
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 30 June 2020
Sec. Cancer Genetics
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.01065
This article is part of the Research Topic Multi-omic Data Integration in Oncology View all 14 articles
 Marco Chierici1*†
Marco Chierici1*† Nicole Bussola1,2†
Nicole Bussola1,2† Alessia Marcolini1†
Alessia Marcolini1† Margherita Francescatto1,3
Margherita Francescatto1,3 Alessandro Zandonà4
Alessandro Zandonà4 Lucia Trastulla5Claudio Agostinelli2
Lucia Trastulla5Claudio Agostinelli2 Giuseppe Jurman1*‡
Giuseppe Jurman1*‡ Cesare Furlanello1,6‡
Cesare Furlanello1,6‡Recent technological advances and international efforts, such as The Cancer Genome Atlas (TCGA), have made available several pan-cancer datasets encompassing multiple omics layers with detailed clinical information in large collection of samples. The need has thus arisen for the development of computational methods aimed at improving cancer subtyping and biomarker identification from multi-modal data. Here we apply the Integrative Network Fusion (INF) pipeline, which combines multiple omics layers exploiting Similarity Network Fusion (SNF) within a machine learning predictive framework. INF includes a feature ranking scheme (rSNF) on SNF-integrated features, used by a classifier over juxtaposed multi-omics features (juXT). In particular, we show instances of INF implementing Random Forest (RF) and linear Support Vector Machine (LSVM) as the classifier, and two baseline RF and LSVM models are also trained on juXT. A compact RF model, called rSNFi, trained on the intersection of top-ranked biomarkers from the two approaches juXT and rSNF is finally derived. All the classifiers are run in a 10x5-fold cross-validation schema to warrant reproducibility, following the guidelines for an unbiased Data Analysis Plan by the US FDA-led initiatives MAQC/SEQC. INF is demonstrated on four classification tasks on three multi-modal TCGA oncogenomics datasets. Gene expression, protein expression and copy number variants are used to predict estrogen receptor status (BRCA-ER, N = 381) and breast invasive carcinoma subtypes (BRCA-subtypes, N = 305), while gene expression, miRNA expression and methylation data is used as predictor layers for acute myeloid leukemia and renal clear cell carcinoma survival (AML-OS, N = 157; KIRC-OS, N = 181). In test, INF achieved similar Matthews Correlation Coefficient (MCC) values and 97% to 83% smaller feature sizes (FS), compared with juXT for BRCA-ER (MCC: 0.83 vs. 0.80; FS: 56 vs. 1801) and BRCA-subtypes (0.84 vs. 0.80; 302 vs. 1801), improving KIRC-OS performance (0.38 vs. 0.31; 111 vs. 2319). INF predictions are generally more accurate in test than one-dimensional omics models, with smaller signatures too, where transcriptomics consistently play the leading role. Overall, the INF framework effectively integrates multiple data levels in oncogenomics classification tasks, improving over the performance of single layers alone and naive juxtaposition, and provides compact signature sizes1.
The challenge of integrating multi-omics data is as old as bioinformatics itself (1, 2), but, despite the wide literature, it remains an open issue nowadays, even worth being funded by major institutions2.
This study introduces Integrative Network Fusion (INF), a reproducible network-based framework for high-throughput omics data integration that leverages machine learning models to extract multi-omics predictive biomarkers. Originally conceptualized and tested on multi-omics metagenomics data in an early preliminary version (3, 4), INF combines the signatures retrieved from both the early-integration approach of variable juxtaposition (juXT) and an intermediate-integration approach [SNF, (5)], to find the optimal set of predictive features. In particular, first a set of top-ranked features is extracted by juXT by a classifier, here Random Forest (RF) and linear Support Vector Machine (LSVM). Then, a feature ranking scheme (rSNF) is computed on SNF-integrated features and finally a RF model (rSNFi) is trained on the intersection of two sets of top-ranked features from juXT and rSNF, obtaining an approach that effectively integrates multiple omics layers and provides compact predictive signatures. Selection bias and data-leakage effects are controlled by performing the experiments within a rigorous Data Analysis Plan (DAP) to warrant reproducibility, following the guidelines of the US FDA-led initiatives MAQC/SEQC (6–8). In particular, to alleviate the computational burden of the full DAP pipeline, an approximated DAP is designed to lighten computing without significantly affecting the results. Further, experiments are run on samples with randomly shuffled labels as a sanity check vs. overfitting effects and, finally, INF robustness is verified by testing on different train/test splits.
We test INF on three datasets retrieved from the TCGA repository, to predict either the estrogen receptor status (ER) or the cancer subtype on the breast invasive carcinoma (BRCA) dataset, and to predict the overall survival (OS) on the kidney renal clear cell carcinoma (KIRC) and acute myeloid leukemia (AML) datasets. Overall, INF improves over the performance of single layers and naive juxtaposition on all four oncogenomics tasks, extracting a biologically meaningful compact set of predictive biomarkers. Notably, the transcriptomics layer is prevalent inside the inferred INF signatures, consistently with published findings (9).
The INF framework is currently designed to integrate an arbitrary number of one-dimensional omics layers. We plan to further extend the framework by enabling the integration of histopathological features extracted from whole slide images (10) or deep features from radiological images (11) extracted by deep neural network architectures, carefully addressing all potential caveats (12).
Three multi-modal cancer datasets generated by The Cancer Genome Atlas (TCGA) Research Network (https://www.cancer.gov/tcga) and four classification tasks are considered in this study. Protein expression (prot), gene expression (gene), and copy number variants (cnv) are used to predict breast invasive carcinoma (BRCA) estrogen receptor status (0: negative; 1: positive) and subtypes (luminal A, luminal B, basal-like, HER2-enriched). Methylation (meth), gene expression (gene), and microRNA expression (mirna) are used to predict acute myeloid leukemia (AML) and kidney renal clear cell carcinoma (KIRC) overall survival (0: alive; 1: deceased). The number of samples and features for each omic layer and classification task are detailed in Table 1; class balance, split by dataset, is reported in Table 2.
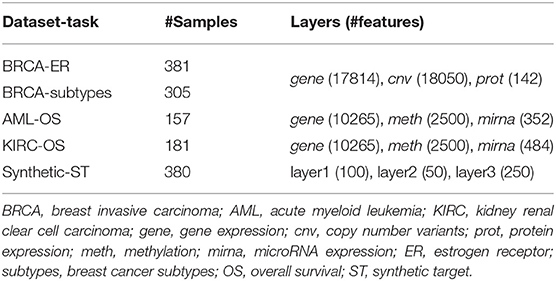
Table 1. Data summary.
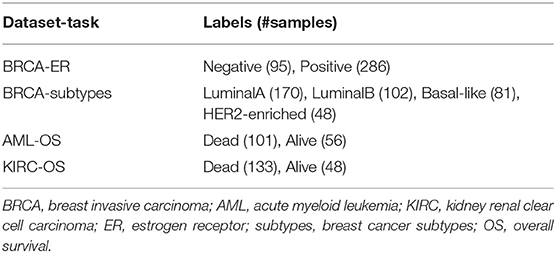
Table 2. Class balance.
For AML (13) and KIRC (14), gene expression is profiled using the Illumina HiSeq2000 and quantified as log2-transformed RSEM normalized counts; miRNA mature strand expression is profiled using the Illumina Genome Analyzer and quantified as reads per million miRNA mapped; and methylation is assessed by Illumina Human Methylation 450K and expressed as beta values. For BRCA (15), gene expression is profiled with Agilent 244K custom gene expression microarrays; protein expression is assessed by reverse phase protein arrays; copy number profiles are measured using Affymetrix Genome-Wide Human SNP Array 6.0 platform, copy number variants are segmented by the TCGA Firehose pipeline using GISTIC2 method, and then mapped to genes.
The original data is publicly accessible on the National Cancer Institute GDC Data Portal (https://portal.gdc.cancer.gov/) and the Broad GDAC Firehose (https://gdac.broadinstitute.org/), where further details on data generation can be found. The data was retrieved in December, 2019 and January, 2020 using the RTCGA R library (16).
Furthermore, the INF pipeline has been tested on a synthetic dataset with 380 observations in two classes (70% class 1 and 30% class 2, defining the synthetic target ST), 3 pseudo-omics layers, and 400 features (layer 1: 100; layer 2: 50; layer 3: 250). The dataset is generated in-house using scikit-learn's make_classification function with the arguments shuffle=False and flip_y=0. The number of informative features and the difficulty of the task were set on a per-layer basis, as summarized in Table 3.

Table 3. Synthetic data summary for each simulated layer.
The INF pipeline integrates two or more omics layers, e.g., gene expression, protein expression, or methylation, in a machine learning framework for improved patient classification and biomarker identification in cancer. The core consists of three main components, structured as in Figure 1, managing the integration of the omics layers and their predictive modeling. A baseline integration method (juXT) is first considered by training a Random Forest (RF) (17) or a linear Support Vector Machine (LSVM) (18) classifier on juxtaposed multi-omics data, ranking features by ANOVA F-value. Secondly, the multi-omics features are integrated by Similarity Network Fusion (SNF) (5), a method that computes a sample similarity network for each data type and fuses them into one network. INF introduces a novel feature ranking scheme (rSNF) that sorts multi-omics features according to their contribution to the SNF-fused network structure. A RF or LSVM classifier is trained on the juxtaposed multi-omics data, ranking features by rSNF. A compact RF model (rSNFi) is finally trained on the juxtaposed dataset restricted on the intersection of top-ranked biomarkers from juXT and rSNF.
