
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol. , 26 August 2020
Sec. Hematologic Malignancies
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.01538
 Naval Daver*†
Naval Daver*† Allyson Price†
Allyson Price† Christopher B. Benton
Christopher B. Benton Keyur Patel
Keyur Patel Weiguo Zhang
Weiguo Zhang Marina Konopleva
Marina Konopleva Naveen Pemmaraju
Naveen Pemmaraju Koichi Takahashi
Koichi Takahashi Michael Andreeff
Michael Andreeff Gautam Borthakur
Gautam BorthakurThe prognostics implications of patients with acute myeloid leukemia harboring non-canonical FLT3 is unknown. The use of tyrosine kinase inhibitors in this patient population has not been previously reported. We report successful targeted therapy against non-ITD, non-D835 driver FLT3 alterations in two patient case studies with acute myeloid leukemia.
The two leading types of FLT3 mutations found in AML include internal tandem duplications in the juxtamembrane domain (ITD, 17–34%) and mutations in the tyrosine kinase domain (TKD) activation loop (~7%) (1). Mechanistically, FLT3-ITDs and FLT3-TKDs induce activation of transduction intermediates, including STAT5, AKT, and ERK1/2 (2). Several small-molecule tyrosine kinase inhibitors (TKIs) act as direct inhibitors of FLT3 via competitive inhibition of ATP-binding sites in the FLT3 receptor KD. These include midostaurin, gilteritinib, quizartinib, sorafenib, lestaurtinib, and crenolanib (3–6). Currently, midostaurin (7) and gilteritinib (8) are USA FDA-approved that have activity in FLT3-ITD and -TKD mutated patients.
However, the prognostic implications and response to TKI therapy of FLT3 sequence alterations outside the known FLT3-ITD and FLT3-TKD remain unknown. Frohling et al. identified non-canonical gain-of-function driver mutations (FLT3 S451F, Y572, V592G, and R834Q) and bystander passenger alleles (T167A, V194M, Y364H, and G831E) in 222 AML patients without a FLT3-ITD or -D835 mutation (9). Functionally, AML cells harboring these driver mutations were amenable to TKI therapy. The overall prevalence of non-synonymous driver mutations in was 1.7% (6 of 349 analyzed samples). Herein, we discuss two patients with AML who acquired non-canonical FLT3 activating alleles FLT3-V592G and FLT3-N676K, respectively at relapse and responded to therapy with sorafenib.
A 66-year-old man was diagnosed with diploid, NPM1-mutated, FLT3 wild-type, CEBPA wild-type AML in June 2013. The patient was induced with a clofarabine-based regimen. He was refractory and was re-induced with clofarabine. A Day 21 aspirate revealed 6% blasts. He began therapy with 5-azacytidine administered Days 1–7 every 4–5 weeks and achieved transfusion independence. After cycle 7 a repeat aspirate revealed relapsed AML with 20% blasts. The patient was referred to our institution.
The initial aspirate revealed 24% blasts. A next generation sequencing (NGS)-based analysis for the detection of somatic mutations in the coding sequences of 28 genes (including FLT3) was performed on DNA from the bone marrow sample. The methodology of our mutation analysis panel and coverage by genes and codons has been previously published (10) (Supplementary Table 1). The AML was found to have (1) somatic mutations: NPM1 (W288fs*12) and TET2 exon 3 duplication (T556fs*11); (2) variants of uncertain significance: DNMT3A exon 17 insertion (V657_D658insE); and (3) a variant of probable germline origin: GATA2 (P161A). Details for these detected variants are provided in the Supplementary Information. PCR-based DNA analysis did not detect ITD or codon 835/836 point mutations in the FLT3 (lower limit of detection ~1% of mutant DNA).
