 Lehang Lin
Lehang Lin Xu Cheng
Xu Cheng- Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation, Medical Research Center, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China
Almost all cancer cells possess multiple epigenetic abnormalities, which cooperate with genetic alterations to enable the acquisition of cancer hallmarks during tumorigenesis. As the most frequently found epigenetic change in human cancers, aberrant DNA methylation manifests at two major forms: global genomic DNA hypomethylation and locus-specific promoter region hypermethylation. It has been recognized as a critical contributor to esophageal squamous cell carcinoma (ESCC) malignant transformation. In ESCC, DNA methylation alterations affect genes involved in cell cycle regulation, DNA damage repair, and cancer-related signaling pathways. Aberrant DNA methylation patterns occur not only in ESCC tumors but also in precursor lesions. It adds another layer of complexity to the ESCC heterogeneity and may serve as early diagnostic, prognostic, and chemo-sensitive markers. Characterization of the DNA methylome in ESCC could help better understand its pathogenesis and develop improved therapies. We herein summarize the current research and knowledge about DNA methylation in ESCC and its clinical significance in diagnosis, prognosis, and treatment.
Introduction
As the ninth most common cancer in the world, esophageal carcinoma is also the sixth leading cause of cancer-related deaths (1). Esophageal squamous cell carcinoma (ESCC) is the predominant subtype of esophageal carcinoma and accounts for ~90% of the cases (2). Although enormous progress has been made in early diagnosis and multimodal therapies, the prognosis of ESCC patients remains dismal, with the overall 5-year survival rate below 30% (2, 3). To achieve better clinical outcomes, a lot more work needs to be done to understand the pathogenesis of this disease thoroughly.
Intensive molecular biological studies have revealed that epigenetic dysregulation is a fundamental characteristic of nearly all human cancers (4). The most widely studied epigenetic modification is DNA methylation, which meanwhile is the most frequently found abnormal epigenetic change in human cancers. In mammals, DNA methylation occurs predominantly at the 5′ position of cytosine forming cytosine guanine dinucleotides. This modification is catalyzed and maintained by enzymes known as DNA methyltransferases (DNMTs) (5). The two major DNA methylation changes that occur in human cancers, including ESCC, are global DNA hypomethylation and site-specific CpG island promoter hypermethylation (6, 7). Experimental studies indicate that DNA hypomethylation of repetitive sequences (i.e., long interspersed nucleotide elements, LINEs) may predispose cells to chromosomal defects and rearrangements that result in genetic instability. Thus, global DNA hypomethylation increases chromosomal instability, leading to cancer development (8, 9). On the other hand, hypermethylation of the CpG islands located in gene promoter regions may involve in carcinogenesis as a result of three possible mechanisms: (1) cytosine methylation facilitates gene mutation, as 5-methylcytosine is deaminated to thymine; (2) aberrant DNA methylation may be associated with allelic loss; (3) tumor suppressor genes (TSGs) may be inactivated by promoter hypermethylation (7, 10–12) (Figure 1).
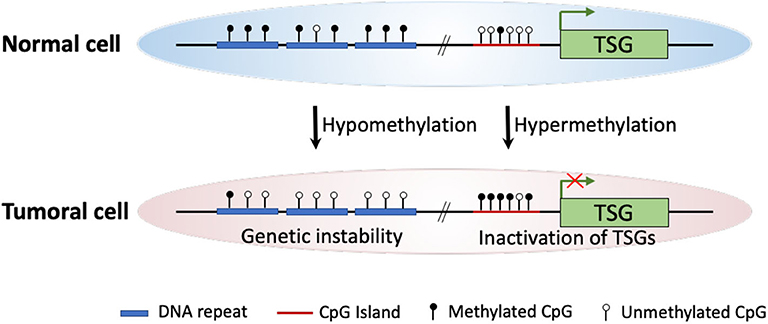
Figure 1. Schematic diagram of major DNA methylation changes in human cancer. In cancer cells, global hypomethylation at repetitive sequences might cause genetic instability, whereas site-specific CpG island promoter hypermethylation could lead to silencing of tumor suppressor genes (TSGs).
In the context of tumor epigenetic heterogeneity, DNA methylation has also been the focus of intense research due to the quantitative nature of DNA methylation assays and the relative ease of obtaining sufficient stable genomic DNA (13, 14). Mechanistically, changes in DNA methylation contribute to the cancer development process through regulation of the spatial chromatin organization and alteration of the transcriptome at the right timing and environment (15). To date, DNA methylation heterogeneity has been quantified and been linked to clinical variables in various cancer types, including prostate cancer (16), chronic lymphocytic leukemia (17), glioblastoma (18), Ewing sarcoma (19), as well as ESCC (20). These findings demonstrate the power of DNA methylation sequencing in analyzing intratumor heterogeneity. At the same time, they suggest the extensive involvement of DNA methylation in tumor development.
In this brief review, we summarize recent advances regarding DNA methylation changes in the initiation and progression of ESCC. We also refer to their possible applications in the clinical management of ESCC patients.
