
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol., 18 August 2020
Sec. Molecular and Cellular Oncology
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.01460
This article is part of the Research TopicMutant p53 in Cancer Progression and Personalized Therapeutic TreatmentsView all 14 articles
 Vitaly Chasov1
Vitaly Chasov1 Regina Mirgayazova1
Regina Mirgayazova1 Ekaterina Zmievskaya1
Ekaterina Zmievskaya1 Raniya Khadiullina1
Raniya Khadiullina1 Aygul Valiullina1
Aygul Valiullina1 Joseph Stephenson Clarke2
Joseph Stephenson Clarke2 Albert Rizvanov1
Albert Rizvanov1 Matthias G. J. Baud2*
Matthias G. J. Baud2* Emil Bulatov1,3*
Emil Bulatov1,3*The transcription factor p53 is a key tumor suppressor that is inactivated in almost all cancers due to either point mutations in the TP53 gene or overexpression of its negative regulators. The p53 protein is known as the “cellular gatekeeper” for its roles in facilitating DNA repair, cell cycle arrest or apoptosis upon DNA damage. Most p53 mutations are missense and result in either structural destabilization of the protein, causing its partial unfolding and deactivation under physiological conditions, or impairment of its DNA-binding properties. Tumor cells with p53 mutations are generally more immunogenic due to “hot spot” neoantigens that instigate the immune system response. In this review, we discuss the key therapeutic strategies targeting mutant p53 tumors, including classical approaches based on small molecule intervention and emerging technologies such as gene editing and T cell immunotherapy.
The transcription factor p53 functions as a tumor suppressor and is considered as one of the most promising molecular targets for cancer therapy, as it regulates a plethora of intracellular metabolic pathways, including DNA damage repair, apoptosis, and senescence. The p53 protein is widely known as the “guardian of the genome” that prevents the proliferation of cells harboring genetic aberrations, notably oncogenic mutations. In both stressed and unstressed cells, the p53 protein is subject to post-translational modifications, including phosphorylation, acetylation, ubiquitination, and methylation that regulate its stability, localization (cytoplasm or nucleus) and transcriptional activity. Phosphorylation of Ser or Thr residues of p53 was shown to correlate with increasing of p53 activity in response to cellular stress (1).
The TP53 gene encoding the p53 protein is the most frequently altered gene in human tumors (2). The loss of transcriptional functions leading to the deactivation of intrinsic tumor suppressive responses associated with wild-type (WT) p53 is the primary outcome of p53 mutations, and is a hallmark of most cancers The majority of p53 mutations are missense, i.e., cause single residue substitutions, and occur within the DNA-binding domain (DBD). These can be classified as either “DNA contact” or “conformational” mutations (3). “DNA contact” mutations occur in regions that make direct contact with target DNA sequences and are critical for DNA binding, whereas “conformational” mutations diminish DNA-binding by distorting the protein structure through destabilization. Most of these mutations are loss-of-function and exert a dominant negative effect on the WT protein functions. Beyond this, cancer cells appear to gain selective advantages by retaining only the mutant form of the protein, associated with enhanced cell proliferation, metastasis and chemoresistance (4).
The intracellular p53 level is tightly regulated by its negative regulator murine double minute 2 (MDM2) ubiquitin ligase, mostly through ubiquitination followed by proteasomal degradation. In most human cancers, p53 is deactivated either due to mutation or because of the overexpression of MDM2. The strategy of enhancing p53 functions by means of small molecule MDM2 inhibitors has long been of interest to the field by its perceived tractability (5). However, despite development of dozens of high-affinity compounds and multiple clinical trials, none have yet produced a registered drug, suggesting that alternative paths should be given greater attention (6). The MDM2-induced degradation of p53 could be regulated by p14ARF that inhibits the oncogenic action of MDM2 and enhances p53-dependent transactivation and apoptosis (7).
The general approaches employed to destroy the p53-mutant tumor cells are implemented either via restoration of its WT oncosuppressor properties, or focus on tumor elimination by manipulating key components of the immune system. In this review we discuss the current and emerging therapeutic strategies against mutant-p53-driven cancers based on small molecule re-activators, gene editing technologies (introduction of WT gene or CRISPR/Cas mediated corrections) and T cell immunotherapy (Figure 1).
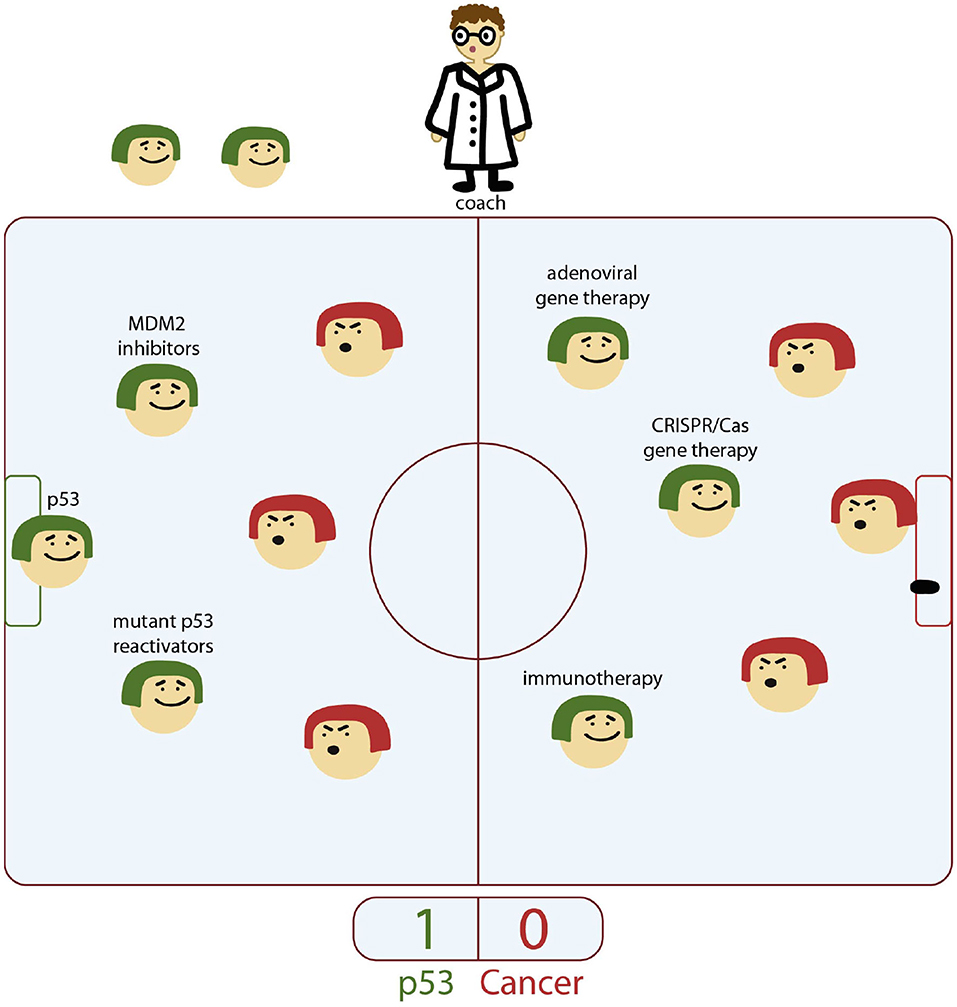
Figure 1. Key players in the p53 team. P53 is the genome “gatekeeper.” MDM2 inhibitors, mutant p53 re-activators are the players of defensive line, whereas adenoviral gene therapy, gene editing tools, and immunotherapy are part of the offensive line in p53 team. New and yet unknown powerful players are expected to enter the game at the forefront of cancer treatment and score a success under the researcher coaching.