The patient received salvage therapy on protocol with E7070, a synthetic sulfonamide cell cycle inhibitor, in combination with idarubicin and cytarabine (ClinicalTrials.gov Identifier: NCT01692197) in June 2014 and achieved a complete remission (CR) with no evidence of minimal-residual disease (MRD) on a multiparametric 19-color flow-cytometry. He received one consolidation cycle and proceeded to allogeneic stem cell transplant from a 10/10 matched unrelated donor in September 2014. The patient achieved 100% donor chimerism by day 28. In February 2015, ~140 days post-transplant, a repeat bone marrow aspirate revealed 66% blasts. The 28-gene NGS panel revealed (1) somatic mutations: in addition to the previously noted NPM1 and TET2 mutations the patient demonstrated a FLT3-exon 14 missense mutation (V592G); (2) variants of uncertain significance: in addition to the previously noted DNMT3A alteration, an EZH2 exon 19 mutation was noted (E726Q); and (3) variants of germline origin: including EZH2, ABL1, IDH1, NOTCH1, TET2 (x3 variants). FLT-ITD and -D835 mutations were not detected by PCR-based DNA analysis. Based on the rapid rate of blast increase, the patient's relapsed disease was more proliferative than the pre-transplant AML.
The acquisition of the non-canonical driver FLT3-V592G mutation, known to be sensitive to FLT3-inhibition in in vitro (9) and known to reside near the juxtamembrane domain, prompted therapy with the multikinase inhibitor sorafenib. The patient started hydroxyurea with sorafenib orally daily continuously at 400 mg twice daily. Three weeks after initiation of the sorafenib the leukocytosis abated and the hydroxyurea was discontinued, in March 2015. An aspirate done 29 days after initiation of the sorafenib revealed CR with no MRD on a multiparametric flow-cytometry. The trend of peripheral blast percentage, white blood cell count, and platelets during sorafenib therapy is shown in Figure 1A. After 3 months of sustained remission with sorafenib monotherapy, 5-azacitidine (75 mg/m2 Days 1–5 repeated every 4–5 weeks) was added with the intent to enhance the durability of remission. At the time of this report, the patient remains on sorafenib (200 mg PO BID continuous) and azacitidine (75 mg/m2 Days 1–3 every) therapy with the cycles repeated every 4–6 weeks based on count while maintaining MRD negative CR for over 60 months (see updated counts trend in Figures 2A–C).
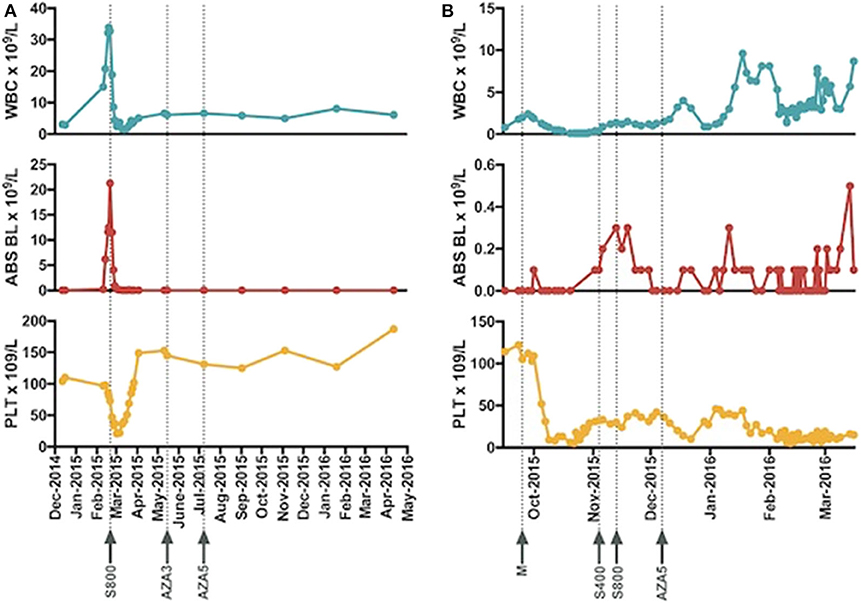
Figure 1. The trend of white blood cell count, absolute peripheral blasts, and platelet and timelines of introduction of sorafenib for patient 1 (A) and patient 2 (B) are presented. Abbreviations: WBC, white blood cell, ABS BL, absolute blasts, PLT, platelets, S800 (sorafenib 800 mg daily), AZA 3 (azacitidine 75 mg/m2 Day 1–3 each cycle), AZA 5 (azacitidine 75 mg/m2 Day 1–3 each cycle), M, mutational profiling, S400 (sorafenib 400 mg daily).
