 Anne Berberich1,2†
Anne Berberich1,2† Frederik Bartels1,2†
Frederik Bartels1,2† Zili Tang1,3†
Zili Tang1,3† Maximilian Knoll1,3Sonja Pusch1,2Nanina Hucke1,2Tobias Kessler1,2
Maximilian Knoll1,3Sonja Pusch1,2Nanina Hucke1,2Tobias Kessler1,2 Zhen Dong4Benedikt Wiestler5
Zhen Dong4Benedikt Wiestler5 Frank Winkler1,2Michael Platten6,7
Frank Winkler1,2Michael Platten6,7 Wolfgang Wick1,2
Wolfgang Wick1,2 Amir Abdollahi1,3Dieter Lemke1,2*
Amir Abdollahi1,3Dieter Lemke1,2*- 1German Cancer Research Center (DKFZ), German Cancer Consortium (DKTK), Heidelberg, Germany
- 2Department of Neurology, University of Heidelberg Medical School and National Center for Tumor Diseases (NCT), Heidelberg, Germany
- 3Division of Molecular and Translational Radiation Oncology, Heidelberg Ion Therapy Center (HIT), German Cancer Research Center (DKFZ), Heidelberg Institute of Radiation Oncology (HIRO), University of Heidelberg Medical School and National Center for Tumor Diseases (NCT), Heidelberg, Germany
- 4Department of Neurosurgery, Tongji Medical College, Tongji Hospital, Huazhong University of Science and Technology, Wuhan, China
- 5Department of Neuroradiology, Klinikum rechts der Isar der Technischen Universität, Munich, Germany
- 6Department of Neurology, Medical Faculty Mannheim, Heidelberg University, Heidelberg, Germany
- 7DKTK Clinical Cooperation Unit Neuroimmunology and Brain Tumor Immunology, German Cancer Research Center (DKFZ), Heidelberg, Germany
Background: Glioma therapy is challenged by the diffuse and invasive growth of glioma. Lysosomal protein transmembrane 5 (LAPTM5) was identified as an invasion inhibitor by an in vivo screen for invasion-associated genes. The aim of this study was to decipher the function of LAPTM5 in glioblastoma and its interaction with the CD40 receptor which is intensively evaluated as a target in the therapy of diverse cancers including glioma.
Methods: Knockdown of LAPTM5 was performed in different glioma cell lines to analyze the impact on clonogenicity, invasiveness, sensitivity to temozolomide chemotherapy, and tumorigenicity in vitro and in vivo. An expression array was used to elucidate the underlying pathways. CD40 knockdown and overexpression were induced to investigate a potential crosstalk of LAPTM5 and CD40. LAPTM5 and CD40 were correlated with the clinical outcome of glioma patients.
Results: Knockdown of LAPTM5 unleashed CD40-mediated NFκB activation, resulting in enhanced invasiveness, clonogenicity, and temozolomide resistance that was overcome by NFκB inhibition. LAPTM5 expression correlated with better overall survival in glioblastoma patients depending on CD40 expression status.
Conclusion: We conclude that LAPTM5 conveyed tumor suppression and temozolomide sensitation in CD40-positive glioblastoma through the inhibition of CD40-mediated NFκB activation. Hence, LAPTM5 may provide a potential biomarker for sensitivity to temozolomide in CD40-positive glioblastoma.
Introduction
Despite relevant improvements in the molecular understanding of glioma, treatment options remain limited for glioma patients. Glioma cells infiltrate diffusely throughout the whole brain, forming a functional tumor network that contribute to therapy resistance and recurrence after standard multimodal therapy including surgery, irradiation, and chemotherapy with temozolomide (1, 2). The complex processes underlying tumor cell migration and invasion have been studied intensively, but effective treatments targeting the pro-invasive mechanisms are still required (3–6).
Screening for invasion-associated genes identified lysosomal protein transmembrane 5 (LAPTM5) as a relevant gene involved in the invasion of glioblastoma. LAPTM5, which is localized in the lysosomal membrane (7), was downregulated in a variety of human cancer cells, suggesting that the inactivation of LAPTM5 plays a role in tumorigenesis (8). Low LAPTM5 expression was significantly correlated with poor prognosis in patients with esophageal squamous cell carcinoma and non-small cell lung cancer (NSCLC) (8). Furthermore, LAPTM5-mediated programmed cell death was involved in the spontaneous regression of neuroblastoma cells (9). In HeLA cells, the ectopic overexpression of LAPTM5 induced apoptosis via LAPTM5-mediated lysosomal pathways (10). In addition, LAPTM5 was rapidly and transiently repressed by treatment with CD40 ligand in B-cells (11), which resulted in the alternative name CD40-ligand activated transcript. Recently, CD40 was discovered as an emerging target in cancer immunotherapy, and CD40 agonistic therapies are being evaluated in antitumor trials for different cancer types including non-Hodgkin's lymphoma, advanced urothelial carcinoma, malignant melanoma, pancreatic adenocarcinoma, renal cells carcinoma, NSCLC, and glioma (12). The interaction of CD40 and the CD40 ligand (CD40L or CD154) has been shown to exert a plethora of immune-stimulating effects in the innate and the adaptive immune system (Supplemental Figure 1) (13–19). In addition, substantial CD40 expression was detected in a variety of common solid cancer types including bladder cancer (102/131, 78%), melanoma (41/71, 57.7%), breast cancer (53%), lung cancer (67/129, 51.9%), colon cancer (87/110, 79%) as well as B-lineage malignancies (14–19). CD40 expression was frequently shown to be associated with prolonged survival; however, contrary results were evident in lung and esophageal cancer where CD40 expression correlated with poor prognosis (17–20). In glioma, only scarce and partially contradictory data exist about LAPTM5 and CD40 (20, 21). Therefore, the aims of this study were to characterize the role of LAPTM5 in glioblastoma, to elucidate its interaction with CD40, and to analyze the underlying signaling pathways involved in tumorigenicity.
Materials and Methods
Glioblastoma Cells and Culture Conditions
All glioblastoma cell lines [LN-229 (RRID:CVCL_0393), LN-308 (RRID:CVCL_0394), U-87MG ATCC (RRID:CVCL_0022), LN-428 (RRID:CVCL_3959), and T98G (RRID:CVCL_0556)] were purchased from the American Type Culture Collection (Manassas, VA, USA) and cultured under standard conditions. Glioblastoma-initiating cells (T325, T269, and S24) were established from freshly dissected glioblastoma tissue from adult patients after informed consent and cultured in neurosphere medium (DMEM/F12 medium, Life Technologies, Carlsbad, CA, USA) supplemented with B27 supplement, heparin (5 μg/ml), basic fibroblast growth factor (20 ng/ml), and epidermal growth factor (20 ng/ml) (22). All cell lines tested have an isocitrate dehydrogenase wild-type status. The SNP profiles matched known profiles or were unique. The cell lines were authenticated using Multiplex Cell Authentication by Multiplexion (Heidelberg, Germany) as previously described (23). Absence of mycoplasma infection is screened for on a regular basis.
Knockdown of Gene Expression
The knockdown of LAPTM5 gene expression was induced using a commercial set of five lentiviral small hairpin RNAs (shRNA) targeting the 3′UTR region of the LAPTM5 transcript. The most effective construct was used for the experiments (Mission®, shRNA TRCN0000123149, Clone ID: NM_006762.1-1614s1c1) (Sigma Aldrich, Taufkirchen, Germany). The non-targeting shRNA lentiviral construct (Mission® SHC002, Sigma Aldrich) was used as a control (24). Furthermore, to reevaluate the experiments with another shRNA system, the knockdown of LAPTM5 was also performed with pZIP-SFFV-RFP (shERWOOD-UltramiR shRNA Target Gene Set, cat. no.: TLHSU1400-7805-pZIP-SFFV-RFP-GVO-TRI, Transomic Technologies, Huntsville, AL 35806, USA). The knockdown of CD40 gene expression was induced using a commercial green fluorescent protein (GFP)-tagged lentiviral shRNA interfering system (Mission shRNA TRCN000038715, ID: NM_011611). After transduction, the cells were selected by treatment with neomycin and fluorescence-activated cell sorting (FACS) of GFP-positive cells. The non-targeting GFP-shRNA lentiviral construct (Mission® SHC004, Sigma Aldrich) was used as a control.
The transient knockdown of CD40 was performed with siRNA transfection (Sigma Aldrich) using lipofectamine (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. All knockdown experiments were controlled with the respective non-targeting shRNA lentiviral construct. In double-knockdown experiments, two respective non-targeting shRNA lentiviral constructs were used as control. All different control knockdown constructs were named v-control (vector control).
CD40 Overexpression
CD40-overexpressing cells were generated after the transfection of cells with pcDNA3 control or pcDNA3 CD40 plasmid, as generously provided by M. Weller (Zurich, Switzerland). The CD40-vector was generated by the amplification of cDNA prepared from EJ cells using CD40-specific primers (forward primer: 5′-CTGGTCTAAGCTTGCCATGGTTC-3′ and reverse primer: 5′-TGGGTGGCGGCCGCCTCACT-3′) which were inserted into the HindIII/NotI sites of pcDNA3 (25). After transfection, the CD40-positive cells were sorted by flow cytometry. In cells with combined CD40 overexpression and LAPTM5 knockdown, the control cells were transfected with empty pcDNA3 and transduced with the respective non-targeting shRNA lentiviral construct. All control constructs were named v-control (vector control).
