
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 28 October 2020
Sec. Thoracic Oncology
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.585292
This article is part of the Research Topic Therapeutic Strategies in EGFR Mutant Lung Cancer View all 26 articles
 Jinrong Liao1†Zeng Chen1†Zongyang Yu2†Tao Huang3Dan Hu4Ying Su1Zhiyong He5Changyan Zou1Lurong Zhang1*Xiandong Lin1,6*
Jinrong Liao1†Zeng Chen1†Zongyang Yu2†Tao Huang3Dan Hu4Ying Su1Zhiyong He5Changyan Zou1Lurong Zhang1*Xiandong Lin1,6*Cancer patients who initially benefit from Erlotinib, a drug targeting EGFR path, eventually develop resistance to the drug. The underlying mechanism is largely unknown. This study investigated the role of ARL4C in Erlotinib resistance development of NSCLC. qRT-PCR and Western blotting were performed to analyze the expression of mRNA and protein of ARL4C in two NSCLC cell lines (HCC827 and PC-9). Several assays (MTS, colony formation, transwell migration, luciferase reporter, and chromatin-immunoprecipitation) were used to explore the role of ARL4C in biofunctional changes of Erlotinib-resistant cells and their associations with Jak2/Stat 5/β-catenin signaling. Results demonstrated that (1) long-term use of Erlotinib resulted in downregulation of ARL4C; (2) overexpression of ARL4C could regain the sensitivity to Erlotinib in the drug-resistant HCC827/ER cells, while downregulation of ARL4C increased HCC827, and PC-9 cells' resistance to the drug; (3) Erlotinib-induced downregulation of ARL4C resulted in phosphorylation of Jak2/Stat5 and upregulation of β-catenin and their related molecules Axin2, CD44, Ccnd1, Lgr-5, and MMP7, which promoted the malignant behaviors of Erlotinib-resistant cells; (4) chromatin immunoprecipitation and luciferase reporter assay revealed that Stat5 could bind to β-catenin promoter to upregulate molecules to maintain the malignant behaviors, which might count for how Erlotinib-resistant cell survived while EGFR path was blocked; (5) the expression of ARL4C was not associated with known EGFR gene mutations in both Erlotinib-resistant cells and NSCLC tissues. Our data suggest that Erlotinib resistance of NSCLCs is associated with downregulation of ARL4C via affecting Jak/Stat/β-catenin signaling. ARL4C could serve as a biomarker to predict the effectiveness of TKI targeting therapy and a potential therapeutic target for overcoming Erlotinib resistance in NSCLC.
Lung cancer is the most commonly diagnosed malignancy and a leading cause of cancer death worldwide (1). Non-small cell lung cancer (NSCLC) accounts for ~80% of all lung cancer cases (2). EGFR (Epidermal Growth Factor Receptor) tyrosine kinase inhibitors (TKIs) are a group of important targeting drugs for the treatment of NSCLCs with EGFR mutations, including exon 19 deletions or L858R substitutions. However, the acquired drug resistance occurs within 1 year after the treatment with the first generation of EGFR-TKI, which is related partially with the T790M secondary gatekeeper mutation (3), or activation of other alternative pathways, such as HGF/Met (hepatocyte growth factor/mesenchymal-epithelial transition factor), HER2 (Erb-B2 Receptor Tyrosine Kinase 2), and PIK3CA (Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha) (4). However, mechanisms of TKI resistance in about 30% cases remain unclear (5). Seeking for the unknown mechanisms is a hot research topic in lung cancer research (6).
With selected methods, we found that downregulation of ARL4C (ADP-ribosylation factor-like 4C) might be associated with TKI resistance. ARL4C, also known as arl7, is a 192-amino-acid protein belonging to a small GTP enzyme. It is one of the subfamily members of ADP ribosylation factor and plays an important role in vesicle transport and signal transduction (7). The biofunction of ARL4C is not very clear yet. A study conducted by Su and his colleagues showed that high-level expression of ARL4C could inhibit the migration of ovarian cancer cells. Patients with high level of ARL4C mRNA had a good prognosis (8). Another study with gastric cancer showed that the expression of ARL4C was abnormal and the molecule was involved in tumor cell growth and cell migration (9). Using immunohistochemical analysis, Fujii et al. showed that ARL4C was abnormally expressed in lung cancer tissues. It was involved in the proliferation and invasion of lung cancer cells in vitro and in vivo (10). However, the role of ARL4C in TKI resistance is unexplored.
In this study, we investigated the role of ARL4C in TKI resistance of NSCLC cells by analyzing its functions with various assays. Our data demonstrated for the first time that ARL4C contributed to TKI resistance by activation of the JAK/STAT (Janus Kinase/signal transducer and activator of transcription) signaling pathway for survival when EGFR path blocked. The results suggest that ARL4C could be a promising biomarker for patients who likely benefit from TKI-based targeting therapy.
NSCLC tissues from 42 patients (22 men and 20 women with a median age of 60.5 years old) who have undergone lung cancer resection in Fujian Cancer Hospital between January 2008 and June 2009 were collected. None of the patients received chemotherapy before surgery, and cancer tissues were obtained immediately after surgical resection. One part of every cancer specimen was frozen at −80°C for measuring ARL4C mRNA level. The remaining part of the tissues was fixed in formalin and embedded in paraffin (FFPE) for detecting EGFR mutations.
HCC827 and PC-9 (EGFR 19del) cell lines purchased from ATCC were cultured in H1640 medium, containing 10% FBS (GIBCO BRL, Rockville, MD, U.S.A. Cat No. 10099233), 100 U/ml penicillin, and 100 μg/ml streptomycin in an incubator containing 5% CO2 at 37°C. The cells were exposed to gradually increased concentration of Erlotinib (Selleck, Houston, TX, USA. Cat No. S1023) from 0, 100, 200, 400, 800, 1,600, and 3,200 nM in their culture medium. After passing 17 generations in 6 months of the selection, Erlotinib was removed from the medium. The cells growing in 3,200 nM of Erlotinib were labeled as TKI Erlotinib-resistant (ER) cell lines HCC827/ER and PC-9/ER, respectively.
Several virus vectors were purchased from Hanheng Biotechnology Co Ltd (Beijing, China). To create cell lines with ARL4C overexpression, ARL4C expression vector HBLV-ARL4C (pHBLV-CMV-mcs-3flag-EF1-ZsGReen-T2A-PURO inserted with ARL4C gene) was used. The original vector was used as vector alone control. For ARL4C knockdown in cells, HBLV-ARL4C-shrna1, HBLV-ARL4C-shrna2, and HBLV-ARL4C-shrna3 were used and their parental vector pHBLV-U6-MCS-CMV-Zsgreen was used as vector alone control. HCC827, HCC827/ER, PC-9, and PC-9/ER cells cultured in six-well-plates (5 × 105/well) were infected with 10 MOI of respective viral vectors in the presence of 6 μg/ml of polyamine. After 48 h, the cells were selected with 2 μg/ml of Puromycin for 2 weeks to obtain stable infected cells. Newly established cells were HCC827/ER/vector, HCC827/ER/ARL4C-OE, HCC827/vector, HCC827/ARL4C-SH, PC-9/ER/vector, PC-9/ER/ARL4C-OE, PC-9/vector, and PC-9/ARL4C-SH. The symbols -OE and -SH means overexpression and knockdown, respectively.
The level of ARL4C mRNA expression was determined in 42 lung cancer tissues by real-time PCR. ARL4C and β-atenin, Axin2 (Axis Inhibition Protein 2), CD44, Ccnd1 (Cyclin D1), Lgr-5 (Leucine Rich Repeat Containing G Protein-Coupled Receptor 5), and MMP-7 (Matrix Metallopeptidase 7) were assessed in NSCLC cell lines (HCC827, PC-9, HCC827/ER, and PC-9/ER) also by real-time PCR. Total RNA was extracted from tissues and cell lines using Trizol reagent (Invitrogen, Grand Island, NY, USA) following the manufacturer's instruction. cDNA was synthesized from 1 μg of total RNA using M-MuLV reverse transcriptase (Promega, Madison, WI, USA). Real-time PCR was performed using SYBR1 Green Dye detection systems (Roche, Switzerland). The primers for amplifying ARL4C, β-catenin, Axin2, CD44, Ccnd1, Lgr-5, and MMP-7 are shown in Supplementary Table 1. Real-time PCR parameters were 95°C for 10 min, followed by 40 cycles of 95°C for 10 s, 55°C for 10 s and 72°C for 20 s, and 40°C for 30 s at the end of the 40 cycles. Relative quantity of mRNA expression was calculated by using the 2−ΔΔCt method. All measurements were repeated in triplicate.
Relative mRNA expression levels of ARL4C were quantified in all lung cancer samples using the comparative 2−ΔΔCt method and lung cancer sample T27 as reference reported previously (11, 12). The housekeeping gene glyceraldehyde phosphate dehydrogenase (GAPDH) was used to normalize expression levels of ARL4C.
