 Georgios Efthymiou1
Georgios Efthymiou1 Angélique Saint1,2†
Angélique Saint1,2† Michaël Ruff1†
Michaël Ruff1† Zeinab Rekad1
Zeinab Rekad1 Delphine Ciais1
Delphine Ciais1 Ellen Van Obberghen-Schilling1,2*
Ellen Van Obberghen-Schilling1,2*- 1Université Côte d'Azur, CNRS, INSERM, iBV, Nice, France
- 2Centre Antoine Lacassagne, Nice, France
Normal tissue homeostasis and architecture restrain tumor growth. Thus, for a tumor to develop and spread, malignant cells must overcome growth-repressive inputs from surrounding tissue and escape immune surveillance mechanisms that curb cancer progression. This is achieved by promoting the conversion of a physiological microenvironment to a pro-tumoral state and it requires a constant dialog between malignant cells and ostensibly normal cells of adjacent tissue. Pro-tumoral reprogramming of the stroma is accompanied by an upregulation of certain extracellular matrix (ECM) proteins and their cognate receptors. Fibronectin (FN) is one such component of the tumor matrisome. This large multidomain glycoprotein dimer expressed over a wide range of human cancers is assembled by cell-driven forces into a fibrillar array that provides an obligate scaffold for the deposition of other matrix proteins and binding sites for functionalization by soluble factors in the tumor microenvironment. Encoded by a single gene, FN regulates the proliferation, motile behavior and fate of multiple cell types, largely through mechanisms that involve integrin-mediated signaling. These processes are coordinated by distinct isoforms of FN, collectively known as cellular FN (as opposed to circulating plasma FN) that arise through alternative splicing of the FN1 gene. Cellular FN isoforms differ in their solubility, receptor binding ability and spatiotemporal expression, and functions that have yet to be fully defined. FN induction at tumor sites constitutes an important step in the acquisition of biological capabilities required for several cancer hallmarks such as sustaining proliferative signaling, promoting angiogenesis, facilitating invasion and metastasis, modulating growth suppressor activity and regulating anti-tumoral immunity. In this review, we will first provide an overview of ECM reprogramming through tumor-stroma crosstalk, then focus on the role of cellular FN in tumor progression with respect to these hallmarks. Last, we will discuss the impact of dysregulated ECM on clinical efficacy of classical (radio-/chemo-) therapies and emerging treatments that target immune checkpoints and explore how our expanding knowledge of the tumor ECM and the central role of FN can be leveraged for therapeutic benefit.
Introduction
Historically, studies addressing the genesis and progression of cancer have focused on the genotype of tumor cells. In the case of carcinomas, nascently transformed epithelial cells progress to an invasive phenotype by the accumulation of mutations. However, this is only part of the story, as schematized in Figure 1. Tumor progression and expansion are accompanied by major changes in the tissue immediately adjacent to premalignant lesions and require reciprocal interactions between malignant epithelial cells and stromal cells. Tumor-induced alterations in the reactive stroma involving modifications in ECM composition, organization, and physical properties, have drawn increasing attention over the past few years as a more holistic view of tumor progression, complexity and heterogeneity of tumor microenvironment (TME) is being embraced and scrutinized for the discovery of novel, clinically relevant therapeutic opportunities.
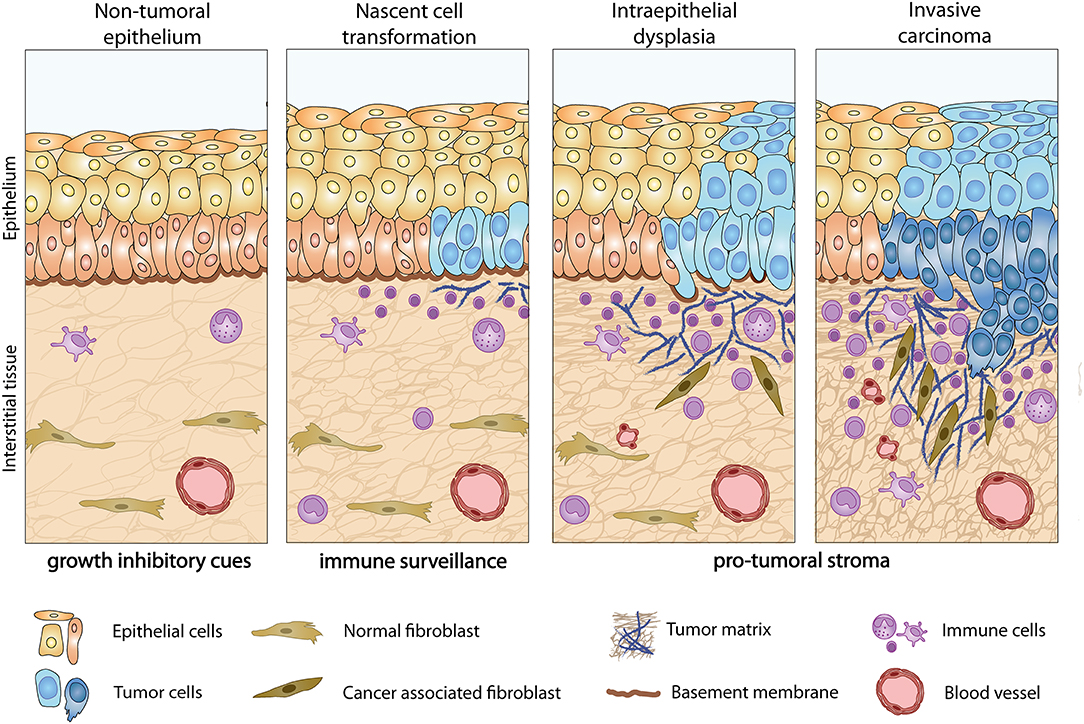
Figure 1. Tumor-induced stromagenic reprogramming during carcinoma progression. In normal epithelial tissue (left) homeostatic processes and the presence of an intact basement membrane restrain tumor growth. During early stages of tumor development, nascently transformed cells release pro-inflammatory cues that recruit immune cells to the dermal-epidermal interface and stimulate a wound healing response characterized by fibroblast activation and recruitment of angiogenic blood vessels. Activated stromal cells in turn promote the invasive phenotype of tumor cells through direct and indirect mechanisms. This tumor-stroma interplay is accompanied by the upregulation of a specific set of ECM components (1) and their receptors (2), in both tumor and stromal cells.
The ECM proteins induced in tumor tissue are often development- and disease-specific isoforms generated by alternative splicing events. Such is the case with fibronectin (FN), as described below. Before focusing on FN and its multi-faceted role in the tumor setting, we will briefly discuss important notions and emerging themes regarding the production, organization and remodeling of ECM in tumor tissue. Numerous outstanding reviews are cited in this section to provide a more comprehensive picture of these themes.
Stromal Reprogramming Through Tumor-Stroma Crosstalk
Tumor Matrisome
Gene expression screens have revealed that many genes encoding ECM components are dysregulated during tumor progression (3, 4). As the ECM is composed of large insoluble components, its protein composition has been detailed only recently. In an effort led by the laboratory of Richard Hynes, proteomics-based methods coupled with bioinformatics were used to define the “matrisome” of several normal and diseased tissues, including multiple tumor types (5). The computationally predicted matrisome corresponds to over 1,000 genes encoding a set of 278 core components and 753 matrisome-associated proteins, of which 86% of the core matrisome proteins and 58% of the matrisome-associated components have been detected in tissues using ECM-focused proteomics strategies [see (6) for the latest Matrisome database]. Examples of upregulated ECM components in cancer include collagens, non-collagen glycoproteins (FN, tenascin C, periostin), proteoglycans (biglycan, decorin), ECM regulators (cathepsin B, LOXL), and secreted factors (TGF-β1) [reviewed in (7)], to name only a few.