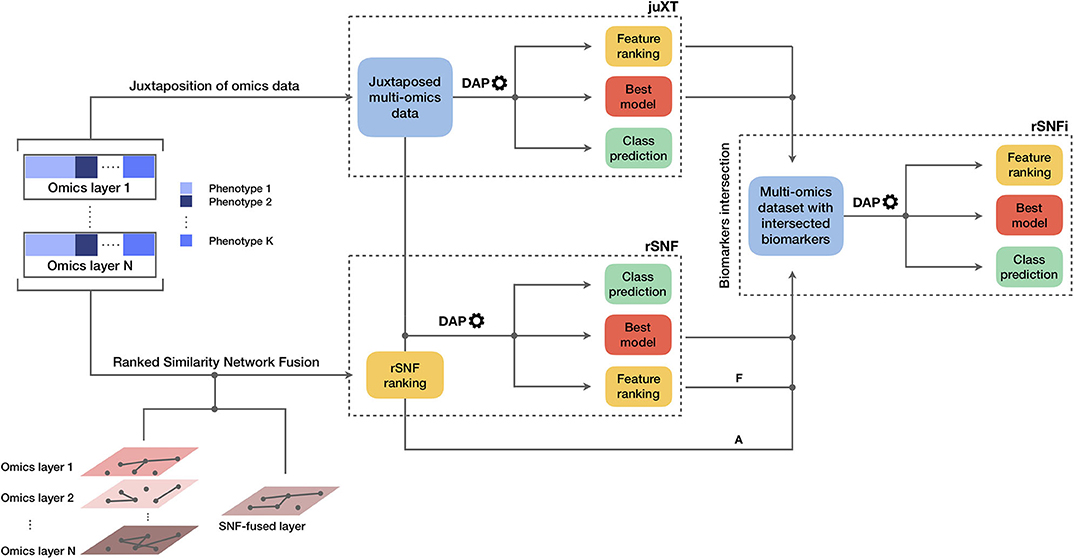
Figure 1. Graphical representation of the INF workflow for N omics datasets with K phenotypes. A first RF or LSVM classifier is trained on the juxtaposed data, ranking features by ANOVA F-value (juXT). The data sets are then integrated by Similarity Network Fusion, the features are ranked by rSNF and a RF or LSVM model is developed on the juxtaposed dataset with the rSNF feature ranking (rSNF). Finally, a RF or LSVM classifier is trained on the juxtaposed dataset restricted to the intersection of juXT and rSNF top discriminant feature lists (rSNFi). The classifier is either RF or LSVM throughout the INF workflow. All the predictive models are developed within the DAP described in the methods and graphically represented in Figure 2. The alternative and mutually exclusive paths A and F are followed by the “accelerated DAP” and the “full DAP” procedures, respectively (see section 2).
In a comparative review of scientific literature, SNF (5) emerged as one of the most reliable alternatives to simple juxtaposition-based integration. SNF is a non-Bayesian network-based method that can be divided into two main steps: the first step builds a sample-similarity network for each omics dataset, where nodes represent samples and edges encode a scaled exponential Euclidean distance kernel computed on each pair of samples; the second step implements a non-linear combination of these networks into a single similarity network through an iterative procedure. The multi-omics datasets are first converted into graphs, and for each graph two matrices are computed: a patient pairwise similarity matrix (“status matrix”), and a matrix with similarity of each patient to the K most similar patients, through K-nearest neighbors (“local affinity matrix”). At each iteration, the status matrix is updated through the local affinity matrix, generating two parallel interchanging processes. The status matrices are finally fused together into a single network. Spectral clustering is performed on the fused network, in order to identify sub-communities of samples, potentially reflecting phenotypes. The clustering performance is evaluated with respect to a ground truth, i.e., the real phenotype each sample belongs to, by the Normalized Mutual Information (NMI) score. SNF integrates multiple omics datasets into a single comprehensive network in the space of samples rather than measurements (e.g., gene expression values).
This work proposes multi-omics integration as an approach to identify robust biomarkers of samples phenotypes or cancer subtypes (e.g., survival status vs. breast cancer subtyping); consequently, it is necessary to extract measurements information from the SNF-fused network of samples. To this aim, we extended SNF by implementing rSNF (ranked SNF), a feature-ranking scheme based on SNF-fused network clustering. In detail, a patient network Wi is built for each feature fi, based on fi alone, and spectral clustering is performed on it. Then, NMI score is computed comparing the samples clusters found inside Wi with those in the fused network; the higher the score, the more similar the clustering between the fused network and Wi. Thus, each feature fi is associated to a consistency score, ranking all multi-omics features with respect to their relative contribution to the whole network structure.
The entire procedure of similarity networks inference and fusion relies on two hyperparameters: α, the scaling variance in the scaled exponential similarity kernel used for similarity networks construction, and K, the number of nearest neighbors in sparse kernel and scaled exponential similarity kernel construction. While the original method (5) assigned fixed values to α and K, in this study the optimal hyperparameters are chosen among the grids αgrid = {0.3, 0.35, 0.4, 0.45, …, 0.8} and Kgrid = {i∈ℕ, 10 ≤ i ≤ 30} in a 10 × 5-fold cross-validation schema.
To ensure the reproducibility of results and limit overfitting, the development of classification models is performed inside a Data Analysis Plan (DAP) (Figure 2), following the guidelines derived by the U.S. Food and Drug Administration MAQC/SEQC studies (6, 19). Data is split in a training set (TR) and two non-overlapping test sets (TS, TS2), preserving the original proportion of patient phenotypes (classes). The TR/TS/TS2 partitions are 50/30/20 of the entire data set, respectively. The data splitting procedure is repeated 10 times so to obtain 10 different TR/TS/TS2 splits. Predictive models are trained and developed on TR and TS for juXT and rSNF; in the case of rSNFi, the models are trained and developed on TS and TS2 to avoid information leakage due to using the same data both for feature selection and model training (see Figure 3). For each split, Random Forest (RF) or linear kernel Support Vector Machine (LSVM) classifiers are trained on the training partition within a stratified 10 × 5-fold cross-validation (10 × 5-CV). The model performance is assessed in terms of average precision, recall and Matthews Correlation Coefficient (MCC) (20, 21). The MCC is generally regarded as a balanced measure of accuracy and precision that can be used both in binary and multiclass problems (22, 23) and even when classes are imbalanced (24). MCC lies in [−1, 1], with 1 meaning perfect prediction, -1 inverse prediction and 0 random guess. For binary classification tasks, MCC is calculated on true and predicted labels considering true positive (TP), true negative (TN), false positive (FP) and false negative (FN) values, as in the following:
At each CV round, features are ranked either by ANOVA F-value (for juXT, rSNFi) or by the rSNF ranking (see section 2.3) and different classification models are trained for increasing numbers of ranked features, namely 5, 10, 25, 50, 75, and 100% of the total features. A unified list of top-ranked features is then obtained by Borda aggregation of all the ranked CV lists (25, 26). The best model is later retrained on the whole training set restricted to the features yielding the maximum MCC in CV, and validated on the test partition. A global list of top-ranked features is derived for juXT, rSNF, and rSNFi by Borda aggregation of the Borda lists of each TR/TS split (Borda of Bordas, “BoB”). The signatures for juXT, rSNF, and rSNFi are defined by the top N features of the corresponding BoB lists, with N being the median size of top features across all experiments.
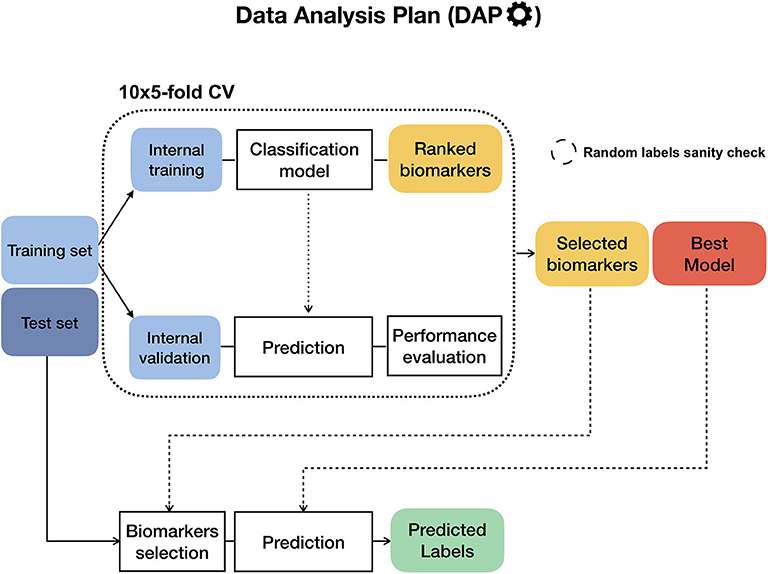
Figure 2. Diagram of the Data Analysis Plan (DAP), originally developed within the FDA-led MAQC/SEQC-II initiatives. If the training set labels are stochastically shuffled beforehand, the DAP runs in “random labels” mode as a sanity check to ensure that the procedure is not affected by systematic bias.
