 Christine J. Kurian
Christine J. Kurian Colin Thomas
Colin Thomas Sarah Houtmann1
Sarah Houtmann1 Thomas Klumpp
Thomas Klumpp Adam Finn Binder
Adam Finn Binder- 1Department of Internal Medicine, Thomas Jefferson University Hospital, Philadelphia, PA, United States
- 2Department of Medical Oncology, Thomas Jefferson University Hospital, Philadelphia, PA, United States
Plasma cell dyscrasias and myeloproliferative neoplasms (MPN) are hematologic malignancies arising from two distinct hematopoietic cell lineages. They rarely occur concomitantly. Here, we report a case of a patient with a recent diagnosis of a JAK2 V617F positive MPN who presented with a new diagnosis of plasma cell leukemia. The patient had presented to the hospital with a leukocytosis predominantly comprised of plasma cells, followed by work-up involving peripheral blood flow cytometry, FISH analysis, and bone-marrow biopsy. FISH analysis was suggestive of a common progenitor cell for these distinct hematologic malignancies. To our knowledge, this case represents the second reported instance of a concomitant JAK2 positive MPN with primary plasma cell leukemia.
Introduction
There are several published reports of patients with concomitant myeloproliferative neoplasms (MPN) and plasma cell dyscrasias (PCD), the peculiarity of which being that these two malignancies arise from separate lineages in the hematopoietic ancestral tree (1, 2). Apart from case reports of patients with MPNs and PCDs, one prospective study found the coexistence of monoclonal gammopathy of undetermined significance (MGUS) in patients with a MPN to be 8.2%, which is higher than the average prevalence of MGUS in the general population (3%) (3, 4).
Other studies, however, have not found a significantly higher proportion of MGUS in patients with MPN—overall clouding the picture of this association (4). When patients are diagnosed with plasma cell dyscrasia and an MPN, based on one case series, it seems the MPN either precedes or is diagnosed concurrently with the plasma cell dyscrasia (5). Although bone marrow fibrosis is common on diagnosis of MM, ~38% of patients, it is likely a reactive process (5). There have been a few reports of myelofibrosis in plasma cell leukemia; however, these have been presumed to be reactive with negative JAK2 mutations (5, 6).
Unlike MGUS, smoldering myeloma, or multiple myeloma, primary plasma cell leukemia (pPCL) has a distinct pathogenesis (7). pPCL is defined as de novo leukemia having >2 × 109/L plasma cells in the peripheral blood or plasma cells making up >20% of peripheral blood leukocytes. It is considered the most aggressive of all plasma cell neoplasms with poor overall survival (8).
Candoni et al. described a similar case in 2004 with a case of plasma cell leukemia occurring in a patient with thrombocythemia. At the time of his ET diagnosis, he was treated with aspirin, hydroxyurea, and busulphan. In this patient's PCL, there was a high expression of P-glycoprotein and notably his karyotype showed a deletion of chromosome 7 typically associated with secondary leukemias. It was thought that his previous cytoreductive therapy exposure with hydroxyurea and busulphan may have contributed to the development of PCL (9).
Here, we report a another case of a patient with JAK2 V617F myeloproliferative syndrome, most likely essential thrombocytosis (ET), and new diagnosis of plasma cell leukemia (PCL): this is the second known reported case in the literature.
Case Description
A 76-year-old Caucasian woman with a history of paroxysmal atrial fibrillation, hypertension, chronic sciatica requiring assistance with a walker, gastroesophageal reflux disease, and a presumed diagnosis of JAK2 V617F positive ET initially presented to an outside hospital with a left clavicular fracture following a mechanical fall. The patient was incidentally found to have a leukocytosis of 71,000 cells/mcL (66% lymphocytic). She was transferred to Thomas Jefferson University Hospital (TJUH) for further hematologic work-up.
