 Bin Zhang†
Bin Zhang† Ping Cheng
Ping Cheng
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 19 February 2021
Sec. Cancer Immunity and Immunotherapy
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.561372
Oncolytic viruses (OVs) are potential antitumor agents with unique therapeutic mechanisms. They possess the ability of direct oncolysis and the induction of antitumor immunity. OV can be genetically engineered to potentiate antitumor efficacy by remodeling the tumor immune microenvironment. The present mini review mainly describes the effect of OVs on remodeling of the tumor immune microenvironment and explores the mechanism of regulation of the host immune system and the promotion of the immune cells to destroy carcinoma cells by OVs. Furthermore, this article focuses on the utilization of OVs as vectors for the delivery of immunomodulatory cytokines or antibodies.
Oncolytic viruses (OVs) are naturally occurring or genetically engineered viruses that can selectively target malignant tissues while reducing the infection intensity in the normal tissues (1). As a potential treatment modality against cancer, the past two decades has witnessed a breakthrough in oncolytic virotherapy. The Food Drug Administration (FDA) approved the first oncolytic agent T-VEC for the treatment of patients with melanoma in 2015. T-VEC is an attenuated herpes simplex virus type 1 (HSV-1) that was genetically modified to encode the granulocyte-macrophage colony-stimulating factor (GM-CSF) (2). Unlike in the general gene therapy, OVs not only serve as vectors for transgenic delivery but also as active pharmaceutical agents.
The anti-tumor activity of OVs involves a variety of mechanisms, including the natural interactions among tumor cells, viruses, and the immune system (3). Although it has not been fully confirmed, the mechanism underlying the anti-tumor activity of OVs can mainly be categorized into two types. One is the selective killing of tumor cells by OVs. However, this effect is influenced by the expression of cell surface receptors and the antiviral response of the host cells. The other mechanism of OV-mediated anti-tumor activity is associated with the induction of systemic anti-tumor immunity. The local delivery of immune regulatory factors by viral vectors is beneficial in creating create a proinflammatory tumor microenvironment, which in turn promotes the systemic antitumor immunity (4). Furthermore, the release of tumor-associated antigens (TAAs) after tumor cell lysis can promote the adaptive immune responses, which in turn mediates tumor regression at distant tumor sites that are unexposed to the virus (4). Presently, a variety of OVs have been developed for the treatment of cancer in pre-clinical and clinical trials, including the Newcastle disease virus (NDV) (5), reovirus (6), adenovirus (7), and vesicular stomatitis virus (VSV) (8), among others. Table 1 lists the several OVs that are currently underway for clinical trials and their indications.
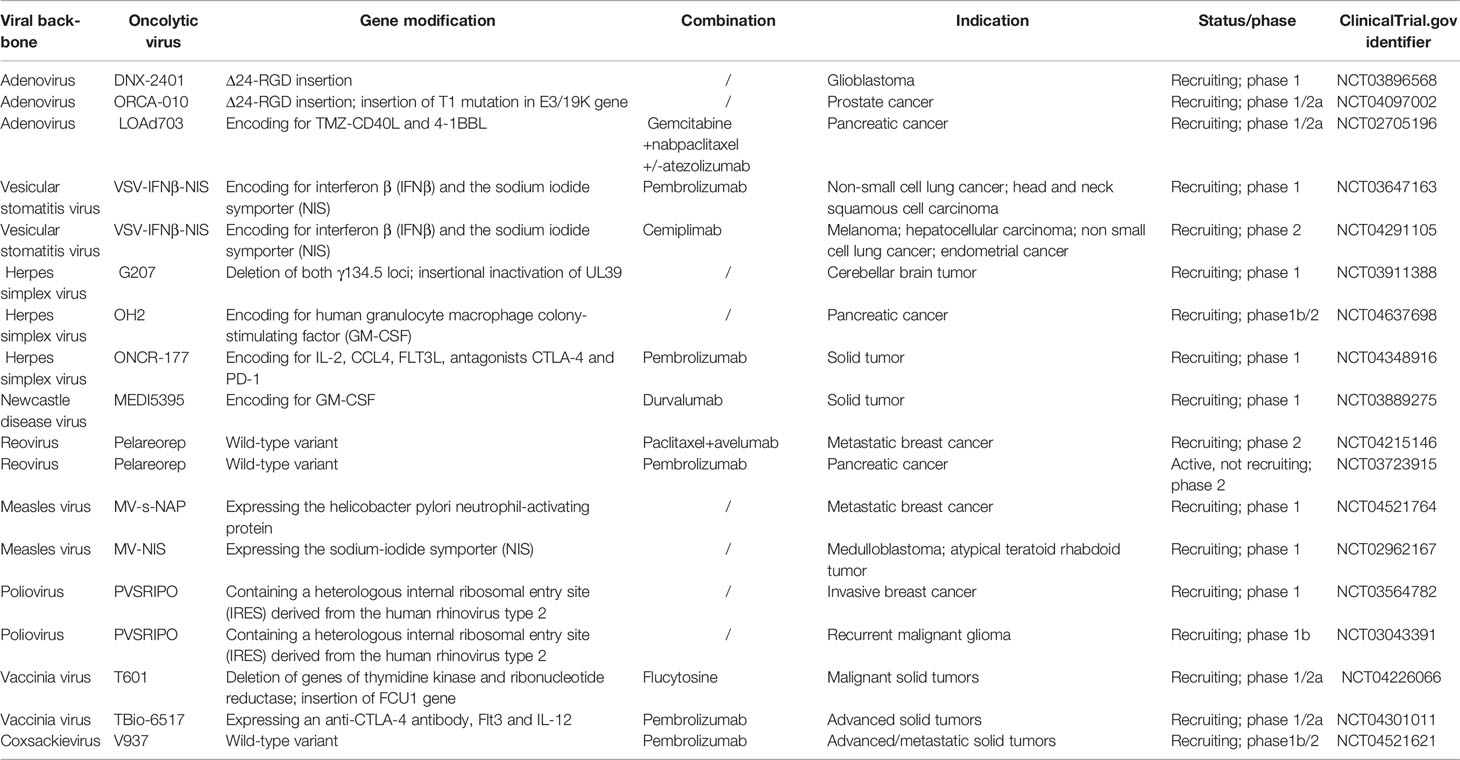
Table 1 List of part of ongoing clinical trials of OVs on Clinical trials.gov.