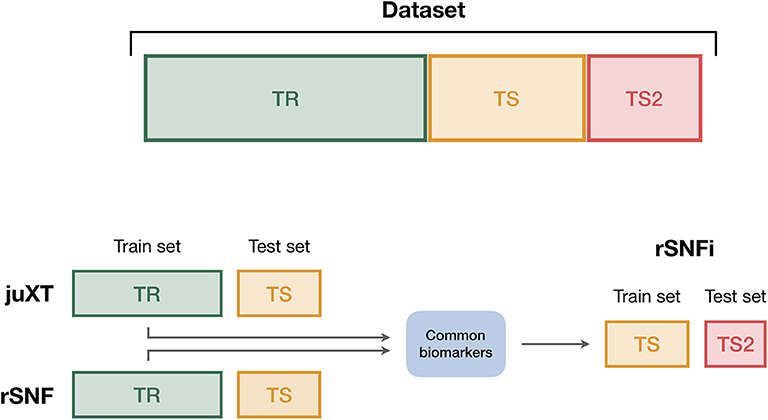
Figure 3. Data splitting procedure. To avoid information leakage due to the use of the same data both for feature selection and model training, we considered different train and test sets according to the integration scheme. In particular, each data set is split into three non-overlapping partitions (TR/TS/TS2), corresponding to the 50/30/20% of the entire data set, respectively. The TR/TS/TS2 partitions preserve the original proportion of patient phenotypes. Predictive models for juXT and rSNF are trained on TR and validated on TS, while for rSNFi the train set is TS (with features restricted to the intersected biomarkers of juXT and rSNF) and TS2 the test set.
In the “full” version of the DAP (fDAP), described above, the rSNF ranking is performed at each CV round on the training portion of the data. Since this procedure is quite demanding in terms of computational time, even if parallelized (≈ 9 feature/min), we devised an “accelerated” version of the DAP (aDAP), where the rSNF ranking is precomputed on the whole TR data and used as is at each CV round. We assessed the fDAP vs. aDAP performance on the synthetic dataset as well as BRCA-ER and BRCA-subtypes by comparing the overall metrics and measuring the dissimilarity of the rSNF BoB of the two DAPs by the Canberra distance (25).
RF models are trained using 500 trees, measuring the quality of a split as mean decrease in the Gini impurity index (17); the regularization parameter C of LSVM models is tuned over the grid within a 10 × stratified Monte Carlo cross-validation (50% training/validation proportion). Results for RF models are summarized in Table 4, while LSVM models performance is detailed in the Supplementary Tables BRCA-ER_LSVM, KIRC-OS_LSVM.
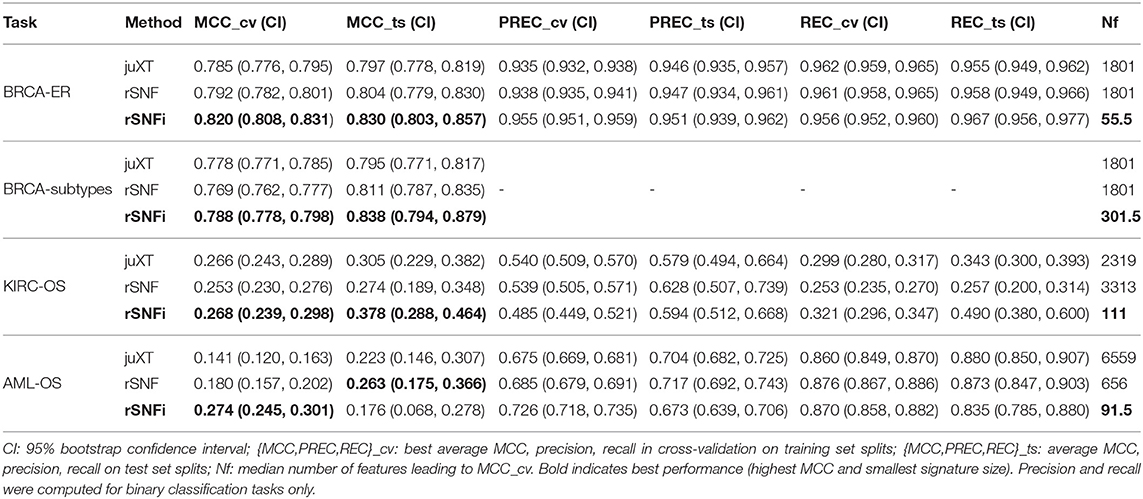
Table 4. Summarized best predictive performances for each classification task using RF model and three omics layers.
To ensure that the predictive profiling procedure is not affected by selection bias, the whole INF workflow, including the rSNF procedure, is also repeated after randomly scrambling the training set labels (“random labels” mode): in this setup, the performance of a classifier unaffected by systematic bias should be close to that of a random predictor, with MCC close to zero.
The complete INF pipeline is implemented through the workflow management tool Snakemake (27, 28), which allows automatic handling of all dependencies required to generate the INF output. The pipeline operates on N omics input files, one for each layer that should be integrated, and a single file describing the patient labels. The omics files are tab-separated text matrices with patients on the rows and features on the columns, with row and column identifiers. The label file is a single column file with patient phenotypes, with no header. This input structure, with one file per omic layer and a label file, simplifies the downstream analysis and reduces to a minimum the preprocessing burden for the end user.
The predictive profiling module, including the DAP, is written in Python 3.6 on top of NumPy (29) and scikit-learn methods (30). The ranked SNF (rSNF) procedure is implemented in R (31) leveraging the original R scripts provided by SNF authors (5), extended by a dedicated script for SNF tuning and a main script for SNF analysis and the post-SNF feature selection procedure, which is parallelized over the features for efficiency using the foreach R library.
The INF computations were run on the FBK Linux high-performance computing facility KORE, on a 8-core i7 3.4 GHz Linux workstation, and on a 72-vCPU 2.7 GHz Platinum Intel Xeon 8168 Microsoft Azure cloud machine (F72s v2 series).
To further foster reproducibility and support users and future developers, the full code of this benchmark is publicly shared on the GitLab repository https://gitlab.fbk.eu/MPBA/INF. Additional information is included in the Supplementary Material available on the publisher's website, while the full set of experimental data can be accessed at http://dx.doi.org/10.6084/m9.figshare.12052995.v1.
The INF workflow was run on all tasks considering 3-layer integration and all 2-layer combinations; the DAP was also run separately on all single-layer datasets in order to obtain a baseline. All results presented here refer to experiments performed with RF classifier. Experiments using LSVM were performed on BRCA-ER and KIRC-OS obtaining similar classification performances, top features and layer contributions (Supplementary Tables BRCA-ER_LSVM, KIRC-OS_LSVM). The classifier performance for 3-layer integration is summarized in Table 4, in terms of average cross-validation MCC on the 10 training set splits (MCC_cv) with 95% Studentized bootstrap confidence intervals (CI) as (MCC_min, MCC_max), average MCC on the 10 test set splits (MCC_ts) with CI, and median number of features (Nf) yielding MCC_cv. Similarly, precision (PREC) and recall (REC) are reported in Table 4 as average cross-validation and test set values with CI. As expected, whenever there is a non-negligible unbalance toward the positive class, the number of false positives tends to increase, with more false positives yielding a comparatively low precision with higher recall, and vice versa. In both cases, the MCC efficiently works in balancing the two effects. The classifier performance on single-layer and 2-layer data is summarized in Figure 4.
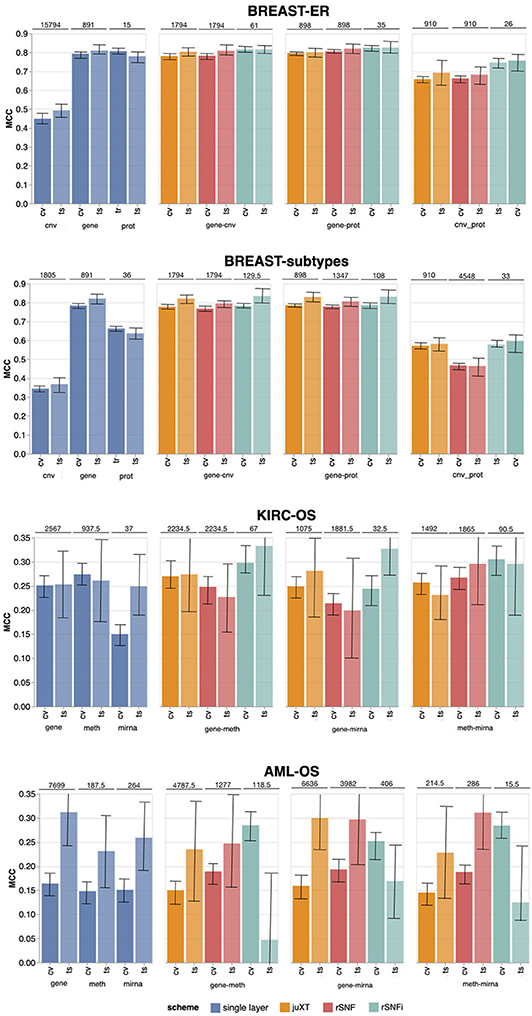
Figure 4. Overview of Random Forest classification performance (MCC, Matthews Correlation Coefficient) on the four tasks in cross validation (“CV”) and test (“ts”), on single-layer (blue shades) and on all two-layer combinations for juXT (orange), rSNF (red) and rSNFi (green). Bars indicate 95% confidence intervals. On top of each CV-ts pair is the median number of features leading to best CV performance.
A comparison between the “accelerated” flavor of the DAP (aDAP) and the full DAP (fDAP) was run on synthetic data, BRCA-ER and BRCA-subtypes data, with aDAP yielding similar performance metrics and top-ranked biomarker lists as fDAP (Supplementary Tables Synthetic_RF, BRCA_RF_fDAP, canberra_distances), while being ≈30 × faster (for BRCA-ER, approx. 2 vs. 64 h, or 300 features/min vs. 9 features/min). All the results presented here were thus obtained using aDAP. Moreover, the INF workflow running in “random labels” mode achieved an average cross-validation MCC ≈0, as expected by a procedure unaffected by systematic bias.
