 Donjete Simnica
Donjete Simnica Harald Ittrich3
Harald Ittrich3
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Oncol. , 11 September 2020
Sec. Pharmacology of Anti-Cancer Drugs
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.540030
Drug-promoted cancers are increasingly recognized as a serious clinical problem in patients receiving BRAF inhibitory treatment. Here we report on a patient with BRAF mutant hairy cell leukemia and monoclonal B-cell lymphocytosis (MBL), who responded durably to BRAF/MEK inhibitors (BRAFi/MEKi) but experienced transformation of a RAS mutant MBL to chronic lymphocytic leukemia (CLL) with accelerated nodal progression. Hypothesizing that BRAFi triggered excessive MEK-ERK signaling in the MBL/CLL clone via the CRAF/RAS complex as previously described for BRAFi-induced cancers, BRAFi was discontinued inducing a rapid remission of the CLL on MEKi alone. Liquid biopsy monitoring showed a continuous increase of the MBL/CLL clone from the start of BRAFi/MEKi treatment followed by a rapid decline upon BRAFi withdrawal. Next-generation sequencing of a cohort of patients with MBL and monoclonal gammopathy of unclear significance (MGUS) revealed that almost one third of these cases harbored RAS mutations. In view of the population frequency of lymphatic pre-malignant conditions and the prevalence of RAS mutations in such cases, vigilant surveillance remains critical in patients treated with BRAF inhibitors.
Activating BRAF mutations (most prominently BRAF V600E), providing oncogenic signaling through the mitogen-activated protein kinase (MAPK) pathway, are the key molecular driver in a variety of solid tumors including roughly 50% of malignant melanomas, 15% of thyroid cancers, 8% of colorectal cancers, 3% of lung cancers, and 2% of pancreatic cancers. In hematological malignancies, BRAF mutations occur in approximately 7% of multiple myeloma and almost 100% of classical hairy cell leukemias (HCL) (1). Small molecule BRAF inhibitors (BRAFi) are increasingly used for therapeutic targeting with and without concomitant MEK inhibitors (MEKi) yielding convincing clinical results.
Despite the fact that BRAF is a key effector downstream of RAS and upstream of MEK, ATP-competitive BRAFi are not effective in RAS mutant tumor models (2). In fact, such inhibitors have opposing effects on MAPK signaling depending on the BRAF mutational status. In BRAF V600E mutant tumors, BRAFi effectively block MAPK signaling and inhibit tumor growth. However, in tumor and normal cells expressing wild-type BRAF, these inhibitors have been found to promote BRAF dimerization, activation and binding to RAS-GTP, ultimately resulting in stimulation of MEK-ERK signaling and proliferative effects (3, 4). This paradoxical activation is thought to explain why this class of drugs may induce or promote RAS mutant neoplasias which emerges as a more and more serious clinical problem the more patients are offered this targeted treatment approach (5–12).
Here we report on a patient treated with BRAFi/MEKi for BRAF V600E mutant HCL. This treatment resulted in the development of chronic lymphocytic leukemia (CLL) from a preexisting KRAS G12D mutant monoclonal B-cell lymphocytosis (MBL). By next-generation sequencing of blood in an independent cohort of individuals with MBL and monoclonal gammopathy of undetermined significance (MGUS) we found that almost one third of these cases harbored clonal or subclonal activating RAS mutations. We conclude that patients with such RAS mutant precursor lesions have to be critically selected for and carefully monitored during tumor treatment with BRAFi.
Informed consent was obtained from the reported patient with HCL and MBL/CLL as well as from eleven MBL and eleven MGUS control patients for the use of their diagnostic material as approved by the institutional review board (Ethikkommission der Ärztekammer Hamburg, Germany, project number PV4767).
Genomic DNA was isolated from peripheral blood mononuclear cells (PBMNCs) using GenElute Mammalian Genomic DNA Miniprep kit (Sigma-Aldrich, St. Louis, United States) according to the supplier’s instructions.
