 Bi Chen1†
Bi Chen1† Peng Ye
Peng Ye Wen-Hao Yang
Wen-Hao Yang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 04 September 2020
Sec. Molecular and Cellular Oncology
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.01657
This article is part of the Research Topic Estrogen and Progesterone Axis in Human Cancers View all 6 articles
Estrogen and progesterone regulate the growth and development of human tissues, including the reproductive system and breasts, through estrogen and progesterone receptors, respectively. These receptors are also important indicators for the clinical prognosis of breast cancer and various reproductive cancers. Many studies have reported that cancer stem cells (CSCs) play a key role in tumor initiation, progression, metastasis, and recurrence. Although the role of estrogen and progesterone in human organs and various cancers has been studied, the molecular mechanisms underlying the action of these hormones on CSCs remain unclear. Therefore, further elucidation of the effects of estrogen and progesterone on CSCs should provide a new direction for developing pertinent therapies. In this review, we summarize the current knowledge on the estrogen and progesterone axis involved in cancer stemness and discuss potential therapeutic strategies to inhibit CSCs by targeting relevant pathways.
Steroid hormones play a major role in the pathogenesis and progression of breast, ovarian, and other types of cancers. They can be simply divided into sex hormones, which include male hormones (androgen) and female hormones (estrogen and progesterone), and corticosteroids, which are grouped into glucocorticoids and mineralocorticoids. These hormones generally regulate cell function by activating nuclear steroid receptors, which can be classified into five types: estrogen receptors (ERs), progesterone receptors (PRs), androgen receptors, glucocorticoid receptors, and mineralocorticoid receptors (1). However, steroid hormones can achieve rapid regulation of cell activity through membrane receptors (2). Emerging research has shown that female hormones are involved in the proliferation, differentiation, and metastasis of cancer stem cells (CSCs), which can self-renew, possess a tumor-initiating ability, and contribute to tumor development and recurrence. Their phenotype is dynamically regulated by cell signaling transduction and the tumor microenvironment (TME) (3, 4). In the TME, tumor-associated fibroblasts (TAFs), macrophages (TAMs) and mesenchymal stem cells secrete interleukin 6 (IL-6), interleukin 8 (IL-8), and chemokine (C-X-C motif) ligand 7 (CXCL7), stimulating the self-renewal of CSCs (3). In addition, the TME comprises stromal cells and secretes various cytokines, growth factors, and proteases, including platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), and matrix metalloproteinases. These proteins promote tumor invasion and eventual metastasis (5, 6). Various cancers of the reproductive organs are particularly vulnerable to estrogen and progesterone through ERs and PRs, respectively. In the classical model for steroid hormone signaling, hormones enter the cells through binding to the compatible receptors on the plasma membrane, leading to the internalization. These complexes then bind directly to DNA response elements, such as estrogen response elements, and regulate the nuclear transcription of target genes, thereby changing the biological response of CSCs (7–9). Estrogen and progesterone also participate in the regulation of CSC populations through a paracrine manner (10, 11). Moreover, microRNAs (miRNAs) are involved in the regulation of the sex hormone axis in CSCs. They are small non-coding RNAs that can silence their cognate target genes by degrading mRNA molecules or inhibiting their translation (12). MiRNAs have been reported to contribute to the regulation of cancer stemness and metastasis, suggesting that they may function as oncogenes or tumor suppressors (13, 14). Both estrogen and progesterone can regulate self-renewal of CSCs by miRNA expression (15–18).
Hormone-related cancers such as breast cancer depend on estrogen signaling and therefore respond to endocrine therapies that block estrogen signaling. Endocrine therapy is a key treatment for hormone-related cancers and has proven clinical benefits (19, 20). However, hormone-related tumors often show endocrine resistance. Numerous mechanisms, including receptor mutations (21, 22) and crosstalk in other signaling pathways (23), have been proposed to explain the emergence of endocrine resistance. Increasing evidence suggests that CSCs play a critical role in endocrine therapy resistance (24–27). During the formation of CSC-like cells, cancer cells undergo cell reprogramming, which resets the differentiated cells to a pluripotent state through nuclear transfer, cell fusion, and overexpression of transcription factors, such as octamer-binding transcription factor 4 (Oct-4), sex-determining region Y-Box 2 (SOX2), Kruppel-like factor 4 (KLF4), and c-MYC (or OSKM) (28). Also known as stemness markers, these transcription factors are associated with endocrine therapy resistance (29, 30). The mechanism of endocrine resistance in CSCs also includes the upregulation of self-renewal signaling pathways such as Wnt and Hedgehog (31–33). Progesterone and estrogen are the most essential hormones that regulate the stem cells of the human reproductive system (34–36). They can induce CSC proliferation and increase the risk of reproductive cancers (17, 37, 38). Therefore, both estrogen and progesterone appear to be key regulators that control the number and function of CSCs. It is thus pivotal to explore the relationship between female hormones and CSCs to develop more cancer therapies that target CSCs.
CSCs, a subpopulation of cancer cells that can self-renew, exist in most cancer types and promote tumorigenesis, tumor drug resistance, metastasis, and relapse. They are also known as tumor-initiating or sphere-forming cells, and they can be isolated from most types of human cancers, including breast, brain, liver, lung, stomach, colon, prostate, pancreas, and head and neck cancers (39). CSCs usually have some characteristics in common with normal stem cells, including relative quiescence, an active DNA repair system, aggressiveness, and drug resistance (40, 41). Tumor resistance to radiotherapy and chemotherapy and recurrence can be attributed to the presence of CSCs (42). The reason why CSCs are inherently resistant to chemotherapy and radiotherapy is mainly because of quiescence, during which CSCs exhibit a slow growth rate, with the cell population maintained in the G0 phase (43). In addition, CSCs have the plasticity to change from the state of quiescence to that of continuous differentiation and proliferation in response to conventional chemotherapy and radiotherapy, leading to tumor recurrence (44). Some studies have also reported that CK5+ breast cancer cells possess CSC-like properties and that progesterone treatment can induce the conversion of ER+/PR+/CK5− cells to ER−/PR−/CK5+ cells, which represent a relatively quiescent state, leading to resistance to endocrine therapy and chemotherapy (45–47). These findings imply that female hormones regulate the fate of CSCs.
Because CSCs display plasticity and are a small population of cells within a tumor, it is difficult to accurately identify and eradicate them. At present, the expression of cell surface markers is often used to identify CSCs in preclinical cell models (48). In patients with colorectal cancer, Lgr5 is an important biomarker of colorectal CSCs, and detecting Lgr5+ CSCs is a crucial indicator to predict tumor recurrence (49). ATP-binding cassette subfamily G member 2 (ABCG2) has been found to play an integral role in the molecular mechanisms underlying multidrug resistance in CSCs (50). B-cell specific Moloney murine leukemia virus integration site 1 (BMI-1) is a stemness-related biomarker that can maintain self-renewal of CSCs (39, 51). CXC chemokine receptor 4 (CXCR4) is an another stemness-related biomarker associated with tumor growth, invasion, metastasis, and relapse (39, 52). Moreover, cell surface markers such as CD24, CD26, CD44, CD90, CD133, CD166, CD177, aldehyde dehydrogenase 1 (ALDH1), and epithelial cell adhesion molecule (EPCAM) have been identified as biomarkers of CSCs in various types of cancers (39, 53–55). Table 1 presents a summary of the currently known CSC biomarkers and their characteristics. CSCs can, however, also be found in single CSC marker–negative cell populations, which means that any single CSC marker does not cover all CSC populations. Therefore, multiple CSC markers are needed to encompass most CSC subsets. For example, CD44+/CD24−/ALDH1+ breast cancer cells represent the most enriched set of breast cancer stem cells (BCSCs). This method of combining multiple CSC markers to define CSC populations is thus highly predictive of tumor malignancy (56).
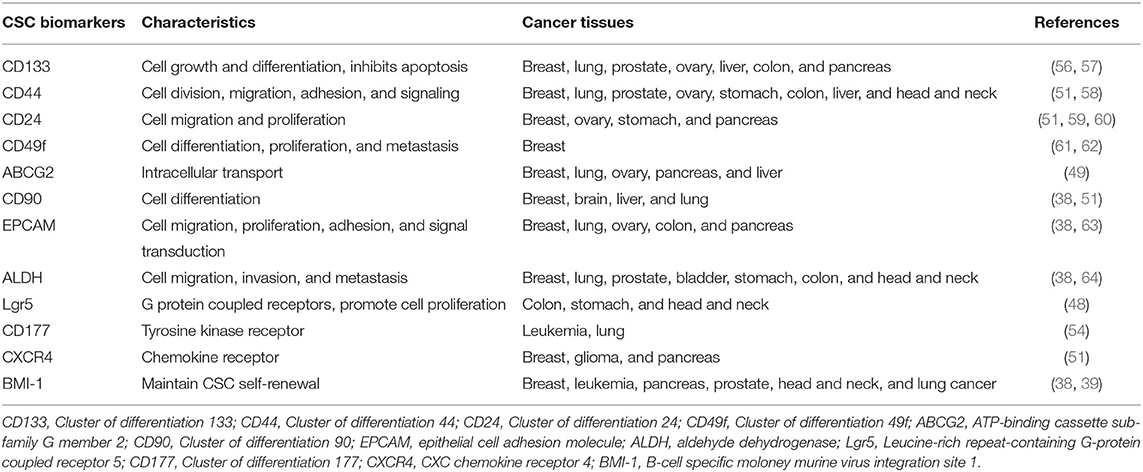
Table 1. CSC biomarkers and their related characteristics.
Because CSCs promote tumor recurrence and progression, it is particularly important to eradicate them. At present, many drugs against CSCs have been developed that target the mechanisms regulating CSCs. For example, drugs targeting CSC-associated surface markers include anti-EPCAM, anti-CD16, and anti-CD47. Clinical trials have also investigated other anti-CSC drugs that target developmental pathways, upregulated apoptotic pathways, the TME, and upregulated drug efflux pumps (44). However, because these cells display plasticity, no specific surface markers have been identified as yet; furthermore, considering the complexity of the regulatory pathways involved in their regulation, it is challenging to completely eradicate CSCs. Therefore, we must obtain a better understanding of the regulatory mechanisms of CSCs to develop more effective therapies for eliminating them. Herein we focus on discussing the effects of estrogen and progesterone on CSCs.
Estrogen exerts its biological function by binding to ERs, which are generally composed of membrane ERs (mostly G protein–coupled receptors) and nuclear ERs (ERα and ERβ) (65, 66). Estrogen primarily regulates CSCs through these receptors.