Quantitative Real-Time PCR
RNA extraction, cDNA synthesis, and quantitative real-time PCR (qRT-PCR) were performed as previously described (26). All results were normalized to glyceraldehyde-3-phosphate dehydrogenase serving as a housekeeping gene (27). The primer sequences are listed in Supplemental Table 1.
Whole-Exome mRNA Analysis
Gene expression analysis was performed to compare the LAPTM5 knockdown in U87MG cells with the respective controls (28). From each cell line, three independent total RNA samples were used for the microarray. The sample analysis was performed on an illumina HT 12 microarray chip at the genomics core facility (German Cancer Research Center, Heidelberg, Germany). The differentially expressed genes were identified after adjustment for multiple testing using the Benjamini–Hochberg correction and a significance threshold of p < 0.05. Ingenuity Pathway Analysis (Ingenuity Systems, Redwood City, CA, USA) was used for protein network and pathway analysis. The experiment ArrayExpress accession code is E-MTAB-6316. For the gene set enrichment analysis (GSEA), the GSEA software tool was downloaded from the homepage of the Broad Institute (http://software.broadinstitute.org/gsea). The hallmark gene sets from the Molecular Signature Database v6.1 were used for the exploratory testing of pathway enrichments in the full dataset of genes in the microarray. The number of gene set permutations was set to 1,000.
Matrigel Invasion Assay
Glioblastoma cell invasion was measured in vitro with the Boyden chamber assay (BD Biosciences, San Jose, CA, USA) (29). The migrated cells were stained with 4,6-diamidino-2-phenylindol (Vector Laboratories, Burlingame, CA, USA), scanned with Cell Observer (Zeiss, Oberkochen, Germany), and counted using ImageJ software (Bethesda, MD, USA). The number of migrated cells was corrected by proliferation control, which was assessed in parallel by crystal violet staining.
Limiting Dilution Assay
The clonogenic potential of glioblastoma cells was evaluated with the Limiting Dilution Assay as previously described (22, 30). Four different dilutions (300, 50, eight, and one cell per well) were seeded in a 96-well plate and cultured for 28 days (24 wells per dilution). The number of wells with colony formation was counted. The cells were treated with 2.5 μg/ml JSH-23 and 10 μM temozolomide (U87MG) or 5 μM temozolomide (LN229 and LN308) depending on the intrinsic sensitivity to the temozolomide treatment of the respective cell lines.
Immunoblot Analysis
Whole cell lysates were prepared as described previously (26). The antibodies used are listed in Supplemental Table 2. Equal protein loading was controlled with mouse α-tubulin (1:5,000, Sigma Aldrich, USA) staining.
Flow Cytometry Analysis
For flow cytometry, the cells were dissociated with accutase, washed, stained with CD40-FITC or the respective isotype control, and analyzed with a BD-FACS Canto II flow cytometer (BD Biosciences, USA); final data were processed with the FlowJo flow cytometry analysis software (Tree Star Inc., USA). Specific fluorescence intensity was calculated by using the mean fluorescence signal of CD40 divided by the mean fluorescence isotype signal.
Xenograft Mouse Model
All works involving animals were approved by the governmental authorities (Aktenzeichen: 35-9185.81 G-136/12 Regierungspräsidium Karlsruhe, Germany) and performed in accordance with the German animal protection law. To test the tumorigenicity in vivo, a 200-μl suspension of 1 × 107 tumor cells in 100 μl phosphate-buffered saline and 100 μl Matrigel (BD Biosciences, San Jose, CA, USA) was injected s.c. in the flank of 8-week-old female CD1 nu/nu mice (Charles River Laboratories, Sulzfeld, Germany) (31). The tumor volume was calculated by (length × width × width)/2. The mice were sacrificed in accordance with the German animal protection law when they showed symptoms of disease or when the maximal tumor diameter reached 1.5 cm. Treatment (oral gavage) with 21 mg/kg temozolomide in methylcellulose vs. methylcellulose alone in controls started on day 14 after tumor implantation and was given for 7 days every 24 h.
Methylation and mRNA Expression Correlation Analysis
The CpG sites in proximity to the LAPTM5 gene location were identified using the UCSC Genome Browser available at https://genome.ucsc.edu/. The dataset including 27k methylation data and mRNA expression data was downloaded from The Cancer Genome Atlas (TCGA), available at http://cancergenome.nih.gov/. The Pearson correlation coefficient of methylation level at the indicated CpG site and LAPTM5 mRNA expression was calculated using the software R.
5-Azacytidin Demethylation
Human glioma-initiating cell lines T325, T269, and T1 were incubated with 5 μM 5-azacytidin (Sigma-Aldrich, St. Louis, MO, USA) dissolved in dimethyl sulfoxide (DMSO) for 5 days with a complete change of the growth medium and with fresh 5-azacytidin added every day. The respective control cells were incubated with DMSO only. The cells were then harvested and LAPTM5 mRNA expression was measured using qRT-PCR.
Clinical Survival Analysis
To assess the prognostic value of LAPTM5 in high CD40-expressing tumors, methylation array (450 k) and RNASeq data from the TCGA-Glioblastoma Multiforme (GBM) project were used. For the selection of a reasonable cutoff for CD40 expression, similarity of patients was derived from methylation-based correlation (cluster boostrap of the correlation matrix, pvclust package) using CpG probes significantly associated with CD40 gene expression (linear model, Bonferroni adjusted p < 0.05, n = 24), resulting in two main clusters. Using a receiver operating characteristic (ROC) analysis, a cutoff between both clusters was selected based on maximization of the Youden index. The respective cutoff was then applied to the RNASeq cohort to select CD40 high-expressing tumors. Survival analysis was performed with parametric survival regressions, assuming a Weibull distribution.
CIBERSORT Analysis
CIBERSORT allows the estimation of immune cell fractions from bulk expression data (32). Preprocessed RNASeq data was used to calculate immune cell fractions.
Statistical Analysis
All in vitro experiments reported represent at least three replicated independent experiments containing triplicates or more. A statistical analysis of differences between mean values was performed using Student's t-test. The differences between estimated clonogenic cell frequencies analyzed by the ELDA web tool were compared using output of tests for pair-wise differences in active cell frequency between groups (33). A two-way ANOVA model was used for interaction analysis between two factors, e.g., gene knockdown and inhibiting agent. If not stated otherwise, all statistical analyses were performed using GraphPad Prism version 6.00 for MacOS X, GraphPad Software, La Jolla, CA, USA. All p-values were two-tailed. A p < 0.05 was considered significant, with asterisks marking the different levels of significance: ns, p ≥ 0.05; *p < 0.05; **p < 0.01. TCGA data analyses were performed in R (v3.6.1) (34). Survival analyses and ROC analyses were performed using the survival (35) and dataAnalysisMisc packages (36).
Results
LAPTM5 Inhibits Tumorigenicity
An in vivo screen for invasion-associated genes in glioblastoma (Figure 6A, further unpublished data) identified LAPTM5 as a highly anti-invasive gene in glioblastoma. Briefly, 47,400 transcripts by ~200,000 shRNA sequences were transduced in U87 MG cells to identify new invasion-associated genes. Here LAPTM5 was found to be downregulated in the invasive front of U87MG-derived orthotopically implanted tumors.
LAPTM5 was relevantly expressed in all glioblastoma cell lines and primary glioblastoma cells tested (Figure 1A). Methylation in silico analysis further revealed that LAPTM5 expression was negatively regulated by methylation of the CpG sites Cg10001720 and Cg12732155, which was substantiated by treatment with the demethylating agent 5-aza-2′-deoxycytidine. This treatment induced > 6-fold upregulation of LAPTM5 mRNA expression in primary glioblastoma cell lines (Supplemental Figure 2).
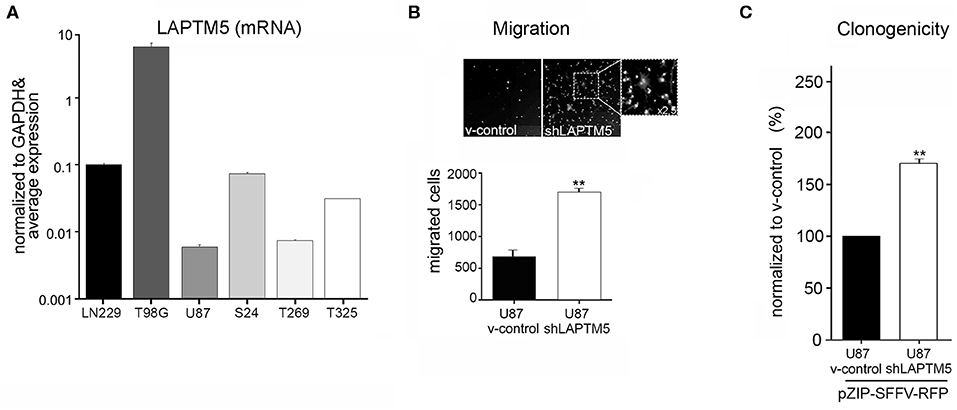
Figure 1. LAPTM5 acts as tumor suppressor gene in glioma. (A) LAPTM5 mRNA expression in glioblastoma cell lines (U87MG, LN229; T98G) and in primary glioma-initiating cells S24, T269, and T325 analyzed by quantitative real-time PCR and presented in relation to the average LAPTM5 expression in the tested cells. (B) LAPTM5 inhibits the invasion of U87MG cells. Representative image fields (×10 magnification) of migrated cells in Boyden Chamber invasion assay (upper figure) as well as total number of migrated cells (lower figure) of three independent experiments demonstrate a significantly reduced migration of LAPTM5 expressing (v-control) compared to LAPTM5 knockdown cells. (C) LAPTM5 inhibits the clonogenicity of U87MG cells. LAPTM5-knockdown U87MG cells show a significantly higher clonogenicity than the respective LAPTM5 expressing (v-control) cells. Figures represent the mean value of three independent experiments. *p < 0.05, **p < 0.01.