Source of Tumor ECM
Fibroblasts are considered to be the major source of ECM in tumor tissue as they are abundant, highly secretory and competent for ECM assembly [reviewed in (8)]. Other stromal cells, including vascular and immune cells, contribute to tumor ECM production as well. In vitro and in vivo studies have established that matrix proteins expressed by malignant cells also become directly incorporated in the matrix. Sets of tumor cell-derived ECM proteins were elegantly identified using xenograft models in which human tumor cells were grafted in murine hosts (5, 9–11). Interestingly, in these models the ECM composition was found to differ depending on the metastatic potential of the malignant cells, their tissue of origin, and whether they were derived from primary tumors or metastases. The multicellular origin of the neoplastic ECM holds true for human tumors as well. In a single-cell transcriptomic analysis of oral squamous cell carcinomas, ECM genes that are often linked with EMT (e.g., TGFBI, LAMC2, tenascin C) were found to be upregulated in carcinoma cells. Interestingly, their expression was enhanced in a subset of tumor cells displaying a partial EMT phenotype and located in close apposition to surrounding stroma, as determined by immunohistochemistry (12). These results indicate that paracrine signals from the stromal compartment trigger ECM gene expression in leading-edge cancer cells and they suggest a role for the upregulated matrix proteins in tumor invasion. Just as stromal mediators can trigger ECM gene expression in malignant cells, malignant cells can increase matrix production in the stromal compartment by promoting the activation of normal fibroblasts, of various origins, to carcinoma-associated fibroblasts (CAFs) as schematized in Figure 2. In addition to reprogramming CAF precursors, cancer cells recruit immune cells to the TME, such as tumor-associated macrophages (TAMs), neutrophils, dendritic cells, natural killer cells, T and B lymphocytes (13, 14). All of these cells represent a potential source of ECM components. This has been shown for TAMs which are extremely abundant in several tumor pathologies [as reviewed in (15, 16)]. TAMs also “enrich” the tumor matrix by secreting high levels of ECM-binding cytokines and growth factors that stimulate fibroblast activation (17).
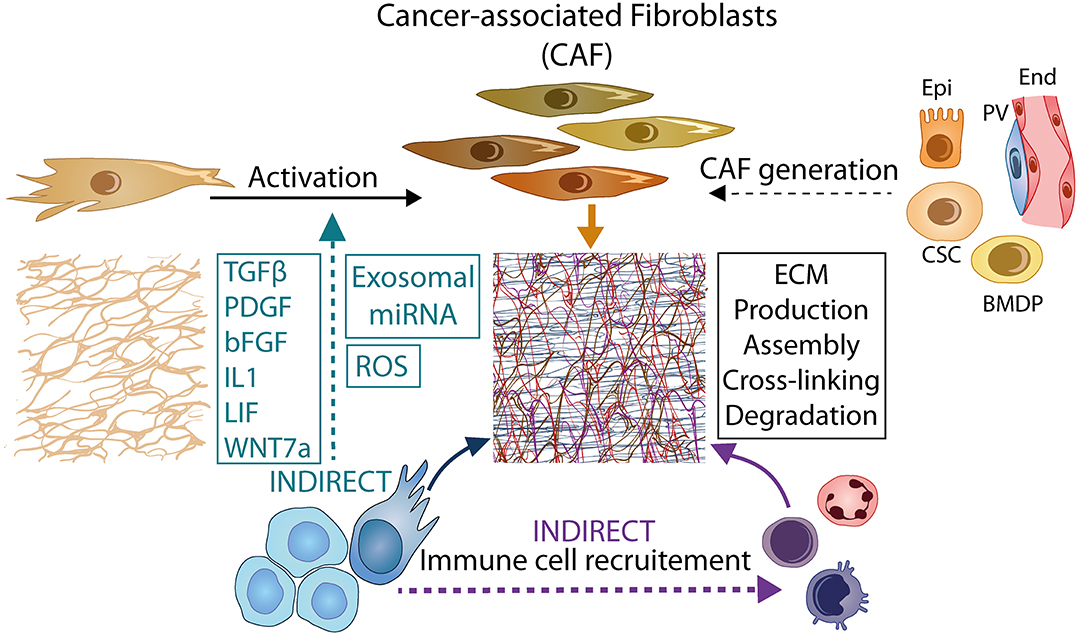
Figure 2. Tumor-CAF crosstalk and molecular mediators of ECM reprogramming. Tumor cells promote the generation of CAFs from resident fibroblasts or cells of different origin through the secretion of cytokines (e.g., TGF-β, PDGF, bFGF, IL1, LIF, WNT7A), the production of ROS, and exosomal delivery of miRNA. CAFs remodel the ECM by producing, assembling, cross-linking, and degrading ECM components. Tumor cells and infiltrating immune cells are also important proponents of ECM remodeling. The complex crosstalk between tumor and stromal cells leads to a global increase in ECM abundance and stiffness which in turn amplifies CAF activation via a positive feedback loop. Epi, epithelial cells; End, endothelial cells; PV, peri-vascular cells; CSC, cancer stem cells; BMDP, bone marrow derived precursor cells.
CAF Heterogeneity
The tumor-promoting effects of CAFs have been widely investigated and include the enhancement of cell proliferation, survival, migration/invasion, angiogenesis, chemoresistance, and immunosuppression, as detailed in recent reviews (18–20). Their activity is mediated through the secretion of a plethora of growth factors, cytokines and exosomes, but also through the production and remodeling of the ECM. CAFs have been equated to myofibroblasts, or activated fibroblasts linked to wound healing and contracture (21), because they often express α-smooth muscle actin (αSMA). However, it is now clear that CAFs exist as a heterogeneous population with distinct, yet overlapping, functions. Precise characterization of CAFs has been difficult as no marker is exclusive or absolute. Various combinations of markers including but not limited to αSMA, FAP, FSP1, CAV1, IL6, VIM, ITGB1, PDGFRα/β have been used to identify CAF subtypes in different tumor tissues using flow cytometry (22, 23) and single cell transcriptomics (12, 24). However, the distinct functions of these CAF subtypes and their associated ECM are not well-characterized to date.
CAFs arise from several different cell types including resident fibroblasts, bone marrow-derived precursor cells, vascular smooth muscle cells, pericytes, cancer stem cells, as well as endothelial and certain epithelial cells via endothelial- and epithelial-to-mesenchymal transition, respectively (25–28). CAF heterogeneity is thus due in part to their diverse origin, which is still under intense investigation and undoubtedly depends on the tumor (sub)type and anatomical localization. In addition, extracellular signals from the microenvironment, in particular mediators from other cells, drive CAF heterogeneity and dynamic changes in biomarker expression. Moreover, the positioning of CAFs in time and space, with respect to tumor cells, is an important determinant underlying the generation of CAF subtypes (23, 29–31).
CAF Generation
Until recently, the activation states of CAFs have been oversimplified and reduced to normal fibroblasts and activated fibroblasts, determined by αSMA expression. However, as indicated above, the activation state of CAFs cannot be solely defined by αSMA expression since certain CAF populations display only minimal αSMA levels (30, 32, 33).