Using a custom gene panel (QIAseq Targeted DNA Panel) gene regions of interest were amplified starting from 100 ng of PBMNC or bone marrow mononuclear cells (BMMNCs) genomic DNA, respectively. The panel covered the following genes: BRAF, HRAS, KRAS, and NRAS. Library preparation was performed according to the supplier’s instructions. Briefly, DNA was fragmented by a fragmentation enzyme mix and Illumina adapters containing 12-base unique molecular identifiers (UMIs) were ligated onto each DNA molecule. During target enrichment PCR, gene and hotspot specific primers amplify the regions of interest followed by a 21-cycle universal PCR, where all fragments of interest are amplified once more for final library construction. The final library was quantified by Qubit (Thermo Fisher Scientific Inc.) and fragment size was analyzed using an Agilent 2100 Bioanalyzer (Agilent technologies). Multiplex sequencing was performed with a 300-cycle dual indexed (8 nucleotides) paired-end run on a NextSeq sequencer (Illumina) at an estimated depth of 26 500 reads. Variant calling was performed using smCounter2 (13). The incorporated UMIs allow for elimination of biases that can be introduced during PCR amplification and therefore offer a highly accurate variant calling at a variant frequency level of ≥1%. For details of multiplex PCR and sequencing approach for the identification of RAS mutations in the MGUS patient cohort please refer to original publication (14).
For sensitive clonal monitoring, the IGH gene locus containing the rearranged VH, DH and JH segments was amplified by multiplex PCR from genomic DNA using previously published protocols (15). Amplicon extension with Illumina adapter sequences and unique indices was achieved through a second PCR reaction. Primers were purchased from Metabion (Martinsried, Germany) and PCRs were performed using Phusion HS II (Thermo Fisher Scientific Inc.) according to the supplier’s instructions. Finally, amplicons with the expected size were purified after agarose gel electrophoresis using the NucleoSpin® Gel and PCR Clean-up kit (Macherey-Nagel). After amplicon quantification and quality control with a Qubit (Thermo Fisher Scientific Inc.) and an Agilent 2100 Bioanalyzer (Agilent technologies), respectively, sequencing was performed on an Illumina MiSeq platform (600–cycle single indexed, paired-end run). Analysis of the IGH locus was computed using the MiXCR analysis tool (16). Only productive sequences with a read count ≥2 were included in the analysis.
Within 2 h of peripheral blood collection, erythrocyte lysis using a standard lysis buffer (ammonium chloride 8.29 g/l, EDTA 0.372 g/l, potassium hydrogen carbonate 1 g/l) was performed followed by flow cytometry using an 5-color flow cytometry panel established for routine clinical analysis of B cell disorders (all directly labeled antibodies purchased from Beckman Coulter). Measurements were performed on a FC500 cytometer (Beckman Coulter, Krefeld, Germany). B and CLL cell counts were calculated from absolute blood counts.
Sequencing data generated for this study can be found in the European Nucleotide Archive (ENA). ID: PRJEB36480.
We report on an instructive case of a patient with HCL and concomitant MBL, who received dual BRAFi/MEKi after his fifth HCL relapse. This patient responded quickly and achieved a durable HCL remission with excellent tolerability of both agents. Yet, 24 months after treatment initiation, he experienced transformation of MBL to overt CLL with accelerated nodal progression (Figure 1A). At this time, the HCL was in ongoing partial remission.
