 Komal Srinivasa
Komal Srinivasa Kevin A. Cross
Kevin A. Cross Sonika Dahiya
Sonika Dahiya- 1Department of Pathology & Immunology, Washington University School of Medicine, St. Louis, MO, United States
- 2Department of Neurosurgery, Washington University School of Medicine, St. Louis, MO, United States
BRAF (alternately referred to as v-raf murine sarcoma viral oncogene homolog B1) is a proto-oncogene involved in the mitogen-activated protein kinase (MAPK) pathway. BRAF alterations are most commonly missense mutations or aberrant fusions. These mutations are observed in numerous primary central nervous system tumors as well as metastases. This review discusses the prevalence of BRAF alteration within select notable CNS tumors, and their prognostic associations. Included are some novel entities such as diffuse leptomeningeal glioneuronal tumor (DLGNT), polymorphous low grade neuroepithelial tumor of the young (PLNTY), and multinodular and vacuolating neuronal tumor (MVNT). Knowledge of this gene’s integrity in CNS and PNS tumors can have profound diagnostic and therapeutic implications. Also reviewed are the current state of targeted therapy against aberrant BRAF as it pertains mostly to the CNS and to a lesser extent in PNS, and certain diagnostic aspects.
Introduction
Personalized medicine has revolutionized cancer care in the 21st century, particularly in the areas of diagnosis and treatment. The application of this knowledge to CNS cancer has been variable. While the incorporation of molecular findings has become routine in diagnosing CNS tumors, particularly with the revised 2016 WHO guidelines, targeted therapies have not become similarly common. Patients with gliomas and other primary CNS tumors have a pressing need for new therapies. Five-year survival for primary central nervous system tumors is an estimated 59–64% (1).
In this review, we focus on the proto-oncogene BRAF and its relevance in these aspects of CNS and PNS tumors. We detail its normal and pathologic function, its prevalence and prognostic relevance in select primary and metastatic neoplasms, and finally challenges and future directions of targeted therapy.
BRAF Function
BRAF, alternately referred to as v-Raf murine sarcoma viral oncogene homolog B1, encodes for one of three members of the rapidly accelerating fibrosarcoma (RAF) serine/threonine kinase family. It is located on the long arm of chromosome 7 at position 34. It normally functions as part of the mitogen activated protein kinase (MAPK) pathway, enabling cells to respond to extracellular growth signals. Classically, these growth signals are recognized by tyrosine kinase receptors residing within the plasma membrane, leading to a cascade of phosphorylation events involving protein kinases of the Ras, Raf, MEK, and MAPK/ERK families. MAP kinases, in turn, activate various signals promoting cell growth and survival (Figure 1).
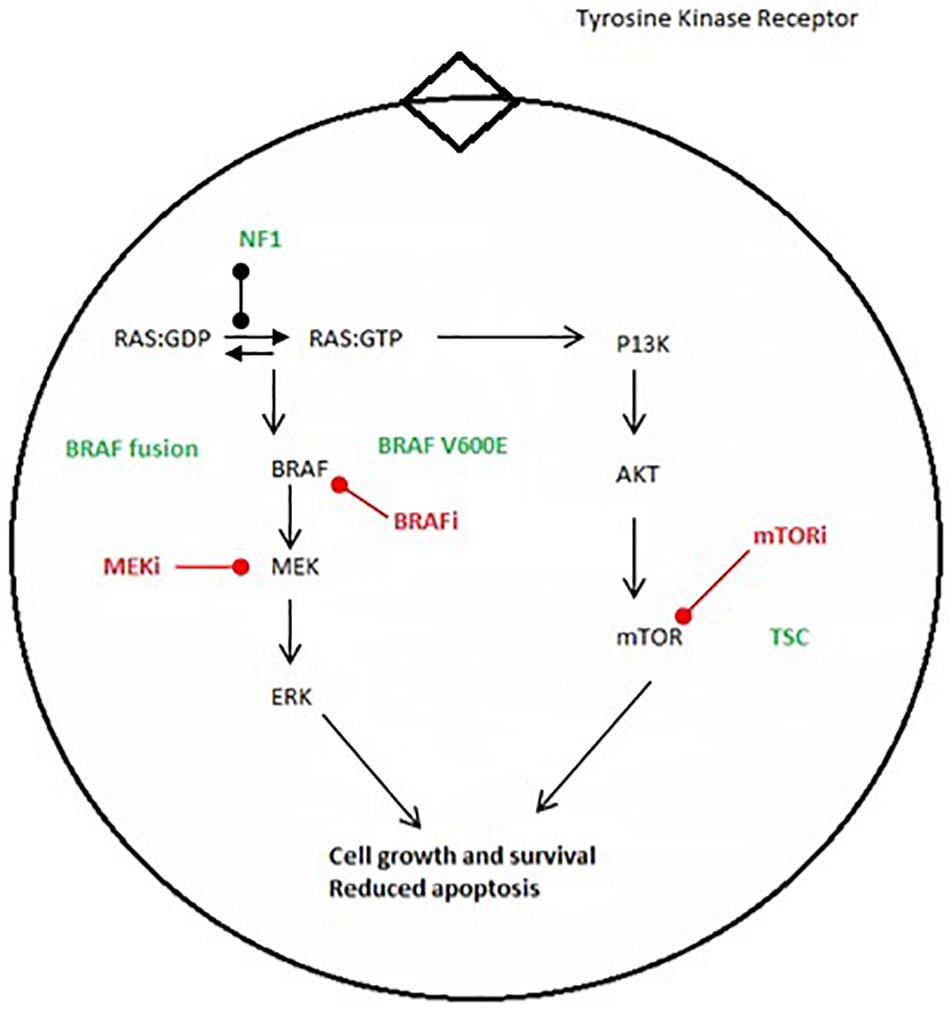
Figure 1. Major oncogenic pathways MAPK and mTOR pathways. Green font shows major genetic alteration. Red font shows proposed molecular therapy.
Owing to this role, the MAPK pathway is frequently implicated in human cancer. Derangement can occur by activating events in its upstream signals, or by loss of function in regulatory proteins, such as neurofibromin-1. Gain-of-function mutations in BRAF commonly result from pathologic fusion or missense mutations, these occurring in near mutually exclusive fashion (2). In addition to its promotion of cell growth and survival, oncogenic BRAF is also known to induce cell senescence (3).
Mechanisms of Pathologic Activation
Gain of function in BRAF results primarily from aberration in the N-terminal portion of the protein, which encodes for an auto-inhibitory domain. Without this domain the protein’s function is uncoupled from upstream signals and remains constitutively active. BRAF fusion proteins frequently lack this auto-inhibitory domain entirely. Fusions typically arise from tandem duplications (70% of cases), and less often from deletions and insertions (<3% cases) (4, 5). In the commonest variant, the 5′ segment of the KIAA1549 gene replaces that of BRAF (6). Numerous KIAA1549:BRAF fusion variants have been described involving different exons (Table 1). However, 80% of fusions involve exons 15 or 16 of KIAA1549 with exon 9 of BRAF (6).

Single nucleotide mutations can lead to similar effects (6). The most frequent and well-studied missense mutation substitutes valine for glutamate at position 600 (V600E) (7). Other, less frequent, variants include V600D, V600R, and V600K (8). These mutations are thought to induce gain of function by mimicking the phosphorylated state of the protein, as they appear in close proximity to regulatory phosphorylation sites at codons 598 and 601.
We detail below how distinguishing between these two mechanisms may impact patient care. And, although rare, it should be noted that BRAF fusions and single nucleotide mutation may occur concurrently, as is the case in an estimated 1–3% of low grade gliomas, 3% of PA and PXA, and 1.6% of pilocytic astrocytomas (5, 9) (Table 2).