MDM2 is mostly known for its oncogenic properties, though its role beyond cancer, notably inflammation, has received increasing attention in recent years (8–10). Numerous synthetic modulators that activate WT p53 by MDM2-dependent, e.g., Nutlin-3a (11–13), and MDM2-independent mechanisms (14–16) have been reported. However, Nutlins and similar inhibitors of MDM2 often demonstrated side effects in clinical trials, such as off-target issues and dose-limiting hematological toxicities, e.g., thrombocytopenia and neutropenia.
Chemoresistant MDM2 mutations were also reported to evolve, although there is evidence that this may be addressed by combination therapies using stapled-peptide MDM2 antagonists. Such mutations occur in N-terminal p53-binding domain, zinc finger and RING domains. “Stapled” peptide inhibitor (PM2) has been reported, which has a covalent hydrocarbon linkage bridging the adjacent turns of an alpha helical peptide for improved stability (17). The peptide recapitulated key p53 signature residues and targeted the N-terminal domain of MDM2. The structural mimicry and extended spatial contacts with the protein allowed PM2 to retain binding (KD = 117 nM) to mutant forms of MDM2 resistant to Nutlins.
Targeting tumors with mutant TP53, both somatic and germline, presents a challenging yet potentially highly rewarding approach as such mutations are the main driver of various types of cancer (18). The equilibrium between the properly folded and misfolded states of p53 can be affected by compounds that interact with mutant p53 and reinstate its native fold and function (Figure 2B). A number of small molecules have been developed to target and stabilize specific mutant forms of p53 and restore WT resembling transcriptional activity, thereby leading to cell cycle arrest or apoptosis of mutant tumor cells. While many tumor suppressor genes are predominantly inactivated in cancer through deletion, truncating mutations or epigenetic mechanisms, the majority of p53 cancer mutations are missense mutations which lead to the expression of functionally altered full-length mutant p53 proteins with single amino acid substitutions. Approximately one third of oncogenic p53 mutants are conformationally unstable due to specific “hot spot” residues that are mutated at a disproportionately high frequency, most of which reside in the structured p53 DNA-binding region (19). The nine most frequent mutations (R175H, R248Q, R273H, R248W, R273C, R282W, G245S, R249S, Y220C), the majority of which are DNA contact mutants, account for about 30% of all p53 cancer driving mutations.
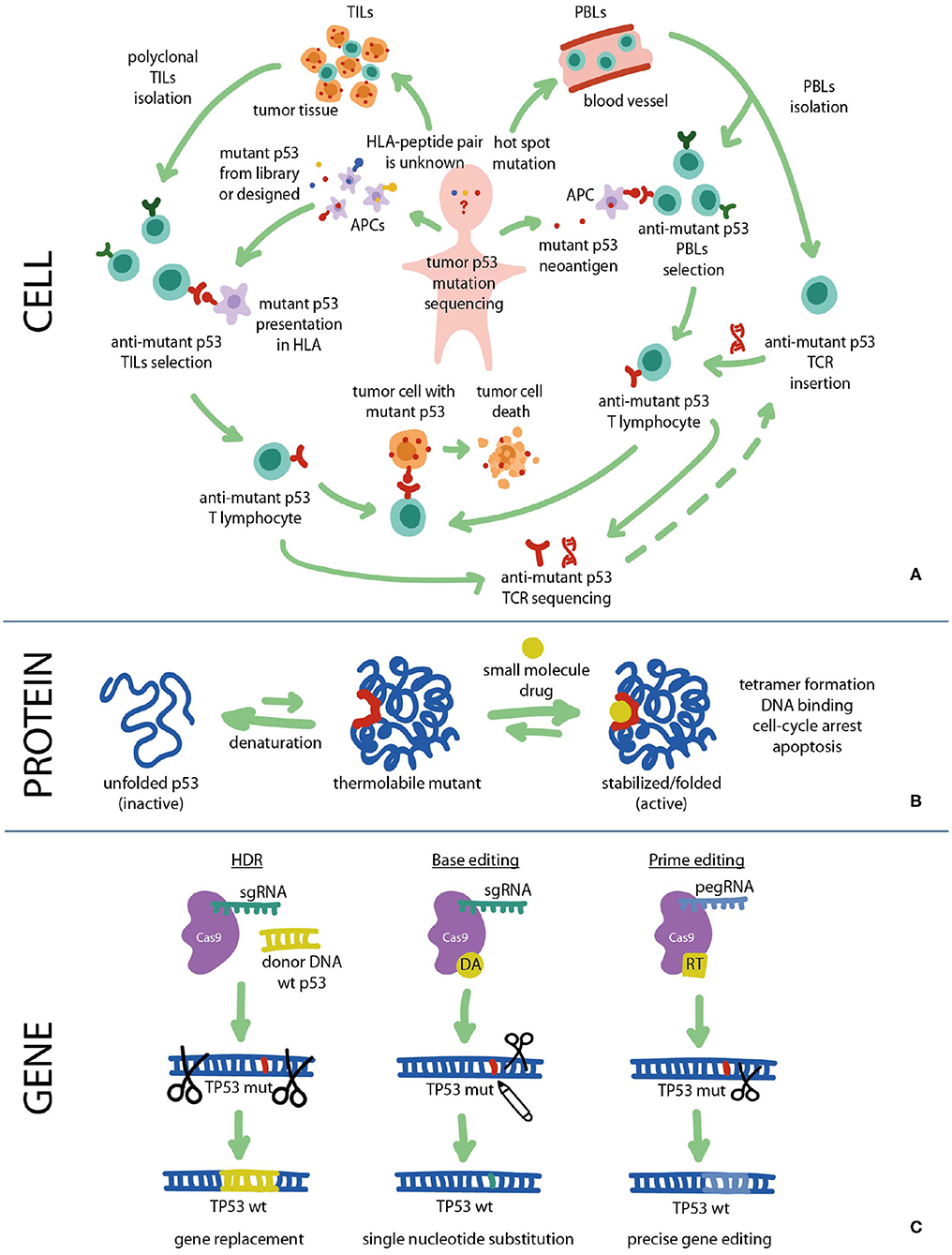
Figure 2. Fighting cancer via p53 pathway can be implemented at all levels: cell, protein, and gene. (A) Cancer cells carrying mutant p53 can be targeted with immunotherapy using mutant p53-specific TILs or TCR-T cells. (B) At the protein level the DNA-binding and transcriptional functions of mutant p53 can be restored using small molecule re-activators that stabilize the protein in its active biological conformation. (C) At the gene level TP53 mutations can be repaired using CRISPR/Cas9 gene editing approaches such as HDR, Base editing and Prime editing. HDR, homology directed repair; HLA, human leukocyte antigen; PBL, peripheral blood lymphocyte; RT, reverse transcriptase; TCR, T cell receptor; TIL, tumor-infiltrating lymphocyte.