Overall, integrating multiple omics layers with INF yields better or comparable classification performance than using only features from a single layer or naïve omics juxtaposition, at the same time with much more compact signature sizes. On 3-layer BRCA-subtypes and 2- or 3-layer KIRC-OS, INF outperforms the single layers, as well as juXT and rSNF (Figure 4, Table 4). On 2-layer BRCA-subtypes, INF performance on gene-cnv and gene-prot is comparable to the best-performing single-layer data (gene) and superior to cnv and prot single layers, while INF on cnv-prot only improves over the cnv single layer. On the BRCA-ER task, the performance with INF integration of 2 or 3 layers is still better than using single layers, nevertheless to a smaller extent, except for cnv-prot integration which performs better than cnv alone but slightly worse than gene and prot single layers. The good performances achieved at the gene and prot single layers do not come unexpected, since the biological nature of the target ER-status is defined at transcriptomics level. On the more difficult AML-OS task, INF has better performance over both rSNF and juXT on gene-mirna and meth-mirna integration, still improving over single-layer performance both in terms of MCC and reduced signature sizes.
For BRCA-ER, three-layer INF (rSNFi) integration performs better than either rSNF or juXT (MCC test 0.830 vs. 0.804, 0.797 for rSNF and juXT, respectively). All two-layer INF integrations perform similarly to, or better than, the corresponding rSNF and juXT integrations, in particular for cnv-prot integration (MCC test 0.746 vs. 0.682, 0.692 resp. for rSNF and juXT).
On BRCA-subtypes, the 3-layer INF integration performs better than either rSNF or juXT (MCC test 0.838 vs. 0.811, 0.795 resp. for rSNF and juXT), nevertheless without improving over the gene single-layer performance (MCC test 0.821). However, the INF median signature size is only 301.5, compared to 1801 for rSNF and juXT, and 891 for the gene layer alone. All two-layer INF integrations yield better performance than their corresponding juXT or rSNF integrations.
Omics integration is particularly effective for KIRC-OS, as all 2- and 3-layer INF integrations outperform juXT, rSNF, and each of the single-layer classifiers. In fact, 3-layer rSNFi achieves MCC test 0.378 vs. 0.274, 0.305 (resp. for juXT, rSNF), 0.296, 0.327, 0.333 (resp. rSNFi meth-mirna, gene-mirna, gene-meth), and 0.253, 0.261, 0.249 (resp. gene, meth, mirna).
For AML-OS, INF feature sets are always more compact than either juXT or rSNF, with three-layer integration giving better MCC than any of the INF two-layer integrations (MCC test 0.176 vs. 0.125, 0.169, 0.047, respectively three-layer vs. meth-mirna, gene-mirna, gene-meth). Moreover, cross-validation MCCs corresponding to INF integration are better than any single layer MCC as well as rSNF and juXT.
For all tasks, INF signatures are markedly more compact with respect to both juXT and rSNF. With 91.5 vs. 6559 (1.4%) median features (rSNFi vs. juXT), the largest reduction in size occurs for AML-OS 3-layer integration, while the least reduction is observed for BRCA-subtypes task, with 301.5 vs. 1801 (16.7%) median features (rSNFi vs. juXT).
In terms of contributions from the omics datasets being integrated, the gene layer generally provides the largest number of features to the signatures identified by the INF workflow. In particular for the BRCA dataset, in both ER and subtypes tasks, the gene layer contributes over 95% of the top features for juXT and rSNFi, with rSNF signatures being slightly more balanced (prot contribution remains marginal, while cnv provides 28.3 and 17.7% of the top features in ER and subtypes tasks respectively). This is expected as the class label is defined mainly at transcriptomics level. In AML-OS experiments, the layer contributing the most is still gene, accounting for ca. 78, 73, and 81% of the top feature sets for RF juXT, rSNF and rSNFi experiments, respectively. In KIRC-OS experiments, gene is the layer contributing the most to the top juXT and rSNF feature sets, while meth is the major contributor for rSNFi. The percentage of features from each omic layer contributing to the top signatures for juXT, rSNF and rSNFi 3-layer integrations are reported in Supplementary Tables layer_contribution. The RF rSNFi signatures for all tasks are available in Supplementary Tables BRCA-ER_RF_rSNFi, BRCA-subtypes_RF_rSNFi, AML-OS_RF_rSNFi and KIRC-OS_RF_rSNFi.
Even though a systematic biological interpretation of the identified signatures is beyond the scope of this work, to ascertain the reliability of our results we compared them with published data. The top features in the BRCA-ER rSNFi signature include multiple genes known to be associated with breast carcinoma progression and outcome such as AGR3, B3GNT, and MLPH (32–34). In addition we find the estrogen receptor gene (ESR1 from the gene and ER-alpha from the prot layer) and the transcription factor GATA3 (from both gene and prot layers) (35). Both the BRCA-ER and BRCA-subtypes signatures include genes previously identified as novel biomarkers for intrinsic breast carcinoma subtype prediction (36). Interestingly there is only partial overlap between the top features identified in BRCA ER vs. subtypes tasks. Considering AML-OS task, it is noteworthy to mention that the top feature identified has been recently reported as a potential biomarker predicting overall survival in a subset of AML patients (37).
Within the mirna features of the AML-OS signature, MIR-203 expression was recently found to be associated with AML patient survival (38); MIR-100 is highly expressed in AML and was found to regulate cell differentiation and survival (39); high expression of miR-504-3p was reported to be associated with favorable AML prognosis (40). Given that the rSNFi signature identified in the KIRC-OS task contains a large percentage of methylation data (86.5%), its direct interpretation is more difficult. It is however interesting to observe that all the 15 gene features in the signature are identified as prognostic markers for renal carcinoma according to the Human Protein Atlas (41).
The features selected by juXT, rSNF and rSNFi are projected on a bi-dimensional space using the UMAP unsupervised multidimensional projection method (42, 43). Here we show an example on the BRCA-subtypes 3-layer dataset, with a UMAP projection of the features selected by juXT (Figure 5) compared to the UMAP projection of the INF signature (Figure 6) for one of the 10 data splits (the UMAP plots for the remaining 9 splits are in Figures S1, S2). Colors represent cancer subtypes and shapes represent training/test partitions. Using the 1801 juXT features, cancer subtypes are roughly clustered, with HER2-enriched and Luminal B being more dispersed (Figure 5). The clusters appear to be more sharply defined in the projection of the 302-feature INF signature: in particular, Basal-like patients form a distinct cluster, while Luminal A, Luminal B and HER2-enriched patient clusters are close to each other, slightly overlapping yet hinting to a trajectory pattern (Figure 6). The HER2/luminal cluster contains two patients classified as basal-like subtype, consistently with the findings of (44).
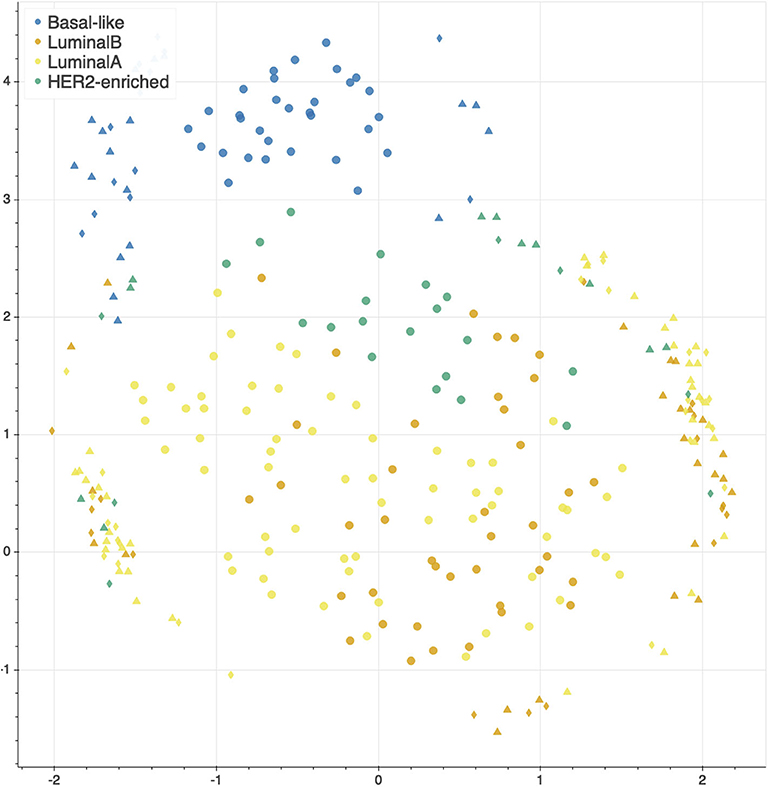
Figure 5. UMAP projection on the BRCA-subtypes task with 3-layer juxtaposed data. Circle, TR set; triangle, TS set; diamond, TS2 set.