The conventional model of estrogen signaling is direct genomic signaling; in this process, estrogen binds to ERα or ERβ to promote DNA transcription in the nucleus (66). ERα and ERβ share common structural features that are characterized by several functional domains, and they maintain receptor-specific signal transduction through exclusive elements (65, 67, 68). Estradiol (E2) is a steroidal estrogen with two subtypes, 17α-estradiol (17α-E2), and 17β-estradiol (17β-E2). 17α-E2 is a naturally occurring enantiomer of 17β-E2 and possesses low activity to activate ERα and ERβ, the classical estrogen receptors. The downstream signaling and physiological functions of endogenous 17α-E2 are unclear (69, 70). Rather than 17α-E2, 17β-E2 is generally considered to be the physiological form of estrogen; it typically activates estrogen receptors due to its high affinity with ERα and ERβ (69). Furthermore, because 17β-E2 has a higher binding affinity for ERα than for ERβ, estrogen mainly functions in concert with ERα to perform its biological functions (71, 72). For example, estrogen induces the binding of ERα to the promoter region of piwi-like RNA-mediated gene silencing 1 (PIWIL1), a critical gene for stem cell self-renewal, leading to the overexpression of PIWIL1 in endometrial cancer cells and stimulating cancer cell proliferation (73, 74). However, most CSCs are ER−; accordingly, the estrogen signal is mediated through paracrine signaling from non-CSCs (expressing ER or PR) to CSCs (10, 75, 76). The fibroblast growth factor (FGF)/Tbx3 signaling, epidermal growth factor (EGF), and Notch signaling pathways operate downstream of estrogen in the regulation of ER− CSCs (77, 78) (Figure 1). In addition, estrogen can reduce the proliferation and self-renewal capacity of CSCs by downregulating the embryonic stem cell genes NANOG, OCT4, and SOX2 (79). This explains why patients with an ER+ tumor tend to have better prognoses than other patients (80, 81). Contradictory results reported by these studies can be attributed to differences in experimental designs. Moreover, a few studies have reported that estrogen itself does not change the stem characteristics of stem or progenitor cells (45, 46, 82). Therefore, further studies are warranted to validate the effects of estrogen on CSCs.
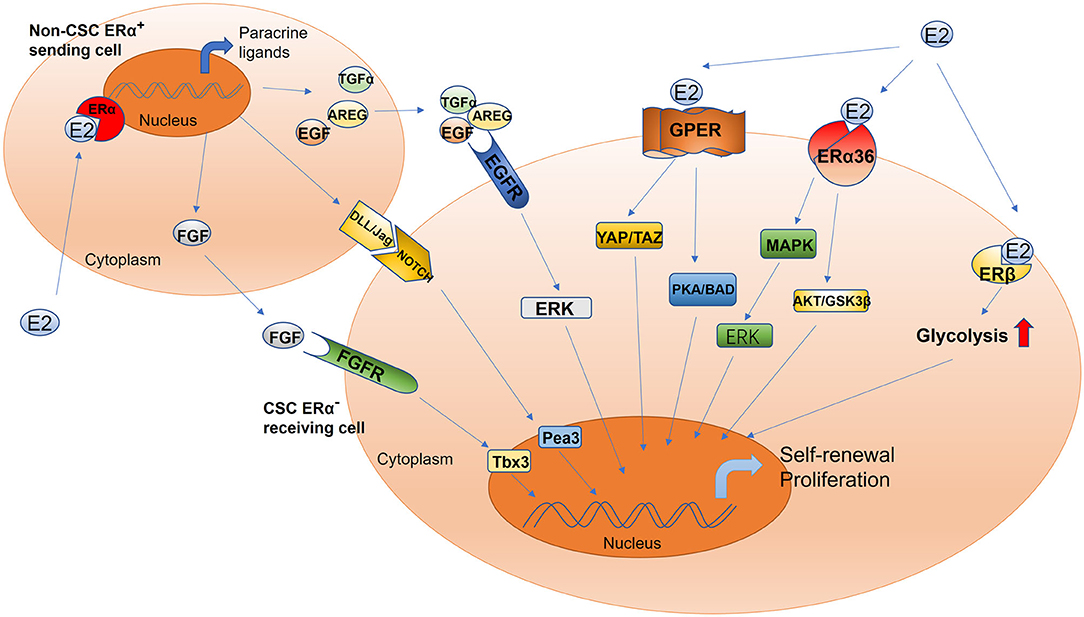
Figure 1. Schematic of estrogen-mediated paracrine signaling and major pathways for the activation of different estrogen receptor isoforms in CSCs. The sending cell (non-CSC) responds to estradiol and initiates the production of the fibroblast growth factor receptor, epidermal growth factor receptor, and Notch ligands. The signal cascade is initiated within the receiving cell (CSC), including but not limited to ERK, Tbx3, and Pea3 signaling. This mechanism drives increased CSC activity. Estrogen promotes the proliferation and self-renewal of CSCs through different estrogen receptor isoforms, including GPER, ERα36, and ERβ. GPER, G-protein coupled receptor; ERα36, estrogen receptor alpha 36; ERβ, estrogen receptor beta; YAP, Yes-associated protein; TAZ, tafazzin; PKA, protein kinase A; BAD, Bcl-2 antagonist of cell death; AKT, also known as protein kinase B; GSK-3β, glycogen synthase kinase-3 beta; ERK, extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinase; Tbx3, T-box transcription factor; Pea3, polyomavirus enhancer activator 3; AREG, amphiregulin; TGFα, transforming growth factor alpha; FGF, fibroblast growth factor; EGF, epidermal growth factor; DLL/JAG, Notch ligands Jagged (Jag) and Delta-like (Dll).
Although estrogen can bind to nuclear ERs to regulate CSCs, this nuclear, transcriptional action cannot account for all the biological functions of ERs (66). Although most CSCs are ER− (10, 75), estradiol can nevertheless increase the number of CSCs, possibly via the non-genomic signaling pathway, which is a regulatory mechanism that does not involve direct binding to DNA and only occurs in the plasma membrane and cytoplasm (66, 83, 84). The non-genomic estrogen signals that regulate CSCs are mainly mediated by cell membrane–associated ERs, including G protein–coupled ER (GPER/GPR30) (85–87), ERα variant ERα36 (88–90), and ERβ (91) (Figure 1).
The estrogen membrane receptor GPER/GPR30 can be activated by estrogen and mediates rapid non-genomic signaling. GPER/GPR30 is a transmembrane receptor expressed by both ER+ and ER− breast cancer cells and plays a role in breast cancer development (92). Hu et al. demonstrated that estrogen can activate tafazzin (TAZ) through GPR30 to regulate the Hippo pathway and promote breast cancer cell proliferation and migration as well as tumor growth (86). TAZ, a GPER-induced transcription coactivator, is also associated with the self-renewal and tumor-initiating capacities of CSCs (93). TAZ is highly expressed in BCSCs; the downregulation of TAZ expression in these cells can significantly reduce their sphere-forming ability and chemotherapy resistance (94, 95). Further, estrogen can reportedly increase the number of CSCs by activating TAZ, thereby promoting chemotherapy resistance. Huang et al. proved that GPER activation by estrogen suppressed triple-negative breast cancer (TNBC) MDA-MB-231 cell proliferation, migration, invasion, and angiogenesis as well as the process of epithelial–mesenchymal transition by regulating the miR-199a-3p/CD151 axis and inactivating the Hippo signaling pathway; they also found that miR-199a-3p overexpression distinctly inhibited the Hippo pathway by downregulating the expression of Yes-associated protein 1 (YAP1) in MDA-MB-231 cells (85). YAP1 is a major downstream effector of the Hippo pathway; it is a recognized oncogene and plays a key role in regulating stem cell self-renewal and differentiation (96–100). Therefore, estrogen's ability to activate GPER to regulate the CD151/miR-199a-3p biological axis and inhibit the Hippo pathway may be related to its role in regulating CSCs. However, further studies are needed to validate the accuracy of this hypothesis. In addition, Chan et al. demonstrated that estrogen activates GPER-mediated protein kinase A (PKA)/Bcl-2 antagonist of cell death (BAD) signaling, and this also plays a crucial role in maintaining the stemness of BCSCs. GPER activation by estrogen reportedly induces PKA and BAD–Ser118 phosphorylation to promote the proliferation of BCSCs (87). In general, estrogen can positively regulate CSCs through GPER/GPR30; although the underlying mechanism is not particularly well-understood, it can still provide clues to facilitate the development of targeted drugs.
The most commonly found isoform of ERα is the 66-kDa protein ERα66; other forms include ERα36, a variant of ERα with a molecular weight of 36 kDa, which is a plasma membrane–based receptor in some human tissues and breast cancer cell lines with or without ERα66 (90, 100, 101). Despite ERα36 lacking both AF-1 and AF-2 transactivation domains of the full-length ERα66 and transcriptional activity, it retains DNA binding, dimerization, and ligand-binding domains (102, 103). In the absence of full-length ERs, estrogen can mediate rapid non-genomic estrogen signals to regulate CSCs through ERα36 (90). ERα36-mediated rapid estrogen signaling via the AKT/GSK3β pathway is known to positively regulate ER+ breast cancer stem/progenitor cells (88). ERα36 can also enhance the self-renewal capacity of CSCs to promote tamoxifen resistance; ERα36 downregulation can significantly reduce the number of CSCs and block an increase in their population and the formation of tumorspheres, thus overcoming tamoxifen resistance (88, 104, 105). Increased expression of ERα36 is one of the underlying mechanisms of tamoxifen resistance, and this indicates that ERα36 can serve as a therapeutic target for inhibiting this resistance (106, 107). In addition, knocking out the expression of ERα36 in the HER2+ breast cancer cell line SKBR3 has been reported to reduce HER2 expression, and the number of ALDHHigh cells also decreases (89). In summary, estrogen-activated ERα36 can positively regulate CSCs, and ERα36 downregulation can suppress cancer stemness (108). Thus, ERα36 can be used to target CSCs (109, 110).
ERβ is mainly expressed in the nucleus, but it can also be found in the cytoplasm and plasma membrane, where it can mediate non-genomic estrogen signals (111). In comparison with ERα, the expression level of ERβ evidently decreases with the progression of cancer. Even so, ERβ participates in regulating CSCs, and thus, it is gradually attracting research attention (110). Through single-cell analysis, human BCSCs sorted by fluorescence-activated cell sorting have been compared with total tumor cells, and the expression of the ERβ gene was found to be significantly upregulated (112). With regard to the potential of ERβ as a stemness marker, Ma et al. found that the expression of ERβ was closely related to that of the CSC markers CD44 and ALDH1 in the absence of ERα and is essential for the growth of mammospheres (113). Moreover, ERβ is reportedly responsible for the upregulation of glycolysis. The maintenance of the phenotype of BCSCs depends on ERβ-mediated glycolysis. Thus, ERβ can be considered a stemness marker in CSCs (113). Human prostate CSCs and papillary thyroid CSCs highly express ERβ (114, 115). ERβ overexpression in CSCs can promote cancer stemness through estrogen signaling (91, 115). Thus, estrogen can directly regulate CSCs by activating ERβ. However, contradictory results have been reported too: some studies have suggested that ERβ is responsible for repressing proliferation and inducing apoptosis (116, 117) and that upregulated ERβ levels are positively correlated with better disease-free survival (118). Moreover, many studies have reported that activated ERβ inhibits cancer stemness and induces apoptosis of CSCs in mouse as well as human tumor models (88, 89). Based on the inhibitory effects of ERβ on CSCs and considering that the current method of managing prostate diseases is to prevent cell regeneration, targeting ERβ appears promising (119, 120). At present, the known ERβ signaling mechanism in breast and other types of cancers is not as clear as the ERα signaling mechanism; therefore, the regulation of CSCs by ERβ signaling demands further explorations.
Estrogen can also affect cancer stemness by regulating the expression of miRNA. For example, estrogen has been reported to increase the expression of miR-21 and reduce self-renewal of cancer cells with stem cell–like properties by inhibiting the translation of the stem cell genes Oct-4, c-Myc, Nanog, and Sox2 (15). In addition, stimulating ERα+ breast cancer cells with estrogen reportedly promotes tumor-initiating cell renewal via the suppression of miR-140 expression (16). Moreover, ovarian cancer may be caused by ovarian cancer-initiating cells characterized by surface antigen CD44 and the ovarian CSC maker c-KIT (CD117). Estrogen-induced expression of the transcription factor E2F6 by inhibiting miRNA-193a activity upregulates c-KIT to promote ovarian tumorigenesis (121, 122). Thus, estrogen can regulate the formation and differentiation of CSCs by affecting the expression level of miRNA.