Tumor microenvironment is a sophisticated niche of developing cancerous cells and various non-cancerous components; and the latter mainly consists of cancer-associated fibroblasts (CAFs), extracellular matrix (ECM), vascular endothelial cells, tumor-associated immune cells, as well as soluble substances such as cytokines and chemokines (9). These components together constitute an environment that is conducive for the maintenance of tumor cell growth. They are involved in immune modulation in the tumor microenvironment to varying degrees, even for non-immune cells such as CAFs. Past studies have suggested that not only do CAFs induce the epithelial-mesenchymal transition (EMT) by secreting multiple growth factors, they also, exhibit immunosuppressive phenotypes because of the expression of the inhibitory surface proteins such as PD-L1 (10).The analysis of the immune profiles of tumor is helpful to predict the disease progression and to customize the treatment regimen. The TIME may exhibit distinguishing immunological status based on the heterogeneity in the cell populations, diseases and patients. Currently, three different classes of TIME have been proposed, including inflamed, excluded and deserted TIME (11). Inflamed TIMEs, or “hot” tumor, are characterized by the abundant accumulation in the tumor core and the stroma of T cells expressing PD-1 and/or CTLA-4, myeloid cells and monocytes. It is however simultaneously accompanied by several proinflammatory cytokines. Inflamed tumors are often positively associated with patients’ responses to cancer immunotherapy (12). The excluded TIMEs are also quite abundant in the immune cells, albeit there is a relative void of immune effector cells in the tumor core. These immune cells are mainly present at the border of the tumor mass possibly due to the absence of specific chemokines and the presence of substantial barriers or specific inhibitors (13). The deserted TIMEs are considered to be immunological “cold” tumor. The typical indication of this status is the lack of immune cells and cytokines in either the core or in the stroma of the tumor mass.
Effectual oncolytic virotherapy is closely linked to the tumor microenvironment. On one hand, tumor microenvironment may limit the efficacy of OVs. For example, viral transmission after intratumoral injection may be weakened by the substantial barriers present at the tumor mass, such as the dense ECM network (14). The degradation of ECM by the use of relaxin or specific enzymes, such as hyaluronidase, has been demonstrated to promote viral spread among tumor tissues (15, 16). In addition, elevated interstitial hydrostatic pressure caused by fibrosis and vascular abnormalities in tumors have been reported to provide another barrier to oncolytic virotherapy (17). There are a few related reviews that have elaborated on the challenges of tumor microenvironment to OVs and have proposed reasoned solutions (18). On the other hand, OVs serve as powerful immunological stimuli and possess the ability of remodeling of the TIME (Figure 1), which is also the focus of this opinion article. OVs influence the entire immunological process via multiple mechanisms and promote the recruitment and activation of immune cells. Thus, the capability of OVs to “hot” TIME can enhance the sensitivity of tumors to immunotherapy (19).
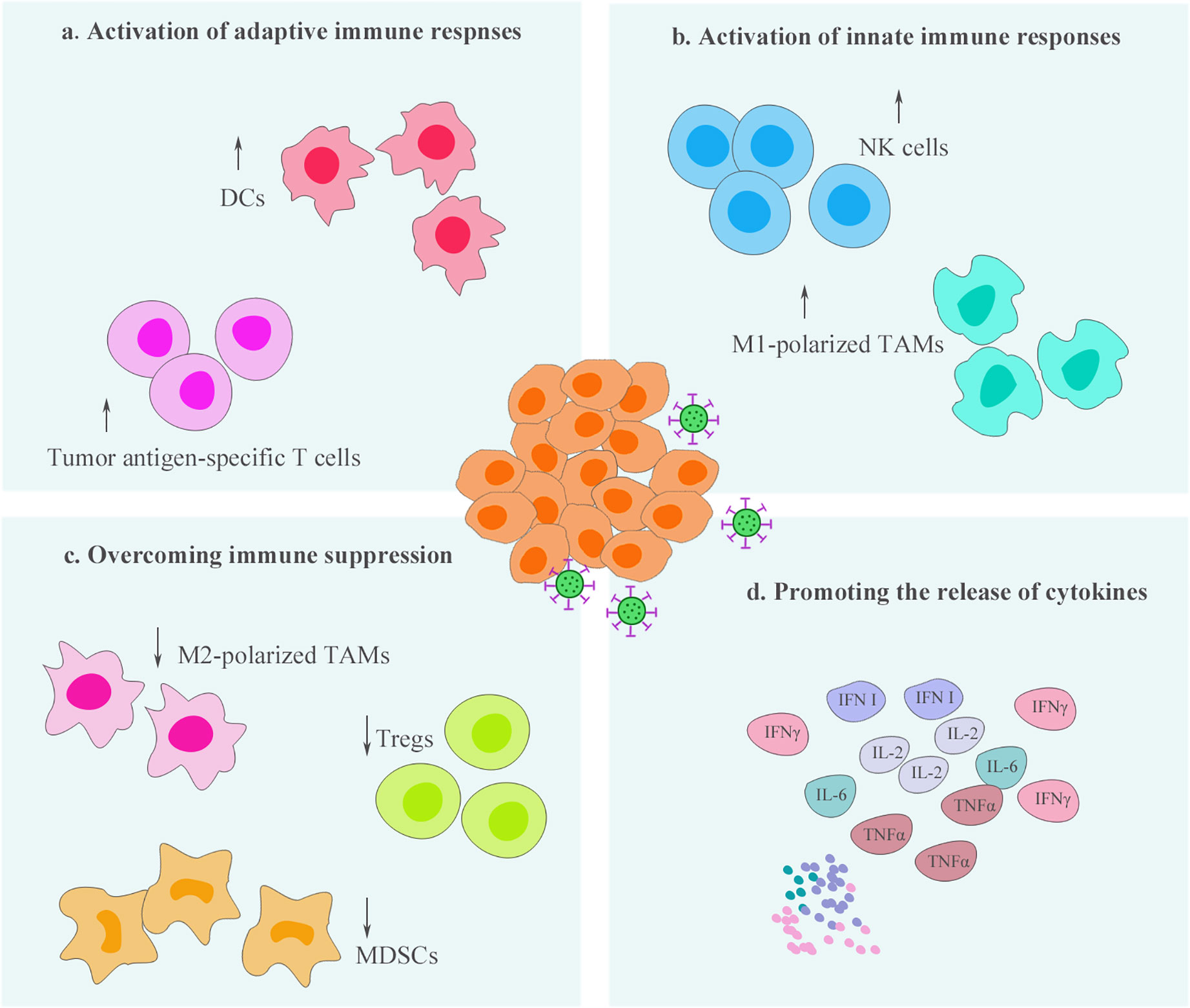
Figure 1 Oncolytic virotherapy process the ability to remodel the tumor immune microenvironment (TIME). Oncolytic viruses (OV) infection can enhance the infiltration and activity of immune cells, including innate and adaptive immune cells, within the TIME (A–B). At the same time, these therapeutic viruses reduce the populations of immunosuppressive cells and promote the immunophenotypes of immune cells shift toward antitumor status, thereby, overcoming immune suppression within the TIME (C). In addition, activation of antitumor immunity by oncolytic virotherapy is often accompanied by the production of a variety of proinflammatory cytokines, which is beneficial to further “hot” the TIME (D).