Silencing of LAPTM5 (Supplemental Figure 3A) confirmed its anti-invasive function in U87MG cells in vitro. The LAPTM5 knockdown (shLAPTM5) cells demonstrated significantly increased invasiveness by a factor of ~3 compared to the respective control cells (v-control) (Figure 1B). Additionally, LAPTM5 inhibited the clonogenicity of U87MG glioblastoma cells as LAPTM5 knockdown cells demonstrated a significantly higher clonogenic capacity compared to the respective vector control cells (Figure 1C). This pro-clonogenic phenotype of LAPTM5 knockdown cells was confirmed with two different knockdown constructs (Supplemental Figure 3B).
LAPTM5 Function Is CD40 Dependent
LAPTM5 was previously shown to be regulated by the CD40 receptor in immune cells and CD40 is highly expressed in up to 40% of glioblastoma (11). In order to identify the underlying molecular mechanisms of LAPTM5 function, we aimed to explore the CD40–LAPTM5 interaction in glioblastoma. For that, established glioblastoma cell lines were examined for membrane CD40 expression by flow cytometry, revealing only U87MG cells to be clearly positive for CD40 expression (Figure 2A). To test whether this is an artifact due to medium-related conditions or a problem of established cell lines in general, the glioma-initiating cells S24, T1, T269, T325, ZH305, ZH161, WJ, KNG002, MM, and PJ (22) were also tested for CD40 expression by flow cytometry but were all negative (data not shown).
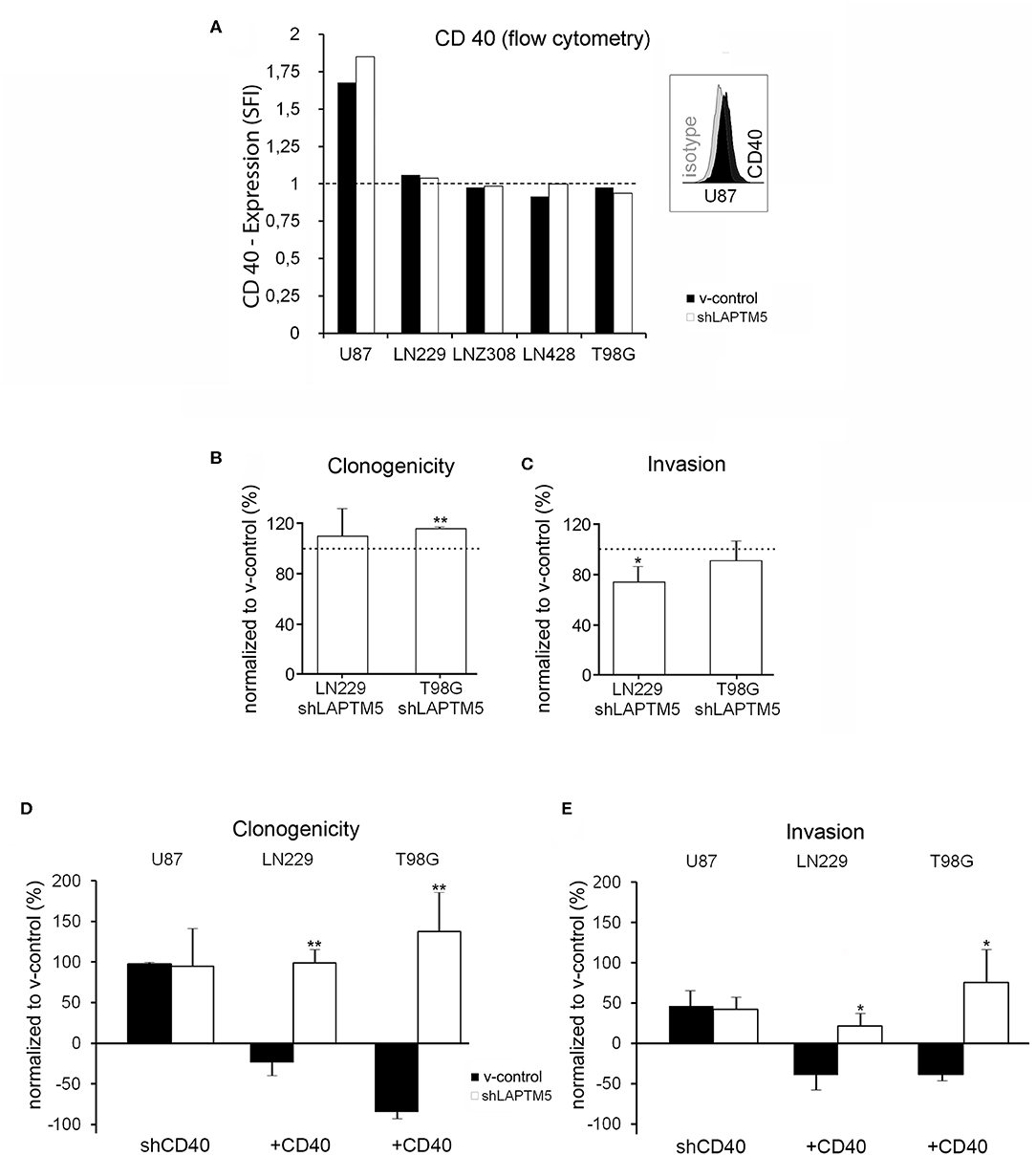
Figure 2. Tumor-suppressive functions of LAPTM5 were dependent on CD40 expression. (A) Flow cytometric analysis after CD40 surface staining illustrates that only U87MG v-control and shLAPTM5 cells are positive for CD40, whereas the other glioblastoma cell lines were negative for CD40 membrane expression and did not show a specific fluorescence index clearly >1 when dividing the mean fluorescence of the CD40 staining by the mean fluorescence of the respective isotype control. (B) Clonogenicity in the CD40-negative LN229 cells did not relevantly differ between v-control control and shLAPTM5 cells and was slightly higher in the CD40-negative T98G cells silenced for LAPTM5 compared to the respective v-control control cells. The graphs show the clonogenicity of shLAPTM5 normalized to the respective v-control control cells. (C) Knockdown of LAPTM5 led to a slightly reduced invasiveness of LN229 cells and did not relevantly alter the invasiveness in T98G cells compared to the respective LAPTM5-expressing (v-control) cells. The graphs visualize the invasiveness of shLAPTM5 cells normalized to the respective v-control control cells. (D,E) Graphs show the clonogenicity (D) and invasiveness (E) depending on CD40 expression status in relation to the respective v-control control cells. The dotted lines signify no change in relation to v-control control cells. The figures represent the mean value of three independent experiments. *p < 0.05, **p < 0.01. In CD40-positive U87MG cells, silencing of CD40 results in an equalized clonogenicity and invasiveness of v-control and LAPTM5 knockdown cells and thereby mirrors the phenotypes of the CD40-negative LN229 and T98G v-control and shLAPTM5 cells (B,C). Exogenous overexpression of CD40 in the CD40-negative LN229 and T98G cells results in a significant higher clonogenicity and invasiveness of LAPTM5 knockdown in relation to the respective v-control control cells and thereby confirms the results observed in the CD40-positive U87MG cells (demonstrated in Figures 1B,C).
Interestingly, in contrast to CD40-expressing U87MG cells, LAPTM5 hardly influenced the clonogenicity and the invasiveness in the CD40-negative LN229 and T98G cells. Clonogenicity was not enhanced in LAPTM5-depleted LN229 cells and only by ~15% in LAPTM5-knockdown T98G cells (Figure 2B). Invasiveness even had a tendency to be reduced in LAPTM5-knockdown LN229 and T98G cells (Figure 2C), suggesting that CD40 expression might be required for LAPTM5-mediated effects. Indeed the knockdown of CD40 in U87MG v-control and shLAPTM5 cells (Supplemental Figure 4A) resulted in enhanced clonogenicity by ~100% and invasiveness by ~50% in U87MG, but the difference between the control and the shLAPTM5 cells vanished (Figures 2D,E, left row) and thereby mirrored the situation of the CD40-negative LN229 and T98G cells (Figures 2B,C).