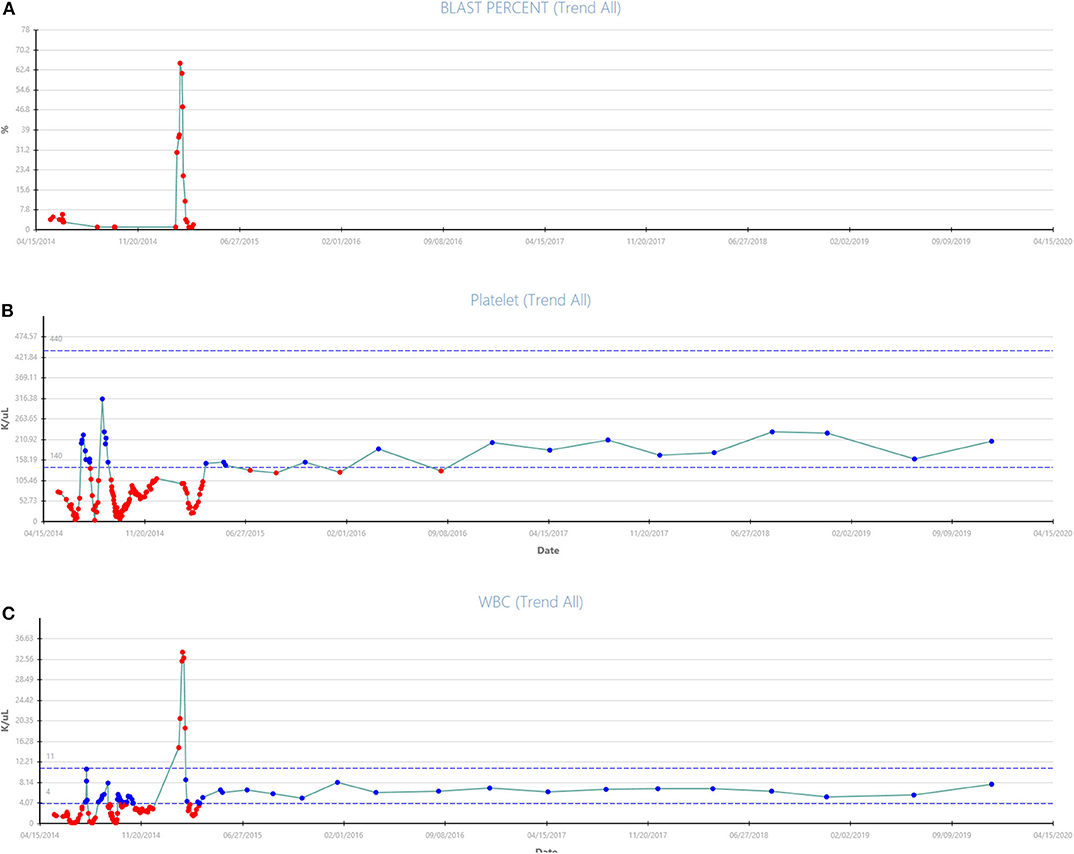
Figure 2. (A–C) Updated trend of blast percentage, white blood count, and platelets from presentation in March 2015 to December 2019 with continuation of treatment with Vidaza + Sorafenib combination therapy for patient #1 are shown.