Most inducers of CAF-like phenotypes (Figure 2) are also involved in fibroblast conversion to myofibroblasts during fibrosis, such as TGF-β. TGF-β is a master regulator of myofibroblast and CAF generation (34, 35) and a powerful inducer of several ECM components including collagen of type I, II, III, IV, and V, FN, thrombospondin, osteopontin, tenascin C, TGFBI, periostin, elastin, hyaluronic acid, osteonectin/SPARC, as well as chondroitin/dermatan sulfate proteoglycans, such as biglycan and decorin (36–38). TGF-β not only activates resident fibroblasts but also promotes the differentiation of CAF precursors including adipose tissue-derived stem cells, endothelial cells, and bone marrow-derived mesenchymal stem cells (39).
Similar to TGF-β, platelet-derived growth factor (PDGF) and fibroblast growth factor 2 (bFGF/FGF2) play critical roles in myofibroblast activation and fibrosis (40–42). In cancer, they were found to regulate CAF activation and αSMA expression as well, although their effects varied depending on the cell types examined (32, 43–46).
Reactive oxygen species (ROS) generated by cancer cells were shown to promote the fibroblast-to-myofibroblast transition by a mechanism involving TGF-β, PDGF, and CXCL12 signaling (47). Certain cytokines are important activators of fibroblasts, such as IL1α, which triggers fibroblast differentiation in inflammatory CAFs by inducing LIF, a cytokine of the IL6 family that activates JAK/STAT signaling (29). Apart from growth factors and cytokines, cancer cells also produce extracellular vesicles containing miRNA (e.g., miR-9, miR-155, miR-211) and proteins (TGF-β, BMP, and tetraspanins) that induce fibroblast activation or CAF generation from mesenchymal stem cells [reviewed in (48)].
ECM Organization and Remodeling
In addition to the stimulation of ECM protein expression, tumor-fibroblast crosstalk profoundly impacts matrix assembly, cross-linking and remodeling. The assembly of matrix macromolecules into a 3D structure is a dynamic process largely carried out in the tumor stroma by CAFs. Fibrillar collagens are the major components of the tumor ECM and collagen architecture is severely altered in tumor tissue [see (49, 50)]. In breast cancer, distinct patterns of fibrillar collagen organization, termed “tumor-associated collagen signatures” (TACS 1–3), have been defined to classify the changes in collagen arrangement that accompany carcinoma progression (51). TACS 3, characterized by straightened and aligned collagen fibers oriented perpendicular to the tumor boundary, was found to be an independent prognostic indicator of poor survival (52). For the present review, it is important to note the interdependence of collagen and FN networks. Fibrillar assembly of FN is required for collagen fibrillogenesis (53–56). Indeed, FN is a provisional matrix molecule (57) that provides a template for deposition of not only collagen, but for several ECM components including LTBP1, fibulin, and thrombospondin [(58) and references therein] as well.
Cellular “Oncofetal” Fibronectin: A Key Multi-Regulatory Component of the Tumor ECM
FN is a major core component of the tumor matrisome. Initially discovered as a “contaminant” in one of the steps of fibrinogen isolation more than 70 years ago (59), it is now one of the most extensively studied proteins, in terms of structural analysis and functional aspects.
Fibronectin Structure
FN is a high molecular weight glycoprotein composed of two similar subunits of 220–250 kDa (60, 61), linked together via disulfide bonding between two carboxy-terminal cysteine residues per subunit. FN is secreted in a soluble form by hepatocytes into the bloodstream (plasma FN, pFN), or expressed in tissues by fibroblasts and other cell types (cellular FN, cFN) forming an insoluble mesh. The primary structure of a FN subunit is characterized by the presence of three distinct types of repeats [reviewed in (62)], as schematized in Figure 3. There are 12 type I repeats (FNI1−12), two type II repeats (FNII1−2), and 15 (up to 17, see below) type III repeats [FNIII1−15 (64)]. Apart from the repetitive domains, there is also a variable region (V or IIICS, type III connecting segment) that lies between FNIII14 and FNIII15. The V region is not addressed in this review. The reader is invited to see other works, such as Xu et al. (62), Schwarzbauer and DeSimone (65), and references therein.
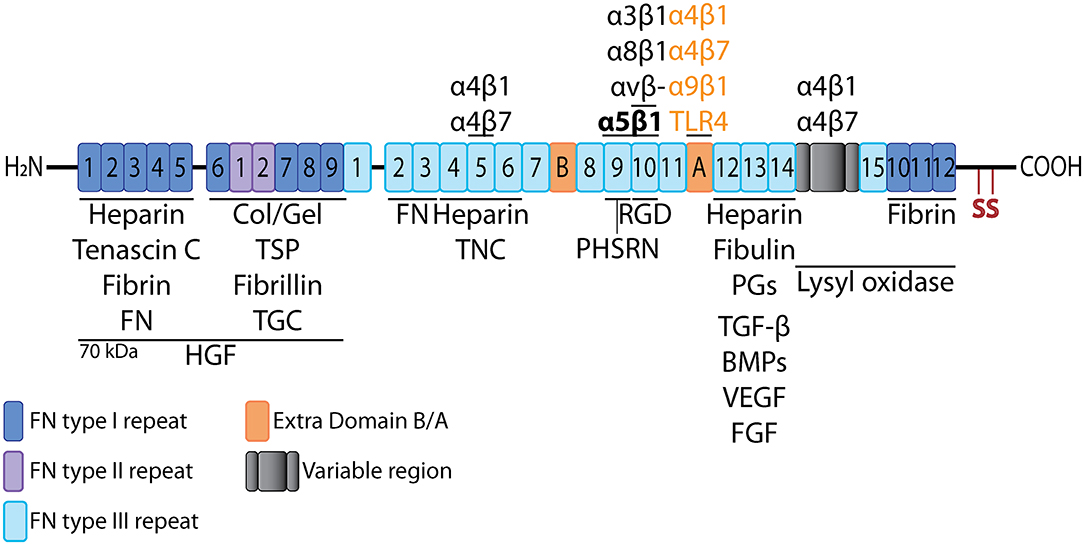
Figure 3. Fibronectin linear structure. Schematic representation of the linear structure of FN molecule, showing the different types of repeats and multiple binding sites for cells and other molecules. Adapted from Xu and Mosher (62) and Van Obberghen-Schilling et al. (63). TSP, thrombospondin; Col/Gel, collagen/gelatin; PGs, proteoglycans; TGC, tissue transglutaminase.
FNI and FNII repeats are composed of 45 and 60 amino acids, respectively, and they contain cysteine residues that form intra-domain disulfide bonds (66, 67). By contrast, FNIII repeats are composed of 90 amino acids, they contain no cysteines, and are organized in two antiparallel β-sheets folded in a sandwich-like conformation with a hydrophobic core (68–71). This structure results in a compact form that can be extended when strain is applied (72). There are 15 FNIII domains present in every FN monomer, and two additional domains termed Extra Domains (EDB and EDA) that are only included in cFN via alternative splicing, as described below.
Fibronectin Interactions and Function
The modular structure of FN and its multiple post-translational modifications result in numerous interactions with a variety of molecules that mediate cell attachment, ECM assembly, cell motility, cytoskeleton contractility, and host-pathogen interactions, to name just a few. Integrins represent the major family of cellular receptors through which FN exerts multiple functions in health and disease (73).
Integrin α5β1 is the “classic” FN receptor, that recognizes the tripeptide cell-binding site Arg-Gly-Asp (RGD) located in the FNIII10 repeat (71, 74–76). This interaction is facilitated and further stabilized by the synergistic effect of the PHSRN site located in FNIII9 (75). Binding of FN to α5β1 results in activation of the integrin, subsequently leading in Rho-mediated acto-myosin contractility that in turn promotes assembly of fibronectin into a fibrillar matrix (77–80). Integrin αvβ3 also binds the RGD site, as do α3β1, α8β1, αIIbβ3, and other αv-based integrins [reviewed in (62)], while α4β1 and α4β7 bind to specific sequences in FNIII5 (81), FNIII14 (76), and in the V region (82–84).