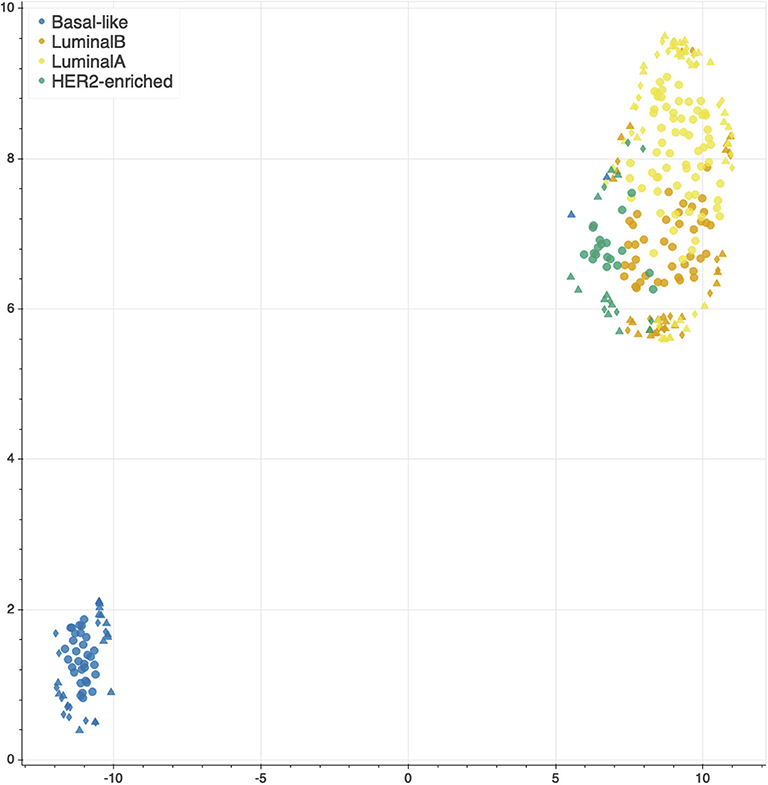
Figure 6. UMAP projection on the BRCA-subtypes task with 3-layer juxtaposed data restricted to the rSNFi signature. Circle, TR set; triangle, TS set; diamond, TS2 set.
Ritchie et al. (45) defined omics data integration as the combination of multiple omics datasets that can be used for the development of models to predict complex traits or phenotypes. The problem of data integration in computational biology is far from having a consolidated and shared solution. Many long-standing obstacles are still far from being overcome, and the increasing availability of data [e.g., TCGA, (46)] and computational tools [see for instance (47–51) and https://github.com/mikelove/awesome-multi-omics], also interactive [e.g., (52)], is raising new issues that need to be addressed. In fact, not only are existing datasets still lacking standardization protocols to deal with their complexity and heterogeneity, but also the reliability, reproducibility and interpretability of new computational methods are emerging as urgent and relevant questions (53). Moreover, modern technologies allow the rapid extraction of high-dimensional, high-throughput features from different sources (e.g., gene expression, DNA sequencing, metabolomics, or high-resolution images), which in turn require collaboration between biologists, computer scientists, physicians and other experts. The lack of common methodologies and terminologies can transform this synergy into a further level of complexity in the process of data integration (54). As observed in (55, 56), specific technological limits, noise levels and variability ranges affect the different omics, and thus confounding the underlying biological signals, yielding that really integrative analysis is still very rare, while different methods often discover different kinds of patterns, as evidenced by the lack of consistency in the published results, although efforts in this direction have started appearing (57, 58).
Indeed, the underlying hypothesis of multi-omics integration is that different omics data can provide complementary information (56) [although sometimes redundant (9)], and thus a broader insight with respect to single-layer analysis, for a better understanding of disease mechanisms (59). This assumption has been confirmed by multiple studies on diverse diseases, such as cardiovascular disease (60), diabetes (61), liver disease (62), or mitochondrial diseases (63), and also longitudinally (64), suggesting that the more complex the disease the more advantageous the integration. As the co-occurrence of multiple causes and correlated events is a well-known characteristic of tumorigenesis and cancer development, the integration of data generated from multiple sources can thus be particularly useful for the identification of cancer hallmarks (65–68).
Many computational strategies have been introduced that combine multiple types of data to identify novel biomarkers and thus to predict a phenotype of interest or drive the development of intervention protocols. Given the heterogeneity of data and tasks, these techniques deal with the data integration at different levels of the learning process: (i) by concatenating the features before fitting a model (early-integration), (ii) by incorporating the integration step into the model training (intermediate-integration), or (iii) by combining the outputs of distinct models for the final prediction (late-integration) (69, 70).
In the early-integration approach, also known as juxtaposition-based, the multi-omics datasets are first concatenated into one matrix. To deal with the high-dimensionality of the joint dataset, these methods generally adopt matrix factorization (55, 56, 58, 71), statistical (47, 49, 58, 60, 62, 72–76), and machine learning tools (58, 76, 77). Alternatively, data models relying on polyglot approaches can be used especially in (bio)informatics applications (78, 79). Although the dimensionality reduction procedure is necessary and may improve the predictive performance, it can also cause the loss of key information (69). Moreover, biomarkers identified purely on a computational statistics rationale from meta-omics features often lack biological plausibility (80).
In order to maximize the contribution of the single-omics layer, the late-integration methods first model each dataset individually, and then merge or average the results; they are also known as model-driven (70, 81). Although these techniques avoid the pre-selection of the features, they do not leverage the hidden correlations between the data, posing again the risk of signal loss (80, 82).
The intermediate-integration strategies aim at developing a joint model that accounts for the correlation between the omics layers, to boost their combined predictive power (83). Among these methods, the network-based models refer to the reconstruction of a graph representing the complex biological interactions (76, 84), known or predicted, between the variables to discover novel informative relationships (85). They have successfully been applied in cancer research for the identification of pan-cancer drug targets (86), the detection of subtype-specific pathways (83, 87) and of genetic aberrations (88), or the stratification of cancer patients (89–91). In particular, Koh et al. (44) predicted breast cancer subtypes by applying a modified shrunken centroid method in the development of their network-based tool, iOmicsPASS. Further, breast cancer datasets in TGCA represent a benchmark for integrative models (92–94), as well as AML (95).
More recently, the success of deep learning algorithms in various bioinformatics fields (96) prompted the adoption of deep neural networks for omics-integration in precision oncology. Autoencoders and convolutional neural networks have been effectively trained for the prediction of prognostic outcomes (9, 97), response to chemotherapeutic drugs (50), and gene targeting (98), by adopting either an early-integration (9, 98) or a late-integration (50, 97). Although deep learning models hold the potential to include image-derived features in the integration workflow, they suffer from interpretability and generalization issues (99).
Although it is clear that no single method is consistently preferable, and that most of the proposed approaches are task and/or data dependent (80), the complexity of tumor analysis suggests that network-based approaches are needed (87, 100).
In this context, it is clear that omics-integration is one of the most promising and demanding challenges of the modern bioinformatics, and that there is an urgent need to prove the reproducibility, interpretability, and generalization capability of the proposed methods (85, 101).
We present the INF framework for the characterization of cancer patient phenotypes by integrated multi-omics signatures, combining an improved version of a state-of-the-art integration technique (5) with predictive models developed inside a Data Analysis Plan (6) for machine learning. The framework is applied to TCGA data to predict clinically relevant patient phenotypes such as the overall survival or cancer subtypes.
The simplest approach for multi-omics data integration consists in juxtaposition of normalized measurements into one joint matrix, followed by the development of a predictive model. Juxtaposition-based integration is considered as a baseline technique, since it is the most naïve approach to combine two datasets; moreover, it enables to identify multi-omics signatures by borrowing discriminatory strength from information derived by all datasets. Juxtaposition further dilutes the already possible low signal-to-noise ratio in each data type, affecting the understanding of the biological interactions at the different omics levels.
Conversely, the INF method for omics data integration is an improvement of the popular Similarity Network Fusion (SNF) approach (5), which has inspired several studies in the scientific literature, specifically in cancer genomics (77, 87, 102–106). SNF maximizes the shared or correlated information between multiple datasets by combining data through inference of a joint network-based model, accounting for how informative each data type is to the observed similarity between samples.
Two innovative solutions have been implemented in this study: (i) we devised a SNF-based procedure to rank variables according to their importance in clustering samples with similar phenotypes; and (ii) predictive models were developed exploiting the SNF-ranked variables, inside a rigorous Data Analysis Plan which ensures reproducibility (6, 19).
The performance of INF was assessed both in terms of statistical properties as well as biological interest. Concerning the statistical aspect, INF was compared with predictive models developed on the juxtaposed datasets (juXT technique), as well as on the single-layer datasets. With INF, smaller signature sizes were systematically derived to achieve comparable or even better performance both in cross-validation and in test. This is an added value for INF, as biological validation of biomarkers can definitely benefit from signatures of small size in terms of both costs and required time. This main achievement is mainly due to the novel rSNF ranking, which increases the signal-to-noise ratio from the combined layers by prioritizing the most discriminant biomarkers in terms of network mutual information. rSNF exploits two main SNF advantages: integration of heterogeneous data and clustering of sample networks. The main peculiarity of the SNF integrative procedure is its robustness to noise (5), because weak similarities among samples (low-weight edges) disappear, except for low-weight edges supported by all networks, which are conserved depending on how tightly connected their neighborhoods are across networks. Moreover, the rSNFi step further increases the signal-to-noise ratio by training a predictive classifier on multi-omics juxtaposed data restricted to the top-ranked biomarkers shared by juXT and rSNF models. The resulting signatures are compact in size (up to 99% reduction w.r.t. juXT) while allowing predictive models to achieve equal or better performance compared to naïve juxtaposition or the single layers alone. While a comprehensive evaluation of the biological meaning of the signatures identified through the INF framework is beyond the scope of this work, we assessed their general validity with a thorough literature search. Our investigation shows that the signatures identified through the INF framework include biological markers that are relevant in the tasks under analysis and are consistent with previously published data. Further, as in (9), the largest contribution in the biomarkers' lists is provided by gene expression, while epigenomics, proteomics and miRNA transcriptomics play a minor role.