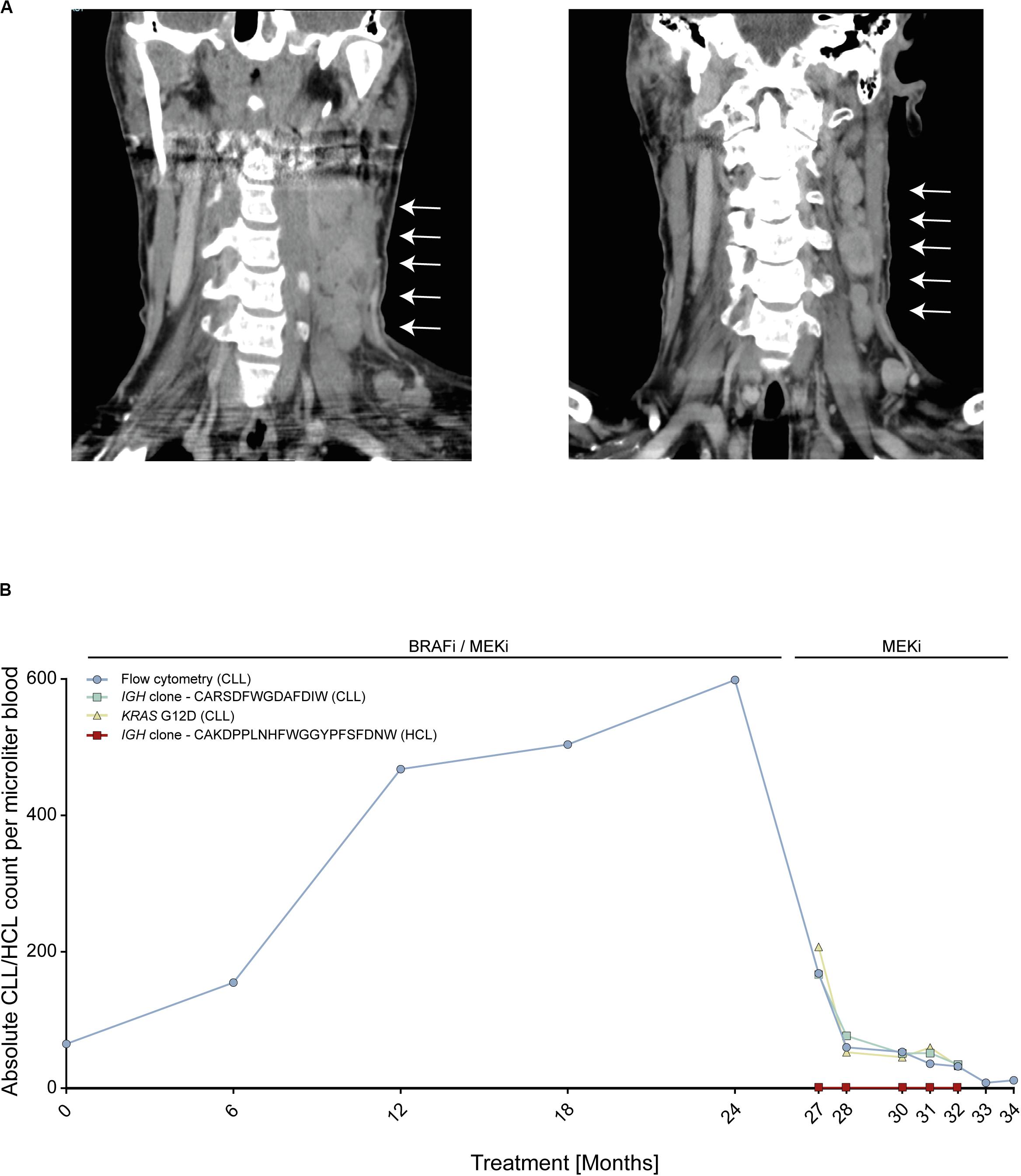
Figure 1. Patient with BRAF V600E-mutant hairy cell leukemia (HCL) and concomitant RAS-mutant B cell lymphocytosis (MBL) experiencing progression to chronic lymphocytic leukemia (CLL). (A) Computed tomography scans of cervical lymph nodes of patient with HCL and concomitant MBL/CLL. On the left, nodal progression on dual BRAFi/MEKi. On the right, nodal remission 3 months after BRAFi withdrawal. (B) Absolute MBL/CLL and HCL cell counts over time during combined BRAFi/MEKi and after BRAFi withdrawal. Cell counts were deduced from flow cytometry, IGH next-generation sequencing and KRAS G12D liquid biopsy analyses of peripheral blood. Clone CARSDFWGDAFDIW (green line) represents the MBL/CLL clone and clone CAKDPPLNHFWGGYPFSFDNW (red line) represents HCL clone. IGH, Immune globulin heavy locus. Arrows indicate cervical lymphnode.