In November of 2018, ~6 months prior to admission to TJUH, the patient received routine outpatient lab-work showing a platelet count of 547,000 cell/mcL. At time of formal hematological evaluation, her platelet count had decreased to 446,000 cell/mcL. She had no evidence of iron deficiency anemia or positive markers for inflammation (Table 1). PCR analysis for the JAK2 V617F point mutation was positive. She had a presumed diagnosis of ET, although she did not have a bone marrow biopsy at the time of diagnosis (10). Hydroxyurea was not started. Repeat lab work in April of 2019, ~1 month before admission to TJUH, revealed a platelet count of 262,000 cells/mcL and white cell count of 14,770 cells/mcL (48.1% lymphocytic). No plasma cells were noted on the differential.
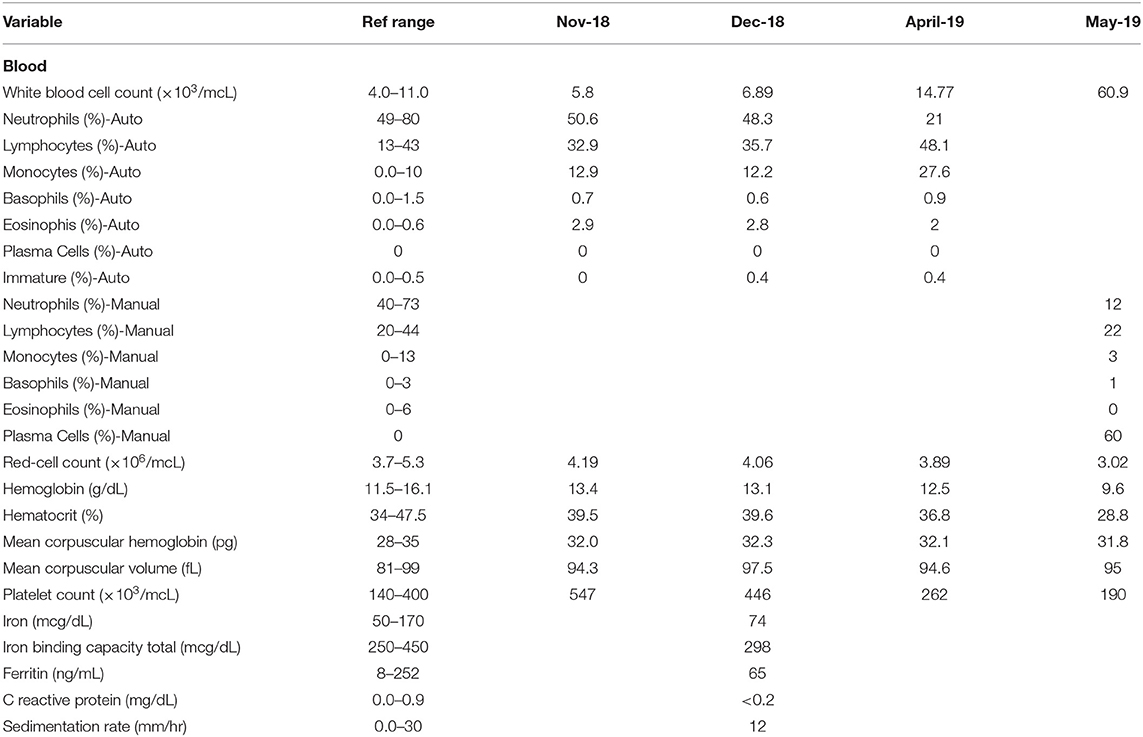
Table 1. Temporal comparison of lab work from November 2018 (Nov-18), December 2018 (Dec-18), April 2019 (April-19), and May 2019 (May-19)—admission to Thomas Jefferson University Hospital.
Given her mild leukocytosis in April of 2019, her physician ordered a peripheral blood flow cytometry. The phenotypes shown were a mixed population of maturing myeloid cells, monocytes, eosinophils, B cells, and T cells. CD34 positive blasts comprised 0.1% of the total nucleated cells. Monocytes and eosinophils comprised 4.7 and 1.6%, respectively. There was no presence of a clonal plasma cell population (CD38 cell population).