DCs are well-known antigen-presenting cells (APCs) that are responsible for processing and presenting antigens to the effector cells in the context of the major histocompatibility (MHCs). They play an important role in the initiation of innate and adaptive immune responses, particularly, matured DCs provide T cells co-stimulatory signals and are essential for T cell priming (20). However, it is often inadequate to induce potent antitumor immune responses in a tumor mass due to the immunosuppressive TIME. Currently, several researchers have proposed anticancer agents that target of DCs, such as the DC vaccines aiming at activation and accumulation of functional DCs (21). Alternatively, activating DCs via oncolytic virotherapy can unleash T cell responses, which is also a potential therapeutic strategy. After OV infection, the innate immune system rapidly recognizes virus particles and promotes DC maturation. Although viral infection initially induces a virus-targeting immune response, DCs process the ability to cross-present tumor antigens to cytotoxic T lymphocytes, thereby initiating a tumor-specific immune response (22).
Oncolysis induced by a recombinant poliovirus-rhinovirus chimera exposed the tumor antigens, while DCs co-cultured with the supernatant from the chimera-infected tumor cells exhibited increased expression of type I IFNs, CD40, CD80, and CD83, suggesting that this virus promotes APC maturation (23). A high proportion of splenic CD11c+CD8+DCs was detected in a mouse model treated with an engineered adenovirus, moreover, tumor-infiltrating plasmacytoid DCs displayed a mature phenotype with the ability to prime tumor-specific cytotoxic T cell responses (24). In addition, several studies have demonstrated that other OVs, such as vaccinia virus (25), measles virus (26), and HSV (27), can also enhance the antigen presentation ability of DCs, which is often accompanied by an increased expression of costimulatory/activation molecules, such as CD80, CD86, and MHC II. OVs assist DCs in its functions in two main ways. First, OVs promote DCs to recognize the tumor antigens by upregulating antigen presentation pathways in tumors. It is already well-established that, to evade immunosurveillance, tumor cells upregulate immune-inhibitory surface receptors and downregulate functional molecules related to antigen processing and presentation (28, 29). In this setting, OVs can overcome some evasion strategies. For example, an ovarian cancer cell line exhibited higher expression of MHC class I and other molecules involved in antigen processing, such as the transporter associated with antigen processing (TAP) and β2-microglobulin (β2M) when exposed to an oncolytic reovirus. This effect promoted DC maturation, which resulted in CD8+ T cell-mediated adaptive immune responses (30). The second one is that OVs facilitate the penetration of proinflammatory cytokines in the TIME, thereby creating a favorable environment for DC activation.
T cells are highly heterogeneous cell populations with different immunophenotypes and play an indispensable role in adaptive cancer immunity. The main T-cell subsets in the TIME consist of regulatory T cells (Treg), helper T cells (Th), cytotoxic T cells, exhausted T cells, and anergic T cells. These T cell subsets are involved in different immune functions, such as immune effects mediated by cytotoxic T cells or immune suppression mediated by Treg cells (31, 32). T cells can only respond to cancer cells effectually when multiple factors are coordinated. They need to be first primed and activated and then trafficked to for infiltration in the tumors while circumventing the immunosuppressive cells and other inhibitory factors in the TIME (33). However, T cells often dysfunction in cancer (34). OVs that serve as immune-modulating platforms can help overcome barriers and strengthen the T cell-mediated antitumor immunity.
Naïve T cells are primed by T cell receptor (TCR)-mediated recognition of antigenic peptides presented in the context of MHC complex. Antigens of sufficient magnitude play a crucial role in the process of T cell priming. OVs can function as in situ vaccines, and, in the appropriate setting, tumors directly serve as an in situ source of neoantigen vaccination. Indeed, local OV infection causes direct lysis of tumor cells together with the release of TAAs. For example, an oncolytic adenovirus has been shown to broaden the cancer-specific neoantigen repertoires and enhance the cytotoxic T cell responses to cancer in a murine model of liver cancer (35). In addition, arming OVs with a tumor antigen can further potentiate the T cell immunity. The heterologous prime-boost regimen involving different OVs encoding for the same TAA can help direct cytotoxic T lymphocytes toward cancer-specific new epitopes and away from the virus antigens. This approach has proven successful. For instance, it has been demonstrated that an enhanced tumor antigen-specific T cell response, following priming with an adenovirus encoding for tumor antigen human dopachrome tautomerase (hDCT), in combination with the utilization of a Maraba virus encoding for the same antigen, acted as a booster dose (36).
Differentiated effector T cells possess the ability for circulation and trafficking. Following recruitment by chemokines, the effector T lymphocytes migrate to the established tumor site in order to induce tumor cell killing. OVs can increase infiltration of the T cells in a variety of ways. Local viral infection causes type I IFN response followed by release of T cell-recruiting cytokines and chemokines in the tumor microenvironment. It is well-established that, after oncolytic virotherapy, the immunophenotype of the tumor can switch from the so-called “cold” state to the “hot” state, thereby allowing cytotoxic T cells to infiltrate in the TIME and perform tumor cell killing (37). More importantly, OVs possess the capacity to enhance T cell activation. Indeed, in a past study, the authors showed that although rotavirus vaccines did not significantly increase the overall frequency of T cells in the TIME, they resulted in a highly significant proportion of T cells with increased expression of the activation markers OX40 and CD137 (38).OVs stimulate the secretion of inflammatory mediators such as interleukin-1β, tumor necrosis factor (TNF), and complement components, causing an increase in the expression of selectin on endothelial cells, which acts as the key signal for T cell infiltration. After infiltrating in the TIME, T cells continue to face interference from immunosuppressive cells and other inhibitory factors. OVs can induce the conversion of immunosuppressive cells to proinflammatory phenotypes and promote the development of T cell responses both in vitro and in vivo (23) Furthermore, exploiting engineered OVs to mediate the direct engagement between T cells and tumor cells, such as OVs armed with a bispecific T cell engager (BiTE), a bispecific affinity reagent that binds to CD3 (or other T cell activators) and target antigens on cancer cells is also a potential strategy. Freedman and colleagues demonstrated that an oncolytic adenovirus expressing EpCAM targeting BiTE led to the activation of both CD4- and CD8-positive T cells to destroy tumor cells (39). Moreover, OVs can be used in conjunction with chimeric antigen receptor-modified T cell therapy (CAR-T). Wing and colleagues have explored the combination of CAR T cells targeting folate receptor alpha (FR-α) with oncolytic adenovirus expressing BiTE targeting EGFR. In the study, they demonstrated that oncolytic adenoviruses carrying BiTE could enhance the CAR T cell activation and proliferation and thereby promoted the re-targeting of CAR-T cells in the absence of FR-α as well as improving the tumor killing by CAR T cells (40).