It should be noted that, especially in computational biology, multicollinearity between pairs of predictors and/or layers is intrinsic in the problem. Nevertheless, most machine learning models are indeed designed to identify the relevant predictors even in the presence of strong linear or non-linear correlations, provided that an appropriate DAP, feature ranking method, and diagnostic tools (e.g., random labels) are adopted against selection bias. To this aim, the application of a DAP derived from the MAQC-II initiative for model selection is a core attribute of the INF framework.
A fair comparison of INF results with other integration methods is currently unfeasible due to the number and variety of computational pipelines with dissimilar datasets, preprocessing methods, data analysis plans, and performance metrics.
This work is based on the original R implementation of the SNF algorithm (5). However, we are aware that Open Source implementations exist in other programming languages, in particular snfpy for Python (107). In a future release of the INF workflow, we plan to migrate the SNF-related parts to snfpy or a similar Python-based implementation, in order to drop the dependency on R and to potentially improve the overall performance.
In its current version, the INF framework supports the integration of two or more one-dimensional omics layers. As part of our future effort we will add support for the integration of medical imaging layers, for example leveraging the extraction of histopathological features from whole slide images by deep learning (10) or using radiomics or deep features from radiological images (11). In both cases, further issues will emerge from the interactions between the omics and the non-omics data, needing particular care in the integration (12).
The original contributions presented in the study are included in the article/supplementary files, further inquiries can be directed to the corresponding author/s.
CA, LT, and GJ: conceptualization. MC, NB, AM, AZ, LT, CA, and GJ: methodology. MF: interpretation. GJ: coordination. MC, NB, AM, MF, GJ, and CF: writing. All authors contributed to the article and approved the submitted version.
AZ was employed by the company NIDEK Technologies Srl. CF was employed by the company HK3 Lab.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors wish to thank Dr. Valerio Maggio for helpful discussions on aspects of the machine learning workflow and for paper proofreading. The results published here are in whole or part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.01065/full#supplementary-material
1. ^INF source code is publicly available on the GitLab repository https://gitlab.fbk.eu/MPBA/INF, while data is archived at http://dx.doi.org/10.6084/m9.figshare.12052995.v1
2. ^European Call Multi-omics for genotype-phenotype associations (RIA) https://ec.europa.eu/info/funding-tenders/opportunities/portal/screen/opportunities/topic-details/biotec-07-2020
1. Benton D. Bioinformatics-principles and potential of a new multidisciplinary tool. Trends Biotechnol. (1996) 14:261–72. doi: 10.1016/0167-7799(96)10037-8
2. Chung SY, Wong L. Kleisli: a new tool for data integration in biology. Trends Biotechnol. (1999) 17:351–5. doi: 10.1016/S0167-7799(99)01342-6
3. Zandoná A. Predictive networks for multi meta-omics data integration (Doctoral Programme in Biomolecular Sciences). University of Trento. Trento (2017). Available online at: http://eprints-phd.biblio.unitn.it/2547/
4. Trastulla L. Techniques of Integration for High-Throughput Omics Data. Department of Mathematics, University of Trento, Trento (2016).
5. Wang B, Mezlini A, Demir F, Fiume M, Tu Z, Brudno M, et al. Similarity network fusion for aggregating data types on a genomic scale. Nat Methods. (2014) 11:333–7. doi: 10.1038/nmeth.2810
6. The MAQC Consortium. The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models. Nat Biotechnol. (2010) 28:827–38. doi: 10.1038/nbt.1665
7. The SEQC/MAQC-III Consortium. A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequence Quality Control consortium. Nat. Biotechnol. (2014) 32:903–14. doi: 10.1038/nbt.2957
8. Shi L, Kusko R, Wolfinger RD, Haibe-Kains B, Fischer M, Sansone SA, et al. The international MAQC Society launches to enhance reproducibility of high-throughput technologies. Nat Biotechnol. (2017) 35:1127–8. doi: 10.1038/nbt.4029
9. Chai H, Zhou X, Cui Z, Rao J, Hu Z, Yang Y. Integrating multi-omics data with deep learning for predicting cancer prognosis. bioRxiv. [Preprint]. (2019) 807214. doi: 10.1101/807214
10. Bizzego A, Bussola N, Chierici M, Cristoforetti M, Francescatto M, Maggio V, et al. Evaluating reproducibility of AI algorithms in digital pathology with DAPPER. PLoS Comput. Biol. (2019) 15:e1006269. doi: 10.1371/journal.pcbi.1006269
11. Bizzego A, Bussola N, Salvalai D, Chierici M, Maggio V, Jurman G, et al. Integrating deep and radiomics features in cancer bioimaging. In: IEEE Conference on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB). Siena (2019) p. 1–8. doi: 10.1101/568170
12. López de Maturana E, Alonso L, Alarcón P, Martín-Antoniano IA, Pineda S, Piorno L, et al. Challenges in the integration of omics and non-omics data. Genes. (2019) 10:238. doi: 10.3390/genes10030238
13. The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. (2013) 368:2059–74. doi: 10.1056/NEJMoa1301689
14. The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. (2013) 499:43. doi: 10.1038/nature12222
15. The Genome Atlas Research Network. Comprehensive molecular portraits of human breast tumours. Nature. (2012) 490:61. doi: 10.1038/nature11412
16. Kosinski M, Biecek P. RTCGA: The Cancer Genome Atlas Data Integration (2019). R package version 1.16.0. Available online at: https://rtcga.github.io/RTCGA
18. Cortes C, Vapnik VN. Support-vector networks. Mach Learn. (1995) 20:273–97. doi: 10.1007/BF00994018
19. Zhang W, Yu Y, Hertwig F, Thierry-Mieg J, Zhang W, Thierry-Mieg D, et al. Comparison of RNA-seq and microarray-based models for clinical endpoint prediction. Genome Biol. (2015) 16:133. doi: 10.1186/s13059-015-0694-1
20. Matthews BW. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim Biophys Acta. (1975) 405:442–451. doi: 10.1016/0005-2795(75)90109-9
21. Baldi P, Brunak S, Chauvin Y, Andersen CAF, Nielsen H. Assessing the accuracy of prediction algorithms for classification: an overview. Bioinformatics. (2000) 16:412–24. doi: 10.1093/bioinformatics/16.5.412
22. Gorodkin J. Comparing two K-category assignments by a K-category correlation coefficient. Comput Biol Chem. (2004) 28:367–74. doi: 10.1016/j.compbiolchem.2004.09.006
23. Jurman G, Riccadonna S, Furlanello C. A comparison of MCC and CEN error measures in multi-class prediction. PLoS ONE. (2012) 7:e41882. doi: 10.1371/journal.pone.0041882
24. Chicco D, Jurman G. The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. BMC Genomics. (2020) 21:6. doi: 10.1186/s12864-019-6413-7
25. Jurman G, Merler S, Barla A, Paoli S, Galea A, Furlanello C. Algebraic stability indicators for ranked lists in molecular profiling. Bioinformatics. (2008) 24:25864. doi: 10.1093/bioinformatics/btm550
26. Jurman G, Riccadonna S, Visintainer R, Furlanello C. Algebraic comparison of partial lists in bioinformatics. PLoS ONE. (2012) 7:e36540. doi: 10.1371/journal.pone.0036540
27. Köster J, Rahmann S. Snakemake–a scalable bioinformatics workflow engine. Bioinformatics. (2012) 28:2520–2. doi: 10.1093/bioinformatics/bts480
28. Köster J, Rahmann S. Snakemake–a scalable bioinformatics workflow engine (Erratum). Bioinformatics. (2018) 34:3600. doi: 10.1093/bioinformatics/bty350
30. Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, et al. Scikit-learn: machine learning in Python. J Mach Learn Res. Trelgol Publishing (2011) 12:2825–30.