One day prior to transfer to TJUH, in May 2019, the patient presented to an outside hospital after a mechanical fall involving her walker, resulting in a fracture of the left clavicle. A complete blood cell count upon admission to TJUH showed a white cell count of 60,900 cells/mcL with a manual differential indicating 60% plasma cells: ~36,540 plasma cells/mcL (36.5 × 109/L), well above 2 × 109/L plasma cells as per PCL diagnostic criteria (Table 1). Peripheral blood flow cytometry showed a monoclonal plasma cell population, comprising 70% of analyzed cells showing the following phenotype: CD5−, CD10−, CD19−, CD20−, CD22−, CD38+ (bright), CD138+, CD45−, CD56−, CD117−, cytoplasmic kappa light chain+ (dim), and cytoplasmic lambda light chain-. On bone marrow examination, the aspirate smear showed sheets of plasma cells comprising 95% of all nucleated cells with an overall hypercellular marrow of 90%. Of note, rare megakaryocytes were seen. Fluorescence in-situ hybridization (FISH) analysis revealed that the plasma cells were positive for loss of centromere 7 (7q), typical and atypical translocations of CCND1/IGH [(11;14)(q13.3;q32.3)], rearrangement of IGH gene involving 14q32.3, deletion of RB1 (13q), and deletion of TP53 (17p); the MPN FISH panel on non-plasma cells was positive for deletion 5q, loss of centromere 7 (7q), deletion of BCR (22q), and deletion of Rb1 (13q) (Table 2).

Table 2. Fluorescence in-situ Hybridization (FISH) result summary: including multiple myeloma (MM) FISH panel on C138+ enriched plasma cells and myeloproliferative neoplasm (MPN) panel on non-enriched unstimulated non-plasma cells.
In late May 2019, the patient underwent treatment with 1 cycle of bortezomib, cyclophosphamide, and dexamethasone (VCD). This was complicated by an admission for volume overload. Her white blood cell count fell from 80,000 to 50,000 during the first few days of treatment, but it was not completely controlled. She was then switched to Dara-VCD. During cycle 1, she was admitted to the hospital for renal failure, during which she became dialysis dependent. After 4 cycles of Dara-VCD, her free kappa light chains decreased from 18,014 mg/L to 8,394 mg/L by September 2019. She was ultimately taken off dialysis. In mid-September, her free kappa light chains increased to 9,396, requiring initiation of cycle 1 of dexamethasone, cyclophosphamide, etoposide, and cisplatin (DCEP). This was performed inpatient, and her free kappa light chains decreased to 4305.2 mg/L. She then began treatment with carfilzomib, lenalidomide, and dexamethasone (Krd) in October 2019. During cycle 2, carfilzomib was lowered to 56 mg/m2 weekly. This cycle was delayed due to admission for neutropenic fever. During initiation of cycle 3, her free kappa decreased to 639 mg/L. For cycle 4 in February 2020, her revlimid was held temporarily, but restarted on day 11. She also had an admission for pneumonia. Her free kappa light chains at this time were 774 mg/L. One year after the patient's initial presentation, the smear from a repeat bone marrow examination in March 2020 showed residual plasma cell myeloma/leukemia (~10% marrow involvement). JAK2 V617F mutation analysis was positive. She unfortunately unexpectedly passed in late March 2020 while at home. A timeline of her course is detailed in Figure 1.

Figure 1. Timeline of patient's course.
Diagnostic Assessment
JAK2 Mutation Analysis
The laboratory test was performed at Integrated Oncology, a business unit of Esoterix Genetics Laboratory, LLC, a wholly owned subsidiary of Laboratory Corporation of America Holdings. Genomic DNA was extracted and amplified by real-time PCR using primers for the appropriate region of the JAK2 gene. The mutant and wild-type JAK2 sequences were detected using specific fluorescent probes. The analytical sensitivity of the assay is 1%: detection of 1 mutant copy of the JAK2 gene per 100 normal copies.