Cells were harvested and lysed using RIPA buffer (50 mM Tris–Cl, pH 8.0, 150 mM NaCl, 5 mM EDTA, 0.1% SDS, and 1% NP-40) supplemented with protease inhibitor cocktail (Abcam, Cambridge, MA, USA. Cat No. ab65621). The cell lysates were centrifuged at 12,000 rpm for 30 min at 4°C. Supernatants were collected and protein concentrations were determined by BCA protein assay (Thermo Scientific, Rockford, Illinois, USA). The supernatants (each with 25 μg proteins) were electrophoresed on 10–12% polyacrylamide gel with sodium dodecyl sulfate (SDS) and then transferred onto nitrocellulose membranes (Millipore, Burlington, MA, USA) at 100 V for 1.5 h. After blocking with 3% BSA in TBST (TBS−1% Tween 20) for 1 h, the membranes were incubated with primary antibodies of ARL4C (1:500. Abcam, Cambridge, UK, Cat No. ab122025), and 1:1,000 diluted antibodies against β-catenin (CST, Danvers, MA, USA. Cat No. 8480), p-JAK 2 (CST, Danvers, MA, USA. Cat No. 4406), and p-STAT5 (Cell Signaling Technology, Danvers, MA, USA. Cat No. 4322) overnight at 4°C, respectively. After wash, the membranes were further incubated with horseradish peroxidase-conjugated anti-rabbit antibody. Finally, protein bands were developed with the enhanced chemiluminescence Western blot detection kit Immobilon ECL Ultra Western HRP Substrate (Millipore, Bedford, MA, USA. Cat No. WBULS0500), and images were captured on image station 4,000 mm pro (Carestream, Canada). Image J program was used to quantify the protein band intensity relative to loading control of β-actin.
Cells were plated into 96-well-plates (5,000 cells/well) and grew overnight. Different concentrations of Erlotinib (0, 100, 200, 400, 800, 1,600, and 3,200 nM) were added into the cell cultures in triplicate. After culturing for 72 h, 20 μl of MTS (Promega, Madison, WI, USA. Cat No. g3582) and 100 μl of serum containing medium were added to each well-followed by incubation at 37°C for 2 h. H1640 only with serum was used as background control. The absorption of the palates at 490 nm was read on a Bio-Rad (Hercules, CA, USA) plate reader (model 680). The proliferation of Erlotinib-treated cells was normalized with the control cells (no Erlotinib). IC50 was calculated with SPSS17.0. The experiment was repeated three times.
For colony-formation assay, cells (about 500 cells/well) were seeded and grew in six-well-plates for 48 h before adding Erlotinib. After 14 days, the cells were fixed in methanol and stained with 0.2% crystal violet. Cell colonies (>50 cells/colony) were pictured with Image Scanner (GE, Piscataway, NJ, USA). The number of colonies in each well was counted using Image J.
Cell concentration was adjusted to 7 × 105/ml, and 100 μl of the cell suspension was placed onto the upper chamber of each well on 24-well-Transwell plates coated with 1 mg/ml fibronectin (Millipore). The lower chamber contained medium with 20% FBS. After incubation at 37°C for 48 h, the cells in the upper chamber were wiped off with cotton swabs, and the cells in the other side of chamber membrane were fixed with methanol for 15 min, dried, stained with 0.1% crystal violet, and randomly pictured with a magnification of ×200 under inverted microscope (Olympus, Japan). The cells were counted with Image J program and the average number of the cells from 15 fields was used. The experiment was repeated three times.
Using the online transcription factor binding sites (TFBS) software (http://alggen.lsi.upc.es), we predicted the CTNNB1 promoter containing a STAT5A binding site. CTNNB1 (β-catenin) promoter (−200 to 0 regions) was inserted into pGL3-basic vector (Promega, Madison, WI, USA) as pGL3-CTNNB1 luciferase reporter plasmid (wild-pGL3-CTNNB1). The STAT5 site of the CTNNB1 promoter was mutated (from 5′-atttttctgtcag-3′ to 5′-taaaaagacagtc-3′) and was cloned into pGL3-basic vector to generate CTNNB1 promoter mutated reporter plasmid mut-pGL3-CTNNB1. All constructs were verified by sequencing. For the luciferase reporter assay, HEK293T cells, HEK293T/ARL4C-SH, HCC827, and HCC827/ARL4C-SH cells were transfected with pGL3-CTNNB1-Luc or mut-pGL3-CTNNB1 using X-treme GENE HP DNA Transfection Reagent (Roche, Basel, Switzerland. Cat No. 6366236001). Renilla luciferase was used as internal control. Forty-eight hours later, the transfected cells were harvested and the luciferase activity was measured by Dual-luciferase Reporter Assay System (Promega Corporation, Madison, WI, USA). The relative firefly luciferase activity was calculated by normalizing transfection efficiency using the Renilla luciferase activity.
ChIP assay was carried out using a SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling Technology, Beverly, MA, USA, Cat No. 9002) following the manufacturer's instruction. Briefly, 5 × 106 cells were fixed with 1% formaldehyde and quenched in 0.125 M glycine. Cells were sonicated by Bioruptor Sonication System UCD-300. DNA was immunoprecipitated by either control IgG or phospho-stat5 antibody. Precipitated DNA samples and inputs were amplified by PCR. The primers used for the amplification of stat5 binding site in β-catenin promoter are 5′-cctcttccccgttgtttcca-3′ (sense) and 5′-ggggtgattctttgctaatttca-3′ (antisense).
Two methods were used to detect EGFR mutations. For 42 NSCLC paraffin specimens, DNA was obtained using a paraffin tissue DNA Extraction kit (Qiagen, Hilden, Germany. Cat No. 56404). The concentration of DNA was adjusted to 1 ng/ml, and EGFR mutations were detected using the amplification refractory mutation system (ARMS) with human EGFR Mutations Detection kit (Amoy Diagnostics, Xiamen, China. Cat No. ADx-EG01) according to the manufacturer's protocol as previously described (13). Briefly, ARMS-PCR assay was performed in a 50-μl volume containing 5 μl of PCR buffer, 10 pM forward and reverse primers, 20 pM probe, and 12.5 mM dNTPs. The thermocycling conditions were as follows: 95°C for 5 min, then 15 cycles of 95°C for 25 s, 64°C for 20 s, and 72°C for 20 s, followed by 31 cycles of 93°C for 25 s, 60°C for 35 s, and 72°C for 20 s.
To determine if there is any association between ARL4C expression and EGFR mutations, eight cell lines with different levels of ARL4C were examined with the next-generation sequencing (NGS). DNA was extracted using the GONOROAD kit (Qiagen, Hilden, Germany) and 200 ng of DNA was used to build the library using NEBNext Ultra II DNA library Prep Kit for Illumina (NEB, Ipswich, MA, USA). Integrated DNA technologies (IDT, Skokie, IL, USA) customized probes were used for hybridization capture. All libraries were performed on an MGISEQ2000 instrument according to the manufacturer's instructions (BGI, Shenzhen, Guangdong, China) with 200 cycles, standing for paired-end 100 bp. Then, IDT 10-hotspot gene panels from all eight libraries were used, which included ALK, BRAF, EGFR, ERBB2, KRAS, MET, NRAS, PIK3CA, RET, and ROS1, and were quantitated using Library Quantification Kit-Illumina/Universal (Kapa Biosystems, Wilmington, MA, USA) on an ABI 7500 Real Time PCR system (Applied Biosystems, Waltham, MA, USA). The mutations were detected by the following methods: the Trimmomatic (version 0.39, parameter: PE -threads 4 -phred33 ILLUMINACLIP: adapter.fa:2:30:10 MINLEN:15), and the adapter sequences were AAGTCGGAGGCCAAGCGGTCTTAGGAAGACAA and AAGTCGGATCGTAGCCATGTCGTTCTGTGAGCCAAGGAGTTG and were used to narrow down the raw sequencing data (Fastq), filtering out the adapter contamination reads and low-quality reads to get clean data. Bwa Aln (Version: 0.7.12-r1039) algorithm was used to align the clean data of the human reference genome (hg19) and to get the Sequence Alignment/Map format (sam) file. For the Binary Alignment/Map format (bam) file, the sam file was sorted and the artificial duplication reads were removed by samtools (Version: 0.1.19-44428cd). According to the bed interval file of the 10-hotspot gene panels, Freebayes (version: v1.0.2-6-g3ce827d, parameter: -j -m 10 -q 30 -F 0.001 -C 1 -t bed.file –f hg19.fa) was used to determine the single-nucleotide polymorphisms (SNPs) and insertions or deletions (indels) and then ANNOVAR was used for the annotation.
All data were presented as mean ± SD. Student's t-test was used for analysis. ANOVA was used to determine the statistical difference between or among different experimental groups. The value P < 0.05 was considered as statistically significant difference.
Two NSCLC cell lines, HCC827 and PC-9, were subjected to Erlotinib selection in culture. After 6 months, IC50 of Erlotinib was significantly increased from 289 to 1,843 nM for HCC827 and from 71.08 to 5232.12 nM for PC-9. These Erlotinib-resistant (ER) cell lines were named HCC827/ER and PC-9/ER, which were 5.37 and 73.60 times more resistant to Erlotinib than their parental cells, respectively (Figures 1A–C). However, results of qPCR and Western blotting demonstrated that the levels of ARL4C were significantly lower in HCC827/ER and PC-9/ER cells than in their parental cells (Figures 1D–G, P < 0.0001). Furthermore, β-Catenin expression significantly increased in HCC827/ER and PC-9/ER, compared with their parental cells (Figures 1D–G, both P < 0.0001). Collectively, these results showed that the expression of ARL4C was reduced in TKI-resistant cells (HCC827/ER, PC-9/ER), while the expression of β-catenin was elevated in TKI-resistant cells.
