 Emine Atas
Emine Atas Monika Oberhuber1,2†‡
Monika Oberhuber1,2†‡ Lukas Kenner
Lukas Kenner
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 15 December 2020
Sec. Cancer Metabolism
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.583217
This article is part of the Research Topic Metabolism Meets Function: The Multifaced Role of Metabolism in Cancer View all 24 articles
A metabolic shift from oxidative phosphorylation (OXPHOS) to glycolysis—known as the Warburg effect—is characteristic for many cancers. It gives the cancer cells a survival advantage in the hypoxic tumor microenvironment and protects them from cytotoxic effects of oxidative damage and apoptosis. The main regulators of this metabolic shift are the pyruvate dehydrogenase complex and pyruvate dehydrogenase kinase (PDK) isoforms 1–4. PDK is known to be overexpressed in several cancers and is associated with bad prognosis and therapy resistance. Whereas the expression of PDK1–3 is tissue specific, PDK4 expression is dependent on the energetic state of the whole organism. In contrast to other PDK isoforms, not only oncogenic, but also tumor suppressive functions of PDK4 have been reported. In tumors that profit from high OXPHOS and high de novo fatty acid synthesis, PDK4 can have a protective effect. This is the case for prostate cancer, the most common cancer in men, and makes PDK4 an interesting therapeutic target. While most work is focused on PDK in tumors characterized by high glycolytic activity, little research is devoted to those cases where PDK4 acts protective and is therefore highly needed.
Besides other hallmarks of cancer, such as sustained proliferative signaling, resistance to cell death, invasiveness and increased angiogenesis, tumors are characterized by their altered metabolic features (1, 2). The two major metabolic pathways providing energy in the form of adenosine triphosphate (ATP) are glycolysis and oxidative phosphorylation (OXPHOS) (1, 3). Under aerobic conditions, glucose is metabolized to pyruvate via glycolysis in the cytosol (4). Pyruvate is then processed to CO2 in the mitochondria via the tricarboxylic acid (TCA) cycle and OXPHOS (1, 3) (Figure 1). On the contrary, under anaerobic conditions glycolysis is favored, where pyruvate is mostly converted to lactate and only minimal amounts enter the TCA cycle (1, 3). Cancer cells typically present a metabolic shift from the TCA cycle/OXPHOS to glycolysis or lactate fermentation regardless of the presence of oxygen, a phenomenon known as the “Warburg effect” (7, 8). Hereby, cancer cells obtain survival advantages in hypoxic tumor microenvironments where OXPHOS is compromised (3). However, the switch to aerobic glycolysis in the cancer cell is not only limited to hypoxia but is also activated by deregulated signals enhancing glycolysis or hindering OXPHOS (3, 9, 10).
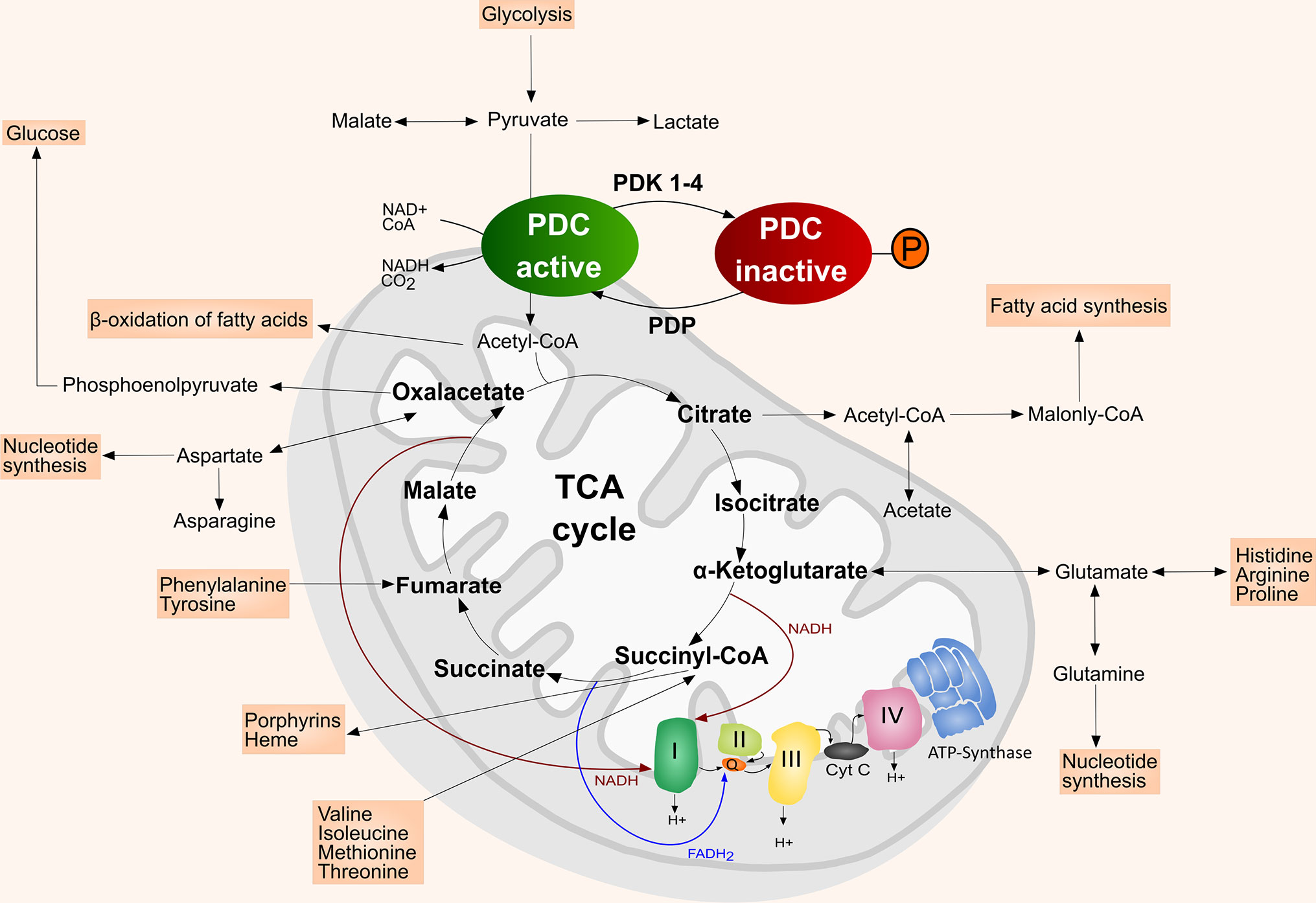
Figure 1 Simplified scheme of the mitochondrion with TCA cycle and the intersecting anaplerotic and cataplerotic reactions, OXPHOS complexes I–IV, and ATP synthase (complex V). In the mitochondrial matrix the PDC catalyzes the irreversible conversion of pyruvate, NAD+ and CoA into acetyl-CoA, NADH and CO2. PDK inactivate the PDC by phosphorylating its E1α subunit, which hinders the entrance of acetyl-CoA into the TCA cycle. The PDC is reactivated upon dephosphorylation by PDP. Adapted from (5). Inspiration (4, 6).