Such “contact” mutants not only lose their transcriptional activity due to impaired DNA binding, but also exhibit dominant-negative (DN) effects on the remaining WT p53 allele in addition to the homologous tumor-suppressors p63 and p73 (20). Mutant p53 proteins can form heterotetramers with WT p53, hampering the function of the latter in tumor suppression (21). The primary outcome of TP53 mutations leading to loss of WT p53 functions is the abrogation of its intrinsic tumor suppressive responses such as senescence and apoptosis, while gain-of-function mutant p53 proteins enhance tumor progression, metastatic potential, and drug resistance, greatly contributing to the malignant cellular phenotype (22–24).
Most p53 mutants lose their transcriptional activity and tumor suppressive function, although approximately a third of p53 mutants are temperature sensitive and display sequence-specific transcriptional activity at sub-physiological temperatures (25, 26). Interestingly, introduction of rationally designed second-site suppressor mutations was shown to stabilize the structure of the p53 DBD and reactivate transcription, providing access to valuable WT like variants for screening and drug discovery (27, 28). At the same time, this suggests that stabilization of such “conformational” mutants may provide an opportunity to reinstate their WT function through the use of modulators of their thermal stability. There is currently enormous interest in the identification of natural or synthetic substances (small molecules, peptides, etc.) that can stabilize mutant p53 in its active biological conformation and restore DNA-binding and transcriptional activity (29).
PRIMA-1 and its methyl analog APR-246 (PRIMA-1MET) are promising small molecules that can restore activity of mutant p53 by interacting with the DNA binding domain, promoting proper folding/function (29). This leads to enhanced expression of pro-apoptotic genes Puma, Noxa, and Bax in p53 mutant cells in addition to activation of cell-cycle genes and PARP cleavage independent of p53 mutation status, as observed in multiple studies that involved various types of cancer such as breast, thyroid, myeloma (30).
Both PRIMA and APR-246 are pro-drugs that are intracellularly converted to the reactive methylene quinuclidinone (MQ), which covalently binds to surface-exposed cysteine residues of mutant p53 as well as WT p53. At the same time, experiments with recombinantly expressed and intracellular p53 proteins have shown that unfolded mutant p53 was modified by PRIMA-1 more efficiently than the correctly folded WT protein (31). MQ may also exert its anticancer effect via an alternative p53-independent mechanism of action based on glutathione (GSH) depletion, leading to upregulation of reactive oxygen species (ROS) levels and modulation of the intracellular redox state (32). Currently, APR-246 in combination with azacitidine has reached Phase III clinical trial for the treatment of TP53 mutant myelodysplastic syndromes (MDS) (NCT03745716) and Phase II for TP53 mutant myeloid neoplasms (NCT03072043, NCT03588078).
Bauer et al. (33) identified a range of 2-sulfonylpyrimidines as mild arylating agents of surface cysteines in both WT p53 and mutant p53 core domains. Cysteine arylation upon treatment with lead molecule PK11007 stabilized the mutant p53 core domain in vitro by up to 3°C in differential scanning fluorimetry experiments. In cells, it induced concentration-dependent upregulation of several p53 target genes (p21, PUMA) in cancer cell lines, although p53-independent cytotoxicity was also observed in p53-null and WT p53 cell lines. Interestingly, PK11007 also induced strong GSH depletion and ROS upregulation in cells, reminiscent of the cellular profile and suggested mode of action of MQ and its derivatives. Altogether, these studies highlighted the important effect of cellular redox modulation and a potential general strategy for the development of covalent anticancer agents targeting mutant p53 and redox pathways synergistically, although the propensity for off-target redoxive cell damage by such agents is high.
The Y220C mutation is the ninth most frequent p53 missense mutant overall and is associated with over 100,000 new cancer cases per year worldwide, predominantly breast and ovarian cancer (18, 34). Behind the most common “contact” mutations (vide infra), it is by far the most frequent “conformational” p53 cancer mutation. This large-to-small residue mutation creates an extended cavity on the protein surface that destabilizes the DBD by ~4 kcal/mol (35), causing denaturation and aggregation. The hydrophobic and “druggable” nature of the Y220C pocket offers a fruitful opportunity for targeting using small-molecule stabilizers. Critically, the mutation-induced crevice is distant from the p53 surfaces involved in DNA recognition or protein-protein interactions, allowing for the development of targeted chemical agents that stabilize the DBD without interfering with binding of its natural substrates.
In recent years, fragment-based and in silico screening methods have led to the identification of several potent lead compound families that bind the Y220C pocket. A range of carbazole derivatives displaying low micromolar affinity increased the melting temperature of p53-Y220C and slowed its rate of aggregation in vitro. PK9328 (KD = 2 μM) induced cell viability reduction of several Y220C cancer cell lines, although some toxicity was also observed in other cell lines not carrying this mutation, possibly suggesting off-target effects (36). Pyrazole derivative PK7088 rescued the folding of p53-Y220C and restored transactivation and downstream upregulation of p21 and Noxa expression, correlating with cell cycle arrest and apoptosis (37).