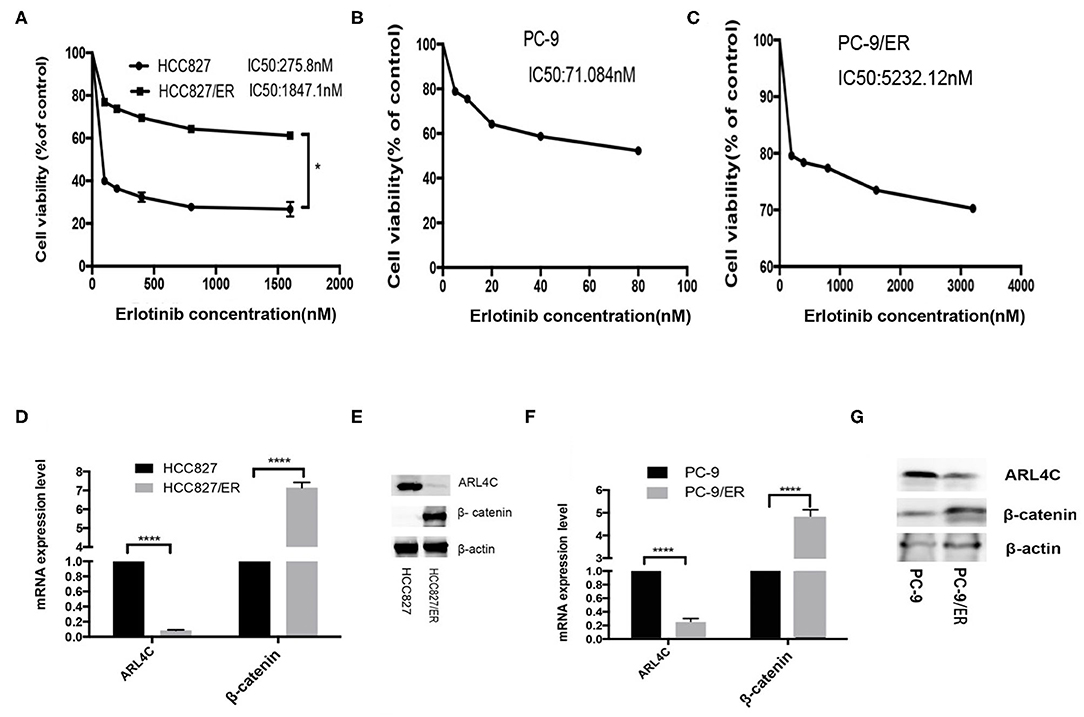
Figure 1. Expression of ARL4C and β-Catenin in TKI-resistant NSCLC cell lines. (A) MTS assay of the proliferation of in Erlotinib-resistant HCC827/ER and Erlotinib-sensitive HCC827 cells in different concentrations of Erlotinib. (B,C) MTS assay of the dose response of Erlotinib-sensitive PC-9 cells (B) and Erlotinib-resistant PC-9/ER (C) to different concentrations of Erlotinib. (D,E) qPCR and Western blotting analyzed ARL4C and β-catenin expression in HCC827/ER and HCC827 cells. (F,G) qPCR and Western blotting detected ARL4C and β-catenin expression in PC-9/ER and PC-9 cells. *P < 0.05; ****P < 0.0001.
To confirm that the downregulation of ARL4C was associated with the increased Erlotinib resistance, sh-ARL4C-knockout lentivirus was used to infect HCC827 and PC-9 cells. Western blotting showed that the expression level of ARL4C in the infected cells HCC827/ARL4C-sh and PC-9/ARL4C-sh were significantly lower than those in control cells infected with lentivirus vector alone (HCC827/Vector and PC-9/Vector) (Figures 2A–C, both P < 0.0001).
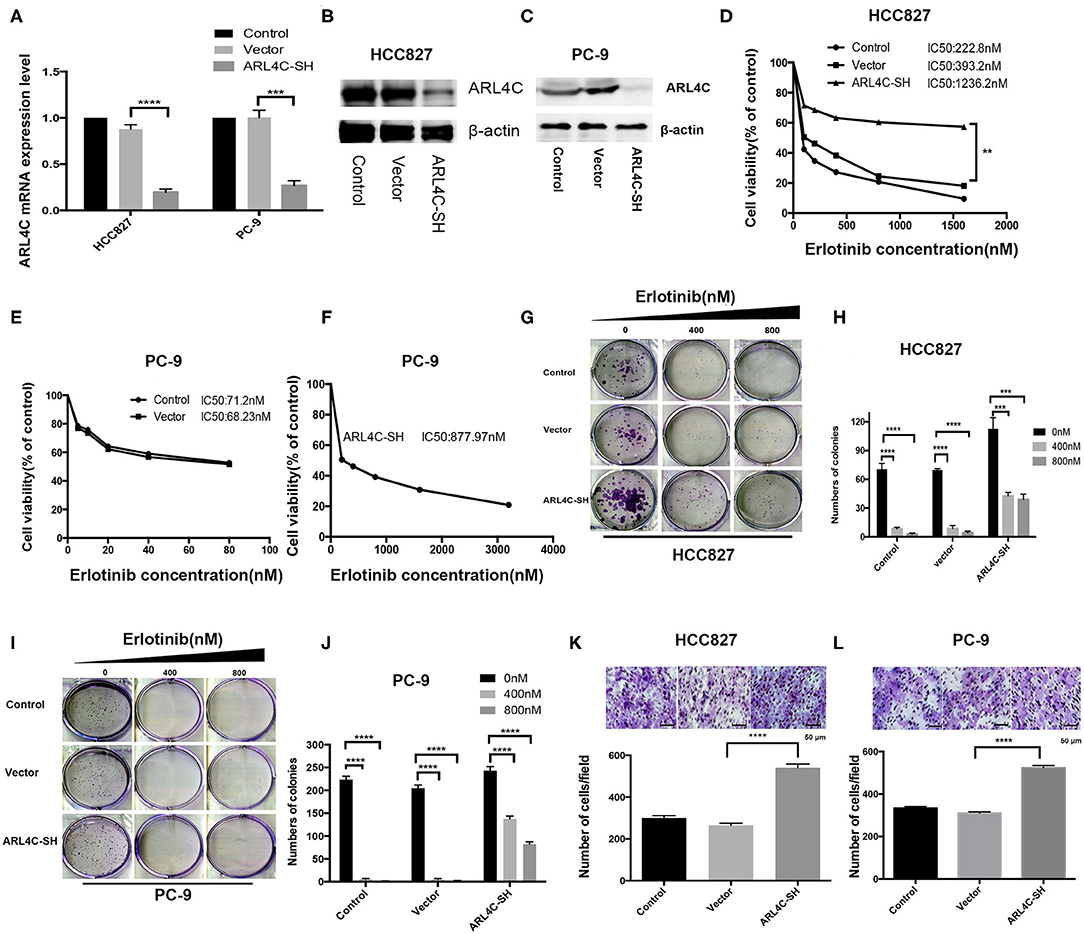
Figure 2. ARL4C knockdown enhances Erlotinib resistance of lung cancer cells. (A) qPCR quantification of ARL4C mRNA levels in ARL4C knockdown cells HCC827 (HCC827-ARL4C-SH) and PC-9 (PC-9-ARL4C-SH), control vector-infected HCC827 (HCC827-Vector) and PC-9 (PC-9-Vector), and their parental HCC827 and PC-9 control cells. (B) Western blotting detection of ARL4C expression in HCC827-ARL4C-SH, HCC827-Vector, and HCC827 (control) cells, and in (C) in PC-9-ARL4C-SH, PC-9-Vector, and PC-9 (control) cells. (D) IC50 of HCC827-ARL4C-SH, HCC827-Vector, and HCC827 (control) cells measured by MTS. (E,F) Erlotinib IC50 of PC-9-ARL4C-SH, PC-9-Vector, and PC-9 (control) cells measured by MTS. (G,H) Colony formation of HCC827-ARL4C-SH, HCC827-Vector, and HCC827 (control) cells. (I,J) Colony formation of PC-9-ARL4C-SH, PC-9-Vector, and PC-9 (control) cells. (K) Transwell assays of the migration of HCC827-ARL4C-SH, HCC827-Vector, and HCC827 (control) cells. (L) Transwell assays of the migration of PC-9-ARL4C-SH, PC-9-Vector, and PC-9 (control) cells. **P < 0.01; ***P < 0.001; ****P < 0.0001.
Effect of low ARL4C on Erlotinib IC50 was examined with MTS assay. Results showed that knockdown of ARL4C significantly increased Erlotinib resistance as compared to their parental cells. IC50 values of Erlotinib for HCC827, HCC827/vector, and HCC827/ARL4C-sh were 222.8, 393.2, and 1236.2 nM, respectively. The values for PC-9, PC-9/vector, and PC-9/ARL4C-sh were 71.2, 68.23, and 877.97 nM, respectively (Figures 2D–F, P < 0.01).
