 Lei Sang
Lei Sang Ying-Jie He
Ying-Jie He Jiaxin Kang1†
Jiaxin Kang1† Hongyi Ye
Hongyi Ye Xiao-Dong Luo
Xiao-Dong Luo Jianwei Sun
Jianwei Sun- 1State Key Laboratory for Conservation and Utilization of Bio-Resources in Yunnan, Center for Life Sciences, School of Life Sciences, Yunnan University, Kunming, China
- 2Key Laboratory of Medicinal Chemistry for Natural Resource, Ministry of Education and Yunnan Province, School of Chemical Science and Technology, Yunnan University, Kunming, China
Overexpression of DGUOK promotes mitochondria oxidative phosphorylation and lung adenocarcinoma progression. However, the role and mechanism of DGUOK in regulation of mitochondria function and lung cancer progression still poorly understood. Here we demonstrated that DGUOK regulated NAD+ biogenesis. Depletion of the DGUOK significantly decreased NAD+ level. Furthermore, knockout of the DGUOK considerably reduced expression of the NMNAT2, a key molecule controlling NAD+ synthesis, at both mRNA and protein levels. Ectopic expression of the NMNAT2 abrogated the effect of knockdown of DGUOK on NAD+. Notably, this regulation is independent of DGUOK -mediated mitochondria complex I activity. We also showed that NMNAT2 was highly expressed in lung adenocarcinoma and negatively correlated with the patient overall survival. Our study suggested that DGUOK regulates NAD+ in a NMNAT2 dependent manner and DGUOK-NMNAT2-NAD+ axis could be a potential therapeutic target in lung adenocarcinoma.
Introduction
Mitochondrial deoxyguanosine kinase (DGUOK) is a rate-limiting enzyme for the salvage pathway of purine deoxynucleotide biogenesis in mitochondria (1). It has been shown that DGUOK phosphorylates purine deoxyribonucleosides in the mitochondrial matrix. In addition, this protein phosphorylates several purine deoxyribonucleoside analogs which were used in the treatment of lymphoproliferative disorders, and this phosphorylation is critical for the effectiveness of the analogs (2). Mutations in the DGUOK led to mitochondrial DNA (mtDNA) depletion typically in the liver and brain, causing a hepatocerebral phenotype (3). We have previously shown that DGUOK was frequently overexpressed in lung adenocarcinoma and aberrant expression of DGUOK correlated with tumor progression and patient overall survival (4). However, the role and mechanism of DGUOK in lung cancer still poorly understood.
Nicotinamide adenine dinucleotide (NAD) is a critical sirtins (SIRT) coenzyme, NAD levels are very important for regulation of SIRT activity and thus are associated with a number of cellular and biological processes including cell survival, senescence, proliferation and Parkinson's disease (5–7). It exists in both oxidized (NAD+) and reduced (NADH) forms. Previous studies have demonstrated that NAD+ was biosynthesized through two major pathways: the de novo and salvage pathways (8). The salvage pathway is important for the maintenance of NAD+ level in cancer cells and involves 2 major enzymes, one of which is phosphoribosyltransferase (NAMPT) and the other is nicotinate phosphoribosyltransferase (NAPRT) (9). NAMPT acts as a rate‐limiting enzyme in the salvage pathway and works by transferring a phosphoribosyl group to nicotinamide (NAM) to form nicotinamide mononucleotide (NMN). NMN is then converted into NAD+ by nicotinamide mononucleotide adenylyltransferase (NMNAT). There are three NMNAT isoforms (NMNAT1–3) with different tissues and subcellular distributions in mammals. NMNAT family members catalyze the synthesis of NAD+ both in the de novo pathway and the salvage pathway (10, 11). Recent studies have shown that NMNAT2 is involved in colorectal cancer progression (12, 13). NAPRT is involved in the synthesis of NAD+ from nicotinic acid (9). Cancer cells were reported to have a high rate of NAD+ turnover and a low ratio of cytosolic NAD+/NADH due to their elevated metabolic needs. However, the role and mechanism of the NAD+ and NMNAT2 in cancer progression is poorly understood.
In this study, we demonstrated that DGUOK regulates NAD+ biogenesis through NMNAT2, and this regulation is independent of DGUOK-mediated mitochondria complex I activity (4). NMNAT2 expression was increased in lung adenocarcinoma and NMNAT2 level was negatively correlated with the overall survival of patients with lung adenocarcinoma. Our results indicate that DGUOK plays a pivotal role in mitochondrial function and tumor progression through regulation of the NMNAT2.
Material and Methods
Cell Culture
Lung cancer cell line H1650, H1299, and immortalized human kidney epithelial cell HEK293T were obtained from the Moffitt Cancer Center Lung Cancer Center of Excellence cell line repository. These cell lines were free of microbial (including mycoplasma) contamination. H1650 and H1299 cells were cultured in RPMI1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. HEK293T cells were maintained in DMEM medium supplemented with 10% FBS and 1% penicillin/streptomycin. All the cells were cultured at 37°C in a humidified 5% CO2 incubator.
Inhibitor Treatment
For Rotenone treatment, H1650 cells were incubated with medium containing 500 nM (Sigma, P5499) at 37°C for 10 h in a 5% CO2 incubator prior to RNA extraction.
Plasmids
The DGUOK knockout was performed using pLenti CRISPR V2 vector (Addgene_52961) encoding sgRNA targeting human DGUOK, and the sequence targeting DGUOK is: 5’-CCCCGAAGGCTCTCCATCGA-3’
NMNAT2 cDNA was subcloned into pLenti-CMV-blasticidin vector (Addgene_17486) between BamH I and XhoI sites. pLKOs encoding shRNAs for the NDFUB8 and the NMNAT2 were purchased from Sigma (ndufb8, TRCN0000318424 and NMNAT2, TRCN0000318425). Retroviral and lentiviral particles were packaged in HEK293T cells using the PEI transfection method and concentrated as previously described (14).
Antibody
The following antibodies were used in this study: anti-NMNAT2 (sc-515206), anti-GAPDH (sc-32233), and anti-DGUOK (sc-398093) antibodies were from Santa Cruz. Anti-NDUFB8 (459210) was from Thermo Fisher. Anti-Mouse, HRP (7076S) were from Cell Signaling.