Apart from cell receptors, FN also interacts with ECM components via distinct sites in the FN molecule. Through the 70 kDa region (see Figure 3), FN binds collagen and gelatin, as well as fibrillin, and thrombospondin (85). This results in the enrichment of the provisional FN mesh with additional components of the matrisome, contributing to ECM maturation, which in turn promotes cell adhesion in vivo, and blood vessel morphogenesis during embryonic development and pathological angiogenesis [reviewed in (62, 63)]. The formation of the provisional meshwork lies on the ability of FN to self-associate at three distinct regions [reviewed in (86)], promoting polymeric assembly and mediating FN fibrillogenesis (87).
FN fibrillogenesis is a multistep process that involves the modular structure of FN, interactions of FN with other molecules, and cytoskeletal rearrangements in cells that assemble it [reviewed in (80)]. In brief, FN in a compact conformation is presented to the cell surface in an autocrine or paracrine manner. FN binding to α5β1 triggers integrin activation, clustering and the recruitment of cytoplasmic partners, including ILK, PINCH, parvin, and tensin. This intracellular machinery drives Rho-mediated stress fiber formation. Cell-generated acto-myosin contractility applies strain on the FN molecule resulting in its switch from a compact to a stretched state, thereby allowing intermolecular interactions required for FN incorporation into fibrils (72, 88, 89). Furthermore, integrin clustering and formation of complexes with additional cell receptors, like syndecan-4 [reviewed in (90)] or urokinase plasminogen activator receptor [uPAR (91)], can enhance FN assembly and strengthen FN-integrin binding (91–93). Finally, longitudinal and lateral association of FN molecules to existing fibrils results in FN polymerization, probably mediated by the protein-disulfide isomerase activity of FN, located in the FNI12 (94).
On this polymerized FN network, many other ECM associated components are assembled, such as heparin sulfate proteoglycans via their respective binding sites (64, 95–98), enhancing adhesion and spreading (92, 99). Similarly, TNC binds to FN and fine tunes cell adhesion and motility during angiogenesis and tumor progression [reviewed in (63)]. Finally, FN acts as a scaffold upon which the bioavailability and activity of several growth factors is orchestrated (100). Interaction of FN with growth factors (e.g., members of the TGF-β superfamily, PDGF, HGF, VEGF, FGF) may impact cell migration, cell proliferation, survival signals, and angiogenesis, as downstream outcomes of their activation through mechanical or enzymatic activation (101).
Fibronectin Splicing: The Oncofetal Fibronectin Variants
The 75 kbp long human FN1 gene is composed of 46 exons, and produces up to 20 distinct isoforms via alternative splicing [(102) and reviewed in (76)]. The first alternatively spliced region identified was the Extra Domain A (EDA, EIIIA, EDI), a FNIII repeat lying between FNIII11 and FNIII12, followed by the discovery of Extra Domain B (EDB, EIIIB, EDII) between FNIII7 and FNIII8 (103–106). Extra Domains are encoded by a single exon each, and they are only present in the cFN. Conversely, pFN lacks both Extra Domains.
Regulation of FN splicing depends strictly on tissue type and developmental stage, and it is tightly coupled to the activity of members of the SR protein family [(e.g., SRSF3, SRSF5) reviewed in (107)]. pFN is expressed throughout the entire lifespan of the organism, though declining with age (108–110). In contrast, cFN expression is elevated during embryonic development but diminishes significantly after birth (111–113). Intriguingly, cFN is re-expressed during the adult life under certain conditions that involve TGF-β signaling. Such conditions include tissue repair, fibrosis, angiogenesis, and cancer (114–117). Accordingly, increased SRSF3 and SRSF5 expression correlates with certain types of cancers [i.e., oral squamous cell carcinoma (118, 119)] and TGF-β signaling has been shown to regulate their expression (120), similar to that of cFNs (121–123). In light of the restricted expression of Extra Domain-containing FN, the hypernym “oncofetal FN” was used in the early 1980s to collectively describe these FN variants.
Functional Roles of the Extra Domains
Despite extensive research, the precise functional properties of EDB and EDA have yet to be fully deciphered. Non-exhaustive lists of in vitro and in vivo studies regarding EDB and EDA functions can be found in Muro et al. (124) and To and Midwood (125).
A series of elegant approaches have shed light in the functions of EDB and EDA. In two independent in vivo studies, mice expressing FN with constitutively included or excluded EDA were generated. All animals were viable and developed normally. However, mice lacking EDA displayed abnormal and delayed skin wound healing, and decreased motor coordination abilities, while mice constitutively expressing EDA showed a pronounced decrease in the level of FN in all tissues and decreased locomotory activity (126, 127). Interestingly, both mouse strains had shorter lifespans compared to control littermates (126). By contrast, deletion of EDB displayed no significant phenotype in mouse development and fertility, but fibroblasts extracted from EDB-null mice grew more slowly in vitro, and were less efficient at depositing and assembling FN (128). Most importantly, absence of both Extra Domains was deleterious for the organism due to severe cardiovascular defects (e.g., vascular leakage, defective angiogenesis) (129) suggesting that the Extra Domains have overlapping functions during embryonic development, and at least one of the two is necessary for normal body growth.
In disease-challenged situations in the adult, when cFN expression reappears, both Extra Domains have been correlated with a pro-fibrotic tissue landscape. More specifically, increased expression of FN-EDA resulted in differentiation of normal lipocytes to myofibroblast-like cells (115) and this phenotype was mediated by TGF-β1 (130). Furthermore, absence of EDA-containing FN in an idiopathic pulmonary lung fibrosis mouse model resulted in less collagen deposition and fewer α-SMA expressing myofibroblasts. This effect correlated with diminished activation of TGF-β suggesting that EDA is implicated in latent TGF-β activation (124). Most importantly, the presence of EDA highly correlated with enhanced matrix remodeling, matrix metalloproteinase (MMP) expression, and re-organization of the actin cytoskeleton (131), pointing toward a pro-fibrotic role for EDA. Similar findings were obtained in a tumoral context when tumor sections were found to be enriched in FN-EDB and FN-EDA in newly formed blood vessels of the tumor (132, 133). Reinforcing the potential tumorigenic role of cFN, EDA-containing FN induced G1-S phase transition by increasing the expression of Cyclin D1 and upregulation of integrin-mediated mitogenic signal transduction (134).
The aforementioned effects may be due in part to the increased cell receptor repertoire of cFN compared to pFN. More specifically, EDA contains an EDHIGEL sequence that has been identified as a binding site for integrins α4β1, α4β7, and α9β1 (135, 136). Furthermore, EDA is a ligand and activator of toll-like receptor 4 (TLR4) (137), thus triggering immune responses, described in subsequent sections. Conversely, no receptor has been identified so far for EDB, though a role has been suggested for EDB in osteoblast differentiation involving a β1-containing integrin (138).
cFN and the Hallmarks of Cancer
A major finding during the early days of FN research was that surface fibroblast antigen (SFA), as FN was named at the time, was significantly reduced in quantity upon malignant transformation of chicken and human fibroblasts infected with Rous Sarcoma Virus (RSV) (139, 140). However, it is now widely acknowledged that FN is strongly upregulated in several different tumor types. As the “malignant cell-centric” view of tumors shifted in recent years to a more inclusive view that encompasses their microenvironment, reports of the tumor suppressive functions of FN have been replaced by reports of its positive role in tumor growth and metastasis [reviewed in (86)]. This can be at least partly explained by the role of FN as a provisional matrix component promoting the formation of a primed TME that sustains cancer cell survival, stimulates proliferation, migration, angiogenesis and immune modulation (Figure 4). In this section, we will comment on the implication of cFN in these processes and highlight how cFN induction at tumor sites regulates various cellular responses that characterize the cancer hallmarks defined by Hanahan and Weinberg in 2001 (141) and amended in 2011 (142).