Altogether, the signal transduction of estrogen in CSCs is complex, and accordingly, more studies need to be conducted to completely elucidate the effects of estrogen on CSCs. Whether ERs can be used as a target to steadily regulate CSCs also demands further investigations.
The effects of estrogen on CSCs are not completely clear. The regulation of CSCs by estrogen requires ERs. At present, anti-estrogen drugs that target and regulate ER activity are mainly divided into three categories: selective ER modulators (SERMs), selective ER downregulators (SERDs), and aromatase inhibitors (123). Using these to treat patients with ER+ tumors has undoubtedly led to great success, but endocrine resistance often occurs post treatment. As most CSCs are ER−, they are not the targets of anti-estrogen therapy. With these treatments, not only are CSCs uninhibited but also significantly enriched. Letrozole, an aromatase inhibitor, can stimulate an increase in the number of CD44+/CD24− breast cancer cells and the formation of mammospheres after treatment (124). In addition, anti-estrogen treatment (tamoxifen) can increase the number of CSCs and promote formation of tumorspheres. Further, it has been reported that the expression of the embryonic stem cell marker SOX2 and subsequent activation of the WNT signaling pathway play a key role in inducing drug resistance after tamoxifen treatment (125). Moreover, treatment with the anti-estrogen drugs tamoxifen or fulvestrant decreases cell proliferation but increases the BCSC population through JAG1–NOTCH4 receptor activation (10). Altogether, these findings confirm that inhibiting estrogen signaling in cancer cells may promote the stemness of CSCs, causing resistance to endocrine therapies. Treating patients with cancer using anti-estrogen drugs alone is not ideal. Identifying additional mechanisms responsible for resistance to endocrine therapy and combining anti-estrogen therapy with blockade of these mechanisms are bound to increase treatment efficacy (78, 126).
Anti-estrogen drugs do not effectively inhibit CSCs according to current research and thus have limited clinical application. These drugs often lead to drug resistance and tumor recurrence in patients with cancer. Therefore, new therapeutic strategies and therapies that can effectively and specifically block estrogen-induced cancer stemness are urgently required. In recent years, other drugs or genes have also been studied for their potential to block estrogen-induced cancer stemness. Oct-4 is a key transcription factor associated with the pluripotent and self-renewal characteristics of embryonic stem cells, germ cells, and adult human stem cells (127, 128). The ablation of Oct-4 expression in MCF-7 breast cancer cells causes apoptosis of CSCs and inhibits tumor growth (129). Metformin, an antidiabetic drug, has recently been reported to reduce tumor risk in some cancers associated with diabetes, including breast cancer (130, 131). Metformin can also inhibit the expression of Oct-4 in estrogen-induced CD44+/CD24−/low MCF-7 cells and significantly reduce the size and number of their mammospheres (132). Melatonin and tocopherols have also been shown to inhibit cancer stemness. These three drugs can inhibit the binding of ER-estrogen complexes to Oct-4 promoter regions to reduce Oct-4 expression, thereby inhibiting the self-renewal of CSCs (132–134). In addition, the let-7 miRNA family is involved in carcinogenesis and tumor progression by inducing CSC differentiation. A study showed that let-7c, a member of the let-7 family, inhibits estrogen-induced Wnt signaling by reducing ERα expression, then downregulates the self-renewal ability of CSCs (14). ABCG2 is used as a surface marker to isolate CSCs from cancer cells (135). Estrogen promotes cell proliferation by upregulating ABCG2, which can be suppressed by reserpine, in endometrial cancer cells (136). Although these drugs have been reported to block estrogen-induced cancer stemness, they have not been tested in vivo, and the studies only used a single cell line. Palbociclib, a cyclin-dependent kinase 4/6 (CDK4/6) inhibitor, has been used to treat patients with ER+ and HER2− advanced breast cancer. Palbociclib can inhibit the expression of cyclin-dependent kinase 4 (CDK4) and reduce the proportion of estrogen-induced CSCs in ER+ and HER2− breast cancer cell lines (137). Furthermore, the importance of phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) signaling for maintaining the CSC phenotype in renal cell cancer (138), prostate cancer (139), and lung cancer (140) has been confirmed. In breast cancer, the increase of CSC populations from tamoxifen treatment can be prevented with mTOR inhibitors (141). Taken together, these anti-CSC drugs are promising, but further research is required before they can be clinically used. Their underlying mechanisms should be thoroughly explored in the future, and their effectiveness and safety carefully evaluated in well-designed clinical trials.
In the classical progesterone signaling pathway, progesterone exerts its biological functions mainly by binding to nuclear PRs. In the regulatory pathways of CSC activity, progesterone plays a role by binding to not only nuclear PRs but also cell membrane PRs (mPRs) (142). Moreover, different isoforms and post-transcriptional modification of PRs are related to CSC activity (143). However, most CSCs are PR−; progesterone regulates CSC activity through paracrine actions between PR+ and PR− cells (17). Some studies have reported that progesterone can also regulate the miRNA expression involved in CSC proliferation and formation (18, 144). In addition, the complex relationship between progesterone and other hormones, such as prolactin and growth hormones (GHs), also affects CSC activity (47, 145). Considering these preclinical mechanisms for regulating CSCs, some treatment strategies can be designed to block cancer stemness by targeting the progesterone signal.
The PR+ phenotype of cancer cells usually indicates a good response to endocrine therapy and better prognosis in clinical tumors. However, in the case of advanced breast cancer, PRs become a critical factor that promotes the generation of CSCs and results in poor prognoses. Nuclear PRs have two isoforms: PR-A (94 kDa) and PR-B (114 kDa). These are transcribed from the same gene by two distinct promoters, resulting in different transcriptional and functional activities (146). The level and ratio of PR-A and PR-B in reproductive tissues vary based on developmental stage and hormonal status (147, 148). In normal breast cells, the isoforms are coexpressed at similar levels, but in breast cancer cells, the ratio is disrupted, with PR-A being overexpressed (35, 149). In a T47D cell model of breast cancer, PR-A was found to dominantly drive CSC expansion, and PR-B enhanced anchorage-independent proliferation. Furthermore, in comparison with PR-B+ tumorspheres, PR-A+ tumorspheres comprise more CSC populations, such as ALDH1+, CD44+/CD24−, and CD49f+/CD24− cell populations, and the expression of CSC-related genes, such as FOXO1, p21, KLF4, PTGES, WNT4, and NOTCH2 is enhanced (143). In addition, high PR-A expression is more likely to cause tumor recurrence after treatment with tamoxifen (150). These results suggest that different isoforms are associated with distinct CSC populations and tumor recurrence. At present, the functional activity of progesterone is mainly assessed by measuring total PR expression, but the expression of PR isoforms is neglected. It is easy to differentiate between PR-A and PR-B using western blotting because their molecular weights markedly differ. Immunohistochemistry (IHC) can also be used to identify them in clinical biopsy specimens (151), but antibodies used for the detection of PR-A have been found to detect not only PR-A but also PR-B (152). Thus, developing new antibodies to distinguish one isoform from the other is pivotal for predicting tumor malignancy and for the clinical prognosis of patients.
The post-translation modification process of PRs can also influence the generation of CSCs. PRs undergo extensive modifications post-translation, including phosphorylation, acetylation, ubiquitination, SUMOylation, and methylation (153, 154). The phosphorylation of PR-A Ser294 is necessary for the characteristics of CSCs; the mutation of PR-A Ser294 to Ala (S294A) can evidently prevent CSC expansion and promote cancer cell proliferation (143, 155). How other post-translational modifications of PRs affect CSC activity remain unknown, and further investigations are thus warranted. Because progesterone exerts its biological function by binding to PRs, the structure and isoform of these receptors can affect progesterone-mediated functions.
PR-A and PR-B are key mediators in the progesterone signal. However, progesterone can also bind to mPRs to exert its biological effects through a non-classical and non-genomic mechanism (156, 157) (Figure 2). This mechanism is characterized by rapid action and does not require much time to induce the transcription and translation of target genes into proteins. The rapid non-nuclear signaling pathways activated by progesterone include the following: the extracellular signal–regulated kinase (ERK) pathway, cyclic AMP/PKA pathway, cyclic GMP/protein kinase G (PKG) pathway, Ca++ influx/protein kinase C (PKC) activation pathway, and phosphoinositide 3-kinase (PI3K)/Akt pathway (158). However, how these pathways affect CSC activity in tumor tissues remains unclear. In the basal-like MCF10A cells lacking nuclear PRs, progesterone activates the PI3K/Akt pathway via mPRs, resulting in the inactivation of FOXO transcriptional activity, downregulation of miRNA-29, and upregulation of KLF4, a transcription factor which is necessary for the maintenance of CSCs (142) (Figure 2). To summarize, the relationship between other non-nuclear signaling pathways and CSCs remains unclear and thus requires further investigations.
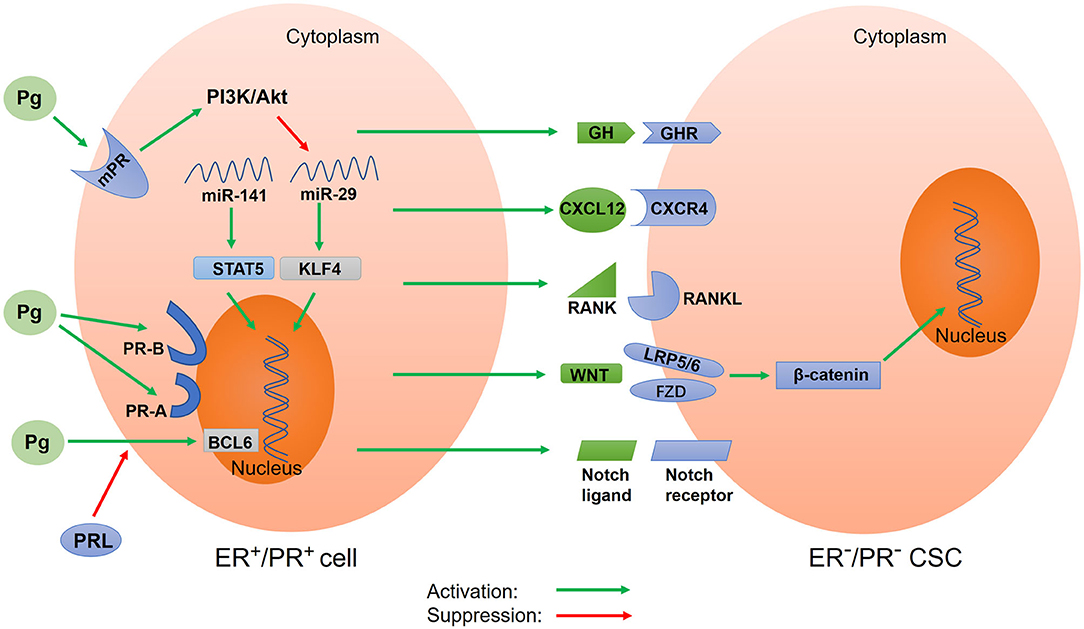
Figure 2. Schematic of progesterone-induced regulatory networks in CSCs. Progesterone binds to nuclear progesterone receptors (PRs) and membrane PRs, regulating the expression of target genes. Different isoforms of nuclear PR (PR-A and PR-B) alter progesterone-mediated functions in CSCs. Progesterone also regulates CSC activity via paracrine signals, including CXCR4/CXCL12, RANK/RANKL, WNT/β-catenin, and Notch signaling. Additionally, progesterone downregulates microRNA to increase the expression of stemness genes. Prolactin inhibits the expression of BCL6 induced by progesterone. In addition, progesterone stimulates the secretion of growth hormone (GH) to promote BCSC properties. Pg, progesterone; mPR, membrane progesterone receptor; PR-A, progesterone receptor isoform A; PR-B, progesterone receptor isoform B; PRL, prolactin; GH, growth hormone; GHR, growth hormone receptor; BCL6, B-cell lymphoma-6; CXCR4, CXC chemokine receptor type 4; CXCL12, CXC chemokine ligand 12; RANK, receptor activator of nuclear factor κB; RANKL, receptor activator of nuclear factor κB ligand; STAT5, activator of transcription 5A; KLF4, Krüppel-like factor 4.
