the BCR-induced PI3K signalling pathway activation are genetic alterations that are common in BL (23, 55, 56).
Fundamental studies by Mitchison in the 1970’s established the essential role of T helper cells in antibody responses. Hapten-protein carrier conjugates revealed that carrier-specific T cells were necessary for the maturation of hapten-specific B cells [(57) and reviewed in Ref (10)]. Then, it was described that this help from T cells consisted in co-stimulatory signals through CD40 ligand (CD40L) to B cells leading them to proliferate, differentiate, and antibody class-switching (10, 58). T cells in the T-B border that have undergone T-B interactions can migrate inside the follicles as Tfh cells, afterwards making cognate interactions with GC B cells within the GC reaction. T cell help into GCs are needed to maintain the reaction (59–63).
At present, a combination of markers is needed to identify Tfh cells as a distinct population. Tfh cells differentiate from the classical CD4+ T cell subpopulation and share plastic characteristics with other CD4+ helper T cells until they engaged in GC reactions. Inside GCs, the expression of typically described Tfh cells associated-molecules—CXCR5, PD-1, BCL6, BTLA4, ICOS (inducible T cell costimulator) and SAP—is upregulated whilst CD127, PSGL1, and EBI2 are downregulated (64–66). When these molecules are low-intermediate expressed, particularly CXCR5, PD-1, ICOS and SAP, define stage known as pre-GC Tfh cells (10, 66–69).
Peripheral T-cell lymphomas (PTCLs) are generally described as diverse and aggressive malignancies with unfavorable therapeutic outcomes (70). Nodal T-cell lymphomas with Tfh-cell phenotype are classified into three diseases: angioimmunoblastic T cell lymphoma (AITL), follicular T-cell lymphoma (FTCL) and nodal PTCL with Tfh cell phenotype. AITL is an aggressive rare tumor with a 5-year survival of only 33%, first described as a distinct clinic-pathologic entity in the 1970s and is the best well-established subtype of mature PTCL (71, 72). The tumors contain neoplastic Tfh cells expressing BCL-6, CD10, CXCL13, PD-1, ICOS, SAP, and CXCR5 (10, 73–75). TET2, DNMT3A and IDH2 mutations have been detected in about 80% (76–78), 20–30% (76, 79, 80), and 20–30% of the cases (81), respectively. The mechanism of action described for these molecules is by dysregulating DNA methylation (70). A missense mutation in RHOA GTPase is detected in 50–70% AITL patients (77, 78, 82, 83). It has been described that some primary cutaneous T cell lymphomas also originate from neoplastic cells that express Tfh cells markers, and can also induce the typical rosettes found in AITL (84).
Representative clinical symptoms of AITL are generalized lymphadenopathy, hepatosplenomegaly, fever, effusion/ascites and skin rash. The incidence is low without sex predilection, affecting advanced-age individuals (median age of diagnosis 65 years) (72). Characteristically, lymph nodes acquire an effaced architecture, with only a few benign follicles been retained. A typical feature is to find infiltration beyond the capsule of the lymph node, with a preserved but enlarged subcapsular sinus; also high endothelial venules (HEV) and FDCs proliferate (71). Infiltration of other cells include: B cells, plasma cells, eosinophils, histiocytes and epithelioid cells. Active EBV infection can be found in most large B cells, whilst the malignant Tfh cells do not (72).
Cytological diagnosis of AITL is usually difficult and both reactive and lymphomatous processes need to be discarded. Combination of conventional cytology, immunocytochemistry and flow cytometry is needed to make an accurate diagnosis (85).
AITL, a lymphoma with poor prognosis, is often refractory to chemotherapy or relapses. Due to the unfavorable outcomes for PTCL patients treated with chemotherapy alone, autologous stem cell transplantation (SCT), as a consolidation treatment for first-line therapy or salvage therapy for relapse/refractory PTCL patients, may be an option. On the other hand, some relapse/refractory AITL patients may benefit from allogenic-SCT, presumably because of graft-versus lymphoma effects (74).
FTCL presents clinical and immunophenotype features of AITL but differs histologically. Two patterns have been described, one shows a GC-like growth with IgD+ B cells surrounding the neoplastic cells and the second resembles a FL-like pattern with malignant cells forming nodules. Another difference with AITL is the absence of proliferation of HEVs and FDCs (86).
While TET2, RHOA and DNMT3A mutations have been shown in both, AITL and FTCL, there is no evidence of IDH2 mutation in FTCL (87). Also, 20% of cases show a t(5;9) (q33;q22) (ITK/SYK) translocation but studies are limited (88).
The study and characterization of normal Tfh cells phenotyping led to the recognition and classification of previously diagnosed PTCLs-NOS (Not otherwise specified) to Nodal PTCLs with Tfh cell phenotype (84). While clinical, phenotypic, pathological, and genetic features overlap with AITL, further research is needed to include this neoplasm within the spectrum of one entity. Differences with AITL include the absence of expansion of HEVs and FDCs while histological differences from FTCL are due to the diffuse pattern of Nodal PTCLs with Tfh cell phenotype (86, 89).
