 Zhou Jiang
Zhou Jiang Jennifer L. Hsu1
Jennifer L. Hsu1 Mien-Chie Hung
Mien-Chie Hung
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 22 July 2020
Sec. Cancer Metabolism
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.01197
This article is part of the Research Topic Cancer Cell Metabolism and Immunomodulation in the Context of Tumor Metastasis View all 30 articles
Immune checkpoint inhibitors (ICIs) targeting immune checkpoint proteins, such as CTLA-4 and PD-1/PD-L1, have demonstrated remarkable and durable clinical responses in various cancer types. However, a considerable number of patients receiving ICIs eventually experience a relapse due to diverse resistance mechanisms. As a result, there have been increasing research efforts to elucidate the molecular mechanisms behind resistance to ICIs and improve patient outcomes. There is growing evidence that the dysregulated metabolic activity of tumor cells generates an immunosuppressive tumor microenvironment (TME) that orchestrates an impaired anti-tumor immune response. Notably, the immunosuppressive TME is characterized by nutrient shortage, hypoxia, an acidic extracellular milieu, and abundant immunosuppressive molecules. A detailed understanding of the TME remains a major challenge in mounting a more effective anti-tumor immune response. Herein, we discuss how tumor cells reprogram metabolism to modulate a pro-tumor TME, driving disease progression and immune evasion; in particular, we highlight potential approaches to target metabolic vulnerabilities in the context of anti-tumor immunotherapy.
Immune checkpoint proteins are paired regulators of immune response. These molecules are crucial for self-tolerance, which blocks immune cells from attacking autologous cells aimlessly. However, by utilizing immune checkpoint pathways, cancer cells can send an “off” signal to anti-tumor immune cells and escape immune surveillance. Immunotherapy drugs called immune checkpoint inhibitors (ICIs) work by blocking checkpoint proteins from binding with their partner proteins, and this in turn decreases the “off” signal and enhances anti-tumor immunity. Immunotherapy promises to be more significant than any other form of treatment, especially for patients whose tumors have already metastasized. Two pathways that control immune inhibitory signals have been successfully targeted in clinical applications: (1) cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and its ligand B7 molecules and (2) programmed cell death 1 (PD-1) and programmed cell death ligand 1 (PD-L1) (1, 2). Antibodies against CTLA-4, PD-1, and PD-L1 have demonstrated durable clinical responses and have been approved by the U.S. Food and Drug Administration for various cancers (Table 1).
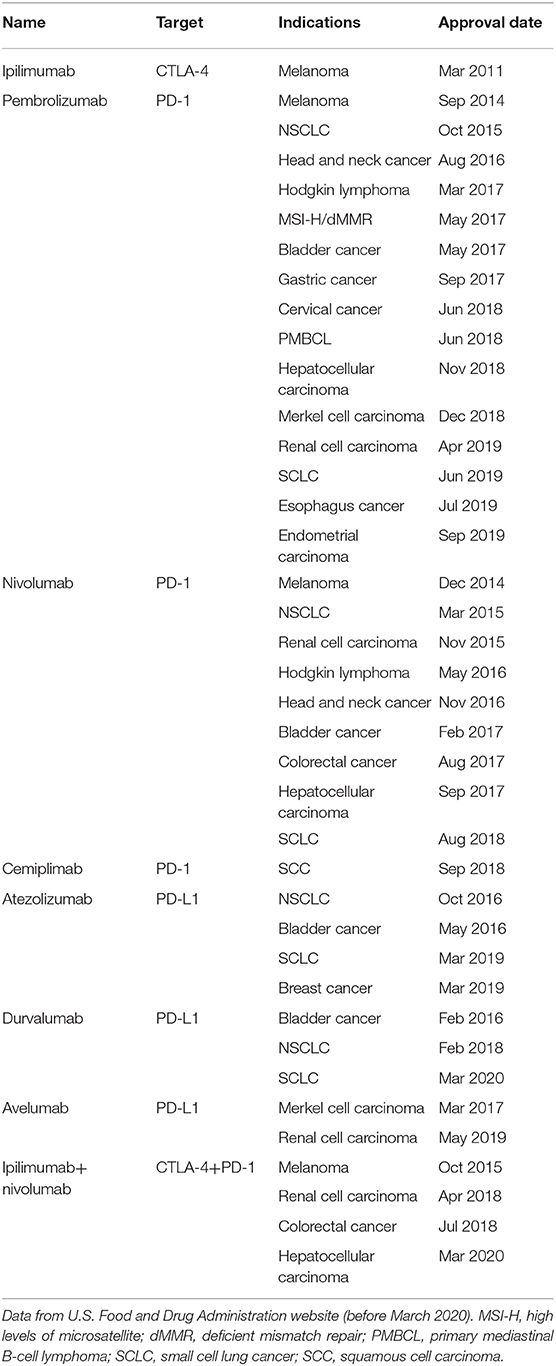
Table 1. U.S. Food and Drug Administration–approved immune checkpoint inhibitors for cancer treatment.
Despite the promising clinical success of ICIs, ~60 to 70% of patients do not respond to immunotherapy as a single agent; in contrast, those who demonstrate initial response eventually develop resistance (3). A number of mechanisms of resistance to ICIs, including interferon (IFN)-γ signaling pathway mutations, JAK1/JAK2-inactivating mutations, the absence of antigen presentation, and the upregulation of IFN pathway-driven inhibitory immune checkpoints, have been well-investigated (4, 5). In addition, in the tumor microenvironment (TME), mounting evidence indicates that abnormal metabolic activities of cancer cells play an essential role in the suppression of the anti-tumor immune response and lead to tumor immune evasion and metastasis (6).
Therefore, a deep understanding of the metabolic differences between tumor and normal tissue and their impact on anti-tumor immune response will not only help expand therapeutic options, but also help overcome the issue of resistance over time. The impact of metabolism on the tumor cell itself, including the generation of redox equivalents, energy, and macromolecules (proteins, lipids, DNA, and RNA) has been well-addressed (7). In this review, we focus on how the abnormal metabolism of tumor cells modulates the TME profile and leads to tumor progression and immune evasion. Furthermore, on the basis of current knowledge of cancer cell metabolism, we highlight potential therapeutic strategies that could restrict tumor progression and enhance the anti-tumor effect of ICIs.
Nutrient competition between different cells can influence cell survival, growth, and function. The demand for nutrients is especially high in the TME, and the competition between tumor cells and immune cells can dampen the anti-tumor response (Figure 1).