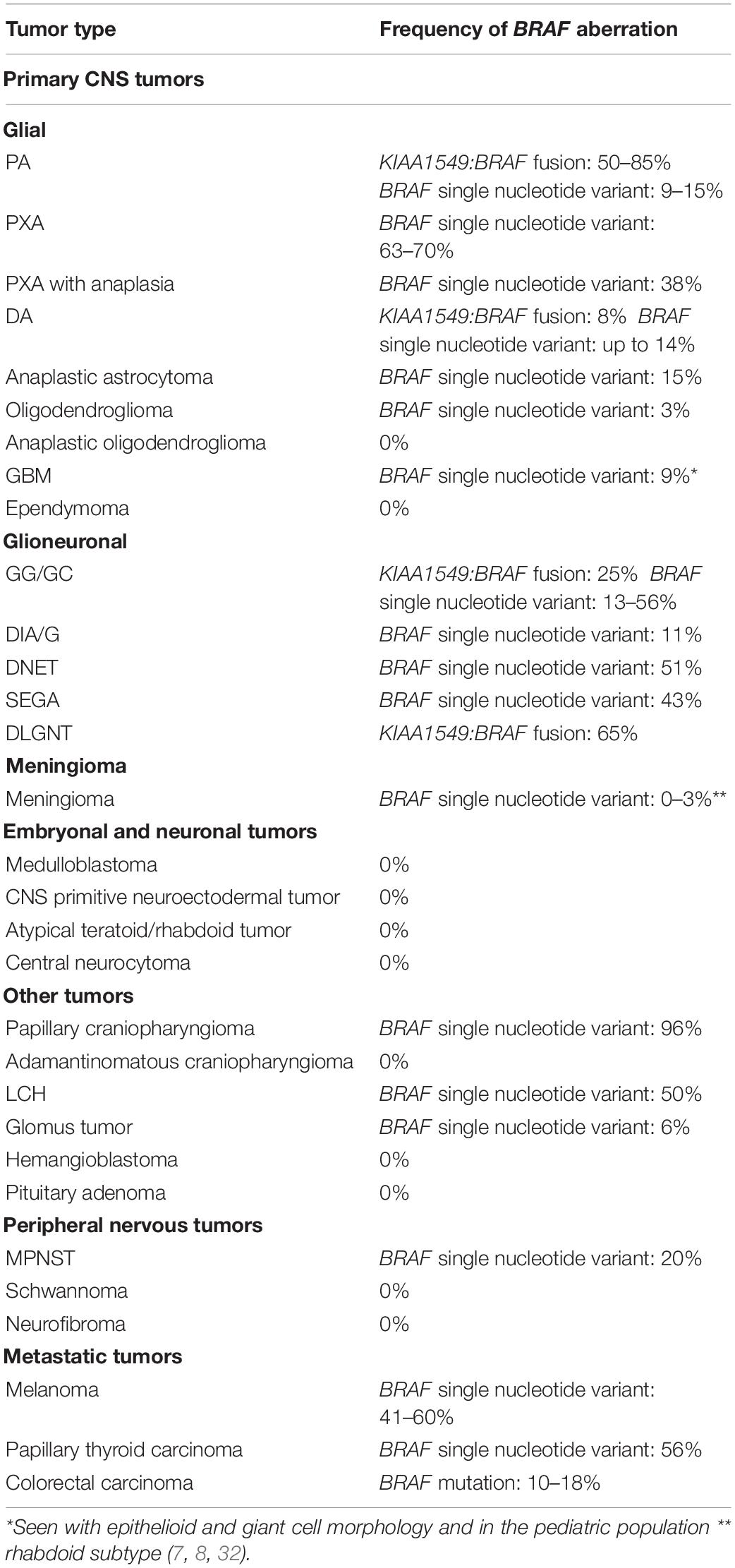
Table 2. The frequency BRAF aberrations in primary and metastatic tumors to the CNS (PA, pilocytic astrocytoma; PXA, pleomorphic xanthoastrocytoma; DA, diffuse astrocytoma; GBM, glioblastoma; GG/GC, ganglioglioma/gangliocytoma; DIA, desmoplastic infantile astrocytoma/ganglioglioma; DNET, dysembryoplastic neuroepithelial tumor; SEGA, subependymal giant cell astrocytoma; DLGNT, diffuse leptomeningeal glioneuronal tumor; LCH, Langerhans cell histiocytosis; MPNST, malignant peripheral nerve sheath tumor).
Primary CNS Tumors
Gliomas
Low Grade Glioma
Pilocytic astrocytoma
Pilocytic astrocytoma (PA) is the commonest primary childhood CNS tumor, accounting for 30% of the total. These tumors are illustrative for the role of BRAF testing. Over two thirds of these tumors arise within the posterior fossa (10). Roughly 85% of cases are sporadic, and 15% are associated with the inherited tumor predisposition syndrome neurofibromatosis type 1 (NF1) (11) (Figure 2).
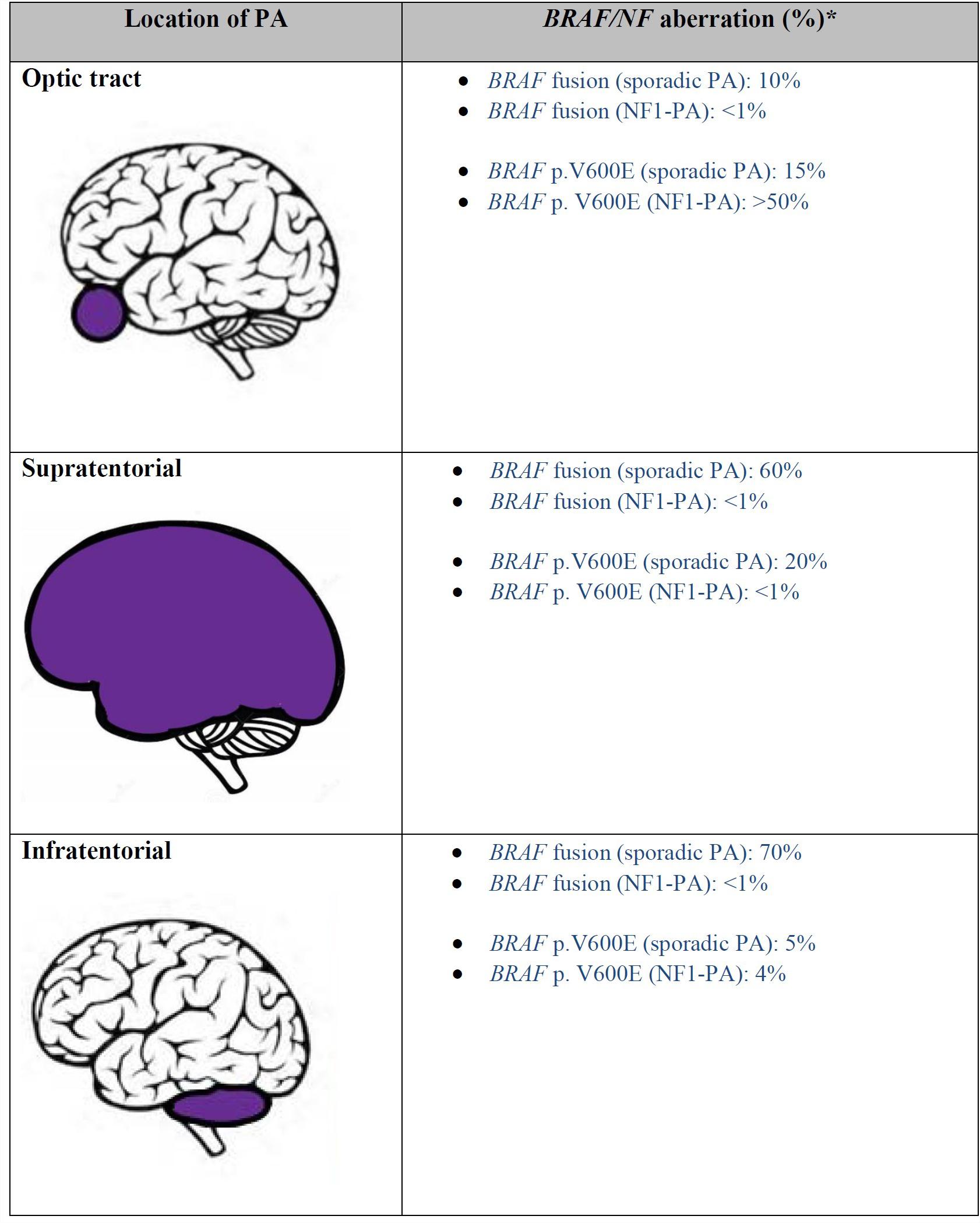
Figure 2. Frequency of BRAF alteration in pilocytic astrocytoma based on location (References: 5, 7, 8, 18, 19, 102). *: all the listed values are approximate numbers. NF1, Neurofibromatosis 1; PA, pilocytic astrocytoma.
Despite histologic similarity, sporadic and NF1 associated pilocytic astrocytomas exhibit different characteristics in terms of location and clinical course. While the vast majority of pilocytic astrocytomas are sporadic and arise in the posterior fossa, in NF1 patients 50–76% occur in the optic pathway and are known as optic pathway gliomas (11). NF-1 patients also frequently develop PA in the cerebral hemispheres. In fact, just 4% of cerebellar PAs are associated with NF1. Nevertheless, both supratentorial and infratentorial locations of PA frequently require treatment. Optic pathway gliomas, which may threaten vision, are most often treated with chemotherapy, while posterior-fossa PAs, which threaten brainstem compression or obstructive hydrocephalus, are most often resected.
These differences are believed to result from differing underlying oncogenic mechanisms, namely BRAF and NF1 (−/−) activation of the MAPK and PI3K/mTOR pathways. Sporadic PAs more commonly demonstrate BRAF fusion rather than single nucleotide mutations, as nearly 80% of cerebellar PAs harbor fusions, which most often involve KIAA1549 (Table 1 and Figure 3). However, in fewer than 5 percent of cases the fusion involves other genes involved in the MAPK pathway such as FAM131B, SRGAP, QK1, RNF130, CLCN6, MKRN1, GNA11, FZR1, and MACF1 (2, 4, 5, 12–14). V600E is not thought to play a major role in their pathogenesis. Its prevalence in all pediatric PAs regardless of location is estimated to be around 6% (15).