Global DNA Hypomethylation and Promoter CPG Island Hypermethylation in ESCC
Global DNA hypomethylation in cancer tissues was first observed more than 3 decades ago and has been recognized as a key mechanism involved in carcinogenesis (21, 22). Nevertheless, its presence and importance remain less well-understood in ESCC. To evaluate the global DNA methylation status in ESCC, several studies have measured the methylation level of the LINE-1. LINE-1 constitutes a substantial portion (~17%) of the human genome and has therefore attracted attention as a useful surrogate marker for global DNA methylation (23–27). Importantly, genome-wide hypomethylation was consistently observed in ESCCs, and LINE-1 hypomethylation was strongly correlated with ESCC progression, leading to a poor prognosis among ESCC patients (23–26). However, this association still awaits further confirmation in larger cohorts, and potential mechanisms by which global DNA hypomethylation affects ESCC behavior have yet to be clearly revealed.
Compared with global genomic DNA hypomethylation, locus-specific DNA hypermethylation, which mostly occurs at promoter CpG islands, has been extensively studied in ESCC by candidate gene approaches. In ESCC, as well as other malignancies, aberrant hypermethylation in promoter CpG islands is involved in the major components of cell cycle regulation, DNA damage repair, and cancer-related signaling pathways. For example, cell cycle-related genes CDKN2A, RASSF1A, and RASSF10 are frequently hypermethylated and transcriptionally silenced in ESCC (28–32). Inactivation of DNA repair genes MGMT, MLH1, and MSH2 in ESCC mainly attributes to the methylation change in their promoter regions (33–35). Promoter hypermethylation in the genes of APC, RUNX3, and ZNF382 respectively affects the Wnt/β-catenin, TGF-β, and NF-κB pathways in ESCC (36–40). Additionally, epigenetic studies on ESCC have discovered many known and putative TSGs that are frequently inactivated by promoter hypermethylation (Table 1). Although growing evidence has demonstrated that tobacco smoking and alcohol consumption are the two major risk factors for ESCC, and correlations between gene promoter methylation and smoking exposure have also been observed in ESCC, the exact triggers and mechanisms responsible for aberrant ESCC DNA methylation changes remain elusive (62–65).

Table 1. Aberrantly hypermethylated genes in ESCC.
DNA Methylation Change is a Critical Event in the Pathogenesis of ESCC
The histologic progression of ESCC is a multi-step process that begins from normal squamous epithelium to low-grade dysplasia, high-grade dysplasia, carcinoma in situ, and invasive carcinoma (66, 67). In addition to genetic alterations, epigenetic abnormalities, in particular DNA methylation changes, have become widely accepted as common molecular features during ESCC development (68–70).
There is growing recognition that aberrant DNA methylation patterns occur in the earliest stage of ESCC and may parallel the histologic changes observed during ESCC carcinogenesis. For example, promoter methylation of TSGs, including p16, p14, FHIT, MGMT, and TFF1 was detected in precancerous dysplastic lesions, indicating their early development in ESCC (42, 43). High in normal-1 (HIN-1) is a tumor suppressor gene that is highly expressed in many epithelial tissues. Methylation of this gene promoter occurs in the early stages of ESCC and accumulates with esophageal progression tendency during carcinogenesis, resulting in HIN-1 silencing in carcinomas-in-situ (44). By analyzing normal esophageal mucosa, esophageal dysplasia, and ESCC, Guo et al. also found clear evidence of accumulated methylation events during ESCC progression, which included accumulation of promoter region methylation of genes MGMT, p16, BRCA1, MLH1, CDH1, RARβ2, DAPK, and APC (28, 71). These findings are reminiscent of the idea that hypermethylation abnormalities may represent a “field cancerization” or “field defect” of preneoplastic changes, which occur early in carcinogenesis and predispose cells to subsequent malignant transformation (72–74).
From the standpoint of epigenetic heterogeneity, DNA methylation change is also involved in the evolutionary trajectory of ESCC. In 1976, Peter C. Nowell firstly proposed the Darwinian-like clonal evolution of tumor cells (75). Since then, models of tumor evolution derived from intratumor heterogeneity have greatly improved our understanding of tumorigenesis (76, 77). Investigations of intratumor heterogeneity at the DNA methylation level have provided unique opportunities in identifying epigenetic alterations associated with tumor evolution.
In ESCC, DNA methylation status within promoters of transcription factors SIM2 and SIX1 is strongly correlated with their heterogeneous expression pattern, which is further associated with ESCC differentiation and progression (78–80). Using the Illumina Human Methylation 450K (HM450) Bead array, Hao et al. performed multi-region global methylation profiling on 3 ESCC patients and found numbers of TSGs, including EPHA7, ABCB4, PCDH10, DOK1, etc., were heterogeneously hypermethylated at their promoters within tumors from the same individual (41). This observation suggests that intratumor heterogeneous DNA methylation may play a role in the subclonal diversification of ESCC tumors. Moreover, the construction of phylogenies independently from DNA methylation (phyloepigenetic tree) and somatic mutations (phylogenetic tree) yielded highly concordant as well as complementary evolutionary histories (41). This finding supports the concept of co-dependency of aberrant DNA methylation and genetic alterations and again demonstrates the involvement of DNA methylation heterogeneity in the ESCC evolutionary process. In an attempt to gain deeper insights into the DNA methylation heterogeneity of ESCC, most recently, Teng et al. generated single-base resolution whole-genome bisulfite sequencing data on 84 ESCCs and paired paraneoplastic tissues (20). Their analysis also identified numerous DNA methylation alterations associated with ESCC carcinogenesis and lymph node metastasis. In general, they found ESCC manifested substantial inter- and intratumor DNA methylation heterogeneity. Strikingly, the degree of intertumor DNA methylation heterogeneity of ESCC was even higher than that of prostate cancer or chronic lymphocytic leukemia, two well-recognized heterogeneous cancer types (16, 17). Furthermore, ESCC patients with a great extent of DNA methylation heterogeneity tended to experience more aggressive disease and worse overall survival (20).