Immunoblot
Cells were lyzed in SDS-NP40 buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1% NP40, 1% SDS, 1 mM protease inhibitors cocktail) on ice for 1 min. Cells were scraped from the plate and sonicated briefly 3 times. Then lysates were heated at 95°C for 5 min and centrifuged at 20,000 × g at 4°C for 10 min. 50 μg total cell lysates were separated on SDS-PAGE and then transferred onto PVDF membrane. The membranes were blocked with non-fat dry milk for 30 min at room temperature. Following washing 3 times, the blots were incubated with primary antibodies and then peroxidase-linked anti-mouse IgG (cell signaling,7076S). The bands were detected by an ECL-plus Western blotting detection system (Tanon-5200Multi).
qRT-PCR
Cells were washed with ice-cold PBS and total RNA was extracted from using TIANGEN RNaesy Mini kit (ER501-01). Reverse transcription was performed using the Transgen cDNA synthesis kit (AT311-03). Quantitative real-time PCR (qRT-PCR) was carried out with the Bio-rad real-time PCR system using Transgene SYBR Green PCR master mix (AQ131-02).
Primers were used as follows:
NMNAT2 Q-PCR NS: 5’-GAGGCAGATATGGAGGTGATTG-3’NMNAT2 Q-PCR CAS: 5’-TTTTGTATTTGCGGAGTATTGAGG-3’;NMNAT1 Q-PCR NS: 5’-TGGGTGGAAGTTGATACATGG-3’NMNAT1 Q-PCR CAS: 5’-TCCAGGCCTTTCTAGAGTAGG -3’NRK1 Q-PCR NS: 5’- GACTCTCCGGGATACTTTGATG-3’NRK1 Q-PCR CAS: 5’- CCTCTTCAGATTTTGTTCCATCC -3’NAMPT Q-PCR NS: 5’- GCTGCCACCTTATCTTAGAGTT -3’NAMPT Q-PCR CAS: 5’- CTTGTCAACTTCTGTAGCAAACC -3’NARPT Q-PCR NS: 5’- GTCCTCATCGTAGTCAGCAAC -3’NARPT Q-PCR CAS: 5’- CACCAGCTTATAGACGCCAC -3’NADSYN1 Q-PCR NS: 5’- CAAGATACAGGCTTGGACCAG-3’NADSYN1 Q-PCR CAS: 5’- CCGCTAGGACTTGAAACGAG -3’GAPDH Q-PCR NS: 5’-TGAAGGTCGGAGTCAACGG-3’,GAPDH Q-PCR CAS:5’-AGAGTTAAAAGCAGCCCTGGTG-3’.
Blue Native Electrophoresis and Immunoblot
Blue native (BN) electrophoresis was performed as previously described (15). The Western blot analysis was carried out as described above.
Cell Migration and Invasion Assay
Cell migration and invasion assays were performed as previously described (16). Migration and invasion assayed were carried out for 5 h and 12 h, respectively.
Measurement of NAD+ Levels: LC/MS Method
Preparation of Cellular Extracts: DGUOK-knockout and control H1650 cells (1x107/well in 10 ml) were seeded in 10 cm plates and cultured overnight. For metabolite analysis, cultural medium was discarded. After washing with PBS twice, the cells were extracted using 1 ml of cold 80% methanol. Following incubation at room temperature for 15 min, the methanol extracts were sonicated (60 W, work 3 s, interval 3 s, for 3 times) at 4°C. After centrifugation (12,000 g) at 4°C for 15 min, the supernatants were dried using the EYELA Centrifugal Concentrator (CVE-3110) at 7,000 rpm, 37°C. The samples were then dissolved with deionized water.
An Agilent 6100 LC/MSD system (Agilent Technologies, Palo Alto, CA, USA) was employed for the quantitative analysis of NAD+. A HILIC column (2.1 × 100 mm, 1.8 µm, Waters Corp., Tokyo, Japan) was used for the chromatographic separation of target. In short, 0.1% formic acid water (phase A) and acetonitrile (phase B) were of the solvent system with a suitable gradient elution procedure at a flow rate of 0.15 ml/min (0–10 min, 98%–2% B; 10–15 min, 2% B). The injection volume was 2.0 µl. The gas temperature was 325°C at a rate of 8 L/min, the nebulizer pressure was 35 psi, the fragmentor was 45 V. the NAD+ mass acquisition was performed using selective ion mode (SIM) at m/z 664.1 in positive condition. The quantitative NAD+ was obtained by analyzing the mass response abundance of NAD+ in standard solution and sample solutions. Metabolite peak areas were manually checked for consistency in retention times, compared with known standards and normalized based on protein concentrations and the resultant peak areas were subjected to metabolomic analyses by utilizing MetaboAnalyst 2.0. Relative significance in metabolite levels were analyzed using Student’s t test.
Data Analysis
All the experiments were repeated three times and each experiment was performed in three replicates per sample. Data were analyzed using GraphPad Prism 6.0 (Graph-Pad Software Inc., San Diego, CA, USA), and all results were expressed as means ± SEM. Statistically significant differences were determined using Student’s t-test for two-group analysis. Statistical significance was defined as *P < 0.05, **P < 0.01 or ***P < 0.001.
Result
DGUOK Regulates NAD+ Biogenesis
We have recently reported that DGUOK overexpression promotes lung adenocarcinoma progression (4). Loss of DGUOK significantly induced mtDNA deletion and inhibited mitochondria complex I activity. Ran Jing et al. reported that NAD+ is a potential therapeutic target for mtDNA depletion syndrome, suggesting that DGUOK could involve in NAD biosynthesis (17). To test our hypothesis, we first assessed DGUOK level in a panel of lung cancer cell lines. After normalization, the results showed that DGUOK levels were higher in H1650, H1299, and A549 cells (Figure 1A).