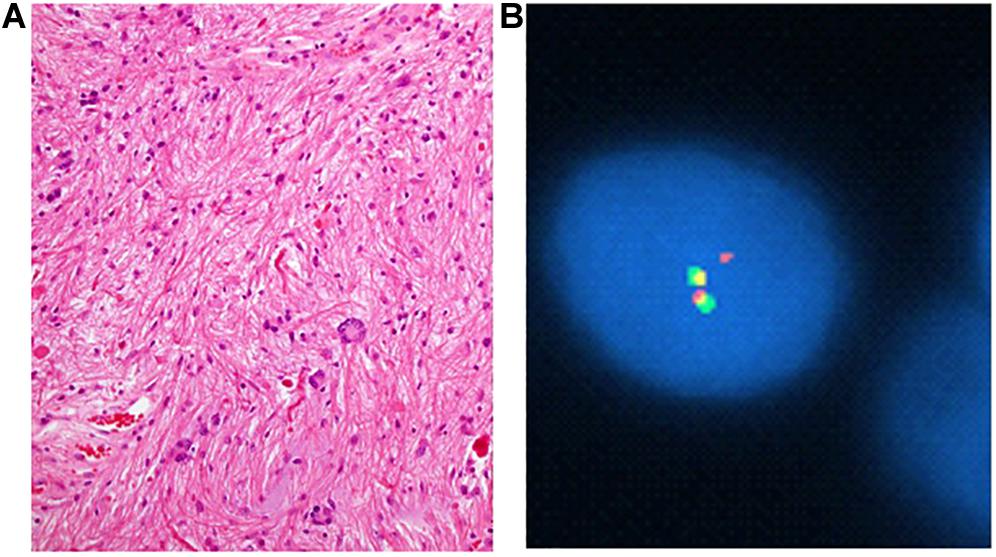
Figure 3. Pilocytic astrocytoma with compact areas harboring piloid cells, abundant Rosenthal material, and some multinucleate cells (“pennies on a plate”) (H&E; A) and florescence in situ probe showing KIAA1549:BRAF fusion. Yellow signal signifies fusion (B).
The signature molecular aberration in NF1-associated PAs, by contrast, is loss of neurofibromin. This product of the NF1 gene is a negative regulator of Ras. Its loss activates both the MAPK and PI3K/mTOR signaling pathways (11, 16).
While cases have been reported of optic gliomas bearing both KIAA1549:BRAF and NF1 loss, these are rare (17). Therefore, these markers define two seemingly mutually exclusive subgroups of this entity.
BRAF genotype may also have prognostic implications. Some have argued KIAA1549:BRAF may lead to a less-aggressive phenotype. In a study of 58 subtotally resected PAs, Hawkins et al. found 5-year progression-free survival to be 65% in fusion patients and 17% in wild type (p = 0.002) (18). While Horbinski et al. found BRAF fusion in PAs to be associated with better prognosis as well, this was not statistically significant (19). It is speculated that the less-aggressive phenotype in BRAF fusion patients may result from the phenomenon of oncogene-induced cell senescence (18).
Overall, survival rates in pediatric pilocytic astrocytoma are excellent, generally exceeding 90% at 5 years (20, 21).
Diffuse glioma
The diagnosis of diffuse glioma has evolved with the incorporation of molecular markers, but remains particularly challenging. The 2016 WHO revised guidelines identified the importance of IDH1/2 and 1p/19q codeletion status for classification of diffuse gliomas into astrocytic or oligodendroglial phenotypes. However, pediatric diffuse gliomas rarely demonstrate these findings (22). BRAF aberrations, by contrast, are more frequent in the pediatric population (6, 22). While studies in adults estimate the prevalence of all BRAF mutation at less than 1% (7, 23), in children and adolescents rates are estimated to be 3% for fusion, and 8–43% for V600E. (6, 15, 24). This might indicate somewhat distinct molecular underpinnings in evolution of diffuse gliomas in adults when compared with the pediatric population.
There is a low incidence of diffuse glioma in children, with less than 10% of pediatric low grade gliomas falling into this category in some studies (20). In addition, they are frequently examined in combination with other subtypes of low-grade glioma in studies. Prognostic predictions in this population are therefore challenging. However, limited data do suggest that V600E may be associated with more aggressive phenotype. In a large series of pediatric low-grade gliomas, Lassaletta et al. revealed V600E mutations in 10 of 23 diffuse astrocytomas (43%), 2 of 15 pilomyxoid astrocytomas (13%), and 14 of 70 low grade gliomas not otherwise specified (20%) (24). They analyzed outcomes from these patients in combination with other low grade gliomas and found V600E was associated with worse 5 year (50.1 vs. 72%) and 10 year PFS (27 vs. 60.2%) (24). The cIMPACT-NOW update four guidelines suggest using a designation “diffuse glioma, BRAF V600E-mutant” for tumors exhibiting a BRAF V600E mutation (25).
In adults, grade II diffuse gliomas account for 24% all CNS cases and are found largely within the cerebral hemispheres. BRAF mutations occur in less than 1%, limiting prognostic assessments. The most important predictors of prognosis in this tumor subset remain extent of resection, tumor location, and patient age (6, 19).
Pleomorphic xanthoastrocytoma
Pleomorphic xanthoastrocytoma (PXA) accounts for less than 1% of all astrocytic tumors, but is more frequent in children (10, 26). These tumors show a predilection for the temporal lobe and superficial cortices (10). BRAF V600E is highly enriched in this population, with 2/3 of adult and pediatric cases bearing it, including a smaller number of cases with anaplasia (33% of such cases) (7).
Overall, prognosis for PXA is favorable, with recurrence-free survival estimated to be 64% following initial resection (27). The prognostic utility of BRAF V600E has been investigated, but remains indeterminate (27–29).
In addition to BRAF, PXA frequently features aberrations in CDKN2A, which encodes for the cyclin dependent kinase inhibitor p16 (9). In a study of 38 primary and anaplastic PXAs, 87% were found to exhibit a homozygous deletion. However, this deletion did not correlate with BRAF status, as it was distributed equally among wild type and mutant tumors (30). In this same cohort, BRAF status did not clearly confer survival benefit.
High Grade Gliomas
The frequency of BRAF mutations in adult GBM is estimated to be 1–3% (31). However, certain cohorts have higher frequencies. Glioblastomas in teenage and young adults are enriched for the V600E mutation, and it may be observed in up to 50% of the epithelioid variant (31, 32, 33) (Figure 4C).
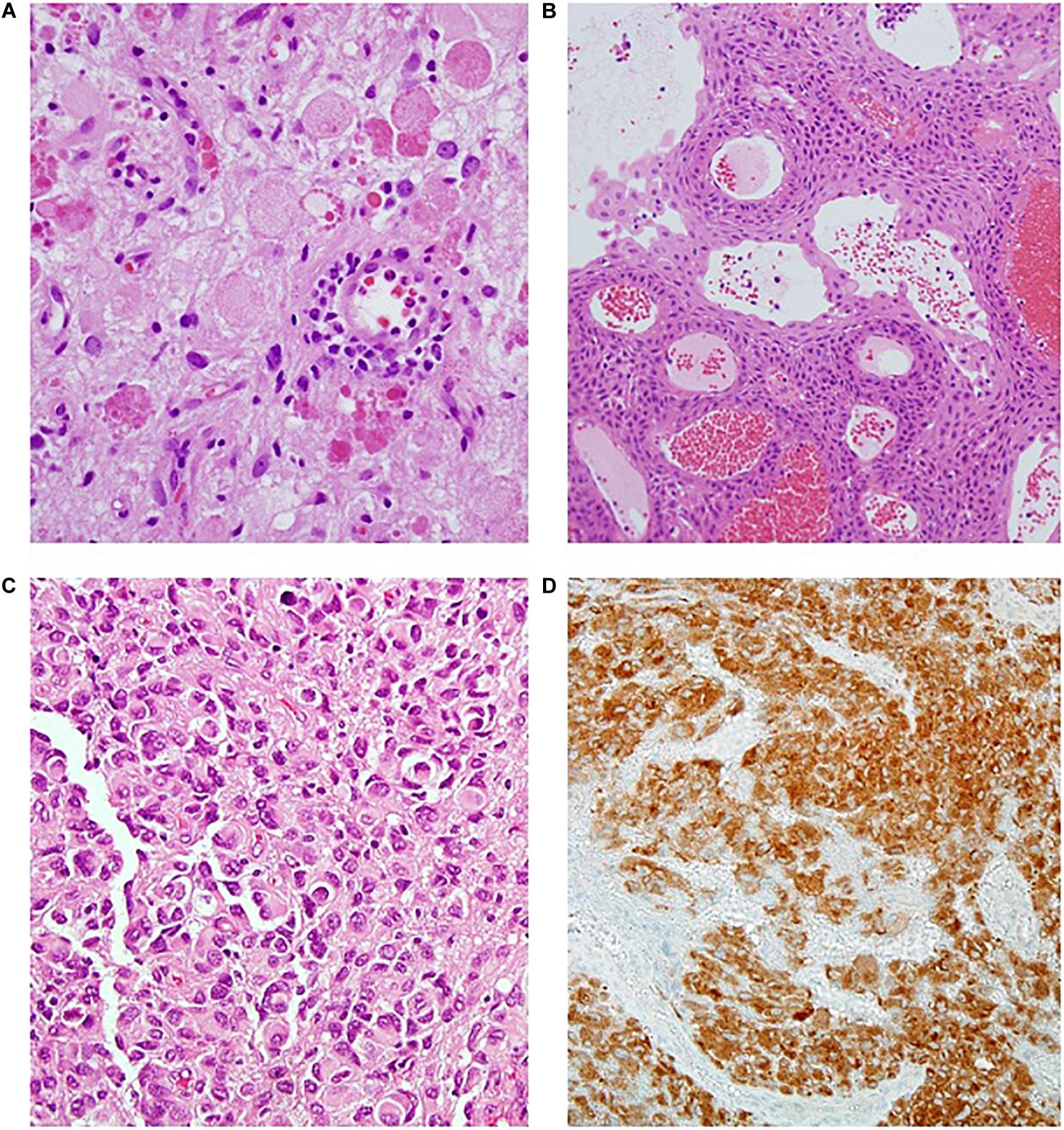
Figure 4. (A) Ganglioghoma (H&E); (B) Papillary craniopharyngioma (H&E); (C) Epithelioid glioblastoma (H&E); (D) BRAF immunostaining showing diffuse positive staining (BRAF IPX).