NK cells, which are one of the key components of the innate immune system, are the first line of defense against cancer and other heterologous pathogenic infection (41). Unlike for T cells, the activation of NK cells did not require TCRs, rather it relied on the balance among the activating, co-stimulatory and inhibitory receptors (41). NK cells possess a powerful cytolytic activity. Activated NK cells exert neoplasm killing in a variety of ways, including via induction of apoptosis (42) and via direct cytolysis by the release of perforin and granzymes (43), Moreover, previous findings have shown that NK cells exhibit intrinsic memory-like properties and possess the ability to undergo rapid clonal expansion in response to a re-challenge (44).
Some preclinical and clinical studies have demonstrated the existence of a cross-talk between OVs and NK cells in cancer immunotherapy. On one hand, NK cells are important immune effectors in the context of oncolytic virotherapy. For instance, a study involving oncolytic NDV combined with immune checkpoint inhibitors demonstrated that the depletion of NK cells could significantly limit their therapeutic effects, which in turn suggests that NK cells are necessary for the regimen including OVs (45). On the other hand, OVs can enhance the proliferation and activity of NK cells. Oncolytic reovirus can promote the anti-tumor activity of NK cells by activating DC in vitro (46). Another study conducted in immune-competent mouse models of melanoma found that oncolytic VSV induced the secretion of IL-28 within the tumor microenvironment, which resulted in the promotion of NK cell activation in vivo and sensitized tumors to the NK cell-recognition and killing (47). OVs stimulate NK cell-mediated immune responses via pattern recognition receptors (PRRs), a class of molecules that are expressed by innate immune cells responsible for sensing heterologous substances. For example, surface toll-like receptor 2 (TLR2), one of the PRRs, mediate NK cell responses stimulated by oncolytic HSV (48). In addition to remodeling the natural occurring NK cells within the TIME, OVs can also promote the homing of NK cells to the tumor site. This homing property correlates with that of the NK cell-recruiting cytokines. For example, it has been demonstrated that oncolytic parvovirus can facilitate the recruitment of NK cells by expressing cytokines IL-2 and MCP-3/CCL7 in a pancreatic ductal adenocarcinoma model (49). Similarly, a combination of the oncolytic adenovirus encoding IL-12 and TRAIL, respectively, was also demonstrated to play an important role in increasing the infiltration of NK cells into the tumors (50). Moreover, OVs possess the ability to potentiate NK cells’ adoptive transfer therapy (51).
TAMs represent the crucial elements involved in tumorigenesis that are characterized by the inhibition of antitumor immunity and the promotion of tumor progress (52). The enrichment of TAMs in the tumor site is linked to the poor prognosis and the short survival time in most tumor types (53–55). TAMs produce high amount of immunosuppressive and proangiogenic factors such as IL-10, arginase, transforming growth factor β (TGFβ), or vascular endothelial growth factors (VEGFs); factors such as these enable the tumor-promoting functions of TAMs (56–58). Macrophages exhibit distinct polarization status in response to different stress factors. It is commonly considered that M1 macrophages are proinflammatory and cytotoxic, whereas M2 macrophages are immunosuppressive. Therefore, agents that can induce polarization of TAMs toward M1 type together with the expression of proinflammatory cytokines might be conducive to cancer therapy.
OVs serve as powerful immunological stimuli and are beneficial to shift the phenotype functions of macrophages, as demonstrated by oncolytic paramyxovirus infection of macrophages (59). In this study, virus-treated co-culture conditions induced an anti-tumor phenotype in macrophage in vitro accompanied by a higher level of immunostimulatory surface markers and cytokines (59). The mechanisms of how OV infection induce the phenotypic changes of macrophages remain uncertain, but it may be related to virus-induced cytokine release. Indeed, Brown and colleagues investigated the effect of recombinant poliovirus (PVSRIPO) on macrophage activity in vitro and found that PVSRIPO infection activated immunosuppressed macrophages in a type I IFN-dominant fashion (23). This OV-mediated remodeling effect is analogously pronounced in solid tumor, wherein OVs create an inflamed milieu that promotes the recruitment and activation of macrophages. For example, it has been reported that the treatment with oncolytic vaccinia virus GLV-1h68 elicited a significant upregulation of proinflammatory cytokines such as IL-3, IL- 6, IFN-γ, and CXCL10, as well as enhancing the infiltration of proinflammatory macrophages to the tumor site in a xenograft colorectal cancer model (60). Similarly, a triple combination therapy including oncolytic HSV increased macrophage infiltration and M1-like polarization, which contributed to glioblastoma eradication (61). Notably, in some cases, OVs may not significantly reduce the amounts of TAMs within the tumor site, instead they remodel TAMs mainly by converting the status of immunosuppressed polarization.
Myeloid cells are a highly heterologous cell population and can differentiate into MDSCs in response to pathologically persistent stimulation such as chronic infection or inflammation associated with the disease (62). MDSCs are mainly enriched in tumor tissues and other pathological sites and not present in the healthy tissues. Accumulating evidence supports that MDSCs in cancer have emerged as the key contributors to tumor growth and metastasis (63). MDSCs possess the properties of immune suppression. They suppress important immunological processes, particularly in T cell-mediated antitumor responses, by the production of inhibitory factors such as TGFβ, indoleamine 2,3 dioxygenase (IDO), and COX2 (64–66). In addition to intrinsic immune-inhibitory characteristics, MDSCs promote tumor angiogenesis via the secretion of diverse growth factors (67). It is now clear that MDSCs reduce the efficacy of cancer immunotherapy; therefore, therapeutics tailored for these cells represent potential therapeutic opportunities.
Previous reports have determined prostaglandin E2 (PGE2) as a critical mediator for MDSCs infiltration by the CXCL-12-CXCR4 pathway (68). In this regard, targeting of PGE2 can help overcome immunosuppression associated with MDSCs, as in the cases of the expression of PGE2 inactivating enzyme 15-hydroxyprostaglandin dehydrogenase (HPGD) by oncolytic vaccinia virus (69). In this study, the engineered OV selectively depleted the MDSCs in the tumor, and, at the same time, the reduction of MDSC populations increased the sensitivity of resistant tumor to oncolytic virotherapy. In addition to serving as a vector for delivery of therapeutic genes specific for MDSCs, OVs per se also possess the capacity for remodeling the frequency and activity of MDSCs. For example, a CpG-rich oncolytic adenovirus can reduce the inhibition of MDSCs by enhancing the TLR9 stimulation in a syngeneic mouse model of melanoma (70). The modulation of MDSCs by OVs may correlate with PRRs in MDSCs. Apart from the abovementioned TLR9 receptor, other OVs limit MDSCs by acting on different PRRs and the relevant signaling molecules, such as in the inhibition of MDSCs by oncolytic reovirus in a TLR3-dependent manner (71) or by oncolytic VSV in a MyD88 signaling-dependent manner (72). MDSCs have intrinsic tumor tropism, which is advantageous, allowing their exploitation as a vehicle for tumor-specific OVs. Eisenstein and colleagues have generated a recombinant oncolytic VSV loaded into MDSCs and found that the MDSCs provided a protective role for the systemic delivery of OVs (73). Importantly, the OVs induced the phenotype of MDSCs switch from the protumor M2 type to antitumor M1 type as a result of virus-mediated inflammatory response; thus, the combination increased the tumor killing as well as the therapeutic index (73).