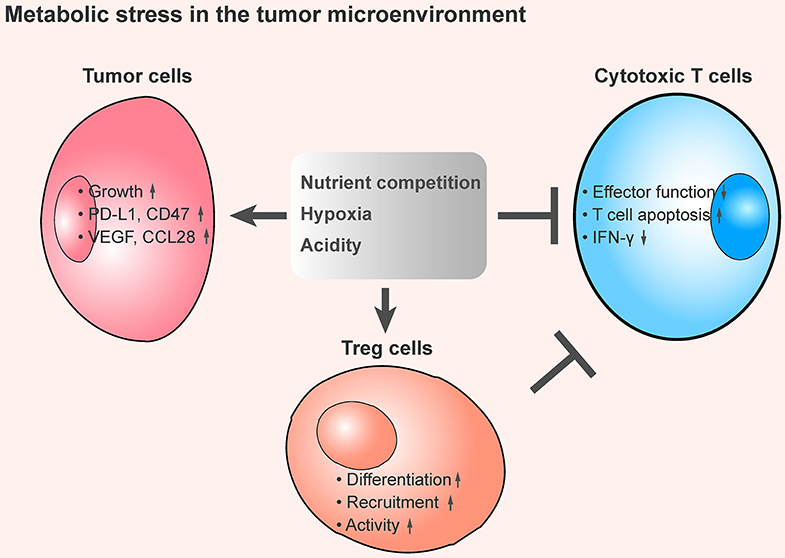
Figure 1. The tumor metabolic stress shapes an immunosuppressive the tumor microenvironment. An overview of metabolic stress in the TME that mediates immune suppression. In the TME, cancer cells exhibit a substantial demand of nutrients, including glucose, amino acids, and fatty acids, and this contributes to the lack of dioxygen and maintains high production of H+. These metabolic stresses promote tumor cell growth, increase the expression of immune checkpoint proteins and immunosuppressive cytokines secretion, enhance the inhibitory function of regulatory T cells, and inhibit the anti-tumor effect of tumor-infiltrating cytotoxic T cells, thereby leading to an immunosuppressive TME.
Anti-tumor immunity is impaired by the competition for carbohydrates. Tumors exhibit high rates of glycolysis; thus, glucose is in high demand in tumor cells. One study showed that upregulated PD-L1 on tumor cells promotes mammalian target of rapamycin (mTOR) activity and glycolytic metabolism, which leads to tumor-mediated glucose restriction, alters CD8+ T-cell metabolism, and dampens the ability of T cells to produce IFN-γ. Even when tumors are highly antigenic, the competition for glucose still inhibits T-cell activity (8). Glucose deprivation also suppresses anti-tumor effector functions of intratumoral Th1 CD4+ T cells by limiting the Ca2+-NF-AT1 signaling pathway in CD4+ T cells (9).
Also damaging to anti-tumor response is the competition for amino acids. The enzyme indoleamine 2,3-dioxygenase (IDO), which catalyzes the essential amino acid tryptophan along the kynurenine pathway, is widely expressed in human cancers, and higher IDO expression is correlated with poorer prognosis in a variety of cancer types (10). Expression of IDO by tumor cells, dendritic cells, and macrophages leads to immune suppression within the TME (11–14). IDO contributes to immune regulation by producing kynurenine, a ligand for the aryl hydrocarbon receptor, and by consuming tryptophan to trigger amino acid-sensing signal transduction pathways. The upregulated kynurenine activates the aryl hydrocarbon receptor, which is a ligand-activated transcription factor (15), and increases immunosuppression by promoting differentiation of T-regulatory cells (Tregs) (16), and this in turn suppresses anti-tumor immune response (17) and decreases the immunogenicity of dendritic cells (16). Moreover, IDO expression leads to a rapid consumption of tryptophan in the tumor milieu (18). Below 0.5 μM tryptophan, T-cell proliferation is inhibited significantly (19). The reduction of tryptophan can also trigger stress-response pathways that respond to amino acid withdrawal, such as, general control non-derepressible 2 (GCN2) and mTOR. GCN2 responds to the presence of uncharged transfer RNA; amino acid insufficiency then activates GCN2, which leads to phosphorylation of its downstream molecule eukaryotic initiation factor 2α (eIF2α) (20). Phosphorylated eIF2α blocks the ribosomal translation of most mRNA species, and leads to cell-cycle arrest and functional anergy in CD8+ T cells (21). Activated GCN2 inhibits Th17 differentiation in CD4+ T cells but promotes immunosuppressive Treg differentiation and activity (22, 23). Thus, the depletion of tryptophan may impair anti-tumor response by inhibiting CD8+ cells function while promoting Treg activities. Targeting IDO-kynurenine-tryptophan axis could be an effective strategy to enhance the efficiency of immunotherapy. The IDO1-specific inhibitor 1-methyltryptophan has been shown to significantly inhibit IDO1 activity, and the effects of 1-methyltryptophan in enhancing T-cell responses against tumor antigens, allograft antigens, and autoantigens in vivo have been validated (24, 25). In addition, some orally available IDO1 inhibitors, epacadostat, and navoximod, have demonstrated safety and could reverse tryptophan depletion and kynurenine accumulation (26).
Similar to competition for tryptophan, competition for L-arginine also triggers GCN2 and mTOR signaling (27, 28). T cells with increased L-arginine levels display improved anti-tumor activity due to a combination of phenotypic changes, including improved survival and maintenance of a T central memory-like phenotype. However, some studies showed that arginine supports tumor cell growth and suppresses anti-tumor immunity (29). In tumor cells, L-arginine is a substrate of nitric oxide synthase and arginase (30). Additional studies indicated that nitric oxide activates cyclooxygenase-2 (31), which suppresses type I interferon–mediated tumor eradication in melanoma, and upregulates tumoral PD-L1 expression (32, 33). Thus, arginine metabolism in tumor cells promotes tumor progression and immune evasion. Depletion of L-arginine in leukemia by the addition of a PEGylated form of the catabolic enzyme arginase I (peg-Arg-I) has demonstrated anti-tumor activity (34, 35). However, L-arginine depletion also suppresses T-cell responses in tumors by inducing myeloid-derived suppressor cell (MDSC) infiltration (36). Therefore, therapeutic strategies that specifically deplete L-arginine metabolism in tumor cells are needed to eradicate tumor without dampening anti-tumor immunity.
Emerging evidence also shows that tumors may engage in high rates of fatty acid uptake. The survival and metastasis of tumor cells depend on fatty acid uptake and consumption, and subsequent catabolism through fatty acid β-oxidation pathway (37). Indeed, limiting low-density lipoprotein uptake reduces the oncogenic properties of pancreatic adenocarcinoma and renders cancer cells more sensitive to chemotherapy (38). Given that CD8+ effector T cells also take up fatty acids at high rates (39), fatty acid may be another environmental nutrient that the CD8+ effector T cells require to compete with tumor cells in the TME.
Hypoxia occurs when there is a shortage of dioxygen and tissues are inadequately oxygenated (40). In the TME, rapidly proliferation of tumor cells results in heterogeneously distributed zones of low oxygen concentration, which leads to hypoxic stress. Under such condition, hypoxia appears to be an important metabolic regulator that contributes to immunosuppression and tumor heterogeneity (41) (Figure 1).
Hypoxia can diminish anti-tumor immunity directly. One study showed that hypoxia abolishes the killing potential of natural killer (NK) cells by decreasing the surface expression of NK cell activating receptors NKG2D and CD16 (42). Hypoxia also induces T-cell apoptosis by inhibiting the expression of C-C motif chemokine receptor 7 (CCR7), which is essential for T-cell differentiation (43). Furthermore, while Treg infiltration in the tumor is associated with poor survival in patients with various cancers (17), hypoxia-activated hypoxia-inducible factor (HIF)-1 has been shown to promote Treg differentiation through upregulation of FoxP3 expression (44).