The interaction of neoplastic Tfh cells and their niches has not been extensively studied. The reported infiltration of different immune cells and the proliferation of FDCs and HEV in AITL are probably caused by a stimulatory niche but the underlying mechanisms are still unknown. Some signals present in a normal counterpart niche like IL-21, IL-4, or/and IL-6 are over-expressed in AITL creating a loop of Tfh cell generation and FDCs growth (84, 90, 91). Also, this microenvironment could explain in part the depletion of Treg cells in AITL, an important population for suppressing Tfh cells in immune responses (92). Regulatory CAR T cells therapy might be a potential treatment to re-establish a favorable microenvironment.
Although neoplastic cells in FTCL show a GC- or follicle-growth pattern, the low incidence of this malignancy has made difficult the in-depth study of their interaction with resident cells. Understanding the crosstalk between neoplastic cells and their niche would definitely potentiate the development of more rationale treatments.
Originally discovered by Alexander Maximow and subsequently termed FDCs by Steinman et al. in 1978, FDCs are critical participants in the GC reaction (93–96).
FDCs are stromal cells residing exclusively in B cell follicles, where they play a key role supporting B cell homeostasis and maintaining the follicular architecture. They are essential promoting robust humoral immune responses through the retention of antigens within immune complexes (ICs) over long periods. For this, FDCs express complement receptors (CRs)-1 and -2 and can be induced to express Fc-gamma receptor (FcyR) IIb (93, 97). Lymphoid organs lacking B cells or tumor necrosis factor (TNF) or lymphotoxin (LT) are devoid of FDCs (98–100). Mice lacking stromal CR1 and CR2 have reduced T-dependent antibody responses (93, 98, 101). ICs are released in FDCs-derived iccosomes, then cognate GC B cells can acquire antigen and present it to Tfh cells. FDCs also support the proliferation of GC B cells enhancing antibody production (93, 98).
FDCs are a subset very different from conventional DCs (cDCs). FDCs originate from stromal cells: it has been shown that in the spleen, FDCs come from vascular mural cells but in the lymph nodes, FDCs come from marginal reticular cells (MRCs) (102, 103). Nowadays, it is suggested that different stromal cells of secondary lymphoid organs—including FDCs and MRCs—are generated from one and the same precursor (93).
Functionally, whereas cDCs activate naïve T cells by presentation of processed antigens via major histocompatibility complex (MHC) molecules, FDCs show unprocessed antigens, trapped in ICs, to GC B cells. In addition, FDCs secrete the signalling molecule Mfge8 which has been shown to be essential in controlling the removal of apoptotic GC B cells. It has been suggested that FDC-mediated phagocytosis of apoptotic GC B cells might play an important role in avoiding autoimmunity (93).
Termed FDC sarcomas, the first reported cases of tumors derived from FDCs occurred in cervical lymph nodes. FDC sarcoma is classified as a distinct entity by the World Health Organization (WHO) under histiocytic and DC neoplasms Classification of Tumours. It is described as a neoplastic proliferation of spindled to ovoid cells with morphologic and immunophenotypic characteristics similar to those of normal FDCs. Despite the fact that their histopathological, morphological and clinical features have been described relatively in detail, their clinical course is unpredictable and no specific treatment is available (95, 104).
While FDC sarcomas do not have gender predilection, it mainly occurs during adulthood (median age in the fifth decade). Interestingly, a very rare and distinct variant of FDC sarcoma consistently associated with the EBV, termed inflammatory pseudotumor-like variant of FDC sarcoma, is more prevalent in females. Approximately 10–20% of FDC sarcoma cases have presented or concur with Castleman disease, a rare and non-malignant lymphoproliferative disorder, typically the hyaline vascular variant (104).
We now know also that FDC sarcomas can involve any anatomical area besides nodal sites. FDC sarcomas generally appear as a slow growing mass, an asymptomatic and painless cervical lymphadenopathy (95, 104, 105). Nearly a third of FDC sarcoma cases arise in extranodal sites: tonsils, skin, mediastinum, gastrointestinal tract and soft tissue (104, 106, 107). Furthermore, it seems there is an association between FDC sarcoma and the autoimmune diseases, paraneoplastic pemphigus and myasthenia gravis (104, 108–111).
Histopathology and cytomorphology of FDC sarcomas are characteristic, however their identification may be difficult and additional confirmation with immunohistochemical studies is frequently necessary. FDC sarcomas generally present the immunophenotype of normal FDCs, being positive for: CD21 (CR2), CD23 (Fc epsilon RII) and CD35 (CR1) (104, 112, 113). Clusterin and podoplanin are other molecules shown to have high sensitivity for FDC sarcomas (114–116). Clusterin shows strong positivity with weak to no expression in other DC tumors (114).
Especially when arising from extranodal sites, FDC sarcoma can often be misdiagnosed (106, 117). Then, differential diagnosis is needed, including interdigitating DC sarcoma, thymoma, spindle cell carcinoma, metastatic undifferentiated carcinomas, malignant melanoma and gastrointestinal stromal tumor (GIST) (104).
Clinical courses of FDC sarcomas are not consistent and consequently, treatment schemes are variable. Complete surgical resection seems to be the treatment of choice for both primary and recurrent lesions, with unclear benefits from radiation and chemical therapies (111, 118).
Being a very uncommon neoplasm, the in-depth study of FDC sarcomas have been difficult and almost neglected. Although it has been described an enrichment of Tfh and Treg cells in FDC sarcomas compared to other mesenchymal tumors (119), the interaction of malignant FDCs with other lymphocytes and other resident cells has not yet been studied. High levels of PD-1 and its ligands PD-L1 and PD-L2 (119) and the B/T cells mixed with the neoplastic population (120, 121) could point to a feedback from these lymphocytes to support the neoplastic niche and the evasion of effector immune cells.