Altogether, these data suggest the biological significance of DNA methylation change in ESCC. However, additional research is required to determine the extent to which intratumor heterogeneity of DNA methylation reflects differences in regional or clone-specific driver or passenger events of ESCC, and to what extent the alterations to DNA methylation and genetics may be functionally related. It is also not yet known whether and how the pattern of DNA methylation change will evolve during ESCC progression.
Clinical Implications of DNA Methylation Change in ESCC
In several cancer types, the methods of detecting aberrant DNA methylation changes have been applied to clinical fields related to early diagnosis, prognostic prediction, and personalized therapy.
In ESCC, as with other cancers, early detection is a prerequisite for treatment success and survival improvement. In fact, aberrantly methylated genes have been detected in the plasma or serum from ESCC patients, implying that they may serve as non-invasive diagnostic biomarkers. Using methylation-specific polymerase chain reaction, Das et al. detected the methylation status of MGMT from 100 newly diagnosed ESCC patients and reported that promoter hypermethylation of MGMT was present in 70% of serum samples (81). Zheng et al. evaluated the diagnostic role of RUNX3 methylation in serum DNA of ESCCs and found that RUNX3 hypermethylation was detectable in 51.4% (36/70) of the cases, which was significantly higher than that of the healthy donors (39). Moreover, Li et al. showed that the serum DNA detection of a panel of methylated genes, including RAR-β, DAPK, CDH1, p16, and RASSF1A, had a high diagnostic sensitivity of 82% and specificity of 100% for ESCC patients. They thereby suggested that it might be more efficient to early diagnose ESCC through integrative analysis of the methylation status of multiple TSGs (33). Of note, the early diagnostic value of aberrant methylation changes in serum DNA has been widely investigated by many other studies in ESCC. Such methylation events include hypermethylation of genes SFRP1, CASZ1, CDH13, ING2, DKK-3, TAC1, etc. (45–47, 82).
The aberrant methylation pattern has also been identified as a prognostic indicator for ESCC patients. Recently, promoter hypermethylation of CHL1 and RHCG, two novel tumor suppressor candidates, has been reported to be associated with poor differentiation and increased invasion of ESCC, as well as advanced tumor stage and decreased overall survival (48, 49). Using DNA from the plasma of ESCC patients, Liu et al. analyzed the methylation status of Wnt antagonists SFRP1, DKK3, and RUNX3 and showed that patients carrying two or three of these hypermethylated genes had a significantly elevated risk of cancer recurrence, compared with those without methylated genes (46). In an attempt to identify DNA methylation markers in predicting lymph node metastasis, the genome-wide methylation of 86 ESCC patients has been recently assessed, and a 10-probe lymph node metastasis-associated methylation signature has been established through stringent bioinformatics analyses (83). All in all, finding genes of prognostic impact aberrantly methylated allows one to customize therapy for ESCC patients: patients with a worse prognosis might benefit from a more aggressive treatment strategy, while patients with low risk can forego radical surgery.
Methylation might result in the deactivation of genes that are responsive or unresponsive to chemotherapy and radiotherapy. Therefore, the methylation status of certain genes can be used to predict a patient's treatment response in advance (84). The best known example is MGMT promoter methylation and the resultant response to DNA alkylating agents in gliomas (85). However, ESCC studies in this regard are very limited. PAX5 gene methylation was identified as an excellent marker for squamous cell carcinoma detection (50). According to Kurimoto et al.'s study, hypermethylation of PAX5 was significantly correlated with increased ESCC cell proliferation and cisplatin resistance, leading to poor recurrence-free survival and overall survival (51). CHFR is an early mitotic checkpoint gene that controls cell cycle progression at the G2/M checkpoint and maintains chromosomal integrity (86). Promoter region methylation of CHFR was found frequently in ESCC and shown to sensitize ESCC to taxane treatment (52).
Discussion
Epigenetic alterations have been recognized as key contributors to cancer initiation and progression. Amongst these, DNA methylation is one of the most extensively studied epigenetic modifications that occurs in the earliest stage of cancers (87). The aim of this review is to discuss the aberrant DNA methylation changes affecting ESCC carcinogenesis. However, many investigations into such epigenomic features of ESCC are still in their infancy.
Depending on the genomic location, DNA methylation may have different biological functions. Methylation in promoter CpG islands is typically associated with gene repression, while methylation in the gene body is usually linked to active gene expression. Accumulating evidence has shown that intergenic regions contain many regulatory elements, such as enhancers, silencers, and non-coding RNAs, and their functions may also be affected by DNA methylation (84). However, there is a current lack of knowledge with regards to these types of changes during ESCC development. Thus, the ESCC methylome landscape awaits further characterization through sequence-based approaches such as whole-genome bisulfite sequencing at single-base levels of resolution.