Hypoxia also suppresses anti-tumor immunity by upregulating immune checkpoint proteins. Tumor cells take advantage of this upregulation to suppress the anti-tumor function of immune cells through the interaction of the inhibitory costimulatory molecules with their ligands. Some studies showed that hypoxia-activated HIF-1α upregulates PD-L1 expression on tumor cells and immune cells by binding directly to a hypoxia response element in the proximal promoter of CD274 (encoding PD-L1) (45–47). The upregulated PD-L1 limits cytotoxic T-cell activity, and thus increases the resistance of tumor cells to cytotoxic T cell-mediated lysis. As potential clinical applications, nitric oxide signaling could block hypoxia-activated HIF-1α function (48). Nitroglycerin (also called GTN), an activator of nitric oxide signaling, blocks PD-L1 expression in hypoxic tumor cells and suppresses hypoxia-driven cytotoxic T-cell apoptosis, thereby increasing the sensitivity of tumor cells to T cell-mediated cytotoxicity (45). As a mechanism to disrupt tumor hypoxia, the hypoxia-activated prodrug TH-302 also demonstrated pre-clinical benefits in improving immunotherapy efficiency by promoting CD8+ T-cell effector function and diminishing MDSCs (47). CD47, also known as integrin-associated protein, interacts with macrophages expressing signal-regulatory protein α (SIRPα) and delivers a “don't eat me” signal to avoid phagocytosis and innate immune surveillance (49). CD47 is overexpressed in various types of cancer, and its overexpression is correlated with poor prognosis in patients (50, 51). One study showed that HIF-1α-dependent expression of CD47 leads to decreased phagocytosis of tumor cells, which promotes cancer progression and immune evasion (52). MHC class I chain-related (MIC) molecules A and B both play important roles in tumor immunosurveillance and are expressed on NK cells, lymphokine-activated killer cells, and cytotoxic T cells (53). MIC molecules are expressed in various types of carcinomas (54), and their interaction with natural killer group 2D receptor on NK, LAK, and effector T cells leads to the activation of those cells and the subsequent lysis of tumor cells (55). One study revealed that hypoxia-activated HIF-1α decreases the surface expression of MIC molecules and leads to immune escape. The study also revealed that activation of nitric oxide signaling interferes with this immune escape mechanism (56).
In addition, hypoxia induces the secretion of immunosuppressive molecules in tumor cells. Through HIF-1α activation, hypoxia induces the expression of vascular endothelial growth factor (VEGF) (57), which promotes tumor angiogenesis. Also, tumor-secreted VEGF promotes the infiltration of MDSCs in the tumor (58), which inhibits the anti-tumor function of T cells and contributes to tumor progression (59). Secreted VEGF restricts the maturation of dendritic cells, blocks antigen presentation, promotes macrophage polarization from anti-tumor M1 to pro-tumor M2, and enables immune escape (58, 60). Thus, VEGF is a promising target for immune therapy. Other hypoxia-induced molecules, such as, CCL28, recruit CC chemokine receptor 10-positive Tregs to the tumor region and facilitate tumor immune evasion (61).
Collectively, hypoxia not only promotes an immunosuppressive TME but also induces the expression of immune checkpoint receptors and immunosuppressive molecules, leading to immune surveillance escape.
The TME of solid tumors is acidic because tumor cells favor aerobic glycolysis, which results in significant production of lactate and H+. With this metabolic preference, also known as the Warburg effect (62), the overabundance of lactate allows fast incorporation of carbon into biomass (e.g., nucleotides, amino acids, and lipids) and facilitates rapid cell proliferation (63). Although tumor cells maintain high production of H+, the major acidic metabolite is exported to the extracellular side. As a result, the TME becomes more acidic (64): the extracellular pH value inside patients' tumors is 6.9–7.0 (in contrast to 7.3–7.4 in normal tissues) (65), and in mouse tumor models, the tumor pH value is around 6.2–6.9 (66). Tumor acidity is important for tumor progression and metastasis (67) and is hypothesized to suppress the anti-tumor functions of immune cells (68) (Figure 1).
Acidic conditions have been found to inhibit the proliferation, differentiation, and cytokine production of cytotoxic T cells (69), and suppress their anti-tumor effects. However, the mechanisms underlying this process remain unclear. One possibility is that the pro-inflammatory cytokine IFN-γ is acid unstable and can be denatured in the acidic environment (70). IFN-γ is well-known to promote the recruitment of T cells through paracrine signaling. Notably, IFN-γ facilitates T cell-mediated killing by upregulating MHC class I expression on tumor cells and directly promotes tumor cell ferroptosis, a type of programmed cell death dependent on iron (70, 71). Furthermore, IFN-γ promotes the activation of M1 macrophages that are involved in anti-tumor immunity (72); assists in the differentiation of pro-inflammatory Th1; and inhibits pro-tumor Th2 differentiation (73). Thus, IFN-γ plays an anti-tumor role by maximizing the anti-tumor efficiency of M1 macrophages and CD8+ T cells. In turn, the denaturation of IFN-γ in the TME can suppress cytotoxic T cell-mediated tumor killing function and facilitate the polarization of the anti-tumor Th1 phenotype toward the pro-tumor Th2 phenotype, as well as prevent the activation of M1 macrophages (74). Therefore, in an acidic TME, tumor cells can easily evade immune surveillance and resist immunotherapy.
The negative impact of acidity on the anti-tumor activity of immune cells may manifest as clinical resistance. Indeed, studies showed that tumor acidity is correlated with poor prognosis in cancer patients who received ICI therapy (74, 75). Therefore, targeting acidity could be a promising strategy to improve immunotherapy efficiency. One option is to neutralize tumor pH by bicarbonate; after the pH value is neutralized in the TME, tumors showed improved response to ICIs as well as to adoptive T-cell therapy (76). Blocking the export of protons could also prevent tumor acidity. Tumor cells export intracellular protons by receptor enzymes, such as, V-ATPase. Esomeprazole, an inhibitor of V-ATPase, promotes the infiltration and effector function of IFN-γ+ cytotoxic T cells, suggesting that V-ATPase inhibition can increase the therapeutic potential of adoptive T cell immunotherapy (77). Collectively, the above findings indicate that tumor acidity induces immune evasion while inhibition of tumor acidity improves immunotherapy efficiency.
Adenosine is a product of the enzymatic breakdown of adenosine 5′-triphosphate (ATP), which can directly influence adenosine receptor-expressing cells and promote tumor growth, survival, and metastasis. Both adenosine and ATP are present at very low levels in extracellular fluids (78). However, ATP can be released into the extracellular milieu by cells under stress due to hypoxia, apoptosis, necrosis, etc. (79). ATP is gradually catalyzed and dephosphorylated by two ectonucleotidases, CD39 and CD73 (80), which generates adenosine (81). High expression of CD39 and CD73 is strongly correlated with poor clinical outcomes in patients with various cancer types (81–83). These ectonucleotidases can be induced by hypoxia and are highly expressed on cells in the TME, including tumor cells, MDSCs, Tregs, and tumor-associated macrophages (81, 84–86).