Tumorigenesis is often aided by the shaping of the TIME by cytokines. Several reports have demonstrated that the oncogene-driven expression of cytokines and/or chemokines is associated with an increase in the number of immunosuppressive cells, such as Gr-1+CD11b+ myeloid cells (74), as well as a decrease in the frequency of immune effector cells such as CD103+ DCs (75). Multifunctional cytokines in the tumor microenvironment are not only involved in tumor progress, but they also act as pivotal mediators for the antitumor responses. Oncolytic virotherapy induce immunogenic cell death (ICD) of cancerous cells together with abound release of danger-associated molecular patterns (DAMPs), such as ATP, nuclear high mobility group box 1 (HMBG1), and calreticulin (76–78). These proinflammatory substances exhibit the intrinsic properties of immune-stimulating, which is beneficial to the recruitment and activation of immune effector cells to the tumor site. Moreover, viral particles are detected by PRRs on the surface and/or cytoplasm of innate immune cells, which culminates into intracellular immune responses against viruses in a type I IFN-dominant fashion (79). This innate antiviral machinery induces the generation of an inflamed TIME by driving the expression of IFN-inducible immunomodulatory cytokines (26). In addition, OVs can serve as a platform for the expression of various cytokines. For example, the first FDA-approved OV agent derived from oncolytic HSV-1 was genetically modified to encode for GM-CSF (80). However, it remains to be understood how OV-induced production of various cytokines by different cell compositions can be coordinated to determine the immune landscapes of the TIME.
Tumor cells can evade immune surveillance by virtue of immune checkpoint molecules that link to ligands expressed on the immune cells. The typical checkpoint molecules consist of programmed cell death protein 1 (PD-1) and its ligand (PD-L1), cytotoxic T lymphocyte- associated protein 4 (CTLA-4), and lymphocyte activation gene 3 (LAG3). In fact, the immune checkpoint blockade can reverse the immune effector cell anergy by targeting the inhibitory signaling pathways and have been investigated with success in multiple tumor types (81–83). However, tumors with low mutation burden often correlates with the resistance of immune checkpoint blockade due to the dearth of sufficient antigen recognition by T lymphocytes. Oncolytic virotherapy can increase the expression of checkpoint molecules within the tumor site. For example, oncolytic Maraba virus induce the upregulation of PD-L1 in ovarian cancers (84) and oncolytic NDV induce the upregulation of CTLA-4 in melanoma (45). A past study demonstrated OV-mediated production of type I IFN as a key mediator for the upregulation of immune checkpoint molecules (85). Moreover, following OV infection, adaptive immune resistance is also involved in the increase of checkpoint molecules, which may correlate with the compensatory immunosuppressive pathways (86).
Accumulating evidence support the potential of OVs as a promising therapeutic agent. The potent antitumor activity of OVs can be attributed to their unique mechanisms of action, including direct oncolysis and the induction of immune responses. OVs remodel the immune landscape of the TIME by regulating immune cells and the relevant cytokines within the tumor microenvironment. These therapeutic viruses possess the ability to recruit and activate immune effector cells, particularly CD8+ T lymphocytes. Simultaneously, they reduce the amounts of immunosuppressive cells and alternatively induce the phenotype of the cells that shift from the protumor status to the antitumor status. OV-induced increased infiltration of immune effector cells is often accompanied by abound secretion of proinflammatory cytokines. It is now evident that oncolytic virotherapy can create an inflamed TIME, or a “hot” tumor, which is expected to yield a superior efficacy in combination with cancer immunotherapy. Further development of oncology and virotherapy is expected to provide a deeper understanding of the interaction between OVs and the TIME in the future.
XLW retrieved the relevant paper and finished the original draft. BZ modified the original draft and added substantial contents. PC provided constructive guidance and critical advice on the framework this review. All authors contributed to the article and approved the submitted version.
This work was supported by the National Science and Technology Major Projects of New Drugs (2018ZX09201018-013), the National Science and Technology Major Project for Infectious Diseases Control (2017ZX10203206-004) and the National Natural Science Foundation of China (81101728).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov (2016) 15(9):660. doi: 10.1038/nrd.2016.178
2. Ott PA, Hodi FS. Talimogene Laherparepvec for the Treatment of Advanced Melanoma. Clin Cancer Res (2016) 22(13):3127–31. doi: 10.1158/1078-0432.CCR-15-2709
3. Pikor LA, Bell JC, Diallo JS. Oncolytic Viruses: Exploiting Cancer’s Deal with the Devil. Trends Cancer (2015) 1(4):266–77. doi: 10.1016/j.trecan.2015.10.004
4. Raja J, Ludwig JM, Gettinger SN, Schalper KA, Kim HS. Oncolytic virus immunotherapy: future prospects for oncology. J Immunother Cancer (2018) 6(1):140. doi: 10.1186/s40425-018-0458-z
5. Altomonte J, Marozin S, Schmid RM, Ebert O. Engineered newcastle disease virus as an improved oncolytic agent against hepatocellular carcinoma. Mol Ther: J Am Soc Gene Ther (2010) 18(2):275–84. doi: 10.1038/mt.2009.231
6. Kennedy BE, Murphy JP, Clements DR, Konda P, Holay N, Kim Y, et al. Inhibition of Pyruvate Dehydrogenase Kinase Enhances the Antitumor Efficacy of Oncolytic Reovirus. Cancer Res (2019) 79(15):3824–36. doi: 10.1158/0008-5472.CAN-18-2414