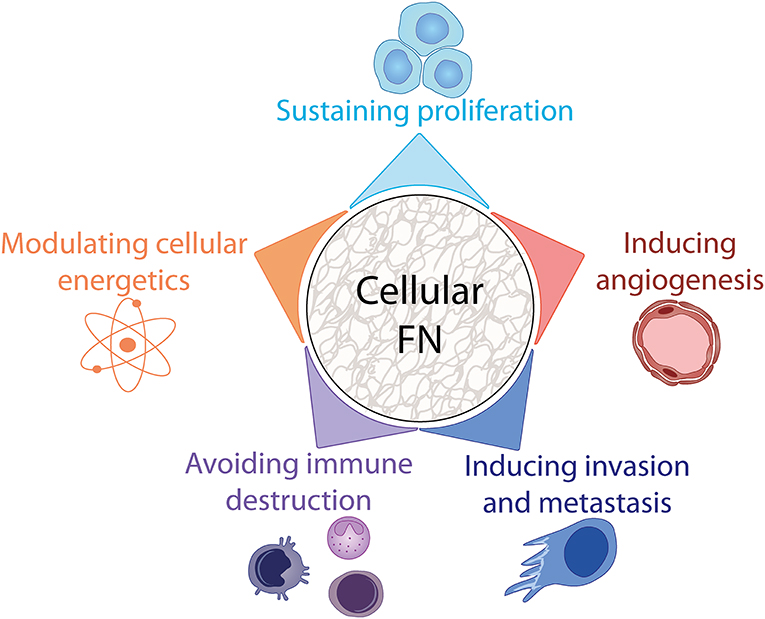
Figure 4. Involvement of cFN in cancer hallmarks. FN participates in tumor progression by impacting several enabling hallmarks of cancer, see text for details.
Sustaining Proliferative Signaling and Evading Growth Suppression
Several lines of evidence have brought FN under the spotlight for its role in cell proliferation. Developmental processes, such as the establishment of antero-posterior polarity, the formation of the neural tube and mesodermally derived tissues are thought to be regulated by FN-mediated cell proliferation (143). Regarding tissue homeostasis, FN choreographs the proliferative phase of wound healing by bringing together different cells and components (144). In cancer, FN is a basic component of the tumor niche that has been shown to facilitate cancer cell proliferation and survival. In vitro studies have underlined the role of FN in promoting cancer cell growth, survival, and invasion in glioma (145), renal cell carcinoma (146), and gall bladder carcinoma (147). In vivo, it was shown that tumor cells injected in mice lacking circulating FN grew more slowly and apoptosis was increased (148). Similarly, tissue-specific depletion of FN resulted in lower tumor cell proliferation and invasion in the bone marrow (148).
One of the earliest studies on cFN variants highlighted the potency of EDA-containing FN to induce expression of cyclin D1, hyperphosphorylation of pRb and activation of ERK2 resulting in cell cycle progression (134). Similar results were obtained 10 years later when Kohan and colleagues described that recombinant EDA-containing peptides were able to induce MAPK-ERK1/2 activation and fibroblast differentiation through α4β7 binding and FAK activation (149). In a 3D cell culture system, blocking of FN-α5β1 interaction induced apoptosis in breast cancer cells via a mechanism that involves Akt, suggesting a protective role of FN for tumor cells (150). Though the authors did not directly assess the anti-apoptotic role of EDA per se, they hypothesized that it is the EDA-mediated strengthening of FN-α5β1 interactions (151) that results in the protective effect of FN against cell death. Finally, in two human tumor cell lines it was shown utilizing CRISPR/Cas9 technology that exclusion of EDA resulted in a pronounced decrease in cell proliferation (152).
In vivo, ovarian cancer cells displayed decreased proliferation and metastasis in mice bearing a tissue-specific deletion of Fn1 in the lining of the peritoneal cavity. The effects were attributed to a tumor-stroma crosstalk and the participation of TGF-β signaling (153). The splicing pattern of the FN produced in control mice was not identified, but given its cellular nature, and the implication of TGF-β in Extra Domain inclusion (see previous section), addressing how normal cells residing in the TME influence tumor cells by cFN expression is a question worth-addressing.
In contrast to EDA, the role of EDB in tumor cell proliferation is largely unknown, in spite of its increased presence in the TME. In vascular endothelial cells, EDB-containing peptides were found to stimulate proliferation (154), while EDB knock-down impaired cell growth (154, 155).
Inducing Angiogenesis
FN clearly occupies a central note in the “angiome,” the global protein connectivity network of genes associated with angiogenesis (156). The importance of FN in angiogenesis was first revealed by genetic studies in the mouse demonstrating that invalidation of the FN gene induces embryonic lethality (around E9.5) with cardiovascular and angiogenesis defects (143). Intriguingly, specific ablation of cFN (including both EDB and EDA domains) in mice that still express pFN also triggered defective angiogenesis leading to hemorrhagic vessels and embryonic lethality at E10.5 (129), attesting to a critical role for these cFN exons in developmental angiogenesis. The source of the cFN is also critical for its role in vascular development. In the neonatal retina, angiogenesis is regulated by endothelium-derived FN in an autocrine manner (157). This is an important notion, as FN production in endothelial cells is tightly coupled to its assembly (155), and assembly of cFN into a three-dimensional fibrillar meshwork is essential for neovessel formation (158).
Concerning the role of FN in tumor angiogenesis, results from animal models (e.g., inducible deletion, tumor xenografts) are less clear. Post-natal deletion of endothelial cFN in a spontaneous RIPTag-driven model of carcinogenesis fails to inhibit tumor angiogenesis (159, 160), suggesting a complex functional role of FN in tumor angiogenesis and partially explaining the disappointing results of targeting FN-binding integrins in the clinic, as discussed below. Nonetheless, cFN has been recognized for quite some time to be a useful marker of cancer-associated vessels (133, 161, 162). Expression of cFN is also upregulated in malignant cells of certain tumors with mesenchymal phenotypes. This is the case for glioblastoma multiforme (GBM) (163), a devastating malignancy in which cFN was shown to be expressed in both blood vessels and tumor cells (164). In addition to cell-autonomous effects of cFN on the invasive behavior of tumor cells, paracrine effects of GBM cell-derived FN enhance the recruitment of blood vessels through integrin-dependent binding to endothelial cells.
Effects of FN on endothelial cell adhesion, spreading and migration have been extensively studied in vitro. However, it is important to consider that beyond its role as ligand for signaling receptors on endothelial cells, FN in perivascular matrices constitutes an obligate scaffold for organization of the vessel-associated ECM and a repository for pro-angiogenic factors [reviewed in (63)]. FN can bind directly to (165) and modulate the function of VEGF (166), one of the most potent angiogenic factors. Moreover, it has been demonstrated that astrocytic derived FN promotes retinal angiogenesis by dual integrin-dependent and -independent functions on endothelial retinal cells, promoting filopodia adhesion or VEGF-induced directional tip cell migration, respectively (167). Thus, there is still much work to be done on several fronts to fully grasp the role of FN in tumor angiogenesis and how this is linked to tumor expansion, as discussed below.