Recently, our group reported several potent iodophenol lead molecules displaying low micromolar binding affinity in vitro, thermal stabilization of up to 2.2°C and selective pro-apoptotic activity in a panel of Y220C cancer cells. Structure-activity studies culminated in aminobenzothiazole derivatives MB710 and MB725, which demonstrated in vitro KD up to 4 μM for p53-Y220C by isothermal titration calorimetry (38). MB725 also showed potent and selective viability reduction of several p53-Y220C cancer cell lines such as BXPC-3 (pancreatic adenocarcinoma), HUH-7 (hepatocellular carcinoma), NUGC3 (gastric adenocarcinoma), while maintaining comparatively low toxicity in WT p53 WI38 (normal fibroblasts), and NUGC4 (gastric adenocarcinoma) in the same concentration range. Importantly, the correlation between in vitro thermal stabilization and selective viability reduction in Y220C cell lines represents an important milestone toward first-in-class anticancer drugs that rescue p53-Y220C function. This provides a compelling rationale for future lead optimization efforts toward potent, non-toxic targeted agents for reactivating the Y220C mutant in anticancer therapy.
ZMC-1 (zinc metallochaperone-1) is a thiosemicarbazone-based small molecule that rescues the WT protein folding and transcriptional activity of p53-R175H mutant by buffering the intracellular Zn2+ levels (39). The underlying rationale is that zinc is required for the correct folding of WT p53 protein and mutations that impair zinc binding strength can hamper protein stability and conformation, leading to impaired sequence-specific DNA binding to p53 response elements (3, 40). ZMC-1 restored site-specific DNA binding and upregulation of p53 target genes (p21, Puma, Mdm2) (41), and inhibited mouse xenograft tumor growth with high allele-specificity for the p53-R175H (p53-R172 in mice) mutant. While zinc buffering alone was insufficient to induce apoptosis (41), ZMC-1 also activated p53 by induction of ROS through its ability to chelate other metal ions (Fe2+, Fe3+, Cu2+) (42). The 3rd-generation thiosemicarbazone COTI-2 functions similarly through both p53-mediated pathways and p53-independent redox homeostatic mechanisms (43) and has entered a Phase II clinical trial (NCT02433626), although it is of note that thiosemicarbazone cancer drug candidates have known nonspecific cytotoxicity and effects on iron metabolic pathways (44).
Gene therapy is a promising therapeutic option and some practical examples have already been studied and successfully applied to re-establish WT p53 expression and activity in cancer cells. Gene therapy involves the replacement or addition of a correct copy of the abnormal gene with a view to restore the genetic information, thus reinstating the WT phenotype.
Currently, gene therapy approaches are based on the combination of genetic material with suitable delivery systems that are often limited by the requirement for efficient nuclear delivery and gene expression. Several primary delivery systems for TP53 gene-based therapeutics have been developed using various viral vectors, including adenoviral, retroviral, vaccine-derived vectors and non-viral ones based on liposomes, polymeric, and gold nanoparticles that allow overcoming systemic delivery hurdles (45). Currently, adenoviral vectors demonstrate minimum side effects among viral vectors used for TP53 gene therapy.
Up until now, several clinical studies using viral vectors for the delivery of p53 have been conducted for experimental medicines, such as Advexin and Gendicine. Advexin (Introgen Therapeutics Inc., TX, USA) is an adenoviral-based experimental therapeutic that provided delivery of WT p53 to cancer cells and demonstrated anticancer activity following amended expression of p53 (46). Gendicine, based on recombinant human p53 adenovirus (Shenzhen SiBiono GeneTech Co. Ltd., China), was approved in 2003 by the China Food and Drug Administration (CFDA) as a first-in-class gene therapy product to treat head and neck cancer, and entered the commercial market in 2004 (47).
Novel adenoviral vectors for cancer gene therapy targeting the p53 pathway were developed to improve the transgene expression levels. Two adenoviral vectors were reported that differ only in the promoter site: the constitutive CMV promoter and the p53-responsive PG promoter where a p53-responsive element is inserted in the viral vector (48). The p53 expression was found to be substantially higher in PCa cells after transduction with AdPGp53 compared to AdCMVp53, and DU145 cells were particularly susceptible to the AdPGp53 tumor suppressor properties.
However, the application of viral vectors can induce high immunogenicity and enhance pre-existing immunity, which limits their clinical use and requires development of new systems with equal efficiency but better safety profiles. Non-viral vectors could present significant advantages when compared with viral ones due to their safety and low cost; nevertheless, viral vectors currently dominate gene therapy clinical trials because of their relatively high delivery efficiency. Thus, viral vectors for the delivery of WT TP53 gene are seen as strong players in the p53 team, however, introduction of other powerful players would increase the firepower of the offensive line.
There are numerous molecular tools for programmable genome editing at a clinical level, including zinc-finger nucleases (ZFNs) (49, 50), transcription activator-like effector nucleases (TALENs) (51, 52), clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) (53). CRISPR/Cas is widely seen as a revolutionary technology for biomedical research with immense clinical opportunities for treating cancer and genetic disorders.
In 2016 the laboratory of David Liu at Harvard University developed an advanced version of CRISPR/Cas enzymes, called Base Editors (BEs), which can mediate specific point mutations in genomic DNA and the resulting amino acid sequence of a target protein (54, 55). BEs constitute enzymatically inactive Cas9 nickase (nCas9) fused to either cytidine deaminase (cytidine BE) or adenosine deaminase (adenosine BE) that result in cytosine-to-thymine or adenine-to-guanine conversion in DNA. In human cells BEs function with high efficiency (15–75%) and low indel rates (<0.1%) compared to classical CRISPR/Cas9 technique based on homology directed repair (HDR). BEs could significantly advance treatment of mutation-associated cancer and genetic diseases by specifically correcting pathogenic mutations in the target gene.
In 2019 the same laboratory reported new gene editing tool, Prime Editors (PEs), based on even more advanced CRISPR/Cas9 “search-and-replace” technology (56). Here, the desired genetic information is directly introduced using nCas9 fused to reverse transcriptase that is directed by prime editing guide RNA (pegRNA) specifying the target DNA sequence and encoding the genetic edits. PEs expand the list of available genome editing tools and together with BEs they can potentially correct ~89% of all known pathogenic human genetic variants.
Several clinical trials are in progress to apply CRISPR/Cas9 for the treatment of patients with mutation-associated disorders, such as β-thalassemia (NCT03655678, NCT03728322) and sickle cell disease (NCT03745287) whereby genetic manipulations with blood cells are carried out ex vivo and then gene-corrected cells are infused back to the patient. A particularly remarkable example is Leber congenital amaurosis 10 (NCT03872479), for which CRISPR-based investigational therapy is administered in vivo via subretinal injection.
Oncogenic or disease-causing mutations represent the primary targets for gene editing therapies. The highest mutation rate of TP53 among other genes makes it a highly desirable target for gene editing tools, e.g., to reverse missense mutation back to the WT state. Chira et al. (57) proposed a CRISPR-based delivery system of a functional TP53 gene. According to the authors, the entire mutated TP53 locus could be deleted and then replaced with a functional copy by homologous recombination. In principle, this might be feasible because the CRISPR/Cas9 system is capable of making such large insertions (58). As a result, the WT phenotype of TP53 could be recovered by replacing the perturbed gene with its functional copy leading to normal p53 expression and tumor regression.