A 68-year-old male with a history of myelodysplastic syndrome with diploid karyotype was on observation for 2 years. A surveillance bone marrow aspirate showed 8% blasts with a trisomy 16 in 1 metaphase and diploid karyotype in 19 metaphases. He received four cycles of 5-azacitidine without response and progressed to AML with 20% blast. He received decitabine for 10 days without response followed by fludarabine, cytarabine, G-CSF (FLAG) therapy without response. The patient was referred to our institution in September 2015.
The initial bone marrow at our institution showed 40% blasts and trisomy 16 in eight metaphases. The 28-gene NGS mutation panel identified (1) somatic mutations: ASXL1 (G629fs), NRAS (G12A), KRAS (G12D), FLT3 (N676K in exon 16), and GATA2 (M388fs); (2) variants of uncertain origin: TP53 (M44V); and (3) variants of probable germline origin: TET2 (P363L). FLT3-ITD and -D835 mutations were not detected by PCR.
The patient received salvage with oral MDM2 inhibitor, DS-3032B, (ClinicalTrials.gov Identifier: NCT02319369) and did not respond. The acquisition of the non-canonical driver FLT3-N676K mutation with demonstrated in vitro sensitivity to TKI therapy (9) prompted the use of oral TKI sorafenib 200 mg twice daily continuously in November 2015. Ten days into therapy the dose was escalated to sorafenib 400 mg twice daily. An aspirate on day 28 revealed 3% blasts in a hemodilute sample with flow cytometry demonstrating 7.9% aberrant blasts. For subsequent cycles 5-azacitidine was added to the sorafenib in December 2015. He completed 4 cycles of the combination and the repeat bone marrow from March 2016 demonstrated 6% blasts in a hypercellular marrow. Changes in blood counts with therapy are shown in Figure 1B.
In this report of 2 cases we discuss the potential for FLT3-inhibitor based targeted therapy against non-ITD, non-D835 driver FLT3 alterations in patients with AML. Codon 592 (p.V592) is a part of juxtamembrane (JM) switch motif responsible for maintenance of auto-inhibitory confirmation of FLT3 in the absence of a ligand and contains a STAT5-binding motif. V592 is a driver mutation that promotes growth and survival of AML cells by activation of downstream signal transduction cascades in MONO-MAC-6 cell lines (11). Down regulation of FLT3 by lentiviral transduction of shRNA targeting the FLT3 transcript decreased FLT3 mRNA levels and downstream STAT5 phosphorylation with reduced cell viability (11). In vitro studies suggest that V592G harboring BaF3 cells are highly sensitive to the FLT3-inhibitor midostaurin (12). The sensitivity of V592G to sorafenib in vitro and in patients with AML remained unknown. Our patient with FLT3-V592G AML had a rapid response to single agent sorafenib.
The N676 mutation was discovered in the ATP-binding region of the TKD of the FLT3 gene (13). FLT3-N676K is predicted to interfere with FLT3 auto-inhibition by reducing the stability of JM domain and induces phosphorylation of FLT3 and downstream pathways. Huang et al. noted that retroviral expression of N676K induced AML in syngeneic mice with a transforming potential similar to FLT3-ITD. Leukemic cells harboring the FLT3-N676K mutation in the absence of an ITD mutation were highly sensitive to FLT3 inhibitors quizartinib and crenolanib (14). Our patient with a non-canonical driver FLT3-N676K responded well to therapy with sorafenib.
One of the limitations of our manuscript is the lack of ex vivo data showing a direct suppression of FLT3 and downstream pathways in these patients by sorafenib. While it may be postulated given the known preclinical activity of sorafenib to FLT3 mutated cells and rapid nature of the responses that the mechanism of action in these patients was likely mediated by the FLT3 inhibitory activity of sorafenib, it is feasible that some of the clinical activity could be driven by multikinase inhibition of prosurvival pathways by sorafenib. This would be an interesting area of investigation for future analysis in non-canonical FLT3 mutations.