To address the molecular driver underlying the suspected BRAFi-induced accelerated progression, we performed next-generation sequencing using a gene panel including activating RAS mutations. We found that the initial MBL clone as well as the emerging CLL carried an activating KRAS G12D mutation, but no BRAF mutation, thus explaining the observed CLL progression. The RAS mutation was confirmed by Sanger sequencing from CLL tumor material (resected lymph node). Since enhanced MAPK signaling was expected to be attenuated upon withdrawal of the BRAFi, treatment was continued with the MEKi only, inducing a nodal CLL decline with maintained HCL control (Figure 1A). Despite the predominantly nodal CLL progression, multicolor flow cytometry and NGS-based blood monitoring of lymphoma clones over time (IGH and KRAS) provided a liquid biopsy window into disease dynamics showing a continuous increase of the MBL/CLL clone from the start of the dual BRAFi/MEKi treatment followed by rapid decline of the CLL clone upon withdrawal of the BRAFi (Figure 1B). MBL represents a common precursor lesion of CLL with relatively high population prevalence with some studies suggesting that up to 10% of individuals >40 years may harbor clonal B cell populations (17). Since activating RAS mutations may be present in lymphatic diseases and precursor states, we wished to explore RAS mutational frequencies in a small cohort of eleven MBL patients from our institution. To this end, we used an NGS targeted sequencing approach with an established sensitivity of ≥1%, since even subclonal RAS mutations in an MBL background are expected to be selected on therapeutic pressure with BRAFi and may give rise to transformation. This analysis showed that two of eleven (18%) MBL cases harbored activating RAS mutations (HRAS G13S and KRAS A146T). While the HRAS mutation was subclonal in MBL003 (present in 2.37% of MBL cells), the KRAS mutation in MBL005 was present in all MBL cells like in our index patient. Since a similar risk for transformation may exist in individuals with RAS mutant MGUS, a precursor lesion for multiple myeloma, we re-analyzed an NGS dataset from a cohort of eleven individuals with MGUS, that was previously published by our group, for the prevalence of RAS mutations (14). In this cohort, four out of eleven (36%) MGUS cases harbored activating NRAS mutations (NRAS G12C, G13 C/R/V, and Q61K). In two of these patients, more than one NRAS mutation was found in different subclones (Table 1).

Table 1. RAS mutations in individuals with MBL and MGUS.