7. Villanueva E, Navarro P, Rovira-Rigau M, Sibilio A, Méndez R, Fillat C. Translational reprogramming in tumour cells can generate oncoselectivity in viral therapies. Nat Commun (2017) 8:14833. doi: 10.1038/ncomms14833
8. Kimpel J, Urbiola C, Koske I, Tober R, Banki Z, Wollmann G, et al. The Oncolytic Virus VSV-GP Is Effective against Malignant Melanoma. Viruses (2018) 10(3):108. doi: 10.3390/v10030108
9. Belli C, Trapani D, Viale G, D’Amico P, Duso BA, Della Vigna P, et al. Targeting the microenvironment in solid tumors. Cancer Treat Rev (2018) 65:22–32. doi: 10.1016/j.ctrv.2018.02.004
10. Pinchuk IV, Saada JI, Beswick EJ, Boya G, Qiu SM, Mifflin RC, et al. PD-1 ligand expression by human colonic myofibroblasts/fibroblasts regulates CD4+ T-cell activity. Gastroenterology (2008) 135(4):1228–37, 37.e1-2. doi: 10.1053/j.gastro.2008.07.016
11. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature (2017) 541(7637):321–30. doi: 10.1038/nature21349
12. Lee HJ, Park IA, Song IH, Shin SJ, Kim JY, Yu JH, et al. Tertiary lymphoid structures: prognostic significance and relationship with tumour-infiltrating lymphocytes in triple-negative breast cancer. J Clin Pathol (2016) 69(5):422–30. doi: 10.1136/jclinpath-2015-203089
13. Beatty GL, Winograd R, Evans RA, Long KB, Luque SL, Lee JW, et al. Exclusion of T Cells From Pancreatic Carcinomas in Mice Is Regulated by Ly6C(low) F4/80(+) Extratumoral Macrophages. Gastroenterology (2015) 149(1):201–10. doi: 10.1053/j.gastro.2015.04.010
14. Harrison D, Sauthoff H, Heitner S, Jagirdar J, Rom WN, Hay JG. Wild-type adenovirus decreases tumor xenograft growth, but despite viral persistence complete tumor responses are rarely achieved–deletion of the viral E1b-19-kD gene increases the viral oncolytic effect. Hum Gene Ther (2001) 12(10):1323–32. doi: 10.1089/104303401750270977
15. Lee WJ, Choi IK, Lee JH, Lee JS, Kim YO, Rah DK, et al. Relaxin-expressing adenovirus decreases collagen synthesis and up-regulates matrix metalloproteinase expression in keloid fibroblasts: in vitro experiments. Plast Reconstr Surg (2012) 130(3):407e–17e. doi: 10.1097/PRS.0b013e31825dbf56
16. Martinez-Quintanilla J, He D, Wakimoto H, Alemany R, Shah K. Encapsulated stem cells loaded with hyaluronidase-expressing oncolytic virus for brain tumor therapy. Mol Ther: J Am Soc Gene Ther (2015) 23(1):108–18. doi: 10.1038/mt.2014.204
17. Heldin CH, Rubin K, Pietras K, Ostman A. High interstitial fluid pressure - an obstacle in cancer therapy. Nat Rev Cancer (2004) 4(10):806–13. doi: 10.1038/nrc1456
18. Zheng M, Huang J, Tong A, Yang H. Oncolytic viruses for cancer therapy: barriers and recent advances. Mol Ther-Oncolytics (2019) 15:234–47. doi: 10.1016/j.omto.2019.10.007
19. Bommareddy PK, Shettigar M, Kaufman HL. Integrating oncolytic viruses in combination cancer immunotherapy. Nat Rev Immunol (2018) 18(8):498–513. doi: 10.1038/s41577-018-0014-6
20. Hansen M, Andersen MH. The role of dendritic cells in cancer. Semin Immunopathol (2017) 39(3):307–16. doi: 10.1007/s00281-016-0592-y
21. Dastmalchi F, Karachi A, Yang C, Azari H, Sayour EJ, Dechkovskaia A, et al. Sarcosine promotes trafficking of dendritic cells and improves efficacy of anti-tumor dendritic cell vaccines via CXC chemokine family signaling. J Immunother Cancer (2019) 7(1):321. doi: 10.1186/s40425-019-0809-4
22. Gasteiger G, Kastenmuller W, Ljapoci R, Sutter G, Drexler I. Cross-priming of cytotoxic T cells dictates antigen requisites for modified vaccinia virus Ankara vector Vaccines. J Virol (2007) 81(21):11925–36. doi: 10.1128/JVI.00903-07
23. Brown MC, Holl EK, Boczkowski D, Dobrikova E, Mosaheb M, Chandramohan V, et al. Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen-specific CTLs. Sci Trans Med (2017) 9(408):eaan4220. doi: 10.1126/scitranslmed.aan4220
24. Lapteva N, Aldrich M, Rollins L, Ren W, Goltsova T, Chen S-Y, et al. Attraction and activation of dendritic cells at the site of tumor elicits potent antitumor immunity. Mol Ther (2009) 17(9):1626–36. doi: 10.1038/mt.2009.111
25. Greiner S, Humrich JY, Thuman P, Sauter B, Schuler G, Jenne L. The highly attenuated vaccinia virus strain modified virus Ankara induces apoptosis in melanoma cells and allows bystander dendritic cells to generate a potent anti-tumoral immunity. Clin Exp Immunol (2006) 146(2):344–53. doi: 10.1111/j.1365-2249.2006.03177.x
26. Guillerme JB, Boisgerault N, Roulois D, Ménager J, Combredet C, Tangy F, et al. Measles virus vaccine-infected tumor cells induce tumor antigen cross-presentation by human plasmacytoid dendritic cells. Clin Cancer Res (2013) 19(5):1147–58. doi: 10.1158/1078-0432.CCR-12-2733
27. Benencia F, Courrèges MC, Fraser NW, Coukos G. Herpes virus oncolytic therapy reverses tumor immune dysfunction and facilitates tumor antigen presentation. Cancer Biol Ther (2008) 7(8):1194–205. doi: 10.4161/cbt.7.8.6216
28. Rotte A, Jin JY, Lemaire V. Mechanistic overview of immune checkpoints to support the rational design of their combinations in cancer immunotherapy. Ann Oncol (2018) 29(1):71–83. doi: 10.1093/annonc/mdx686
29. Cai L, Michelakos T, Yamada T, Fan S, Wang X, Schwab JH, et al. Defective HLA class I antigen processing machinery in cancer. Cancer Immunol Immunother (2018) 67(6):999–1009. doi: 10.1007/s00262-018-2131-2
30. Gujar S, Dielschneider R, Clements D, Helson E, Shmulevitz M, Marcato P, et al. Multifaceted therapeutic targeting of ovarian peritoneal carcinomatosis through virus-induced immunomodulation. Mol Ther (2013) 21(2):338–47. doi: 10.1038/mt.2012.228
31. Basu R, Whitlock BM, Husson J, Le Floc’h A, Jin W, Oyler-Yaniv A, et al. Cytotoxic T Cells Use Mechanical Force to Potentiate Target Cell Killing. Cell (2016) 165(1):100–10. doi: 10.1016/j.cell.2016.01.021
32. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res (2017) 27(1):109–18. doi: 10.1038/cr.2016.151