31. R Core Team. R: A Language and Environment for Statistical Computing. Vienna (2019). Available online at: https://www.R-project.org/
32. Garczyk S, von Stillfried S, Antonopoulos W, Hartmann A, Schrauder MG, Fasching PA, et al. AGR3 in breast cancer: prognostic impact and suitable serum-based biomarker for early cancer detection. PLoS ONE. (2015) 10:e0122106. doi: 10.1371/journal.pone.0122106
33. Potapenko IO, Lüders T, Russnes HG, Helland, Sørlie T, Kristensen VN, et al. Glycan-related gene expression signatures in breast cancer subtypes; relation to survival. Mol Oncol. (2015) 9:861–76. doi: 10.1016/j.molonc.2014.12.013
34. Thakkar A, Raj H, Ravishankar, Muthuvelan B, Balakrishnan A, Padigaru M. High expression of three-gene signature improves prediction of relapse-free survival in estrogen receptor-positive and node-positive breast tumors. Biomarker Insights. (2015) 10:BMI.S30559. doi: 10.4137/BMI.S30559
35. Guo Y, Yu P, Liu Z, Maimaiti Y, Chen C, Zhang Y, et al. Prognostic and clinicopathological value of GATA binding protein 3 in breast cancer: a systematic review and meta-analysis. PLoS ONE. (2017) 12:e0174843. doi: 10.1371/journal.pone.0174843
36. Milioli HH, Vimieiro R, Riveros C, Tishchenko I, Berretta R, Moscato P. The discovery of novel biomarkers improves breast cancer intrinsic subtype prediction and reconciles the labels in the METABRIC data set. PLoS ONE. (2015) 10:e0129711. doi: 10.1371/journal.pone.0129711
37. Bai H, Zhou M, Zeng M, Han L. PLA2G4A is a potential biomarker predicting shorter overall survival in patients with Non-M3/NPM1 wildtype acute myeloid leukemia. DNA Cell Biol. (2020) 39:700–708. doi: 10.1089/dna.2019.5187
38. Guo Y. Clinical significance of serum MicroRNA-203 in patients with acute myeloid leukemia. Bioengineered. (2019) 10:345–352. doi: 10.1080/21655979.2019.1652490
39. Zheng YS, Zhang H, Zhang XJ, Feng DD, Luo XQ, Zeng CW, et al. MiR-100 regulates cell differentiation and survival by targeting RBSP3, a phosphatase-like tumor suppressor in acute myeloid leukemia. Oncogene. (2012) 31:80–92. doi: 10.1038/onc.2011.208
40. Li SM, Zhao YQ, Hao YL, Liang YY. Upregulation of miR-504-3p is associated with favorable prognosis of acute myeloid leukemia and may serve as a tumor suppressor by targeting MTHFD2. Eur Rev Med Pharmacol Sci. (2019) 23:1203–13. doi: 10.26355/eurrev_201902_17013
41. Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, et al. A pathology atlas of the human cancer transcriptome. Science. (2017) 357:eaan2507. doi: 10.1126/science.aan2507
42. McInnes L, Healy J, Saul N, Großberger L. UMAP: uniform manifold approximation and projection. J Open Source Softw. (2018) 3:861. doi: 10.21105/joss.00861
43. McInnes L, Healy J, Melville J. UMAP: uniform manifold approximation and projection for dimension reduction. arXiv [preprint.] arXiv:1802.03426 (2018).
44. Koh HWL, Fermin D, Vogel C, Pui Choi K, Ewing RM, Choi H. iOmicsPASS: network-based integration of multiomics data for predictive subnetwork discovery. NPJ Syst Biol Appl. (2019) 5:22. doi: 10.1038/s41540-019-0099-y
45. Ritchie MD, Holzinger ER, Li R, Pendergrass SA, Kim D. Methods of integrating data to uncover genotype-phenotype interactions. Nat Rev Genet. (2015) 16:85–97. doi: 10.1038/nrg3868
46. Conesa A, Beck S. Making multi-omics data accessible to researchers. Sci Data. (2019) 6:251. doi: 10.1038/s41597-019-0258-4
47. Rohart F, Gautier B, Singh A, Lê Cao KA. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput Biol. (2017) 13:e1005752. doi: 10.1371/journal.pcbi.1005752
48. Ulfenborg B. Vertical and horizontal integration of multiomics data with miodin. BMC Bioinformatics. (2019) 20:649. doi: 10.1186/s12859-019-3224-4
49. Meng C, Basunia A, Peters B, Gholami AM, Kuster B, Culhane AC. MOGSA: integrative single sample gene-set analysis of multiple omics data. Mol Cell Proteomics. (2019) 18(8 Suppl 1):S153–68. doi: 10.1074/mcp.TIR118.001251
50. Sharifi-Noghabi H, Zolotareva O, Collins CC, Ester M. MOLI: multi-omics late integration with deep neural networks for drug response prediction. Bioinformatics. (2019) 35:i501–9. doi: 10.1093/bioinformatics/btz318
51. Zanfardino M, Franzese M, Pane K, Cavaliere C, Monti S, Esposito G, et al. Bringing radiomics into a multi-omics framework for a comprehensive genotype-phenotype characterization of oncological diseases. J Transl Med. (2019) 17:337. doi: 10.1186/s12967-019-2073-2
52. Netanely D, Stern N, Laufer I, Shamir R. PROMO: an interactive tool for analyzing clinically-labeled multi-omic cancer datasets. BMC Bioinformatics. (2019) 20:732. doi: 10.1186/s12859-019-3142-5
53. Lionelli S. Philosophy of biology: the challenges of big data biology. eLife. (2019) 8:e47381. doi: 10.7554/eLife.47381
54. Schneider MV, Jimenez RC. Teaching the fundamentals of biological data integration using classroom games. PLoS Comput Biol. (2012) 8:e1002789. doi: 10.1371/journal.pcbi.1002789
55. Pucher BM, Zeleznik OA, Thallinger GG. Comparison and evaluation of integrative methods for the analysis of multilevel omics data: a study based on simulated and experimental cancer data. Brief Bioinform. (2018) 28:1–11. doi: 10.1093/bib/bby027
56. Cantini L, Zakeri P, Hernandez C, Naldi A, Thieffry D, Remy E, et al. Benchmarking joint multi-omics dimensionality reduction approaches for cancer study. bioRxiv. [Preprint]. (2020) 905760. doi: 10.1101/2020.01.14.905760
57. McCabe SD, Lin DY, Love MI. Consistency and overfitting of multi-omics methods on experimental data. Brief Bioinform. (2019) bbz070. doi: 10.1093/bib/bbz070
58. Wu C, Zhou F, Ren J, Li X, Jiang Y, Ma S. A selective review of multi-level omics data integration using variable selection. High Throughput. (2019) 8:4. doi: 10.3390/ht8010004
59. Karczewski K, Snyder M. Integrative omics for health and disease. Nat Rev Genet. (2018) 19:299–310. doi: 10.1038/nrg.2018.4
60. Leon-Mimila P, Wang J, Huertas-Vazquez A. Relevance of multi-omics studies in cardiovascular diseases. Front Cardiovasc Med. (2019) 6:91. doi: 10.3389/fcvm.2019.00091
61. Prélot L, Draisma H, Anasanti M, Balkhiyarova Z, Wielscher M, Yengo L, et al. Machine learning in multi-omics data to assess longitudinal predictors of glycaemic trait levels. bioRxiv. [Preprint]. (2018) 358390. doi: 10.1101/358390
62. Del Chierico F, Nobili V, Vernocchi P, Russo A, De Stefanis C, Gnani D, et al. Gut microbiota profiling of pediatric nonalcoholic fatty liver disease and obese patients unveiled by an integrated meta-omics-based approach. Hepatology. (2017) 65:451–64. doi: 10.1002/hep.28572
63. Khan S, Ince-Dunn G, Suomalainen A, Elo LL. Integrative omics approaches provide biological and clinical insights: examples from mitochondrial diseases. J Clin Invest. (2020) 130:20–8. doi: 10.1172/JCI129202
64. Tarazona S, Balzano-Nogueira L, Conesa A. Chapter eighteen - multiomics data integration in time series experiments. In: Jaumot J, Bedia C, Tauler R, editors. Comprehensive Analytical Chemistry, Vol. 82. Elsevier (2018). p. 505–32. doi: 10.1016/bs.coac.2018.06.005
65. Chakraborty S, Hosen MI, Ahmed M, Shekhar HU. Onco-Multi-OMICS approach: a new frontier in cancer research. BioMed Res Int. (2018) 2018:9836256. doi: 10.1155/2018/9836256
66. Gallo Cantafio ME, Grillone K, Caracciolo D, Scionti F, Arbitrio M, Barbieri V, et al. From single level analysis to multi-omics integrative approaches: a powerful strategy towards the precision oncology. High Throughput. (2018) 7:33. doi: 10.3390/ht7040033
67. Sathyanarayanan A, Gupta R, Thompson EW, Nyholt DR, Bauer DC, Nagaraj SH. A comparative study of multi-omics integration tools for cancer driver gene identification and tumour subtyping. Brief Bioinform. (2019) bbz121. doi: 10.1093/bib/bbz121
68. Liu SH, Shen PC, Chen CY, Hsu AN, Cho YC, Lai YL, et al. DriverDBv3: a multi-omics database for cancer driver gene research. Nucleic Acids Res. (2019) 48:D863–70. doi: 10.1093/nar/gkz964
69. Li Y, Wu FX, Ngom A. A review on machine learning principles for multi-view biological data integration. Brief Bioinform. (2018) 19:325–40. doi: 10.1093/bib/bbw113
70. Vlahou A, Magni F, Mischak H, Zoidakis J. Integration of Omics Approaches and Systems Biology for Clinical Applications. Hoboken, NJ: John Wiley & Sons (2018). doi: 10.1002/9781119183952
71. Stein-O'Brien GL, Arora R, Culhane AC, Favorov AV, Garmire LX, Greene CS, et al. Enter the matrix: factorization uncovers knowledge from omics. Trends Genet. (2018) 34:790–805. doi: 10.1016/j.tig.2018.07.003
72. Argelaguet R, Velten B, Arnol D, Dietrich S, Zenz T, Marioni JC, et al. Multi-Omics Factor Analysis-a framework for unsupervised integration of multi-omics data sets. Mol Syst Biol. (2018) 14:e8124. doi: 10.15252/msb.20178124