Using a targeted drug that works on a specific molecular cancer lesion may appear a simple therapeutic concept. Yet, this concept must be considered oversimplified since systemic inhibition of oncogenic signaling not only operates on the tumor cell’s mutational landscape, but also on other (e.g., pre-malignant) tissue’s occult mutational landscapes. We currently face a growing body of evidence showing that BRAF inhibition (with and without concurrent MEK inhibition) may trigger the development of cancer from pre-neoplastic lesions (e.g., melanoma and non-melanoma skin cancer, pancreatic cancer) or drive progression of existing but unrecognized cancers (e.g., colon cancer) (5–12).
We show that not only pre-cancerous skin lesions, but also hematological pre-cancerous lesions may progress upon BRAF inhibition. A mechanistic scheme illustrating this concept is shown in Figure 2.
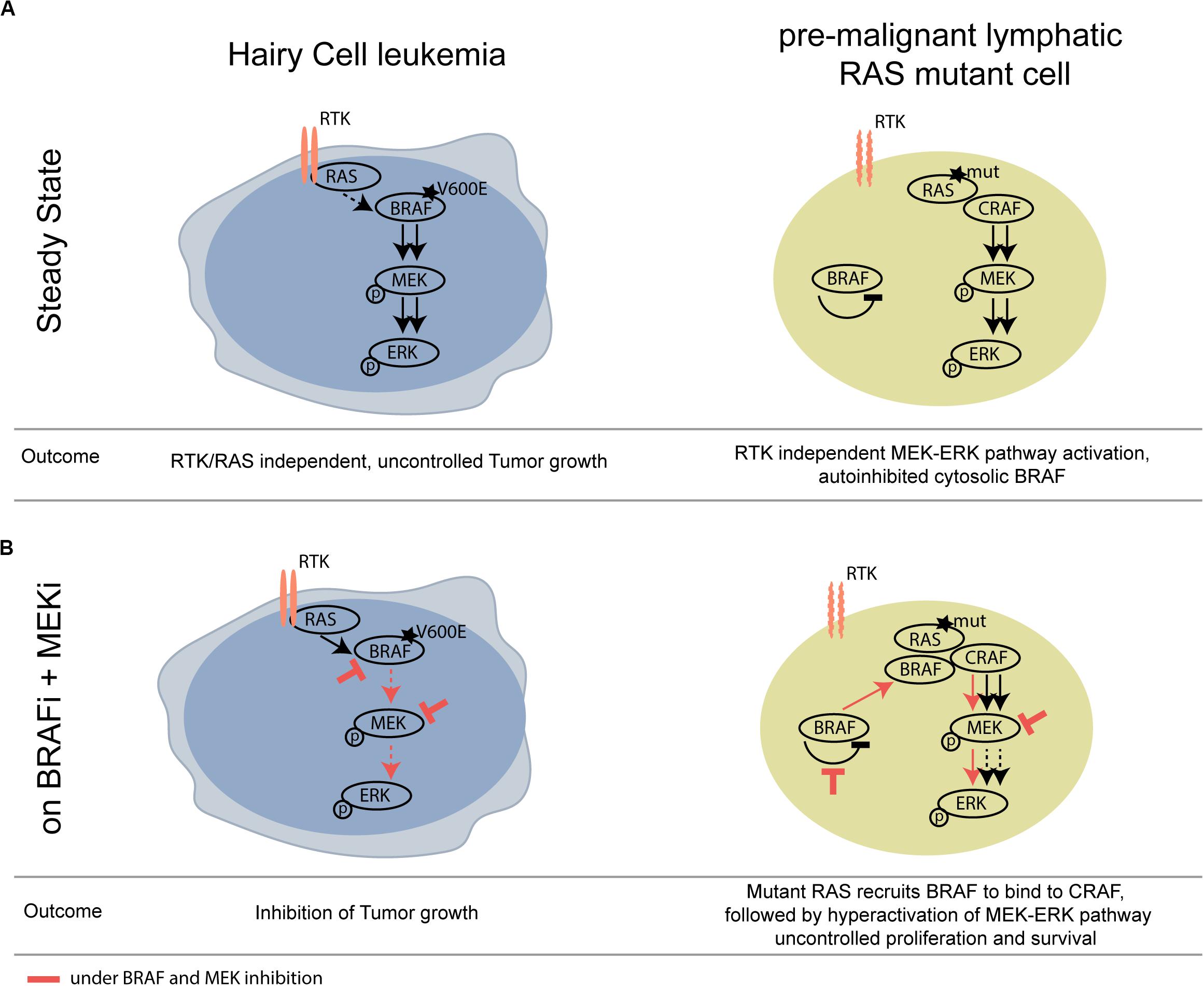
Figure 2. Schematic overview of differential activity of BRAFi/MEKi in BRAF V600E HCL versus RAS mutant pre-malignant lymphoid cells. (A) At steady state, mutant BRAF signals RTK/RAS independently and thereby drives tumor growth of HCL. (B) Upon BRAF and MEK inhibition, the activity of the MAPK pathway is strongly compromised in the HCL context, however, leading to paradoxical activation of the MAPK pathway in the BRAF wildtype, RAS mutant lymphoid clone. RTK, receptor tyrosine kinase.
In this case, we observed a steady increase of the MBL/CLL clone over time on BRAFi/MEKi treatment followed – after 2 years – by more important nodal progression. The rather slow dynamics over 2 years may be attributed by the concomitant MEKi, which, however, was not able to fully block paradoxical stimulation.
While in CLL, the overall frequency of RAS mutations appears to be below 10% (in low-resolution studies), in multiple myeloma and its precursor MGUS, deregulation of MAPK signaling via activating RAS mutations is a more common finding present on a clonal or subclonal level in 36–49% of patients depending on the resolution of the detection technology (14, 18). Our own previous data on a hospital cohort of non-hematooncological patients show that the overall frequencies of MBL and MGUS in individuals between 50 and over 80 years of age range from 0.9 up to 5% for MBL and 5.1 up to 11.8% for MGUS (19). Considering the RAS mutational frequencies of these precursor lesions (according to our data almost one third of cases with RAS mutations), this may result in significant numbers of patients at risk for the development of treatment-requiring hematological cancers on BRAF inhibition.