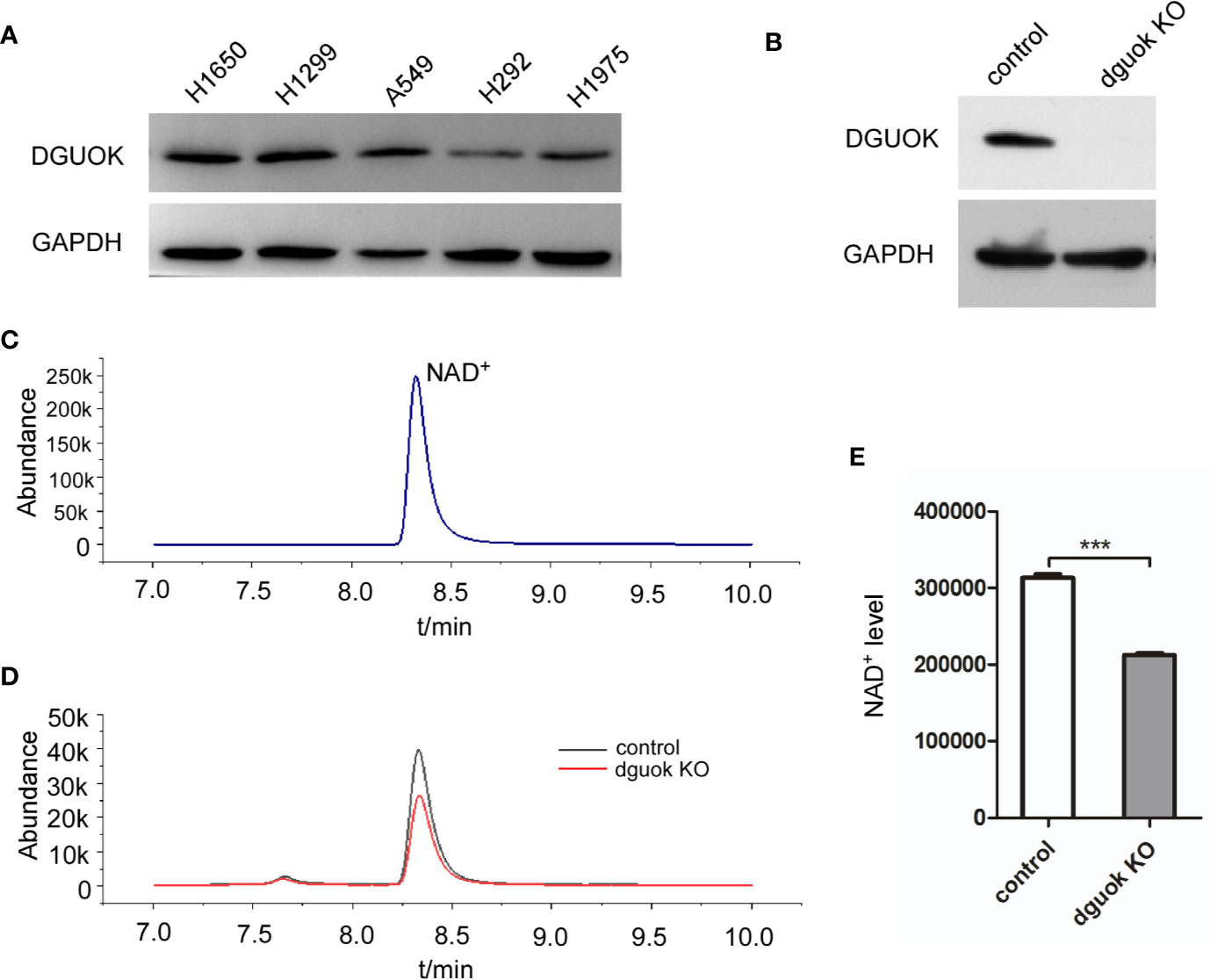
Figure 1 DGUOK regulates NAD+ biogenesis. (A) Western blot analysis of DGUOK level in different lung cancer cell lines. (B) Western blot analysis of DGUOK expression in DGUOK-KO and control H1650 cells. (C) NAD+ standard curve revealed by LC-MS. (D) LC-MS analysis of NAD+ level in DGUOK-KO and control H1650 cells. (E) Quantification of NAD+ level in DGUOK-KO and control H1650 cells. ***p < 0001.
To examine if DGUOK regulates NAD+ level, we first employed LC-MS to examine the content of NAD+ in DGUOK-KO and control H1650 cells. Figure 1B showed that the DGUOK protein expression was completely abrogated after infection of cells with DGUOK-KO lentivirus. To identify the level of NAD+, we first examined NAD+ LC-MS curve (Figure 1B). LC-MS analysis revealed a significant decrease in the NAD+ level in H1650 DGUOK-KO cells (Figures 1C–E). These results indicate a critical role of DGUOK in regulation of NAD+ biogenesis.
DGUOK Regulates NMNAT2 Expression
NAD+ is synthesized through two known pathways, i.e., eight-step de novo cascade and the salvage pathway, in which several enzymes are involved including NAMPT, NAPRT, NRK1/2, and NMNAT1-3 (8). To investigate how DGUOK regulates NAD+ biogenesis, we examined the mRNA levels of these enzymes by quantitative real‐time PCR. There was no significant difference in the expression of NAMPT, NAPRT, NRK1/2, and NMNAT1-3 between DGUOK-KO and control H1650 cells (Figure 2A). Notably, we found that the mRNA levels of the NMNAT2 were dramatically decreased in DGUOK-KO cells (Figure 2A). Accordingly, we observed the low expression of NMNAT2 protein level in DGUOK-KO cells (Figure 2B). To further verify our results, we analyzed the mRNA level of the NMNAT2 in DGUOK-KO and control H1299 cells. The results showed that depletion of DGUOK significantly inhibited the expression of the NMNAT2 (Figure 2C). As NMNAT2 catalyzes the synthesis of NAD+ both in the de novo and salvage pathways. Our data indicated that NMNAT2 is likely to be a key player that bridges DGUOK and NAD+ biogenesis.