As discussed in this review, although lymphomas from GC B cells are explored in more detail and better understood, Tfh lymphomas and FDCs sarcomas need more attention.
Tfh lymphomas diagnosis is challenging, requiring multimodality methods including conventional cytology, immunohistochemistry, and flow cytometry. Usually with a poor prognosis, treatments need to be combined, frequently with unfavorable outcomes.
On the other hand, FDC sarcomas can often be misdiagnosed and differential diagnoses are needed. With variable clinical courses and unspecific and heterogeneous treatment at present, surgical resection is the treatment of choice.
Greater knowledge of the normal GC microuniverse will undoubtedly provide insights on its neoplastic side, allowing us better diagnosis, treatment, prognosis, and monitoring, the better to improve the quality of life of patients.
Dedicated to the memory of LF-R, a brilliant Mexican immunologist who inspired many generations of scientists through his passion. His legacy will last forever.
Conceptualization, LF-R and JY-P. Writing—Original Draft Preparation, RM-F, RM-A, and JY-P. Writing—Review and Editing, RM-F, RM-A, RC-S, LF-R, and JY-P. Supervision, LF-R and JY-P. All authors contributed to the article and approved the submitted version.
This work was supported by a grant from the National Council for Science and Technology-CONACYT Mexico (221102) to LF-R.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Iris Estrada-García for helpful discussions and thoughtful comments on the manuscript. RM-A is a CONACYT fellow. RC-S and JY-P are members of the National System of Researchers, SNI-CONACYT.
1. Berek C, Berger A, Apel M. Maturation of the immune response in germinal centers. Cell (1991) 67(6):1121–9. doi: 10.1016/0092-8674(91)90289-B
2. Jacob J, Kelsoe G, Rajewsky K, Weiss U. Intraclonal generation of antibody mutants in germinal centres. Nature (1991) 354(6352):389–92. doi: 10.1038/354389a0
3. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol (2012) 30:429–57. doi: 10.1146/annurev-immunol-020711-075032
4. MacLennan IC. Germinal centers. Annu Rev Immunol (1994) 12:117–39. doi: 10.1146/annurev.iy.12.040194.001001
5. Kuppers R, Klein U, Hansmann ML, Rajewsky K. Cellular origin of human B-cell lymphomas. N Engl J Med (1999) 341(20):1520–9. doi: 10.1056/NEJM199911113412007
6. Stevenson F, Sahota S, Zhu D, Ottensmeier C, Chapman C, Oscier D, et al. Insight into the origin and clonal history of B-cell tumors as revealed by analysis of immunoglobulin variable region genes. Immunol Rev (1998) 162:247–59. doi: 10.1111/j.1600-065X.1998.tb01446.x
7. Klein U, Dalla-Favera R. Germinal centres: role in B-cell physiology and malignancy. Nat Rev Immunol (2008) 8(1):22–33. doi: 10.1038/nri2217
8. Shulman Z, Gitlin AD, Targ S, Jankovic M, Pasqual G, Nussenzweig MC, et al. T follicular helper cell dynamics in germinal centers. Science (2013) 341(6146):673–7. doi: 10.1126/science.1241680
9. Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, et al. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell (2010) 143(4):592–605. doi: 10.1016/j.cell.2010.10.032
10. Vinuesa CG, Linterman MA, Yu D, MacLennan IC. Follicular Helper T Cells. Annu Rev Immunol (2016) 34:335–68. doi: 10.1146/annurev-immunol-041015-055605
11. Garside P, Ingulli E, Merica RR, Johnson JG, Noelle RJ, Jenkins MK. Visualization of specific B and T lymphocyte interactions in the lymph node. Science (1998) 281(5373):96–9. doi: 10.1126/science.281.5373.96
12. Van den Eertwegh AJ, Noelle RJ, Roy M, Shepherd DM, Aruffo A, Ledbetter JA, et al. In vivo CD40-gp39 interactions are essential for thymus-dependent humoral immunity. Ivivo expression of CD40 ligand, cytokines, and antibody production delineates sites of cognate T-B cell interactions. J Exp Med (1993) 178(5):1555–65. doi: 10.1084/jem.178.5.1555
13. MacLennan IC, Toellner KM, Cunningham AF, Serre K, Sze DM, Zuniga E, et al. Extrafollicular antibody responses. Immunol Rev (2003) 194:8–18. doi: 10.1034/j.1600-065X.2003.00058.x
14. Coffey F, Alabyev B, Manser T. Initial clonal expansion of germinal center B cells takes place at the perimeter of follicles. Immunity (2009) 30(4):599–609. doi: 10.1016/j.immuni.2009.01.011