Activating Invasion and Metastasis
Elevated FN expression is associated with invasive tumors and poor prognosis in many cancers [as reviewed in (55, 150, 163, 168–172), to cite a few]. However, this is not the case in all tumor pathologies and the role of FN in tumor invasion and metastasis has been controversial [(86) and references therein]. Lin and colleagues recently analyzed 72 studies published over the past four decades that address the role of cancer cell-derived FN (termed cancerous FN) and stromal FN in tumor progression (86). Interestingly, a tumor-suppressive function for cancerous FN was reported in 57.7% of the articles prior to 2000, yet only 15% since that date. Conversely, reports of a tumor- and metastasis-promoting role for cancerous FN increased from 11.5% before 2000 to 25% after 2000. Publications describing the implication of stromal FN in early tumor progression, but not late metastasis, remained constant at 30%.
These results raise the question of how FN came to be pro-tumoral. The increase in tumor-promoting effects of FN observed over the past several years is likely due to improved biological tools and approaches as well as a more holistic view of cancer. Indeed, since 2000 the field has evolved from the study of 2D cultured tumor lines and xenografts in immunocompromised mice models, to multimodal analysis of human tumors in their tissue context. The emergence of single cell transcriptomics (vs. transcriptomic studies that measure tumors in bulk) has refined the molecular signatures of tumor cells and different stromal cell populations thus providing a closer look at FN expression and function in different cell types, and across cancer types. Moreover, former studies on tumors were largely centered on malignant cells. However, in most carcinomas FN is produced and assembled by stromal cells (173). Indeed, recent studies have shown that stromal FN participates not only in the early steps of tumor formation but also in the promotion of tumor cell motility and invasive behavior. Remodeling the matrix by CAFs is a key feature of cancer cell invasion (174). The ability of fibroblasts to induce cancer cell invasion was found to depend on the amount of FN that they produce and assemble (175). Assembly of the protein is required, as addition of soluble FN failed to promote invasion of the cancer cells through collagen gels. Once aligned by CAFs, linear arrays of FN fibers can promote directional migration of carcinoma cells (55, 176). In the case of head and neck squamous cell carcinomas, migration of tumor cell cohorts on fibroblast-derived matrices involves the cFN-binding integrin αvβ6 and is associated with the activation of latent TGF-β at the tumor-stroma interface (55, 177). Treatment of lung tumor cells with soluble FN stimulated migration and invasion via FAK/Src/PI3K/ERK as well as activation of MMP expression (178). Thus, FN can act both as a physical scaffold laying the path for tumor cell invasion, a platform for latent TGF-β activation and a ligand for activation of intra-cellular signaling pathways and subsequent induction of matrix-degrading proteases. Finally, given that FN is an exquisitely extensible molecule, tensile forces and FN-dependent mechano-signaling in the TME play a decisive role in invasion and metastasis. The topic of ECM stiffness and tissue mechanics is excellently reviewed in (89).
More Than a Marker of Epithelial to Mesenchymal Transition (EMT)
FN is a mesenchymal marker par excellence. In mesenchymal-like tumors, such as glioblastomas, FN expression and assembly by tumor cells has been shown to facilitate intercellular cohesion and collective invasion through a basement membrane-like ECM (164). FN knock-down in glioma xenografts reduced tumor growth and improved survival of implanted animals (164, 179). In epithelial tumors its expression is often used to detect mesenchymal transition (180). EMT is a complex program whereby epithelial cells loose polarity and cell-cell adhesion to acquire a mesenchymal phenotype and invasive properties [reviewed in (25, 180, 181)]. Early studies described the role of FN in EMT during chick embryo gastrulation and neurulation (182). Later, FN was associated with EMT during tumorigenesis. However, it is more than just a marker of EMT. FN can contribute to mesenchymal transition by providing a platform for integrin-dependent activation of latent TGF-β (183). Thus, at the leading edge of invasive tumors paracrine interactions between CAFs and malignant epithelial cells can promote a so-called partial EMT (pEMT) phenotype characterized by the expression of EMT-related genes in tumor cells that retain their epithelial phenotype (12). This is the case for leading edge tumor cells that express αvβ6, a cellular receptor for EDA-containing cFN. In ovarian cancer, TGF-β produced by tumor cells stimulates mesenchymal transition in mesothelial cells resulting in the upregulation of FN in the ECM of mesothelial cells which increases adhesion and invasion of the sub-mesothelial basement membrane by ovarian carcinoma cells (153).
FN in Metastasis
Circulating FN also contributes to tumor angiogenesis and metastatic spread of malignant cells. In pFN-deficient mice, obtained by conditional KO of the Fn1 gene in the liver, von Au and colleagues showed that a decrease in pFN reduces tumor angiogenesis, tumor growth and bone metastasis through an apparent feed forward upregulation of its own production and by modulating the response to VEGF (148). Plasma FN is one of the most abundant adhesion proteins in the blood. However, it is functionally invisible to the apical surface of endothelial cells in mature blood vessels. Following injury or angiogenic stimulation, endothelial cells upregulate cFN production and become responsive to the pro-adhesive and integrin-mediated angiogenic functions of FN. In a study by Barbazan et al., FN deposits were detected on the luminal side of hepatic blood vessels in human colorectal cancer patients (184). Using a mouse model of intestinal tumor metastasis, they demonstrated that FN deposits in the hepatic vasculature facilitate the arrest of circulating tumor cells and extravasation via a mechanism involving talin-dependent integrin signaling in the tumor cells. pFN can also promote lung metastasis by forming pFN-fibrin clots that retain circulating tumor cells via integrin αvβ3 (177).
The final step of cancer progression is colonization of secondary organs. This highly rate-limiting step is critically affected by the matrix microenvironment (185). For making a hospitable home, cancer cells need to prepare the local microenvironment before they arrive at the distant secondary site referred to as the pre-metastatic niche (186). Pioneering studies from David Lyden's lab revealed the importance of bone marrow-derived cells (BMDC) in the establishment of a pre-metastatic niche for tumor cell metastasis (187). In response to soluble factors, such as VEGF or PDGF, VEGFR1-positive BMDCs are mobilized to colonize sites of future metastasis, prior to the arrival of tumor cells, by interacting with tumor-induced EDA-containing FN through α4β1 integrin. In turn, VEGFR1-positive BMDCs secrete chemokines, such as SDF1 to attract CXCR4+ tumor cells to the newly formed metastatic niche. EDA-containing FN, together with tenascin C, versican and periostin have also been found in other secondary sites prior to tumor cell arrival, and may be important for recruitment of stromal cells as well as for circulating tumor cells to the pre-metastatic niche (153, 188–190).
Avoiding Immune Destruction
Tumor Promoting Inflammation
The acquisition of functional capabilities allowing survival, proliferation and spread of cancer cells, defined as hallmarks most recently by Hanahan and Weinberg (142), are rendered possible by so-called enabling characteristics. One important enabling feature that has drawn much attention over the past 20 years is the inflammatory state of tumor lesions. Chronic inflammation driven by infiltrating immune cells can empower multiple cancer hallmarks.
ECM remodeling in the tumor stroma is associated to the release of proteolytic fragments, termed “matrikines,” into the microenvironment. Some of these ECM domains retain secondary structure and can display bioactivity (191) as Damage-Associated Molecular Pattern (DAMP) molecules, endogenous activators of innate immunity (192). Toll-like receptor 4 (TLR4), a DAMP receptor initially thought to be restricted to immune cells, is present and functional on a variety of non-immune normal cells and tumor cells. TLR4 has been implicated in the development of several types of cancer and fibrosis (193, 194). As part of the anti-tumor immune response, DAMP-induced TLR4 activation triggers the production of pro-inflammatory cytokines, chemokines, and effector molecules. However, continuous TLR4 signaling results in chronic inflammation. Recombinant fragments of FN containing the EDA domain, but not the full length (soluble) protein were shown to bind and activate the TLR4 (137). Binding of EDA results in TLR4-mediated NF-κB pathway activation and subsequent production of pro-inflammatory, pro-fibrotic cytokines and MMPs (137, 195, 196).