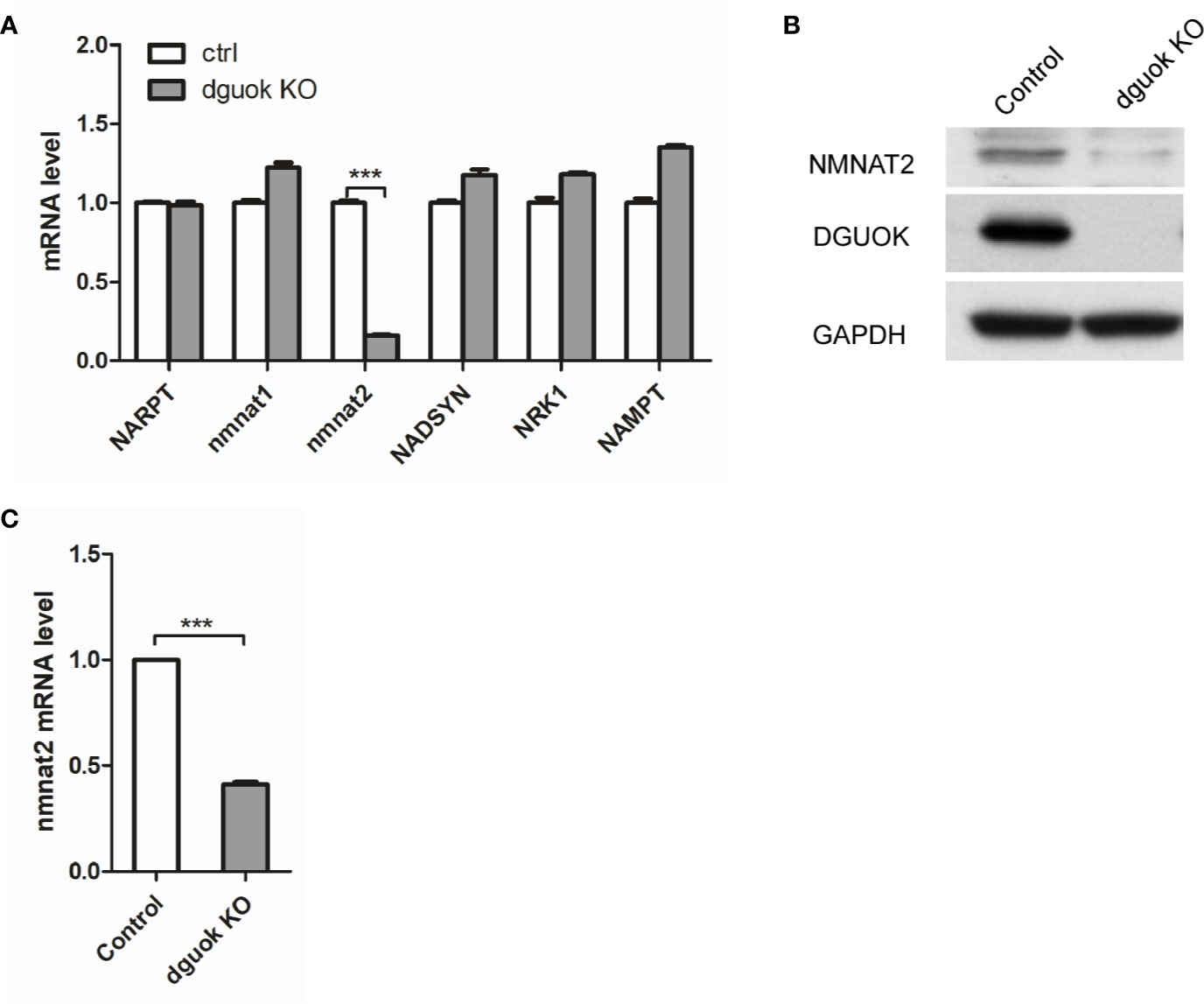
Figure 2 DGUOK controls NMNAT2 expression at mRNA and protein levels. (A) Quantitative RT-PCR analysis of the NARPT, NMNAT1/2, NADSYN, NRK1, NAMPT mRNA levels in DGUOK-KO and control H1650 cells. (B) Western blot analysis of DGUOK and NMNAT2 protein levels in DGUOK-KO and control H1650 cells. (C) Quantitative RT-PCR analysis of the NMNAT2 level in DGUOK-KO and control H1299 cells. ***p < 0001.
DGUOK Regulates NAD+ Biogenesis Through NMNAT2
To further confirm our hypothesis, we first evaluated the effect of the NMNAT2 knockdown (KD) on NAD+ level. As expected, NAD+ content was reduced by 60% in NMNAT2-KD H1650 cells compared to their controls (Figures 3A–C). To investigate if DGUOK regulates NAD+ biogenesis through NMNAT2, we examined whether ectopic expression of NMNAT2 could rescue NAD+ content in DGUOK-KO H1650 cells. Western blot analysis revealed that NMNAT2 protein was efficiently expressed in NMNAT2-transfected cells (Figure 3D). NAD+ levels in DGUOK-KO cells were restored after ectopic expression of NMNAT2 (Figures 3D–F). Taken together, these data suggested that NMNAT2 mediates DGUOK regulated NAD+ biogenesis.
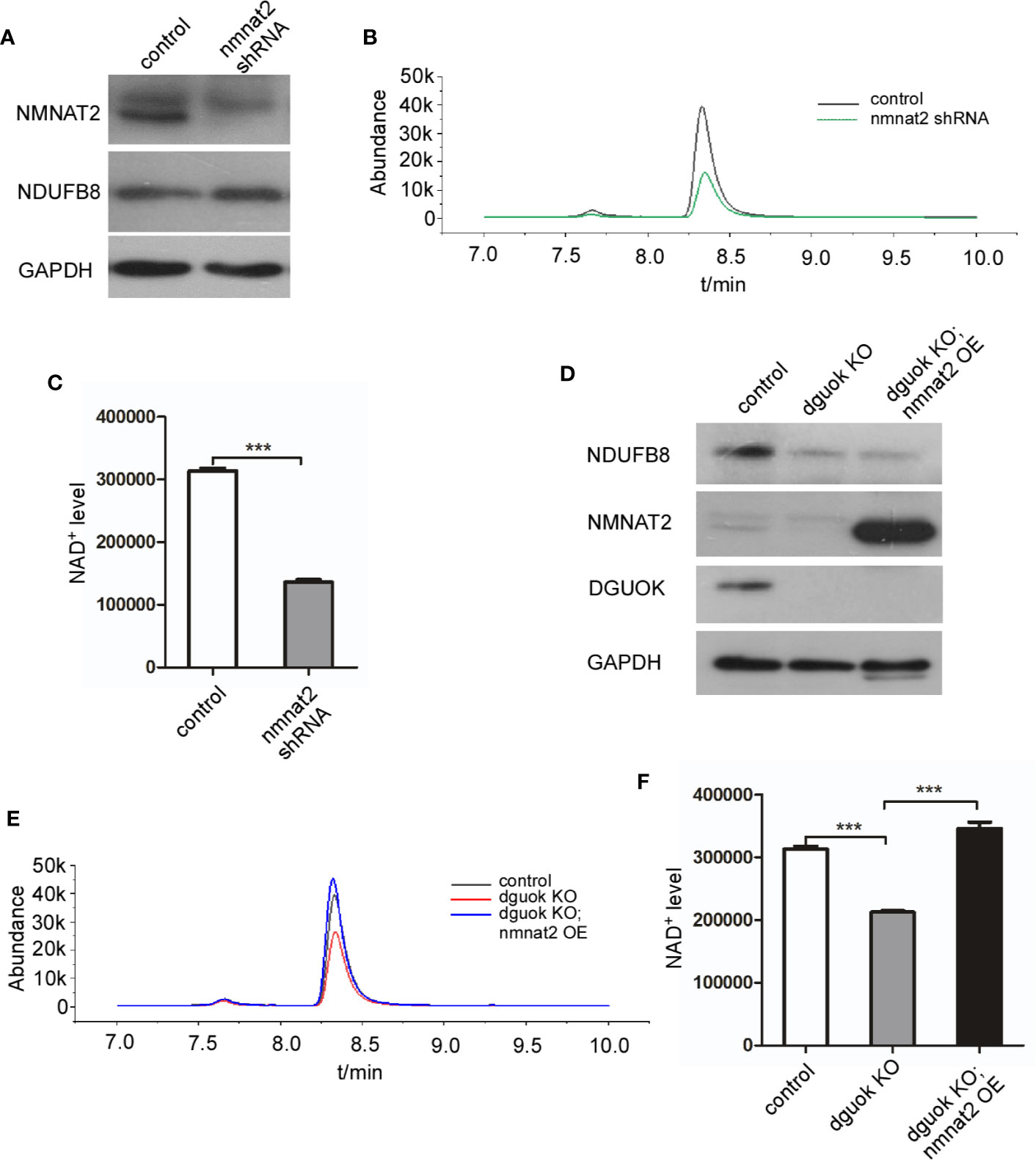
Figure 3 DGUOK regulates NAD+ biosynthesis through NMNAT2. (A) Western blot analysis of NMNAT2 and NDUFB8 expression level in NMNAT2-KD and control H1650 cells. (B) LC-MS analysis of NAD+ level in NMNAT2-KD and control H1650 cells. (C) Quantification of NAD+ level in NMNAT2-KD and control H1650 cells. (D) Western blot analysis of NMNAT2, NDUFB8 and DGUOK expression levels in DGUOK-KO, DGUOK-KO/NMNAT2-OE and control H1650 cells. (E) LC-MS analysis of NAD+ level in DGUOK-KO, DGUOK-KO/NMNAT2-OE and control H1650 cells. (F) Quantification of NAD+ level in DGUOK-KO, DGUOK-KO/NMNAT2-OE and control H1650 cells. ***p < 0001.
NMNAT2 Expression Regulated by DGUOK Is Independent of Mitochondria Respiratory Complex I Activity