15. Camacho SA, Kosco-Vilbois MH, Berek C. The dynamic structure of the germinal center. Immunol Today (1998) 19(11):511–4. doi: 10.1016/S0167-5699(98)01327-9
16. Wang Y, Carter RH. CD19 regulates B cell maturation, proliferation, and positive selection in the FDC zone of murine splenic germinal centers. Immunity (2005) 22(6):749–61. doi: 10.1016/j.immuni.2005.04.012
17. Stratigopoulou M, van Dam TP, Guikema JEJ. Base Excision Repair in the Immune System: Small DNA Lesions With Big Consequences. Front Immunol (2020) 11:1084. doi: 10.3389/fimmu.2020.01084
18. Stevenson FK, Sahota SS, Ottensmeier CH, Zhu D, Forconi F, Hamblin TJ. The occurrence and significance of V gene mutations in B cell-derived human malignancy. Adv Cancer Res (2001) 83:81–116. doi: 10.1016/S0065-230X(01)83004-9
19. Berglund M, Thunberg U, Amini RM, Book M, Roos G, Erlanson M, et al. Evaluation of immunophenotype in diffuse large B-cell lymphoma and its impact on prognosis. Mod Pathol (2005) 18(8):1113–20. doi: 10.1038/modpathol.3800396
20. Dave SS, Fu K, Wright GW, Lam LT, Kluin P, Boerma EJ, et al. Molecular diagnosis of Burkitt’s lymphoma. N Engl J Med (2006) 354(23):2431–42. doi: 10.1056/NEJMoa055759
21. Mamessier E, Broussais-Guillaumot F, Chetaille B, Bouabdallah R, Xerri L, Jaffe ES, et al. Nature and importance of follicular lymphoma precursors. Haematologica (2014) 99(5):802–10. doi: 10.3324/haematol.2013.085548
22. Mlynarczyk C, Fontan L, Melnick A. Germinal center-derived lymphomas: The darkest side of humoral immunity. Immunol Rev (2019) 288(1):214–39. doi: 10.1111/imr.12755
23. Basso K, Dalla-Favera R. Germinal centres and B cell lymphomagenesis. Nat Rev Immunol (2015) 15(3):172–84. doi: 10.1038/nri3814
24. Heward JA, Kumar EA, Korfi K, Okosun J, Fitzgibbon J. Precision medicine and lymphoma. Curr Opin Hematol (2018) 25(4):329–34. doi: 10.1097/MOH.0000000000000437
25. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature (2000) 403(6769):503–11. doi: 10.1038/35000501
26. Korfi K, Ali S, Heward JA, Fitzgibbon J. Follicular lymphoma, a B cell malignancy addicted to epigenetic mutations. Epigenetics (2017) 12(5):370–7. doi: 10.1080/15592294.2017.1282587
27. Krysiak K, Gomez F, White BS, Matlock M, Miller CA, Trani L, et al. Recurrent somatic mutations affecting B-cell receptor signaling pathway genes in follicular lymphoma. Blood (2017) 129(4):473–83. doi: 10.1182/blood-2016-07-729954
28. Li H, Kaminski MS, Li Y, Yildiz M, Ouillette P, Jones S, et al. Mutations in linker histone genes HIST1H1 B, C, D, and E; OCT2 (POU2F2); IRF8; and ARID1A underlying the pathogenesis of follicular lymphoma. Blood (2014) 123(10):1487–98. doi: 10.1182/blood-2013-05-500264
29. Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell (2017) 171(2):481–94.e15. doi: 10.1016/j.cell.2017.09.027
30. Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Delabie J, Ott G, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood (2004) 103(1):275–82. doi: 10.1182/blood-2003-05-1545
31. Lacy SE, Barrans SL, Beer PA, Painter D, Smith AG, Roman E, et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: a Haematological Malignancy Research Network report. Blood (2020) 135(20):1759–71. doi: 10.1182/blood.2019003535
32. Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med (2018) 24(5):679–90. doi: 10.1038/s41591-018-0016-8
33. Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med (2018) 378(15):1396–407. doi: 10.1056/NEJMoa1801445
34. Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature (2009) 459(7247):717–21. doi: 10.1038/nature07968
35. Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature (2010) 463(7277):88–92. doi: 10.1038/nature08638
36. Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science (2008) 319(5870):1676–9. doi: 10.1126/science.1153629
37. Lenz G, Wright GW, Emre NC, Kohlhammer H, Dave SS, Davis RE, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci U.S.A. (2008) 105(36):13520–5. doi: 10.1073/pnas.0804295105
38. Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature (2011) 470(7332):115–9. doi: 10.1038/nature09671