In mesenchymal cells, TLR4 signaling leads to the stimulation of a pro-fibrotic gene program with augmented expression of tissue repair, wound healing and ECM remodeling genes, while induction of inflammatory genes is relatively weak (193). A second FN Type III domain, FNIII-1c, was shown to activate TLR4-mediated inflammatory cytokine release in human fibroblasts in synergy with the EDA domain (197). Type III domains of FN can be released from the matrix by proteolysis, or become exposed in response to mechanical forces (198). Therefore, the presence of EDA-containing cFN fragments or mechanically strained fibers in the tumor matrix landscape can trigger and sustain innate immunity, inflammation, and myofibroblast generation driven by one or more EDA-dependent inflammatory feedback loops (199).
Regulation of Anti-tumoral Immunity
The immune system is an important barrier against tumor progression. How tumor cells develop immune system-evading mechanisms and how the different immune cells interact with tumor cells is a field of intense research. In general, the presence of tumor infiltrating CD8-expressing lymphocytes in the TME is associated with an improved prognosis and a better response to therapy in a broad range of tumor types (200–205). However, the presence of immune cells with inhibitory function, such as myeloid-derived suppressor cells and regulatory T cells (Tregs) that dampen the immune control of cancer, can be associated with worse outcome [(204) and references therein]. Analysis of histological sections of tumor samples led to the segregation of cancer immune phenotypes into three distinct profiles: immune-inflamed, immune-dessert and immune-excluded (206). Immune-inflamed and immune-dessert phenotypes are generally characterized by an abundant or sparse immune infiltrate, respectively. In immune-inflamed tumors, the immune cells are positioned in proximity to the tumor cells. Immune-excluded tumors also display an abundant infiltrate but the immune cells fail to effectively penetrate the tumor parenchyma and they remain in the stroma surrounding tumor cell nests. This immune phenotype is characterized by an excessive deposition of ECM components, including dense aligned bundles of collagen and FN around tumor islets. Live-cell imaging studies on patient-derived lung tumor tissue sections revealed active T cell motility in regions of loose FN and collagen I, whereas T cells migrated poorly in dense matrix areas surrounding tumor nests (207, 208). Thus, the ECM can promote tumor evasion from the immune system by limiting the anti-tumor activity of T cells, either directly by inhibiting the contact of infiltrating immune cells with cancer cell nests (207, 209) or indirectly through the recruitment of TAMs that cause lymphocyte retention in the stroma (210). The latter mechanism, using live imaging techniques in a mouse model and on fresh human carcinoma slices, demonstrated that TAMs impede CD8-expressing T cells from reaching tumor cells by lymphocyte trapping in the stroma and consequently limit the efficacy of immune check point inhibitor (anti-PD-1) treatment. TAMs are key components of the tumor ECM microenvironment directly affecting its production and remodeling. TAMs isolated from human ovarian carcinomas (15) and from an orthotopic colorectal cancer model (16) display a gene expression profile in which matrix glycoproteins, including FN, are highly upregulated (211). Clearly, the ECM is emerging as an important component of stromal-based immunomodulatory mechanisms that alter the trafficking, maturation and function of immune cells through multiple mechanisms, many of which have yet to be uncovered. cFN is a prominent component as a provider of DAMPs, by virtue of its obligate role in activation of ECM-tethered latent TGF-β and its function as a mechanically-tuned repository of immunomodulatory cytokines and growth factors, as discussed in previous sections.
Deregulating Cellular Energetics
The metabolic shift toward aerobic glycolysis in cancer cells is a well-established phenomenon that is currently used in tumor diagnosis. During the past decade, numerous studies underlined a potential metabolic crosstalk between normal and transformed cells in the tumor niche (212). The link between the ECM and cell metabolic activity presents an emerging, compelling field of scientific research. Recently, several groups have provided evidence pinpointing toward shifts in cell metabolic processes mediated by matrix composition, stiffness, and remodeling (213–215). Moreover, physical properties of the TME, like pH regulation, are shaped by increased expression of pumps and transporters, and their transportation to the plasma membrane [reviewed in (216)]. Thus, the tumor landscape is sculpted and primed in order to promote cancer cell growth, motility and invasion. To our knowledge, no EDB- or EDA-oriented studies regarding cellular energetics have been reported. The handful of studies describing the role of FN Extra Domains in regulating metabolic processes has only been performed in non-tumoral contexts, thus whether the presence of the Extra Domains reflects reprogramming of cell energetics is an open question.
In an in vivo study, investigators used a diabetes-impaired endothelial vasodilation mouse model to study the effect of EDA-containing FN. Mice constitutively excluding EDA displayed increased endothelial dysfunction, and the underlying mechanism involved increased superoxide anion levels, NADPH oxidase (NOX4) expression, and TGF-β, suggesting a protective role of FN-EDA against vascular oxidative stress (217). Conversely, overexpression of FN-EDA or pFN in a mouse monocyte macrophage cell line, dysregulated the endogenous sterol response pathway through ER stress response, though no difference was observed between cells expressing the different isoforms (218). EDB was not included in the study, and its potential involvement is thought provoking and constitutes an interesting perspective.
Targeting the Dysregulated Tumor ECM
Potential Therapeutic Implications of FN
The striking implication of tumor ECM components and their cellular receptors in tumor progression and response to conventional and emerging therapies has led to the quest for novel TME-directed therapeutic strategies. Different approaches have been employed to target FN, or its receptors (219). The first approach pioneered by Neri et al. is based on antibody-mediated delivery of therapeutic agents to cFN isoforms present in the TME. Both EDA and EDB domains have been used for specific delivery of cytokines, cytotoxic agents, chemotherapy drugs and imaging agents to tumors expressing cFN variants [reviewed in (161, 220)]. The tumor-targeting immunocytokine L19-IL2 is a good example. It is composed of the human scFv antibody fragment (L19), specific to the EDB domain of FN, fused to recombinant human IL2. Following extensive preclinical studies, L19-IL2 immunocytokine has demonstrated therapeutic activity in advanced solid cancer (221). In combination with darcabazine, it displayed encouraging clinical activity in patients with metastatic melanoma in a phase I and II studies (222, 223). A randomized Phase IIb study was completed but not yet reported (NCT01055522). L19-IL2 was also tested in combination with radiotherapy. In a preclinical study, that combination enhance the radiotherapy-induced antitumor immune reaction and provided a long-lasting antitumor effect dependent on EDB expression and infiltration of cytotoxic T cells (224). A Phase I clinical study combining L19-IL2 with Stereotactic Ablative Body Radiotherapy (SABR) in oligometastic tumors (NCT02086721) was completed but results are not published. Additional clinical trials involving L19-derived targeting agents (L19-IL2, L19-TNF) alone or combined with other therapies are ongoing in several tumor types (see the NIH website identifier: https://clinicaltrials.gov).