Fluorescence in-situ Hybridization (FISH) Study: MM and MPN Panels
The MM FISH panel study was performed on CD138+ enriched plasma cells; the MPN FISH study was performed on non-enriched unstimulated cells, results of which likely represent abnormalities from non-plasma cells or a mixture of different cell populations. Probe sets from CytoTest, Inc. The cutoff value for the 5q, 7q, BCR/ABL1 translocation, RB1, 17p13.1, 20q12, IGH rearrangement, CCND1/IGH translocation, and MYC gene rearrangement were <5, <6, <2, <8, <8, <7, <7, <2, and <4%, respectively.
There were no overt diagnostic challenges in this case.
Discussion
To our knowledge, this case is one of the few known reports of a patient with a concomitant MPN and pPCL. The literature of reported cases of patients with concomitant MPN and plasma cell dyscrasias raises much interest, namely because these malignancies originate from two separate cellular lineages in hematopoiesis.
Given the literature demonstrating this phenomenon, it has been proposed that in these instances of concomitant malignancies, a common cellular origin might exist. Wang et al. showed that both myeloid and lymphoid cells recovered from BM and spleens of mice with myelofibrosis both had positive JAK2 V617F mutations, suggesting the mutation occurred initially in a common hematopoietic stem cell (HSC) (11). Some evidence suggests that CML hematopoietic stem cell ancestry is shared with lymphocyte stem cell ancestry of the B-cell lineage (12). In addition, acute leukemia can be of mixed lineage origin or even undergo lineage switch suggesting a common hematopoietic stem cell (13). Hou et al. had found a novel human B cell/myeloid common progenitor cell based on absence of CXC chemokine receptor expression reinforcing the concept of shared ancestry (14). A case of a woman with chronic lymphocytic leukemia (CLL) and JAK-2 V617F mutation was reported showing that the JAK2 mutation existed in the B lymphocytes, but not in T lymphocytes. In this case, it was postulated that the JAK-2 mutation was a secondary event to which a primary gene mutation occurred at the common B lymphoid and myeloid stem cell—further suggesting the possibility of a common, pluripotent, myeloid, and B lymphoid progenitor cell (15). Another report assessing the presence of JAK2 V617F mutation in different cell lines in eight patients with PV found that six of these patients solely had the JAK2 mutation in the myeloid lineage of cells; however, one PV patient was found to have the mutation present in both B and T lymphocytes (as well as the myeloid cells), while another PV patient was found to have the mutation present in the B lymphocytes in addition to myeloid cells (16). This indicates that the JAK2 mutation can occur as early as the multipotent HSC level in some MPN cases.
On the FISH analysis utilizing a MM panel on the patient's plasma cells (Table 2), certain common chromosomal abnormalities were present that are common in pPCL. Common pPCL chromosomal abnormalities present included IgH translocations, loss of 13q, and loss of 17p (TP53). For the MPN FISH panel conducted, the deletion of 5q, 7q, and 13q are chromosomal abnormalities notable in MPNs; 13q deletion was also seen in the plasma cells, which is a deletion also found to be prevalent in CLL, MM, and pPCL, as previously mentioned (17). The positive deletion of 13q in the MPN panel is less common for typical MPN chromosomal abnormalities (17). The shared abnormalities on the FISH panel for the plasma cells and non-plasma cells included the 13q deletion and 7q deletion. Interestingly, the 7q deletion is not a typical finding in pPCL FISH analysis, since it is more associated with myeloid disorders (18, 19). The shared 7q deletion suggests that the patient's MPN and plasma cell leukemia may have arisen from a common cellular origin. The 7q deletion, despite being a predominantly myeloid chromosomal abnormality, is also a prognosticator of poor outcome, especially for leukemic transformation in MPN (20, 21).