The metabolic shift from TCA cycle/OXPHOS to aerobic glycolysis is tightly regulated (11). The following review focuses on the key players in this regulation—the mitochondrial pyruvate dehydrogenase complex (PDC) and the pyruvate dehydrogenase kinases (PDK). PDK have been associated with tumor aggressiveness, proliferation, anti-apoptotic effects and therapy resistance in numerous malignancies (12–16). We here provide a compact overview on the latest research on the cancer specific levels of PDK isoforms and their associations with tumor aggressiveness and therapy resistance. We will also address the remarkable energy metabolism of prostate cancer (PCa) and the resulting effects of PDK on tumor growth. A detailed discussion of metabolic pathways intersecting with the TCA cycle and their interplay with the PDC/PDK axis is beyond the scope of this review but can be found here: Gray et al., Hirschey et al., Martínez-Reyes and Chandel et al., Vander Heiden and DeBerardinis et al. (4, 17–19).
One of the main enzymes regulating the metabolic shift in mammals is the mitochondrial PDC (20, 21). It is composed of the pyruvate dehydrogenase (E1), dihydrolipoamide acetyl-transferase (E2), dihydrolipoamide dehydrogenase (E3) and the E3 binding protein (E3BP) (4, 22). The PDC catalyzes the irreversible conversion of pyruvate, nicotinamide adenine dinucleotide (NAD+) and coenzyme-A (CoA) into acetyl-CoA, NADH and CO2 (4). The converted acetyl-CoA then enters the TCA cycle (4). Thus, PDC represents an important link between glycolysis and TCA cycle/OXPHOS (20, 21, 23) (Figure 1). PDC is more active in the healthy and well-nourished state, whereas its activity is decreased during fasting or low glucose levels, but also in diabetes and most cancer types (21, 24). The activity of the PDC is mainly regulated by four PDK isoenzymes (PDK1–4) that are located in the mitochondrial matrix (20, 25). PDK1–4 achieve a reversible downregulation of the PDC by phosphorylating specific serine residues (Ser293, Ser300, and Ser232) of its E1α subunit, thereby reducing the metabolic flux through the PDC and downstream pathways (4, 20, 23, 25). The E1α subunit of the PDC can be dephosphorylated by pyruvate dehydrogenase phosphatase (PDP), which leads to the reactivation of the PDC (4, 22, 23) (Figure 1). In addition, PDC can also be reversely acetylated and succinylated (26, 27). Acetylation of the PDC E1α subunit by acetyl-CoA acetyltransferase 1 (ACAT1) results in dissociation of PDP1 from the PDC and PDK1 recruitment, thereby suppressing PDC activity (26, 28, 29). Lysine desuccinylation of PDC subunits by sirtuin (SIRT) 3 and SIRT5 also results in suppression of PDC activity (26).
Besides generating reductive equivalents for OXPHOS, the TCA cycle provides precursors for biosynthetic processing of lipids, amino acids, and nucleotides (17, 19). Anaplerotic (carbon replenishing) and cataplerotic (carbon expending) pathways intersecting the TCA cycle balance the carbon flux (4). Pyruvate provides carbon either via the PDC or alternatively via conversion to oxaloacetate (4, 17, 30). Furthermore, glutamine contributes glutamate, α-ketoglutarate, aspartate, CO2, pyruvate, lactate, alanine and citrate to the TCA cycle, which makes it a key player in the mitochondrial metabolism supporting proliferation of cancer cells (Figure 1) (4, 17, 19, 30). A detailed discussion of the role of glutamine metabolism in cancer was published by Masisi et al. (31).
PDK1–4 are differentially expressed in several metabolic tissues (32). PDK1 is abundant in the cardiac muscle, pancreatic islets, and skeletal muscle and is expressed at lower levels in other tissues (20, 22, 23, 33–35). PDK2 on the contrary is ubiquitously expressed, with the highest expression levels in the heart, diaphragm, kidney, and red skeletal muscles (22). Other tissues such as liver, brain, testis, ovaries, and lung show lower PDK2 protein levels (22). While PDK3 has a weak expression pattern in kidney, brain, testis, and lung, PDK4 is mainly expressed in the heart, skeletal muscle, pancreatic islets and at intermediate levels in the liver, lung, and kidney (20, 22, 23, 33–35).
PDK1, a downstream target of hypoxia inducible factor 1 alpha (HIF1α), is upregulated in a number of cancers including ovarian cancer (OCa) (36), gastric cancer (GCa) (37, 38), colorectal cancer (CRCa) (39), PCa (40), and acute myeloid leukemia (AML) (41). Involvement of PDK1 has also been implicated in cancer cell epithelial–mesenchymal transition (EMT) and metastasis, for example in metastasis of liver aggressive 4T1 breast cancer (BrCa) cells to the liver, which implies an oncogenic role (12) (Table 1). PDK1 can be tyrosine phosphorylated and thereby activated by tyrosine kinase fibroblast growth factor receptor 1 (FGFR1), which localizes to mitochondria (64). Interestingly, both the PDC and PDK1 were also detected in the outer mitochondrial matrix, where PDK1 can be directly phosphorylated by tyrosine kinases (64).
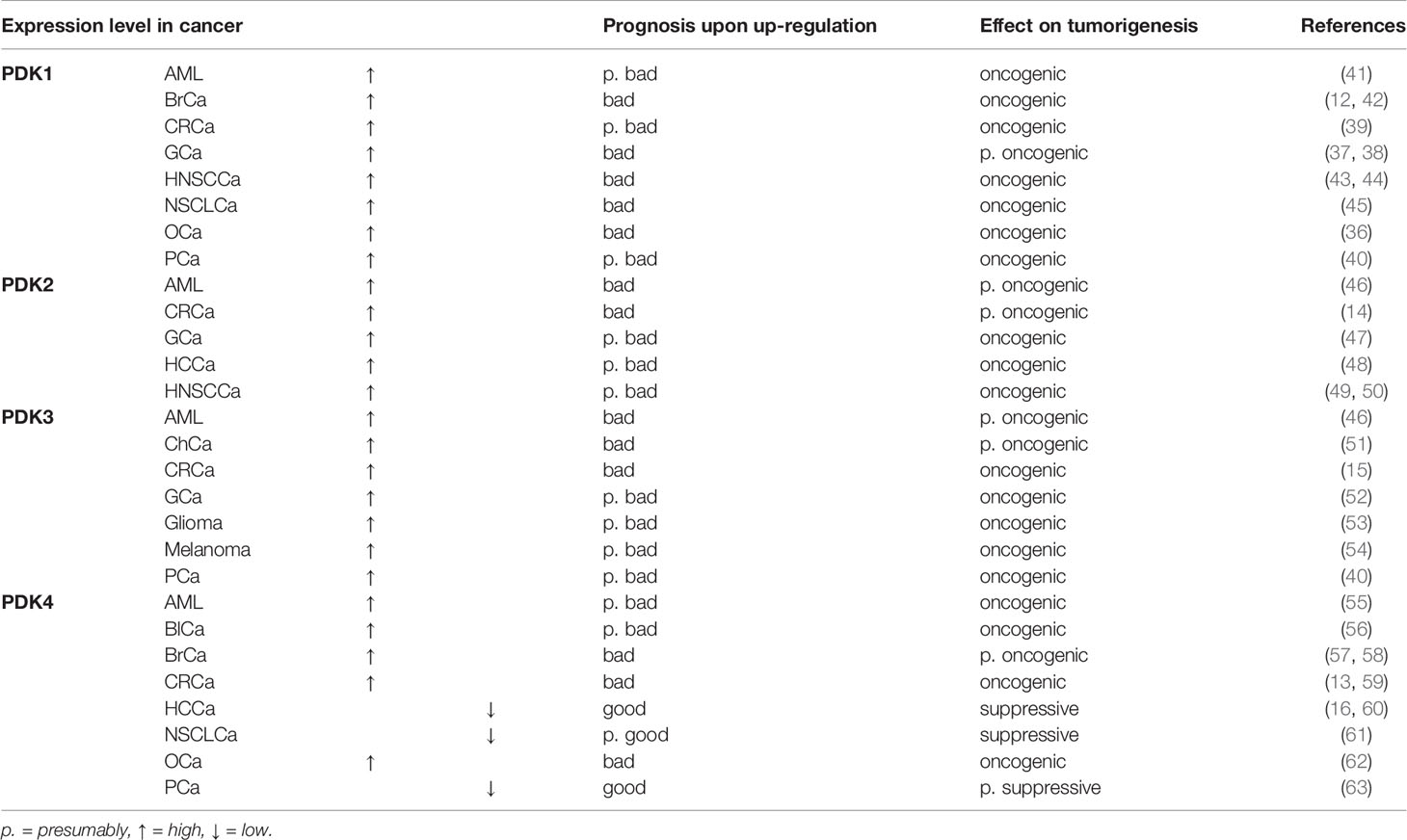
Table 1 Overview of PDK1–4 expression levels in different cancer types, their effect on prognosis upon up-regulation and tumorigenesis.