The cells with knockdown of ARL4C (HCC827/ARL4C-sh and PC-9/ARL4C-sh) had a marked enhancement of proliferation and colony formation, compared to their parental HCC827 and PC-9 cells when treated with Erlotinib (Figures 2G–J, both P < 0.001). These ARL4C low-expressing cell lines HCC827/ARL4C-sh and PC-9/ARL4C-sh cells also had a significantly increased capability of the migration, passing through the polyester membrane, compared with control cells HCC827/vector and PC-9/vector (Figures 2K,L, both P < 0.001).
These results suggest that knockdown ARL4C enhances TKI Erlotinib tolerance, colony formation, and migration of NSCLC cells.
To further confirm the relationship between Erlotinib resistance and ARL4C expression, both Erlotinib-resistant HCC827/ER and PC-9/ER cells were infected with ARL4C overexpression lentivirus to regain the ARL4C expression. Results of qPCR and Western blot showed that the expression levels of ARL4C in HCC827/ER/ARL4C-OE and PC-9/ER/ARL4C-OE were significantly higher than those of vector alone control cells, HCC827/ER/Vector, and PC-9/ER/Vector (Figures 3A–C, P < 0.001).
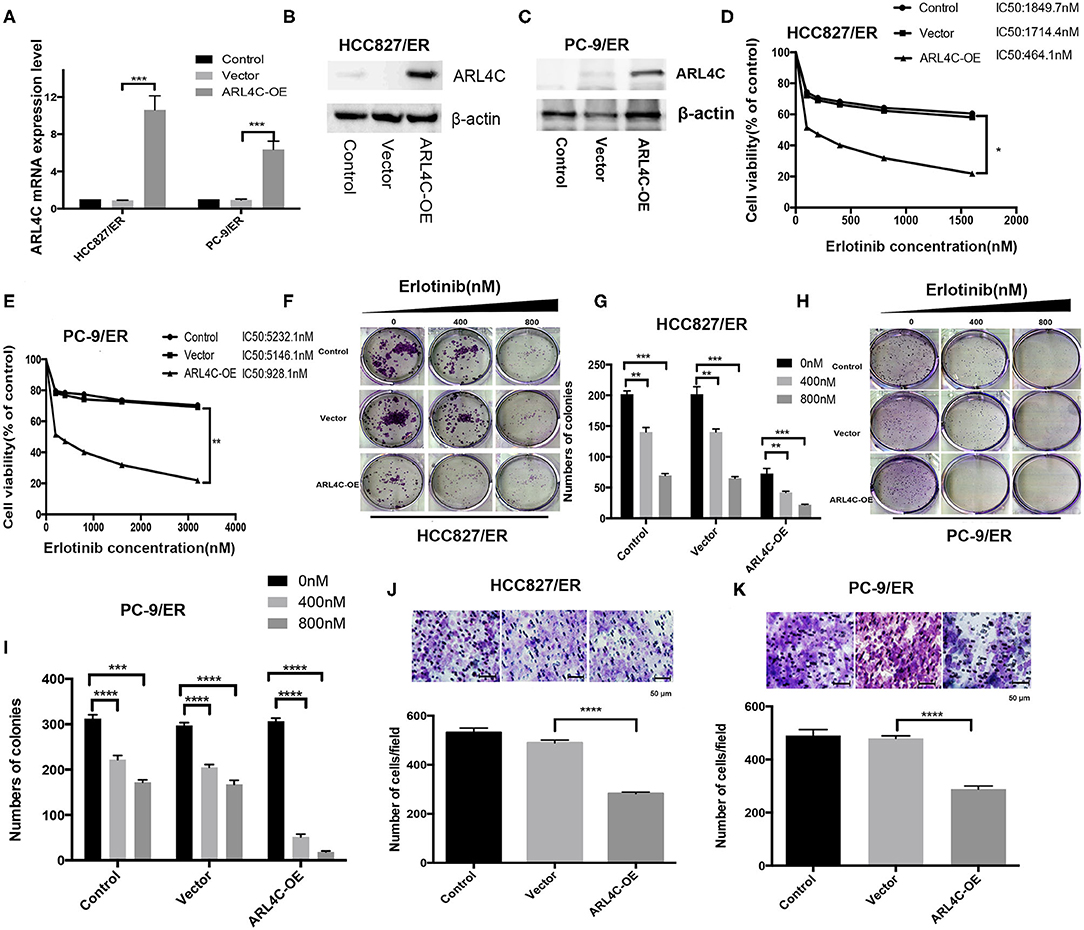
Figure 3. ARL4C overexpression decreases Erlotinib resistance of lung cancer cells. (A) Left: qPCR quantification of ARL4C expression in HCC827/ER ARL4C-overexpressing (HCC827/ER-ARL4C-OE), HCC827/ER-Vector (HCC827/ER-Vector), and HCC827/ER (control) cells. Right: qPCR quantification of ARL4C expression in PC-9/ER ARL4C-overexpressing (PC-9/ER-ARL4C-SH), PC-9/ER-ARL4C-control (PC-9/ER-Vector), and PC-9/ER (control) cells. (B) Western blotting detection of ARL4C expression in HCC827/ER-ARL4C-OE, HCC827/ER-Vector, and HCC827/ER (control) cells. (C) Western blotting detection of ARL4C expression in PC-9/ER-ARL4C-OE, PC-9/ER vector, and PC-9/ER (control) cells. (D,E) Erlotinib IC50 for HCC827/ER and PC-9 /ER cells ARL4C overexpressing with HBLV-ARL4C-SH3 and Vector measured by MTS. (F,G) Colony formation of HCC827/ER-ARL4C-OE, HCC827/ER-Vector, and HCC827/ER (control) cell. (H,I) Colony formation of PC-9/ER-ARL4C-OE, PC-9/ER-vector, and PC-9/ER (control) cells. (J) Transwell assay of the migration of HCC827/ER-ARL4C-OE, HCC827/ER-Vector, and HCC827/ER (control) cells. (K) Transwell assay of the migration of PC-9/ER-ARL4C-OE, PC-9/ER-vector, and PC-9/ER (control) cells. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
To examine the effect of high level of ARL4C on cell susceptibility to Erlotinib, the IC50 was measured with MTS assay. Results showed that the overexpression of ARL4C significantly decreased Erlotinib resistance of both HCC827/ER and PC-9/ER cells. IC50 values of Erlotinib for HCC827/ER, HCC827/ER/Vector, and HCC827/ER/ARL4C-OE cells were 1.85, 1.71, and 0.47 μM, respectively. The values for PC-9/ER, PC-9/ER/Vector, and PC-9/ER/ARL4C-OE cells were 5232.1, 5146.1, and 928.1 nM, respectively (Figures 3D,E, both P < 0.05).
Similarly, the effect of high level of ARL4C on cell colony formation and migration in the presence of Erlotinib was tested. Results showed that the overexpression of ARL4C in HCC827/ER/ARL4C-OE and PC-9/ER/ARL4C-OE cells resulted in an reduced sensitivity to Erlotinib as compared to HCC827/ER and PC-9/ER cells (Figures 3F–I, P < 0.01, P < 0.001, and P < 0.0001). The migration of HCC827/ER/ARL4C-OE and PC-9/ER/ARL4C-OE was significantly decreased. The number of cells passing through the polyester membrane decreased, compared to control cells HCC827/ER-vector and PC-9/ER-vector (Figures 3J,K, both P < 0.0001).
These data suggest that overexpression of ARL4C in Erlotinib-resistant lung cancer cells increases the sensitivity of the cells to TKI Erlotinib and inhibits cell migration.
It has been shown that cells resistant to Erlotinib expressed a high level of β-catenin (12). Our results also showed that ARL4C was expressed at a significantly low level in Erlotinib-resistant HCC827/ER and PC-9/ER cells, compared to Erlotinib-sensitive HCC827 and PC-9 cells (Figures 1D–G, P < 0.0001), while β-Catenin expression significantly increased in Erlotinib-resistant cells (Figures 1D–G, P < 0.0001). Therefore, we speculate that β-catenin might contribute to ARL4C-associated Erlotinib resistance.
To test our hypothesis, the effect of ARL4C on the β-catenin expression in HCC827 and HCC827/ER cells was examined. As expected, knockdown of ARL4C with lentivirus vectors HBLV-ARL4C-shrna1s significantly increased the expression of β-catenin in HCC827 cells. Furthermore, the expression of β-catenin-regulated target genes, Axin2, CD44, Ccnd1, Lgr-5, and MMP7, was also significantly increased in ARL4C shRNA transfected cells (Figures 4A,B, P < 0.05). In contrast, overexpression of ARL4C significantly suppressed the expression of β-catenin as well as β-catenin-regulated target genes, Axin2, CD44, Ccnd1, Lgr-5, and MMP7, in HCC827/ER cells (Figures 4C,D, P < 0.01; P < 0.001).