DNA methylation changes have emerged as clinically-relevant disease markers due to their chemical stability and measurable feasibility (88). As mentioned previously, there are numerous candidate methylation events that may serve as diagnostic, prognostic, and chemo-sensitive markers in ESCC. However, the cancer type-specificity of many of these methylation changes remains to be determined in larger ESCC cohorts. Their predictive and prognostic value is also challenged by the property of tumor heterogeneity, especially when phenotypically diversified cancerous cells are unequally distributed and asynchronously evolving over space and time. This entails personalized and longitudinal studies assessing methylation changes in the course of ESCC development. It is also worth investigating whether the extent of DNA methylation heterogeneity by itself could be used as a clinically useful biomarker in ESCC.
Unlike genetic alterations that are essentially fixed, epigenetic changes, including DNA hypermethylation, are intriguingly dynamic and theoretically reversible. This makes them attractive targets for cancer therapy or chemoprevention (89). Indeed, several studies have demonstrated that TSG expression could be restored after treatment of cells with demethylating agents, and DNMT inhibitors, such as azacytidine and decitabine, are currently under preclinical and clinical investigations in various cancer types (e.g., ClinicalTrials.gov Identifier: NCT01193517, NCT03666559, and NCT04187703) (90–93). Nonetheless, up until now, very limited data have been shown concerning the effectiveness of DNMT inhibitors in ESCC. Future research on this field are required to gain a more in-depth insight into ESCC development and open a new area for ESCC treatment. Ideally, we will finally reach our goal of “epigenetic precision medicine” in ESCC.
Author Contributions
LL conceived and wrote the review. XC and DY helped the revision. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Natural Science Foundation of China (81802692 and 81872306), Fundamental Research Funds for the Central Universities (19ykpy118), China Postdoctoral Science Foundation (2018M640867 and 2019T120774), and Guangdong Science and Technology Department (2017B030314026).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a shared affiliation, though no other collaboration, with the authors.
Acknowledgments
We thank Meng Zeng for his discussion regarding the manuscript.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
2. Pennathur A, Gibson MK, Jobe BA, Luketich JD. Oesophageal carcinoma. Lancet. (2013) 381:400–12. doi: 10.1016/S0140-6736(12)60643-6
3. Holmes RS, Vaughan TL. Epidemiology and pathogenesis of esophageal cancer. Semin Radiat Oncol. (2007) 17:2–9. doi: 10.1016/j.semradonc.2006.09.003
4. Jones PA, Baylin SB. The epigenomics of cancer. Cell. (2007) 128:683–92. doi: 10.1016/j.cell.2007.01.029
5. Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer. (2003) 3:253–66. doi: 10.1038/nrc1045
6. Ahrens TD, Werner M, Lassmann S. Epigenetics in esophageal cancers. Cell Tissue Res. (2014) 356:643–55. doi: 10.1007/s00441-014-1876-y
7. Baba Y, Watanabe M, Baba H. Review of the alterations in DNA methylation in esophageal squamous cell carcinoma. Surg Today. (2013) 43:1355–64. doi: 10.1007/s00595-012-0451-y
8. Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. (2009) 1:239–59. doi: 10.2217/epi.09.33
9. Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science. (2003) 300:455. doi: 10.1126/science.1083557
10. Shen JC, Rideout WM III, Jones PA. High frequency mutagenesis by a DNA methyltransferase. Cell. (1992) 71:1073–80. doi: 10.1016/S0092-8674(05)80057-1
11. Robertson KD. DNA methylation, methyltransferases, and cancer. Oncogene. (2001) 20:3139–55. doi: 10.1038/sj.onc.1204341
12. Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet. (1999) 21:163–7. doi: 10.1038/5947
13. Mazor T, Pankov A, Song JS, Costello JF. Intratumoral heterogeneity of the epigenome. Cancer Cell. (2016) 29:440–51. doi: 10.1016/j.ccell.2016.03.009
14. Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. (2012) 12:323–34. doi: 10.1038/nrc3261
15. Liu J, Dang H, Wang XW. The significance of intertumor and intratumor heterogeneity in liver cancer. Exp Mol Med. (2018) 50:e416. doi: 10.1038/emm.2017.165
16. Fraser M, Sabelnykova VY, Yamaguchi TN, Heisler LE, Livingstone J, Huang V, et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature. (2017) 541:359–64. doi: 10.1038/nature20788