Glioneuronal Tumors
Ganglioglioma (GG)
Ganglioglioma accounts for 7% of childhood and adolescent primary CNS tumors (26). These are slow growing, circumscribed tumors that may harbor both cystic and solid components (10). Histologically they show neuronal (comprised by generally ganglionic) and glial elements in variable proportions (Figure 4A). BRAF missense mutations are estimated to be found in half of specimens, with BRAF fusions seen in another 10–25% (6, 7, 34). In a series of 53 pediatric gangliogliomas, Dahiya et al. found BRAF V600E to be associated with shorter recurrence free survival (35).
Diffuse Leptomeningeal Glioneuronal Tumor
Diffuse leptomeningeal glioneuronal tumor (DLGNT) is a rare entity added only recently to the WHO classification of CNS tumors, and these have not been assigned a WHO grade as their natural history is not clearly delineated yet (10). It is characterized by leptomeningeal spread of histologically monomorphic, oligodendroglial-like cells, usually in children and adolescents. Molecularly, it features loss of 1p, without an abnormality in IDH (10, 36, 37). In a review of 30 cases, Deng et al. demonstrated pathologic activation of the MAPK pathway in 80%, and suggested this to be a hallmark molecular feature of this tumor (37). In 66% of their specimens, this was a result of KIAA1549:BRAF (37). They further defined and characterized two molecular subclasses using DNA methylation profiling. Class 1 was found more commonly in younger patients (median age 5), with frequent 1p/19q codeletions (47%), and was associated with 100% 5-year progression free survival. Subclass 2 tended to arise in older patients (median age 14), who experienced 43% 5 year overall survival. BRAF fusions were observed equally in the two groups (76% in class 1 vs. 77% in class 2).
Polymorphous Low Grade Neuroepithelial Tumor of the Young (PLNTY)
In 2017, Huse et al. described a new entity of low-grade, oligodendroglioma-like neuroepithelial tumor, naming this polymorphous low grade neuroepithelial tumor of the young (PLNTY) (38). These are, generally, epileptogenic tumors located in the subcortical temporal lobe and associated with dystrophic calcifications (39, 40). Histologically, these tumors show a diverse astrocytic and ependymal appearance, though frequently there is an oligodendroglioma-like component (38). There is immunoreactivity for CD34 and OLIG2. Tumor cells lack IDH1 R132H or 1p/19q co-deletion. Nearly all, however, featured overactivation of the MAPK pathway (38). In Huse’s original series, 3 of 7 cases were BRAF V600E mutant and the remaining cases exhibited FGFR2/3 fusion events (no BRAF fusion was detected) (38).
Multinodular and Vacuolating Neuronal Tumor (MVNT)
Multinodular and vacuolating neuronal tumor (MVNT) is a provisional entity included in the 2016 WHO classification of CNS tumors (10). Due to their indolent behavior and ease of surgical excision, Huse et al. suggest assigning this entity a WHO grade I designation, however, a formal WHO grade has not yet been assigned (41). MVNT is a supratentorial tumor which generally appears on MRI as multiple subcortical, FLAIR-hyperintense nodules (42). Histologically the tumor cells arrange into clusters and have conspicuous intracytoplasmic and stromal vacuolation (41, 43). They express OLIG2, CD34, and are also labeled by anti-HuC/HuD with variable expression of synaptophysin, and no reactivity for chromogranin, NeuN and/or neurofilament (10, 41). In their original description Pekmezci et al., analyzed eight samples using phospho-ERK immunohistochemistry and demonstrated uniform MAPK pathway overactivation in this tumor (43). With next-generation sequencing, they subsequently revealed mutations in BRAF, MAP2K1, and FGFR2 as candidate drivers (43). Although the sample size in this study was small, interestingly, the two BRAF mutations observed were L597R and G469S and not V600E. Though uncommon, these have previously been described in langerhans cell histiocytosis, Erdheim-Chester disease, and melanoma (43).
Non-neuroepithelial Tumors
Langerhans Cell Histiocytosis
Langerhans cell histiocytosis (LCH) is the most common histiocytic tumor affecting children. Within the CNS, these are usually found in the skull or hypothalamus. BRAF point mutation is found in an estimated 69% of cases, with 47% of those representing V600E (44). Other mutations include T599A, 600DLAT insertion and BRAF V600D (45).
BRAF alteration portends worse clinical course, as shown by Heritier et al. where they studied 315 patients, of which 55% were harboring V600E, and found this was associated with a seven-fold increased risk of resistance to standard treatment (21.9 vs. 3.3%) (46). These patients also reactivated at a higher rate over a 5 year follow-up (42.8 vs. 28.1%), and experienced more permanent and severe sequelae (46).
Craniopharyngioma
Craniopharyngioma is a benign, WHO grade I tumor that has relatively low risk of mortality, but causes significant morbidity in terms of endocrine and visual function (Figure 4B). Interestingly, though the two subtypes (i.e., adamantinomatous and papillary) are associated with strongly dichotomous molecular features, they behave largely similarly in patients, aside from age of initial presentation. In terms of morbidity and mortality, there are to our knowledge no studies correlating tumor subtype with long term outcome. Karavitaki, for example, reviewed 121 cases of craniopharyngioma and found no difference in overall survival at 10 years (47).
In 2014 Brastianos et al. revealed the V600E substitution in 95% of 39 papillary specimens (48). Meanwhile, 96% of adamantinomatous subtypes were shown to harbor mutations in CTNNB1 and all adamantinomatous specimens were BRAF wild-type. The authors therefore proposed these markers defined two distinct and mutually exclusive clonal patterns (48). Since the original account, rare adamantinomatous specimens have been discovered to bear BRAF V600E (49). However, the specificity of distinct alterations overall remains very high.
Glomus Tumor
Glomus tumors are seldomly encountered mesenchymal neoplasms, and typically follow a benign clinical course (50, 51). Most glomus tumors are superficial, less than 2 cm, and surgically curable (52). BRAF V600E appears with a frequency of 6–11% (50, 51, 53). Its presence in glomus tumors may predict a more aggressive phenotype (53). Karamzadeh et al. reviewed 102 cases, largely originating from the extremities (53). In this series BRAF V600E was not observed in any of 57 benign cases, but was enriched in tumors of uncertain malignant potential (3/14, 21%) and malignant varieties (3/24, 12%). These tumors showed atypical histological features including deep location, size more than 2 cm, infiltrative growth, and mitotic count of ≥5/50 HPFs (53).
Malignant Peripheral Nerve Sheath Tumors
Malignant peripheral nerve sheath tumors (MPNST) comprise 5–10% of all soft tissue sarcomas, are usually deep seated, and arise from a peripheral nerve trunk (54). 50% are associated with NF1. Overall, NF1 patients have an 8–13% lifetime risk of developing it (55). The rate of BRAF V600E in all MPNSTs is estimated to be 6.5% (2.9% in NF1, 11.9% sporadic) (56). BRAF fusions have also been reported, though these are less frequent. (56) Little is currently known about the prognosis of a BRAF mutation in MPNSTs (55, 56), though one group found no statistically significant differences in survival or time to between mutant and wild-type cohorts (55).
Metastatic Tumors to the CNS
Brain metastases are the most common intracranial neoplasm in adults, with an incidence estimated at between 8.3 and 11 per 100,000 (57). The most frequent primary tumors are lung, melanoma, breast, renal, and colorectal (57). The role of BRAF is under intense study in several of these contexts. CNS-specific outcomes, however, are ill-defined.
Estimates of BRAF aberration in non-small-cell lung carcinoma are less than 10% (58). In one large series of 1,046 cases, rates were 4.9% in adenocarcinoma and 0.3% in squamous cell carcinoma (58). V600E accounted for 57% of the total aberrant BRAF in cases of adenocarcinoma and squamous cell carcinoma in this study, and was independently associated with poorer prognosis. The study did not examine CNS specific outcomes, however.
In melanoma, BRAF mutations are found in approximately 50% of stage IV disease, and of those roughly 70% are V600E and 20% are V600K (59). BRAF mutation is associated with slightly younger age at primary diagnosis, but does not appear to influence overall survival (60, 61). Kotecha et al. found that for patients who had stereotactic radiosurgery, the 12 month local failure rate was higher amongst patients with BRAF wildtype metastatic melanoma compared with BRAF mutant cases (22% compared with 6% (62). Prevalence of BRAF mutation in brain-specific metastases is similar to prevalence in other metastatic sites (63).
BRAF mutations are rare in breast and renal carcinoma, but they do occur in 5–10% of patients with metastatic colon carcinoma (64). Over 95% of these are V600E (64). These patients exhibit shorter overall survival as compared to their wild type counterparts (65). CNS-specific outcomes are unfortunately lacking.
Approximately 25–83% of papillary thyroid carcinoma harbor BRAF mutation, where it is associated with a more aggressive phenotype (66). However, this tumor rarely metastasizes to the brain and in one meta-analysis, BRAF status was not related to distant metastasis (66).
Successes and Challenges in Targeting BRAF
To date, oncogenic BRAF has been targeted with varying success scale in humans in melanoma, non-small cell lung cancer, colorectal carcinoma, and thyroid carcinoma, as well as gliomas and glioneuronal tumors. By far, the majority of clinical experience has been gained in metastatic melanoma. This experience has highlighted several challenges that will inform treatment of patients with BRAF-associated CNS or PNS disease. Additionally, if these treatments are to gain clinical relevance, CNS-specific obstacles must be considered.