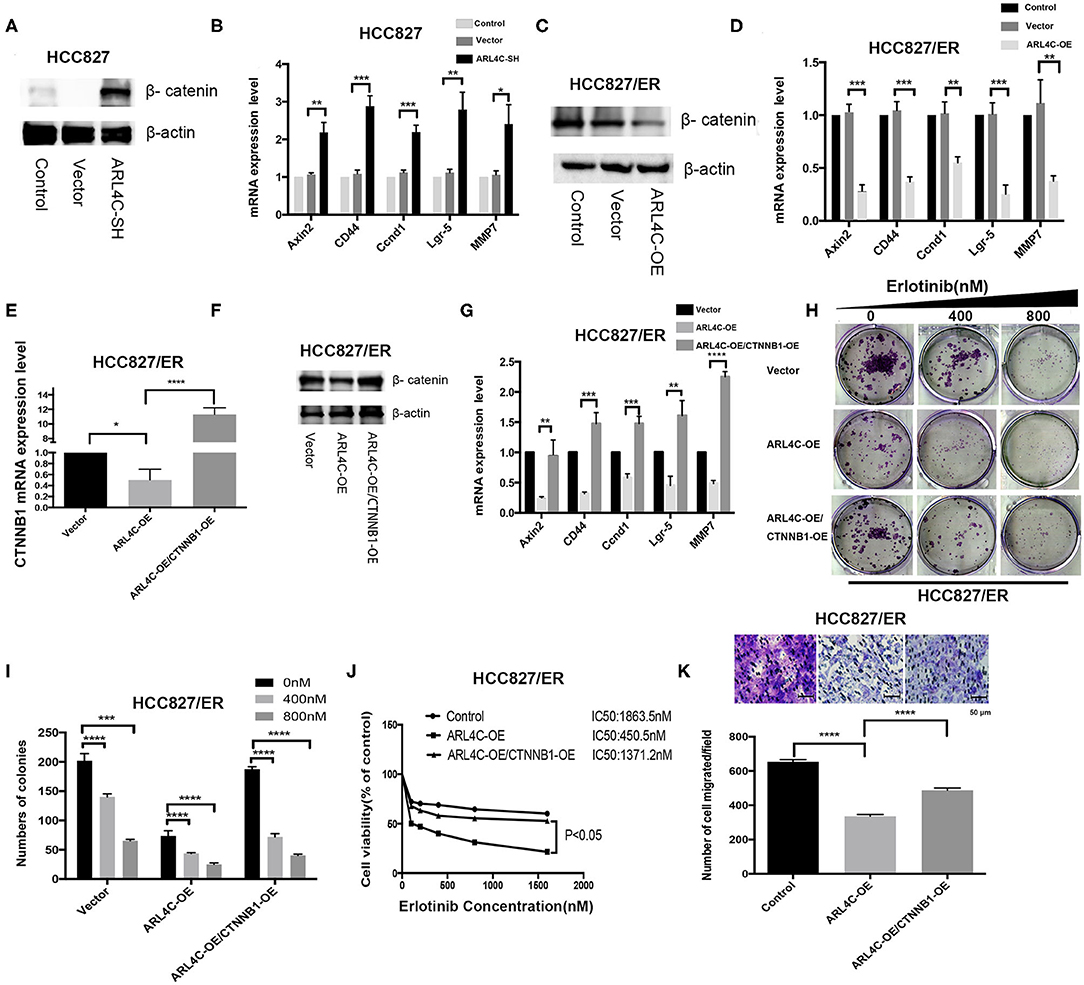
Figure 4. Enhancement of β-catenin is critical for ARL4C to reduce TKI Erlotinib resistance. (A) Western blot showed β-catenin protein expression in HCC827-ARL4C-SH, HCC827-Vector, and HCC827 (control) cells. (B) The expression of β-catenin-target genes Axin, CD44, Ccnd1, Lgr-5, and MMP7 in HCC827-ARL4C-SH, HCC827-Vector, and HCC827 (control) cells detected by real-time PCR. (C) Detection of β-catenin protein expression in HCC827/ER-ARL4C-OE, HCC827/ER-Vector, and HCC827/ER (control) cells by Western blot. (D) The expression of β-catenin-target genes Axin2, CD44, Ccnd1, Lgr-5, and MMP7 in HCC827/ER-ARL4C-OE, HCC827/ER-Vector, and HCC827/ER (control) cells measured by real-time PCR. (E,F) β-catenin (CTNNB1) expression in HCC827/ER/ARL4C-OE, HCC827/ER/ARL4C-OE–vector, and HCC827/ER/ARL4C-OE-CTNNB1-OE in the rescue assay analyzed by real-time PCR and Western blot. (G) Real-time PCR quantified the expression of β-catenin-target genes, Axin2, CD44, Ccnd1, Lgr-5, and MMP7 in HCC827/ER/ARL4C-OE, HCC827/ER/ARL4C-OE–vector, and HCC827/ER/ARL4C-OE-CTNNB1-OE. (H,I) Colony formation of HCC827/ER/ARL4C-OE, HCC827/ER/ARL4C-OE–vector, and HCC827/ER/ARL4C-OE-CTNNB1-OE. (J) Erlotinib IC50 for HCC82/ER/ARL4C-OE, HCC827/ER/ARL4C-OE–vector, and HCC827/ER/ARL4C-OE-CTNNB1-OE measured by MTS assay. (K) Transwell assays on the migration of HCC827/ER/ARL4C-OE, HCC827/ER/ARL4C-OE–vector, and HCC827/ER/ARL4C-OE-CTNNB1-OE. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
To further confirm the relationship between ARL4C and β-catenin, a rescue assay was performed by overexpressing β-catenin in HCC827/ER/ARL4C-OE. qPCR and Western blotting showed that the expression level of β-catenin in HCC827/ER/ARL4C-OE/CTNNB1-OE was significantly higher than that in HCC827/ER/ARL4C-OE (Figures 4E,F, P < 0.05 and P < 0.0001). Moreover, the expression of ARL4C-inhibited β-catenin-target genes Axin2, CD44, Ccnd1, Lgr-5, and MMP7 was increased (Figure 4G, P < 0.01, P < 0.001, and P < 0.0001), which was positively correlated to the expression of β-catenin.
In clonogenic assays, it was found that the overexpression of β-catenin significantly increased the proliferation of ARL4C-overexpressed HCC827/ER cells when treated with Erlotinib (Figures 4H,I, P < 0.001 and P < 0.0001). More importantly, the overexpression of β-catenin by lentivirus significantly decreased ARL4C-induced Erlotinib sensitivity in HCC827/ER cells. IC50 values of Erlotinib for HCC827/Vector, HCC827/ER/ARL4C-OE, and HCC827/ER/ARL4C-OE/CTNNB1-OE were 1863.5, 450.5, and 1371.2 nM, respectively (Figure 4J, P < 0.05). Transwell migration assay also showed that the overexpression of β-catenin increased the migration of ARL4C-overexpressed HCC827/ER cells (Figure 4K, P < 0.0001). These results suggest that β-catenin has an opposite effect to ARL4C on the development of TKI Erlotinib resistance of NSCLC cells.
Previous studies showed that several relevant downstream signals, such as ERK, JAK, and AKT, might be involved in tumor-promoting effects of ARL4C (14). To further explore potential mechanisms related to the acquired TKI resistance, phosphorylation levels of JAK were examined in ARL4C knockdown-HCC827 cells. Results showed that the phosphorylation level of JAK2 was significantly increased, compared to that in control cells (Figure 5A). Consistently, the overexpression of ARL4C significantly decreased the phosphorylation of JAK2 in HCC827/ER cells, compared to that in control cells (Figure 5B). The JAK-STAT signaling pathway not only regulates tumor development but also is closely related to TKI resistance (14). These results indicated that ARL4C abnormal expression affected the phosphorylation of JAK2.