Extracellular adenosine activates signaling pathways through G protein-coupled adenosine receptors (AR), A1, A2a, A2b, and A3, all of which are widely expressed in both immune cells and tumor cells in the TME (87). Adenosine-AR signals enables tumors to escape immune surveillance by suppressing the activity of multiple anti-tumor immune cells, including CD8+ T cells, dendritic cells, natural killer cells, and M1 macrophages, while enhancing the activity of immunosuppressive cell types, including MDSCs and Tregs. The adenosine-AR signals in dendritic cells upregulate IL-10, IDO-1, TGFβ, and arginase-2, thus facilitating naïve T-cell differentiation toward Th2 lineages and promoting tumor growth (87). Adenosine also limits natural killer cell differentiation, proliferation, and production of the pro-inflammatory cytokines IFN-γ and TNFα (88, 89). In macrophages, adenosine induces pro-tumor M2 macrophage polarization by reducing the expression of IL-2, TNFα, and nitric oxide but upregulating arginase-1, IL-10, and VEGF (87, 90).
The adenosine-AR axis also promotes immunosuppressive cell functionality. One study showed that adenosine-AR promotes MDSC expansion and facilitates their immunosuppressive activity (91). A2BR activation on MDSCs and tumor cells promotes tumor progression by inducing VEGF secretion and angiogenesis (83, 92). Adenosine accumulation also boosts cancer cell survival and proliferation by activation of AKT, ERK1/2, JNK, and protein kinase C δc (93).
Unlike adenosine receptor A2AR, A2BR, and A3, one study showed adenosine receptor A1 (ADORA1) signaling axis suppresses tumor PD-L1-mediated immune evasion (94). Downregulation and inhibition of adenosine receptor A1 (ADORA1) significantly induces tumor PD-L1 expression by promoting ATF3 transcriptional activity. Although the role of adenosine signaling in tumor immunity is different from that in immune cells, the antagonist of the adenosine receptor 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) has demonstrated synergistic effect with PD-1 antibody in melanoma and non-small cell lung cancer (NSCLC) (94).
Generally, adenosine exhibits a pro-tumor role in the TME (Figure 2). Therefore, targeting adenosine signaling pathways could be a promising anti-tumor strategy. CPI-444 is an orally available AR inhibitor; in a mouse model, it showed anti-tumor effect as a single agent and synergistic anti-tumor activity when combined with anti-PD-1/PD-L1 antibodies (95). Similarly, several AR antagonists in combination with anti-PD-1/PD-L1 are being investigated in the clinical trial stage (NCT02655822, NCT03207867, and NCT02740985). Depleting extracellular adenosine by blocking the activity of CD39 and CD73 could also restrict adenosine signaling pathways. Treatment with the CD39 inhibitor POM-1 or a blocking antibody demonstrated enhanced anti-tumor immunity by increasing cytotoxic T cell- and natural killer cell-mediated killing function in a mouse model (96). CD73 is in turn targeted by the monoclonal antibody MEDI9447, which inhibits CD73 ectonucleotidase activity. The combination of MEDI9447 and PD-1 antibodies in tumor models showed additive activity against adenosine-mediated immunosuppression, and a phase I study of MEDI9447 in cancer patients was initiated accordingly (NCT02503774) (97).
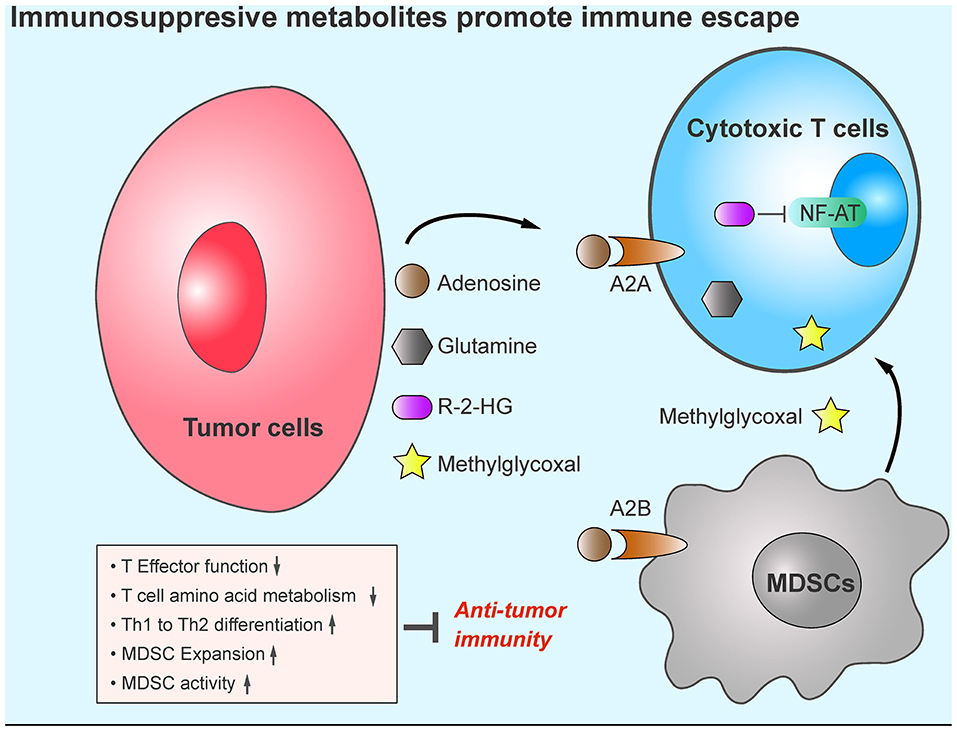
Figure 2. Tumor metabolites promote tumor immune evasion. Compared with other cell types, tumor cells display increased production of immunosuppressive metabolites, such as, adenosine, R-2-HG, glutamine, and methylglyoxal in the TME. These metabolites cause cytotoxic T cell anergy, increase the infiltration of MDSCs, and impair anti-tumor immunity.
Mutations in the IDH1 gene (encoding isocitrate dehydrogenase 1) have been found in more than 70% of grade 2 and 3 astrocytomas and oligodendrogliomas, and in glioblastomas developed from these lower-grade brain tumors (98). The IDH1 mutations occur arginine 132 in IDH1 and arginine 172 in IDH2 (99), and have been shown to induce the production of R-2-hydroxyglutarate (98). One study showed that tumor cells export R-2-hydroxyglutarate into the TME. The released R-2-hydroxyglutarate is taken up by activated T cells, where it significantly suppresses NFAT and NF-κB nuclear translocation and inhibits T-cell receptor signaling and polyamine biosynthesis, thus directly impairing T-cell activation (100) (Figure 2). Co-treatment with IDH1-mutant inhibitors (BAY-1436032 and AGI-5198) enhances the efficacy of immunotherapy against IDH1-mutant tumors in vivo (100–102).