To further substantiate the CD40 dependency of LAPTM5-mediated anti-tumorigenic effects, CD40 was overexpressed in the CD40-negative LN229 and T98G v-control and shLAPTM5 cells (Supplemental Figures 4B,C). Indeed CD40 overexpression resulted in higher clonogenicity and enhanced invasiveness of shLAPTM5 LN229 (clonogenicity + ~100%; invasiveness + ~25%) and T98G cells (clonogenicity + ~140%; invasiveness + ~75%) compared to the respective v-control vector control cells which became less clonogenic and invasive (Figures 2D,E). These results were consistent with the effects observed in U87MG cells. In summary, LAPTM5-mediated anti-tumorigenic effects were dependent on CD40-expression: CD40 knockdown blocked and CD40 overexpression unleashed LAPTM5-mediated anti-tumorigenic effects in three different cell lines.
LAPTM5 Sensitizes to Temozolomide in CD40-Expressing Glioblastoma Cells
To examine the relevance of LAPTM5 in the mediation of treatment resistance to standard therapy for glioblastoma patients, the efficacy of temozolomide chemotherapy was further analyzed according to LAPTM5 status in CD40-expressing U87MG cells. The LAPTM5-expressing U87MG cells were more sensitive to temozolomide treatment compared to the LAPTM5 knockdown cells. Temozolomide reduced clonogenicity by 68% in v-control control cells but only by 23% in shLAPTM5 U87MG cells (Figure 3A).
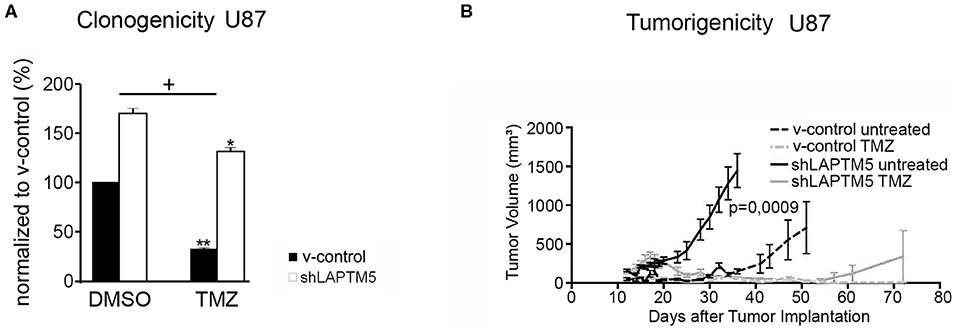
Figure 3. LAPTM5-sensitized CD40-positive U87MG cells to temozolomide chemotherapy. (A) LAPTM5 sensitizes U87MG cells to temozolomide treatment, whereas LAPTM5 knockdown cells were significantly more resistant against temozolomide chemotherapy. Figures elucidate the differences in clonogenicity in relation to v-control control cells treated with DMSO control. The asterisk shows a significant effect of the treatment on the cell line (*p < 0.05, **p < 0.01); the plus sign points out that the shLAPTM5 cells are more resistant to temozolomide compared with control cells as the difference in clonogenicity is more prominent after treatment as measured by ANOVA. (B) LAPTM5 relevantly delays the tumor growth of U87MG glioblastoma cells in vivo and sensitizes tumors to temozolomide treatment. The figure represents the changes in mean tumor volume of five mice per group over an observation time of 100 days.
These LAPTM5-mediated effects were further confirmed in vivo. LAPTM5 inhibited the tumorigenicity of implanted U87MG cells. The tumor volumes of shLAPTM5 tumors already exceeded the size of 500 mm3 after 27 days, which was taken as an undoubted evidence of tumor growth, whereas tumors with LAPTM5 expression did not reach this tumor size earlier than 56 days after tumor cell implantation. Furthermore, temozolomide treatment relevantly and sustainably inhibited tumor growth in LAPTM5-expressing tumors without any evidence for tumor recurrence within the observation time of 100 days. In contrast, tumor recurrence was observed in one out of five shLAPTM5 tumors after initial shrinkage due to temozolomide treatment (Figure 3B), supporting that LAPTM5 sensitized the CD40-positive U87MG tumor cells to temozolomide.
LAPTM5 Conveys Tumor-Suppressive Effects via Inhibition of CD40-Mediated NFκB Signaling
To understand the molecular mechanisms by which LAPTM5 inhibited invasion and tumorigenicity and sensitized to temozolomide treatment, a gene set enrichment analysis of whole-exome expression profiled U87MG cells was performed and compared with U87MG cells silenced for LAPTM5 expression. Here gene network and pathway analysis showed a strong upregulation of TNF-α signaling via the NFκB pathway in shLAPTM5 cells (Figure 4A; Supplemental Table 3), suggesting an inhibitory effect of LAPTM5 on NFκB signaling. An immunoblot analysis confirmed the activation of the NFκB pathway in shLAPTM5 U87MG cells, demonstrated by an increased expression of phosphorylated IκBα (Figure 4B). Of note is that CD40 expression was mandatory in inducing NFκB signaling in shLAPTM5 U87MG cells as pathway activation was no longer observed after the additional knockdown of CD40 in these cells (Figure 4B). On the functional level, JSH-23, a specific inhibitor of the NFκB pathway, particularly reduced the cell invasiveness in shLAPTM5 but did not alter the invasiveness of v-control U87MG cells, indicating that LAPTM5 knockdown promotes higher cell invasiveness via NFκB pathway activation (Figure 4C).
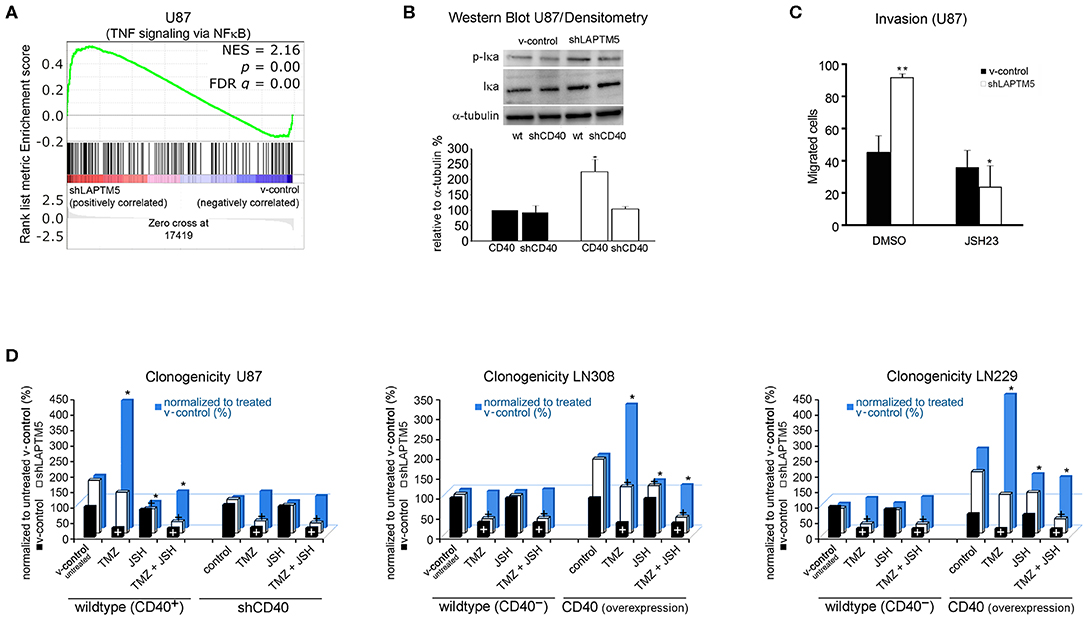
Figure 4. LAPTM5 conveys tumor-suppressive effects by inhibition of NFκB-pathway via CD40. (A) Gene set enrichment analysis revealed a significantly higher activation of TNF-α signaling via the NFκB pathway in LAPTM5 knockdown compared to v-control U87MG cells. (B) Immunoblot analysis confirmed the activation of NFκB pathway in shLAPTM5 cells as demonstrated by the higher phosphorylation of p-Iκa in LAPTM5 knockdown compared to the respective LAPTM5-expressing (v-control) U87MG cells. NFκB pathway activation could not be substantiated after additional knockdown of CD40 in shLAPTM5 and v-control U87MG cells. The figure demonstrates one representative immunoblot analysis (upper panel) and the quantification of immunoblot analysis performed with image J of three independent experiments (lower panel). The asterisk signifies that shLAPTM5 wild type has a significantly higher p-Iκa-level than v-control wild-type cells (*p < 0.05). (C) The functional relevance of NFκB activation in LAPTM5 knockdown cells was further substantiated by treatment with the NFκB inhibitor JSH-23, which specifically reduced the increased invasiveness of LAPTM5 knockdown cells, whereas it did not relevantly alter the invasiveness in v-control cells (*p < 0.05, ** p < 0.01). (D) The graphs demonstrate the effects of temozolomide treatment, NFκB inhibition with JSH-23, and the combination of both treatments depending on the CD40 expression status of the respective cell lines. For that, CD40 was silenced in the CD40-positive U87MG cells or exogenously overexpressed in the CD40-negative LN229 and LN308 cell lines, and clonogenicity was analyzed after the respective treatments. The clonogenicity of v-control (white columns) and shLAPTM5 (black columns) cells are demonstrated in relation to the v-control wild-type cells treated with DMSO control. The plus sign signifies that there was a significant therapy effect measured by ANOVA compared with DMSO (untreated) control (at least p < 0.05). The blue bars show the ratio of shLAPTM5 relative to v-control cells for every single treatment (black in relation to white column for each treatment) to compare and illustrate the efficacy of each therapy in shLAPTM5 and control cells; the asterisk points out that the shLAPTM5 cells react differently from v-control cells when compared to the ratio shLAPTM5/v-control in DMSO control treatment as measured by ANOVA (at least p < 0.05). The LAPTM5 knockdown led to a higher clonogenicity in CD40-positive glioblastoma cells and to a higher resistance against temozolomide treatment. The LAPTM5 knockdown cells were particularly sensitive to NFκB inhibition by JSH-23 in CD40-positive glioblastoma cells. In addition, NFκB inhibition re-sensitized the resistant LAPTM5 knockdown cells to treatment with temozolomide. In the CD40-negative glioblastoma cells, LN229 and LN308 as well as U87MG cells silenced for CD40; no differences were observed between the respective v-control and LAPTM5 knockdown cells, and JSH-23 had no effect on clonogenicity.