One of the limitations of our work consists in the fact that our data imply, but not directly proof that the RAS mutation is involved in paradoxical pathway activation in this case. The failure to directly show this is due to a lack of sufficient biological material from this case taken on BRAFi treatment. Nonetheless, the mechanistic concept of paradoxical BRAFi induced cancer progression in RAS mutant cells is well established and should therefore also apply to this case (3, 4, 20). Another limitation of our work is related to the fact that we don’t show any clinical case experiencing the development of myeloma from MGUS. Nevertheless, we chose to include RAS mutational analyzes for MGUS as well since this pre-malignant condition also concerns the B lineage and may result in similar problems. Clearly, these results at this point do not prove an increased risk for myeloma development in patients with RAS mutant MGUS on BRAFi, but in our view they should sound a note of caution.
Taken together, our study highlights several important points. First, caution must be exercised when using BRAFis in patients or populations that might harbor RAS mutant cells in skin, bowel, or other sites. Vigilant surveillance will remain critical in clinical protocols using these agents and even screening for such mutations (e.g., in circulating blood DNA) should be considered. Second, drug safety is likely to vary depending on the targeted molecule within a given signaling pathway, and although paradoxical activation of the MAPK pathway occurs with BRAF inhibition, the addition of a MEKi to a BRAFi might not be sufficient to overcome the paradoxical and unwanted ERK phosphorylation. Third, to prevent paradoxical stimulation in patients vitally requiring BRAFi treatment while harboring a pre-cancerous lesion with activating RAS mutation, a new generation of BRAFi termed “paradox breakers,” such as PLX8394 (Plexxikon®), is under development and will hopefully allow safe application of BRAFi in these vulnerable patients (21, 22).
Careful clinical monitoring and translational studies will be required to foster our understanding of safe application of BRAF inhibitors in routine clinical practice.
The datasets generated for this study can be found in the European Nucleotide Archive (ENA), ID: PRJEB36480.
The studies involving human participants were reviewed and approved by the Ethikkommission der Ärztekammer Hamburg. The patients/participants provided their written informed consent to participate in this study.
AS and MB: conception and design. DS and MB: development of methodology. DS, HI, AS, MB, and CB: acquisition and interpretation of data, writing, review, and/or revision of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grant BI 1711/4-1 of the German Research Foundation to MB.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Tiacci E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MP, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med. (2011) 364:2305–15.
2. Hoeflich KP, Herter S, Tien J, Wong L, Berry L, Chan J, et al. Antitumor efficacy of the novel RAF inhibitor GDC-0879 is predicted by BRAFV600E mutational status and sustained extracellular signal-regulated kinase/mitogen-activated protein kinase pathway suppression. Cancer Res. (2009) 69:3042–51. doi: 10.1158/0008-5472.can-08-3563
3. Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. (2010) 464:431–5. doi: 10.1038/nature08833
4. Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. (2010) 464:427–30. doi: 10.1038/nature08902
5. Oberholzer PA, Kee D, Dziunycz P, Sucker A, Kamsukom N, Jones R, et al. RAS mutations are associated with the development of cutaneous squamous cell tumors in patients treated with RAF inhibitors. J Clin Oncol. (2012) 30:316–21. doi: 10.1200/jco.2011.36.7680
6. Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. (2012) 366:207–15.