PDK2 is the only PDK enzyme that has been confirmed as p53 target (65). P53 downregulates and controls PDK2 expression on transcriptional and posttranscriptional level and thereby reduces the Warburg effect (25, 65). In hepatocellular carcinoma (HCCa) (48) and GCa cells (47) proliferation and migration were suppressed after downregulation of PDK2. PDK2 has also been associated with therapy resistance in CRCa cells (14), in head and neck squamous cell carcinoma (HNSCCa) cells (49), and in non-small cell lung cancer (NSCLCa) patients (66) (Table 1).
PDK3, which has the highest binding affinity to the PDC, is the least studied isoenzyme of the PDK (25). Similarly to PDK1, PDK3 is induced by HIF-1α, and higher expression is associated with higher tumor stage in many cancers (15, 67). In GCa (52), glioma (53), PCa (40), AML (46), and melanoma (54), high expression of PDK3 has been shown. Knockdown of PDK3 in the GCa cell lines SGC7901 and AGS (52), and the PCa cell line LNCaP (40) inhibited proliferation and induced apoptosis. Moreover, elevated expression of PDK3 is associated with chemo resistance in GCa cells (68), increased drug resistance in CoCa cells (15) and correlates with poor prognosis in cholangiocarcinoma (ChCa) (51), and AML (46) (Table 1).
While the regulation of PDK1–3 reflects the immediate energy demands of the cell, PDK4 reflects whole organism energy balance and is upregulated during excessive exercise (69), starvation (70), in insulin resistant states and diabetes (6, 57). PDK4 is also involved in the control of muscle size in cancer stages or after chemotherapy treatment, which renders it interesting as a target to combat cancer-associated cachexia (20). Based on the metabolic function of the respective tissue, the cancer type and stage, high PDK4 expression can act either oncogenic or tumor suppressive, as described below (Table 1).
The overexpression of PDK4 is associated with poorer prognosis in BrCa patients, irrespective of their molecular or histological subtype (58) and is associated with antiestrogen resistance (57). Duan et al. showed that PDK4 expression induced by benzyl butyl phthalate promotes glycolysis and proliferation in AML cells (55). In human metastatic CoCa cells, knockdown of PDK4 reduced their migratory and invasive properties (13). Furthermore, HIF1α expression was reduced in PDK4 knockdown cells, suggesting a correlation between PDK4 and HIF1α (13). PDK4 is also linked to enhanced cell proliferation and invasion in OCa (62) and bladder cancer (BlCa) (56). In addition, PDK4 has been identified as a positive regulator and activator of mechanistic target of rapamycin complex 1 (mTORC1) by cAMP response element binding protein (CREB)-mediated transcriptional regulation of the small GTPase Ras homologue enriched in brain (RHEB) (71). Additionally, Wu et al. suggested that PDK4 is essential for tumor necrosis factor alpha (TNF-α) to execute its pro-survival function via nuclear factor ‘kappa-light-chain-enhancer’ of activated B-cells (NF-kB), and consequently PDK4 deficiency in HCCa cells results in apoptosis (72).
In contrast, a tumor suppressive effect of PDK4 was observed in lung cancer (61, 73) and HCCa (16, 60). Sun et al. described that a metabolic switch from glycolysis to OXPHOS was observed in NSCLCa cells that underwent EMT, which was induced by knockdown of PDK4 (73). In the liver, PDK4 expression is associated with increased survival and liver function of patients undergoing liver resection due to colorectal liver metastases, and its downregulation predicted poor prognosis in HCCa patients (59). Besides that, loss of PDK4 resulted in enhanced lipogenesis and more aggressive tumors in HCCa (16). In PCa, Oberhuber et al. showed the association of low PDK4 with a risk of earlier disease recurrence in PCa, independent of tumor grading and tumor stage (63).
PDK1–4 have been associated with therapy resistance in several cancers. Qian et al. revealed that miR-4290 improved the sensitivity of GCa cells to cisplatin and induced apoptosis by downregulating PDK1 expression (38). Moreover, genetic knockdown of PDK1 abolished hypoxia-induced 5-fluorouracil (5-FU) resistance in GCa cells (74) and sensitized resistant OCa cells to cisplatin-induced cell death and apoptosis (75). Recently, PDK2 has been shown to induce resistance to 5-FU in chemo resistant CRCa cells (14), to be associated with cisplatin resistance in HNSCCa cells (49) and acquired paclitaxel-resistance in NSCLCa patients (66). PDK3 is associated with chemo resistance in GCa cells (68) and increased drug resistance in CoCa cells (15). Altered regulation of PDK4 is suggested to play a role in antiestrogen resistance in BrCa cells (57). In tamoxifen resistant MCF-7 breast cancer cells, PDK4 mRNA overexpression, but not enhanced protein levels, have been shown (57). Wang et al. described PDK4-induced chemo resistance in OCa (62). Sun et al. showed that downregulation of PDK4 in lung cancer drives EMT and promotes erlotinib resistance in EGFR mutant lung cancer cells (73). The combination of chemotherapeutic drugs with dichloroacetate (DCA), a PDK inhibitor, have been shown to enhance therapeutic efficacy (76). In addition, DCA has been described to increase radiosensitivity by increasing tumor oxygenation and reactive oxygen species (ROS) activity (76).
The association of PDK to therapy resistance can be explained by the anti-apoptotic and ROS protective benefits of the Warburg effect, which results in proliferative advantages (27, 76). The Warburg effect is supported by activated oncogenes and HIF1α, which induce the expression of glycolytic enzymes and transporters, such as glucose transporters (GLUTs) or lactate dehydrogenase A (LDHA) that are involved in glucose uptake, lactate production, and lactate secretion (27, 77). As a result, tumors are characterized by high levels of glycolysis and lactate production, and low levels of PDC activity and OXPHOS (27, 77). High accumulation of lactate and low OXPHOS activity lead to reduced activation of the apoptotic cascade and ROS, which protect cancer cells from cytotoxic effects of oxidative damage and apoptosis (3, 9, 10, 27, 76). Although aerobic glycolysis generates less energy (2 ATP per glucose molecule) than OXPHOS (36 ATP per oxidized glucose molecule) a high rate of glucose uptake of the tumor can compensate the tumor’s energetic demands (10, 11). DeBerardinis et al. suppose that generation of energy via glycolysis is faster and therefore more attractive than the more energy efficient but slower OXPHOS (77).