Similarly, only U87MG LAPTM5 knockdown cells were highly sensitive to treatment with the NFκB inhibitor JSH-23, resulting in a significant inhibition of clonogenicity, which was not demonstrated in control cells (Figure 4D, left panel).
As LAPTM5 was shown to sensitize CD40-positive U87MG glioblastoma cells to temozolomide and inhibited the NFκB pathway, the functional role of potential NFκB-mediated temozolomide resistance in LAPTM5 knockdown cells as well as the connection with CD40 expression was further investigated.
Consistent with the previous data (Figure 3A), temozolomide relevantly reduced clonogenicity in v-control U87MG cells, whereas shLAPTM5 cells were more resistant against temozolomide treatment. Moreover, NFκB inhibition specifically sensitized shLAPTM5 U87MG cells to temozolomide treatment, resulting in a comparable inhibitory efficacy of temozolomide on clonogenicity in LAPTM5 knockdown and v-control control cells (Figure 4D, left panel).
Importantly, the differences in response to temozolomide and JSH-23 treatment vanished after knockdown of CD40 in U87MG v-control and shLAPTM5 cells. After CD40 knockdown, not only the higher clonogenicity but also the temozolomide resistance effect of the LAPTM5 knockdown cells could not be observed anymore. Furthermore, no relevant response to the inhibition of the NFκB pathway with JSH-23 treatment was seen in LAPTM5 and CD40 double-knockdown cells (Figure 4D, left panel).
Aiming to exclude potential cell line artifacts, CD40-negative LN229 as well as LN308 control and shLAPTM5 cells were further examined with respect to responsiveness to temozolomide and NFκB inhibition. Consistent with the data in CD40-silenced U87MG cells, the knockdown of LAPTM5 did not confer resistance against temozolomide. Moreover, NFκB inhibition had no relevant effect on shLAPTM5 or v-control control cells regardless of co-treatment with temozolomide. However, the overexpression of CD40 induced a marked resistance against temozolomide in LN229 and LN308 shLAPTM5 cells, but not in v-control control cells. Of note is that again only CD40-positive shLAPTM5 cells showed relevant sensitivity to NFκB inhibition, which was able to level the shLAPTM5-induced temozolomide resistance (Figure 4D, middle and right panel). In conclusion, these data demonstrate that LAPTM5 mediates the sensitivity to temozolomide by the inhibition of CD40-induced NFκB pathway activation in CD40-positive glioblastoma cells.
The Prognostic Value of LAPTM5 Is Dependent on CD40 Abundance
To validate our hypothesis that LAPTM5 acts as a potential tumor suppressor in CD40 high-expressing tumors, we evaluated its prognostic value in the TCGA-GBM cohort (Figure 5A). Methylation array data were used to identify similar patients based on associations between methylation patterns and CD40 expression (Figures 5B–D), which were then used to define reasonable cutoffs for CD40 expression based on a ROC analysis (Figure 5F). For the cohort with both methylation and RNASeq data, a survival analysis showed a significant interaction term between the methylation-derived CD40 cluster and LAPTM5 expression: increasing expression leads to a risk decrease in CD40 tumors in contrast to CD40 low tumors (Figure 5E). Next, the complete RNASeq cohort was evaluated by utilizing the previously derived classification into CD40 low/high tumors. Importantly, neither CD40 nor LAPTM5 expression alone was able to prognostically separate the patients (Figure 5G, Supplemental Figure 5). In CD40 high tumors, a higher LAPTM5 expression is associated with better prognosis, while it is associated with a worse prognosis in CD40 low-expressing tumors (Figure 5I, Supplemental Figure 5). To exclude confounding of survival analysis by immune effects, CD40 and LAPTM5 expression was correlated with the signature of different immune cells. Here higher LAPTM5 expression was associated with a higher amount of M2-polarized macrophage levels as compared to LAPTM5 low tumors in CD40 high tumors analyzed by CIBERSORT (32) (Figure 5H).
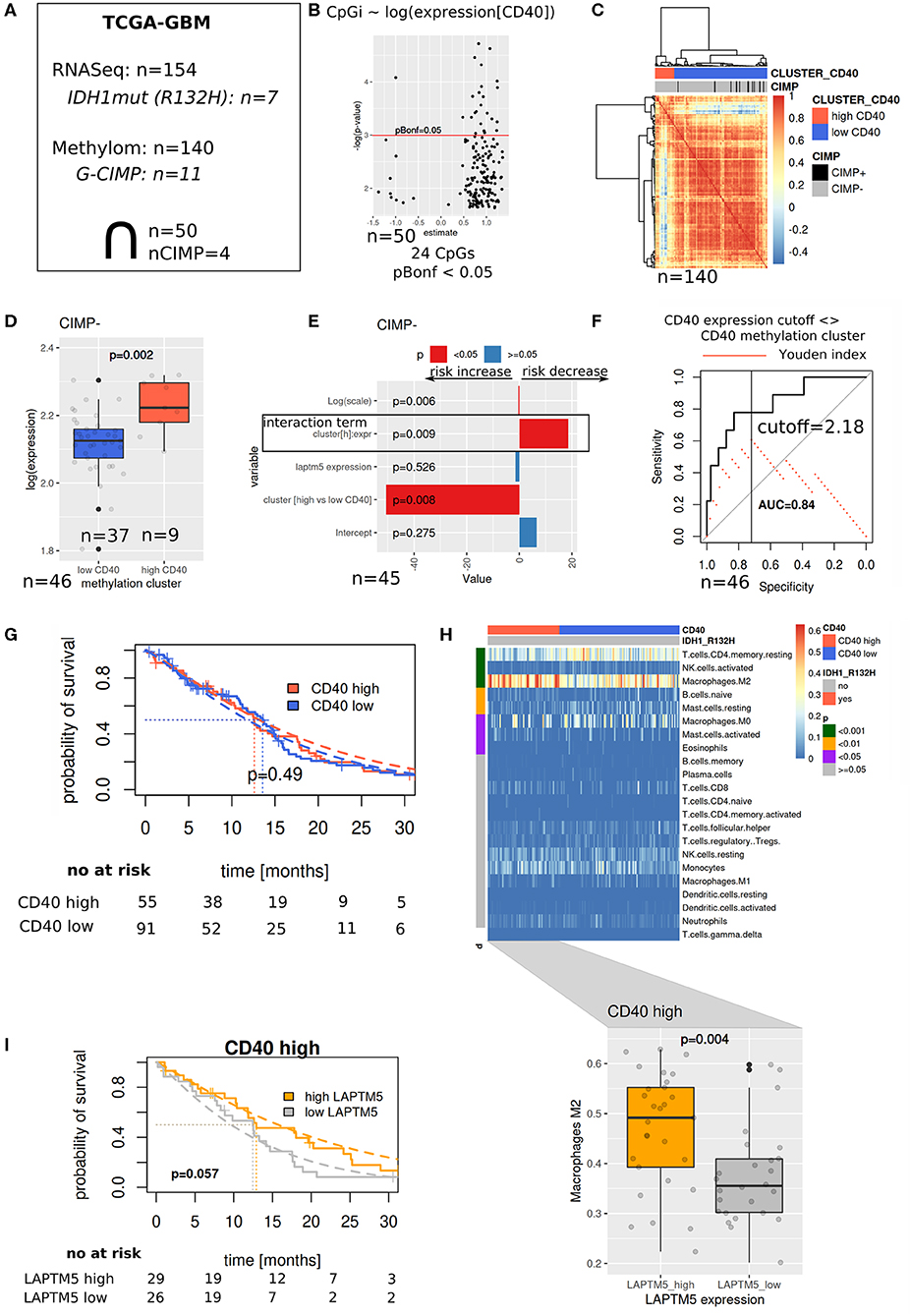
Figure 5. The prognostic value of LAPTM5 is dependent on CD40 abundance. (A) Evaluated The Cancer Genome Atlas (TCGA)-glioblastoma multiforme methylome (450 k array) and RNASeq datasets. (B) Associations between CD40 expression and methylation array probes (linear model) with Bonferroni adjusted p < 0.05. (C) Patient similarity (Pearson correlation matrix) based on CpGs selected in (B) and used for CD40 cutoff identification in the complete TCGA methylome (450 k) cohort. (D) CD40 and expression in methylation derived clusters from (C). (E) Survival analysis of overlapping samples (parametric survival regression, Weibull distribution). (F) Cutoff selection of CD40 expression for the separation of clusters. (G) Kaplan–Meier survival curve for CD40 expression data with the applied cutoff from (F). (H) CIBERSORT estimated immune cell fractions for the RNASeq cohort (upper part, p-values: Wilcoxon test) and M2 fractions on CD40 high tumors (lower part). (I) Prognostic value of LAPTM5 in CD40 high samples (p-value: parametric survival regression).