The first BRAF inhibitors (BRAFi) described were non-specific “multikinase” inhibitors. These were developed before recognition of widespread BRAF mutation, including V600E, in human cancer. Sorafenib (Bayer/Onyx) is the archetypal agent of this family. This drug was FDA approved for use in advanced renal cell carcinoma in 2005, for hepatocellular carcinoma in 2007, and for metastatic thyroid carcinoma in 2013. In a phase III trial in metastatic melanoma, the addition of sorafenib to carboplatin/paclitaxel failed to improve overall survival (67). This was likely a result of poor efficacy against V600E. Importantly, sorafenib was also the first BRAFi to be associated with paradoxical MAPK activation in BRAF wild-type and BRAF fusion cells. This was reported in vitro in 1999 (68), but its clinical significance was recognized only later, with the observation that patients treated for renal cell carcinoma developed keratoacanthomas and squamous cell carcinomas (69). In 2011 this adverse effect was highlighted in a phase II trial of children with low-grade astrocytoma, during which 9 of 11 patients unexpectedly and rapidly progressed following three cycles of treatment with the drug (70). This led to a premature termination of the trial. Subsequent experience has reinforced opinion that this unexpected result was due to paradoxical ERK activation (70, 71).
Second generation inhibitors include vemurafenib, dabrafenib, and encorafenib, all of which are currently FDA approved for use in metastatic melanoma. Unlike sorafenib, these were specifically engineered to target V600E-bearing BRAF. Vemurafenib (PLX4032, Zelboraf, Plexxikon/Genentech) was developed in 2008 by a structure-guided approach and rapidly underwent phase I and II trials (72). In 2011 data from a phase III randomized controlled trial comparing vemurafenib to dacarbazine in patients with unresectable stage IIIc or IV disease were reported (73). The vemurafenib cohort experienced 63% relative risk reduction in death and 74% relative risk reduction in tumor progression (71). Results of a trial comparing dabrafenib (GSK2118436, Tafinlar, GlaxoSmithKline) to dacarbazine were similarly positive (74). Encorafenib was granted approval in 2018 for use in combination with binimetinib, a MEKi.
Second-generation inhibitors also induce MAPK activation in non-V600E cells, leading to the adverse effect of secondary cutaneous neoplasm in 14–26% of patients (75). Similarly to sorafenib, this includes cells bearing BRAF fusion protein. In vitro, cortical neurospheres containing KIAA1549-BRAF treated with an analog of vemurafenib exhibit paradoxical growth (71).
While second generation inhibitors target V600E, an FDA-approved therapy specific to KIAA1549:BRAF is lacking. Current strategy for these tumors is toward targeting other molecules in the MAPK pathway. In vitro, cells bearing BRAF fusion are susceptible to MEK inhibitors such as selumetinib, binimetinib, trametinib, and cobimetinib (76, 77). In vivo, success has also been achieved in some patients. One phase II trial studied selumetinib in recurrent or refractory BRAF-aberrant or NF1 associated low-grade gliomas (78). In a subset of 18 patients harboring the KIAA1549:BRAF fusion, seven (39%) exhibited partial response (78). Two randomized, controlled trials are currently comparing selumetinib with standard chemotherapy in pediatric low-grade glioma (78). In addition, next-generation Raf inhibitors in phase I/II trials may also have efficacy against BRAF fusion proteins (79, 80).
Acquired resistance has proved to be the major obstacle to durable response in patients with a variety of cancers treated with BRAFi. In melanoma, median response duration to BRAFi monotherapy is just 5–8 months (81). It is generally accepted that acquired resistance to BRAFi occurs via reactivation of the MAPK and, to a lesser extent, PI3K/mTOR pathways. This can result, for example, from overexpression or mutation in Ras that bypasses Raf, or by secondary activating mutations in MEK (82, 83). Combination BRAFi + MEKi has improved clinical responses, likely by addressing this escape mechanism. In melanoma, BRAFi + MEKi extends duration of response to 9.5 months (81). Consequently, combination therapy is now standard-of-care. Resistance to combination therapy is being investigated, but appears to involve similar mechanisms of upstream activation and parallel activation of the PI3K/mTOR pathway.
One CNS-specific obstacle to BRAF targeted therapy may be the blood-brain or blood-tumor barrier. There is some suggestion of this effect in data from trials in metastatic melanoma. The phase II COMBI-MB trial evaluated the efficacy of dabrafenib plus trametinib in patients with BRAF V600E positive melanoma with asymptomatic brain metastases (84). Whereas median response duration was 10.2 months for extracranial disease, it was just 6.5 month for intracranial disease (84). Some preclinical data do suggest that brain bioavailability of BRAFi may be limited by this barrier. In vitro, Durmus et al. found vemurafenib was efficiently effluxed by transporters of the ATP-binding cassette family (ABC), which are active in the blood-brain barrier (85). In vivo, those mice coadministered vemurafenib with elacridar, an inhibitor of ABCG2, experienced increased brain concentrations of drug (85).
In glioma, clinical trial data of BRAFi are thus far limited. VE-BASKET was a non-randomized, open-label study that evaluated vemurafenib monotherapy in patients with WHO grades I-IV glioma harboring the V600E mutation (86). It enrolled 24 subjects: 7 PXA, 2 PA, and 15 grade III/IV gliomas. 20% of these patients had partial response, 40% had stable disease and one complete response was observed in a PXA. Overall, responses in lower-grade tumors were better, though sample sizes were small and this difference did not reach statistical significance (86). Hargrave et al. more recently reported the results of dabrafenib monotherapy in a subset of pediatric patients with recurrent, BRAF mutant, low grade glioma (87). Of their 32 patients, 44% experienced objective response by response assessment in neuro-oncology (RANO) criteria, as compared to the apparent rate of 10% in historical controls treated with standard of care. Mean duration of response was 11 months. Other case series and reports have confirmed similar findings (31, 88–90).
There is reason to believe BRAFi + MEKi may be superior to BRAFi monotherapy in glioma as in other cancers. In an animal model of V600E high-grade glioma, Grossaeur et al. documented longer treatment effects when analyzing the frequency of proliferative tumor cells with combination therapy (91). They also suggested combined BRAFi + MEKi therapy may prevent secondary RAS driven cancers, such as squamous cell carcinomas, which can be seen with monotherapy (91). Small series of combination in both pediatric and adult high grade glioma have also demonstrated occasional rapid and durable responses (31, 92). A phase II trial (NCT02684058) is currently underway studying dabrafenib and trametinib in children and adolescent patients with BRAF V600E low grade glioma and adults with relapsed or refractory high grade glioma (92).
Papillary subtype craniopharyngioma is another obvious target for investigation given its high (95%) prevalence of V600E. Several groups have described cases of dramatic and sometimes-sustained response to BRAFi with or without MEKi (93–98). A multi-institutional phase II trial (Alliance A071601) is currently underway to test the combination of vemurafenib and cobimetinib) (98).
BRAF Testing in Neuropathology
In testing for BRAF aberration, knowledge of the broader clinical context, as well as the limitations of tissue and available tests, will help to optimize testing algorithms.
Molecular platforms are the “gold standard” of BRAF analysis. These include Sanger Sequencing, allele specific polymerase chain reaction (PCR), pyrosequencing, High Resolution Melting curve and Next Generation Sequencing (NGS) (99, 100). High fidelity sequencing is the most sensitive test for a wide variety of possible aberrations. However, the ability to perform these tests may be limited by the quality and quantity of tissue and the ability to extract high-quality DNA. These tests may also be cost-prohibitive.
Such circumstances thus call for immunohistochemistry as a triage before performing the molecular testing. It has been shown that the most frequent and clinically relevant point mutation is V600E, against which a commercially available monoclonal antibody currently exists (Figure 4D). Easy to perform and relatively inexpensive, this stain may be used either as a screen to be followed by more dedicated testing, or alternately in isolation. The sensitivity of this test has been shown to be near, or at, 100% (101). However, false negatives may still result from freezing or surgical cautery artifact, prompting some caution (2).
Fluorescence in situ hybridization fusion and break apart probes for KIAA1549-BRAF provide a reliable method for diagnosis of fusion genes (Figure 3B). Because of the heterogeneity of observed KIAA1549-BRAF fusions, the tester must be attuned to whether the probe is a fusion or break apart probe, and in the case of a break apart probe, the specific exon (Table 1).
Even with detailed molecular analysis available, the limitations to apply this knowledge must be recognized as our experience is still evolving. An example from the pediatric context is instructive. Given a limited sample of a pediatric brain tumor featuring piloid glial cells with or without Rosenthal fibers, and eosinophilic granular bodies, the presence of a BRAF fusion may be highly suggestive, but not pathognomonic, of a pilocytic astrocytoma (36, 102). Molecular findings always require correlation with the patient’s clinical, histological, and radiologic details.
Conclusion
BRAF alterations are identifiable using current diagnostic techniques, and at present play an important role in the pathologic workup of CNS tumors and to a lesser degree in PNS. BRAF targeted therapies, particularly newer generation and in combination, hold promise for use against several subtypes of tumors.