17. Landau DA, Clement K, Ziller MJ, Boyle P, Fan J, Gu H, et al. Locally disordered methylation forms the basis of intratumor methylome variation in chronic lymphocytic leukemia. Cancer Cell. (2014) 26:813–25. doi: 10.1016/j.ccell.2014.10.012
18. Klughammer J, Kiesel B, Roetzer T, Fortelny N, Nemc A, Nenning KH, et al. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space. Nat Med. (2018) 24:1611–24. doi: 10.1038/s41591-018-0156-x
19. Sheffield NC, Pierron G, Klughammer J, Datlinger P, Schonegger A, Schuster M, et al. DNA methylation heterogeneity defines a disease spectrum in Ewing sarcoma. Nat Med. (2017) 23:386–95. doi: 10.1038/nm.4273
20. Teng H, Xue M, Liang J, Wang X, Wang L, Wei W, et al. Inter- and intratumor DNA methylation heterogeneity associated with lymph node metastasis and prognosis of esophageal squamous cell carcinoma. Theranostics. (2020) 10:3035–48. doi: 10.7150/thno.42559
21. Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. (1983) 301:89–92. doi: 10.1038/301089a0
22. Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, et al. Induction of tumors in mice by genomic hypomethylation. Science. (2003) 300:489–92. doi: 10.1126/science.1083558
23. Kawano H, Saeki H, Kitao H, Tsuda Y, Otsu H, Ando K, et al. Chromosomal instability associated with global DNA hypomethylation is associated with the initiation and progression of esophageal squamous cell carcinoma. Ann Surg Oncol. (2014) 21:S696–702. doi: 10.1245/s10434-014-3818-z
24. Hoshimoto S, Takeuchi H, Ono S, Sim MS, Huynh JL, Huang SK, et al. Genome-wide hypomethylation and specific tumor-related gene hypermethylation are associated with esophageal squamous cell carcinoma outcome. J Thorac Oncol. (2015) 10:509–17. doi: 10.1097/JTO.0000000000000441
25. Baba Y, Watanabe M, Murata A, Shigaki H, Miyake K, Ishimoto T, et al. LINE-1 hypomethylation, DNA copy number alterations, and CDK6 amplification in esophageal squamous cell carcinoma. Clin Cancer Res. (2014) 20:1114–24. doi: 10.1158/1078-0432.CCR-13-1645
26. Iwagami S, Baba Y, Watanabe M, Shigaki H, Miyake K, Ishimoto T, et al. LINE-1 hypomethylation is associated with a poor prognosis among patients with curatively resected esophageal squamous cell carcinoma. Ann Surg. (2013) 257:449–55. doi: 10.1097/SLA.0b013e31826d8602
27. Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet. (2009) 10:691–703. doi: 10.1038/nrg2640
28. Guo M, Ren J, House MG, Qi Y, Brock MV, Herman JG. Accumulation of promoter methylation suggests epigenetic progression in squamous cell carcinoma of the esophagus. Clin Cancer Res. (2006) 12:4515–22. doi: 10.1158/1078-0432.CCR-05-2858
29. Du Z, Ma K, Sun X, Li A, Wang H, Zhang L, et al. Methylation of RASSF1A gene promoter and the correlation with DNMT1 expression that may contribute to esophageal squamous cell carcinoma. World J Surg Oncol. (2015) 13:141. doi: 10.1186/s12957-015-0557-y
30. Kuroki T, Trapasso F, Yendamuri S, Matsuyama A, Alder H, Mori M, et al. Promoter hypermethylation of RASSF1A in esophageal squamous cell carcinoma. Clin Cancer Res. (2003) 9:1441–5.
31. Mao WM, Li P, Zheng QQ, Wang CC, Ge MH, Hu FJ, et al. Hypermethylation-modulated downregulation of RASSF1A expression is associated with the progression of esophageal cancer. Arch Med Res. (2011) 42:182–8. doi: 10.1016/j.arcmed.2011.04.002
32. Lu D, Ma J, Zhan Q, Li Y, Qin J, Guo M. Epigenetic silencing of RASSF10 promotes tumor growth in esophageal squamous cell carcinoma. Discov Med. (2014) 17:169–78.
33. Ling ZQ, Li P, Ge MH, Hu FJ, Fang XH, Dong ZM, et al. Aberrant methylation of different DNA repair genes demonstrates distinct prognostic value for esophageal cancer. Dig Dis Sci. (2011) 56:2992–3004. doi: 10.1007/s10620-011-1774-z
34. Fang MZ, Jin Z, Wang Y, Liao J, Yang GY, Wang LD, et al. Promoter hypermethylation and inactivation of O(6)-methylguanine-DNA methyltransferase in esophageal squamous cell carcinomas and its reactivation in cell lines. Int J Oncol. (2005) 26:615–22. doi: 10.3892/ijo.26.3.615
35. Su Y, Yin L, Liu R, Sheng J, Yang M, Wang Y, et al. Promoter methylation status of MGMT, hMSH2, and hMLH1 and its relationship to corresponding protein expression and TP53 mutations in human esophageal squamous cell carcinoma. Med Oncol. (2014) 31:784. doi: 10.1007/s12032-013-0784-4