33. Dugnani E, Pasquale V, Bordignon C, Canu A, Piemonti L, Monti P. Integrating T cell metabolism in cancer immunotherapy. Cancer Lett (2017) 411:12–8. doi: 10.1016/j.canlet.2017.09.039
34. Crespo J, Sun H, Welling TH, Tian Z, Zou W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol (2013) 25(2):214–21. doi: 10.1016/j.coi.2012.12.003
35. Woller N, Guerlevik E, Fleischmann-Mundt B, Schumacher A, Knocke S, Kloos AM, et al. Viral infection of tumors overcomes resistance to PD-1-immunotherapy by broadening neoantigenome-directed T-cell responses. Mol Ther (2015) 23(10):1630–40. doi: 10.1038/mt.2015.115
36. Pol JG, Zhang L, Bridle BW, Stephenson KB, Resseguier J, Hanson S, et al. Maraba virus as a potent oncolytic vaccine vector. Mol Ther (2014) 22(2):420–9. doi: 10.1038/mt.2013.249
37. Yang Y, Xu W, Peng D, Wang H, Zhang X, Wang H, et al. An oncolytic adenovirus targeting transforming growth factor β inhibits protumorigenic signals and produces immune activation: a novel approach to enhance anti-PD-1 and anti-CTLA-4 therapy. Hum Gene Ther (2019) 30(9):1117–32. doi: 10.1089/hum.2019.059
38. Shekarian T, Sivado E, Jallas AC, Depil S, Kielbassa J, Janoueix-Lerosey I, et al. Repurposing rotavirus vaccines for intratumoral immunotherapy can overcome resistance to immune checkpoint blockade. Sci Trans Med (2019) 11(515):eaat5025. doi: 10.1126/scitranslmed.aat5025
39. Freedman JD, Hagel J, Scott EM, Psallidas I, Gupta A, Spiers L, et al. Oncolytic adenovirus expressing bispecific antibody targets T-cell cytotoxicity in cancer biopsies. EMBO Mol Med (2017) 9(8):1067–87. doi: 10.15252/emmm.201707567
40. Wing A, Fajardo CA, Posey AD Jr., Shaw C, Da T, Young RM, et al. Improving CART-cell therapy of solid tumors with oncolytic virus-driven production of a bispecific T-cell engager. Cancer Immunol Res (2018) 6(5):605–16. doi: 10.1158/2326-6066.CIR-17-0314
41. Shimasaki N, Jain A, Campana D. NK cells for cancer immunotherapy. Nat Rev Drug Discov (2020) 19(3):200–18. doi: 10.1038/s41573-019-0052-1
42. Takeda K, Hayakawa Y, Smyth MJ, Kayagaki N, Yamaguchi N, Kakuta S, et al. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat Med (2001) 7(1):94–100. doi: 10.1038/83416
43. Kim N, Kim M, Yun S, Doh J, Greenberg PD, Kim TD, et al. MicroRNA-150 regulates the cytotoxicity of natural killers by targeting perforin-1. J Allergy Clin Immunol (2014) 134(1):195–203. doi: 10.1016/j.jaci.2014.02.018
44. Cooper MA, Elliott JM, Keyel PA, Yang L, Carrero JA, Yokoyama WM. Cytokine-induced memory-like natural killer cells. Proc Natl Acad Sci U S A (2009) 106(6):1915–9. doi: 10.1073/pnas.0813192106
45. Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Trans Med (2014) 6(226):12. doi: 10.1126/scitranslmed.3008095
46. Errington F, Steele L, Prestwich R, Harrington KJ, Pandha HS, Vidal L, et al. Reovirus activates human dendritic cells to promote innate antitumor immunity. J Immunol (2008) 180(9):6018–26. doi: 10.4049/jimmunol.180.9.6018
47. Wongthida P, Diaz RM, Galivo F, Kottke T, Thompson J, Pulido J, et al. Type III IFN interleukin-28 mediates the antitumor efficacy of oncolytic virus VSV in immune-competent mouse models of cancer. Cancer Res (2010) 70(11):4539–49. doi: 10.1158/0008-5472.CAN-09-4658
48. Kim M, Osborne NR, Zeng W, Donaghy H, McKinnon K, Jackson DC, et al. Herpes simplex virus antigens directly activate NK cells via TLR2, thus facilitating their presentation to CD4 T lymphocytes. J Immunol (2012) 188(9):4158–70. doi: 10.4049/jimmunol.1103450
49. Dempe S, Lavie M, Struyf S, Bhat R, Verbeke H, Paschek S, et al. Antitumoral activity of parvovirus-mediated IL-2 and MCP-3/CCL7 delivery into human pancreatic cancer: implication of leucocyte recruitment. Cancer Immunol Immunother CII (2012) 61(11):2113–23. doi: 10.1007/s00262-012-1279-4
50. El-Shemi AG, Ashshi AM, Na Y, Li Y, Basalamah M, Al-Allaf FA, et al. Combined therapy with oncolytic adenoviruses encoding TRAIL and IL-12 genes markedly suppressed human hepatocellular carcinoma both in vitro and in an orthotopic transplanted mouse model. J Exp Clin Cancer Res CR (2016) 35:74. doi: 10.1186/s13046-016-0353-8
51. Yoo JY, Jaime-Ramirez AC, Bolyard C, Dai H, Nallanagulagari T, Wojton J, et al. Bortezomib Treatment Sensitizes Oncolytic HSV-1-Treated Tumors to NK Cell Immunotherapy. Clin Cancer Res (2016) 22(21):5265–76. doi: 10.1158/1078-0432.CCR-16-1003
52. Ostuni R, Kratochvill F, Murray PJ, Natoli G. Macrophages and cancer: from mechanisms to therapeutic implications. Trends Immunol (2015) 36(4):229–39. doi: 10.1016/j.it.2015.02.004
53. Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med (2010) 362(10):875–85. doi: 10.1056/NEJMoa0905680
54. Tiainen S, Tumelius R, Rilla K, Hämäläinen K, Tammi M, Tammi R, et al. High numbers of macrophages, especially M2-like (CD163-positive), correlate with hyaluronan accumulation and poor outcome in breast cancer. Histopathology (2015) 66(6):873–83. doi: 10.1111/his.12607
55. Hu Y, He MY, Zhu LF, Yang CC, Zhou ML, Wang Q, et al. Tumor-associated macrophages correlate with the clinicopathological features and poor outcomes via inducing epithelial to mesenchymal transition in oral squamous cell carcinoma. J Exp Clin Cancer Res CR (2016) 35:12. doi: 10.1186/s13046-015-0281-z
56. Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res (2006) 66(23):11238–46. doi: 10.1158/0008-5472.CAN-06-1278
57. Biswas SK, Gangi L, Paul S, Schioppa T, Saccani A, Sironi M, et al. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-kappaB and enhanced IRF-3/STAT1 activation). Blood (2006) 107(5):2112–22. doi: 10.1182/blood-2005-01-0428
58. Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, DeNardo DG, et al. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res (2010) 70(19):7465–75. doi: 10.1158/0008-5472.CAN-10-1439