It has been shown that DGUOK is required for mtDNA maintain and mitochondria respiratory complex I activity (4). To determine whether the regulation of NAD+ biogenesis and NMNAT2 expression by DGUOK is due to the mitochondrial complex I activity, we investigated the effects of Rotenone, the complex I inhibitor, on NMNAT2 and DGUOK level. As shown in Figures 4A, B, Rotenone treatment did not significantly reduce the expression of DGUOK protein and NMNAT2 mRNA, respectively (Figures 4A, B), suggesting that regulation of NMNAT2 by DGUOK is independent of mitochondria Complex I activity. To further confirm the result, we knocked down the NDUFB8, a nuclear genome encoded complex I subunit, to inhibit complex I level and activity (Figure 4C). Immunoblotting and qRT-PCR analyses showed that depletion of the NDUFB8 severely damaged the assembly of the complex I. However, inhibition of mitochondria complex I activity by NDUFB8 knockdown had no effect on the NMNAT2 mRNA level (Figure 4D). At the same time, while NAD+ is necessary for the mitochondrial complex I function, we did not observe that reduction of NAD+ level by NMNAT2 knockdown affected the level of mitochondrial complex I (Figure 4E). These results indicated that the regulation of NMNAT2 by DGUOK is independent of mitochondria respiratory complex I activity and that DGUOK controls mitochondrial function through regulation of two parallel pathways.
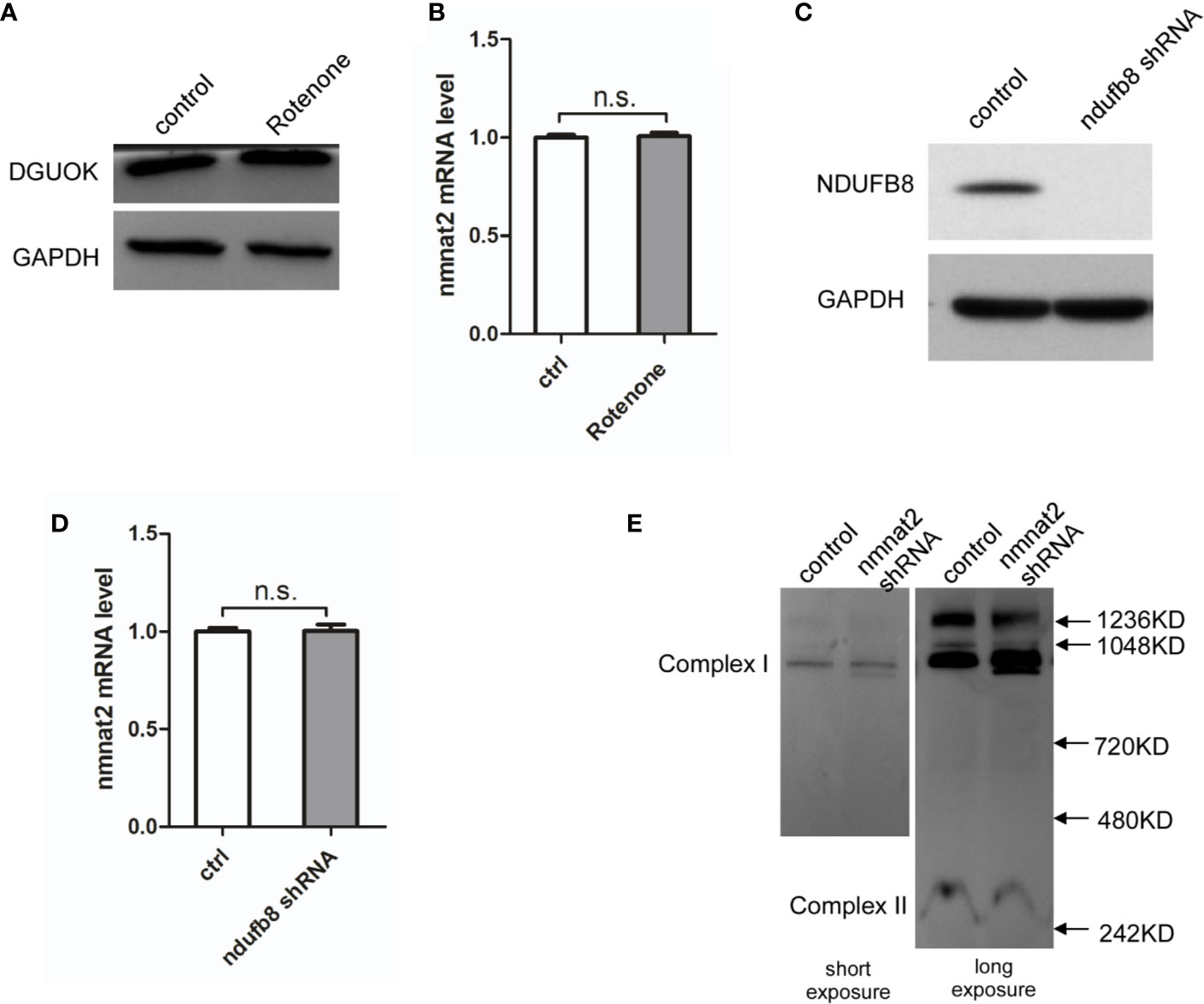
Figure 4 DGUOK regulates NMNAT2 independent of mitochondria complex I activity. (A) Western blot analysis of DGUOK in control and Rotenone treated H1650 cells. (B) qRT-PCR analysis of the NMNAT2 mRNA level in Rotenone treated and control H1650 cells. (C) Western blot analysis of NDUFB8 in NDUFB8-KD and control H1650 cells. (D) qRT-PCR analysis of the NMNAT2 mRNA level in NDUFB8-KD and control H1650 cells. (E) BN-PAGE gel analysis of mitochondria complex I level in NMNAT2-KD and control H1650 cells. n.s. not significant.