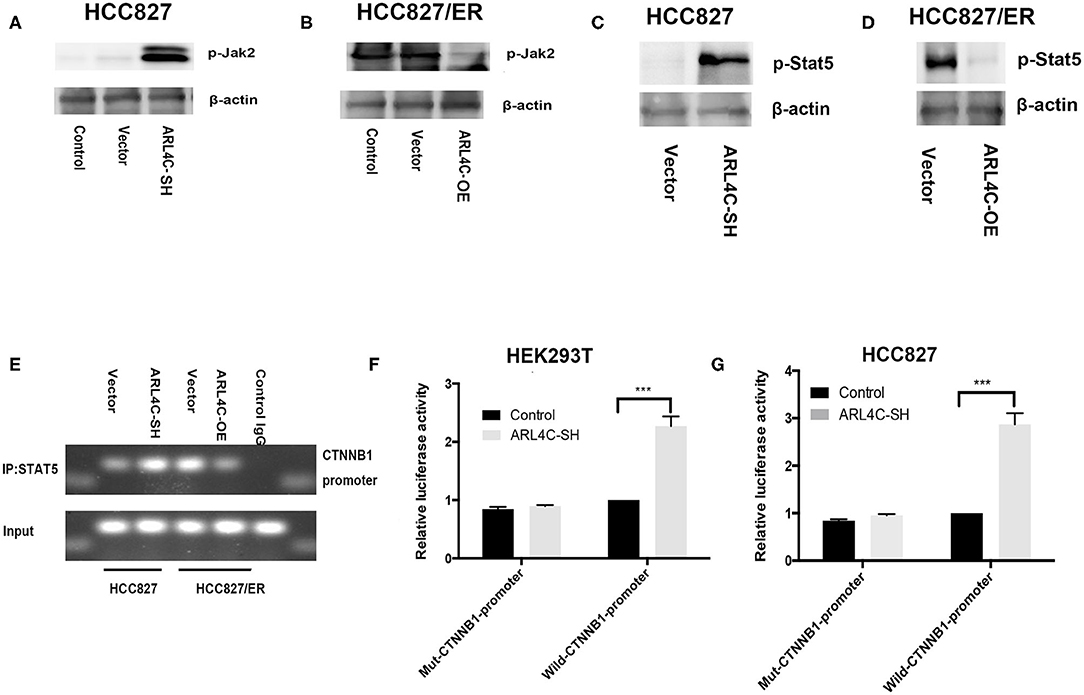
Figure 5. ARL4C regulates β-catenin expression through the JAK2/STAT5A signal pathway. (A–D) The expression of p-JAK2 in HCC827-ARL4C-SH, HCC827-Vector, and HCC827 (control) cells (A) and in HCC827/ER-ARL4C-OE, HCC827/ER-Vector, and HCC827/ER (control) cells (B) detected by Western blot. STAT5 protein expression in HCC827-ARL4C-SH, HCC827-Vector cells (C) and in HCC827/ER-ARL4C-OE, HCC827/ER-Vector cells (D) also detected by Western blot. (E) The enrichment of the β-catenin (CTNNB1) promoter in immunoprecipitated (IP) STAT5 and the input of STAT5 in HCC827/ER-ARL4C-OE, HCC827/ER-Vector, HCC827-ARL4C-SH, and HCC827-Vector and followed by Western blot. Unrelavant IgG was used as a negative control staining. (F,G) Luciferase activity in HEK293T cells (F) and HCC827 cells (G) co-transfected with pGL3-CTNNB1-Luc or mut-pGL3-CTNNB1-Luc and HBLV-ARL4C-SH3 by the Dual-Luciferase Reporter Assay System, ***P < 0.001.
We further studied if ARL4C regulated STAT5A. In HCC827 cells, the knockdown of ARL4C upregulated the phosphorylation of STAT5A as detected by Western blot analysis (Figure 5C). In contrast, the overexpression of ARL4C in HCC827/ER cells downregulated the phosphorylation of STAT5A (Figure 5D).
Chromatin immunoprecipitation showed that the overexpression of ARL4C resulted in the loss of the promoter of STAT5 binding to CTNNB1 (Figure 5E). We found that luciferase expression directed by a 200-bp fragment of the β-catenin promoter containing this STAT5A site was increased in ARL4C downregulated HEK293T cell (Figure 5F). Similarly, downregulation of ARL4C resulted in a significant enhancement of β-catenin promoter activity in HCC827 cells (Figure 5G). These results indicated that the β-catenin promoter contained a conserved STAT5A binding site. STAT5A acts as a transcription factor in regulating the mRNA expression of β-catenin by binding and activating the β-catenin promoter.
These data suggest that the regulation of β-catenin expression by ARL4C might be through the JAK2/STAT5A signal pathway in the regulating TKI resistance of NSCLC.
To explore the association between EGFR mutation and ARL4C, EGFR mutations in 42 NSCLC paraffin samples were detected with amplification refractory mutation system (ARMS) with human EGFR Mutations Detection kit. Among 42, 20 were found with EGFR mutations and only 1 had a T790M mutation, while the other 21 had no EGFR mutation (in Supplementary Table 2). In these 42 tissues, mRNA expression level of ARL4C in the EGFR mutation group was −0.064 ± 0.091 (lgRQ, RQ = 2−ΔΔCt), while the level in the group without EGFR mutation was −0.023 ± 0.099, indicating that the expression of ARL4C was not associated with EGFR gene mutations (Figure 6C, P = 0.7668).
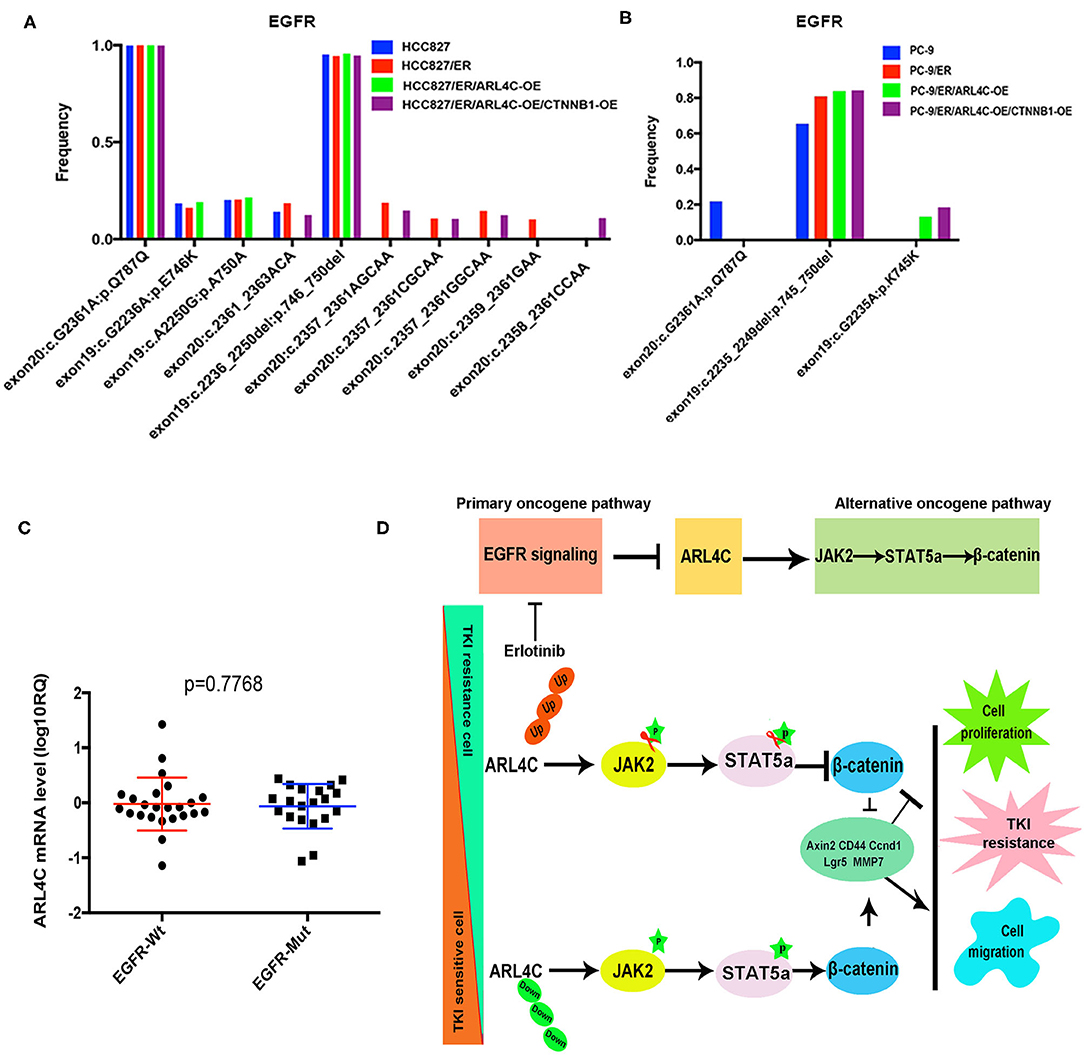
Figure 6. ARL4C alteration is not associated with known-mutated EGFR genes but affects JAK/STAT/β-Catenin path to stimulate the malignant behaviors of NSCLC cells: EGFR mutations were detected by ARMS with human EGFR Mutations Detection kit in 42 NSCLC samples. Sensitive EGFR mutations: Exon19:c.2236_2250del: p.746_750del; Synonymous mutation: Exon20:c.G2361A:p.Q787Q. (A) No differences of EGFR mutations among HCC827, HCC827/ER, HCC827/ER/ARL4C-OE, and HCC827/ER/ARL4C-OE /CTNNB1-OE cell lines. (B) No differences of EGFR mutations among PC-9, PC-9/ER, PC-9/ER/ARL4C-OE, and PC-9/ER/ARL4C-OE/CTNNB1-OE cell lines. (C) Distribution of ARL4C mRNA levels in NSCLC patients with EGFR mutations and without EGFR mutation(s). Bold lines represent the mean value for each patient cohort, RQ = 2−ΔΔCt. Similar expression level of ARL4C was found in 42 cases of NSCLC tissues, regardless of their EGFR mutation status. (D) A graphic illustration of the role of ARL4C in Erlotinib resistance. In TKI-resistant ARL4C-reduced NSCLC cells, the overexpression of ARL4C inhibited the expression of β-catenin by phosphorylation of JAK/STAT, resulting in an inhibitory effect on TKI resistance, cell proliferation, and migration, which was related to Axin2, CD44, Ccnd1, Lgr-5, and MMP7. Likewise, in the downregulation of ARL4C in sensitive cell lines, the results were vice versa.