Glutamine is the most abundant free amino acid in the blood, and its circulating concentration is around 0.5 mmol/L (102). Glutamine plays an essential role in supplying both carbon and nitrogen sources for anabolic growth and proliferation (103). A recent study suggested that glutamine metabolic programing in tumor cells renders anti-tumor immune response less effective. Blocking glutamine metabolism by the glutaminase inhibitor JHU083 inhibits tumor growth by disabling “Warburg” physiology (104). JHU083 thus increases glutamine and glucose content in the TME. Although glutamine metabolism is also essential for CD8+ T cell activity (105) glutamine blockade shapes CD8+ T cells toward a highly proliferative, activated, and long-lived phenotype by upregulating glucose anaplerosis in CD8+ T cells (104). The role of glutamine metabolism in myeloid-derived suppressor cells (MDSCs) has also been reported; JHU083 reduces the recruitment of MDSCs to TME, induces MDSC apoptosis, promotes the conversion of MDSC to pro-inflammatory M1 macrophage, and renders ICI-resistant tumors sensitive to immunotherapy (106). Collectively, blockade of glutamine metabolism reshapes the TME and enhances anti-tumor immunity (Figure 2).
Emerging evidence indicates that extracellular glutamine promotes ferroptosis. One study demonstrated that glutaminolysis is essential for ferroptosis, and glutaminolysis inhibitor compound-968 prevents ferroptosis in mouse embryonic fibroblasts (107). Another study showed that miR-137 suppresses the expression of glutamine transporter SLC1A5 to decrease tumor cell glutamine uptake and inhibits ferroptosis, suggesting a critical role of glutaminolysis in promoting ferroptosis (108). However, ferroptosis is an important prerequisite of immune checkpoint therapy-mediated tumor eradiation (71). Thus, an inhibitor of glutaminolysis may suppress immune checkpoint therapy-induced ferroptosis. Detailed mechanisms of glutaminolysis and ferroptosis should be investigated to prevent this antagonism effect.
Methylglyoxal (MG), a by-product of glycolysis, is present ubiquitously in living cells (109–111). MG induces the production of advanced glycation end products and promotes tumor proliferation in vivo (112, 113). Because tumors exhibit high rates of aerobic glycolysis, enhanced aerobic glycolysis in tumors leads to the accumulation of MG in the TME, raising concerns about the role of MG in promoting immune evasion. One study indicated that MDSCs display high levels of MG production in the TME, which promotes the transfer of MG into cytotoxic T cells in a cell-cell contact-dependent manner and leads to the accumulation of MG in cytotoxic T cells. For the reason that MG can easily bind with L-arginine and L-glutamine, MG depletes these amino acids in those T cells and paralyzes anti-tumor immunity. N-N-dimethylbiguanide (DMBG), which neutralizes the glycation activity of MG, can reverse the suppressive effects of MDSCs on cytotoxic T cells and sensitize immunotherapy-resistant tumor to ICI treatment (114). Moreover, as an immunosuppressive metabolite, MG can be easily detected in peripheral blood. Therefore, it could serve as a biomarker of immunotherapy resistance (Figure 2). Although MG scavenger and ICIs have demonstrated encouraging synergistic therapeutic effects, the detailed mechanisms of how MG causes anti-tumor cell fatigue while facilitating the survival of pro-tumor cells need to be further investigated.
Although immune checkpoint blockade has demonstrated durable anti-tumor activity in a variety of cancers, the overall response rate is far from being satisfactory, necessitating a comprehensive understanding of the mechanisms of decreased anti-tumor immunity under a hostile TME and the identification of predictive biomarkers for patient selection. Tumor cells enhance nutrient uptake, deplete oxygen, increase acidity in the TME, and upregulate pro-tumor metabolite production to create an immunosuppressive TME, which promotes tumor progression and immune evasion. Therefore, targeting cancer cell metabolic pathways could restrict tumor growth and invasion as well as restore an anti-tumor TME. Also limiting to patient welfare has been the inadequacy of predictive biomarkers of response to immunotherapy. The metabolites secreted into the TME by cancer cells could be promising biomarkers owing to their accessible measurability. Thus, furthering our understanding of cancer metabolism will not only broaden the current knowledge of interplay in the tumor microenvironment but also overcome immunotherapy resistance by expanding therapeutic options so that more patients can benefit from immunotherapy.
ZJ collected the data and wrote the review. JH contributed to writing and discussion. YL and GH contributed to scientific discussion. M-CH edited this manuscript and supervised this work. All authors contributed to the article and approved the submitted version.
The 2019 AACR-Pfizer Immuno-oncology Research Fellowship (Grant No. 19-40-49-JIAN); Breast Cancer Research Foundation (Grant No. BCRF-19-070); National Institutes of Health (CCSG CA016672); the Cancer Prevention and Research Institute of Texas (MIRA Grant No. RP160710); Patel Memorial Breast Cancer Endowment Fund; and The University of Texas MD Anderson–China Medical University Sister Institution Fund.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We apologize to the many authors and colleagues whose works are not cited due to limited space. We thank the members of M-CH's laboratory for helpful discussion. We acknowledge Sarah Bronson of Scientific Publications, Research Medical Library, The University of Texas MD Anderson Cancer Center, for editing the manuscript.
1. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. (1996) 271:1734–6. doi: 10.1126/science.271.5256.1734
2. Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. (2005) 65:1089–96.
3. Yan Y, Kumar AB, Finnes H, Markovic SN, Park S, Dronca RS, et al. Combining immune checkpoint inhibitors with conventional cancer therapy. Front Immunol. (2018) 9:1739. doi: 10.3389/fimmu.2018.01739
4. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. (2017) 168:707–23. doi: 10.1016/j.cell.2017.01.017
5. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. (2018) 359:1350–5. doi: 10.1126/science.aar4060
6. Martinez-Outschoorn UE, Peiris-Pages M, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. (2017) 14:11–31. doi: 10.1038/nrclinonc.2016.60
7. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. (2011) 11:85–95. doi: 10.1038/nrc2981
8. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
9. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell. (2015) 162:1217–28. doi: 10.1016/j.cell.2015.08.012
10. Munn DH. Blocking IDO activity to enhance anti-tumor immunity. Front Biosci. (2012) 4:734–45. doi: 10.2741/e414
11. Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. (2003) 9:1269–74. doi: 10.1038/nm934
12. Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med. (2011) 17:1094–100. doi: 10.1038/nm.2438
13. Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. (2011) 478:197–203. doi: 10.1038/nature10491
14. Prendergast GC, Smith C, Thomas S, Mandik-Nayak L, Laury-Kleintop L, Metz R, et al. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol Immunother. (2014) 63:721–35. doi: 10.1007/s00262-014-1549-4
15. Stockinger B, Hirota K, Duarte J, Veldhoen M. External influences on the immune system via activation of the aryl hydrocarbon receptor. Semin Immunol. (2011) 23:99–105. doi: 10.1016/j.smim.2011.01.008
16. Nguyen NT, Kimura A, Nakahama T, Chinen I, Masuda K, Nohara K, et al. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci USA. (2010) 107:19961–6. doi: 10.1073/pnas.1014465107
17. Wang WL, Chang WL, Yang HB, Chang IW, Lee CT, Chang CY, et al. Quantification of tumor infiltrating Foxp3+ regulatory T cells enables the identification of high-risk patients for developing synchronous cancers over upper aerodigestive tract. Oral Oncol. (2015) 51:698–703. doi: 10.1016/j.oraloncology.2015.04.015
18. Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res. (2012) 72:5435–40. doi: 10.1158/0008-5472.CAN-12-0569
19. Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med. (1999) 189:1363–72. doi: 10.1084/jem.189.9.1363
20. Wek RC, Jiang HY, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans. (2006) 34:7–11. doi: 10.1042/BST0340007
21. Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. (2005) 22:633–42. doi: 10.1016/j.immuni.2005.03.013
22. Sharma MD, Baban B, Chandler P, Hou DY, Singh N, Yagita H, et al. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest. (2007) 117:2570–82. doi: 10.1172/JCI31911
23. Keller TL, Zocco D, Sundrud MS, Hendrick M, Edenius M, Yum J, et al. Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat Chem Biol. (2012) 8:311–7. doi: 10.1038/nchembio.790
24. Grohmann U, Fallarino F, Bianchi R, Belladonna ML, Vacca C, Orabona C, et al. IL-6 inhibits the tolerogenic function of CD8 alpha+ dendritic cells expressing indoleamine 2,3-dioxygenase. J Immunol. (2001) 167:708–14. doi: 10.4049/jimmunol.167.2.708
25. Grohmann U, Fallarino F, Silla S, Bianchi R, Belladonna ML, Vacca C, et al. CD40 ligation ablates the tolerogenic potential of lymphoid dendritic cells. J Immunol. (2001) 166:277–83. doi: 10.4049/jimmunol.166.1.277
26. Liu X, Shin N, Koblish HK, Yang G, Wang Q, Wang K, et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood. (2010) 115:3520–30. doi: 10.1182/blood-2009-09-246124
27. Rodriguez PC, Quiceno DG, Ochoa AC. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood. (2007) 109:1568–73. doi: 10.1182/blood-2006-06-031856
28. Wang R, Jiao H, Zhao J, Wang X, Lin H. L-Arginine enhances protein synthesis by phosphorylating mTOR (Thr 2446) in a nitric oxide-dependent manner in C2C12 cells. Oxid Med Cell Longev. (2018) 2018:7569127. doi: 10.1155/2018/7569127
29. Kim SH, Roszik J, Grimm EA, Ekmekcioglu S. Impact of l-arginine metabolism on immune response and anticancer immunotherapy. Front Oncol. (2018) 8:67. doi: 10.3389/fonc.2018.00067
30. Popolo A, Adesso S, Pinto A, Autore G, Marzocco S. L-Arginine and its metabolites in kidney and cardiovascular disease. Amino Acids. (2014) 46:2271–86. doi: 10.1007/s00726-014-1825-9
31. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. (2016) 165:35–44. doi: 10.1016/j.cell.2016.02.065
32. Feun L, You M, Wu CJ, Kuo MT, Wangpaichitr M, Spector S, et al. Arginine deprivation as a targeted therapy for cancer. Curr Pharm Des. (2008) 14:1049–57. doi: 10.2174/138161208784246199
33. Prima V, Kaliberova LN, Kaliberov S, Curiel DT, Kusmartsev S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc Natl Acad Sci USA. (2017) 114:1117–22. doi: 10.1073/pnas.1612920114
34. Hernandez CP, Morrow K, Lopez-Barcons LA, Zabaleta J, Sierra R, Velasco C, et al. Pegylated arginase I: a potential therapeutic approach in T-ALL. Blood. (2010) 115:5214–21. doi: 10.1182/blood-2009-12-258822
35. Morrow K, Hernandez CP, Raber P, Del Valle L, Wilk AM, Majumdar S, et al. Anti-leukemic mechanisms of pegylated arginase I in acute lymphoblastic T-cell leukemia. Leukemia. (2013) 27:569–77. doi: 10.1038/leu.2012.247
36. Fletcher M, Ramirez ME, Sierra RA, Raber P, Thevenot P, Al-Khami AA, et al. l-Arginine depletion blunts antitumor T-cell responses by inducing myeloid-derived suppressor cells. Cancer Res. (2015) 75:275–83. doi: 10.1158/0008-5472.CAN-14-1491
37. Kamphorst JJ, Cross JR, Fan J, de Stanchina E, Mathew R, White EP, et al. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci USA. (2013) 110:8882–7. doi: 10.1073/pnas.1307237110
38. Guillaumond F, Bidaut G, Ouaissi M, Servais S, Gouirand V, Olivares O, et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc Natl Acad Sci USA. (2015) 112:2473–8. doi: 10.1073/pnas.1421601112
39. O'Sullivan D, van der Windt GJ, Huang SC, Curtis JD, Chang CH, Buck MD, et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity. (2014) 41:75–88. doi: 10.1016/j.immuni.2014.06.005
40. Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. (2012) 148:399–408. doi: 10.1016/j.cell.2012.01.021
41. Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell. (2007) 129:465–72. doi: 10.1016/j.cell.2007.04.019
42. Sarkar S, Germeraad WT, Rouschop KM, Steeghs EM, van Gelder M, Bos GM, et al. Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PLoS ONE. (2013) 8:e64835. doi: 10.1371/journal.pone.0064835
43. Sun J, Zhang Y, Yang M, Zhang Y, Xie Q, Li Z, et al. Hypoxia induces T-cell apoptosis by inhibiting chemokine C receptor 7 expression: the role of adenosine receptor A(2). Cell Mol Immunol. (2010) 7:77–82. doi: 10.1038/cmi.2009.105
44. Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci USA. (2012) 109:E2784–93. doi: 10.1073/pnas.1202366109
45. Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. (2014) 74:665–74. doi: 10.1158/0008-5472.CAN-13-0992
46. Palsson-McDermott EM, Dyck L, Zaslona Z, Menon D, McGettrick AF, Mills KHG, et al. Pyruvate kinase M2 is required for the expression of the immune checkpoint PD-L1 in immune cells and tumors. Front Immunol. (2017) 8:1300. doi: 10.3389/fimmu.2017.01300
47. Jayaprakash P, Ai M, Liu A, Budhani P, Bartkowiak T, Sheng J, et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J Clin Invest. (2018) 128:5137–49. doi: 10.1172/JCI96268
48. Brune B, Zhou J. Nitric oxide and superoxide: interference with hypoxic signaling. Cardiovasc Res. (2007) 75:275–82. doi: 10.1016/j.cardiores.2007.03.005
49. Matlung HL, Szilagyi K, Barclay NA, van den Berg TK. The CD47-SIRPalpha signaling axis as an innate immune checkpoint in cancer. Immunol Rev. (2017) 276:145–64. doi: 10.1111/imr.12527
50. Yoshida K, Tsujimoto H, Matsumura K, Kinoshita M, Takahata R, Matsumoto Y, et al. CD47 is an adverse prognostic factor and a therapeutic target in gastric cancer. Cancer Med. (2015) 4:1322–33. doi: 10.1002/cam4.478
51. Li Y, Lu S, Xu Y, Qiu C, Jin C, Wang Y, et al. Overexpression of CD47 predicts poor prognosis and promotes cancer cell invasion in high-grade serous ovarian carcinoma. Am J Transl Res. (2017) 9:2901–10.