There have, however, been studies published that argue against the theory of a common cellular origin for these two malignancies. Kuroda et al. showed independent genetic mutations in a patient with the coexistence of multiple myeloma and ET; the JAK2 V617F was identified in peripheral white blood cells and bone marrow-derived non-myeloma cells, but not in CD138+ myeloma cells from the bone marrow (22). Additionally, despite previously stated, there have been reports of patients with concurrent CLL and JAK2 V617F positive MPN that did not show the JAK2 mutation present in the lymphoid cells (4). Overall, it appears there is mixed evidence in support of a common cellular lineage for when these two separate malignancies appear in the same patient; however, it remains possible that in some instances of concomitant MPNs and plasma cell dyscrasias, there might be a common cellular origin.
It is also possible that the bone marrow microenvironment (BMM) could influence concomitant disease. Schofield first demonstrated that bone marrow cells transplanted from wild-type mice into W/Wv mice (mutation in kit) could continue hematopoiesis indefinitely. In comparison, cells that formed colonies in the spleen (colony-forming units-spleen cells) and were subsequently transplanted lost that ability (23, 24). While early studies focused on the role of the microenvironment in promoting normal hematopoiesis, more recent data has shown that alterations in the BMM may support malignant conditions including MPNs and myeloid leukemia (25–27). Lundberg et al. found that bone marrow vascularity was increased and disorganized in patients with MPN and correlated with JAK-2 burden (28). The endothelial cells of the BMM can also be stimulated by proangiogenic and pro-inflammatory factors to release VEGF, which causes increased blast survival and proliferation (29, 30). Plasma cell interactions with the BMM are not as well-studied, but there is evidence that various factors secreted by endothelial cells and stromal cells affect myeloma cell migration (26). While there are no specific studies in relation to the BMM and concomitant disease from a myeloid progenitor and lymphoid progenitor, it appears not unlikely that these individually studied phenomena could coexist in the described patient.
Speculation for the incidence of coexistence for these two malignancies has also been focused on other shared precipitating factors. Interleukin-6 (IL-6) has been postulated as a possible culprit in certain instances linking the association of these two malignancies. IL-6 is known as a promoter of platelet production as well as being a player in the pathogenesis of MM, given the fact that it is a potent human myeloma cell growth factor (4). Radiation or drug exposure, namely hydroxyurea, has been postulated as another shared precipitating factor for both malignancies, although not relevant in this particular case.
Strengths of this case report include its novelty, the availability of patient data and ability to assess this patient's outcome, and its educational value. While there is a similar case detailed from 2004 of a patient from 1997, there is still value in reporting this case, which is more than 15 years later. The patient detailed by Candoni et al. had previous exposure to cytoreductive therapy (hydroxyurea and busulphan), which may have contributed to the development of PCL. Our patient was not started on any similar treatment prior to her diagnosis of PCL. Further research is needed in the association of these diseases to understand whether these two cases reflect similar or divergent mechanisms of pathogenesis. Limitations included the lack of literature of this exact case given its novelty, which leads to an inability to generalize. While previous associations between MPNs and plasma cell dyscrasias have been noted in the literature, this is one of the few published reports of a case of a concomitant JAK-2 positive MPN and primary plasma cell leukemia. The exact mechanism by which this phenomenon occurs remains under debate due to their distinct lineages. As awareness of these cases grows, our hope is that the understanding of these disease states will be further elucidated.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Ethics Statement
At time of writing of the manuscript the individual in the case described had passed away. Written informed consent was obtained from the individual's healthcare proxy and next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
CK organized and wrote manuscript. CT and SH helped with writing of manuscript. TK and AB reviewed and edited manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.01497/full#supplementary-material
References
1. Maerki J, Katava G, Siegel D, Silberberg J, Bhattacharyya PK. Unusual case of simultaneous presentation of plasma cell myeloma, chronic myelogenous leukemia, and a Jak2 positive myeloproliferative disorder. Case Rep Hematol. (2014) 2014:738428. doi: 10.1155/2014/738428
2. Badelita S, Dobrea C, Colita A, Dogaru M, Dragomir M, Jardan C, et al. The simultaneous occurrence of multiple myeloma and JAK2 positive myeloproliferative neoplasms - Report on two cases. J Med Life. (2015) 8:55–61. http://www.ncbi.nlm.nih.gov/pubmed/25914740.