In addition to regulating CSC activity by binding to nuclear PRs and mPRs, progesterone can exert its effects through paracrine activity. Progesterone acts on PR+ cells to cause changes in the surrounding tumor microenvironment, thereby affecting nearby PR− cells. The paracrine signals of progesterone-induced CSC expansion include the receptor activator of nuclear factor-kappa B ligand (RANKL) and WNT4 (Figure 2). The downstream component of the RANKL/RANK signal is the NF-κB pathway, which is an important pathway involved in CSC activity regulation (11). In cases of breast cancer with BRCA1 mutations, activated RANKL/RANK signaling has been reported to increase CSC expansion (159). Further, WNT4, the most typical WNT ligand, can promote the action of progesterone. The WNT/β-catenin signaling pathway is a classical pathway for regulating CSCs. Progesterone stimulates PR− cells through paracrine signals to produce WNT4 receptors, such as the coreceptor LRP5/6 and the cognate receptor frizzled protein (FZD) (34, 35, 160). The WNT4 ligand binds to the cysteine-rich domain of FZD and simultaneously binds to coreceptor LRP5/6 to activate the WNT/β-catenin pathway (32, 161). β-catenin is then released from the degradation complex, entering the nucleus and causing the expression of target genes (162). In T47D breast cancer cells, progesterone activates and upregulates the Notch pathway, which participates in CSC self-renewal (163). A recent study suggested that fallopian tube epithelial cells (FTECs) are the origin of epithelial ovarian cancer and that they can increase the expression of stemness genes (WNT and Notch) post treatment with estradiol or progesterone (164). Interestingly, in a study involving a mouse model, the activation of the Notch pathway in ovaries inhibited progesterone and estrogen secretion (165). This result was also confirmed by another study, which reported that the inhibition of the Notch signal stimulated progesterone secretion (166). This could be because there exists a protective negative feedback mechanism in normal tissues to prevent the overactivation of the Notch pathway. Progesterone-mediated CXC chemokine receptor type 4 (CXCR4) signaling is another paracrine pathway that regulates CSC expansion (Figure 2). The CXCR4 receptor and its CXC chemokine ligand 12 (CXCL12) are key mediators of progesterone-induced normal breast stem/progenitor cell functions (167). It has been confirmed that the CXCR4/CXCL12 pathway can maintain and promote prostate CSCs and enhance radiotherapy resistance (168).
miRNA is a type of non-coding small RNA that affects cancer stemness by regulating gene expression at the transcriptional level (169). Estrogen upregulates the expression of miR-29, which inhibits self-renewal and promotes differentiation; by contrast, progesterone downregulates the expression of miR-29 to enhance CSC characteristics (17) (Figure 2). This downregulation of miR-29 expression by progesterone leads to an increase in the protein level of the transcription factor KLF4, which is necessary for maintaining the pluripotency of CSCs and embryonic stem cells (18). Further, the downregulation of the expression of miR-29 enhances the expansion of CK5+ and CD44+ cancer cells, resulting in increased stem-like properties in vitro and in vivo (170). Progesterone has also been reported to downregulate the expression of miR-141, a member of the miR-200 family of tumor suppressors, leading to an increase in the number of stem-like breast cancer cells (CK5+ and CD44+ cells) (Figure 2). Further, the downregulation of miR-141 expression can upregulate the activator of transcription 5A (STAT5A), which is important for mammary stem cell expansion (144).
The regulation of CSCs by progesterone is affected by prolactin and GH (Figure 2). The B-cell lymphoma-6 (BCL6) gene is an oncogene and a transcriptional repressor that plays a key role in maintaining leukemia stem cells. The upregulation of BCL6 induced by progesterone can promote the generation of the CK5+ stem cell population (47, 171). However, prolactin can inhibit the expression of BCL6 induced by progesterone (47). Further, progesterone stimulates the secretion of GH in human breast epithelial cells to increase the proliferation of GH receptor+ stem/progenitor breast cells (145). In addition, GH can reportedly promote BCSC properties and enhance cancer migration and invasion (172).
Considering the mechanisms by which progesterone promotes CSCs, targeting PRs or downstream effectors of paracrine pathways is a reasonable treatment strategy. A study reported that mifepristone, an anti-progesterone drug that competes with progesterone for PRs, can be used for treating chemotherapy-resistant TNBC (173). Moreover, mifepristone reduces the number of CSCs in cases of TNBC (174). Another study found that mifepristone inhibits the proliferation, migration, and invasion of endometrial cancer cells by blocking the PI3K/AKT pathways (175). Further, a recent genome-wide RNAi study demonstrated that mifepristone is one of the best drugs for inhibiting CSCs (176). In the same study, by integrating RNAi screening results and functional mapping of CSC processes, the authors uncovered some potential therapeutic targets that could regulate the fate of BCSCs. They used a panel of 15 drugs to test these targets and found that mifepristone, salinomycin, and JQ1 showed the best anti-CSC activity. Onapristone is another selective PR antagonist that prevents PR-mediated transcription. It inhibits the nuclear translocation of phosphorylated PR (S294) (177). Herein, we earlier discussed that PR-A Ser294 phosphorylation is necessary for CSC development. Therefore, onapristone can disrupt the activities of CSCs by inhibiting the phosphorylation of PR-A Ser294. This conclusion was verified by a study that reported that the combination of onapristone and the FOXO1 inhibitor AS1842856 prevented the formation of tumorspheres in breast cancer cells (143). Besides, as mentioned earlier, activated RANKL/RANK signaling can affect CSC expansion. Therefore, denosumab, an anti-RANKL monoclonal antibody, can inhibit the progression of lung cancer by blocking the RANKL/RANK signaling pathway (178). It has also been demonstrated that the inhibition of RANK signaling markedly reduces the CSC pool and reduces tumor recurrence in the case of breast cancer (179). Hence, treatment with denosumab may inhibit progesterone-mediated CSC activity. In breast cancer with BRCA1 mutations, metformin inhibits RANKL and sensitizes CSCs to denosumab (180). Therefore, the combination of metformin and denosumab appears to be an effective treatment strategy.
To summarize, the potential of targeting the progesterone-induced axis to inhibit CSCs has been proved in preclinical models; future clinical studies should validate pertinent preclinical data. Progesterone is produced by the human body and cannot be targeted by drugs. The present anti-progesterone drugs function by binding to PRs. However, such progesterone antagonists cannot completely block progesterone-mediated CSC activity. Thus, using a combination of drugs that target PRs and the downstream mediators of PR signaling appears to be a better strategy.
Targeting CSCs requires a better understanding of pertinent regulatory mechanisms. Both estrogen and progesterone signals have been shown to regulate CSCs, but many mechanisms remain to be comprehensively understood. Although conventional estrogen antagonists, progesterone modulators, and blocking agents of downstream pathways have been found to inhibit CSC activity in preclinical models, several issues still need to be resolved. First, many regulatory pathways are common between CSCs and normal stem cells, and thus, it is difficult to control side effects when targeting these pathways in a clinical setting. In addition, targeting one of the pathways in the regulatory networks of CSCs may activate other pathways, leading to the persistent generation of more CSCs. To comprehensively elucidate the downstream regulatory mechanisms of the estrogen and progesterone axis, sequencing a single tumor cell with stemness properties for the complete CSC gene map may be a better approach to discover a relatively complete regulatory network (181).
The manuscript was written and edited by BC, PY, YC, XY, and W-HY. TL and J-HC reviewed and edited the manuscript. XY and W-HY supervised the entire work. All authors agree to be responsible for the publication.
This work was supported by Guangzhou Key Medical Discipline Construction Project Fund, National Natural Science Foundation of China (81872138), National Natural Science Foundation of China Youth Project (81802647), Natural Science Foundation of Heilongjiang Province of China (QC2017111), YingTsai Young Scholar Award (CMU108-YTY-04), the Ministry of Science and Technology (MOST 109-2314-B-039-054), and Inha University Research Grant (to J-HC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Due to limited space, we apologize for those publications that may have been ignored in citations.
1. Mahfouz A, Lelieveldt BP, Grefhorst A, van Weert LT, Mol IM, Sips HC, et al. Genome-wide coexpression of steroid receptors in the mouse brain: identifying signaling pathways and functionally coordinated regions. Proc Natl Acad Sci USA. (2016) 113:2738–43. doi: 10.1073/pnas.1520376113
2. Norman AW, Mizwicki MT, Norman DP. Steroid-hormone rapid actions, membrane receptors and a conformational ensemble model. Nat Rev Drug Disc. (2004) 3:27–41. doi: 10.1038/nrd1283
3. Korkaya H, Liu S, Wicha MS. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Investig. (2011) 121:3804–9. doi: 10.1172/JCI57099
4. Ye J, Wu D, Wu P, Chen Z, Huang J. The cancer stem cell niche: cross talk between cancer stem cells and their microenvironment. Tumour Biol. (2014) 35:3945–51. doi: 10.1007/s13277-013-1561-x
5. Koontongkaew S. The tumor microenvironment contribution to development, growth, invasion and metastasis of head and neck squamous cell carcinomas. J Cancer. (2013) 4:66–83. doi: 10.7150/jca.5112
6. Das M, Law S. Role of tumor microenvironment in cancer stem cell chemoresistance and recurrence. Int J Biochem Cell Biol. (2018) 103:115–24. doi: 10.1016/j.biocel.2018.08.011
7. Ranganathan P, Nadig N, Nambiar S. Non-canonical estrogen signaling in endocrine resistance. Front Endocrinol. (2019) 10:708. doi: 10.3389/fendo.2019.00708
8. Yaşar P, Ayaz G, User SD, Güpür G, Muyan M. Molecular mechanism of estrogen-estrogen receptor signaling. Reproductive Med Biol. (2017) 16:4–20. doi: 10.1002/rmb2.12006
9. Levin ER, Hammes SR. Nuclear receptors outside the nucleus: extranuclear signalling by steroid receptors. Nat Rev Mol Cell Biol. (2016) 17:783–97. doi: 10.1038/nrm.2016.122
10. Simões BM, O'Brien CS, Eyre R, Silva A, Yu L, Sarmiento-Castro A, et al. Anti-estrogen resistance in human breast tumors is driven by JAG1-NOTCH4-dependent cancer stem cell activity. Cell Rep. (2015) 12:1968–77. doi: 10.1016/j.celrep.2015.08.050
11. Chen C, Cao F, Bai L, Liu Y, Xie J, Wang W, et al. IKKβ enforces a LIN28B/TCF7L2 positive feedback loop that promotes cancer cell stemness and metastasis. Cancer Res. (2015) 75:1725–35. doi: 10.1158/0008-5472.CAN-14-2111
12. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. (2004) 116:281–97. doi: 10.1016/S0092-8674(04)00045-5
13. Song SJ, Poliseno L, Song MS, Ala U, Webster K, Ng C, et al. MicroRNA-antagonism regulates breast cancer stemness and metastasis via TET-family-dependent chromatin remodeling. Cell. (2013) 154:311–24. doi: 10.1016/j.cell.2013.06.026
14. Sun X, Xu C, Tang SC, Wang J, Wang H, Wang P, et al. Let-7c blocks estrogen-activated Wnt signaling in induction of self-renewal of breast cancer stem cells. Cancer Gene Therapy. (2016) 23:83–9. doi: 10.1038/cgt.2016.3
15. Zhang Y, Eades G, Yao Y, Li Q, Zhou Q. Estrogen receptor α signaling regulates breast tumor-initiating cells by down-regulating miR-140 which targets the transcription factor SOX2. J Biol Chem. (2012) 287:41514–22. doi: 10.1074/jbc.M112.404871
16. Bhat-Nakshatri P, Wang G, Collins NR, Thomson MJ, Geistlinger TR, Carroll JS, et al. Estradiol-regulated microRNAs control estradiol response in breast cancer cells. Nucl Acids Res. (2009) 37:4850–61. doi: 10.1093/nar/gkp500
17. Finlay-Schultz J, Sartorius CA. Steroid hormones, steroid receptors, and breast cancer stem cells. J Mammary Gland Biol Neoplasia. (2015) 20:39–50. doi: 10.1007/s10911-015-9340-5
18. Zhang P, Andrianakos R, Yang Y, Liu C, Lu W. Kruppel-like factor 4 (Klf4) prevents embryonic stem (ES) cell differentiation by regulating Nanog gene expression. J Biol Chem. (2010) 285:9180–9. doi: 10.1074/jbc.M109.077958
19. Ma CX, Sanchez CG, Ellis MJ. Predicting endocrine therapy responsiveness in breast cancer. Oncology. (2009) 23:133–42.