Discussion
This study describes the inhibiting effects of LAPTM5 on tumorigenicity in CD40-positive glioblastoma and its role as a negative regulator of CD40-mediated NFκB signaling. LAPTM5 relevantly inhibits the clonogenicity and the invasiveness of CD40-positive glioblastoma cells. In addition, as shown in vitro and in vivo, LAPTM5 sensitizes CD40-positive glioblastoma cells to temozolomide treatment, the standard chemotherapy for glioblastoma patients. These LAPTM5-induced tumor-suppressing effects are mediated by the inhibition of the CD40-dependent NFκB pathway activation. Finally, a TCGA analysis revealed that a higher expression of LAPTM5 has a positive effect on survival in CD40-positive glioblastoma patients.
Previous studies in other tumor entities already suggested the potential role of LAPTM5 in tumorigenesis (8). However, the role of LAPTM5 had not been assessed in the prototypical invasive tumor glioblastoma so far. We here demonstrate that LAPTM5 inhibits tumorigenicity in glioblastoma based on functional in vitro and in vivo experiments and in silico analysis. Importantly, we show that the observed LAPTM5-mediated tumor-suppressive and temozolomide-sensitizing effects are exclusively observed in CD40-expressing glioblastoma cells, indicating an intact CD40–LAPTM5 axis as a prerequisite for the tumor-suppressive activity of LAPTM5. Silencing of CD40 in CD40-positive U87MG cells abolished the pro-tumorigenic and temozolomide-resistant properties of LAPTM5 knockdown cells, whereas CD40 overexpression in primarily CD40-negative glioblastoma cells recreated the aggressive phenotype and temozolomide resistance of LAPTM5 knockdown cells. An interaction between LAPTM5 and the CD40 receptor was previously shown in B cells (11). Here, for the first time, we could demonstrate the link between CD40 and LAPTM5 in glioblastoma and characterize the role of the CD40–LAPTM5 axis in tumorigenicity.
CD40 represents an emerging immune-modulating target in cancer treatment (12). In glioblastoma, CD40-based immunotherapy showed an inhibitory effect on tumor growth in preclinical glioblastoma models in vivo (21). Beyond these immune-modulating effects, CD40 signaling was shown to be involved in apoptosis and the neovascularization of glioma cells (37, 38). In this study, we demonstrated that CD40 signaling plays a key role for LAPTM5-mediated effects on invasion, clonogenicity, and sensitivity to temozolomide treatment. A further pathway analysis demonstrated that LAPTM5 mediates these tumor-suppressing and temozolomide-sensitizing effects by the inhibition of CD40-induced NFκB pathway activation. The activation of the NFκB pathway by CD40 signaling has been previously reported in B cells and is consistent with our data (39, 40). Our results indicate that CD40 activates the NFκB pathway in glioblastoma cells only in the absence of LAPTM5 expression, leading to enhanced tumorigenicity and resistance against temozolomide. As CD40-mediated NFκB activation seems to be required for specific LAPTM5-mediated effects, these molecular mechanisms also explain the absence of LAPTM5-dependent effects in CD40-negative glioblastoma cells as summarized in Figure 6. In addition, the results are in line with previous studies supporting the role of NFκB pathway activation in promoting glioblastoma cell invasiveness and resistance to alkylating chemotherapies (41, 42).
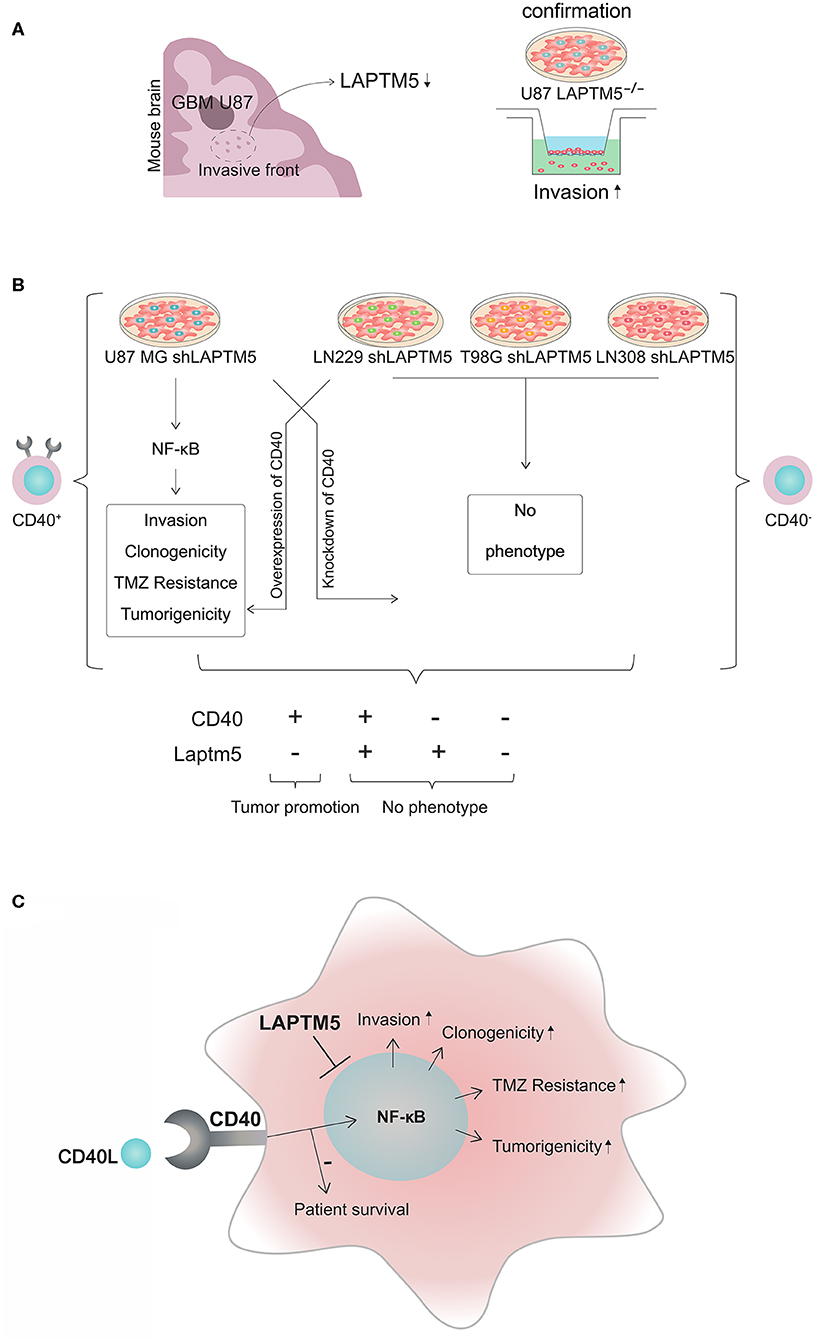
Figure 6. CD40–LAPTM5 crosstalk in glioblastoma invasion and temozolomide resistance. (A) LAPTM5 was found to be downregulated in the invasive front of U87MG cells in an in vivo screen for invasion-associated genes. A total of 47,400 transcripts by ~200,000 shRNA sequences were transduced in U87MG cells to identify new invasion-associated genes. In vitro experiments confirmed that LAPTM5-knockdown U87MG cells were significantly more invasive than the respective vector control cells. (B) Overview about CD40-dependent LAPTM5-mediated tumor-suppressive effects. LAPTM5 knockdown resulted in the activation of NFkB pathway in CD40-positive U87MG cells, leading to increased invasion, clonogenicity, tumorigenicity, and temozolomide resistance, which were no longer observed after additional knockdown of CD40 in these cells. Accordingly, the LAPTM5 knockdown cells had no different phenotype compared to the respective LAPTM5-expressing control cells in CD40-negative LN229, T98G, and LN308 cells. Overexpression of CD40 in primarily CD40-negative cells recreated the pro-tumorigenic phenotype and temozolomide resistance of LAPTM5 knockdown cells. (C) Graphic illustration of the CD40–LAPTM5 crosstalk and its effects in tumorigenicity and temozolomide resistance in glioblastoma. TMZ, temozolomide.