73. Dao MC, Sokolovska N, Brazeilles R, Affeldt S, Pelloux V, Prifti E, et al. A data integration multi-omics approach to study calorie restriction-induced changes in insulin sensitivity. Front Physiol. (2019) 9:1958. doi: 10.3389/fphys.2018.01958
74. Zeng ISL, Lumley T. Review of statistical learning methods in integrated omics studies (an integrated information science). Bioinform Biol Insights. (2018) 12:1–16. doi: 10.1177/1177932218759292
75. Qiu C, Yu F, Su K, Zhao Q, Zhang L, Xu C, et al. Multi-omics data integration for identifying osteoporosis biomarkers and their biological interaction and causal mechanisms. IScience. (2020) 23:100847. doi: 10.1016/j.isci.2020.100847
76. Misra BB, Langefeld C, Olivier M, Cox LA. Integrated omics: tools, advances and future approaches. J Mol Endocrinol. (2019) 62:R21–45. doi: 10.1530/JME-18-0055
77. Speicher NK, Pfeifer N. Integrating different data types by regularized unsupervised multiple kernel learning with application to cancer subtype discovery. Bioinformatics. (2015) 31:i268–75. doi: 10.1093/bioinformatics/btv244
78. Chromiak M, Stencel K. A data model for heterogeneous data integration architecture. In: Kozielski S, Mrozek D, Kasprowski P, Ma?ysiak-Mrozek B, Kostrzewa D, editors. Beyond Databases, Architectures, and Structures. BDAS 2014. Communications in Computer and Information Science, Vol. 424. Springer (2014). p. 547–56. doi: 10.1007/978-3-319-06932-6_53
79. Reisman S, Hatzopoulos T, Läufer K, Thiruvathukal GK, Putonti C. A polyglot approach to bioinformatics data integration: a phylogenetic analysis of HIV-1. Evol Bioinform Online. (2016) 12:23–7. doi: 10.4137/EBO.S32757
80. Rappoport N, Shamir R. Multi-omic and multi-view clustering algorithms: review and cancer benchmark. Nucleic Acids Res. (2018) 46:10546–62. doi: 10.1093/nar/gky889
81. Marín de Mas I. Chapter sixteen - multiomic data integration and analysis via model-driven approaches. In: Jaumot J, Bedia C, Tauler R, editors. Comprehensive Analytical Chemistry, Vol. 82. Elsevier (2018). p. 447–76. doi: 10.1016/bs.coac.2018.07.005
82. Gadepally V, Mattson T, Stonebraker M, Wang F, Luo G, Laing Y, et al. Heterogeneous data management, polystores, and analytics for healthcare: VLDB 2019. In: Workshops, Poly and DMAH. Los Angeles, CA: Springer Nature (2019). doi: 10.1007/978-3-030-33752-0
83. Vantaku V, Dong J, Ambati CR, Perera D, Donepudi SR, Amara CS, et al. Multi-omics integration analysis robustly predicts high-grade patient survival and identifies CPT1B effect on fatty acid metabolism in bladder cancer. Clin Cancer Res. (2019) 25:3689–701. doi: 10.1158/1078-0432.CCR-18-1515
84. Zhou G, Li S, Xia J. Network-based approaches for multi-omics integration. In: Li S, editor. Computational Methods and Data Analysis for Metabolomics. Methods in Molecular Biology, Vol. 2104. New York, NY: Humana (2020). p. 469–87. doi: 10.1007/978-1-0716-0239-3_23
85. Bersanelli M, Mosca E, Remondini D, Giampieri E, Sala C, Castellani G, et al. Methods for the integration of multi-omics data: mathematical aspects. BMC Bioinformatics. (2016) 17:S15. doi: 10.1186/s12859-015-0857-9
86. do Valle ÍF, Menichetti G, Simonetti G, Bruno S, Zironi I, Fernandes Durso D, et al. Network integration of multi-tumour omics data suggests novel targeting strategies. Nat Commun. (2018) 9:4514. doi: 10.1038/s41467-018-06992-7
87. Verbeke LPC, Van den Eynden J, Fierro AC, Demeester P, Fostier J, Marchal K. Pathway relevance ranking for tumor samples through network-based data integration. PLoS ONE. (2015) 10:e0133503. doi: 10.1371/journal.pone.0133503
88. Dimitrakopoulos C, Kumar Hindupur S, Häfliger L, Behr J, Montazeri H, Hall MN, et al. Network-based integration of multi-omics data for prioritizing cancer genes. Bioinformatics. (2018) 34:2441–8. doi: 10.1093/bioinformatics/bty148
89. Zhao L, Yan H. MCNF: a novel method for cancer subtyping by integrating multi-omics and clinical data. IEEE/ACM Trans Comput Biol Bioinform. (2019). doi: 10.1109/TCBB.2019.2910515. [Epub ahead of print].
90. Rappoport N, Shamir R. NEMO: cancer subtyping by integration of partial multi-omic data. Bioinformatics. (2019) 35:3348–56. doi: 10.1093/bioinformatics/btz058
91. Yang B, Zhang Y, Pang S, Shang X, Zhao X, Han M. Integrating multi-omic data with deep subspace fusion clustering for cancer subtype prediction. IEEE/ACM Trans Comput Biol Bioinform. (2019). doi: 10.1109/TCBB.2019.2951413. [Epub ahead of print].
92. Xu A, Chen J, Peng H, Han GQ, Cai H. Simultaneous interrogation of cancer omics to identify subtypes with significant clinical differences. Front Genet. (2019) 10:236. doi: 10.3389/fgene.2019.00236
93. Kechavarzi BD, Wu H, Doman TN. Bottom-up, integrated -omics analysis identifies broadly dosage-sensitive genes in breast cancer samples from TCGA. PLoS ONE. (2019) 14:e0210910. doi: 10.1371/journal.pone.0210910
94. Kalecky K, Modisette R, Pena S, Cho YR, Taube J. Integrative analysis of breast cancer profiles in TCGA by TNBC subgrouping reveals novel microRNA-specific clusters, including miR-17-92a, distinguishing basal-like 1 and basal-like 2 TNBC subtypes. BMC Cancer. (2020) 20:141. doi: 10.1186/s12885-020-6600-6
95. Mehtonen J, Pölönen P, Häyrynen S, Dufva O, Lin J, Liuksiala T, et al. Data-driven characterization of molecular phenotypes across heterogeneous sample collections. Nucleic Acids Res. (2019) 47:e76. doi: 10.1093/nar/gkz281
96. Li Y, Huang C, Ding L, Li Z, Pan Y, Gao X. Deep learning in bioinformatics: introduction, application, and perspective in the big data era. Methods. (2019) 166:4–21. doi: 10.1016/j.ymeth.2019.04.008
97. Poirion O, Chaudhary K, Huang S, Garmire LX. Multi-omics-based pan-cancer prognosis prediction using an ensemble of deep-learning and machine-learning models. medRXiv. [Preprint]. (2019) 19010082. doi: 10.1101/19010082
98. Peng C, Zheng Y, Huang DS. Capsule Network based Modeling of Multi-omics Data for Discovery of Breast Cancer-related Genes. IEEE/ACM Trans Comput Biol Bioinform. (2019). doi: 10.1109/TCBB.2019.2909905. [Epub ahead of print].
99. Hériché JK, Alexander S, Ellenberg J. Integrating imaging and omics: computational methods and challenges. Annu Rev Biomed Data Sci. (2019) 2:175–97. doi: 10.1146/annurev-biodatasci-080917-013328
100. Traag VA, Waltman L, van Eck NJ. From Louvain to Leiden: guaranteeing well-connected communities. Sci Rep. (2019) 9:5233. doi: 10.1038/s41598-019-41695-z
101. Yu XT, Zeng T. Integrative analysis of omics big data. In: Huang T, editor. Computational Systems Biology. Methods in Molecular Biology, Vol. 1754. New York, NY: Humana Press (2018). p. 109–35. doi: 10.1007/978-1-4939-7717-8_7
102. Chiu AM, Mitra M, Boymoushakian L, Coller HA. Integrative analysis of the inter-tumoral heterogeneity of triple-negative breast cancer. Sci Rep. (2018) 8:11807. doi: 10.1038/s41598-018-29992-5
103. Cavalli FMG, Remke M, Rampasek L, Peacock J, Shih DJH, Luu B, et al. Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell. (2017) 31:737–54.e6. doi: 10.1016/j.ccell.2017.05.005
104. Jiang YZ, Ma D, Suo C, Shi J, Xue M, Hu X, et al. Genomic and transcriptomic landscape of triple-negative breast cancers: subtypes and treatment strategies. Cancer Cell. (2019) 35:428–40.e5. doi: 10.1016/j.ccell.2019.02.001
105. Pitroda SP, Weichselbaum RR. Integrated molecular and clinical staging defines the spectrum of metastatic cancer. Nat Rev Clin Oncol. (2019) 16:581–8. doi: 10.1038/s41571-019-0220-6
106. Ma T, Zhang A. Affinity network fusion and semi-supervised learning for cancer patient clustering. Methods. (2018) 145:16–24. doi: 10.1016/j.ymeth.2018.05.020
107. Markello R. snfpy: Similarity Network Fusion in Python. (2019). Available online at: https://snfpy.readthedocs.io/en/latest/
Keywords: multi-omics, classification, network, oncogenomics, predictive modeling
Citation: Chierici M, Bussola N, Marcolini A, Francescatto M, Zandonà A, Trastulla L, Agostinelli C, Jurman G and Furlanello C (2020) Integrative Network Fusion: A Multi-Omics Approach in Molecular Profiling. Front. Oncol. 10:1065. doi: 10.3389/fonc.2020.01065
Received: 31 March 2020; Accepted: 28 May 2020;
Published: 30 June 2020.
Edited by:
Chiara Romualdi, University of Padova, ItalyReviewed by:
Prashanth N. Suravajhala, Birla Institute of Scientific Research, IndiaCopyright © 2020 Chierici, Bussola, Marcolini, Francescatto, Zandonà, Trastulla, Agostinelli, Jurman and Furlanello. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marco Chierici, Y2hpZXJpY2lAZmJrLmV1; Giuseppe Jurman, anVybWFuQGZiay5ldQ==
†These authors share joint first authorship
‡These authors share joint last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.