3. Economopoulos T, Economidou J, Papageorgiou E, Dervenoulas J, Christodoulides C, Pappa V, et al. Monoclonal gammopathy in chronic myeloproliferative disorders. Blut. (1989) 58:7–9. doi: 10.1007/BF00320228
4. Malhotra J, Kremyanskaya M, Schorr E, Hoffman R, Mascarenhas J. Coexistence of myeloproliferative neoplasm and plasma-cell dyscrasia. Clin Lymph Myel Leuk. (2014) 14:31–36. doi: 10.1016/j.clml.2013.09.015
5. Murayama T, Matsui T, Hayashi Y, Taniguchi T, Ito M, Natazuka T, et al. Plasma cell leukemia with myelofibrosis. Ann Hematol. (1994) 69:151–152. doi: 10.1007/BF01695697
6. Kasahara S, Tsurumi H, Yoshikawa T, Hara T, Shimizu M, Oyama M, et al. Plasma cell leukemia with myelofibrosis. J Clin Exp Hematopathol. (2008) 48:71–3. doi: 10.3960/jslrt.48.71
7. Rojas EA, Corchete LA, Mateos MV, García-Sanz R, Misiewicz-Krzeminska I, Gutiérrez NC. Transcriptome analysis reveals significant differences between primary plasma cell leukemia and multiple myeloma even when sharing a similar genetic background. Blood Cancer J. (2019) 9:1–13. doi: 10.1038/s41408-019-0253-1
8. Gundesen MT, Lund T, Moeller HEH, Abildgaard N. Plasma cell leukemia: definition, presentation, and treatment. Curr Oncol Rep. (2019) 21:8. doi: 10.1007/s11912-019-0754-x
9. Candoni A, Tiribelli M, Fanin R. Plasma cell leukemia occuring in a patient with thrombocythemia treated with hyroxyurea and busulphan. Leuk Lymphoma. (2004) 45:821–24. doi: 10.1080/10428190310001615710
10. Barbui T, Thiele J, Gisslinger H, Kvasnicka HM, Vannucchi AM, Guglielmelli P, et al. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: document summary and in-depth discussion. Blood Cancer J. (2018) 8:15. doi: 10.1038/s41408-018-0054-y
11. Wang X, Prakash S, Lu M, Tripodi J, Ye F, Najfeld V, et al. Spleens of myelofibrosis patients contain malignant hematopoietic stem cells. J Clin Invest. (2012) 122:3888–99. doi: 10.1172/JCI64397
12. Vogler LB, Crist WM, Vinson PC, Sarrif A, Brattain MG, Coleman MS. Philadelphia-chromosome-positive pre-B-cell leukemia presenting as blast crisis of chronic myelogenous leukemia. Blood. (1979) 54:1164–70. doi: 10.1182/blood.V54.5.1164.1164
13. Stass S, Mirro J, Melvin S, Pui CH, Murphy SB, Williams D. Lineage switch in acute leukemia. Blood. (1984) 64:701–6. doi: 10.1182/blood.V64.3.701.701
14. Hou YH, Srour EF, Ramsey H, Dahl R, Broxmeyer HE, Hromas R. Identification of a human B-cell/myeloid common progenitor by the absence of CXCR4. Blood. (2005) 105:3488–92. doi: 10.1182/blood-2004-07-2839
15. Kodali S, Chen C, Rathnasabapathy C, Wang JC. JAK2 mutation in a patient with CLL with coexistent myeloproliferative neoplasm (MPN). Leukem Res. (2009) 33:e236–9. doi: 10.1016/j.leukres.2009.06.027
16. Ishii T, Bruno E, Hoffman R, Xu M. Involvement of various hematopoietic-cell lineages by the JAK2 V617F mutation in polycythemia vera. Blood. (2006) 108:3128–34. doi: 10.1182/blood-2006-04-017392
17. Adeyinka A, Dewald GW. Cytogenetics of chronic myeloproliferative disorders and related myelodysplastic syndromes. Hematol Oncol Clin North Am. (2003) 17: 1129–49. doi: 10.1016/S0889-8588(03)00087-X