NGS was used to detect the EGFR mutation in eight cell lines (HCC827, HCC827/ER, HCC827/ER/ARL4C-OE, HCC827/ER/ARL4C-OE/CTNNB1-OE, PC-9, PC-9/ER, PC-9/ER/ARL4C-OE, and PC-9/ER/ARL4C-OE/CTNNB1-OE) with different levels of ARL4C expression. Results (Figures 6A,B, Supplementary Table 3) showed that there was a high frequency of sensitive mutation (exon19:c.2236_2250del:p.746_750del) and no T790M and C797S mutations associated with Erlotinib-induced drug-resistant cells of HCC827/ER and PC-9/ER was found. There was a high frequency of synonymous mutation (exon20:c.G2361A:p.Q787Q) in HCC827, and no new mutation was caused by the overexpression of ARL4C in HCC827/ER and PC-9/ER or by the overexpression of CTNNB1 in HCC827/ER/ARL4C-OE and PC-9/ER/ARL4C-OE.
In this study, the underlying mechanism by which the resistance of Erlotinib, an EGFR-TKI, in NSCLC was explored. The study revealed for the first time that ARL4C was downregulated and β-catenin was upregulated in Erlotinib-resistant cells via JAK2/STAT5A signaling pathway. By manipulating the expression of these two genes (overexpression or knocking down expression), the study provided evidences that the inhibition of ARL4C selectively enhanced the resistance of HCC827 and PC-9 cells to Erlotinib, while the overexpression of ARL4C enhanced the sensitivity of the cancer cells to the drug. The regulation of the drug resistance of lung cancer cells by ARL4C was through activating the β-catenin/JAK2/STAT5A signaling pathway. These data indicate for the first time that ARL4C plays an important role in the resistance of NSCLC cells to Erlotinib.
Several mechanisms that mediate TKI resistance have been identified: (A) EGFR exon 20 T790M mutation (14); (B) Met gene amplification (15); (C) HER2 gene amplification (16); and (D) PIK3CA mutation (4), which counts for the majority of gene mutations responsible for the TKI resistance. However, one third of causes of the TKI resistance remains unknown. Our data (Figure 6) showed that Erlotinib-induced ARL4C reduction did not associate with any currently known mutations of EGFR, indicating that ARL4C is an independent factor for the development of Erlotinib resistance.
EGFR path is one of the major oncogenic pathways via Ras/Raf/MEK/ERK signaling to promote the cancer cell proliferation, migration, and invasion. When the EGFR path is blocked by Erlotinib, the cancer cells have to develop another oncogenic path for their continuous survival. Our data supported that the downregulation of ARL4C upregulated β-catenin via activation of JAK2/STAT5, which could help the EGFR-path-blocked cancer cells to regain their malignant behaviors (Figure 6D).
ARL4C is a member of the ADP-ribosylation factor family of GTP-binding proteins. The abnormal expression of ARL4C possesses carcinogenic effect on many types of tumors (17). Recent studies have indicated that ARL4C could be induced by EGF and promotes the motility of cancer cells by remodeling actin cytoskeleton through Arf6 activation (18), which might be involved in the migration, invasion, and proliferation of cancer cells (18). Consistent with this, our data showed that when EGF path was blocked by Erlotinib, the ARL4C was reduced, which triggered the upregulation of the JAK2/STAT5/β-catenin path to replace the EGFR oncogenic signal path. Similarly, our data supported the idea that the inhibition of ARL4C in wild-type HCC827 and PC-9 cells via sh-RNA could selectively enhance the Erlotinib tolerance, while the overexpression of ARL4C could enhance the Erlotinib sensitivity. The results indicate that ARL4C could be a “switch” between the EGFR path and the JAK2/STAT5/β-catenin path to maintain the proliferation, migration, and invasion of cancer cells when cells are exposed to Erlotinib and EGFR path is blocked.
It is well-known that Wnt/β-catenin signaling is a classic pathway with a crucial role in NSCLC progression. β-catenin is a key component in the Wnt signaling cascade and is involved in cancer cell proliferation and tumor progression (19). In the present study, a significant upregulation of β-catenin was observed in Erlotinib-resistant HCC827/ER and PC-9/ER cells compared with their parental cells HCC827 and PC-9. The result is in accordance with a previous observation that Wnt/β-catenin not only participates in the proliferation, invasion, and metastasis of tumor cells but also induces drug resistance (20). Nuclear accumulation of β-catenin was associated with EGFR mutations (21) and β-catenin overexpression was associated with NSCLC cell resistance to gefitinib (20). DDX17 nucleocytoplasmic shuttling promotes acquired gefitinib resistance in NSCLC cells via activation of β-catenin (22). Rab25 promotes Erlotinib resistance by activating the β1 integrin/AKT/β-catenin pathway in NSCLC (23). Alone this line, our finding added another path, ARL4C/JAK2/STAT5/β-catenin, as a new means for cancer cells to escape the survival inhibition by Erlotinib. Our data showed that in the presence of Erlotinib, the overexpression of β-catenin significantly increased the proliferation and decreased Erlotinib sensitivity in HCC827/ER-OE and PC-9/ER-OE (Figures 4H–K). Thus, ARL4C-induced Erlotinib resistance was via β-catenin signaling.
It is well-known that the JAK-STAT signaling pathway serves a crucial role in cell immunity, division, death, and tumor formation (24). The two families involved in this pathway are Janus kinases (JAKs) and signal transducer and activator of transcription proteins (STATs), encoded by the genes JAK (JAK1, JAK2, JAK3, and TYK2) and STAT (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6), respectively (25). The continuous activation of JAK/STAT could promote malignant transformation of cells, leading to the development of cancers including NSCLC (21). The inappropriate activation of Stat3 or IL-7-mediated IL-7R-JAK3/STAT5 pathway is associated with an unfavorable prognosis in NSCLC patients and correlated with chemoresistance and radioresistance (24). Inhibition of the JAK/STAT3 signaling pathway has therefore been recognized as a promising therapeutic strategy for NSCLC (25). In addition, it was reported that (1) the inhibition of gp130-Jak-Stat3 signaling could partially inhibit Wnt–β-catenin–mediated intestinal tumor growth and regeneration (26); (2) Stat3 and β-catenin were involved in tumorigenesis (27). By using chromatin immunoprecipitation and luciferase reporter assays, this study revealed for the first time that the promoter region of β-catenin contained a conserved STAT5A binding site. STAT5A acted as a transcription factor regulating the mRNA expression of β-Catenin by binding to and activating the β-catenin promoter, forming ARL4C/JAK2/STAT5/β-catenin axis for the survival of Erlotinib-resistant cells.
Our data also supported that the biofunctions of ARL4C/JAK2/STAT5/β-catenin axis were carried out by a set of molecules, such as Axin2, CD44, Ccnd1, Lgr-5, and MMP7. Axin2 is responsible for the stability of β-catenin (28); CD44 is a famous stem cell marker and adhesion molecule to enhance the proliferation, migration, and invasion (29); Ccnd1 enhances the proliferation of cancer cells via promoting the cell's G1/S transition (30); Lgr-5 is a biomarker for stem cells, involved in tumor development (31). MMP7 promotes the migration and invasion of cancer cells (32). All these functional molecules were upregulated in Erlotinib-resistant cells when ARL4C was suppressed and JAK2/STAT5/β-catenin axis was activated (Figure 6D), serving as executors for the malignant behaviors when the EGFR path was blocked in NSCLC cells.
The Erlotinib stress-related ARL4C/JAK2/STAT5/β-catenin axis and the downstream Axin2, CD44, Ccnd1, Lgr-5, and MMP7 could be therapeutic targets when considering how to reverse Erlotinib resistance and how to enhance the Erlotinib killing effects (Figure 6D).
This study demonstrated for the first time that the ARL4C/JAK2/STAT5/β-catenin axis and their downstream molecules Axin2, CD44, Ccnd1, Lgr-5, and MMP7 could help to bypass EGFR oncogenic path and serve as an alternative new signal/function path to maintain the survival and malignant behaviors for HCC827 and PC9 NSCLC cells under the stress of Erlotinib.
The datasets generated for this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: NCBI BioProject (PRJNA648039) (https://www.ncbi.nlm.nih.gov/Traces/study/?acc=PRJNA648039).
The studies involving human participants were reviewed and approved by the Research Ethics Committee of the Fujian Provincial Tumor Hospital. The patients/participants provided their written informed consent to participate in this study.
XL, LZ, ZY, and YS designed the study, analyzed data, and wrote the manuscript. ZY, DH, and ZH collected and analyzed the NSCLC lung tissues for ARL4C expression level. JL, ZC, and CZ performed all the experiments, collected data, and created the figures. TH carried out the bioinformatics and statistical analysis. All authors contributed to the article and approved the submitted version.