Sorafenib is an orally active multi-kinase inhibitor that is 1,000-fold more active against ITD mutant FLT3 than wild-type FLT3 in cell based assays (15). Sorafenib has been used for FLT3-inhibition as a single-agent and in combination with other antileukemic drugs in patients with FLT3-ITD AML. Sorafenib is also effective in vitro against other ATP-binding mutations including N676D and G697R. Commercial availability and a broad spectrum of activity against numerous driver FLT3 allele sequences made sorafenib a logical choice for clinical use in our patients with the non-synonymous driver FLT3 allele sequences. It is important to note that sorafenib and other type II FLT3-inhibitors have demonstrated no preclinical or clinical activity in FLT3-TKD mutated AML (including FLT3-D835) and is generally not recommended to target FLT3-TKD mutant AML. FLT3-TKD mutations are more effectively targeted with type I FLT3-inhibitors such as gilteritinib, midostaurin, and crenolanib.
Routine NGS sequencing of all FLT3 coding exons will lead to more frequent identification of non-synonymous FLT3 alleles. After removing known germline variants based on publically available SNP databases we identified 21 patients with non-synonymous FLT3 allele sequences at our institution since implementation of the 28-gene NGS panel between July 2013 through July 2015 (unpublished data). These include 10 in newly diagnosed untreated AML among 495 evaluated patients (prevalence in new AML = 2%), nine in patients with relapsed AML, 1 in a patient with chronic myelomonocytic leukemia, and one in a patient with myelodysplastic syndrome/myeloproliferative disease. The 21 mutations identified included 12 allele sequences known to have a driver function based on in vitro functional studies including S451F (n = 2), Y572C (n = 2), V592G (n = 2), Y842C (n = 2), N841K (n = 2), N676K (n = 1), and F691L (n = 1) (9, 13, 16, 17). None of these other patients received FLT3 inhibitor therapy. Our experience with sorafenib in the patient with V592G and N676K suggests that patients harboring known activating non-synonymous FLT3-mutations may benefit from TKI therapy either as a single-agent or in combination with other anti-leukemic agents.
Written informed consent was obtained from the individual(s)/legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
ND, AP, CB, KP, NP, WZ, MA, MK, KT, and GB collected and reviewed the data, treated the patients, performed the experiments, and wrote the paper. All authors participated in the discussion, have reviewed and approved the current version of the manuscript.
This study was supported in part by the MD Anderson Cancer Centre Leukemia Support Grant (CCSG) CA016672.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.01538/full#supplementary-material
1. Thiede C, Steudel C, Mohr B, Schaich M, Schakel U, Platzbecker U, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. (2002) 99:4326–35. doi: 10.1182/blood.V99.12.4326
2. Parcells BW, Ikeda AK, Simms-Waldrip T, Moore TB, Sakamoto KM. FMS-like tyrosine kinase 3 in normal hematopoiesis and acute myeloid leukemia. Stem Cells. (2006) 24:1174–84. doi: 10.1634/stemcells.2005-0519
3. Stone RM, Mandrekar S, Sanford BL, Geyer S, Bloomfield CD, Dohner K, et al. The multi-kinase inhibitor midostaurin (M) prolongs survival compared with placebo (P) in combination with daunorubicin (D)/cytarabine (C) induction (ind), high-dose C consolidation (consol), and as maintenance (maint) therapy in newly diagnosed acute myeloid leukemia (AML) patients (pts) age 18-60 with FLT3 mutations (muts): an international prospective randomized (rand) P-Controlled double-blind trial (CALGB 10603/RATIFY [Alliance]). Blood. (2015) 126:6 doi: 10.1182/blood.V126.23.6.6