36. Zare M, Jazii FR, Alivand MR, Nasseri NK, Malekzadeh R, Yazdanbod M. Qualitative analysis of adenomatous polyposis coli promoter: hypermethylation, engagement and effects on survival of patients with esophageal cancer in a high risk region of the world, a potential molecular marker. BMC Cancer. (2009) 9:24. doi: 10.1186/1471-2407-9-24
37. Kim YT, Park JY, Jeon YK, Park SJ, Song JY, Kang CH, et al. Aberrant promoter CpG island hypermethylation of the adenomatosis polyposis coli gene can serve as a good prognostic factor by affecting lymph node metastasis in squamous cell carcinoma of the esophagus. Dis Esophagus. (2009) 22:143–50. doi: 10.1111/j.1442-2050.2008.00862.x
38. Ishii T, Murakami J, Notohara K, Cullings HM, Sasamoto H, Kambara T, et al. Oesophageal squamous cell carcinoma may develop within a background of accumulating DNA methylation in normal and dysplastic mucosa. Gut. (2007) 56:13–9. doi: 10.1136/gut.2005.089813
39. Zheng Y, Zhang Y, Huang X, Chen L. Analysis of the RUNX3 gene methylation in serum DNA from esophagus squamous cell carcinoma, gastric and colorectal adenocarcinoma patients. Hepatogastroenterology. (2011) 58:2007–11. doi: 10.5754/hge10016
40. Cheng Y, Geng H, Cheng SH, Liang P, Bai Y, Li J, et al. KRAB zinc finger protein ZNF382 is a proapoptotic tumor suppressor that represses multiple oncogenes and is commonly silenced in multiple carcinomas. Cancer Res. (2010) 70:6516–26. doi: 10.1158/0008-5472.CAN-09-4566
41. Hao JJ, Lin DC, Dinh HQ, Mayakonda A, Jiang YY, Chang C, et al. Spatial intratumoral heterogeneity and temporal clonal evolution in esophageal squamous cell carcinoma. Nat Genet. (2016) 48:1500–7. doi: 10.1038/ng.3683
42. Li JS, Ying JM, Wang XW, Wang ZH, Tao Q, Li LL. Promoter methylation of tumor suppressor genes in esophageal squamous cell carcinoma. Chin J Cancer. (2013) 32:3–11. doi: 10.5732/cjc.011.10381
43. Lima SC, Hernandez-Vargas H, Simao T, Durand G, Kruel CD, Le Calvez-Kelm F, et al. Identification of a DNA methylome signature of esophageal squamous cell carcinoma and potential epigenetic biomarkers. Epigenetics. (2011) 6:1217–27. doi: 10.4161/epi.6.10.17199
44. Guo M, Ren J, Brock MV, Herman JG, Carraway HE. Promoter methylation of HIN-1 in the progression to esophageal squamous cancer. Epigenetics. (2008) 3:336–41. doi: 10.4161/epi.3.6.7158
45. Jin Z, Olaru A, Yang J, Sato F, Cheng Y, Kan T, et al. Hypermethylation of tachykinin-1 is a potential biomarker in human esophageal cancer. Clin Cancer Res. (2007) 13:6293–300. doi: 10.1158/1078-0432.CCR-07-0818
46. Liu JB, Qiang FL, Dong J, Cai J, Zhou SH, Shi MX, et al. Plasma DNA methylation of Wnt antagonists predicts recurrence of esophageal squamous cell carcinoma. World J Gastroenterol. (2011) 17:4917–21. doi: 10.3748/wjg.v17.i44.4917
47. Liu C, Li N, Lu H, Wang Z, Chen C, Wu L, et al. Circulating SFRP1 promoter methylation status in gastric adenocarcinoma and esophageal square cell carcinoma. Biomed Rep. (2015) 3:123–7. doi: 10.3892/br.2014.388
48. Tang H, Jiang L, Zhu C, Liu R, Wu Y, Yan Q, et al. Loss of cell adhesion molecule L1 like promotes tumor growth and metastasis in esophageal squamous cell carcinoma. Oncogene. (2019) 38:3119–33. doi: 10.1038/s41388-018-0648-7
49. Ming XY, Zhang X, Cao TT, Zhang LY, Qi JL, Kam NW, et al. RHCG suppresses tumorigenicity and metastasis in esophageal squamous cell carcinoma via inhibiting NF-kappaB signaling and MMP1 expression. Theranostics. (2018) 8:185–98. doi: 10.7150/thno.21383
50. Guerrero-Preston R, Michailidi C, Marchionni L, Pickering CR, Frederick MJ, Myers JN, et al. Key tumor suppressor genes inactivated by “greater promoter” methylation and somatic mutations in head and neck cancer. Epigenetics. (2014) 9:1031–46. doi: 10.4161/epi.29025
51. Kurimoto K, Hayashi M, Guerrero-Preston R, Koike M, Kanda M, Hirabayashi S, et al. PAX5 gene as a novel methylation marker that predicts both clinical outcome and cisplatin sensitivity in esophageal squamous cell carcinoma. Epigenetics. (2017) 12:865–74. doi: 10.1080/15592294.2017.1365207
52. Yun T, Liu Y, Gao D, Linghu E, Brock MV, Yin D, et al. Methylation of CHFR sensitizes esophageal squamous cell cancer to docetaxel and paclitaxel. Genes Cancer. (2015) 6:38–48. doi: 10.18632/genesandcancer.46
53. Wang C, Pu W, Zhao D, Zhou Y, Lu T, Chen S, et al. Identification of hyper-methylated tumor suppressor genes-based diagnostic panel for esophageal squamous cell carcinoma (ESCC) in a Chinese han population. Front Genet. (2018) 9:356. doi: 10.3389/fgene.2018.00356