Personalized therapies based on molecular alterations are increasingly in the focus of neuro-oncological treatment strategies requiring optimal patient selection guided by biomarkers for effective treatment allocation. In this regard, the presented data reasonably suggest LAPTM5 expression status as a potential biomarker for sensitivity to temozolomide treatment in CD40-positive glioblastoma.
It has been shown that most high-grade gliomas express CD40 with an observed high CD40 expression in about 40% of the tumor samples in patient-derived tumor tissue (37). Although the screening of our glioblastoma cell lines and primary glioblastoma-initiating cell lines revealed only U87MG glioblastoma cells to be positive for CD40 membrane expression, which is in line with previously published data and might be due to a cell culture artifact (37), the expression data of the tumor tissue suggest that CD40 signaling might be relevant for a large subgroup of patients with high-grade glioma. Furthermore, we demonstrated that high LAPTM5 expression is associated with improved overall survival in CD40 high-expressing glioblastoma. As CD40 is known to be involved in several immune effects, we excluded that the observed association of CD40 high-expressing and LAPTM5 high-expressing gliomas with better prognosis is related to differences in the signature of immune cells. Here we only found a stronger gene signature for M2-polarized macrophages which are known to be associated with even a worse prognosis (43, 44). Therefore, the positive prognostic value of high CD40 and LAPTM5 expression is attributed to the tumor tissue and not to the immune cells. Previous studies demonstrated contradicting results about the correlation of CD40 expression with the survival of glioma patients (20, 21). This might be explained by the differences of LAPTM5 expression in the tumors which were not analyzed in these studies. Of note is that the in silico analysis did not reveal a relevant association of CD40 or LAPTM5 alone with overall survival.
In addition, our data indicate that enhancing CD40 signaling might even have undesirable pro-tumorigenic effects in LAPTM5-negative tumors or parts of the tumors such as the invasive front where low LAPTM5-expressing cells might cluster due to the pro-invasive phenotype. Here CD40 induction might lead to a further activation of the NFκB pathway and thereby to resistance toward temozolomide treatment and fostering the aggressiveness of gliomas.
In conclusion, we identified LAPTM5 as a negative regulator of CD40-mediated NFκB signaling in glioblastoma. CD40 activated the NFκB pathway in cells depleted for LAPTM5 expression, promoting tumor growth and resistance to temozolomide, which could be overcome by NFκB inhibition. The in silico analysis revealed that CD40 high-expressing and LAPMT5 high-expressing gliomas had a better prognosis, which was attributed to the tumor tissue and not to the immune cells. Based on patient survival data and functional in vitro and in vivo experiments, we concluded that LAPTM5 acts as a tumor suppressor in CD40-positive gliomas (Figure 6). In addition, we demonstrated an important role of CD40 in LAPTM5-mediated effects on tumorigenicity and sensitivity to alkylating chemotherapy beyond the known immune-modulating effects of CD40. Therefore, the data indicated that the focus of CD40-stimulating anti-tumor therapies should not exclusively be on the immune compartment.
As CD40 expression was frequently detected in glioblastoma tissue samples and is a prerequisite for the tumor-suppressive activity of LAPTM5, these data provide relevant molecular insights into a potentially new mechanism of resistance against temozolomide in a relevant proportion of glioblastoma patients. Furthermore, based on the presented results, the LAPTM5 expression status provides a potential new biomarker for response to temozolomide treatment in CD40-positive glioblastoma, which merits further evaluation in clinical practice.
Data Availability Statement
The datasets generated for this study can be found in the Experiment ArrayExpress accession is E-MTAB-6316.
Ethics Statement
The animal study was reviewed and approved by Regierungspräsidium Karlsruhe, Karlsruhe, Germany.
Author Contributions
AB, FB, TK, ZD, and DL provided substantial contributions to the conception, design, acquisition, analysis, and interpretation of data, drafting of the article or revising it critically for important intellectual content, and final approval of the version to be published. ZT provided substantial contributions to the acquisition, analysis, and interpretation of data, and final approval of the version to be published. MK and BW provided substantial contributions to the acquisition of data, analysis of biostatistical data, interpretation of data, and final approval of the version to be published. SP and NH provided substantial contributions to the acquisition, analysis, and interpretation of data and final approval of the version to be published. FW and MP drafted the article or revised it critically for important intellectual content and gave final approval of the version to be published. WW and AA provided substantial contributions to the conception and design, analysis and interpretation of data, drafting of the article or revising it critically for important intellectual content, and final approval of the version to be published.
Funding
The German Cancer Aid (Deutsche Krebshilfe, Molecular classification of anaplastic gliomas in the NOA-04 trial, project 110624) provided support to WW and by DFG SFB 1389 TP A03 to WW and TK. The publication fee was supported by Ruprecht-Karls-Universität Heidelberg.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We want to thank the microarray unit of the DKFZ Genomics and Proteomics Core Facility for providing the Illumina Whole-Genome Expression Beadchips and related services. We thank M. Weller for providing us the pcDNA3 CD40 plasmid. We want to thank Katrin Rein for her support in the design of Supplemental Figure 1 and Figure 6.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.00747/full#supplementary-material
Abbreviations
LAPTM5, lysosomal protein transmembrane 5; CD40, cluster of differentiation 40; NFκB, nuclear factor (NF)κB; GIC, glioblastoma-initiating cells; qRT-PCR, quantitative real-time PCR; GSEA, gene set enrichment analysis; LDA, limiting dilution assay; SFI, specific fluorescence intensity; DFS, disease-free survival; TNF-α, tumor necrosis factor alpha.
References
1. Osswald M, Jung E, Sahm F, Solecki G, Venkataramani V, Blaes J, et al. Brain tumour cells interconnect to a functional and resistant network. Nature. (2015) 528:93–8. doi: 10.1038/nature16071
2. Weller M, van den Bent M, Hopkins K, Tonn JC, Stupp R, Falini A, et al. EANO guideline for the diagnosis and treatment of anaplastic gliomas and glioblastoma. Lancet Oncol. (2014) 15:e395–403. doi: 10.1016/S1470-2045(14)70011-7
3. Giese A, Bjerkvig R, Berens ME, Westphal M. Cost of migration: invasion of malignant gliomas and implications for treatment. J Clin Oncol. (2003) 21:1624–36. doi: 10.1200/JCO.2003.05.063
4. Paw I, Carpenter RC, Watabe K, Debinski W, Lo HW. Mechanisms regulating glioma invasion. Cancer Lett. (2015) 362:1–7. doi: 10.1016/j.canlet.2015.03.015
5. Rao JS. Molecular mechanisms of glioma invasiveness: the role of proteases. Nat Rev Cancer. (2003) 3:489–501. doi: 10.1038/nrc1121
6. Cuddapah VA, Robel S, Watkins S, Sontheimer H. A neurocentric perspective on glioma invasion. Nat Rev Neurosci. (2014) 15:455–65. doi: 10.1038/nrn3765
7. Adra CN, Zhu S, Ko JL, Guillemot JC, Cuervo AM, Kobayashi H, et al. LAPTM5: a novel lysosomal-associated multispanning membrane protein preferentially expressed in hematopoietic cells. Genomics. (1996) 35:328–37. doi: 10.1006/geno.1996.0364
8. Nuylan M, Kawano T, Inazawa J, Inoue J. Down-regulation of LAPTM5 in human cancer cells. Oncotarget. (2016) 7:28320–8. doi: 10.18632/oncotarget.8614
9. Inoue J, Misawa A, Tanaka Y, Ichinose S, Sugino Y, Hosoi H, et al. Lysosomal-associated protein multispanning transmembrane 5 gene (LAPTM5) is associated with spontaneous regression of neuroblastomas. PLoS One. (2009) 4:e7099. doi: 10.1371/journal.pone.0007099
10. Jun DY, Kim H, Jang WY, Lee JY, Fukui K, Kim YH. Ectopic overexpression of LAPTM5 results in lysosomal targeting and induces Mcl-1 down-regulation, bak activation, and mitochondria-dependent apoptosis in human HeLa cells. PLoS One. (2017) 12:e0176544. doi: 10.1371/journal.pone.0176544
11. Seimiya M, O-Wang J, Bahar R, Kawamura K, Wang Y, Saisho H, et al. Stage-specific expression of Clast6/E3/LAPTM5 during B cell differentiation: elevated expression in human B lymphomas. Int J Oncol. (2003) 22:301–4. doi: 10.3892/ijo.22.2.301
12. Piechutta M, Berghoff AS. New emerging targets in cancer immunotherapy: the role of cluster of differentiation 40 (CD40/TNFR5). ESMO Open. (2019) 4(Suppl. 3):e000510. doi: 10.1136/esmoopen-2019-000510
13. van den Oord JJ, Maes A, Stas M, Nuyts J, Battocchio S, Kasran A, et al. CD40 is a prognostic marker in primary cutaneous malignant melanoma. Am J Pathol. (1996) 149:1953–61.