7. Zimmer L, Hillen U, Livingstone E, Lacouture ME, Busam K, Carvajal RD, et al. Atypical melanocytic proliferations and new primary melanomas in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. (2012) 30:2375–83. doi: 10.1200/jco.2011.41.1660
8. Carlino MS, Kwan V, Miller DK, Saunders CA, Yip D, Nagrial AM, et al. New RAS-mutant pancreatic adenocarcinoma with combined BRAF and MEK inhibition for metastatic melanoma. J Clin Oncol. (2015) 33:e52–6. doi: 10.1200/jco.2013.51.5783
9. Grey A, Cooper A, McNeil C, O‘Toole S, Thompson J, Grimison P. Progression of KRAS mutant pancreatic adenocarcinoma during vemurafenib treatment in a patient with metastatic melanoma. Int Med J. (2014) 44:597–600. doi: 10.1111/imj.12415
10. Callahan MK, Rampal R, Harding JJ, Klimek VM, Chung YR, Merghoub T, et al. Progression of RAS-mutant leukemia during RAF inhibitor treatment. N Engl J Med. (2012) 367:2316–21. doi: 10.1056/nejmoa1208958
11. Andrews MC, Behren A, Chionh F, Mariadason J, Vella LJ, Do H, et al. BRAF inhibitor-driven tumor proliferation in a KRAS-mutated colon carcinoma is not overcome by MEK1/2 inhibition. J Clin Oncol. (2013) 31:e448–51. doi: 10.1200/jco.2013.50.4118
12. Amaravadi RK, Hamilton KE, Ma X, Piao S, Portillo AD, Nathanson KL, et al. Multiple gastrointestinal polyps in patients treated with BRAF inhibitors. Clin Cancer Res. (2015) 21:5215–21. doi: 10.1158/1078-0432.CCR-15-0469
13. Xu C, Gu X, Padmanabhan R, Wu Z, Peng Q, DiCarlo J, et al. smCounter2: an accurate low-frequency variant caller for targeted sequencing data with unique molecular identifiers. Bioinformatics. (2019) 35:1299–09. doi: 10.1093/bioinformatics/bty790
14. Rossi A, Voigtlaender M, Janjetovic S, Thiele B, Alawi M, Marz M, et al. Mutational landscape reflects the biological continuum of plasma cell dyscrasias. Blood Cancer J. (2017) 7:e537. doi: 10.1038/bcj.2017.19
15. Oberle A, Brandt A, Voigtlaender M, Thiele B, Radloff J, Schulenkorf A, et al. Monitoring multiple myeloma by next-generation sequencing of V(D)J rearrangements from circulating myeloma cells and cell-free myeloma DNA. Haematologica. (2017) 102:1105–11. doi: 10.3324/haematol.2016.161414
16. Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV, et al. MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods. (2015) 12:380–1. doi: 10.1038/nmeth.3364
17. Angelillo P, Capasso A, Ghia P, Scarfò L. Monoclonal B-cell lymphocytosis: does the elderly patient need a specialistic approach? Eur J Int Med. (2018) 58:2–6. doi: 10.1016/j.ejim.2018.09.006
18. Walker BA, Boyle EM, Wardell CP, Murison A, Begum DB, Dahir NM, et al. Mutational spectrum, copy number changes, and outcome: results of a sequencing study of patients with newly diagnosed myeloma. J Clin Oncol. (2015) 33:3911–20. doi: 10.1200/jco.2014.59.1503
19. Voigtlaender M, Vogler B, Trepel M, Panse J, Jung R, Bokemeyer C, et al. Hospital population screening reveals overrepresentation of CD5(-) monoclonal B-cell lymphocytosis and monoclonal gammopathy of undetermined significance of IgM type. Ann Hematol. (2015) 94:1559–65. doi: 10.1007/s00277-015-2409-9
20. Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. (2010) 140:209–21. doi: 10.1016/j.cell.2009.12.040
21. Tutuka CSA, Andrews MC, Mariadason JM, Ioannidis P, Hudson C, Cebon J, et al. PLX8394, a new generation BRAF inhibitor, selectively inhibits BRAF in colonic adenocarcinoma cells and prevents paradoxical MAPK pathway activation. Mol Cancer. (2017) 16:112. doi: 10.1186/s12943-017-0684-x
Keywords: chronic lymphocytic leukemia, monoclonal B cell lymphocytosis, BRAF inhibition, hairy cell leukemia, RAS mutation
Citation: Simnica D, Ittrich H, Bockemeyer C, Stein A and Binder M (2020) Targeting the Mutational Landscape of Bystander Cells: Drug-Promoted Blood Cancer From High-Prevalence Pre-neoplasias in Patients on BRAF Inhibitors. Front. Oncol. 10:540030. doi: 10.3389/fonc.2020.540030
Received: 03 March 2020; Accepted: 13 August 2020;
Published: 11 September 2020.
Edited by:
Olivier Feron, Université catholique de Louvain, BelgiumReviewed by:
Ricardo Martinez, Eli Lilly, United StatesCopyright © 2020 Simnica, Ittrich, Bockemeyer, Stein and Binder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mascha Binder, bWFzY2hhLmJpbmRlckB1ay1oYWxsZS5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.