4. Rollig C, Serve H, Huttmann A, Noppeney R, Muller-Tidow C, Krug U, et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): a multicentre, phase 2, randomised controlled trial. Lancet Oncol. (2015) 16:1691–9. doi: 10.1016/S1470-2045(15)00362-9
5. Ravandi F, Alattar ML, Grunwald MR, Rudek MA, Rajkhowa T, Richie MA, et al. Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood. (2013) 121:4655–62. doi: 10.1182/blood-2013-01-480228
6. Cortes JE, Kantarjian H, Foran JM, Ghirdaladze D, Zodelava M, Borthakur G, et al. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J Clin Oncol. (2013) 31:3681–7. doi: 10.1200/JCO.2013.48.8783
7. Levis M. Midostaurin approved for FLT3-mutated AML. Blood. (2017) 129:3403–6. doi: 10.1182/blood-2017-05-782292
8. Dhillon S. Gilteritinib: first global approval. Drugs. (2019) 79:331–9. doi: 10.1007/s40265-019-1062-3
9. Frohling S, Scholl C, Levine RL, Loriaux M, Boggon TJ, Bernard OA, et al. Identification of driver and passenger mutations of FLT3 by high-throughput DNA sequence analysis and functional assessment of candidate alleles. Cancer Cell. (2007) 12:501–13. doi: 10.1016/j.ccr.2007.11.005
10. Luthra R, Patel KP, Reddy NG, Haghshenas V, Routbort MJ, Harmon MA, et al. Next-generation sequencing-based multigene mutational screening for acute myeloid leukemia using MiSeq: applicability for diagnostics and disease monitoring. Haematologica. (2014) 99:465–73. doi: 10.3324/haematol.2013.093765
11. Spiekermann K, Dirschinger RJ, Schwab R, Bagrintseva K, Faber F, Buske C, et al. The protein tyrosine kinase inhibitor SU5614 inhibits FLT3 and induces growth arrest and apoptosis in AML-derived cell lines expressing a constitutively activated FLT3. Blood. (2003) 101:1494–504. doi: 10.1182/blood-2002-04-1045
12. Whitman SP, Archer KJ, Feng L, Baldus C, Becknell B, Carlson BD, et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: a cancer and leukemia group B study. Cancer Res. (2001) 61:7233−9.
13. Heidel F, Solem FK, Breitenbuecher F, Lipka DB, Kasper S, Thiede MH, et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood. (2006) 107:293–300. doi: 10.1182/blood-2005-06-2469
14. Huang K, Yang M, Pan Z, Heidel FH, Scherr M, Eder M, et al. Leukemogenic potency of the novel FLT3-N676K mutant. Ann Hematol. (2016) 95:783–91. doi: 10.1007/s00277-016-2616-z
15. Zhang W, Konopleva M, Shi YX, McQueen T, Harris D, Ling X, et al. Mutant FLT3: a direct target of sorafenib in acute myelogenous leukemia. J Natl Cancer Inst. (2008) 100:184–98. doi: 10.1093/jnci/djm328
16. Cools J, Mentens N, Furet P, Fabbro D, Clark JJ, Griffin JD, et al. Prediction of resistance to small molecule FLT3 inhibitors: implications for molecularly targeted therapy of acute leukemia. Cancer Res. (2004) 64:6385–9. doi: 10.1158/0008-5472.CAN-04-2148
Keywords: sorafenib, FLT3 mutation, FLT3-V592, FLT3-N676, AML, TKI
Citation: Daver N, Price A, Benton CB, Patel K, Zhang W, Konopleva M, Pemmaraju N, Takahashi K, Andreeff M and Borthakur G (2020) First Report of Sorafenib in Patients With Acute Myeloid Leukemia Harboring Non-Canonical FLT3 Mutations. Front. Oncol. 10:1538. doi: 10.3389/fonc.2020.01538
Received: 12 May 2020; Accepted: 17 July 2020;
Published: 26 August 2020.
Edited by:
Alessandro Isidori, AORMN Hospital, ItalyReviewed by:
Mark Levis, Johns Hopkins University, United StatesCopyright © 2020 Daver, Price, Benton, Patel, Zhang, Konopleva, Pemmaraju, Takahashi, Andreeff and Borthakur. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Naval Daver, bmRhdmVyQG1kYW5kZXJzb24ub3Jn
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.