NMNAT2 Is Upregulated and Correlates With Overall Survival in Patients With Lung Adenocarcinoma
We have previously shown frequent overexpression of DGUOK in lung adenocarcinoma and close association of elevated expression of DGUOK with tumor progression and patient survival (4). Knockout of the DGUOK in H1650 cells significantly inhibited cell migration and invasion (Figures 5A–D). Because DGUOK regulates NMNAT2, we further investigated if DGUOK regulates cell migration and invasion through NMNAT2 and NAD+. NMNAT2 was ectopically expressed in H1650 DGUOK-KO cells. We found that the expression of NMNAT2 largely rescued the effects of depletion of DGUOK on cell migration and invasion (Figures 5A–D), suggesting that NMNAT2 and NAD+ play an important role in DGUOK-mediated cell migration and invasion. Subsequently, we evaluated mRNA level of the DGUOK and the NMNAT2 in lung adenocarcinoma compared to paired adjacent normal tissues (http://gepia.cancer-pku.cn/detail.php?gene=&clicktag=survival).
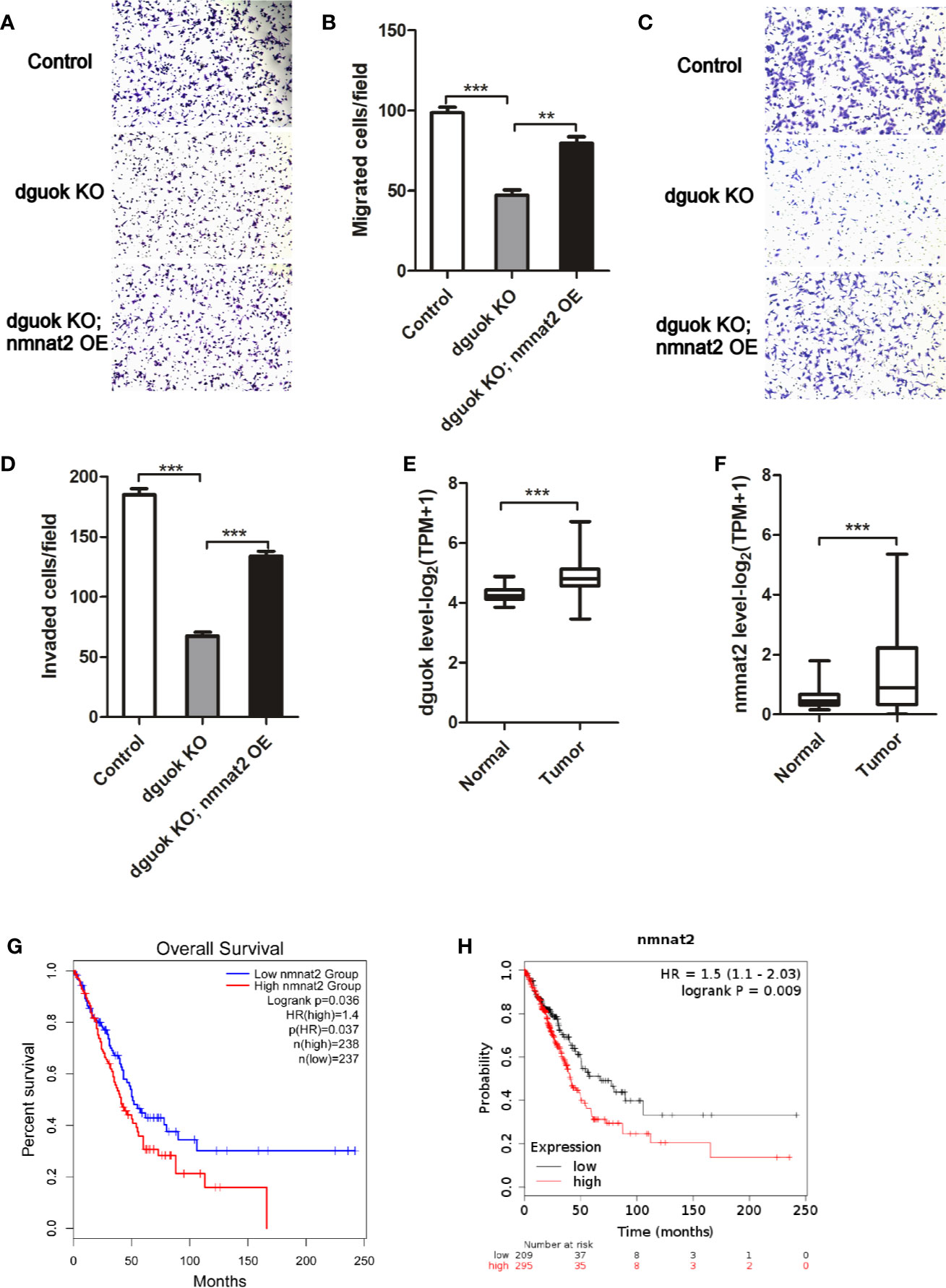
Figure 5 NMNAT2 was up-regulated and correlates with poor overall survival in patients with lung adenocarcinoma. (A) Representative images of in vitro migration in control, DGUOK-KO and DGUOK-KO/NMNAT2 H1650 cells. (B) Ectopic expression of NMNAT2 largely rescued cell migration in DGUOK-KO H1650 cells. (C) Representative images of in vitro invasion in control, DGUOK-KO and DGUOK-KO/NMNAT2 H1650 cells. (D) Ectopic expression of NMNAT2 largely rescued cell invasion in DGUOK-KO H1650 cells. (E) DGUOK level was up-regulated in lung adenocarcinoma when compared to adjacent normal tissue. (F) NMNAT2 level was up-regulated in lung adenocarcinoma when compared to adjacent normal tissue. (G, H). NMNAT2 expression negatively correlates with overall survival of patients with in lung adenocarcinoma in two databases: GEPIA (G, http://gepia.cancer-pku.cn/detail.php?gene=&clicktag=survival) and KM-Express (H, https://kmplot.com/analysis/). **p < 001, ***p < 0001.