39. Kridel R, Sehn LH, Gascoyne RD. Pathogenesis of follicular lymphoma. J Clin Invest (2012) 122(10):3424–31. doi: 10.1172/JCI63186
40. Montoto S, Fitzgibbon J. Transformation of indolent B-cell lymphomas. J Clin Oncol (2011) 29(14):1827–34. doi: 10.1200/JCO.2010.32.7577
41. Choi SM, Betz BL, Perry AM. Follicular Lymphoma Diagnostic Caveats and Updates. Arch Pathol Lab Med (2018) 142(11):1330–40. doi: 10.5858/arpa.2018-0217-RA
42. Randall C, Fedoriw Y. Pathology and diagnosis of follicular lymphoma and related entities. Pathology (2020) 52(1):30–9. doi: 10.1016/j.pathol.2019.09.010
43. McCann KJ, Johnson PW, Stevenson FK, Ottensmeier CH. Universal N-glycosylation sites introduced into the B-cell receptor of follicular lymphoma by somatic mutation: a second tumorigenic event? Leukemia (2006) 20(3):530–4. doi: 10.1038/sj.leu.2404095
44. Green MR, Gentles AJ, Nair RV, Irish JM, Kihira S, Liu CL, et al. Hierarchy in somatic mutations arising during genomic evolution and progression of follicular lymphoma. Blood (2013) 121(9):1604–11. doi: 10.1182/blood-2012-09-457283
45. Li B, Chng WJ. EZH2 abnormalities in lymphoid malignancies: underlying mechanisms and therapeutic implications. J Hematol Oncol (2019) 12(1):118. doi: 10.1186/s13045-019-0814-6
46. Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature (2011) 476(7360):298–303. doi: 10.1038/nature10351
47. Limpens J, Stad R, Vos C, de Vlaam C, de Jong D, van Ommen GJ, et al. Lymphoma-associated translocation t(14;18) in blood B cells of normal individuals. Blood (1995) 85(9):2528–36. doi: 10.1182/blood.V85.9.2528.bloodjournal8592528
48. Kuppers R. Mechanisms of B-cell lymphoma pathogenesis. Nat Rev Cancer (2005) 5(4):251–62. doi: 10.1038/nrc1589
49. Tsujimoto Y, Cossman J, Jaffe E, Croce CM. Involvement of the bcl-2 gene in human follicular lymphoma. Science (1985) 228(4706):1440–3. doi: 10.1126/science.3874430
50. Mulder TA, Wahlin BE, Osterborg A, Palma M. Targeting the Immune Microenvironment in Lymphomas of B-Cell Origin: From Biology to Clinical Application. Cancers (Basel) (2019) 11(7). doi: 10.3390/cancers11070915
51. Ame-Thomas P, Tarte K. The yin and the yang of follicular lymphoma cell niches: role of microenvironment heterogeneity and plasticity. Semin Cancer Biol (2014) 24:23–32. doi: 10.1016/j.semcancer.2013.08.001
52. Robbiani DF, Deroubaix S, Feldhahn N, Oliveira TY, Callen E, Wang Q, et al. Plasmodium Infection Promotes Genomic Instability and AID-Dependent B Cell Lymphoma. Cell (2015) 162(4):727–37. doi: 10.1016/j.cell.2015.07.019
53. Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature (2012) 490(7418):116–20. doi: 10.1038/nature11378
54. Victora GD, Dominguez-Sola D, Holmes AB, Deroubaix S, Dalla-Favera R, Nussenzweig MC. Identification of human germinal center light and dark zone cells and their relationship to human B-cell lymphomas. Blood (2012) 120(11):2240–8. doi: 10.1182/blood-2012-03-415380
55. Sander S, Calado DP, Srinivasan L, Kochert K, Zhang B, Rosolowski M, et al. Synergy between PI3K signaling and MYC in Burkitt lymphomagenesis. Cancer Cell (2012) 22(2):167–79. doi: 10.1016/j.ccr.2012.06.012
56. Schmitz R, Ceribelli M, Pittaluga S, Wright G, Staudt LM. Oncogenic mechanisms in Burkitt lymphoma. Cold Spring Harb Perspect Med (2014) 4(2). doi: 10.1101/cshperspect.a014282
57. Mitchison NA. The carrier effect in the secondary response to hapten-protein conjugates. II. Cellular cooperation. Eur J Immunol (1971) 1(1):18–27. doi: 10.1002/eji.1830010104
58. Noelle RJ, Roy M, Shepherd DM, Stamenkovic I, Ledbetter JA, Aruffo A. A 39-kDa protein on activated helper T cells binds CD40 and transduces the signal for cognate activation of B cells. Proc Natl Acad Sci USA (1992) 89(14):6550–4. doi: 10.1073/pnas.89.14.6550
59. Ansel KM, McHeyzer-Williams LJ, Ngo VN, McHeyzer-Williams MG, Cyster JG. In vivo-activated CD4 T cells upregulate CXC chemokine receptor 5 and reprogram their response to lymphoid chemokines. J Exp Med (1999) 190(8):1123–34. doi: 10.1084/jem.190.8.1123
60. Fuller KA, Kanagawa O, Nahm MH. T cells within germinal centers are specific for the immunizing antigen. J Immunol (1993) 151(9):4505–12.