Primary PCa is lacking the Warburg effect and has a very distinctive energy metabolism compared to most other cancer types, showing high TCA cycle/OXPHOS activity (78, 79). The energy metabolism of the normal prostate cell is a result of its biological function, where the glandular epithelial cells secrete prostatic fluid and its main component—citrate—into the lumen (79, 80). Prostate epithelial cells accumulate extensive amounts of zinc, which inhibit the TCA cycle enzyme m-aconitase (78–80). Thereby citrate cannot be converted to isocitrate, the TCA cycle is truncated, and citrate is secreted by the prostatic epithelial cells (Figure 2A). As the prostatic epithelial cells have low OXPHOS activity, they mainly rely on aerobic glycolysis and are therefore energetically inefficient (78–80). In contrast, PCa cells no longer present zinc-accumulation and citrate-secretion, but activated TCA cycle/OXPHOS, thereby generating additional ATP (78, 80) (Figure 2B). Latonen et al. and Xue et al. show an increase in aconitase expression in PCa cells compared to non-cancerous tissue, which indicates their citrate oxidizing ability (81, 82). Acetyl-CoA provided by the TCA cycle serves as substrate for lipogenesis, which is known to be hyper-activated in PCa and associated with androgen resistance and tumor aggressiveness (17, 83–85). Lipogenic enzymes, as well as genes involved in cholesterol synthesis, have been shown to be regulated by androgen signaling (84). In turn, inhibition of fatty acid synthase (FASN), a key enzyme of de novo fatty acid synthesis, led to reduced androgen receptor (AR) expression in castration-resistant PCa (CRPCa) (85). PDK4 is not only a regulator of PDC activity, but can also alter fatty acid metabolism, as has been shown in HCCa cells (16). Here, knockdown of PDK4 did not alter OXPHOS, but resulted in enhanced expression of FASN and stearoyl-CoA desaturase (SCD) (16). Similarly, PDK4 was shown to enhance lipogenesis in lung cancer cells (61).
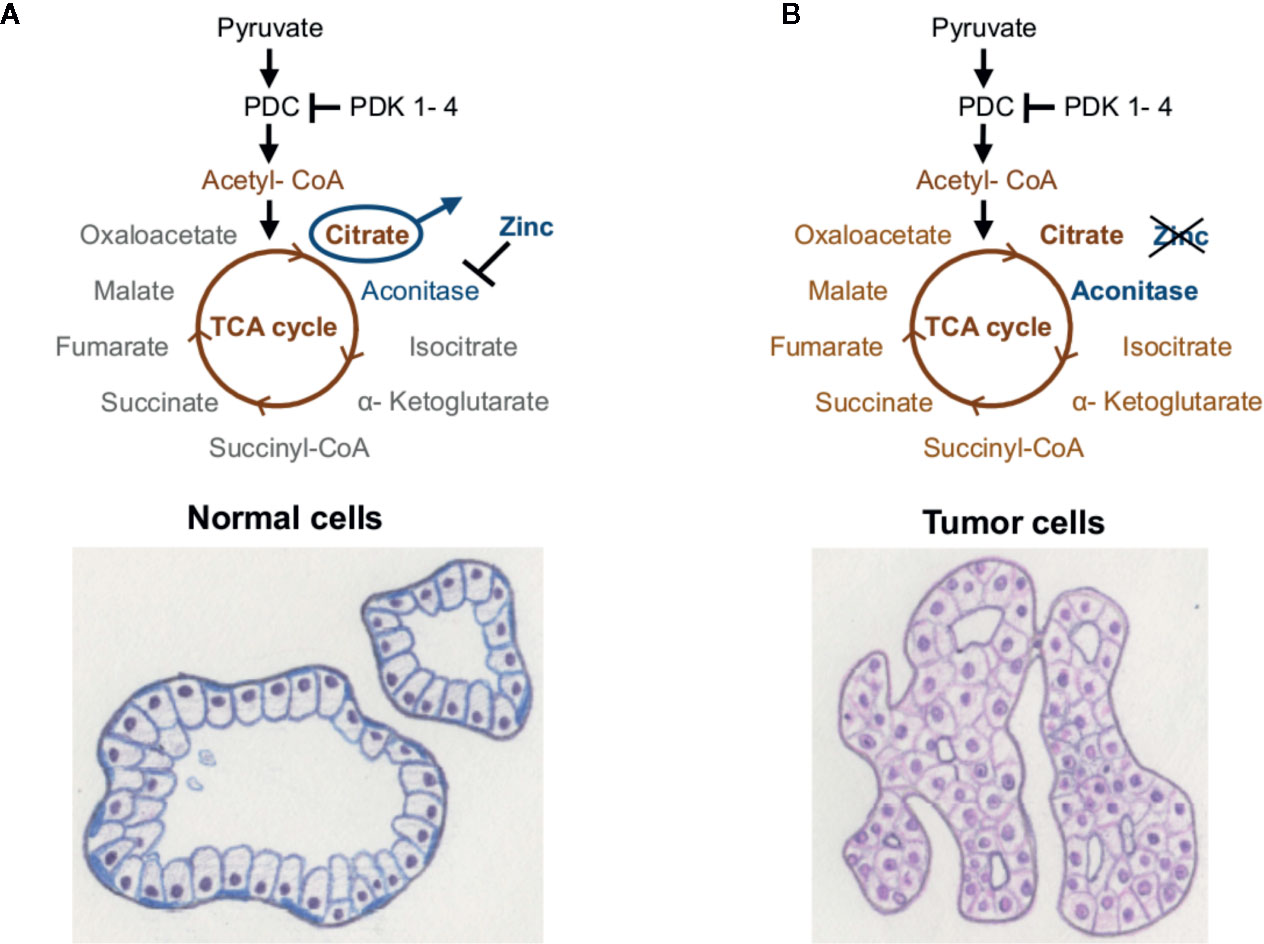
Figure 2 Energy metabolism of the prostate. (A) Healthy prostate cells accumulate high amounts of zinc, which inhibit the enzyme m-aconitase and thereby truncate the TCA cycle. (B) Prostate tumor cells show lower levels of zinc, whereby the enzyme aconitase remains active and citrate can be metabolized via the TCA cycle and OXPHOS. Taken from (5), inspired by (78).
Since low levels of PDK4 result in enhanced OXPHOS and/or enhanced lipogenesis, both of which are associated with poor prognosis in PCa, PDK4 should have a tumor suppressive effect in primary PCa. Recently, a protective effect of high PDK4- expression in PCa in a transcriptomic patient dataset was demonstrated (63). In accordance with these data, augmented gene expression and protein levels of the PDC subunit E1 (PDHA1) and the PDC activator PDP1 were identified in PCa (86). In contrast, Wang et al. reported lower proliferation and increased apoptosis in PCa cells upon knockdown of all PDK isoforms (40). While PDK4 and PDK2 are expressed at lower levels, PDK1 and PDK3 are supposedly overexpressed in PCa and associated with advanced tumor stages (40).
The specific effects of PDK1–4 on PCa energy and fatty acid metabolism have not been investigated yet. In addition to their direct implications on pyruvate provided carbon use and OXPHOS, also compensative mechanisms must be considered. These were shown to be active in metformin treated PCa cells (87). Here, metformin reduces entry of glucose-derived carbon into the TCA cycle due to complex I inhibition (87). The loss of glucose as carbon source was compensated by increased reductive glutamine metabolism, which provides α-ketoglutarate to the TCA cycle (87). Additional inhibition of the reductive glutamine pathway resulted in enhanced PCa cell sensitivity to metformin (87).