CRIPSR/Cas9 gene editing, including Base Editing, Prime Editing and upcoming technologies have set a high expectations bar for future clinical applications (Figure 2C). BEs, PEs and similar approaches that allow introduction of precise genetic corrections into a target locus without deleting the whole gene could potentially be used to correct TP53 missense mutations as a prospective anticancer therapy (59). Given the rapid advancement of CRISPR/Cas9 technologies and their inevitable introduction to clinical practice, both ex vivo and in vivo target gene modifications in a wide range of cancers, including solid tumors, does not seem to be a distant future anymore.
However, efficient intracellular delivery remains one of the main barriers on the path for wider clinical application of CRISPR/Cas9 technology, including for the purposes of therapeutic editing of TP53 gene. There are three primary strategies for intracellular delivery of CRISPR/Cas9 components: viral vectors, lipid nanoparticles and Cas9-sgRNA complexes. Among these the viral gene delivery strategy seems to be the closest to clinical practice because it has been used in classical gene therapy for decades (60). CRISPR/Cas9-induced double strand breaks (DSBs) of the genomic DNA can result in cell cycle arrest or cell death through p53 pathway that induces DNA damage response and activates expression of downstream effector proteins, e.g., cell cycle inhibitor p21CIP1/WAF1. Functioning of the cellular DNA repair mechanisms that get activated upon DSB, which is often an integral initial step of the gene-editing mechanism, explains one of the reasons for low efficiency of the classical CRISPR/Cas9 system (61, 62).
The rapid development of CRISPR/Cas9-based technologies for therapeutic gene editing of the TP53-associated pathologies is expected to enhance precision, enable improved correction of point mutations, provide better delivery, reduce side effects and facilitate wider clinical applications.
TP53 mutations as part of the overall tumor mutational burden (TMB) can be considered an important factor in predicting response to immunotherapy. TP53 missense mutation-associated p53 nuclear accumulation results in a higher local density of tumor-infiltrating lymphocytes (TILs) within the primary tumor (63). The p53 protein can regulate the immune landscape by modulating inflammation, senescence and immunity in the surrounding tumor microenvironment (TME), including tumor stroma, extracellular matrix (ECM) and associated immune cells infiltrate (64). Mutation in p53 can lead to enhanced neo-angiogenesis and ECM remodeling, disruption of innate tumor immunity, genotoxic stress response of the Toll-like receptor (TLR) pathway, favor pro-tumor macrophage signature and alter cell-mediated immunity in cancer (65).
Some pathways leading to T cell exhaustion are upregulated in such tumors, therefore making them a good target for immunotherapeutic treatment based on genetically modified T cells, e.g., T cell receptor (TCR)-T cells or chimeric antigen receptor (CAR)-T cells (66).
Tumor cells elicit immunogenic responses due to “hot spot” mutant p53 epitopes (neoantigens) produced via proteasomal degradation of intracellular protein and presented by major histocompatibility complex (MHC) (Figure 2A). Initial studies showed that tumors with mutated TP53 could be recognized by peripheral blood lymphocytes (PBLs) upon in vitro stimulation and in vivo immunization (67–69). Cancer vaccines based on primed autologous dendritic cells (DCs) reactive to neoepitopes lead to enhanced antitumor T cell responses in ovarian cancer patients and were associated with better survival prognosis (70).
Tumor-specific adoptive cell therapy (ACT) using antigen-experienced T cells, e.g., patient's own autologous TILs, is a novel approach for targeting p53 mutant cancers. In this approach a HLA/neoantigen complex is recognized by T cell receptors (TCRs) of cytotoxic T cells that effect tumor lysis. Particularly interesting are genetically-engineered T cell receptor (TCR)-T cells with known HLA/neoantigen combination generated by transduction or transposition of specific TCRs into autologous or allogeneic T cells (71). Limitations of this method include differentiation status and proliferative potential of TILs/TCR-Ts, and most importantly potential loss of HLA on tumor cells that would restrict the efficiency of T cell-mediated cytotoxicity.
Deniger et al. (72) prospectively evaluated intratumoral T cell responses to autologous somatic mutant p53 neoantigens expressed by human metastatic ovarian cancers. T cells with specificity to mutated neoantigens found in high frequencies in TILs were expanded from resected metastases and then co-cultured with autologous antigen-presenting cells (APCs) expressing mutated p53 epitopes (Y220C and G245S). Immunogenicity of T cell response was confirmed by upregulation of 4-1BB or secretion of IFNg.
Lo et al. (73) screened TILs for recognition of mutated neoantigens in metastatic colorectal cancer patients and observed T cell mediated recognition of immunogenic p53-R175H mutant. Several TCRs were also identified that could be transduced into allogeneic PBLs for ACT application as an off-the-shelf TCR-T cell product targeting cancer cell lines with a wide range of TP53 mutations.
Malekzadeh et al. (74) developed a TP53-specific screening assay to evaluate T cell responses to “hot spot” mutant p53 neoantigens introduced to autologous APCs intracellularly (tandem minigenes) or extracellularly (pulsed peptides). TCRs from CD4+ and CD8+ T cells reactive to mutant p53 neoantigens were identified in lung cancer patients and then TCR-T cells were engineered that recognize the same HLA/neoantigen complex. In follow-up experiments they isolated PBLs from patients with mutant p53 (R175H, Y220C, R248W) tumors by sorting antigen-experienced CD4+ and CD8+ T cells (75). The T cells were then stimulated with p53 neoantigens (naturally occurring processed and presented peptides) in vitro to confirm the recognition and specificity of the immune response.
Future studies will reveal detailed mechanisms of the complex regulatory interplay between the tumor TP53 status and the immune landscape, including p53-mediated innate anti-tumor response and presentation of mutant p53 neoantigens for eliciting immune recognition by T cell receptors.
The set of available molecular tools arming scientists to battle somatic mutation-associated tumors and hereditary diseases has expanded significantly in recent years. Traditional approaches such as rational structure- and fragment-based drug discovery targeting protein interfaces have been successfully complemented with innovative gene- and cell-based technologies. Adenoviral gene therapy and CRISPR/Cas gene editing are advancing in clinical trials for the treatment of mutation-linked diseases, and the expansion of their applications for therapeutic targeting of TP53 mutations inevitably also approaches. Immunotherapy based on genetically engineered T cells (either autologous or allogeneic) complement cancer treatment by providing unique specificity and efficiency. Therefore, the key players in the mutant p53 team—small molecules, adenoviruses, CRISPR/Cas gene editing enzymes, T cell-based therapies and combinations thereof—broaden the therapeutic scope and provide enormous clinical potential for targeting p53 mutant tumors at all levels (gene, protein and cell). We believe that these approaches have truly encouraging opportunities for clinical applications and that major advancements based on them are approaching in the near future. Together they will fuel challenging, but highly rewarding new developments in the field of mutant p53 cancer therapy.