Author Contributions
KS, KC, and SD contributed to writing and editing of this manuscript. SD provided supervision of the writing process. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Yolcu Y, Wahood W, Kerezoudis P, Alvi MA, Habermann EB, Bydon M. Primary central nervous system tumors: comparing two national cancer registries. World Neurosurg. (2019) 128:e719–31. doi: 10.1016/j.wneu.2019.04.247
2. Gessi M, Pietsch T. The diagnostic role and clinical relevance of determination of BRAF status in brain tumors. Pers Med. (2013) 10:405. doi: 10.2217/pme.13.27
3. Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. (2008) 132:363–74. doi: 10.1016/j.cell.2007.12.032
4. Tateishi K, Nakamura T, Yamamoto T. Molecular genetics and therapeutic targets of pediatric low-grade gliomas. Brain Tumor Pathol. (2019) 36:74–83. doi: 10.1007/s10014-019-00340-3
5. Cin H, Meyer C, Herr R, Janzarik W, Lambert S, Jones D, et al. Oncogenic FAM131B–BRAF fusion resulting from 7q34 deletion comprises an alternative mechanism of MAPK pathway activation in pilocytic astrocytoma. Acta Neuropathol. (2011) 121:763–74. doi: 10.1007/s00401-011-0817-z
6. Penman CL, Faulkner C, Lowis SP, Kurian KM. Current understanding of BRAF alterations in diagnosis prognosis, and therapeutic targeting in pediatric low-grade gliomas. Front Oncol. (2015) 5:54. doi: 10.3389/fonc.2015.00054
7. Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold-Mende C, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. (2011) 121:397–405. doi: 10.1007/s00401-011-0802-6
9. Horbinski C, Nikiforova MN, Hagenkord JM, Hamilton RL, Pollack IF. Interplay among BRAF, p16, p53, and MIB1 in pediatric low-grade gliomas. Neuro Oncol. (2012) 14:777–89. doi: 10.1093/neuonc/nos077
10. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. WHO Classification of Tumours of the Central Nervous System. Lyon: World Health Organization, International Agency for Research on Cancer. (2016).
11. Helfferich J, Nijmeijer R, Brouwer OF, Boon M, Fock A, Hoving EW, et al. Neurofibromatosis type 1 associated low grade gliomas: a comparison with sporadic low grade gliomas. Crit Rev Oncol Hematol. (2016) 104:30–41. doi: 10.1016/j.critrevonc.2016.05.008
12. Horbinski C. To BRAF or not to BRAF: is that even a question anymore? J Neuropathol Exp Neurol. (2013) 72:2–7. doi: 10.1097/NEN.0b013e318279f3db
13. Forshew T, Tatevossian RG, Lawson AR, Ma J, Neale G, Ogunkolade BW, et al. Activation of the ERK/MAPK pathway: a signature genetic defect in posterior fossa pilocytic astrocytomas. J Pathol. (2009) 218:172–81. doi: 10.1002/path.2558
14. Jones DTW, Kocialkowski S, Liu L, Pearson DM, Bäcklund LM, Ichimura K, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. (2008) 68:8673–7. doi: 10.1158/0008-5472.CAN-08-2097
15. Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet. (2013) 45:602–12. doi: 10.1038/ng.2611
16. Kaul A, Chen Y-H, Emnett RJ, Dahiya S, Gutmann DH. Pediatric glioma-associated KIAA1549:BRAF expression regulates neuroglial cell growth in a cell type-specific and mTOR-dependent manner.(Research Communication)(Report). Genes Dev. (2012) 26:2561–6. doi: 10.1101/gad.200907.112
17. Rodriguez FJ, Ligon AH, Horkayne-Szakaly I, Rushing EJ, Ligon KL, Vena N, et al. BRAF duplications and MAPK pathway activation are frequent in gliomas of the optic nerve proper. J Neuropathol Exp Neurol. (2012) 71:789–94. doi: 10.1097/NEN.0b013e3182656ef8
18. Hawkins C, Walker E, Mohamed N, Zhang C, Jacob K, Shirinian M, et al. BRAF-KIAA1549 fusion predicts better clinical outcome in pediatric low-grade astrocytoma. Clin Cancer Res. (2011) 17:4790–8. doi: 10.1158/1078-0432.Ccr-11-0034
19. Horbinski C, Hamilton R, Nikiforov Y, Pollack I. Association of molecular alterations, including BRAF, with biology and outcome in pilocytic astrocytomas. Acta Neuropathol. (2010) 119:641–9. doi: 10.1007/s00401-009-0634-9
20. Stokland T, Liu JF, Ironside JW, Ellison DW, Taylor R, Robinson KJ, et al. A multivariate analysis of factors determining tumor progression in childhood low-grade glioma: a population-based cohort study (CCLG CNS9702). Neuro Oncol. (2010) 12:1257–68. doi: 10.1093/neuonc/noq092
21. Gnekow AK, Falkenstein F, von Hornstein S, Zwiener I, Berkefeld S, Bison B, et al. Long-term follow-up of the multicenter, multidisciplinary treatment study HIT-LGG-1996 for low-grade glioma in children and adolescents of the German speaking society of pediatric oncology and hematology. Neuro Oncol. (2012) 14:1265–84. doi: 10.1093/neuonc/nos202
22. Gierke M, Sperveslage J, Schwab D, Beschorner R, Ebinger M, Schuhmann MU, et al. Analysis of IDH1-R132 mutation, BRAF V600 mutation and KIAA1549-BRAF fusion transcript status in central nervous system tumors supports pediatric tumor classification. J Cancer Res Clin Oncol. (2016) 142:89–100. doi: 10.1007/s00432-015-2006-2
23. Kim YH, Nonoguchi N, Paulus W, Brokinkel B, Keyvani K, Sure U, et al. Frequent BRAF gain in low-grade diffuse gliomas with 1p/19q loss. Brain Pathol. (2012) 22:834–40. doi: 10.1111/j.1750-3639.2012.00601.x
24. Lassaletta A, Zapotocky M, Mistry M, Ramaswamy V, Honnorat M, Krishnatry R, et al. Therapeutic and prognostic implications of BRAF V600E in pediatric low-grade gliomas. J Clin Oncol. (2017) 35:2934–41. doi: 10.1200/JCO.2016.71.8726
25. Ellison DW, Hawkins C, Jones DTW, Onar-Thomas A, Pfister SM, Reifenberger G, et al. cIMPACT-NOW update 4: diffuse gliomas characterized by MYB, MYBL1, or FGFR1 alterations or BRAFV600E mutation. Acta Neuropathol. (2019) 137:683–7. doi: 10.1007/s00401-019-01987-0
26. Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007-2011. Neuro Oncol. (2014) 16(Suppl. 4):iv1–63. doi: 10.1093/neuonc/nou223
27. Ida CM, Rodriguez FJ, Burger PC, Caron AA, Jenkins SM, Spears GM, et al. Pleomorphic xanthoastrocytoma: natural history and long-term follow-up. Brain Pathol. (2015) 25:575–86. doi: 10.1111/bpa.12217
28. Tabouret E, Bequet C, Denicolai E, Barrie M, Nanni I, Metellus P, et al. BRAF mutation and anaplasia may be predictive factors of progression-free survival in adult pleomorphic xanthoastrocytoma. Eur J Surg Oncol. (2015) 41:1685–90. doi: 10.1016/j.ejso.2015.09.012
29. Ma C, Feng R, Chen H, Hameed NUF, Aibaidula A, Song Y, et al. BRAF V600E TERT, IDH2 mutations in pleomorphic xanthoastrocytoma: observations from a large case-series study. World Neurosurg. (2018) 120:e1225–33. doi: 10.1016/j.wneu.2018.09.050
30. Vaubel RA, Caron AA, Yamada S, Decker PA, Eckel Passow JE, Rodriguez FJ, et al. Recurrent copy number alterations in low-grade and anaplastic pleomorphic xanthoastrocytoma with and without BRAF V600E mutation. Brain Pathol. (2018) 28:172–82. doi: 10.1111/bpa.12495
31. Johanns TM, Ferguson CJ, Grierson PM, Dahiya S, Ansstas G. Rapid clinical and radiographic response with combined dabrafenib and trametinib in adults with BRAF-mutated high-grade glioma. J Natl Compr Cancer Netw. (2018) 16:4–10. doi: 10.6004/jnccn.2017.7032
32. Behling F, Barrantes-Freer A, Skardelly M, Nieser M, Christians A, Stockhammer F, et al. Frequency of BRAF V600E mutations in 969 central nervous system neoplasms.(Report). Diagn Pathol. (2016) 11:55. doi: 10.1186/s13000-016-0506-2
33. Dahiya S, Emnett RJ, Haydon DH, Leonard JR, Phillips JJ, Perry A, et al. BRAF-V600E mutation in pediatric and adult glioblastoma. Neuro Oncol. (2013) 16:318–9. doi: 10.1093/neuonc/not146
34. Pekmezci M, Villanueva-Meyer JE, Goode B, Van Ziffle J, Onodera C, Grenert JP, et al. The genetic landscape of ganglioglioma. Acta Neuropathol Commun. (2018) 6:47. doi: 10.1186/s40478-018-0551-z
35. Dahiya S, Haydon D, Alvarado D, Gurnett C, Gutmann D, Leonard J. BRAF V600E mutation is a negative prognosticator in pediatric ganglioglioma. Acta Neuropathol. (2013) 125:901–10. doi: 10.1007/s00401-013-1120-y
36. Brandner S, Jaunmuktane Z. Neurological update: gliomas and other primary brain tumours in adults. J Neurol. (2018) 265:717–27. doi: 10.1007/s00415-017-8652-3
37. Deng M, Sill M, Chiang J, Schittenhelm J, Ebinger M, Schuhmann M, et al. Molecularly defined diffuse leptomeningeal glioneuronal tumor (DLGNT) comprises two subgroups with distinct clinical and genetic features. Acta Neuropathol. (2018) 136:239–53. doi: 10.1007/s00401-018-1865-4
38. Huse JTSM, Jones DT, Brathwaite CD, Altman N, Lavi E, Saffery R, et al. Polymorphous low-grade neuroepithelial tumor of the young (PLNTY): an epileptogenic neoplasm with oligodendroglioma-like components, aberrant CD34 expression, and genetic alterations involving the MAP kinase pathway. Acta Neuropathol. (2017) 133:417–29.