Given the importance of the hyperactive FASN and the unique dependence on OXPHOS in primary PCa, we are convinced that the action of PDK4 in PCa has a profound clinical significance and therefore requires immediate research.
PDK1–3 are described as oncogenes in different cancer types where their high expression is associated with EMT and metastasis, higher proliferation and migration, and most relevantly with therapy resistances, such as to 5-FU in CRCa and GCa, cisplatin in HNSCCa and OCa or paclitaxel in NSCLCa. In contrast to PDK1–3, data suggest either oncogenic or tumor suppressive function of PDK4, dependent on the metabolic profile of the tumor. It acts as an oncogene and is linked to therapy resistance in tumors that benefit from high glycolytic activity, such as in BrCa, AML, CoCa, OCa and BlCa. However, PDK4 can act as tumor suppressor in cancers that depend on high OXPHOS activity and/or high amounts of TCA cycle intermediates, as has been shown in PCa. Also in NSCLCa and HCCa low PDK4 levels are described to lead to more aggressive tumors and therapy resistance. We conclude that the combinatorial treatment of DCA with chemotherapeutic drugs might enable overcoming therapy resistances only in cancer types with a fitting metabolic profile (76). A large body of research is directed to tumors that profit from high glycolysis/lactate accumulation, whereas far less is known about those cases, where high OXPHOS contributes to tumor aggressiveness. Especially for PCa, where only little research is available on the mechanistic regulation and effects of PDK4, more research is needed in this regard.
All authors contributed to the conception and design of the review. EA and MO conducted literature research. EA wrote the first draft of the manuscript. All authors contributed to editing and rewriting of the manuscript. EA and MO created the figures for the manuscript. All authors contributed to the article and approved the submitted version.
EA and LK are funded by the Austrian Science Fund (FWF): IPPTO project number DOC 59-B33. MO and LK were funded by the COMET Competence Center CBmed-Center for Biomarker Research in Medicine (FA791A0906.FFG). The COMET Competence Center CBmed is funded by the Austrian Federal Ministry for Transport, Innovation and Technology (BMVIT); the Austrian Federal Ministry for Digital and Economic Affairs (BMDW); Land Steiermark (Department 12, Business and Innovation); the Styrian Business Promotion Agency (SFG); and the Vienna Business Agency. The COMET program is executed by the FFG. LK was in addition funded by the FWF grant P26011 and the Christian-Doppler Lab for Applied Metabolomics. The financial support by the Austrian Federal Ministry for Transport, Innovation and Technology and the National Foundation for Research, Technology and Development is gratefully acknowledged.
LK is a member of the scientific advisory board of CBmed-Center for Biomarker Research in Medicine GmbH. Author MO was employed by COMET centre (K1) CBmed—Center for Biomarker Research in Medicine GmbH.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Assoc. Prof. Dr. Brigitte Hantusch for proof-reading the manuscript.
5-FU, 5-fluorouracil; ACAT1, Acetyl-CoA acetyltransferase 1; AML, Acute myeloid leukemia; AR, Androgen receptor; ATP, Adenosine triphosphate; BrCa, Breast cancer; BlCa, Bladder cancer; ChCa, Cholangiocarcioma; CoA, Coenzyme-A; CoCa, Colon cancer; CRCa, Colorectal cancer; CRPCa, Castration resistant prostate cancer; CREB, CAMP response element binding protein; DCA, Dichloroacetate; E1, Pyruvate dehydrogenase; E2, Dihydrolipoamide acetyl-transferase; E3, Dihydrolipoamide dehydrogenase; E3BP, E3 binding protein; EMT, Epithelial–mesenchymal transition; FASN, Fatty acid synthase; FGFR1, Fibroblast growth factor receptor 1; GCa, Gastric cancer; GLUT, Glucose transporters; HCCa, Hepatocellular carcinoma; HIF1α, Hypoxia inducible factor 1 alpha; HNSCCa, Head and neck squamous cell carcinoma; LDHA, Lactate dehydrogenase A; mTORC1, Mechanistic target of rapamycin complex 1; NAD+, Nicotinamide adenine dinucleotide; NADH, Reduced form of NAD+; NF-kB, Nuclear factor kappa-light-chain-enhancer; NSCLCa, Non-small cell lung cancer; OCa, Ovarian cancer; OXPHOS, Oxidative phosphorylation; PCa, Prostate cancer; PDK, Pyruvate dehydrogenase kinase; PDP, Pyruvate dehydrogenase phosphatase; RHEB, Ras homolog enriched in brain; ROS, Reactive oxygen species; SIRT, Sirtuin; STAT3, Signal transducer and activator of transcription 3; SCD, Stearoyl-CoA desaturase; TCA cycle, Tricarboxylic acid cycle; TNF, Tumor necrosis factor.
1. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
2. Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab (2016) 23(1):27–47. doi: 10.1016/j.cmet.2015.12.006
3. Bhattacharya B, Mohd Omar MF, Soong R. The Warburg effect and drug resistance: The Warburg effect and drug resistance. Br J Pharmacol (2016) 173(6):970–9. doi: 10.1111/bph.13422
4. Gray LR, Tompkins SC, Taylor EB. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci (2014) 71(14):2577–604. doi: 10.1007/s00018-013-1539-2
5. Oberhuber M. Prostate cancer biomarker identification through proteomic and transcriptomic analyses. Doctoral thesis. Vienna, Austria: Medical University of Vienna (2020).