We found that the DGUOK and the NMNAT2 were dramatically up-regulated in lung adenocarcinoma (Figures 5E, F). Similar to the DGUOK, high levels of NMNAT2 were associated with the poor overall survival of patients with lung adenocarcinoma (Figure 5G). To confirm this finding, we further analyzed the data from https://kmplot.com/analysis/and noticed that the NMNAT2 expression was also negatively correlated with the patients’ overall survival (Figure 5H). Collectively, these results indicated that DGUOK-NMNAT2 axis is frequently elevated in lung adenocarcinoma and could be served as poor prognostic biomarker in this malignancy. Since NMNAT2 involved lung adenocarcinoma progression, targeting NMNAT2 may benefit in the treatments of lung adenocarcinoma. As DGUOK-NMNAT2 induces NAD+ biogenesis, the DGUOK-NMNAT2-NAD+ pathway could be potential therapeutic target in lung adenocarcinoma.
Discussion
We have recently reported frequent overexpression of DGUOK lung adenocarcinoma (4). The upregulation of DGUOK was associated with poor overall survival of the patients with lung carcinoma. A well-known function of DGUOK is to regulate mtDNA maintenance and mitochondria complex I activity. It has been shown that DGUOK promotes the lung adenocarcinoma cancer progression and cancer stemness through mitochondria complex I activity (4). In this report, we demonstrated that DGUOK induced NMNAT2 expression at mRNA and protein levels which led to activation of NAD+ biogenesis. More significantly, DGUOK regulation of NMNAT2 is independent of mitochondria complex I activity. Thus, our data revealed a novel function of DGUOK, i.e., link DGUOK to NMNAT2/NAD+ cascade.
NAD+ level has been shown to be up-regulated in human cancer (18). NAD+ is an important coenzyme for SIRT, which involves in different cancers (19–21). Recently, accumulated evidence indicates that the NAD+ biosynthesis pathway plays an important role in tumor progression and metastasis (22). Ye et al reported that targeting the NAD+ salvage pathway suppressed APC mutation-driven colorectal cancer growth (23). Nicotinamide phosphoribosyl transferase (NAMPT), which is a rate-limiting enzyme for NAD+ synthesis in the salvage pathway, was shown to be overexpressed in many types of cancer, suggesting that NAMPT acts as a regulator of cancer invasion and metastasis (7, 24). NMNAT2, also a rate-limiting enzyme for NAD+ synthesis, catalyzes the synthesis of NAD+ both in the de novo pathway and the salvage pathway. NMNAT2 was shown to be a promising diagnostic and therapeutic target for colorectal cancer (12, 13) However the role of NMNAT2 in lung adenocarcinoma is unclear. In this study, we found that NMNAT2 mediates NAD+ biogenesis induced by DGUOK and that the NMNAT2 expression negatively correlates with overall survival in the patients with lung adenocarcinoma.
In summary, our results revealed a new pathway, i.e. DGUOK-NMNAT2-NAD+. DGUOK induces NAD+ level through NMNAT2. The regulation of NMNAT2 and NAD+ by DGUOK is independent of mitochondria respiratory complex I activity. Furthermore, we have shown frequent overexpression of DGUOK-NMNAT2 in lung adenocarcinoma and the association of this pathway with poor prognosis of this malignancy. Furthermore, we recently reported DGUOK promote cancer cell stemness in lung adenocarcinoma. Depletion of DGUOK significantly reduced the sphere formation in H1650 and A549 cells (4). In current study, we showed that knockout of DGUOK reduced cell migration and invasion and that ectopic expression of NMNAT2 overrode these phenotypes resulted from DGUOK knockdown (Figure 5). These findings suggest pro-tumorigenic potential of DGUOK. Collectively, our data indicate that the DGUOK-NMNAT2-NAD+ axis could be a prognostic marker and a critical therapeutic target in lung adenocarcinoma.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Author Contributions
Study concept and design: JK, YH, and JS. Acquisition of data: LS, JK, and YH. Drafting and editing of the manuscript: LS, JK, YH, and JS. Analysis and interpretation of data: WB, HY, and JS. Critical revision of the manuscript: XL. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Natural Science Foundation of China (No.31671448), Yunnan Applicative and Basic Research Program (No.2019FY003030) and a grant (No.2020DAMOP-005) from NHC key Laboratory of Drug Addiction medicine.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Taanman JW, Muddle JR, Muntau AC. Mitochondrial DNA depletion can be prevented by dGMP and dAMP supplementation in a resting culture of deoxyguanosine kinase-deficient fibroblasts. Hum Mol Genet (2003) 12(15):1839–45. doi: 10.1093/hmg/ddg192
2. Rodriguez CO Jr, Mitchell BS, Ayres M, Eriksson S, Gandhi V. Arabinosylguanine is phosphorylated by both cytoplasmic deoxycytidine kinase and mitochondrial deoxyguanosine kinase. Cancer Res (2002) 62(11):3100–5.