VC, JS, and MB contributed to section about small molecules. RM and RK contributed to section about gene editing. AV and EZ contributed to section about immunotherapy. EZ and RM prepared the figures. VC, AR, MB, and EB contributed to introduction and conclusion. VC, MB, and EB conceived the idea and coordinated the writing. All authors contributed to the article and approved the submitted version.
Section on small molecule re-activators was funded by RSF grant 19-74-10022 to EB, and by start-up funds from the Southampton School of Chemistry, EPSRC, and Royal Society International Exchanges Cost Share (IEC\R2\181097) funds to MB. Section on gene editing was funded by RFBR grant 19-54-10005 to EB. Work was performed according to the Russian Government Program of Competitive Growth of Kazan Federal University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ACT, adoptive cell therapy; APC, antigen-presenting cells; BE, base editor; CAR, chimeric antigen receptor; CFDA, China Food and Drug Administration; CRISPR/Cas, clustered regularly interspaced short palindromic repeats/CRISPR-associated; DBD, DNA-binding domain; DC, dendritic cell; DSB, double strand break; ECM, extracellular matrix; HDR, homology directed repair; MDM2, murine double minute 2; MDS, mutant myelodysplastic syndrome; MHC, major histocompatibility complex; MQ, methylene quinuclidinone; nCas9, Cas9 nickase; PBLs, peripheral blood lymphocytes; PE, prime editor; pegRNA, prime editing guide RNA; ROS, reactive oxygen species; TALEN, transcription activator-like effector nuclease; TCR, T cell receptor; TILs, tumor-infiltrating lymphocytes; TMB, tumor mutational burden; TME, tumor microenvironment; TLR, Toll-like receptor; WT, wild-type; ZFN, zing-finger nuclease; ZMC, zinc metallochaperone.
1. Kruse J-P, Gu W. Modes of p53 regulation. Cell. (2009) 137:609–22. doi: 10.1016/j.cell.2009.04.050
2. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature. (2013) 502:333–9. doi: 10.1038/nature12634
3. Joerger AC, Fersht AR. Structure–function–rescue: the diverse nature of common p53 cancer mutants. Oncogene. (2007) 26:2226–42. doi: 10.1038/sj.onc.1210291
4. Amelio I, Mancini M, Petrova V, Cairns RA, Vikhreva P, Nicolai S, et al. p53 mutants cooperate with HIF-1 in transcriptional regulation of extracellular matrix components to promote tumor progression. PNAS. (2018) 115:E10869–78. doi: 10.1073/pnas.1808314115
5. Zhang B, Golding BT, Hardcastle IR. Small-molecule MDM2-p53 inhibitors: recent advances. Future Med Chem. (2015) 7:631–45. doi: 10.4155/fmc.15.13
6. Burgess A, Chia KM, Haupt S, Thomas D, Haupt Y, Lim E. Clinical overview of MDM2/X-targeted therapies. Front Oncol. (2016) 6:7. doi: 10.3389/fonc.2016.00007
7. Stott FJ, Bates S, James MC, McConnell BB, Starborg M, Brookes S, et al. The alternative product from the human CDKN2A locus, p14: participates in a regulatory feedback loop with p53 and MDM2. EMBO J. (1998) 17:5001–14. doi: 10.1093/emboj/17.17.5001
8. Ebrahim M, Mulay SR, Anders H-J, Thomasova D. MDM2 beyond cancer: podoptosis, development, inflammation, and tissue regeneration. Histol Histopathol. (2015) 30:1271–82. doi: 10.14670/HH-11-636
9. Bulatov E, Khaiboullina S, Reis dos HJ, Palotás A, Venkataraman K, Vijayalakshmi M, et al. Ubiquitin-proteasome system: promising therapeutic targets in autoimmune and neurodegenerative diseases. BioNanoSci. (2016) 6:341–4. doi: 10.1007/s12668-016-0233-x
10. Bulatov E, Valiullina A, Sayarova R, Rizvanov A. Promising new therapeutic targets for regulation of inflammation and immunity: RING-type E3 ubiquitin ligases. Immunol Lett. (2018) 202:44–51. doi: 10.1016/j.imlet.2018.08.001
11. Beloglazkina A, Zyk N, Majouga A, Beloglazkina E. Recent small-molecule inhibitors of the p53-MDM2 protein-protein interaction. Molecules. (2020) 25:1211. doi: 10.3390/molecules25051211
12. Bulatov E, Zagidullin A, Valiullina A, Sayarova R, Rizvanov A. Small molecule modulators of RING-Type E3 ligases: MDM and cullin families as targets. Front Pharmacol. (2018) 9:450. doi: 10.3389/fphar.2018.00450
13. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. (2004) 303:844–8. doi: 10.1126/science.1092472
14. Brooks CL, Gu W. p53 ubiquitination: Mdm2 and beyond. Mol Cell. (2006) 21:307–15. doi: 10.1016/j.molcel.2006.01.020
15. Bulatov E, Sayarova R, Mingaleeva R, Miftakhova R, Gomzikova M, Ignatyev Y, et al. Isatin-Schiff base-copper (II) complex induces cell death in p53-positive tumors. Cell Death Discov. (2018) 4:103. doi: 10.1038/s41420-018-0120-z
16. Davidovich P, Aksenova V, Petrova V, Tentler D, Orlova D, Smirnov S, et al. Discovery of novel isatin-based p53 inducers. ACS Med Chem Lett. (2015) 6:856–60. doi: 10.1021/acsmedchemlett.5b00011
17. Wei SJ, Chee S, Yurlova L, Lane D, Verma C, Brown C, et al. Avoiding drug resistance through extended drug target interfaces: a case for stapled peptides. Oncotarget. (2016) 7:32232–46. doi: 10.18632/oncotarget.8572
18. Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, et al. TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum Mutat. (2016) 37:865–76. doi: 10.1002/humu.23035
19. Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. (1994) 265:346–55. doi: 10.1126/science.8023157
20. Gaiddon C, Lokshin M, Ahn J, Zhang T, Prives C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol Cell Biol. (2001) 21:1874–87. doi: 10.1128/MCB.21.5.1874-1887.2001
21. Milner J, Medcalf EA. Cotranslation of activated mutant p53 with wild type drives the wild-type p53 protein into the mutant conformation. Cell. (1991) 65:765–74. doi: 10.1016/0092-8674(91)90384-B
22. Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol. (2010) 2:a001107. doi: 10.1101/cshperspect.a001107
23. Amelio I, Melino G. Context is everything: extrinsic signalling and gain-of-function p53 mutants. Cell Death Discov. (2020) 6:16. doi: 10.1038/s41420-020-0251-x
24. Pitolli C, Wang Y, Mancini M, Shi Y, Melino G, Amelio I. Do mutations turn p53 into an oncogene? Int J Mol Sci. (2019) 20:6241. doi: 10.3390/ijms20246241
25. Müller P, Ceskova P, Vojtesek B. Hsp90 is essential for restoring cellular functions of temperature-sensitive p53 mutant protein but not for stabilization and activation of wild-type p53: implications for cancer therapy. J Biol Chem. (2005) 280:6682–91. doi: 10.1074/jbc.M412767200
26. Dearth LR, Qian H, Wang T, Baroni TE, Zeng J, Chen SW, et al. Inactive full-length p53 mutants lacking dominant wild-type p53 inhibition highlight loss of heterozygosity as an important aspect of p53 status in human cancers. Carcinogenesis. (2007) 28:289–98. doi: 10.1093/carcin/bgl132
27. Brachmann RK, Yu K, Eby Y, Pavletich NP, Boeke JD. Genetic selection of intragenic suppressor mutations that reverse the effect of common p53 cancer mutations. EMBO J. (1998) 17:1847–59. doi: 10.1093/emboj/17.7.1847
28. Joerger AC, Ang HC, Veprintsev DB, Blair CM, Fersht AR. Structures of p53 cancer mutants and mechanism of rescue by second-site suppressor mutations. J Biol Chem. (2005) 280:16030–7. doi: 10.1074/jbc.M500179200
29. Bykov VJN, Wiman KG. Mutant p53 reactivation by small molecules makes its way to the clinic. FEBS Lett. (2014) 588:2622–7. doi: 10.1016/j.febslet.2014.04.017
30. Perdrix A, Najem A, Saussez S, Awada A, Journe F, Ghanem G, et al. PRIMA-1 and PRIMA-1Met (APR-246): from mutant/wild type p53 reactivation to unexpected mechanisms underlying their potent anti-tumor effect in combinatorial therapies. Cancers. (2017) 9:172. doi: 10.3390/cancers9120172
31. Lambert JMR, Gorzov P, Veprintsev DB, Söderqvist M, Segerbäck D, Bergman J, et al. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell. (2009) 15:376–88. doi: 10.1016/j.ccr.2009.03.003
32. Tessoulin B, Descamps G, Moreau P, Maïga S, Lodé L, Godon C, et al. PRIMA-1Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance. Blood. (2014) 124:1626–36. doi: 10.1182/blood-2014-01-548800
33. Bauer MR, Joerger AC, Fersht AR. 2-Sulfonylpyrimidines: mild alkylating agents with anticancer activity toward p53-compromised cells. PNAS. (2016) 113:E5271–80. doi: 10.1073/pnas.1610421113
34. Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. (2007) 28:622–9. doi: 10.1002/humu.20495
35. Bullock AN, Henckel J, Fersht AR. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy. Oncogene. (2000) 19:1245–56. doi: 10.1038/sj.onc.1203434
36. Bauer MR, Jones RN, Tareque RK, Springett B, Dingler FA, Verduci L, et al. A structure-guided molecular chaperone approach for restoring the transcriptional activity of the p53 cancer mutant Y220C. Future Med Chem. (2019) 11:2491–504. doi: 10.4155/fmc-2019-0181
37. Liu X, Wilcken R, Joerger AC, Chuckowree IS, Amin J, Spencer J, et al. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. (2013) 41:6034–44. doi: 10.1093/nar/gkt305
38. Baud MGJ, Bauer MR, Verduci L, Dingler FA, Patel KJ, Horil Roy D, et al. Aminobenzothiazole derivatives stabilize the thermolabile p53 cancer mutant Y220C and show anticancer activity in p53-Y220C cell lines. Eur J Med Chem. (2018) 152:101–14. doi: 10.1016/j.ejmech.2018.04.035
39. Blanden AR, Yu X, Loh SN, Levine AJ, Carpizo DR. Reactivating mutant p53 using small molecules as zinc metallochaperones: awakening a sleeping giant in cancer. Drug Discov Today. (2015) 20:1391–7. doi: 10.1016/j.drudis.2015.07.006
40. Loh SN. The missing zinc: p53 misfolding and cancer. Metallomics. (2010) 2:442–9. doi: 10.1039/c003915b
41. Yu X, Vazquez A, Levine AJ, Carpizo DR. Allele-specific p53 mutant reactivation. Cancer Cell. (2012) 21:614–25. doi: 10.1016/j.ccr.2012.03.042
42. Puca R, Nardinocchi L, Porru M, Simon AJ, Rechavi G, Leonetti C, et al. Restoring p53 active conformation by zinc increases the response of mutant p53 tumor cells to anticancer drugs. Cell Cycle. (2011) 10:1679–89. doi: 10.4161/cc.10.10.15642
43. Lindemann A, Patel AA, Silver NL, Tang L, Liu Z, Wang L, et al. COTI-2, a novel thiosemicarbazone derivative, exhibits antitumor activity in HNSCC through p53-dependent and -independent mechanisms. Clin Cancer Res. (2019) 25:5650–62. doi: 10.1158/1078-0432.CCR-19-0096
44. Heffeter P, Pape VFS, Enyedy ÉA, Keppler BK, Szakács G, Kowol CR. Anticancer thiosemicarbazones: chemical properties, interaction with iron metabolism, and resistance development. Antioxid Redox Signal. (2019) 30:1062–82. doi: 10.1089/ars.2017.7487
45. Caffery B, Lee JS, Alexander-Bryant AA. Vectors for Glioblastoma Gene Therapy: Viral & Non-Viral Delivery Strategies. Nanomaterials (Basel). (2019) 9:105. doi: 10.3390/nano9010105
46. Nemunaitis JM, Nemunaitis J. Potential of Advexin: a p53 gene-replacement therapy in Li-Fraumeni syndrome. Future Oncol. (2008) 4:759–68. doi: 10.2217/14796694.4.6.759
47. Zhang W-W, Li L, Li D, Liu J, Li X, Li W, et al. The first approved gene therapy product for cancer Ad-p53 (Gendicine): 12 years in the clinic. Hum Gene Ther. (2018) 29:160–79. doi: 10.1089/hum.2017.218
48. Tamura RE, da Silva Soares RB, Costanzi-Strauss E, Strauss BE. Autoregulated expression of p53 from an adenoviral vector confers superior tumor inhibition in a model of prostate carcinoma gene therapy. Cancer Biol Ther. (2016) 17:1221–30. doi: 10.1080/15384047.2016.1235655
49. Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci. (1996) 93:1156–60. doi: 10.1073/pnas.93.3.1156
50. Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. (2010) 11:636–46. doi: 10.1038/nrg2842
51. Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. (2009) 326:1509–12. doi: 10.1126/science.1178811
52. Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol. (2013) 14:49–55. doi: 10.1038/nrm3486
53. Jiang F, Doudna JA. CRISPR-Cas9 structures and mechanisms. Annu Rev Biophys. (2017) 46:505–29. doi: 10.1146/annurev-biophys-062215-010822
54. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. (2016) 533:420–4. doi: 10.1038/nature17946
55. Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. (2017) 551:464–71. doi: 10.1038/nature24644
56. Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. (2019) 576:149–57. doi: 10.1038/s41586-019-1711-4
57. Chira S, Gulei D, Hajitou A, Berindan-Neagoe I. Restoring the p53 “Guardian” phenotype in p53-deficient tumor cells with CRISPR/Cas9. Trends Biotechnol. (2018) 36:653–60. doi: 10.1016/j.tibtech.2018.01.014
58. Zhang L, Jia R, Palange NJ, Satheka AC, Togo J, An Y, et al. Large genomic fragment deletions and insertions in mouse using CRISPR/Cas9. PLoS One. (2015) 10:e0120396. doi: 10.1371/journal.pone.0120396
59. Mirgayazova R, Khadiullina R, Chasov V, Mingaleeva R, Miftakhova R, Rizvanov A, et al. Therapeutic editing of the TP53 gene: is CRISPR/Cas9 an option? Genes. (2020) 11:704–17. doi: 10.3390/genes11060704
60. Wilson RC, Gilbert LA. The promise and challenge of in vivo delivery for genome therapeutics. ACS Chem Biol. (2018) 13:376–82. doi: 10.1021/acschembio.7b00680
61. Ihry RJ, Worringer KA, Salick MR, Frias E, Ho D, Theriault K, et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat Med. (2018) 24:939–46. doi: 10.1038/s41591-018-0050-6
62. Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med. (2018) 24:927–30. doi: 10.1038/s41591-018-0049-z
63. Kaur HB, Lu J, Guedes LB, Maldonado L, Reitz L, Barber JR, et al. TP53 missense mutation is associated with increased tumor-infiltrating T-cells in primary prostate Cancer. Hum Pathol. (2019) 87:95–102. doi: 10.1016/j.humpath.2019.02.006
64. Blagih J, Buck MD, Vousden KH. p53, cancer and the immune response. J Cell Sci. (2020) 133: doi: 10.1242/jcs.237453
65. Agupitan AD, Neeson P, Williams S, Howitt J, Haupt S, Haupt Y. P53: a guardian of immunity becomes its saboteur through mutation. Int J Mol Sci. (2020) 21:3452. doi: 10.3390/ijms21103452
66. Titov A, Valiullina A, Zmievskaya E, Zaikova E, Petukhov A, Miftakhova R, et al. Advancing CAR T-cell therapy for solid tumors: lessons learned from lymphoma treatment. Cancers. (2020) 12:125. doi: 10.3390/cancers12010125
67. Carbone DP, Ciernik IF, Kelley MJ, Smith MC, Nadaf S, Kavanaugh D, et al. Immunization with mutant p53- and K-ras-derived peptides in cancer patients: immune response and clinical outcome. J Clin Oncol. (2005) 23:5099–107. doi: 10.1200/JCO.2005.03.158
68. Ito D, Visus C, Hoffmann TK, Balz V, Bier H, Appella E, et al. Immunological characterization of missense mutations occurring within cytotoxic T cell-defined p53 epitopes in HLA-A*0201+ squamous cell carcinomas of the head and neck. Int J Cancer. (2007) 120:2618–24. doi: 10.1002/ijc.22584
69. Couch ME, Ferris RL, Brennan JA, Koch WM, Jaffee EM, Leibowitz MS, et al. Alteration of cellular and humoral immunity by mutant p53 protein and processed mutant peptide in head and neck cancer. Clin Cancer Res. (2007) 13:7199–206. doi: 10.1158/1078-0432.CCR-07-0682
70. Tanyi JL, Bobisse S, Ophir E, Tuyaerts S, Roberti A, Genolet R, et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci Transl Med. (2018) 10:eaao5931. doi: 10.1126/scitranslmed.aao5931
71. Deniger DC, Pasetto A, Tran E, Parkhurst MR, Cohen CJ, Robbins PF, et al. Stable, nonviral expression of mutated tumor neoantigen-specific T-cell receptors using the sleeping beauty Transposon/Transposase system. Mol Ther. (2016) 24:1078–89. doi: 10.1038/mt.2016.51
72. Deniger DC, Pasetto A, Robbins PF, Gartner JJ, Prickett TD, Paria BC, et al. T-cell responses to TP53 “Hotspot” mutations and unique neoantigens expressed by human ovarian cancers. Clin Cancer Res. (2018) 24:5562–73. doi: 10.1158/1078-0432.CCR-18-0573
73. Lo W, Parkhurst M, Robbins PF, Tran E, Lu Y-C, Jia L, et al. Immunologic recognition of a shared p53 mutated neoantigen in a patient with metastatic colorectal cancer. Cancer Immunol Res. (2019) 7:534–43. doi: 10.1158/2326-6066.CIR-18-0686
74. Malekzadeh P, Pasetto A, Robbins PF, Parkhurst MR, Paria BC, Jia L, et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J Clin Invest. (2019) 129:1109–14. doi: 10.1172/JCI123791
Keywords: p53, mutation, small molecules, adenoviral gene therapy, CRISPR/Cas gene editing, immunotherapy
Citation: Chasov V, Mirgayazova R, Zmievskaya E, Khadiullina R, Valiullina A, Stephenson Clarke J, Rizvanov A, Baud MGJ and Bulatov E (2020) Key Players in the Mutant p53 Team: Small Molecules, Gene Editing, Immunotherapy. Front. Oncol. 10:1460. doi: 10.3389/fonc.2020.01460
Received: 23 April 2020; Accepted: 09 July 2020;
Published: 18 August 2020.
Edited by:
Oleg Timofeev, Philipps University of Marburg, GermanyReviewed by:
Magnus Olsson, Karolinska Institutet (KI), SwedenCopyright © 2020 Chasov, Mirgayazova, Zmievskaya, Khadiullina, Valiullina, Stephenson Clarke, Rizvanov, Baud and Bulatov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthias G. J. Baud, bS5iYXVkQHNvdG9uLmFjLnVr; Emil Bulatov, Y2hlbWJpby5rYXphbkBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.