6. Lee I-K. The Role of Pyruvate Dehydrogenase Kinase in Diabetes and Obesity. Diabetes Metab J (2014) 38(3):181. doi: 10.4093/dmj.2014.38.3.181
7. Warburg O. On the Origin of Cancer Cells. Science (1956) 24123(3191):309–14. doi: 10.1126/science.123.3191.309
8. Weinhouse S, Warburg O, Burk D, Schade AL. On Respiratory Impairment in Cancer Cells. Science (1956) 10124(3215):267–72. doi: 10.1126/science.124.3215.267
9. Orang AV, Petersen J, McKinnon RA, Michael MZ. Micromanaging aerobic respiration and glycolysis in cancer cells. Mol Metab (2019) 23:98–126. doi: 10.1016/j.molmet.2019.01.014
10. Zhang W, Zhang S-L, Hu X, Tam KY. Targeting Tumor Metabolism for Cancer Treatment: Is Pyruvate Dehydrogenase Kinases (PDKs) a Viable Anticancer Target? Int J Biol Sci (2015) 11(12):1390–400. doi: 10.7150/ijbs.13325
11. Jang M, Kim SS, Lee J. Cancer cell metabolism: implications for therapeutic targets. Exp Mol Med (2013) 45(10):e45–5. doi: 10.1038/emm.2013.85
12. Dupuy F, Tabariès S, Andrzejewski S, Dong Z, Blagih J, Annis MG, et al. PDK1-Dependent Metabolic Reprogramming Dictates Metastatic Potential in Breast Cancer. Cell Metab (2015) 22(4):577–89. doi: 10.1016/j.cmet.2015.08.007
13. Leclerc D, Pham DNT, Lévesque N, Truongcao M, Foulkes WD, Sapienza C, et al. Oncogenic role of PDK4 in human colon cancer cells. Br J Cancer (2017) 116(7):930–6. doi: 10.1038/bjc.2017.38
14. Liang Y, Hou L, Li L, Li L, Zhu L, Wang Y, et al. Dichloroacetate restores colorectal cancer chemosensitivity through the p53/miR-149-3p/PDK2-mediated glucose metabolic pathway. Oncogene (2020) 39(2):469–85. doi: 10.1038/s41388-019-1035-8
15. Lu C-W, Lin S-C, Chien C-W, Lin S-C, Lee C-T, Lin B-W, et al. Overexpression of Pyruvate Dehydrogenase Kinase 3 Increases Drug Resistance and Early Recurrence in Colon Cancer. Am J Pathol (2011) 179(3):1405–14. doi: 10.1016/j.ajpath.2011.05.050
16. Yang C, Wang S, Ruan H, Li B, Cheng Z, He J, et al. Downregulation of PDK4 Increases Lipogenesis and Associates with Poor Prognosis in Hepatocellular Carcinoma. J Cancer (2019) 10(4):918–26. doi: 10.7150/jca.27226
17. Hirschey MD, DeBerardinis RJ, Diehl AME, Drew JE, Frezza C, Green MF, et al. Dysregulated metabolism contributes to oncogenesis. Semin Cancer Biol (2015) 35:S129–50. doi: 10.1016/j.semcancer.2015.10.002
18. Martínez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun (2020) 11(1):102. doi: 10.1038/s41467-019-13668-3
19. Vander Heiden MG, DeBerardinis RJ. Understanding the Intersections between Metabolism and Cancer Biology. Cell (2017) 168(4):657–69. doi: 10.1016/j.cell.2016.12.039
20. Pin F, Novinger LJ, Huot JR, Harris RA, Couch ME, O’Connell TM, et al. PDK4 drives metabolic alterations and muscle atrophy in cancer cachexia. FASEB J (2019) 33(6):7778–90. doi: 10.1096/fj.201802799R
21. Zhang S, Hulver MW, McMillan RP, Cline MA, Gilbert ER. The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr Metab (Lond) (2014) 11(1):10. doi: 10.1186/1743-7075-11-10
22. Klyuyeva A, Tuganova A, Kedishvili N, Popov KM. Tissue-specific kinase expression and activity regulate flux through the pyruvate dehydrogenase complex. J Biol Chem (2019) 18294(3):838–51. doi: 10.1074/jbc.RA118.006433
23. Kolobova E, Tuganova A, Boulatnikov I, Popov KM. Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. (2001) 9:69–77. doi: 10.1042/0264-6021:3580069
24. Connaughton S, Chowdhury F, Attia RR, Song S, Zhang Y, Elam MB, et al. Regulation of pyruvate dehydrogenase kinase isoform 4 (PDK4) gene expression by glucocorticoids and insulin. Mol Cell Endocrinol (2010) 315(1–2):159–67. doi: 10.1016/j.mce.2009.08.011
25. Woolbright BL, Rajendran G, Harris RA, Taylor JA. Metabolic Flexibility in Cancer: Targeting the Pyruvate Dehydrogenase Kinase:Pyruvate Dehydrogenase Axis. Mol Cancer Ther (2019) 18(10):1673–81. doi: 10.1158/1535-7163.MCT-19-0079
26. Saunier E, Benelli C, Bortoli S. The pyruvate dehydrogenase complex in cancer: An old metabolic gatekeeper regulated by new pathways and pharmacological agents: Pyruvate dehydrogenase complex in cancer. Int J Cancer (2016) 15138(4):809–17. doi: 10.1002/ijc.29564
27. Stacpoole PW. Therapeutic Targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) Axis in Cancer. JNCI: J Natl Cancer Institute (2017) 109(11):djx071. doi: 10.1093/jnci/djx071
28. Fan J, Shan C, Kang H-B, Elf S, Xie J, Tucker M, et al. Tyr Phosphorylation of PDP1 Toggles Recruitment between ACAT1 and SIRT3 to Regulate the Pyruvate Dehydrogenase Complex. Mol Cell (2014) Feb53(4):534–48. doi: 10.1016/j.molcel.2013.12.026
29. Shan C, Kang H-B, Elf S, Xie J, Gu T-L, Aguiar M, et al. Tyr-94 Phosphorylation Inhibits Pyruvate Dehydrogenase Phosphatase 1 and Promotes Tumor Growth. J Biol Chem (2014) 289(31):21413–22. doi: 10.1074/jbc.M114.581124
30. Luengo A, Gui DY, Vander Heiden MG. Targeting Metabolism for Cancer Therapy. Cell Chem Biol (2017) 24(9):1161–80. doi: 10.1016/j.chembiol.2017.08.028
31. Masisi BK, El Ansari R, Alfarsi L, Rakha EA, Green AR, Craze ML. The role of glutaminase in cancer. Histopathology (2020) 76(4):498–508. doi: 10.1111/his.14014
32. Yang R, Guo C. Discovery of potent pyruvate dehydrogenase kinase inhibitors and evaluation of their anti-lung cancer activity under hypoxia. Med Chem Commun (2018) 9(11):1843–9. doi: 10.1039/C8MD00453F
33. Kato M, Li J, Chuang JL, Chuang DT. Distinct Structural Mechanisms for Inhibition of Pyruvate Dehydrogenase Kinase Isoforms by AZD7545, Dichloroacetate, and Radicicol. Structure (2007) 5(8):992–1004. doi: 10.1016/j.str.2007.07.001
34. Park B-Y, Jeon J-H, Go Y, Ham HJ, Kim J-E, Yoo EK, et al. PDK4 Deficiency Suppresses Hepatic Glucagon Signaling by Decreasing cAMP Levels. Diabetes (2018) 67(10):2054–68. doi: 10.2337/db17-1529
35. Tao R, Xiong X, Harris RA, White MF, Dong XC. Genetic Inactivation of Pyruvate Dehydrogenase Kinases Improves Hepatic Insulin Resistance Induced Diabetes. Zang M editor PloS One (2013) 8(8):e71997. doi: 10.1371/journal.pone.0071997
36. Siu MKY, Jiang Y, Wang J, Leung THY, Ngu SF, Cheung ANY, et al. PDK1 promotes ovarian cancer metastasis by modulating tumor-mesothelial adhesion, invasion, and angiogenesis via α5β1 integrin and JNK/IL-8 signaling. Oncogenesis (2020) 9(2):1–16. doi: 10.1038/s41389-020-0209-0