59. Tan DQ, Zhang L, Ohba K, Ye M, Ichiyama K, Yamamoto N. Macrophage response to oncolytic paramyxoviruses potentiates virus-mediated tumor cell killing. Eur J Immunol (2016) 46(4):919–28. doi: 10.1002/eji.201545915
60. Ehrig K, Kilinc MO, Chen NG, Stritzker J, Buckel L, Zhang Q, et al. Growth inhibition of different human colorectal cancer xenografts after a single intravenous injection of oncolytic vaccinia virus GLV-1h68. J Transl Med (2013) 11:79. doi: 10.1186/1479-5876-11-79
61. Saha D, Martuza RL, Rabkin SD. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell (2017) 32(2):253–67.e5. doi: 10.1016/j.ccell.2017.07.006
62. Gabrilovich DI. Myeloid-Derived Suppressor Cells. Cancer Immunol Res (2017) 5(1):3–8. doi: 10.1158/2326-6066.CIR-16-0297
63. Condamine T, Ramachandran I, Youn JI, Gabrilovich DI. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu Rev Med (2015) 66:97–110. doi: 10.1146/annurev-med-051013-052304
64. Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res (2006) 66(2):1123–31. doi: 10.1158/0008-5472.CAN-05-1299
65. Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, et al. Tumor-Expressed IDO Recruits and Activates MDSCs in a Treg-Dependent Manner. Cell Rep (2015) 13(2):412–24. doi: 10.1016/j.celrep.2015.08.077
66. Mao Y, Poschke I, Wennerberg E, Pico de Coaña Y, Egyhazi Brage S, Schultz I, et al. Melanoma-educated CD14+ cells acquire a myeloid-derived suppressor cell phenotype through COX-2-dependent mechanisms. Cancer Res (2013) 73(13):3877–87. doi: 10.1158/0008-5472.CAN-12-4115
67. Shojaei F, Wu X, Qu X, Kowanetz M, Yu L, Tan M, et al. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci U S A (2009) 106(16):6742–7. doi: 10.1073/pnas.0902280106
68. Obermajer N, Muthuswamy R, Odunsi K, Edwards RP, Kalinski P. PGE(2)-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Res (2011) 71(24):7463–70. doi: 10.1158/0008-5472.CAN-11-2449
69. Hou W, Sampath P, Rojas JJ, Thorne SH. Oncolytic Virus-Mediated Targeting of PGE2 in the Tumor Alters the Immune Status and Sensitizes Established and Resistant Tumors to Immunotherapy. Cancer Cell (2016) 30(1):108–19. doi: 10.1016/j.ccell.2016.05.012
70. Cerullo V, Diaconu I, Romano V, Hirvinen M, Ugolini M, Escutenaire S, et al. An oncolytic adenovirus enhanced for toll-like receptor 9 stimulation increases antitumor immune responses and tumor clearance. Mol Ther J Am Soc Gene Ther (2012) 20(11):2076–86. doi: 10.1038/mt.2012.137
71. Katayama Y, Tachibana M, Kurisu N, Oya Y, Terasawa Y, Goda H, et al. Oncolytic Reovirus Inhibits Immunosuppressive Activity of Myeloid-Derived Suppressor Cells in a TLR3-Dependent Manner. J Immunol (2018) 200(8):2987–99. doi: 10.4049/jimmunol.1700435
72. Wongthida P, Diaz RM, Galivo F, Kottke T, Thompson J, Melcher A, et al. VSV oncolytic virotherapy in the B16 model depends upon intact MyD88 signaling. Mol Ther J Am Soc Gene Ther (2011) 19(1):150–8. doi: 10.1038/mt.2010.225
73. Eisenstein S, Coakley BA, Briley-Saebo K, Ma G, Chen HM, Meseck M, et al. Myeloid-derived suppressor cells as a vehicle for tumor-specific oncolytic viral therapy. Cancer Res (2013) 73(16):5003–15. doi: 10.1158/0008-5472.CAN-12-1597
74. Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell (2012) 21(6):836–47. doi: 10.1016/j.ccr.2012.04.024
75. Spranger S, Dai D, Horton B, Gajewski TF. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell (2017) 31(5):711–23.e4. doi: 10.1016/j.ccell.2017.04.003
76. Koks CA, Garg AD, Ehrhardt M, Riva M, Vandenberk L, Boon L, et al. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int J Cancer (2015) 136(5):E313–E25. doi: 10.1002/ijc.29202
77. Donnelly OG, Errington-Mais F, Steele L, Hadac E, Jennings V, Scott K, et al. Measles virus causes immunogenic cell death in human melanoma. Gene Ther (2013) 20(1):7–15. doi: 10.1038/gt.2011.205
78. Miyamoto S, Inoue H, Nakamura T, Yamada M, Sakamoto C, Urata Y, et al. Coxsackievirus B3 is an oncolytic virus with immunostimulatory properties that is active against lung adenocarcinoma. Cancer Res (2012) 72(10):2609–21. doi: 10.1158/0008-5472.CAN-11-3185
79. Stetson DB, Medzhitov R. Type I interferons in host defense. Immunity (2006) 25(3):373–81. doi: 10.1016/j.immuni.2006.08.007
80. Andtbacka RHI, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J Clin Oncol (2015) 33(25):2780–U98. doi: 10.1200/JCO.2014.58.3377
81. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med (2015) 372(4):320–30. doi: 10.1056/NEJMoa1412082
82. Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WEE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med (2015) 373(2):123–35. doi: 10.1056/NEJMoa1504627
83. Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med (2015) 373(19):1803–13. doi: 10.1056/NEJMoa1510665
84. McGray AJR, Huang R-Y, Battaglia S, Eppolito C, Miliotto A, Stephenson KB, et al. Oncolytic Maraba virus armed with tumor antigen boosts vaccine priming and reveals diverse therapeutic response patterns when combined with checkpoint blockade in ovarian cancer. J Immunother Cancer (2019) 7(1):189–. doi: 10.1186/s40425-019-0641-x
85. Samson A, Scott KJ, Taggart D, West EJ, Wilson E, Nuovo GJ, et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci Trans Med (2018) 10(422):eaam7577. doi: 10.1186/s40425-019-0641-x
Keywords: oncolytic virus, tumor immune microenvironment, immune cells, cytokines, immune checkpoints
Citation: Zhang B, Wang XL and Cheng P (2021) Remodeling of Tumor Immune Microenvironment by Oncolytic Viruses. Front. Oncol. 10:561372. doi: 10.3389/fonc.2020.561372
Received: 12 May 2020; Accepted: 30 December 2020;
Published: 19 February 2021.
Edited by:
Nicola Giuliani, University of Parma, ItalyCopyright © 2021 Zhang, Wang and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ping Cheng, cGluZy5jaGVuZ0Bmb3htYWlsLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.