54. Wang X, Wang J, Jia Y, Wang Y, Han X, Duan Y, et al. Methylation decreases the Bin1 tumor suppressor in ESCC and restoration by decitabine inhibits the epithelial mesenchymal transition. Oncotarget. (2017) 8:19661–73. doi: 10.18632/oncotarget.14914
55. Jiang D, He Z, Wang C, Zhou Y, Li F, Pu W, et al. Epigenetic silencing of ZNF132 mediated by methylation-sensitive Sp1 binding promotes cancer progression in esophageal squamous cell carcinoma. Cell Death Dis. (2018) 10:1. doi: 10.1038/s41419-018-1236-z
56. Wang JX, He YL, Zhu ST, Yang S, Zhang ST. Aberrant methylation of the 3q25 tumor suppressor gene PTX3 in human esophageal squamous cell carcinoma. World J Gastroenterol. (2011) 17:4225–30. doi: 10.3748/wjg.v17.i37.4225
57. Tong M, Chan KW, Bao JY, Wong KY, Chen JN, Kwan PS, et al. Rab25 is a tumor suppressor gene with antiangiogenic and anti-invasive activities in esophageal squamous cell carcinoma. Cancer Res. (2012) 72:6024–35. doi: 10.1158/0008-5472.CAN-12-1269
58. Li LW, Yu XY, Yang Y, Zhang CP, Guo LP, Lu SH. Expression of esophageal cancer related gene 4 (ECRG4), a novel tumor suppressor gene, in esophageal cancer and its inhibitory effect on the tumor growth in vitro and in vivo. Int J Cancer. (2009) 125:1505–13. doi: 10.1002/ijc.24513
59. Yue D, Fan Q, Chen X, Li F, Wang L, Huang L, et al. Epigenetic inactivation of SPINT2 is associated with tumor suppressive function in esophageal squamous cell carcinoma. Exp Cell Res. (2014) 322:149–58. doi: 10.1016/j.yexcr.2013.11.009
60. Luo M, Li Y, Shi X, Yang W, Zhou F, Sun N, et al. Aberrant methylation of EYA4 promotes epithelial-mesenchymal transition in esophageal squamous cell carcinoma. Cancer Sci. (2018) 109:1811–24. doi: 10.1111/cas.13615
61. Dong Z, Liang X, Wu X, Kang X, Guo Y, Shen S, et al. Promoter hypermethylation-mediated downregulation of tumor suppressor gene SEMA3B and lncRNA SEMA3B-AS1 correlates with progression and prognosis of esophageal squamous cell carcinoma. Clin Exp Metastasis. (2019) 36:225–41. doi: 10.1007/s10585-019-09964-3
62. Tanaka F, Yamamoto K, Suzuki S, Inoue H, Tsurumaru M, Kajiyama Y, et al. Strong interaction between the effects of alcohol consumption and smoking on oesophageal squamous cell carcinoma among individuals with ADH1B and/or ALDH2 risk alleles. Gut. (2010) 59:1457–64. doi: 10.1136/gut.2009.205724
63. Anantharaman D, Marron M, Lagiou P, Samoli E, Ahrens W, Pohlabeln H, et al. Population attributable risk of tobacco and alcohol for upper aerodigestive tract cancer. Oral Oncol. (2011) 47:725–31. doi: 10.1016/j.oraloncology.2011.05.004
64. Pelucchi C, Gallus S, Garavello W, Bosetti C, La Vecchia C. Cancer risk associated with alcohol and tobacco use: focus on upper aero-digestive tract and liver. Alcohol Res Health. (2006) 29:193–8.
65. Huang Y, Chang X, Lee J, Cho YG, Zhong X, Park IS. Cigarette smoke induces promoter methylation of single-stranded DNA-binding protein 2 in human esophageal squamous cell carcinoma. Int J Cancer. (2011) 128:2261–73. doi: 10.1002/ijc.25569
66. Lin L, Lin DC. Biological significance of tumor heterogeneity in esophageal squamous cell carcinoma. Cancers. (2019) 11:1156. doi: 10.3390/cancers11081156
67. Lin DC, Wang MR, Koeffler HP. Genomic and epigenomic aberrations in esophageal squamous cell carcinoma and implications for patients. Gastroenterology. (2018) 154:374–89. doi: 10.1053/j.gastro.2017.06.066
68. Toh Y, Egashira A, Yamamoto M. Epigenetic alterations and their clinical implications in esophageal squamous cell carcinoma. Gen Thorac Cardiovasc Surg. (2013) 61:262–9. doi: 10.1007/s11748-013-0235-3
69. You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell. (2012) 22:9–20. doi: 10.1016/j.ccr.2012.06.008
70. Sawan C, Vaissiere T, Murr R, Herceg Z. Epigenetic drivers and genetic passengers on the road to cancer. Mutat Res. (2008) 642:1–13. doi: 10.1016/j.mrfmmm.2008.03.002
71. Vogelstein B, Kinzler KW. The multistep nature of cancer. Trends Genet. (1993) 9:138–41. doi: 10.1016/0168-9525(93)90209-Z
72. Guo M, House MG, Hooker C, Han Y, Heath E, Gabrielson E, et al. Promoter hypermethylation of resected bronchial margins: a field defect of changes? Clin Cancer Res. (2004) 10:5131–6. doi: 10.1158/1078-0432.CCR-03-0763
73. Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. (1953) 6:963–8. doi: 10.1002/1097-0142(195309)6:5<963::aid-cncr2820060515>3.0.co;2-q