37. HUR H, XUAN Y, KIM YB, LEE G, SHIM W, YUN J, et al. Expression of pyruvate dehydrogenase kinase-1 in gastric cancer as a potential therapeutic target. Int J Oncol (2012) 42(1):44–54. doi: 10.3892/ijo.2012.1687
38. Qian Y, Wu X, Wang H, Hou G, Han X, Song W. MicroRNA-4290 suppresses PDK1-mediated glycolysis to enhance the sensitivity of gastric cancer cell to cisplatin. Braz J Med Biol Res (2020) 53(5):e9330. [cited 2020 Jun 18]. doi: 10.1590/1414-431x20209330
39. Qin W, Tian Y, Zhang J, Liu W, Zhou Q, Hu S, et al. The double inhibition of PDK1 and STAT3-Y705 prevents liver metastasis in colorectal cancer. Sci Rep (2019) 9(1):12973. doi: 10.1038/s41598-019-49480-8
40. Wang L-Y, Hung C-L, Chen Y-R, Yang JC, Wang J, Campbell M, et al. KDM4A Coactivates E2F1 to Regulate the PDK-Dependent Metabolic Switch between Mitochondrial Oxidation and Glycolysis. Cell Rep (2016) 16(11):3016–27. doi: 10.1016/j.celrep.2016.08.018
41. Qin L, Tian Y, Yu Z, Shi D, Wang J, Zhang C, et al. Targeting PDK1 with dichloroacetophenone to inhibit acute myeloid leukemia (AML) cell growth. Oncotarget (2015) 7(2):1395–407. doi: 10.18632/oncotarget.6366
42. Peng F, Wang J-H, Fan W-J, Meng Y-T, Li M-M, Li T-T, et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene (2018) 37(8):1119. doi: 10.1038/onc.2017.407
43. McFate T, Mohyeldin A, Lu H, Thakar J, Henriques J, Halim ND, et al. Pyruvate Dehydrogenase Complex Activity Controls Metabolic and Malignant Phenotype in Cancer Cells. J Biol Chem (2008) 283(33):22700–8. doi: 10.1074/jbc.M801765200
44. Wigfield SM, Winter SC, Giatromanolaki A, Taylor J, Koukourakis ML, Harris AL. PDK-1 regulates lactate production in hypoxia and is associated with poor prognosis in head and neck squamous cancer. Br J Cancer (2008) 98(12):1975–84. doi: 10.1038/sj.bjc.6604356
45. Liu T, Yin H. PDK1 promotes tumor cell proliferation and migration by enhancing the Warburg effect in non-small cell lung cancer. Oncol Rep (2017) 37(1):193–200. doi: 10.3892/or.2016.5253
46. Cui L, Cheng Z, Liu Y, Dai Y, Pang Y, Jiao Y, et al. Overexpression of PDK2 and PDK3 reflects poor prognosis in acute myeloid leukemia. Cancer Gene Ther (2020) 27(1–2):15–21. doi: 10.1038/s41417-018-0071-9
47. He Z, Li Z, Zhang X, Yin K, Wang W, Xu Z, et al. MiR-422a regulates cellular metabolism and malignancy by targeting pyruvate dehydrogenase kinase 2 in gastric cancer. Cell Death Dis (2018) 9(5):505. doi: 10.1038/s41419-018-0564-3
48. Yu Q, Zhou J, Jian Y, Xiu Z, Xiang L, Yang D, et al. MicroRNA-214 suppresses cell proliferation and migration and cell metabolism by targeting PDK2 and PHF6 in hepatocellular carcinoma. Cell Biol Int (2020) 44(1):117–26. doi: 10.1002/cbin.11207
49. Roh J-L, Park JY, Kim EH, Jang HJ, Kwon M. Activation of mitochondrial oxidation by PDK2 inhibition reverses cisplatin resistance in head and neck cancer. Cancer Lett (2016) 371(1):20–9. doi: 10.1016/j.canlet.2015.11.023
50. Sun W, Zhou S, Chang SS, McFate T, Verma A, Califano JA. Mitochondrial Mutations Contribute to HIF1α Accumulation via Increased Reactive Oxygen Species and Up-regulated Pyruvate Dehydrogenease Kinase 2 in Head and Neck Squamous Cell Carcinoma. Clin Cancer Res (2009) 15(2):476–84. doi: 10.1158/1078-0432.CCR-08-0930
51. Sanmai S, Proungvitaya T, Limpaiboon T, Chua-On D, Seubwai W, Roytrakul S, et al. Serum pyruvate dehydrogenase kinase as a prognostic marker for cholangiocarcinoma. Oncol Lett (2019) 17:5275–82. cited 2020 Apr 24. doi: 10.3892/ol.2019.10185
52. Feng L, Cheng K, Zang R, Wang Q, Wang J. miR-497-5p inhibits gastric cancer cell proliferation and growth through targeting PDK3. Biosci Rep (2019) 39(9):BSR20190654. doi: 10.1042/BSR20190654
53. Xie Z, Li X, Chen H, Zeng A, Shi Y, Tang Y. The lncRNA-DLEU2/miR-186-5p/PDK3 axis promotes the progress of glioma cells. Am J Transl Res (2019) 11(8):4922–34.
54. Kluza J, Corazao-Rozas P, Touil Y, Jendoubi M, Maire C, Guerreschi P, et al. Inactivation of the HIF-1 /PDK3 Signaling Axis Drives Melanoma toward Mitochondrial Oxidative Metabolism and Potentiates the Therapeutic Activity of Pro-Oxidants. Cancer Res (2012) 72(19):5035–47. doi: 10.1158/0008-5472.CAN-12-0979
55. Duan X-L, Ma C-C, Hua J, Xiao T-W, Luan J. Benzyl butyl phthalate (BBP) triggers the malignancy of acute myeloid leukemia cells via upregulation of PDK4. Toxicol Vitro (2020) 62:104693. doi: 10.1016/j.tiv.2019.104693
56. Woolbright BL, Choudhary D, Mikhalyuk A, Trammel C, Shanmugam S, Abbott E, et al. The Role of Pyruvate Dehydrogenase Kinase-4 (PDK4) in Bladder Cancer and Chemoresistance. Mol Cancer Ther (2018) 17(9):2004–12. doi: 10.1158/1535-7163.MCT-18-0063
57. Walter W, Thomalla J, Bruhn J, Fagan DH, Zehowski C, Yee D, et al. Altered regulation of PDK4 expression promotes antiestrogen resistance in human breast cancer cells. SpringerPlus (2015) 4(1):689. doi: 10.1186/s40064-015-1444-2
58. Guda MR, Asuthkar S, Labak CM, Tsung AJ, Alexandrov I, Mackenzie MJ, et al. Targeting PDK4 inhibits breast cancer metabolism. Am J Cancer Res (2018) 8(9):1725–38.
59. Strowitzki MJ, Radhakrishnan P, Pavicevic S, Scheer J, Kimmer G, Ritter AS, et al. High hepatic expression of PDK4 improves survival upon multimodal treatment of colorectal liver metastases. Br J Cancer (2019) 120(7):675–88. doi: 10.1038/s41416-019-0406-9
60. Choiniere J, Wu J, Wang L. Pyruvate Dehydrogenase Kinase 4 Deficiency Results in Expedited Cellular Proliferation through E2F1-Mediated Increase of Cyclins. Mol Pharmacol (2017) 91(3):189–96. doi: 10.1124/mol.116.106757
61. Li G, Li M, Hu J, Lei R, Xiong H, Ji H, et al. The microRNA-182-PDK4 axis regulates lung tumorigenesis by modulating pyruvate dehydrogenase and lipogenesis. Oncogene (2017) 36(7):989–98. doi: 10.1038/onc.2016.265
62. Wang J, Qian Y, Gao M. Overexpression of PDK4 is associated with cell proliferation, drug resistance and poor prognosis in ovarian cancer. CMAR (2018) Volume 11:251–62. doi: 10.2147/CMAR.S185015
63. Oberhuber M, Pecoraro M, Rusz M, Oberhuber G, Wieselberg M, Haslinger P, et al. STAT 3 -dependent analysis reveals PDK 4 as independent predictor of recurrence in prostate cancer. Mol Syst Biol (2020) 16(4):e9247. doi: 10.15252/msb.20199247 cited 2020 May 15.