61. Gulbranson-Judge A, MacLennan I. Sequential antigen-specific growth of T cells in the T zones and follicles in response to pigeon cytochrome c. Eur J Immunol (1996) 26(8):1830–7. doi: 10.1002/eji.1830260825
62. Qi H, Cannons JL, Klauschen F, Schwartzberg PL, Germain RN. SAP-controlled T-B cell interactions underlie germinal centre formation. Nature (2008) 455(7214):764–9. doi: 10.1038/nature07345
63. Zheng B, Han S, Kelsoe G. T helper cells in murine germinal centers are antigen-specific emigrants that downregulate Thy-1. J Exp Med (1996) 184(3):1083–91. doi: 10.1084/jem.184.3.1083
64. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity (2014) 41(4):529–42. doi: 10.1016/j.immuni.2014.10.004
65. Suan D, Nguyen A, Moran I, Bourne K, Hermes JR, Arshi M, et al. T follicular helper cells have distinct modes of migration and molecular signatures in naive and memory immune responses. Immunity (2015) 42(4):704–18. doi: 10.1016/j.immuni.2015.03.002
66. Yu D, Vinuesa CG. The elusive identity of T follicular helper cells. Trends Immunol (2010) 31(10):377–83. doi: 10.1016/j.it.2010.07.001
67. Kerfoot SM, Yaari G, Patel JR, Johnson KL, Gonzalez DG, Kleinstein SH, et al. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity (2011) 34(6):947–60. doi: 10.1016/j.immuni.2011.03.024
68. Kitano M, Moriyama S, Ando Y, Hikida M, Mori Y, Kurosaki T, et al. Bcl6 protein expression shapes pre-germinal center B cell dynamics and follicular helper T cell heterogeneity. Immunity (2011) 34(6):961–72. doi: 10.1016/j.immuni.2011.03.025
69. Lee SK, Rigby RJ, Zotos D, Tsai LM, Kawamoto S, Marshall JL, et al. B cell priming for extrafollicular antibody responses requires Bcl-6 expression by T cells. J Exp Med (2011) 208(7):1377–88. doi: 10.1084/jem.20102065
70. Lemonnier F, Gaulard P, de Leval L. New insights in the pathogenesis of T-cell lymphomas. Curr Opin Oncol (2018) 30(5):277–84. doi: 10.1097/CCO.0000000000000474
71. Federico M, Rudiger T, Bellei M, Nathwani BN, Luminari S, Coiffier B, et al. Clinicopathologic characteristics of angioimmunoblastic T-cell lymphoma: analysis of the international peripheral T-cell lymphoma project. J Clin Oncol (2013) 31(2):240–6. doi: 10.1200/JCO.2011.37.3647
72. Lunning MA, Vose JM. Angioimmunoblastic T-cell lymphoma: the many-faced lymphoma. Blood (2017) 129(9):1095–102. doi: 10.1182/blood-2016-09-692541
73. de Leval L, Gisselbrecht C, Gaulard P. Advances in the understanding and management of angioimmunoblastic T-cell lymphoma. Br J Haematol (2010) 148(5):673–89. doi: 10.1111/j.1365-2141.2009.08003.x
74. Fujisawa M, Chiba S, Sakata-Yanagimoto M. Recent Progress in the Understanding of Angioimmunoblastic T-cell Lymphoma. J Clin Exp Hematop (2017) 57(3):109–19. doi: 10.3960/jslrt.17019
75. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood (2016) 127(20):2375–90. doi: 10.1182/blood-2016-01-643569
76. Odejide O, Weigert O, Lane AA, Toscano D, Lunning MA, Kopp N, et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood (2014) 123(9):1293–6. doi: 10.1182/blood-2013-10-531509
77. Palomero T, Couronne L, Khiabanian H, Kim MY, Ambesi-Impiombato A, Perez-Garcia A, et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet (2014) 46(2):166–70. doi: 10.1038/ng.2873
78. Sakata-Yanagimoto M, Enami T, Yoshida K, Shiraishi Y, Ishii R, Miyake Y, et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet (2014) 46(2):171–5. doi: 10.1038/ng.2872
79. Cairns RA, Iqbal J, Lemonnier F, Kucuk C, de Leval L, Jais JP, et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood (2012) 119(8):1901–3. doi: 10.1182/blood-2011-11-391748
80. Wang C, McKeithan TW, Gong Q, Zhang W, Bouska A, Rosenwald A, et al. IDH2R172 mutations define a unique subgroup of patients with angioimmunoblastic T-cell lymphoma. Blood (2015) 126(15):1741–52. doi: 10.1182/blood-2015-05-644591
81. Couronne L, Bastard C, Bernard OA. TET2 and DNMT3A mutations in human T-cell lymphoma. N Engl J Med (2012) 366(1):95–6. doi: 10.1056/NEJMc1111708
82. Vallois D, Dobay MP, Morin RD, Lemonnier F, Missiaglia E, Juilland M, et al. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T-cell-derived lymphomas. Blood (2016) 128(11):1490–502. doi: 10.1182/blood-2016-02-698977
83. Yoo HY, Sung MK, Lee SH, Kim S, Lee H, Park S, et al. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat Genet (2014) 46(4):371–5. doi: 10.1038/ng.2916
84. Ahearne MJ, Allchin RL, Fox CP, Wagner SD. Follicular helper T-cells: expanding roles in T-cell lymphoma and targets for treatment. Br J Haematol (2014) 166(3):326–35. doi: 10.1111/bjh.12941