20. Ellis MJ, Tao Y, Luo J, A'Hern R, Evans DB, Bhatnagar AS, et al. Outcome prediction for estrogen receptor-positive breast cancer based on postneoadjuvant endocrine therapy tumor characteristics. J Natl Cancer Institute. (2008) 100:1380–8. doi: 10.1093/jnci/djn309
21. Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. (2013) 4:1116–30. doi: 10.1016/j.celrep.2013.08.022
22. Toy W, Weir H, Razavi P, Lawson M, Goeppert AU, Mazzola AM, et al. Activating ESR1 mutations differentially affect the efficacy of ER antagonists. Cancer Discov. (2017) 7:277–87. doi: 10.1158/2159-8290.CD-15-1523
23. Massarweh S, Schiff R. Resistance to endocrine therapy in breast cancer: exploiting estrogen receptor/growth factor signaling crosstalk. Endocrine-Related Cancer. (2006) 13(Suppl.1):S15–24. doi: 10.1677/erc.1.01273
24. Zhou L, Jiang Y, Yan T, Di G, Shen Z, Shao Z, et al. The prognostic role of cancer stem cells in breast cancer: a meta-analysis of published literatures. Breast Cancer Res Treat. (2010) 122:795–801. doi: 10.1007/s10549-010-0999-4
25. Szostakowska M, Trebińska-Stryjewska A, Grzybowska EA, Fabisiewicz A. Resistance to endocrine therapy in breast cancer: molecular mechanisms and future goals. Breast Cancer Res Treat. (2019) 173:489–97. doi: 10.1007/s10549-018-5023-4
26. Araki K, Miyoshi Y. Mechanism of resistance to endocrine therapy in breast cancer: the important role of PI3K/Akt/mTOR in estrogen receptor-positive, HER2-negative breast cancer. Breast Cancer. (2018) 25:392–401. doi: 10.1007/s12282-017-0812-x
27. Howell A, Wardley AM. Overview of the impact of conventional systemic therapies on breast cancer. Endocrine-Related Cancer. (2005) 12(Suppl.1):S9–16. doi: 10.1677/erc.1.01003
28. Yamanaka S, Blau HM. Nuclear reprogramming to a pluripotent state by three approaches. Nature. (2010) 465:704–12. doi: 10.1038/nature09229
29. Piva M, Domenici G, Iriondo O, Rábano M, Simões BM, Comaills V, et al. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol Med. (2014) 6:66–79. doi: 10.1002/emmm.201303411
30. Loh YN, Hedditch EL, Baker LA, Jary E, Ward RL, Ford CE. The Wnt signalling pathway is upregulated in an in vitro model of acquired tamoxifen resistant breast cancer. BMC Cancer. (2013) 13:174. doi: 10.1186/1471-2407-13-174
31. Yu QC, Verheyen EM, Zeng YA. Mammary development and breast cancer: a Wnt perspective. Cancers. (2016) 8:70065. doi: 10.3390/cancers8070065
32. Roarty K, Rosen JM. Wnt and mammary stem cells: hormones cannot fly wingless. Curr Opin Pharmacol. (2010) 10:643–9. doi: 10.1016/j.coph.2010.07.004
33. Flemban A, Qualtrough D. The potential role of hedgehog signaling in the luminal/basal phenotype of breast epithelia and in breast cancer invasion and metastasis. Cancers. (2015) 7:1863–84. doi: 10.3390/cancers7030866
34. Joshi PA, Jackson HW, Beristain AG, Di Grappa MA, Mote PA, Clarke CL, et al. Progesterone induces adult mammary stem cell expansion. Nature. (2010) 465:803–7. doi: 10.1038/nature09091
35. Graham JD, Mote PA, Salagame U, van Dijk JH, Balleine RL, Huschtscha LI, et al. DNA replication licensing and progenitor numbers are increased by progesterone in normal human breast. Endocrinology. (2009) 150:3318–26. doi: 10.1210/en.2008-1630
36. Asselin-Labat ML, Vaillant F, Sheridan JM, Pal B, Wu D, Simpson ER, et al. Control of mammary stem cell function by steroid hormone signalling. Nature. (2010) 465:798–802. doi: 10.1038/nature09027
37. Sun YS, Zhao Z, Yang ZN, Xu F, Lu HJ, Zhu ZY, et al. Risk factors and preventions of breast cancer. Int J Biol Sci. (2017) 13:1387–97. doi: 10.7150/ijbs.21635
38. Hilton HN, Clarke CL, Graham JD. Estrogen and progesterone signalling in the normal breast and its implications for cancer development. Mol Cell Endocrinol. (2018) 466:2–14. doi: 10.1016/j.mce.2017.08.011
39. Zhao W, Li Y, Zhang X. Stemness-related markers in cancer. Cancer Transl Med. (2017) 3:87–95. doi: 10.4103/ctm.ctm_69_16
40. Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. (2017) 23:1124–34. doi: 10.1038/nm.4409
41. Lytle NK, Barber AG, Reya T. Stem cell fate in cancer growth, progression and therapy resistance. Nat Rev Cancer. (2018) 18:669–80. doi: 10.1038/s41568-018-0056-x
42. Zhao J. Cancer stem cells and chemoresistance: the smartest survives the raid. Pharmacol Therapeutics. (2016) 160:145–58. doi: 10.1016/j.pharmthera.2016.02.008
43. Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells—what challenges do they pose? Nat Rev Drug Discov. (2014) 13:497–512. doi: 10.1038/nrd4253
44. Saygin C, Matei D, Majeti R, Reizes O, Lathia JD. Targeting cancer stemness in the clinic: from hype to hope. Cell Stem Cell. (2019) 24:25–40. doi: 10.1016/j.stem.2018.11.017
45. Goodman CR, Sato T, Peck AR, Girondo MA, Yang N, Liu C, et al. Steroid induction of therapy-resistant cytokeratin-5-positive cells in estrogen receptor-positive breast cancer through a BCL6-dependent mechanism. Oncogene. (2016) 35:1373–85. doi: 10.1038/onc.2015.193
46. Axlund SD, Yoo BH, Rosen RB, Schaack J, Kabos P, Labarbera DV, et al. Progesterone-inducible cytokeratin 5-positive cells in luminal breast cancer exhibit progenitor properties. Hormones Cancer. (2013) 4:36–49. doi: 10.1007/s12672-012-0127-5
47. Sato T, Tran TH, Peck AR, Girondo MA, Liu C, Goodman CR, et al. Prolactin suppresses a progestin-induced CK5-positive cell population in luminal breast cancer through inhibition of progestin-driven BCL6 expression. Oncogene. (2014) 33:2215–24. doi: 10.1038/onc.2013.172
48. Najafi M, Farhood B, Mortezaee K. Cancer stem cells (CSCs) in cancer progression and therapy. J Cell Physiol. (2019) 234:8381–95. doi: 10.1002/jcp.27740
49. Merlos-Suarez A, Barriga FM, Jung P, Iglesias M, Cespedes MV, Rossell D, et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell. (2011) 8:511–24. doi: 10.1016/j.stem.2011.02.020
50. Begicevic RR, Falasca M. ABC transporters in cancer stem cells: beyond chemoresistance. Int J Mol Sci. (2017) 18:112362. doi: 10.3390/ijms18112362
51. Codony-Servat J, Codony-Servat C, Cardona AF, Giménez-Capitán A, Drozdowskyj A, Berenguer J, et al. Cancer stem cell biomarkers in EGFR-mutation-positive non-small-cell lung cancer. Clin Lung Cancer. (2019) 20:167–77. doi: 10.1016/j.cllc.2019.02.005
52. Kim WT, Ryu CJ. Cancer stem cell surface markers on normal stem cells. BMB Rep. (2017) 50:285–98. doi: 10.5483/BMBRep.2017.50.6.039
53. Dianat-Moghadam H, Heidarifard M, Jahanban-Esfahlan R, Panahi Y, Hamishehkar H, Pouremamali F, et al. Cancer stem cells-emanated therapy resistance: implications for liposomal drug delivery systems. Off J Control Release Soc. (2018) 288:62–83. doi: 10.1016/j.jconrel.2018.08.043
54. da Silva-Diz V, Lorenzo-Sanz L, Bernat-Peguera A, Lopez-Cerda M, Munoz P. Cancer cell plasticity: impact on tumor progression and therapy response. Semin Cancer Biol. (2018) 53:48–58. doi: 10.1016/j.semcancer.2018.08.009
55. Foster BM, Zaidi D, Young TR, Mobley ME, Kerr BA. CD117/c-kit in cancer stem cell-mediated progression and therapeutic resistance. Biomedicines. (2018) 6:256099. doi: 10.1101/256099
56. Neumeister V, Agarwal S, Bordeaux J, Camp RL, Rimm DL. In situ identification of putative cancer stem cells by multiplexing ALDH1, CD44, and cytokeratin identifies breast cancer patients with poor prognosis. Am J Pathol. (2010) 176:2131–8. doi: 10.2353/ajpath.2010.090712
57. Barzegar Behrooz A, Syahir A, Ahmad S. CD133: beyond a cancer stem cell biomarker. J Drug Targeting. (2019) 27:257–69. doi: 10.1080/1061186X.2018.1479756
58. Tirino V, Desiderio V, Paino F, De Rosa A, Papaccio F, La Noce M, et al. Cancer stem cells in solid tumors: an overview and new approaches for their isolation and characterization. FASEB J. (2013) 27:13–24. doi: 10.1096/fj.12-218222
59. Skandalis SS, Karalis TT, Chatzopoulos A, Karamanos NK. Hyaluronan-CD44 axis orchestrates cancer stem cell functions. Cell Signal. (2019) 63:109377. doi: 10.1016/j.cellsig.2019.109377
60. Tarhriz V, Bandehpour M, Dastmalchi S, Ouladsahebmadarek E, Zarredar H, Eyvazi S. Overview of CD24 as a new molecular marker in ovarian cancer. J Cell Physiol. (2019) 234:2134–42. doi: 10.1002/jcp.27581
61. Sin WC, Lim CL. Breast cancer stem cells-from origins to targeted therapy. Stem Cell Investig. (2017) 4:96. doi: 10.21037/sci.2017.11.03
62. Krebsbach PH, Villa-Diaz LG. The role of integrin α6 (CD49f) in stem cells: more than a conserved biomarker. Stem Cells Dev. (2017) 26:1090–9. doi: 10.1089/scd.2016.0319
63. Haraguchi N, Ishii H, Mimori K, Ohta K, Uemura M, Nishimura J, et al. CD49f-positive cell population efficiently enriches colon cancer-initiating cells. Int J Oncol. (2013) 43:425–30. doi: 10.3892/ijo.2013.1955
64. Yahyazadeh Mashhadi SM, Kazemimanesh M, Arashkia A, Azadmanesh K, Meshkat Z, Golichenari B, et al. Shedding light on the EpCAM: an overview. J Cell Physiol. (2019) 234:12569–80. doi: 10.1002/jcp.28132
65. Kumar V, Green S, Stack G, Berry M, Jin JR, Chambon P. Functional domains of the human estrogen receptor. Cell. (1987) 51:941–51. doi: 10.1016/0092-8674(87)90581-2
66. Arnal JF, Lenfant F, Metivier R, Flouriot G, Henrion D, Adlanmerini M, et al. Membrane and nuclear estrogen receptor α actions: from tissue specificity to medical implications. Physiol Rev. (2017) 97:1045–87. doi: 10.1152/physrev.00024.2016
67. Osborne CK, Schiff R, Fuqua SA, Shou J. Estrogen receptor: current understanding of its activation and modulation. Clin Cancer Res. (2001) 7(12 Suppl.):4338s−42.