64. Hitosugi T, Fan J, Chung T-W, Lythgoe K, Wang X, Xie J, et al. Tyrosine Phosphorylation of Mitochondrial Pyruvate Dehydrogenase Kinase 1 Is Important for Cancer Metabolism. Mol Cell (2011) 44(6):864–77. doi: 10.1016/j.molcel.2011.10.015
65. Contractor T, Harris CR. p53 Negatively Regulates Transcription of the Pyruvate Dehydrogenase Kinase Pdk2. Cancer Res (2012) 72(2):560–7. doi: 10.1158/0008-5472.CAN-11-1215
66. Sun H, Zhu A, Zhou X, Wang F. Suppression of pyruvate dehydrogenase kinase-2 re-sensitizes paclitaxel-resistant human lung cancer cells to paclitaxel. Oncotarget (2017) 8(32):52642–50. doi: 10.18632/oncotarget.16991
67. Lu C-W, Lin S-C, Chen K-F, Lai Y-Y, Tsai S-J. Induction of Pyruvate Dehydrogenase Kinase-3 by Hypoxia-inducible Factor-1 Promotes Metabolic Switch and Drug Resistance. J Biol Chem (2008) 283(42):28106–14. doi: 10.1074/jbc.M803508200
68. Xu J, Shi Q, Xu W, Zhou Q, Shi R, Ma Y, et al. Metabolic enzyme PDK3 forms a positive feedback loop with transcription factor HSF1 to drive chemoresistance. Theranostics (2019) 9(10):2999–3013. doi: 10.7150/thno.31301
69. Wang L, Sahlin K. The effect of continuous and interval exercise on PGC-1α and PDK4 mRNA in type I and type II fibres of human skeletal muscle: Exercise and single fibres genes expression. Acta Physiol (2012) 204(4):525–32. doi: 10.1111/j.1748-1716.2011.02354.x
70. Wu P, Blair PV, Sato J, Jaskiewicz J, Popov KM, Harris RA. Starvation Increases the Amount of Pyruvate Dehydrogenase Kinase in Several Mammalian Tissues. Arch Biochem Biophys (2000) 381(1):1–7. doi: 10.1006/abbi.2000.1946
71. Liu Z, Chen X, Wang Y, Peng H, Wang Y, Jing Y, et al. PDK4 Protein Promotes Tumorigenesis through Activation of cAMP-response Element-binding Protein (CREB)-Ras Homolog Enriched in Brain (RHEB)-mTORC1 Signaling Cascade. J Biol Chem (2014) 289(43):29739–49. doi: 10.1074/jbc.M114.584821
72. Wu J, Zhao Y, Park Y-K, Lee J-Y, Gao L, Zhao J, et al. Loss of PDK4 switches the hepatic NF-κB/TNF pathway from pro-survival to pro-apoptosis. Hepatology (2018) 68(3):1111–24. doi: 10.1002/hep.29902
73. Sun Y, Daemen A, Hatzivassiliou G, Arnott D, Wilson C, Zhuang G, et al. Metabolic and transcriptional profiling reveals pyruvate dehydrogenase kinase 4 as a mediator of epithelial-mesenchymal transition and drug resistance in tumor cells. Cancer Metab (2014) 2(1):20. doi: 10.1186/2049-3002-2-20
74. Xuan Y, Hur H, Ham I-H, Yun J, Lee J-Y, Shim W, et al. Dichloroacetate attenuates hypoxia-induced resistance to 5-fluorouracil in gastric cancer through the regulation of glucose metabolism. Exp Cell Res (2014) 321(2):219–30. doi: 10.1016/j.yexcr.2013.12.009
75. Zhang M, Cong Q, Zhang X-Y, Zhang M-X, Lu Y-Y, Xu C-J. Pyruvate dehydrogenase kinase 1 contributes to cisplatin resistance of ovarian cancer through EGFR activation: ZHANG et al. J Cell Physiol (2019) 234(5):6361–70. doi: 10.1002/jcp.27369
76. Tataranni T, Piccoli C. Dichloroacetate (DCA) and Cancer: An Overview towards Clinical Applications. Oxid Med Cell Longevity (2019) 2019:1–14. doi: 10.1155/2019/8201079
77. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The Biology of Cancer: Metabolic Reprogramming Fuels Cell Growth and Proliferation. Cell Metab (2008) 7(1):11–20. doi: 10.1016/j.cmet.2007.10.002
78. Cutruzzolà F, Giardina G, Marani M, Macone A, Paiardini A, Rinaldo S, et al. Glucose Metabolism in the Progression of Prostate Cancer. Front Physiol (2017) 8:97. cited 2020 Mar 27. doi: 10.3389/fphys.2017.00097
79. Eidelman E, Twum-Ampofo J, Ansari J, Siddiqui MM. The Metabolic Phenotype of Prostate Cancer. Front Oncol (2017) 7:131. doi: 10.3389/fonc.2017.00131
80. Costello LC, Franklin RB. The clinical relevance of the metabolism of prostate cancer; zinc and tumor suppression: connecting the dots Leslie. Mol Cancer (2006) 5(1):17. doi: 10.1186/1476-4598-5-17
81. Latonen L, Afyounian E, Jylhä A, Nättinen J, Aapola U, Annala M, et al. Integrative proteomics in prostate cancer uncovers robustness against genomic and transcriptomic aberrations during disease progression. Nat Commun (2018) 9(1):1176. doi: 10.1038/s41467-018-03573-6
82. Xue Y, Liu Y, Su J, Li J, Wu Y, Guo R, et al. Zinc cooperates with p53 to inhibit the activity of mitochondrial aconitase through reactive oxygen species accumulation. Cancer Med (2019) 8(5):2462–73. doi: 10.1002/cam4.2130
83. Grunt TW. Interacting Cancer Machineries: Cell Signaling, Lipid Metabolism, and Epigenetics. Trends Endocrinol Metab (2018) 29(2):86–98. doi: 10.1016/j.tem.2017.11.003
84. Swinnen JV, Roskams T, Joniau S, Van Poppel H, Oyen R, Baert L, et al. Overexpression of fatty acid synthase is an early and common event in the development of prostate cancer. Int J Cancer (2002) 98(1):19–22. doi: 10.1002/ijc.10127
85. Zadra G, Ribeiro CF, Chetta P, Ho Y, Cacciatore S, Gao X, et al. Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer. Proc Natl Acad Sci USA (2019) 116(2):631–40. doi: 10.1073/pnas.1808834116
86. Chen J, Guccini I, Di Mitri D, Brina D, Revandkar A, Sarti M, et al. Compartmentalized activities of the pyruvate dehydrogenase complex sustain lipogenesis in prostate cancer. Nat Genet (2018) 50(2):219–28. doi: 10.1038/s41588-017-0026-3
Keywords: pyruvate dehydrogenase kinase, tricarboxylic acid cycle, oxidative phosphorylation, Warburg effect, aerobic glycolysis, prostate cancer, cancer metabolism, therapy resistance
Citation: Atas E, Oberhuber M and Kenner L (2020) The Implications of PDK1–4 on Tumor Energy Metabolism, Aggressiveness and Therapy Resistance. Front. Oncol. 10:583217. doi: 10.3389/fonc.2020.583217
Received: 14 July 2020; Accepted: 13 November 2020;
Published: 15 December 2020.
Edited by:
Monica Montopoli, University of Padua, ItalyReviewed by:
Cinzia Domenicotti, Università di Genova, ItalyCopyright © 2020 Atas, Oberhuber and Kenner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lukas Kenner, bHVrYXMua2VubmVyQG1lZHVuaXdpZW4uYWMuYXQ=
†ORCID: Monika Oberhuber, orcid.org/0000-0002-5691-3605
‡These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.