85. Miller , Rana DN, Narine N. Angioimmunoblastic T Cell lymphoma mimics reactive lymphoid tissue on cytomorphology: A multimodality approach utilising cytology, immunocytochemistry and flow cytometry to resolve this diagnostic dilemma. Cytopathology (2017) 28(3):239–41. doi: 10.1111/cyt.12422
86. Attygalle AD. Nodal T-cell lymphomas with a T-follicular helper cell phenotype. Diagn Histopathol (2018) 24(7):227–36. doi: 10.1016/j.mpdhp.2018.05.006
87. Dobay MP, Lemonnier F, Missiaglia E, Bastard C, Vallois D, Jais JP, et al. Integrative clinicopathological and molecular analyses of angioimmunoblastic T-cell lymphoma and other nodal lymphomas of follicular helper T-cell origin. Haematologica (2017) 102(4):e148–51. doi: 10.3324/haematol.2016.158428
88. Streubel B, Vinatzer U, Willheim M, Raderer M, Chott A. Novel t(5;9)(q33;q22) fuses ITK to SYK in unspecified peripheral T-cell lymphoma. Leukemia (2006) 20(2):313–8. doi: 10.1038/sj.leu.2404045
89. Suzuki T, Miyoshi H, Yanagida E, Kawamoto K, Yamada K, Takeuchi M, et al. Clinicopathological differences of nodal ptcl with tfh phenotype from aitl and ptcl, nos, and detection of prognostic marker of nodal ptcl with tfh phenotype. 15th International Conference on Malignant Lymphoma. Lugano, Switzerland: Hematological Oncology (2019). p. 276–7. doi: 10.1002/hon.87_2630
90. Crotty S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity (2019) 50(5):1132–48. doi: 10.1016/j.immuni.2019.04.011
91. Iqbal J, Weisenburger DD, Greiner TC, Vose JM, McKeithan T, Kucuk C, et al. Molecular signatures to improve diagnosis in peripheral T-cell lymphoma and prognostication in angioimmunoblastic T-cell lymphoma. Blood (2010) 115(5):1026–36. doi: 10.1182/blood-2009-06-227579
92. Bruneau J, Canioni D, Renand A, Marafioti T, Paterson JC, Martin-Garcia N, et al. Regulatory T-cell depletion in angioimmunoblastic T-cell lymphoma. Am J Pathol (2010) 177(2):570–4. doi: 10.2353/ajpath.2010.100150
93. Aguzzi A, Kranich J, Krautler NJ. Follicular dendritic cells: origin, phenotype, and function in health and disease. Trends Immunol (2014) 35(3):105–13. doi: 10.1016/j.it.2013.11.001
94. Chen LL, Adams JC, Steinman RM. Anatomy of germinal centers in mouse spleen, with special reference to “follicular dendritic cells”. J Cell Biol (1978) 77(1):148–64. doi: 10.1083/jcb.77.1.148
95. Facchetti F, Lorenzi L. Follicular dendritic cells and related sarcoma. Semin Diagn Pathol (2016) 33(5):262–76. doi: 10.1053/j.semdp.2016.05.002
96. Szakal AK, Gieringer RL, Kosco MH, Tew JG. Isolated follicular dendritic cells: cytochemical antigen localization, Nomarski, SEM, and TEM morphology. J Immunol (1985) 134(3):1349–59.
97. Aguzzi A, Krautler NJ. Characterizing follicular dendritic cells: A progress report. Eur J Immunol (2010) 40(8):2134–8. doi: 10.1002/eji.201040765
98. Allen CD, Cyster JG. Follicular dendritic cell networks of primary follicles and germinal centers: phenotype and function. Semin Immunol (2008) 20(1):14–25. doi: 10.1016/j.smim.2007.12.001
99. Mackay F, Majeau GR, Lawton P, Hochman PS, Browning JL. Lymphotoxin but not tumor necrosis factor functions to maintain splenic architecture and humoral responsiveness in adult mice. Eur J Immunol (1997) 27(8):2033–42. doi: 10.1002/eji.1830270830
100. Ware CF. Network communications: lymphotoxins, LIGHT, and TNF. Annu Rev Immunol (2005) 787–819. doi: 10.1146/annurev.immunol.23.021704.115719
101. Carroll MC, Isenman DE. Regulation of humoral immunity by complement. Immunity (2012) 37(2):199–207. doi: 10.1016/j.immuni.2012.08.002
102. Jarjour M, Jorquera A, Mondor I, Wienert S, Narang P, Coles MC, et al. Fate mapping reveals origin and dynamics of lymph node follicular dendritic cells. J Exp Med (2014) 211(6):1109–22. doi: 10.1084/jem.20132409
103. Krautler NJ, Kana V, Kranich J, Tian Y, Perera D, Lemm D, et al. Follicular dendritic cells emerge from ubiquitous perivascular precursors. Cell (2012) 150(1):194–206. doi: 10.1016/j.cell.2012.05.032
104. Wu A, Pullarkat S. Follicular Dendritic Cell Sarcoma. Arch Pathol Lab Med (2016) 140(2):186–90. doi: 10.5858/arpa.2014-0374-RS
105. Choe JY, Go H, Jeon YK, Yun JY, Kim YA, Kim HJ, et al. Inflammatory pseudotumor-like follicular dendritic cell sarcoma of the spleen: a report of six cases with increased IgG4-positive plasma cells. Pathol Int (2013) 63(5):245–51. doi: 10.1111/pin.12057
106. Duan GJ, Wu F, Zhu J, Guo DY, Zhang R, Shen LL, et al. Extranodal follicular dendritic cell sarcoma of the pharyngeal region: a potential diagnostic pitfall, with literature review. Am J Clin Pathol (2010) 133(1):49–58. doi: 10.1309/AJCP7U8YISBUAVNW
107. Perez-Ordonez B, Rosai J. Follicular dendritic cell tumor: review of the entity. Semin Diagn Pathol (1998) 15(2):144–54.