68. Renoir JM, Marsaud V, Lazennec G. Estrogen receptor signaling as a target for novel breast cancer therapeutics. Biochem Pharmacol. (2013) 85:449–65. doi: 10.1016/j.bcp.2012.10.018
69. Anstead GM, Carlson KE, Katzenellenbogen JA. The estradiol pharmacophore: ligand structure-estrogen receptor binding affinity relationships and a model for the receptor binding site. Steroids. (1997) 62:268–303. doi: 10.1016/S0039-128X(96)00242-5
70. Littlefield BA, Gurpide E, Markiewicz L, McKinley B, Hochberg RB. A simple and sensitive microtiter plate estrogen bioassay based on stimulation of alkaline phosphatase in Ishikawa cells: estrogenic action of delta 5 adrenal steroids. Endocrinology. (1990) 127:2757–62. doi: 10.1210/endo-127-6-2757
71. Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. (2005) 122:33–43. doi: 10.1016/j.cell.2005.05.008
72. Arendt LM, Kuperwasser C. Form and function: how estrogen and progesterone regulate the mammary epithelial hierarchy. J Mammary Gland Biol Neoplasia. (2015) 20:9–25. doi: 10.1007/s10911-015-9337-0
73. Cox DN, Chao A, Baker J, Chang L, Qiao D, Lin H. A novel class of evolutionarily conserved genes defined by piwi are essential for stem cell self-renewal. Genes Dev. (1998) 12:3715–27. doi: 10.1101/gad.12.23.3715
74. Chen Z, Yang HJ, Lin Q, Zhu MJ, Yu YY, He XY, et al. Estrogen-ERα signaling and DNA hypomethylation co-regulate expression of stem cell protein PIWIL1 in ERα-positive endometrial cancer cells. Cell Commun Signal. (2020) 18:84. doi: 10.1186/s12964-020-00563-4
75. Morimoto K, Kim SJ, Tanei T, Shimazu K, Tanji Y, Taguchi T, et al. Stem cell marker aldehyde dehydrogenase 1-positive breast cancers are characterized by negative estrogen receptor, positive human epidermal growth factor receptor type 2, and high Ki67 expression. Cancer Sci. (2009) 100:1062–8. doi: 10.1111/j.1349-7006.2009.01151.x
76. Clayton H, Titley I, Vivanco M. Growth and differentiation of progenitor/stem cells derived from the human mammary gland. Exp Cell Res. (2004) 297:444–60. doi: 10.1016/j.yexcr.2004.03.029
77. Harrison H, Simões BM, Rogerson L, Howell SJ, Landberg G, Clarke RB. Oestrogen increases the activity of oestrogen receptor negative breast cancer stem cells through paracrine EGFR and Notch signalling. Breast Cancer Res. (2013) 15:R21. doi: 10.1186/bcr3396
78. Fillmore CM, Gupta PB, Rudnick JA, Caballero S, Keller PJ, Lander ES, et al. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proc Natl Acad Sci USA. (2010) 107:21737–42. doi: 10.1073/pnas.1007863107
79. Simões BM, Piva M, Iriondo O, Comaills V, López-Ruiz JA, Zabalza I, et al. Effects of estrogen on the proportion of stem cells in the breast. Breast Cancer Res Treat. (2011) 129:23–35. doi: 10.1007/s10549-010-1169-4
80. Huang ZJ, You J, Luo WY, Chen BS, Feng QZ, Wu BL, et al. Reduced tumorigenicity and drug resistance through the downregulation of octamer-binding protein 4 and Nanog transcriptional factor expression in human breast stem cells. Mol Med Rep. (2015) 11:1647–54. doi: 10.3892/mmr.2014.2972
81. Fisher B, Redmond C, Fisher ER, Caplan R. Relative worth of estrogen or progesterone receptor and pathologic characteristics of differentiation as indicators of prognosis in node negative breast cancer patients: findings from National Surgical Adjuvant Breast and Bowel Project Protocol B-06. J Clin Oncol. (1988) 6:1076–87. doi: 10.1200/JCO.1988.6.7.1076
82. McGinn O, Ward AV, Fettig LM, Riley D, Ivie J, Paul KV, et al. Cytokeratin 5 alters β-catenin dynamics in breast cancer cells. Oncogene. (2020) 39:2478–92. doi: 10.1038/s41388-020-1164-0
83. Mendelsohn ME, Karas RH. Rapid progress for non-nuclear estrogen receptor signaling. J Clin Investigation. (2010) 120:2277–9. doi: 10.1172/JCI43756
84. Fuentes N, Silveyra P. Estrogen receptor signaling mechanisms. Adv Protein Chem Struct Biol. (2019) 116:135–70. doi: 10.1016/bs.apcsb.2019.01.001
85. Huang R, Li J, Pan F, Zhang B, Yao Y. The activation of GPER inhibits cells proliferation, invasion and EMT of triple-negative breast cancer via CD151/miR-199a-3p bio-axis. Am J Transl Res. (2020) 12:32–44.
86. Hu Y, Guo R, Wei J, Zhou Y, Ji W, Liu J, et al. Effects of PI3K inhibitor NVP-BKM120 on overcoming drug resistance and eliminating cancer stem cells in human breast cancer cells. Cell Death Dis. (2015) 6:e2020. doi: 10.1038/cddis.2015.363
87. Chan YT, Lai AC, Lin RJ, Wang YH, Wang YT, Chang WW, et al. GPER-induced signaling is essential for the survival of breast cancer stem cells. Int J Cancer. (2020) 146:1674–85. doi: 10.1002/ijc.32588
88. Deng H, Zhang XT, Wang ML, Zheng HY, Liu LJ, Wang ZY. ER-α36-mediated rapid estrogen signaling positively regulates ER-positive breast cancer stem/progenitor cells. PLoS ONE. (2014) 9:e88034. doi: 10.1371/journal.pone.0088034
89. Kang L, Guo Y, Zhang X, Meng J, Wang ZY. A positive cross-regulation of HER2 and ER-α36 controls ALDH1 positive breast cancer cells. J Steroid Biochem Mol Biol. (2011) 127:262–8. doi: 10.1016/j.jsbmb.2011.08.011
90. Zhang XT, Kang LG, Ding L, Vranic S, Gatalica Z, Wang ZY. A positive feedback loop of ER-α36/EGFR promotes malignant growth of ER-negative breast cancer cells. Oncogene. (2011) 30:770–80. doi: 10.1038/onc.2010.458
91. Majumdar S, Rinaldi JC, Malhotra NR, Xie L, Hu DP, Gauntner TD, et al. Differential actions of estrogen receptor α and β via nongenomic signaling in human prostate stem and progenitor cells. Endocrinology. (2019) 160:2692–708. doi: 10.1210/en.2019-00177
92. Wei W, Chen ZJ, Zhang KS, Yang XL, Wu YM, Chen XH, et al. The activation of G protein-coupled receptor 30 (GPR30) inhibits proliferation of estrogen receptor-negative breast cancer cells in vitro and in vivo. Cell Death Dis. (2014) 5:e1428. doi: 10.1038/cddis.2014.398
93. Skinner M. Cancer stem cells: TAZ takes centre stage. Nat Rev Cancer. (2012) 12:82–3. doi: 10.1038/nrc3210
94. Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell. (2011) 147:759–72. doi: 10.1016/j.cell.2011.09.048
95. Bartucci M, Dattilo R, Moriconi C, Pagliuca A, Mottolese M, Federici G, et al. TAZ is required for metastatic activity and chemoresistance of breast cancer stem cells. Oncogene. (2015) 34:681–90. doi: 10.1038/onc.2014.5
96. Lian I, Kim J, Okazawa H, Zhao J, Zhao B, Yu J, et al. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. (2010) 24:1106–18. doi: 10.1101/gad.1903310
97. Zhao AY, Dai YJ, Lian JF, Huang Y, Lin JG, Dai YB, et al. YAP regulates ALDH1A1 expression and stem cell property of bladder cancer cells. OncoTargets Therapy. (2018) 11:6657–63. doi: 10.2147/OTT.S170858
98. Pan Z, Tian Y, Zhang B, Zhang X, Shi H, Liang Z, et al. YAP signaling in gastric cancer-derived mesenchymal stem cells is critical for its promoting role in cancer progression. Int J Oncol. (2017) 51:1055–66. doi: 10.3892/ijo.2017.4101
99. Lai CJ, Lin CY, Liao WY, Hour TC, Wang HD, Chuu CP. CD44 promotes migration and invasion of docetaxel-resistant prostate cancer cells likely via induction of hippo-yap signaling. Cells. (2019) 8:40295. doi: 10.3390/cells8040295
100. Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF. A variant of estrogen receptor-{alpha}, hER-{alpha}36: transduction of estrogen- and antiestrogen-dependent membrane-initiated mitogenic signaling. Proc Natl Acad Sci USA. (2006) 103:9063–8. doi: 10.1073/pnas.0603339103
101. Shi L, Dong B, Li Z, Lu Y, Ouyang T, Li J, et al. Expression of ER-{alpha}36, a novel variant of estrogen receptor {alpha}, and resistance to tamoxifen treatment in breast cancer. J Clin Oncol. (2009) 27:3423–9. doi: 10.1200/JCO.2008.17.2254
102. Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF. Identification, cloning, and expression of human estrogen receptor-α36, a novel variant of human estrogen receptor-α66. Biochem Biophys Res Commun. (2005) 336:1023–7. doi: 10.1016/j.bbrc.2005.08.226
103. Gelsomino L, Panza S, Giordano C, Barone I, Gu G, Spina E, et al. Mutations in the estrogen receptor alpha hormone binding domain promote stem cell phenotype through notch activation in breast cancer cell lines. Cancer Lett. (2018) 428:12–20. doi: 10.1016/j.canlet.2018.04.023
104. Yin L, Wang ZY. Roles of the ER-α36-EGFR/HER2 positive regulatory loops in tamoxifen resistance. Steroids. (2016) 111:95–9. doi: 10.1016/j.steroids.2016.01.019
105. Deng H, Yin L, Zhang XT, Liu LJ, Wang ML, Wang ZY. ER-α variant ER-α36 mediates antiestrogen resistance in ER-positive breast cancer stem/progenitor cells. J Steroid Biochem Mol Biol. (2014) 144:417–26. doi: 10.1016/j.jsbmb.2014.08.017
106. Li G, Zhang J, Xu Z, Li Z. ERα36 as a potential therapeutic target for tamoxifen-resistant breast cancer cell line through EGFR/ERK signaling pathway. Cancer Manag Res. (2020) 12:265–75. doi: 10.2147/CMAR.S226410