14. Slobodova Z, Ehrmann J, Krejci V, Zapletalova J, Melichar B. Analysis of CD40 expression in breast cancer and its relation to clinicopathological characteristics. Neoplasma. (2011) 58:189–97. doi: 10.4149/neo_2011_03_189
15. Wu Y, Wang L, He X, Xu H, Zhou L, Zhao F, et al. Expression of CD40 and growth-inhibitory activity of CD40 ligand in colon cancer ex vivo. Cell Immunol. (2008) 253:102–9. doi: 10.1016/j.cellimm.2008.05.005
16. Ishikawa K, Miyamoto M, Yoshioka T, Kato T, Kaji M, Ohbuchi T, et al. Up-regulation of CD40 with juxtacrine activity in human nonsmall lung cancer cells correlates with poor prognosis. Cancer. (2008) 113:530–41. doi: 10.1002/cncr.23618
17. Cooke PW, James ND, Ganesan R, Wallace M, Burton A, Young LS. CD40 expression in bladder cancer. J Pathol. (1999) 188:38–43. doi: 10.1002/(SICI)1096-9896(199905)188:1<38::AID-PATH315>3.0.CO;2-B
18. Uckun FM, Gajl-Peczalska K, Myers DE, Jaszcz W, Haissig S, Ledbetter JA. Temporal association of CD40 antigen expression with discrete stages of human B-cell ontogeny and the efficacy of anti-CD40 immunotoxins against clonogenic B-lineage acute lymphoblastic leukemia as well as B-lineage non-Hodgkin's lymphoma cells. Blood. (1990) 76:2449–56. doi: 10.1182/blood.V76.12.2449.bloodjournal76122449
19. Matsumura Y, Hiraoka K, Ishikawa K, Shoji Y, Noji T, Hontani K, et al. CD40 expression in human esophageal squamous cell Carcinoma is associated with tumor progression and lymph node metastasis. Anticancer Res. (2016). 36:4467–76. doi: 10.21873/anticanres.10991
20. Werner J-M, Kuhl S, Ulrich K, Krischek B, Stavrinou P, Goldbrunne R, et al. Expression of CD40 correlates negatively with overall and progression-free survival of low- and high-grade gliomas. World Neurosurg. (2019) 130:e17–25. doi: 10.1016/j.wneu.2019.05.112
21. Chonan M, Saito R, Shoji T, Shibahara I, Kanamori M, Sonoda Y, et al. CD40/CD40L expression correlates with the survival of patients with glioblastomas and an augmentation in CD40 signaling enhances the efficacy of vaccinations against glioma models. Neuro Oncol. (2015) 17:1453–62. doi: 10.1093/neuonc/nov090
22. Lemke D, Weiler M, Blaes J, Wiestler B, Jestaedt L, Klein AC, et al. Primary glioblastoma cultures: can profiling of stem cell markers predict radiotherapy sensitivity? J Neurochem. (2014) 131:251–64. doi: 10.1111/jnc.12802
23. Castro F, Dirks WG, Fähnrich S, Hotz-Wagenblatt A, Pawlita M, Schmitt M. High-throughput SNP-based authentication of human cell lines. Int J Cancer. (2013) 132:308–14. doi: 10.1002/ijc.27675
24. Tönjes M, Barbus S, Park YJ, Wang W, Schlotter M, Lindroth AM, et al. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat Med. (2013) 19:901–8. doi: 10.1038/nm.3217
25. Eliopoulos AG, Young LS. Activation of the cJun N-terminal kinase (JNK) pathway by the epstein-Barr virus-encoded latent membrane protein 1 (LMP1). Oncogene. (1998) 16:1731–42. doi: 10.1038/sj.onc.1201694
26. Hertenstein A, Schumacher T, Litzenburger U, Opitz CA, Falk CS, Serafini T, et al. Suppression of human CD4+ T cell activation by 3,4-dimethoxycinnamonyl-anthranilic acid (tranilast) is mediated by CXCL9 and CXCL10. Biochem Pharmacol. (2011) 82:632–41. doi: 10.1016/j.bcp.2011.06.013
27. Lemke D, Pledl H-W, Zorn M, Jugold M, Green E, Blaes J, et al. Slowing down glioblastoma progression in mice by running or the anti-malarial drug dihydroartemisinin? Induction of oxidative stress in murine glioblastoma therapy. Oncotarget. (2016) 7:56713–25. doi: 10.18632/oncotarget.10723
28. Kessler T, Sahm F, Blaes J, Osswald M, Rübmann P, Milford D, et al. Glioma cell VEGFR-2 confers resistance to chemotherapeutic and antiangiogenic treatments in PTEN-deficient glioblastoma. Oncotarget. (2015) 6:31050–68. doi: 10.18632/oncotarget.2910
29. Lemke D, Pfenning PN, Sahm F, Klein A-C, Kempf T, Warnken U, et al. Costimulatory protein 4IgB7H3 drives the malignant phenotype of glioblastoma by mediating immune escape and invasiveness. Clin Cancer Res. (2012) 18:105–17. doi: 10.1158/1078-0432.CCR-11-0880
30. Eirew P, Stingl J, Raouf A, et al. A method for quantifying normal human mammary epithelial stem cells with in vivo regenerative ability. Nat Med. (2008) 14:1384–9. doi: 10.1038/nm.1791
31. Opitz CA, Litzenburger UM, Sahm F, Turashvili G, Aparicio S, Emerman JT, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. (2011) 478:197–203. doi: 10.1038/nature10491
32. Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. (2015) 12:453–7. doi: 10.1038/nmeth.3337
33. Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. (2009) 347:70–8. doi: 10.1016/j.jim.2009.06.008
34. Team RC. A language and environment for statistical computing. Computing. 1. doi: 10.1890/0012-9658(2002)083[3097:CFHIWS]2.0.CO;2
35. Therneau TM, Grambsch PM. Modeling Survival Data: Extending the Cox Model. New York, NY: Springer (2000). doi: 10.1007/978-1-4757-3294-8
36. Paterson I, Doughty VA, McLeod MD, Trieselmann T. dataAnalysisMisc: Collection of Functions for Daily Tasks. R package version 0.99.6.
37. Wischhusen J, Schneider D, Mittelbronn M, Meyermann R, Engelmann H, Jung G, et al. Death receptor-mediated apoptosis in human malignant glioma cells: modulation by the CD40/CD40L system. J Neuroimmunol. (2005) 162:28–42. doi: 10.1016/j.jneuroim.2005.01.005
38. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. CD40 is a regulator for vascular endothelial growth factor in the tumor microenvironment of glioma. J Neuroimmunol. (2010) 222:62–9. doi: 10.1016/j.jneuroim.2009.12.004
39. Annunziata CM, Safiran YJ, Irving SG, Kasid UN, Cossman J. Hodgkin disease: pharmacologic intervention of the CD40-NF kappa B pathway by a protease inhibitor. Blood. (2000) 96:2841–8. doi: 10.1182/blood.V96.8.2841.h8002841_2841_2848
40. Chen D, Ireland SJ, Remington G, Alvarez E, Racke MK, Greenberg B, et al. CD40-mediated NF-κB activation in B cells is increased in multiple sclerosis and modulated by therapeutics. J Immunol. (2016) 197:4257–65. doi: 10.4049/jimmunol.1600782
41. Westhoff M-A, Zhou S, Nonnenmacher L, Karpel-Massler G, Jennewein C, Schneider M, et al. Inhibition of NF-κB signaling ablates the invasive phenotype of glioblastoma. Mol Cancer Res. (2013) 11:1611–23. doi: 10.1158/1541-7786.MCR-13-0435-T
42. Puliyappadamba VT, Hatanpaa KJ, Chakraborty S, Habib AA. The role of NF-κB in the pathogenesis of glioma. Mol Cell Oncol. (2014) 1:e963478. doi: 10.4161/23723548.2014.963478
43. Geisenberger C, Mock A, Warta R, Rapp C, Schwager C, Korshunov A, et al. Molecular profiling of long-term survivors identifies a subgroup of glioblastoma characterized by chromosome 19/20 co-gain. Acta Neuropathol. (2015) 130:419–34. doi: 10.1007/s00401-015-1427-y
Keywords: LAPTM5, CD40, NFκB, temozolomide, glioblastoma
Citation: Berberich A, Bartels F, Tang Z, Knoll M, Pusch S, Hucke N, Kessler T, Dong Z, Wiestler B, Winkler F, Platten M, Wick W, Abdollahi A and Lemke D (2020) LAPTM5–CD40 Crosstalk in Glioblastoma Invasion and Temozolomide Resistance. Front. Oncol. 10:747. doi: 10.3389/fonc.2020.00747
Received: 09 January 2020; Accepted: 20 April 2020;
Published: 05 June 2020.
Edited by:
Massimo Nabissi, University of Camerino, ItalyReviewed by:
Maite Verreault, INSERM U1127 Institut du Cerveau et de la Moelle épinière (ICM), FranceNhan Le Tran, Mayo Clinic, United States
Copyright © 2020 Berberich, Bartels, Tang, Knoll, Pusch, Hucke, Kessler, Dong, Wiestler, Winkler, Platten, Wick, Abdollahi and Lemke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dieter Lemke, d.lemke@dkfz.de; dieter.lemke@med.uni-heidelberg.de
†These authors have contributed equally to this work