3. Mandel H, Szargel R, Labay V, Elpeleg O, Saada A, Shalata A, et al. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet (2001) 29(3):337–41. doi: 10.1038/ng746
4. Lin S, Huang C, Sun J, Bollt O, Wang X, Martine E, et al. The mitochondrial deoxyguanosine kinase is required for cancer cell stemness in lung adenocarcinoma. EMBO Mol Med (2019) 11(12):e10849. doi: 10.15252/emmm.201910849
5. Carafa V, Rotili D, Forgione M, Cuomo F, Serretiello E, Hailu GS, et al. Sirtuin functions and modulation: from chemistry to the clinic. Clin Epigenet (2016) 8:61. doi: 10.1186/s13148-016-0224-3
6. Singh P, Hanson PS, Morris CM. SIRT1 ameliorates oxidative stress induced neural cell death and is down-regulated in Parkinson’s disease. BMC Neurosci (2017) 18(1):46. doi: 10.1186/s12868-017-0364-1
7. Lucena-Cacace A, Otero-Albiol D, Jimenez-Garcia MP, Munoz-Galvan S, Carnero A. NAMPT Is a Potent Oncogene in Colon Cancer Progression that Modulates Cancer Stem Cell Properties and Resistance to Therapy through Sirt1 and PARP. Clin Cancer Res (2018) 24p(5):1202–15. doi: 10.1158/1078-0432.CCR-17-2575
8. Canto C, Menzies KJ, Auwerx J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab (2015) 22(1):31–53. doi: 10.1016/j.cmet.2015.05.023
9. Li XQ, Lei J, Mao LH, Wang QL, Xu F, Ran T, et al. NAMPTand NAPRT, Key Enzymes in NAD Salvage Synthesis Pathway, Are of Negative Prognostic Value in Colorectal Cancer. Front Oncol (2019) 9:736. doi: 10.3389/fonc.2019.00736
10. Zhai RG, Zhang F, Hiesinger PR, Cao Y, Haueter CM, Bellen HJ. NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature (2008) 452(7189):887–91. doi: 10.1038/nature06721
11. Brazill JM, Li C, Zhu Y, Zhai RG. NMNAT: It’s an NAD(+) synthase... It’s a chaperone... It’s a neuroprotector. Curr Opin Genet Dev (2017) 44:156–62. doi: 10.1016/j.gde.2017.03.014
12. Cui C, Qi J, Deng Q, Chen R, Zhai D, Yu J. Nicotinamide Mononucleotide Adenylyl Transferase 2: A Promising Diagnostic and Therapeutic Target for Colorectal Cancer. BioMed Res Int (2016) 2016:1804137. doi: 10.1155/2016/1804137
13. Qi J, Cui C, Deng Q, Wang L, Chen R, Zhai D, et al. Downregulated SIRT6 and upregulated NMNAT2 are associated with the presence, depth and stage of colorectal cancer. Oncol Lett (2018) 16(5):5829–37. doi: 10.3892/ol.2018.9400
14. Sun J, Lu F, He H, Shen J, Messina J, Mathew R, et al. STIM1- and Orai1-mediated Ca(2+) oscillation orchestrates invadopodium formation and melanoma invasion. J Cell Biol (2014) 207(4):535–48. doi: 10.1083/jcb.201407082
15. Jha P, Wang X, Auwerx J. Analysis of Mitochondrial Respiratory Chain Supercomplexes Using Blue Native Polyacrylamide Gel Electrophoresis (BN-PAGE). Curr Protoc Mouse Biol (2016) 6(1):1–14. doi: 10.1002/9780470942390.mo150182
16. Wang J, Shen J, Zhao K, Hu J, Dong J, Sun J. STIM1 overexpression in hypoxia microenvironment contributes to pancreatic carcinoma progression. Cancer Biol Med (2019) 16(1):100–8. doi: 10.20892/j.issn.2095-3941.2018.0304
17. Jing R, Corbett JL, Cai J, Beeson GC, Beeson CC, Chan SS, et al. A Screen Using iPSC-Derived Hepatocytes Reveals NAD(+) as a Potential Treatment for mtDNA Depletion Syndrome. Cell Rep (2018) 25(6):1469–84.e5. doi: 10.1016/j.celrep.2018.10.036
18. Li D, Bi FF, Chen NN, Cao JM, Sun WP, Zhou YM, et al. A novel crosstalk between BRCA1 and poly (ADP-ribose) polymerase 1 in breast cancer. Cell Cycle (2014) 13(21):3442–9. doi: 10.4161/15384101.2014.956507
19. Fang EF, Scheibye-Knudsen M, Brace LE, Kassahun H, SenGupta T, Nilsen H, et al. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell (2014) 157(4):882–96. doi: 10.1016/j.cell.2014.03.026
20. Liu TF, McCall CE. Deacetylation by SIRT1 Reprograms Inflammation and Cancer. Genes Cancer (2013) 4(3-4):135–47. doi: 10.1177/1947601913476948
21. Santolla MF, Avino S, Pellegrino M, De Francesco EM, De Marco P, Lappano R, et al. SIRT1 is involved in oncogenic signaling mediated by GPER in breast cancer. Cell Death Dis (2015) 6:e1834. doi: 10.1038/cddis.2015.201
22. Chowdhry S, Zanca C, Rajkumar U, Koga T, Diao Y, Raviram R, et al. NAD metabolic dependency in cancer is shaped by gene amplification and enhancer remodelling. Nature (2019) 569(7757):570–5. doi: 10.1038/s41586-019-1150-2
23. Ye C, Qi L, Li X, Wang J, Yu J, Zhou B, et al. Targeting the NAD(+) salvage pathway suppresses APC mutation-driven colorectal cancer growth and Wnt/beta-catenin signaling via increasing Axin level. Cell Commun Signal (2020) 18(1):16. doi: 10.1186/s12964-020-0513-5
Keywords: deoxyguanosine kinase, NAD+, NMNAT2, mitochondria complex I, lung adenocarcinoma
Citation: Sang L, He Y-J, Kang J, Ye H, Bai W, Luo X-D and Sun J (2020) Mitochondrial Deoxyguanosine Kinase Regulates NAD+ Biogenesis Independent of Mitochondria Complex I Activity. Front. Oncol. 10:570656. doi: 10.3389/fonc.2020.570656
Received: 08 June 2020; Accepted: 17 November 2020;
Published: 18 December 2020.
Edited by:
Qiongzhu Dong, Fudan University, ChinaCopyright © 2020 Sang, He, Kang, Ye, Bai, Luo and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao-Dong Luo, eGRsdW9AbWFpbC5raWIuYWMuY24=; Jianwei Sun, andzdW5AeW51LmVkdS5jbg==
†These authors have contributed equally to this work