107. Wang Q, Jiang J, Ying G, Xie XQ, Zhang X, Xu W, et al. Tamoxifen enhances stemness and promotes metastasis of ERα36(+) breast cancer by upregulating ALDH1A1 in cancer cells. Cell Res. (2018) 28:336–58. doi: 10.1038/cr.2018.15
108. Guo M, Wang M, Deng H, Zhang X, Wang ZY. A novel anticancer agent Broussoflavonol B downregulates estrogen receptor (ER)-α36 expression and inhibits growth of ER-negative breast cancer MDA-MB-231 cells. Eur J Pharmacol. (2013) 714:56–64. doi: 10.1016/j.ejphar.2013.05.047
109. Pan X, Zhao B, Song Z, Han S, Wang M. Estrogen receptor-α36 is involved in epigallocatechin-3-gallate induced growth inhibition of ER-negative breast cancer stem/progenitor cells. J Pharmacol Sci. (2016) 130:85–93. doi: 10.1016/j.jphs.2015.12.003
110. Huang B, Omoto Y, Iwase H, Yamashita H, Toyama T, Coombes RC, et al. Differential expression of estrogen receptor α, β1, and β2 in lobular and ductal breast cancer. Proc Natl Acad Sci USA. (2014) 111:1933–8. doi: 10.1073/pnas.1323719111
111. Chambliss KL, Yuhanna IS, Anderson RG, Mendelsohn ME, Shaul PW. ERbeta has nongenomic action in caveolae. Mol Endocrinol. (2002) 16:938–46. doi: 10.1210/mend.16.5.0827
112. Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature. (2015) 526:131–5. doi: 10.1038/nature15260
113. Ma R, Karthik GM, Lövrot J, Haglund F, Rosin G, Katchy A, et al. Estrogen receptor β as a therapeutic target in breast cancer stem cells. J Natl Cancer Institute. (2017) 109:1–14. doi: 10.1093/jnci/djw236
114. Fixemer T, Remberger K, Bonkhoff H. Differential expression of the estrogen receptor beta (ERβ) in human prostate tissue, premalignant changes, and in primary, metastatic, and recurrent prostatic adenocarcinoma. Prostate. (2003) 54:79–87. doi: 10.1002/pros.10171
115. Li M, Chai HF, Peng F, Meng YT, Zhang LZ, Zhang L, et al. Estrogen receptor β upregulated by lncRNA-H19 to promote cancer stem-like properties in papillary thyroid carcinoma. Cell Death Dis. (2018) 9:1120. doi: 10.1038/s41419-018-1077-9
116. Thomas C, Gustafsson J. The different roles of ER subtypes in cancer biology and therapy. Nat Rev Cancer. (2011) 11:597–608. doi: 10.1038/nrc3093
117. Reese JM, Bruinsma ES, Monroe DG, Negron V, Suman VJ, Ingle JN, et al. ERβ inhibits cyclin dependent kinases 1 and 7 in triple negative breast cancer. Oncotarget. (2017) 8:96506–21. doi: 10.18632/oncotarget.21787
118. Myers E, Fleming FJ, Crotty TB, Kelly G, McDermott EW, O'Higgins NJ, et al. Inverse relationship between ER-beta and SRC-1 predicts outcome in endocrine-resistant breast cancer. Br J Cancer. (2004) 91:1687–93. doi: 10.1038/sj.bjc.6602156
119. McPherson SJ, Hussain S, Balanathan P, Hedwards SL, Niranjan B, Grant M, et al. Estrogen receptor-beta activated apoptosis in benign hyperplasia and cancer of the prostate is androgen independent and TNFalpha mediated. Proc Natl Acad Sci USA. (2010) 107:3123–8. doi: 10.1073/pnas.0905524107
120. Hussain S, Lawrence MG, Taylor RA, Lo CY, Frydenberg M, Ellem SJ, et al. Estrogen receptor β activation impairs prostatic regeneration by inducing apoptosis in murine and human stem/progenitor enriched cell populations. PLoS ONE. (2012) 7:e40732. doi: 10.1371/journal.pone.0040732
121. Cheng FH, Aguda BD, Tsai JC, Kochańczyk M, Lin JM, Chen GC, et al. A mathematical model of bimodal epigenetic control of miR-193a in ovarian cancer stem cells. PLoS ONE. (2014) 9:e116050. doi: 10.1371/journal.pone.0116050
122. Cheng FHC, Lin HY, Hwang TW, Chen YC, Huang RL, Chang CB, et al. E2F6 functions as a competing endogenous RNA, and transcriptional repressor, to promote ovarian cancer stemness. Cancer Sci. (2019) 110:1085–95. doi: 10.1111/cas.13920
123. Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. (2011) 62:233–47. doi: 10.1146/annurev-med-070909-182917
124. Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci USA. (2009) 106:13820–5. doi: 10.1073/pnas.0905718106
125. Shea MP, O'Leary KA, Fakhraldeen SA, Goffin V, Friedl A, Wisinski KB, et al. Antiestrogen therapy increases plasticity and cancer stemness of prolactin-induced ERα(+) mammary carcinomas. Cancer Res. (2018) 78:1672–84. doi: 10.1158/0008-5472.CAN-17-0985
126. Siegfried JM, Farooqui M, Rothenberger NJ, Dacic S, Stabile LP. Interaction between the estrogen receptor and fibroblast growth factor receptor pathways in non-small cell lung cancer. Oncotarget. (2017) 8:24063–76. doi: 10.18632/oncotarget.16030
127. Pan GJ, Chang ZY, Schöler HR, Pei D. Stem cell pluripotency and transcription factor Oct4. Cell Res. (2002) 12:321–9. doi: 10.1038/sj.cr.7290134
128. Trosko JE. From adult stem cells to cancer stem cells: Oct-4 Gene, cell-cell communication, and hormones during tumor promotion. Ann NY Acad Sci. (2006) 1089:36–58. doi: 10.1196/annals.1386.018
129. Hu T, Liu S, Breiter DR, Wang F, Tang Y, Sun S. Octamer 4 small interfering RNA results in cancer stem cell-like cell apoptosis. Cancer Res. (2008) 68:6533–40. doi: 10.1158/0008-5472.CAN-07-6642
130. Bosco JL, Antonsen S, Sørensen HT, Pedersen L, Lash TL. Metformin and incident breast cancer among diabetic women: a population-based case-control study in Denmark. Cancer Epidemiol Biomarkers Prev. (2011) 20:101–11. doi: 10.1158/1055-9965.EPI-10-0817
131. Noto H, Goto A, Tsujimoto T, Noda M. Cancer risk in diabetic patients treated with metformin: a systematic review and meta-analysis. PLoS ONE. (2012) 7:e33411. doi: 10.1371/journal.pone.0033411
132. Jung JW, Park SB, Lee SJ, Seo MS, Trosko JE, Kang KS. Metformin represses self-renewal of the human breast carcinoma stem cells via inhibition of estrogen receptor-mediated OCT4 expression. PLoS ONE. (2011) 6:e28068. doi: 10.1371/journal.pone.0028068
133. Lopes J, Arnosti D, Trosko JE, Tai MH, Zuccari D. Melatonin decreases estrogen receptor binding to estrogen response elements sites on the OCT4 gene in human breast cancer stem cells. Genes Cancer. (2016) 7:209–17. doi: 10.18632/genesandcancer.107
134. Bak MJ, Furmanski P, Shan NL, Lee HJ, Bao C, Lin Y, et al. Tocopherols inhibit estrogen-induced cancer stemness and OCT4 signaling in breast cancer. Carcinogenesis. (2018) 39:1045–55. doi: 10.1093/carcin/bgy071
135. Prasanphanich AF, White DE, Gran MA, Kemp ML. Kinetic modeling of ABCG2 transporter heterogeneity: a quantitative, single-cell analysis of the side population assay. PLoS Comp Biol. (2016) 12:e1005188. doi: 10.1371/journal.pcbi.1005188
136. Yaguchi T, Onishi T. Estrogen induces cell proliferation by promoting ABCG2-mediated efflux in endometrial cancer cells. Biochem Biophys Rep. (2018) 16:74–8. doi: 10.1016/j.bbrep.2018.10.005
137. Kishino E, Ogata R, Saitoh W, Koike Y, Ohta Y, Kanomata N, et al. Anti-cell growth and anti-cancer stem cell activity of the CDK4/6 inhibitor palbociclib in breast cancer cells. Breast Cancer. (2020) 27:415–25. doi: 10.1007/s12282-019-01035-5
138. Czarnecka AM, Kornakiewicz A, Lian F, Szczylik C. Future perspectives for mTOR inhibitors in renal cell cancer treatment. Future Oncol. (2015) 11:801–17. doi: 10.2217/fon.14.303
139. Dubrovska A, Kim S, Salamone RJ, Walker JR, Maira S-M, Garcia-Echeverria C, et al. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc Natl Acad Sci USA. (2009) 106:268–73. doi: 10.1073/pnas.0810956106
140. Yang Y, Iwanaga K, Raso MG, Wislez M, Hanna AE, Wieder ED, et al. Phosphatidylinositol 3-kinase mediates bronchioalveolar stem cell expansion in mouse models of oncogenic K-ras-induced lung cancer. PLoS ONE. (2008) 3:e2220. doi: 10.1371/journal.pone.0002220
141. Karthik GM, Ma R, Lovrot J, Kis LL, Lindh C, Blomquist L, et al. mTOR inhibitors counteract tamoxifen-induced activation of breast cancer stem cells. Cancer Lett. (2015) 367:76–87. doi: 10.1016/j.canlet.2015.07.017
142. Vares G, Sai S, Wang B, Fujimori A, Nenoi M, Nakajima T. Progesterone generates cancer stem cells through membrane progesterone receptor-triggered signaling in basal-like human mammary cells. Cancer Lett. (2015) 362:167–73. doi: 10.1016/j.canlet.2015.03.030
143. Truong TH, Dwyer AR, Diep CH, Hu H, Hagen KM, Lange CA. Phosphorylated progesterone receptor isoforms mediate opposing stem cell and proliferative breast cancer cell fates. Endocrinology. (2019) 160:430–46. doi: 10.1210/en.2018-00990
144. Finlay-Schultz J, Cittelly DM, Hendricks P, Patel P, Kabos P, Jacobsen BM, et al. Progesterone downregulation of miR-141 contributes to expansion of stem-like breast cancer cells through maintenance of progesterone receptor and Stat5a. Oncogene. (2015) 34:3676–87. doi: 10.1038/onc.2014.298