108. Hsu C, Vega F, Grimes LM, Hunt KK. Follicular dendritic cell sarcoma and associated myasthenia gravis: true, true, related? J Clin Oncol (2011) 29(13):e369–71. doi: 10.1200/JCO.2010.32.7932
109. Lee IJ, Kim SC, Kim HS, Bang D, Yang WI, Jung WH, et al. Paraneoplastic pemphigus associated with follicular dendritic cell sarcoma arising from Castleman’s tumor. J Am Acad Dermatol (1999) 40(2 Pt 2):294–7. doi: 10.1016/S0190-9622(99)70468-8
110. Meijs M, Mekkes J, van Noesel C, Nijhuis E, Leeksma O, Jonkman M, et al. Paraneoplastic pemphigus associated with follicular dendritic cell sarcoma without Castleman’s disease; treatment with rituximab. Int J Dermatol (2008) 47(6):632–4. doi: 10.1111/j.1365-4632.2008.03444.x
111. Saygin C, Uzunaslan D, Ozguroglu M, Senocak M, Tuzuner N. Dendritic cell sarcoma: a pooled analysis including 462 cases with presentation of our case series. Crit Rev Oncol Hematol (2013) 88(2):253–71. doi: 10.1016/j.critrevonc.2013.05.006
112. Chan JK, Fletcher CD, Nayler SJ, Cooper K. Follicular dendritic cell sarcoma. Clinicopathologic analysis of 17 cases suggesting a malignant potential higher than currently recognized. Cancer (1997) 79(2):294–313. doi: 10.1002/(SICI)1097-0142(19970115)79:2<294::AID-CNCR13>3.0.CO;2-W
113. Soriano AO, Thompson MA, Admirand JH, Fayad LE, Rodriguez AM, Romaguera JE, et al. Follicular dendritic cell sarcoma: a report of 14 cases and a review of the literature. Am J Hematol (2007) 82(8):725–8. doi: 10.1002/ajh.20852
114. Grogg KL, Lae ME, Kurtin PJ, Macon WR. Clusterin expression distinguishes follicular dendritic cell tumors from other dendritic cell neoplasms: report of a novel follicular dendritic cell marker and clinicopathologic data on 12 additional follicular dendritic cell tumors and 6 additional interdigitating dendritic cell tumors. Am J Surg Pathol (2004) 28(8):988–98. doi: 10.1097/01.pas.0000112536.76973.7f
115. Grogg KL, Macon WR, Kurtin PJ. A survey of clusterin and fascin expression in sarcomas and spindle cell neoplasms: strong clusterin immunostaining is highly specific for follicular dendritic cell tumor. Mod Pathol (2005) 18(2):260–6. doi: 10.1038/modpathol.3800294
116. Yu H, Gibson JA, Pinkus GS, Hornick JL. Podoplanin (D2-40) is a novel marker for follicular dendritic cell tumors. Am J Clin Pathol (2007) 128(5):776–82. doi: 10.1309/7P8U659JBJCV6EEU
117. Shia J, Chen W, Tang LH, Carlson DL, Qin J, Guillem JG, et al. Extranodal follicular dendritic cell sarcoma: clinical, pathologic, and histogenetic characteristics of an underrecognized disease entity. Virchows Arch (2006) 449(2):148–58. doi: 10.1007/s00428-006-0231-4
118. Perez-Ordonez B, Erlandson RA, Rosai J. Follicular dendritic cell tumor: report of 13 additional cases of a distinctive entity. Am J Surg Pathol (1996) 20(8):944–55. doi: 10.1097/00000478-199608000-00003
119. Laginestra MA, Tripodo C, Agostinelli C, Motta G, Hartmann S, Doring C, et al. Distinctive Histogenesis and Immunological Microenvironment Based on Transcriptional Profiles of Follicular Dendritic Cell Sarcomas. Mol Cancer Res (2017) 15(5):541–52. doi: 10.1158/1541-7786.MCR-16-0301
120. Huang W, Qiu T, Zeng L, Zheng B, Ying J, Feng X. High frequency of clonal IG and T-cell receptor gene rearrangements in histiocytic and dendritic cell neoplasms. Oncotarget (2016) 7(48):78355–62. doi: 10.18632/oncotarget.13058
121. Pileri SA, Grogan TM, Harris NL, Banks P, Campo E, Chan JK, et al. Tumours of histiocytes and accessory dendritic cells: an immunohistochemical approach to classification from the International Lymphoma Study Group based on 61 cases. Histopathology (2002) 41(1):1–29. doi: 10.1046/j.1365-2559.2002.01418.x
Keywords: peripheral T-cell lymphomas, angioimmunoblastic T cell lymphoma, follicular T-cell lymphoma, follicular dendritic cell sarcomas, follicular lymphoma, Burkitt lymphoma, diffuse large B cell lymphoma
Citation: Munguía-Fuentes R, Maqueda-Alfaro RA, Chacón-Salinas R, Flores-Romo L and Yam-Puc JC (2021) Germinal Center Cells Turning to the Dark Side: Neoplasms of B Cells, Follicular Helper T Cells, and Follicular Dendritic Cells. Front. Oncol. 10:587809. doi: 10.3389/fonc.2020.587809
Received: 27 July 2020; Accepted: 26 November 2020;
Published: 15 January 2021.
Edited by:
Jérôme Paggetti, Luxembourg Institute of Health, LuxembourgReviewed by:
Alan G. Ramsay, King’s College London, United KingdomCopyright © 2021 Munguía-Fuentes, Maqueda-Alfaro, Chacón-Salinas, Flores-Romo and Yam-Puc. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Carlos Yam-Puc, jcyampuc@gmail.com
†Deceased
‡These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.