39. Johnson DR, Giannini C, Jenkins RB, Kim DK, Kaufmann TJ. Plenty of calcification: imaging characterization of polymorphous low-grade neuroepithelial tumor of the young. Neuroradiology. (2019) 61:1327–32. doi: 10.1007/s00234-019-02269-y
40. Gupta K, Harreld JH, Sabin ND, Qaddoumi I, Kurian K, Ellison DW. Massively calcified low-grade glioma – a rare and distinctive entity. Neuropathol Appl Neurobiol. (2014) 40:221–4. doi: 10.1111/nan.12080
41. Huse JT, Edgar M, Halliday J, Mikolaenko I, Lavi E, Rosenblum MK. Multinodular and vacuolating neuronal tumors of the cerebrum: 10 cases of a distinctive seizure-associated lesion. Brain Pathol. (2013) 23:515–24. doi: 10.1111/bpa.12035
42. Nunes RH, Hsu CC, da Rocha AJ, do Amaral LLF, Godoy LFS, Watkins TW, et al. Multinodular and vacuolating neuronal tumor of the cerebrum: a new “Leave Me Alone” lesion with a characteristic imaging pattern. Am J Neuroradiol. (2017) 38:1899–904. doi: 10.3174/ajnr.A5281
43. Pekmezci M, Stevers M, Phillips JJ, Van Ziffle J, Bastian BC, Tsankova NM, et al. Multinodular and vacuolating neuronal tumor of the cerebrum is a clonal neoplasm defined by genetic alterations that activate the MAP kinase signaling pathway. Acta Neuropathol. (2018) 135:485–8. doi: 10.1007/s00401-018-1820-4
45. Tran G, Huynh TN, Paller AS. Langerhans cell histiocytosis: a neoplastic disorder driven by Ras-ERK pathway mutations. J Am Acad Dermatol. (2018) 78:579–90.e4. doi: 10.1016/j.jaad.2017.09.022
46. Héritier S, Emile J-F, Barkaoui M-A, Thomas C, Fraitag S, Boudjemaa S, et al. BRAF mutation correlates with high-risk langerhans cell histiocytosis and increased resistance to first-line therapy. J Clin Oncol. (2016) 34:3023–30. doi: 10.1200/jco.2015.65.9508
47. Karavitaki N, Brufani C, Warner JT, Adams CB, Richards P, Ansorge O, et al. Craniopharyngiomas in children and adults: systematic analysis of 121 cases with long-term follow-up. Clin Endocrinol. (2005) 62:397–409. doi: 10.1111/j.1365-2265.2005.02231.x
48. Brastianos PK, Taylor-Weiner A, Manley PE, Jones RT, Dias-Santagata D, Thorner AR, et al. Exome sequencing identifies BRAF mutations in papillary craniopharyngiomas. Nat Genet. (2014) 46:161–5. doi: 10.1038/ng.2868
49. Larkin SJ, Preda V, Karavitaki N, Grossman A, Ansorge O. BRAF V600E mutations are characteristic for papillary craniopharyngioma and may coexist with CTNNB1-mutated adamantinomatous craniopharyngioma. Acta Neuropathol. (2014) 127:927–9. doi: 10.1007/s00401-014-1270-6
50. Cuviello A, Goyal A, Zick A, Ahlawat S, Rodriguez FJ, Belzberg AJ, et al. Sporadic malignant glomus tumor of the brachial plexus with response to targeted therapy directed against oncogenic BRAF. JCO Precis Oncol. (2018) 2:1–7. doi: 10.1200/po.17.00261
51. Chakrapani A, Warrick A, Nelson D, Beadling C, Corless CL. BRAF and KRAS mutations in sporadic glomus tumors. Am J Dermatopathol. (2012) 34:533–5. doi: 10.1097/DAD.0b013e31823931b4
52. Goldblum J, Weiss S, Folpe AL. Enzinger and Weiss’s Soft Tissue Tumors. 7th ed. Philadelphia, PA: Saunders. (2019).
53. Karamzadeh Dashti JN, Bahrami MA, Lee JS, Jenkins LS, Rodriguez MF, Folpe MA, et al. BRAF V600E mutations occur in a subset of glomus tumors, and are associated with malignant histologic characteristics. Am J Surg Pathol. (2017) 41:1532–41. doi: 10.1097/PAS.0000000000000913
54. Love S, Perry A, Ironside J, Budka H. Greenfield’s Neuropathology. 9th ed. London: CRC Press. (2015).
55. Hirbe AC, Pekmezci M, Dahiya S, Apicelli AJ, Van Tine BA, Perry A, et al. BRAF V600E mutation in sporadic and neurofibromatosis type 1-related malignant peripheral nerve sheath tumors. Neuro Oncol. (2014) 16:466–7. doi: 10.1093/neuonc/not248
56. Kaplan HG, Rostad S, Ross JS, Ali SM, Millis SZ. Genomic profiling in patients with malignant peripheral nerve sheath tumors reveals multiple pathways with targetable mutations. J Natl Compr Cancer Netw. (2018) 16:967–74. doi: 10.6004/jnccn.2018.7033
57. Barnholtz-Sloan JS, Sloan AE, Davis FG, Vigneau FD, Lai P, Sawaya RE. Incidence proportions of brain metastases in patients diagnosed (1973 to 2001) in the metropolitan detroit cancer surveillance system. J Clin Oncol. (2004) 22:2865–72. doi: 10.1200/jco.2004.12.149
58. Marchetti A, Felicioni L, Malatesta S, Grazia Sciarrotta M, Guetti L, Chella A, et al. Clinical features and outcome of patients with non-small-cell lung cancer harboring BRAF mutations. J Clin Oncol. (2011) 29:3574–9. doi: 10.1200/jco.2011.35.9638
59. Long GV, Menzies AM, Nagrial AM, Haydu LE, Hamilton AL, Mann GJ, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. (2011) 29:1239–46. doi: 10.1200/jco.2010.32.4327
60. Ekedahl H, Cirenajwis H, Harbst K, Carneiro A, Nielsen K, Olsson H, et al. The clinical significance of BRAF and NRAS mutations in a clinic-based metastatic melanoma cohort. Br J Dermatol. (2013) 169:1049–55. doi: 10.1111/bjd.12504
61. Carlino MS, Haydu LE, Kakavand H, Menzies AM, Hamilton AL, Yu B, et al. Correlation of BRAF and NRAS mutation status with outcome, site of distant metastasis and response to chemotherapy in metastatic melanoma. Br J Cancer. (2014) 111:292–9. doi: 10.1038/bjc.2014.287
62. Kotecha R, Miller JA, Venur VA, Mohammadi AM, Chao ST, Suh JH, et al. Melanoma brain metastasis: the impact of stereotactic radiosurgery, BRAF mutational status, and targeted and/or immune-based therapies on treatment outcome. J Neurosurg. (2017) 129:50. doi: 10.3171/2017.1.Jns162797
63. Hannan EJ, O’Leary DP, MacNally SP, Kay EW, Farrell MA, Morris PG, et al. The significance of BRAF V600E mutation status discordance between primary cutaneous melanoma and brain metastases: the implications for BRAF inhibitor therapy. Medicine. (2017) 96:e8404. doi: 10.1097/md.0000000000008404
64. Cohn AL, Day BM, Abhyankar S, McKenna E, Riehl T, Puzanov I. BRAF(V600) mutations in solid tumors, other than metastatic melanoma and papillary thyroid cancer, or multiple myeloma: a screening study. Onco Targets Ther. (2017) 10:965–71. doi: 10.2147/ott.S120440
65. Taieb J, Lapeyre-Prost A, Laurent Puig P, Zaanan A. Exploring the best treatment options for BRAF-mutant metastatic colon cancer. Br J Cancer. (2019) 121:434–42. doi: 10.1038/s41416-019-0526-2
66. Liu C, Chen T, Liu Z. Associations between BRAF(V600E) and prognostic factors and poor outcomes in papillary thyroid carcinoma: a meta-analysis. World J Surg Oncol. (2016) 14:241. doi: 10.1186/s12957-016-0979-1
67. Flaherty KT, Lee SJ, Zhao F, Schuchter LM, Flaherty L, Kefford R, et al. Phase III trial of carboplatin and paclitaxel with or without sorafenib in metastatic melanoma. J Clin Oncol. (2013) 31:373–9. doi: 10.1200/jco.2012.42.1529
68. Hall-Jackson CA, Eyers PA, Cohen P, Goedert M, Boyle FT, Hewitt N, et al. Paradoxical activation of Raf by a novel Raf inhibitor. Chem Biol. (1999) 6:559–68. doi: 10.1016/s1074-5521(99)80088-x
69. Dubauskas Z, Kunishige J, Prieto VG, Jonasch E, Hwu P, Tannir NM. Cutaneous squamous cell carcinoma and inflammation of actinic keratoses associated with sorafenib. Clin Genitourin Cancer. (2009) 7:20–3. doi: 10.3816/CGC.2009.n.003
70. Karajannis MA, Legault G, Fisher MJ, Milla SS, Cohen KJ, Wisoff JH, et al. Phase II study of sorafenib in children with recurrent or progressive low-grade astrocytomas. Neuro Oncol. (2014) 16:1408–16. doi: 10.1093/neuonc/nou059
71. Sievert AJ, Lang SS, Boucher KL, Madsen PJ, Slaunwhite E, Choudhari N, et al. Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas. Proc Natl Acad Sci USA. (2013) 110:5957–62. doi: 10.1073/pnas.1219232110
72. Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci USA. (2008) 105:3041–6. doi: 10.1073/pnas.0711741105
73. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. (2011) 364:2507–16. doi: 10.1056/NEJMoa1103782
74. Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. (2012) 380:358–65. doi: 10.1016/s0140-6736(12)60868-x
75. Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. (2012) 366:207–15. doi: 10.1056/NEJMoa1105358
76. Jain P, Silva A, Han HJ, Lang SS, Zhu Y, Boucher K, et al. Overcoming resistance to single-agent therapy for oncogenic BRAF gene fusions via combinatorial targeting of MAPK and PI3K/mTOR signaling pathways. Oncotarget. (2017) 8:84697–713. doi: 10.18632/oncotarget.20949
77. Turner JA, Bemis JGT, Bagby SM, Capasso A, Yacob BW, Chimed TS, et al. BRAF fusions identified in melanomas have variable treatment responses and phenotypes. Oncogene. (2019) 38:1296–308. doi: 10.1038/s41388-018-0514-7
78. Fangusaro J, Onar-Thomas A, Young Poussaint T, Wu S, Ligon AH, Lindeman N, et al. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. Lancet Oncol. (2019) 20:1011–22. doi: 10.1016/s1470-2045(19)30277-3
79. Sullivan RJ, Hollebecque A, Flaherty KT, Shapiro GI, Rodon Ahnert J, Millward MJ, et al. A phase I study of LY3009120, a Pan-RAF inhibitor, in patients with advanced or metastatic cancer. Mol Cancer Ther. (2020) 19:460–7. doi: 10.1158/1535-7163.Mct-19-0681
80. Botton T, Talevich E, Mishra VK, Zhang T, Shain AH, Berquet C, et al. Genetic heterogeneity of BRAF fusion kinases in melanoma affects drug responses. Cell Rep. (2019) 29:573–88.e7. doi: 10.1016/j.celrep.2019.09.009
81. Kakadia S, Yarlagadda N, Awad R, Kundranda M, Niu J, Naraev B, et al. Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration-approved targeted therapy in advanced melanoma. Onco Targets Ther. (2018) 11:7095–107. doi: 10.2147/ott.S182721
82. Solit DB, Rosen N. Resistance to BRAF inhibition in melanomas. N Engl J Med. (2011) 364:772–4. doi: 10.1056/NEJMcibr1013704
83. Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. (2014) 4:94–109. doi: 10.1158/2159-8290.Cd-13-0617
84. Davies MA, Saiag P, Robert C, Grob JJ, Flaherty KT, Arance A, et al. Dabrafenib plus trametinib in patients with BRAF(V600)-mutant melanoma brain metastases (COMBI-MB): a multicentre, multicohort, open-label, phase 2 trial. Lancet Oncol. (2017) 18:863–73. doi: 10.1016/s1470-2045(17)30429-1
85. Durmus S, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Oral availability and brain penetration of the B-RAFV600E inhibitor vemurafenib can be enhanced by the P-GLYCOprotein (ABCB1) and breast cancer resistance protein (ABCG2) inhibitor elacridar. Mol Pharm. (2012) 9:3236–45. doi: 10.1021/mp3003144
86. Kaley T, Touat M, Subbiah V, Hollebecque A, Rodon J, Lockhart AC, et al. BRAF Inhibition in BRAF(V600)-mutant gliomas: results from the VE-BASKET study. J Clin Oncol. (2018) 36:3477–84. doi: 10.1200/JCO.2018.78.9990
87. Hargrave DR, Bouffet E, Tabori U, Broniscer A, Cohen KJ, Hansford JR, et al. Efficacy and safety of dabrafenib in pediatric patients with BRAF V600 mutation-positive relapsed or refractory low-grade glioma: results from a phase I/IIa study. Clin Cancer Res. (2019) 25:7303–11. doi: 10.1158/1078-0432.Ccr-19-2177
88. Bautista F, Paci A, Minard-Colin V, Dufour C, Grill J, Lacroix L, et al. Vemurafenib in pediatric patients with BRAFV600E mutated high-grade gliomas. Pediatr Blood Cancer. (2014) 61:1101–3. doi: 10.1002/pbc.24891
89. Lassaletta A, Guerreiro Stucklin A, Ramaswamy V, Zapotocky M, McKeown T, Hawkins C, et al. Profound clinical and radiological response to BRAF inhibition in a 2-month-old diencephalic child with hypothalamic/chiasmatic glioma. Pediatr Blood Cancer. (2016) 63:2038–41. doi: 10.1002/pbc.26086
90. Robinson GW, Orr BA, Gajjar A. Complete clinical regression of a BRAF V600E-mutant pediatric glioblastoma multiforme after BRAF inhibitor therapy. BMC Cancer. (2014) 14:258. doi: 10.1186/1471-2407-14-258
91. Grossauer S, Koeck K, Murphy NE, Meyers ID, Daynac M, Truffaux N, et al. Concurrent MEK targeted therapy prevents MAPK pathway reactivation during BRAFV600E targeted inhibition in a novel syngeneic murine glioma model. Oncotarget. (2016) 7:75839–53. doi: 10.18632/oncotarget.12419
92. Toll SA, Tran HN, Cotter J, Judkins AR, Tamrazi B, Biegel JA, et al. Sustained response of three pediatric BRAF(V600E) mutated high-grade gliomas to combined BRAF and MEK inhibitor therapy. Oncotarget. (2019) 10:551–7. doi: 10.18632/oncotarget.26560
93. Aylwin SJ, Bodi I, Beaney R. Pronounced response of papillary craniopharyngioma to treatment with vemurafenib, a BRAF inhibitor. Pituitary. (2016) 19:544–6. doi: 10.1007/s11102-015-0663-4
94. Brastianos PK, Shankar GM, Gill CM, Taylor-Weiner A, Nayyar N, Panka DJ, et al. Dramatic response of BRAF V600E mutant papillary craniopharyngioma to targeted therapy. J Natl Cancer Inst. (2016) 108:djv310. doi: 10.1093/jnci/djv310
95. Roque A, Odia Y. BRAF-V600E mutant papillary craniopharyngioma dramatically responds to combination BRAF and MEK inhibitors. CNS Oncol. (2017) 6:95–9. doi: 10.2217/cns-2016-0034
96. Rostami E, Witt Nystrom P, Libard S, Wikstrom J, Casar-Borota O, Gudjonsson O. Recurrent papillary craniopharyngioma with BRAFV600E mutation treated with neoadjuvant-targeted therapy. Acta Neurochir. (2017) 159:2217–21. doi: 10.1007/s00701-017-3311-0
97. Himes BT, Ruff MW, Van Gompel JJ, Park SS, Galanis E, Kaufmann TJ, et al. Recurrent papillary craniopharyngioma with BRAF V600E mutation treated with dabrafenib: case report. J Neurosurg. (2018) 130:1–5. doi: 10.3171/2017.11.Jns172373
98. Juratli TA, Jones PS, Wang N, Subramanian M, Aylwin SJB, Odia Y, et al. Targeted treatment of papillary craniopharyngiomas harboring BRAF V600E mutations. Cancer. (2019) 125:2910–4. doi: 10.1002/cncr.32197
99. Kakavand H, Wilmott JS, Long GV, Scolyer RA. Targeted therapies and immune checkpoint inhibitors in the treatment of metastatic melanoma patients: a guide and update for pathologists. Pathology. (2016) 48:194–202. doi: 10.1016/j.pathol.2015.12.010
100. Curry JL, Torres-Cabala C, Tetzlaff MT, Bowman C, Prieto V. Molecular platforms utilized to detect BRAF V600E mutation in melanoma. Semin Cutan Med Surg. (2012) 31:267–73.
101. Capper D, Preusser M, Habel A, Sahm F, Ackermann U, Schindler G, et al. Assessment of BRAF V600E mutation status by immunohistochemistry with a mutation-specific monoclonal antibody. Acta Neuropathol. (2011) 122:11–9.
102. Velázquez Vega EJ, Brat JD. Incorporating advances in molecular pathology into brain tumor diagnostics. Adv Anat Pathol. (2018) 25:143–71. doi: 10.1097/PAP.0000000000000186
103. Collins VP, Jones DTW, Giannini C. Pilocytic astrocytoma: pathology, molecular mechanisms and markers. Acta Neuropathol. (2015) 129:775–88. doi: 10.1007/s00401-015-1410-7
Keywords: BRAF, CNS, PNS, tumor, diagnosis, prognosis, targeted-therapy
Citation: Srinivasa K, Cross KA and Dahiya S (2020) BRAF Alteration in Central and Peripheral Nervous System Tumors. Front. Oncol. 10:574974. doi: 10.3389/fonc.2020.574974
Received: 22 June 2020; Accepted: 20 August 2020;
Published: 15 September 2020.
Edited by:
Leonora Balaj, Harvard Medical School, United StatesReviewed by:
Debanjan Bhattacharya, University of Cincinnati, United StatesSwapna Chaudhuri, Chittaranjan National Cancer Institute, India
Copyright © 2020 Srinivasa, Cross and Dahiya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sonika Dahiya, c2RhaGl5YUB3dXN0bC5lZHU=