74. Leedham SJ, Graham TA, Oukrif D, McDonald SA, Rodriguez-Justo M, Harrison RF, et al. Clonality, founder mutations, and field cancerization in human ulcerative colitis-associated neoplasia. Gastroenterology. (2009) 136:542–50.e6. doi: 10.1053/j.gastro.2008.10.086
75. Nowell PC. The clonal evolution of tumor cell populations. Science. (1976) 194:23–8. doi: 10.1126/science.959840
76. Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res. (2012) 72:4875–82. doi: 10.1158/0008-5472.CAN-12-2217
77. Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. (2012) 366:883–92. doi: 10.1056/NEJMoa1113205
78. Komatsu M, Sasaki H. DNA methylation is a key factor in understanding differentiation phenotype in esophageal squamous cell carcinoma. Epigenomics. (2014) 6:567–9. doi: 10.2217/epi.14.56
79. Nishimura T, Tamaoki M, Komatsuzaki R, Oue N, Taniguchi H, Komatsu M, et al. SIX1 maintains tumor basal cells via transforming growth factor-beta pathway and associates with poor prognosis in esophageal cancer. Cancer Sci. (2017) 108:216–25. doi: 10.1111/cas.13135
80. Tamaoki M, Komatsuzaki R, Komatsu M, Minashi K, Aoyagi K, Nishimura T, et al. Multiple roles of single-minded 2 in esophageal squamous cell carcinoma and its clinical implications. Cancer Sci. (2018) 109:1121–34. doi: 10.1111/cas.13531
81. Das M, Sharma SK, Sekhon GS, Saikia BJ, Mahanta J, Phukan RK. Promoter methylation of MGMT gene in serum of patients with esophageal squamous cell carcinoma in North East India. Asian Pac J Cancer Prev. (2014) 15:9955–60. doi: 10.7314/APJCP.2014.15.22.9955
82. Wang HQ, Yang CY, Wang SY, Wang T, Han JL, Wei K, et al. Cell-free plasma hypermethylated CASZ1, CDH13 and ING2 are promising biomarkers of esophageal cancer. J Biomed Res. (2018) 32:424–33. doi: 10.7555/JBR.32.20170065
83. Roy R, Kandimalla R, Sonohara F, Koike M, Kodera Y, Takahashi N, et al. A comprehensive methylation signature identifies lymph node metastasis in esophageal squamous cell carcinoma. Int J Cancer. (2019) 144:1160–9. doi: 10.1002/ijc.31755
84. Yan W, Herman JG, Guo M. Epigenome-based personalized medicine in human cancer. Epigenomics. (2016) 8:119–33. doi: 10.2217/epi.15.84
85. Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. (2000) 343:1350–4. doi: 10.1056/NEJM200011093431901
86. Scolnick DM, Halazonetis TD. Chfr defines a mitotic stress checkpoint that delays entry into metaphase. Nature. (2000) 406:430–5. doi: 10.1038/35019108
87. Kulis M, Esteller M. DNA methylation and cancer. Adv Genet. (2010) 70:27–56. doi: 10.1016/B978-0-12-380866-0.60002-2
88. Guo M, Peng Y, Gao A, Du C, Herman JG. Epigenetic heterogeneity in cancer. Biomark Res. (2019) 7:23. doi: 10.1186/s40364-019-0174-y
89. Feinberg AP. The key role of epigenetics in human disease prevention and mitigation. N Engl J Med. (2018) 378:1323–34. doi: 10.1056/NEJMra1402513
90. Schrump DS, Fischette MR, Nguyen DM, Zhao M, Li X, Kunst TF, et al. Phase I study of decitabine-mediated gene expression in patients with cancers involving the lungs, esophagus, or pleura. Clin Cancer Res. (2006) 12:5777–85. doi: 10.1158/1078-0432.CCR-06-0669
91. Martin I, Navarro B, Solano C, Calabuig M, Hernandez-Boluda JC, Amat P, et al. Synergistic antioncogenic activity of azacitidine and curcumin in myeloid leukemia cell lines and patient samples. Anticancer Res. (2019) 39:4757–66. doi: 10.21873/anticanres.13659
92. Giri AK, Aittokallio T. DNMT inhibitors increase methylation in the cancer genome. Front Pharmacol. (2019) 10:385. doi: 10.3389/fphar.2019.00385
Keywords: esophageal squamous cell carcinoma, aberrant DNA methylation, global DNA hypomethylation, promoter hypermethylation, heterogeneity, clinical significance
Citation: Lin L, Cheng X and Yin D (2020) Aberrant DNA Methylation in Esophageal Squamous Cell Carcinoma: Biological and Clinical Implications. Front. Oncol. 10:549850. doi: 10.3389/fonc.2020.549850
Received: 07 April 2020; Accepted: 18 September 2020;
Published: 23 October 2020.
Edited by:
Zhi Xie, Sun Yat-sen University, ChinaReviewed by:
Michele Ghidini, Operative Unit of Medical Oncology Gastrointestinal Unit Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, ItalyGiacomo Bregni, Jules Bordet Institute, Belgium
Copyright © 2020 Lin, Cheng and Yin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lehang Lin, bGVoYW5nbGluQGdtYWlsLmNvbQ==; Dong Yin, eWluZDNAbWFpbC5zeXN1LmVkdS5jbg==