This study was supported by the Natural Science Foundation of Fujian Province (No. 2019J01196), the Joint Funds for the Innovation of Science and Technology, Fujian province (Nos. 2017Y9082, 2018Y9113) and Medical Innovation Program of Fujian Province (No. 2019-CX-5), National Natural Science Foundation of China (31701151), National Key R&D Program of China (2018YFC0910403), Shanghai Municipal Science and Technology Major Project (2017SHZDZX01), Shanghai Sailing Program (16YF1413800), and The Youth Innovation Promotion Association of Chinese Academy of Sciences (CAS) (2016245). Funders did not have any role in study design, data collection, and data analysis.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank all researchers in the Laboratory of Radiation Oncology and Radiobiology, Fujian Cancer Hospital for supporting this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.585292/full#supplementary-material
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. (2020) 70:7–30. doi: 10.3322/caac.21590
2. Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. (2018) 553:446–54. doi: 10.1038/nature25183
3. Wu SG, Shih JY. Management of acquired resistance to EGFR TKI-targeted therapy in advanced non-small cell lung cancer. Mol Cancer. (2018) 17:38. doi: 10.1186/s12943-018-0777-1
4. Andrews Wright NM, Goss GD. Third-generation epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of non-small cell lung cancer. Transl Lung Cancer Res. (2019) 8(Suppl. 3):S247–64. doi: 10.21037/tlcr.2019.06.01
5. Majem M, Remon J. Tumor heterogeneity.evolution through space and time in EGFR mutant non small cell lung cancer patients. Transl Lung Cancer Res. (2013) 2:226–37. doi: 10.3978/j.issn.2218-6751.2013.03.09
6. Berger AH, Brooks AN, Wu X, Shrestha Y, Chouinard C, Piccioni F, et al. High-throughput phenotyping of lung cancer somatic mutations. Cancer Cell. (2016) 30:214–28. doi: 10.1016/j.ccell.2016.06.022
7. Matsumoto S, Fujii S, Kikuchi A. Arl4c is a key regulator of tubulogenesis and tumourigenesis as a target gene of Wnt-beta-catenin and growth factor-Ras signalling. J Biochem. (2017) 161:27–35. doi: 10.1093/jb/mvw069
8. Su D, Katsaros D, Xu S, Xu H, Gao Y, Biglia N, et al. ADP-ribosylation factor-like 4C (ARL4C), a novel ovarian cancer metastasis suppressor, identified by integrated genomics. Am J Transl Res. (2015) 7:242–56.
9. Chen X, Su Z, Wang S, Xu H. Clinical and prognostic significance of Arl4c expression in colorectal cancer. Cancer Biomark. (2016) 16:253–7. doi: 10.3233/CBM-150562
10. Kimura K, Matsumoto S, Harada T, Morii E, Nagatomo I, Shintani Y, et al. ARL4C is associated with initiation and progression of lung adenocarcinoma and represents a therapeutic target. Cancer Sci. (2020) 111:951–61. doi: 10.1111/cas.14303
11. Yuan JS, Reed A, Chen F, Stewart CN Jr. Statistical analysis of real-time PCR data. BMC Bioinform. (2006) 7:85. doi: 10.1186/1471-2105-7-85
12. Lin XD, Chen SQ, Qi YL, Zhu JW, Tang Y, Lin JY. Overexpression of thrombospondin-1 in stromal myofibroblasts is associated with tumor growth and nodal metastasis in gastric carcinoma. J Surg Oncol. (2012) 106:94–100. doi: 10.1002/jso.23037
13. Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. (2009) 361:947–57. doi: 10.1056/NEJMoa0810699
14. Lu X, Yu L, Zhang Z, Ren X, Smaill JB, Ding K. Targeting EGFR(L858R/T790M) and EGFR(L858R/T790M/C797S) resistance mutations in NSCLC: current developments in medicinal chemistry. Med Res Rev. (2018) 38:1550–81. doi: 10.1002/med.21488
15. Wang Q, Yang S, Wang K, Sun SY. MET inhibitors for targeted therapy of EGFR TKI-resistant lung cancer. J Hematol Oncol. (2019) 12:63. doi: 10.1186/s13045-019-0759-9
16. de Langen AJ, Jebbink M, Hashemi SMS, Kuiper JL, de Bruin-Visser J, Monkhorst K, et al. Trastuzumab and paclitaxel in patients with EGFR mutated NSCLC that express HER2 after progression on EGFR TKI treatment. Br J Cancer. (2018) 119:558–64. doi: 10.1038/s41416-018-0194-7
17. Fujii S, Matsumoto S, Nojima S, Morii E, Kikuchi A. Arl4c expression in colorectal and lung cancers promotes tumorigenesis and may represent a novel therapeutic target. Oncogene. (2015) 34:4834–44. doi: 10.1038/onc.2014.402
18. Harada T, Matsumoto S, Hirota S, Kimura H, Fujii S, Kasahara Y, et al. Chemically modified antisense oligonucleotide against ARL4C inhibits primary and metastatic liver tumor growth. Mol Cancer Ther. (2019) 18:602–12. doi: 10.1158/1535-7163.MCT-18-0824
19. Stewart DJ. Wnt signaling pathway in non-small cell lung cancer. J Natl Cancer Inst. (2014) 106:djt356. doi: 10.1093/jnci/djt356
20. Cao S, Wang Z, Gao X, He W, Cai Y, Chen H, et al. FOXC1 induces cancer stem cell-like properties through upregulation of beta-catenin in NSCLC. J Exp Clin Cancer Res. (2018) 37:220. doi: 10.1186/s13046-018-0894-0
21. Suzuki M, Shigematsu H, Nakajima T, Kubo R, Motohashi S, Sekine Y, et al. Synchronous alterations of Wnt and epidermal growth factor receptor signaling pathways through aberrant methylation and mutation in non small cell lung cancer. Clin Cancer Res. (2007) 13:6087–92. doi: 10.1158/1078-0432.CCR-07-0591
22. Li K, Mo C, Gong D, Chen Y, Huang Z, Li Y, et al. DDX17 nucleocytoplasmic shuttling promotes acquired gefitinib resistance in non-small cell lung cancer cells via activation of beta-catenin. Cancer Lett. (2017) 400:194–202. doi: 10.1016/j.canlet.2017.02.029
23. Wang J, Zhou P, Wang X, Yu Y, Zhu G, Zheng L, et al. Rab25 promotes erlotinib resistance by activating the beta1 integrin/AKT/beta-catenin pathway in NSCLC. Cell Prolif. (2019) 52:e12592. doi: 10.1111/cpr.12592
24. Groner B, von Manstein V. Jak Stat signaling and cancer: opportunities, benefits and side effects of targeted inhibition. Mol Cell Endocrinol. (2017) 451:1–14. doi: 10.1016/j.mce.2017.05.033
25. Chai EZ, Shanmugam MK, Arfuso F, Dharmarajan A, Wang C, Kumar AP, et al. Targeting transcription factor STAT3 for cancer prevention and therapy. Pharmacol Ther. (2016) 162:86–97. doi: 10.1016/j.pharmthera.2015.10.004
26. Phesse TJ, Buchert M, Stuart E, Flanagan DJ, Faux M, Afshar-Sterle S, et al. Partial inhibition of gp130-Jak-Stat3 signaling prevents Wnt-beta-catenin-mediated intestinal tumor growth and regeneration. Sci Signal. (2014) 7:ra92. doi: 10.1126/scisignal.2005411
27. Wu J, Keng VW, Patmore DM, Kendall JJ, Patel AV, Jousma E, et al. Insertional Mutagenesis Identifies a STAT3/Arid1b/beta-catenin Pathway Driving Neurofibroma Initiation. Cell Rep. (2016) 14:1979–90. doi: 10.1016/j.celrep.2016.01.074
28. Song X, Wang S, Li L. New insights into the regulation of Axin function in canonical Wnt signaling pathway. Protein Cell. (2014) 5:186–93. doi: 10.1007/s13238-014-0019-2
29. Bhattacharya R, Mitra T, Ray Chaudhuri S, Roy SS. Mesenchymal splice isoform of CD44 (CD44s) promotes EMT/invasion and imparts stem-like properties to ovarian cancer cells. J Cell Biochem. (2018) 119:3373–83. doi: 10.1002/jcb.26504
30. Gennaro VJ, Stanek TJ, Peck AR, Sun Y, Wang F, Qie S, et al. Control of CCND1 ubiquitylation by the catalytic SAGA subunit USP22 is essential for cell cycle progression through G1 in cancer cells. Proc Natl Acad Sci USA. (2018) 115:E9298–307. doi: 10.1073/pnas.1807704115
31. Choi YJ, Kim N, Lee HS, Park SM, Park JH, Yoon H, et al. Expression of leucine-rich repeat-containing G-protein coupled receptor 5 and CD44: potential implications for gastric cancer stem cell marker. J Cancer Prevent. (2016) 21:279–87. doi: 10.15430/JCP.2016.21.4.279
Keywords: non-small cell lung cancer, ARL4C, TKI resistance, β-catenin, Jak, Stat
Citation: Liao J, Chen Z, Yu Z, Huang T, Hu D, Su Y, He Z, Zou C, Zhang L and Lin X (2020) The Role of ARL4C in Erlotinib Resistance: Activation of the Jak2/Stat 5/β-Catenin Signaling Pathway. Front. Oncol. 10:585292. doi: 10.3389/fonc.2020.585292
Received: 20 July 2020; Accepted: 01 September 2020;
Published: 28 October 2020.
Edited by:
Yaxiong Zhang, Sun Yat-sen University Cancer Center (SYSUCC), ChinaReviewed by:
Pasquale Pisapia, University of Naples Federico II, ItalyCopyright © 2020 Liao, Chen, Yu, Huang, Hu, Su, He, Zou, Zhang and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiandong Lin, linxdong1970@yeah.net; Lurong Zhang, lz8506@163.com
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.