145. Lombardi S, Honeth G, Ginestier C, Shinomiya I, Marlow R, Buchupalli B, et al. Growth hormone is secreted by normal breast epithelium upon progesterone stimulation and increases proliferation of stem/progenitor cells. Stem Cell Rep. (2014) 2:780–93. doi: 10.1016/j.stemcr.2014.05.005
146. Kastner P, Krust A, Turcotte B, Stropp U, Tora L, Gronemeyer H, et al. Two distinct estrogen-regulated promoters generate transcripts encoding the two functionally different human progesterone receptor forms A and B. EMBO J. (1990) 9:1603–14. doi: 10.1002/j.1460-2075.1990.tb08280.x
147. Patel B, Elguero S, Thakore S, Dahoud W, Bedaiwy M, Mesiano S. Role of nuclear progesterone receptor isoforms in uterine pathophysiology. Human Reprod Update. (2015) 21:155–73. doi: 10.1093/humupd/dmu056
148. Lamb CA, Fabris VT, Jacobsen B, Molinolo AA, Lanari C. Biological and clinical impact of imbalanced progesterone receptor isoform ratios in breast cancer. Endocrine-Related Cancer. (2018) 25:R605–24. doi: 10.1530/ERC-18-0179
149. Graham JD, Yager ML, Hill HD, Byth K, O'Neill GM, Clarke CL. Altered progesterone receptor isoform expression remodels progestin responsiveness of breast cancer cells. Mol Endocrinol. (2005) 19:2713–35. doi: 10.1210/me.2005-0126
150. Mote PA, Gompel A, Howe C, Hilton HN, Sestak I, Cuzick J, et al. Progesterone receptor A predominance is a discriminator of benefit from endocrine therapy in the ATAC trial. Breast Cancer Res Treat. (2015) 151:309–18. doi: 10.1007/s10549-015-3397-0
151. Sletten ET, Arnes M, Lysa LM, Larsen M, Orbo A. Significance of progesterone receptors (PR-A and PR-B) expression as predictors for relapse after successful therapy of endometrial hyperplasia: a retrospective cohort study. BJOG. (2019) 126:936–43. doi: 10.1111/1471-0528.15579
152. Stanczyk FZ, Kannan N, Sherman ME, Trabert B. Progesterone and breast cancer. Endocrine Rev. (2020) 41:bnz001. doi: 10.1210/endrev/bnz001
153. Trevino LS, Weigel NL. Phosphorylation: a fundamental regulator of steroid receptor action. Trends Endocrinol Metab. (2013) 24:515–24. doi: 10.1016/j.tem.2013.05.008
154. Abdel-Hafiz HA, Horwitz KB. Post-translational modifications of the progesterone receptors. J Steroid Biochem Mol Biol. (2014) 140:80–9. doi: 10.1016/j.jsbmb.2013.12.008
155. Knutson TP, Truong TH, Ma S, Brady NJ, Sullivan ME, Raj G, et al. Posttranslationally modified progesterone receptors direct ligand-specific expression of breast cancer stem cell-associated gene programs. J Hematol Oncol. (2017) 10:7. doi: 10.1186/s13045-017-0462-7
156. Thomas P, Pang Y. Membrane progesterone receptors: evidence for neuroprotective, neurosteroid signaling and neuroendocrine functions in neuronal cells. Neuroendocrinology. (2012) 96:162–71. doi: 10.1159/000339822
157. Dressing GE, Goldberg JE, Charles NJ, Schwertfeger KL, Lange CA. Membrane progesterone receptor expression in mammalian tissues: a review of regulation and physiological implications. Steroids. (2011) 76:11–7. doi: 10.1016/j.steroids.2010.09.006
158. Singh M, Su C, Ng S. Non-genomic mechanisms of progesterone action in the brain. Front Neurosci. (2013) 7:159. doi: 10.3389/fnins.2013.00159
159. Rao S, Cronin SJF, Sigl V, Penninger JM. RANKL and RANK: from mammalian physiology to cancer treatment. Trends Cell Biol. (2018) 28:213–23. doi: 10.1016/j.tcb.2017.11.001
160. Gonzalez-Suarez E, Jacob AP, Jones J, Miller R, Roudier-Meyer MP, Erwert R, et al. RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature. (2010) 468:103. doi: 10.1038/nature09495
161. Mohammed MK, Shao C, Wang J, Wei Q, Wang X, Collier Z, et al. Wnt/beta-catenin signaling plays an ever-expanding role in stem cell self-renewal, tumorigenesis and cancer chemoresistance. Genes Dis. (2016) 3:11–40. doi: 10.1016/j.gendis.2015.12.004
162. Matsui WH. Cancer stem cell signaling pathways. Medicine. (2016) 95(1Suppl.1):S8–19. doi: 10.1097/MD.0000000000004765
163. Clarke CL, Graham JD. Non-overlapping progesterone receptor cistromes contribute to cell-specific transcriptional outcomes. PLoS ONE. (2012) 7:e35859. doi: 10.1371/journal.pone.0035859
164. Chang YH, Ding DC, Chu TY. Estradiol and progesterone induced differentiation and increased stemness gene expression of human fallopian tube epithelial cells. J Cancer. (2019) 10:3028–36. doi: 10.7150/jca.30588
165. Wang Y, Lu E, Bao R, Xu P, Feng F, Wen W, et al. Notch signalling regulates steroidogenesis in mouse ovarian granulosa cells. Reprod Fertil Dev. (2019) 31:1091–103. doi: 10.1071/RD18281
166. Guo RH, Chen F, Shi ZD. Suppression of notch signaling stimulates progesterone synthesis by enhancing the expression of NR5A2 and NR2F2 in porcine granulosa cells. Genes. (2020) 11:12. doi: 10.3390/genes11020120
167. Shiah Y-J, Tharmapalan P, Casey AE, Joshi PA, McKee TD, Jackson HW, et al. A progesterone-CXCR4 axis controls mammary progenitor cell fate in the adult gland. Stem Cell Rep. (2015) 4:313–22. doi: 10.1016/j.stemcr.2015.01.011
168. Trautmann F, Cojoc M, Kurth I, Melin N, Bouchez LC, Dubrovska A, et al. CXCR4 as biomarker for radioresistant cancer stem cells. Int J Radiat Biol. (2014) 90:687–99. doi: 10.3109/09553002.2014.906766
169. Zhang Y, Xu B, Zhang XP. Effects of miRNAs on functions of breast cancer stem cells and treatment of breast cancer. OncoTargets Therapy. (2018) 11:4263–70. doi: 10.2147/OTT.S165156
170. Cittelly DM, Finlay-Schultz J, Howe EN, Spoelstra NS, Axlund SD, Hendricks P, et al. Progestin suppression of miR-29 potentiates dedifferentiation of breast cancer cells via KLF4. Oncogene. (2013) 32:2555–64. doi: 10.1038/onc.2012.275
171. Hurtz C, Hatzi K, Cerchietti L, Braig M, Park E, Kim Y, et al. BCL6-mediated repression of p53 is critical for leukemia stem cell survival in chronic myeloid leukemia. J Exp Med. (2011) 208:2163–74. doi: 10.1084/jem.20110304
172. Chen YJ, Zhang X, Wu ZS, Wang JJ, Lau AYC, Zhu T, et al. Autocrine human growth hormone stimulates the tumor initiating capacity and metastasis of estrogen receptor-negative mammary carcinoma cells. Cancer Lett. (2015) 365:182–9. doi: 10.1016/j.canlet.2015.05.031
173. Skor MN, Wonder EL, Kocherginsky M, Goyal A, Hall BA, Cai Y, et al. Glucocorticoid receptor antagonism as a novel therapy for triple-negative breast cancer. Clin Cancer Res. (2013) 19:6163–72. doi: 10.1158/1078-0432.CCR-12-3826
174. Liu R, Shi P, Nie Z, Liang H, Zhou Z, Chen W, et al. Mifepristone suppresses basal triple-negative breast cancer stem cells by down-regulating KLF5 expression. Theranostics. (2016) 6:533–44. doi: 10.7150/thno.14315
175. Sang L, Lu D, Zhang J, Du S, Zhao X. Mifepristone inhibits proliferation, migration and invasion of HUUA cells and promotes its apoptosis by regulation of FAK and PI3K/AKT signaling pathway. OncoTargets Therapy. (2018) 11:5441–9. doi: 10.2147/OTT.S169947
176. Arfaoui A, Rioualen C, Azzoni V, Pinna G, Finetti P, Wicinski J, et al. A genome-wide RNAi screen reveals essential therapeutic targets of breast cancer stem cells. EMBO Mol Med. (2019) 11:e9930. doi: 10.15252/emmm.201809930
177. Huang Y, Hu W, Huang J, Shen F, Sun Y, Ivan C, et al. Inhibiting nuclear phospho-progesterone receptor enhances antitumor activity of onapristone in uterine cancer. Mol Cancer Ther. (2018) 17:464–73. doi: 10.1158/1535-7163.MCT-17-0006
178. Rao S, Sigl V, Wimmer RA, Novatchkova M, Jais A, Wagner G, et al. RANK rewires energy homeostasis in lung cancer cells and drives primary lung cancer. Genes Dev. (2017) 31:2099–112. doi: 10.1101/gad.304162.117
179. Yoldi G, Pellegrini P, Trinidad EM, Cordero A, Gomez-Miragaya J, Serra-Musach J, et al. RANK signaling blockade reduces breast cancer recurrence by inducing tumor cell differentiation. Cancer Res. (2016) 76:5857–69. doi: 10.1158/0008-5472.CAN-15-2745
180. Cuyas E, Martin-Castillo B, Bosch-Barrera J, Menendez JA. Metformin inhibits RANKL and sensitizes cancer stem cells to denosumab. Cell Cycle. (2017) 16:1022–8. doi: 10.1080/15384101.2017.1310353
Keywords: cancer stem cells, cancer stemness, estrogen, female hormone, progesterone
Citation: Chen B, Ye P, Chen Y, Liu T, Cha J-H, Yan X and Yang W-H (2020) Involvement of the Estrogen and Progesterone Axis in Cancer Stemness: Elucidating Molecular Mechanisms and Clinical Significance. Front. Oncol. 10:1657. doi: 10.3389/fonc.2020.01657
Received: 06 June 2020; Accepted: 28 July 2020;
Published: 04 September 2020.
Edited by:
Xing Huang, Zhejiang University, ChinaReviewed by:
Giovanni Nassa, University of Salerno, ItalyCopyright © 2020 Chen, Ye, Chen, Liu, Cha, Yan and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiuwen Yan, c3VyZTgzQGd6aG11LmVkdS5jbg==; Wen-Hao Yang, d2h5MDMzMUBnbWFpbC5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.