18. Albarracin F, Fonseca R. Plasma cell leukemia. Blood Rev. (2011) 25:107–12. doi: 10.1016/j.blre.2011.01.005
19. Tiedemann RE, Gonzalez-Paz N, Kyle RA, Santana-Davila R, Price-Troska T, Van Wier SA, et al. Genetic aberrations and survival in plasma cell leukemia. Leukemia. (2008) 22:1044–52. doi: 10.1038/leu.2008.4
20. Strasser-Weippl K, Steurer M, Kees M, Augustin F, Tzankov A, Dirnhofer S, et al. Chromosome 7 deletions are associated with unfavorable prognosis in myelofibrosis with myeloid metaplasia. Blood. (2005) 105:4146. doi: 10.1182/blood-2004-11-4319
21. Mesa RA, Li CY, Ketterling RP, Schroeder GS, Knudson RA, Tefferi A. Leukemic transformation in myelofibrosis with myeloid metaplasia: a single-institution experience with 91 cases. Blood. (2005) 105:973–977. doi: 10.1182/blood-2004-07-2864
22. Kuroda J, Matsumoto Y, Tanaka R, Kurita K, Kobayashi T, Shimizu D, et al. JAK2V617F-positive essential thrombocythemia and multiple myeloma with IGH/CCND1 gene translocation coexist, but originate from separate clones. Acta Haematol. (2009) 120:177–181. doi: 10.1159/000187645
23. R S. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells. (1978) 4:7–25.
24. Papayannopoulou T, Scadden DT. Stem-cell ecology and stem cells in motion. Blood. (2008) 111:3923–30. doi: 10.1182/blood-2007-08-078147
25. Kumar R, Godavarthy PS, Krause DS. The bone marrow microenvironment in health and disease at a glance. J Cell Sci. (2018) 131:jcs201707. doi: 10.1242/jcs.201707
26. Walkley CR, Olsen GH, Dworkin S, Fabb SA, Swann J, McArthur GAA, et al. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor γ deficiency. Cell. (2007) 129:1097–110. doi: 10.1016/j.cell.2007.05.014
27. Schmitt-Graeff AH, Nitschke R, Zeiser R. The hematopoietic niche in myeloproliferative neoplasms. Mediators Inflamm. (2015) 2015:347270. doi: 10.1155/2015/347270
28. Lundberg LG, Lerner R, Sundelin P, Rogers R, Folkman J, Palmblad J. Bone marrow in polycythemia vera, chronic myelocytic leukemia, and myelofibrosis has an increased vascularity. Am J Pathol. (2000) 157:15–9. doi: 10.1016/S0002-9440(10)64511-7
29. Dias S, Choy M, Alitalo K, Rafii S. Vascular endothelial growth factor (VEGF)-C signaling through FLT-4 (VEGFR-3) mediates leukemic cell proliferation, survival, and resistance to chemotherapy. Blood. (2002) 99:2179–84. doi: 10.1182/blood.V99.6.2179
Keywords: case report, plasma cell leukemia, myeloproliferative neoplasm, JAK2 mutation, essential thrombocytosis
Citation: Kurian CJ, Thomas C, Houtmann S, Klumpp T and Binder AF (2020) Case Report: Concomitant Diagnosis of Plasma Cell Leukemia in Patient With JAK2 Positive Myeloproliferative Neoplasm. Front. Oncol. 10:1497. doi: 10.3389/fonc.2020.01497
Received: 11 May 2020; Accepted: 13 July 2020;
Published: 27 August 2020.
Edited by:
Francesco Passamonti, University of Insubria, ItalyCopyright © 2020 Kurian, Thomas, Houtmann, Klumpp and Binder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christine J. Kurian, Y2hyaXN0aW5lLmt1cmlhbiYjeDAwMDQwO2plZmZlcnNvbi5lZHU=