52. Zhang H, Lu H, Xiang L, Bullen JW, Zhang C, Samanta D, et al. HIF-1 regulates CD47 expression in breast cancer cells to promote evasion of phagocytosis and maintenance of cancer stem cells. Proc Natl Acad Sci USA. (2015) 112:E6215–23. doi: 10.1073/pnas.1520032112
53. Doubrovina ES, Doubrovin MM, Vider E, Sisson RB, O'Reilly RJ, Dupont B, et al. Evasion from NK cell immunity by MHC class I chain-related molecules expressing colon adenocarcinoma. J Immunol. (2003) 171:6891–9. doi: 10.4049/jimmunol.171.12.6891
54. Gonzalez S, Groh V, Spies T. Immunobiology of human NKG2D and its ligands. Curr Top Microbiol Immunol. (2006) 298:121–38. doi: 10.1007/3-540-27743-9_6
55. Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. (2002) 419:734–8. doi: 10.1038/nature01112
56. Barsoum IB, Hamilton TK, Li X, Cotechini T, Miles EA, Siemens DR, et al. Hypoxia induces escape from innate immunity in cancer cells via increased expression of ADAM10: role of nitric oxide. Cancer Res. (2011) 71:7433–41. doi: 10.1158/0008-5472.CAN-11-2104
57. Al-Anazi A, Parhar R, Saleh S, Al-Hijailan R, Inglis A, Al-Jufan M, et al. Intracellular calcium and NF-kB regulate hypoxia-induced leptin, VEGF, IL-6 and adiponectin secretion in human adipocytes. Life Sci. (2018) 212:275–84. doi: 10.1016/j.lfs.2018.10.014
58. Gabrilovich D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol. (2004) 4:941–52. doi: 10.1038/nri1498
59. Khaled YS, Ammori BJ, Elkord E. Myeloid-derived suppressor cells in cancer: recent progress and prospects. Immunol Cell Biol. (2013) 91:493–502. doi: 10.1038/icb.2013.29
60. Wheeler KC, Jena MK, Pradhan BS, Nayak N, Das S, Hsu CD, et al. VEGF may contribute to macrophage recruitment and M2 polarization in the decidua. PLoS ONE. (2018) 13:e0191040. doi: 10.1371/journal.pone.0191040
61. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. (2011) 475:226–30. doi: 10.1038/nature10169
62. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. (1927) 8:519–30. doi: 10.1085/jgp.8.6.519
63. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science. (2009) 324:1029–33. doi: 10.1126/science.1160809
64. Zhang X, Lin Y, Gillies RJ. Tumor pH and its measurement. J Nucl Med. (2010) 51:1167–70. doi: 10.2967/jnumed.109.068981
65. Wike-Hooley JL, Haveman J, Reinhold HS. The relevance of tumour pH to the treatment of malignant disease. Radiother Oncol. (1984) 2:343–66. doi: 10.1016/S0167-8140(84)80077-8
66. Gillies RJ, Raghunand N, Karczmar GS, Bhujwalla ZM. MRI of the tumor microenvironment. J Magn Reson Imaging. (2002) 16:430–50. doi: 10.1002/jmri.10181
67. Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A, et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. (2013) 73:1524–35. doi: 10.1158/0008-5472.CAN-12-2796
68. Lardner A. The effects of extracellular pH on immune function. J Leukoc Biol. (2001) 69:522–30. doi: 10.1189/jlb.69.4.522
69. Bosticardo M, Ariotti S, Losana G, Bernabei P, Forni G, Novelli F. Biased activation of human T lymphocytes due to low extracellular pH is antagonized by B7/CD28 costimulation. Eur J Immunol. (2001) 31:2829–38. doi: 10.1002/1521-4141(200109)31:9<2829::AID-IMMU2829>3.0.CO;2-U
70. Nathan I, Groopman JE, Quan SG, Bersch N, Golde DW. Immune (gamma) interferon produced by a human T-lymphoblast cell line. Nature. (1981) 292:842–4. doi: 10.1038/292842a0
71. Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. (2019) 569:270–4. doi: 10.1038/s41586-019-1170-y
72. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. (2002) 23:549–55. doi: 10.1016/S1471-4906(02)02302-5
73. de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. (2006) 6:24–37. doi: 10.1038/nrc1782
74. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. (2016) 24:657–71. doi: 10.1016/j.cmet.2016.08.011
75. Weide B, Martens A, Hassel JC, Berking C, Postow MA, Bisschop K, et al. Baseline biomarkers for outcome of melanoma patients treated with pembrolizumab. Clin Cancer Res. (2016) 22:5487–96. doi: 10.1158/1078-0432.CCR-16-0127
76. Pilon-Thomas S, Kodumudi KN, El-Kenawi AE, Russell S, Weber AM, Luddy K, et al. Neutralization of tumor acidity improves antitumor responses to immunotherapy. Cancer Res. (2016) 76:1381–90. doi: 10.1158/0008-5472.CAN-15-1743
77. Calcinotto A, Filipazzi P, Grioni M, Iero M, De Milito A, Ricupito A, et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. (2012) 72:2746–56. doi: 10.1158/0008-5472.CAN-11-1272
78. Blay J, White TD, Hoskin DW. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res. (1997) 57:2602–5.
79. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. (2013) 31:51–72. doi: 10.1146/annurev-immunol-032712-100008
80. Yegutkin GG. Nucleotide- and nucleoside-converting ectoenzymes: important modulators of purinergic signalling cascade. Biochim Biophys Acta. (2008) 1783:673–94. doi: 10.1016/j.bbamcr.2008.01.024
81. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev. (2017) 276:121–44. doi: 10.1111/imr.12528
82. Aliagas E, Vidal A, Texido L, Ponce J, Condom E, Martin-Satue M. High expression of ecto-nucleotidases CD39 and CD73 in human endometrial tumors. Mediators Inflamm. (2014) 2014:509027. doi: 10.1155/2014/509027
83. Mittal D, Sinha D, Barkauskas D, Young A, Kalimutho M, Stannard K, et al. Adenosine 2B receptor expression on cancer cells promotes metastasis. Cancer Res. (2016) 76:4372–82. doi: 10.1158/0008-5472.CAN-16-0544
84. d'Almeida SM, Kauffenstein G, Roy C, Basset L, Papargyris L, Henrion D, et al. The ecto-ATPDase CD39 is involved in the acquisition of the immunoregulatory phenotype by M-CSF-macrophages and ovarian cancer tumor-associated macrophages: regulatory role of IL-27. Oncoimmunology. (2016) 5:e1178025. doi: 10.1080/2162402X.2016.1178025
85. Gourdin N, Bossennec M, Rodriguez C, Vigano S, Machon C, Jandus C, et al. Autocrine adenosine regulates tumor polyfunctional CD73(+)CD4(+) effector T cells devoid of immune checkpoints. Cancer Res. (2018) 78:3604–18. doi: 10.1158/0008-5472.CAN-17-2405
86. Li L, Wang L, Li J, Fan Z, Yang L, Zhang Z, et al. Metformin-induced reduction of CD39 and CD73 blocks myeloid-derived suppressor cell activity in patients with ovarian cancer. Cancer Res. (2018) 78:1779–91. doi: 10.1158/0008-5472.CAN-17-2460
87. Jacob F, Perez Novo C, Bachert C, Van Crombruggen K. Purinergic signaling in inflammatory cells: P2 receptor expression, functional effects, and modulation of inflammatory responses. Purinergic Signal. (2013) 9:285–306. doi: 10.1007/s11302-013-9357-4
88. Lokshin A, Raskovalova T, Huang X, Zacharia LC, Jackson EK, Gorelik E. Adenosine-mediated inhibition of the cytotoxic activity and cytokine production by activated natural killer cells. Cancer Res. (2006) 66:7758–65. doi: 10.1158/0008-5472.CAN-06-0478
89. Beavis PA, Divisekera U, Paget C, Chow MT, John LB, Devaud C, et al. Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proc Natl Acad Sci USA. (2013) 110:14711–6. doi: 10.1073/pnas.1308209110
90. Ramanathan M, Pinhal-Enfield G, Hao I, Leibovich SJ. Synergistic up-regulation of vascular endothelial growth factor (VEGF) expression in macrophages by adenosine A2A receptor agonists and endotoxin involves transcriptional regulation via the hypoxia response element in the VEGF promoter. Mol Biol Cell. (2007) 18:14–23. doi: 10.1091/mbc.e06-07-0596
91. Ryzhov S, Novitskiy SV, Goldstein AE, Biktasova A, Blackburn MR, Biaggioni I, et al. Adenosinergic regulation of the expansion and immunosuppressive activity of CD11b+Gr1+ cells. J Immunol. (2011) 187:6120–9. doi: 10.4049/jimmunol.1101225
92. Sorrentino C, Miele L, Porta A, Pinto A, Morello S. Myeloid-derived suppressor cells contribute to A2B adenosine receptor-induced VEGF production and angiogenesis in a mouse melanoma model. Oncotarget. (2015) 6:27478–89. doi: 10.18632/oncotarget.4393
93. Gessi S, Bencivenni S, Battistello E, Vincenzi F, Colotta V, Catarzi D, et al. Inhibition of A2A adenosine receptor signaling in cancer cells proliferation by the novel antagonist TP455. Front Pharmacol. (2017) 8:888. doi: 10.3389/fphar.2017.00888
94. Liu H, Kuang X, Zhang Y, Ye Y, Li J, Liang L, et al. ADORA1 inhibition promotes tumor immune evasion by regulating the ATF3-PD-L1 axis. Cancer Cell. (2020) 37:324–39 e328. doi: 10.1016/j.ccell.2020.02.006
95. Fong L, Hotson A, Powderly JD, Sznol M, Heist RS, Choueiri TK, et al. Adenosine 2A receptor blockade as an immunotherapy for treatment-refractory renal cell cancer. Cancer Discov. (2020) 10:40–53. doi: 10.1158/2159-8290.CD-19-0980
96. Bastid J, Regairaz A, Bonnefoy N, Dejou C, Giustiniani J, Laheurte C, et al. Inhibition of CD39 enzymatic function at the surface of tumor cells alleviates their immunosuppressive activity. Cancer Immunol Res. (2015) 3:254–65. doi: 10.1158/2326-6066.CIR-14-0018
97. Hay CM, Sult E, Huang Q, Mulgrew K, Fuhrmann SR, McGlinchey KA, et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology. (2016) 5:e1208875. doi: 10.1080/2162402X.2016.1208875
98. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. (2009) 462:739–44. doi: 10.1038/nature08617
99. Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. (2010) 17:225–34. doi: 10.1016/j.ccr.2010.01.020
100. Bunse L, Pusch S, Bunse T, Sahm F, Sanghvi K, Friedrich M, et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat Med. (2018) 24:1192–203. doi: 10.1038/s41591-018-0095-6
101. Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. (2013) 340:626–30. doi: 10.1126/science.1236062
102. Pusch S, Krausert S, Fischer V, Balss J, Ott M, Schrimpf D, et al. Pan-mutant IDH1 inhibitor BAY 1436032 for effective treatment of IDH1 mutant astrocytoma in vivo. Acta Neuropathol. (2017) 133:629–44. doi: 10.1007/s00401-017-1677-y
103. Zhang J, Pavlova NN, Thompson CB. Cancer cell metabolism: the essential role of the nonessential amino acid, glutamine. EMBO J. (2017) 36:1302–15. doi: 10.15252/embj.201696151
104. Leone RD, Zhao L, Englert JM, Sun IM, Oh MH, Sun IH, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science. (2019) 366:1013–21. doi: 10.1126/science.aav2588
105. Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol. (2013) 14:500–8. doi: 10.1038/ni.2556
106. Oh MH, Sun IH, Zhao L, Leone RD, Sun IM, Xu W, et al. Targeting glutamine metabolism enhances tumor specific immunity by modulating suppressive myeloid cells. J Clin Invest. (2020) 130:3865–84. doi: 10.1172/JCI131859
107. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. (2015) 59:298–308. doi: 10.1016/j.molcel.2015.06.011
108. Luo M, Wu L, Zhang K, Wang H, Zhang T, Gutierrez L, et al. miR-137 regulates ferroptosis by targeting glutamine transporter SLC1A5 in melanoma. Cell Death Differ. (2018) 25:1457–72. doi: 10.1038/s41418-017-0053-8
109. Kang Y, Edwards LG, Thornalley PJ. Effect of methylglyoxal on human leukaemia 60 cell growth: modification of DNA G1 growth arrest and induction of apoptosis. Leuk Res. (1996) 20:397–405. doi: 10.1016/0145-2126(95)00162-X
110. Du J, Suzuki H, Nagase F, Akhand AA, Yokoyama T, Miyata T, et al. Methylglyoxal induces apoptosis in Jurkat leukemia T cells by activating c-Jun N-terminal kinase. J Cell Biochem. (2000) 77:333–44. doi: 10.1002/(SICI)1097-4644(20000501)77:2<333::AID-JCB15>3.0.CO;2-Q
111. Antognelli C, Talesa VN. Glyoxalases in urological malignancies. Int J Mol Sci. (2018) 19:415. doi: 10.3390/ijms19020415
112. Nokin MJ, Durieux F, Bellier J, Peulen O, Uchida K, Spiegel DA, et al. Hormetic potential of methylglyoxal, a side-product of glycolysis, in switching tumours from growth to death. Sci Rep. (2017) 7:11722. doi: 10.1038/s41598-017-12119-7
113. Antognelli C, Moretti S, Frosini R, Puxeddu E, Sidoni A, Talesa VN. Methylglyoxal acts as a tumor-promoting factor in anaplastic thyroid cancer. Cells. (2019) 8:547. doi: 10.3390/cells8060547
Keywords: immune checkpoint inhibitors, cancer metabolism, tumor microenvironment, immune evasion, cancer cell metabolite
Citation: Jiang Z, Hsu JL, Li Y, Hortobagyi GN and Hung M-C (2020) Cancer Cell Metabolism Bolsters Immunotherapy Resistance by Promoting an Immunosuppressive Tumor Microenvironment. Front. Oncol. 10:1197. doi: 10.3389/fonc.2020.01197
Received: 17 April 2020; Accepted: 12 June 2020;
Published: 22 July 2020.
Edited by:
Baoli Hu, University of Pittsburgh, United StatesReviewed by:
Daria Capece, Imperial College London, United KingdomCopyright © 2020 Jiang, Hsu, Li, Hortobagyi and Hung. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mien-Chie Hung, bWh1bmdAY211LmVkdS50dw==; bWh1bmc3NzAzMEBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.