EDA and EDB based vaccines appear to be promising for the treatment and prevention of cancer. Regarding the EDA domain, a fusion protein between streptavidin and the endogenous TLR4 ligand EDA showed the capacity to target biotinylated antigens to dendritic cells and induce T cell responses in vivo (225). As EDA is known to activate the NF-κB pathway leading to the activation of innate immune system and release of inflammatory cytokines, it has been explored as a cancer vaccine adjuvant in mouse models (195, 226, 227). Immunological activity of EDA depends upon its local intramolecular context within the FN chain. Immobilizing EDA-containing FN fragments within a fibrin matrix model along with antigenic peptides stimulates cytotoxic CD8+ T cell responses in two murine cancer models (228). Thus, delivering ECM-bound FN EDA fragments in combination with antigens could be an attractive option for anti-tumoral immunotherapies.
The second approach consists of directly interrupting the pro-tumoral effects of FN by using antibodies and small-molecule inhibitors that interrupt interactions between FN and its integrin partners. Integrins as therapeutic targets have been a focus of drug development for over 3 decades, with some successes in preclinical studies in cancer. Developments in integrin-directed therapeutics in cancer and other pathologies have been reviewed in Raab-Westphal et al. (2). Integrin α5β1, the “prototype” FN receptor, is implicated in different aspects of tumor progression, and it appears particularly overexpressed in the most aggressive tumor grades. It is a pertinent therapeutic target in solid tumors and appears safe for the patients in a phase I clinical study (229). Unfortunately, in a phase II clinical study, anti-α5β1 integrin antibody volociximab tested as monotherapy in patients with platinum-resistant advanced epithelial ovarian or primary peritoneal cancer, showed insufficient clinical activity (230). In patients with refractory or relapsed metastatic clear cell renal carcinoma, volociximab led to stable decrease in 80% of patients but no randomized controlled trial has been reported so far. Integrin α4β1, a FN receptor that binds to the EDA domain and variable region of the protein, promotes the homing of monocytes to tumors, and is essential for the participation of myeloid cells in angiogenesis and tumor growth. Specific antagonists of integrin α4β1 prevented monocyte stimulation of angiogenesis in vivo, macrophage colonization of tumors, and tumor angiogenesis (231). However, whereas suppression of myeloid cell homing to tumors using α4β1 antagonists appears to be an effective approach to impede tumor angiogenesis and growth, depletion of the integrin in a mouse model of colon adenocarcinoma resulted in an age-dependent effect and accelerated tumor growth in mature mice (232). These findings support a central role for α4β1 in tumor growth control but call for more in depth studies of its cellular expression pattern and function in the TME prior to its use as a pharmacological target.
The αv integrins represent an interesting class of adhesion receptors that recognize RGD-containing ligands, including FN, and have multiple roles in cancer hallmarks (e.g., angiogenesis, growth and dissemination, and immunomodulation) (233). αv-based integrins are overexpressed in several tumor pathologies and their expression can be found on both tumor cells and stromal cells. In addition to being promiscuous receptors, with FN being only one of numerous ligands, the αv-based integrins have been linked to local TGF-β activation, which compounds the complexity of their effects in the TME (234).
Impact of FN on Treatment Response
Integrin-mediated cell-ECM and in particular cell-FN interactions confer resistance to chemotherapy as well as to ionizing radiation (235–241). In an in vitro and in vivo model of human non-small lung cancer, cetuximab promoted FN expression via p38-MAPK-ATF2 signaling (237). Cell adhesion to FN enhanced tumor cell resistance to radiotherapy, and attenuated the cytotoxic and radiosensitizing effects of cetuximab (237). More recently, chemotherapy resistance in esophageal cancer cell lines increased in cells growing in a three-dimensional environment enriched in collagen and FN (242). These results illustrate that FN plays a key role in the response of cancer cells to treatment. Conversely, anti-tumor therapies modify the tumor ECM environment, as exemplified by radiation-induced fibrosis (243).
Concluding Remarks
Our understanding of stromal reprogramming in the TME has advanced at a rapid pace in recent years, driven by technological advances and the integration of massive amounts of information from different fields, spanning multiple scales. As illustrated above, cFN is recurrently a central component of the tumor stroma that contributes to several cancer hallmarks and enabling characteristics. Further understanding of cFN production, assembly and remodeling in primary tumor beds, draining lymph nodes and at metastatic sites is needed to grasp the full complexity of tumor progression and metastatic spread. This knowledge should provide valuable insights into cell-ECM interactions and the physical and functional interplay between different cellular components of the TME. Elucidating the role of cFN variants in modulation of immune cell trafficking, phenotype and functional maturation is of particular importance for improving the current armatorium of immunomodulatory agents and for providing therapeutic alternatives that target the stroma.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The pioneering and insightful scientists whose biochemical, molecular, and genetic studies laid the foundation for the enormous corpus of work on FN were gratefully acknowledged. We also extend our apologies to colleagues whose original contributions could not be cited due to space limitations. EV acknowledges funding from the French National Agency for Research (ANR-16-CE93-0005-01), the Fondation ARC (PJA20151203207, fellowship GE), The Fondation Recherche Médicale (fellowship AS), the Ligue National Contre le Cancer (LNCC/EVO Programme Priorité Tabac), the LabEx SIGNALIFE program (ANR-11-LABX-0028-01), the Fédération Hospitalo-Universitaire OncoAge and the 3IA Côte d'Azur (ANR-19-P3IA-0002).
References
1. Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. (2012) 196:395–406. doi: 10.1083/jcb.201102147
2. Raab-Westphal S, Marshall J, Goodman S. Integrins as therapeutic targets: successes and cancers. Cancers. (2017) 9:110. doi: 10.3390/cancers9090110
3. Eckhardt BL, Parker BS, Laar RK van, Restall CM, Natoli AL, Tavaria MD, et al. Genomic analysis of a spontaneous model of breast cancer metastasis to bone reveals a role for the extracellular matrix. Mol Cancer Res. (2005) 3:1–13.
4. Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. (2003) 33:49–54. doi: 10.1038/ng1060
5. Naba A, Clauser KR, Hoersch S, Carr SA, Hynes RO. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol Cell Proteomics. (2012) 11:M111.014647. doi: 10.1074/mcp.M111.014647
6. Shao X, Taha IN, Clauser KR, Gao Y, (Tom) Naba A. MatrisomeDB: the ECM-protein knowledge database. Nucleic Acids Res. (2020) 48:D1136–44. doi: 10.1093/nar/gkz849
7. Xiong G-F, Xu R. Function of cancer cell-derived extracellular matrix in tumor progression. JCMT. (2016) 2:357. doi: 10.20517/2394-4722.2016.08
8. Karagiannis GS, Poutahidis T, Erdman SE, Kirsch R, Riddell RH, Diamandis EP. Cancer-associated fibroblasts drive the progression of metastasis through both paracrine and mechanical pressure on cancer tissue. Mol Cancer Res. (2012) 10:1403–18. doi: 10.1158/1541-7786.MCR-12-0307
9. Naba A, Clauser KR, Lamar JM, Carr SA, Hynes RO. Extracellular matrix signatures of human mammary carcinoma identify novel metastasis promoters. eLife. (2014) 3:e01308. doi: 10.7554/eLife.01308
10. Naba A, Clauser KR, Whittaker CA, Carr SA, Tanabe KK, Hynes RO. Extracellular matrix signatures of human primary metastatic colon cancers and their metastases to liver. BMC Cancer. (2014) 14:518. doi: 10.1186/1471-2407-14-518
11. Tian C, Clauser KR, Öhlund D, Rickelt S, Huang Y, Gupta M, et al. Proteomic analyses of ECM during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. Proc Natl Acad Sci USA. (2019) 116:19609–18. doi: 10.1073/pnas.1908626116
12. Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell. (2017) 171:1611–24.e24. doi: 10.1016/j.cell.2017.10.044
13. Hill BS, Sarnella A, D'Avino G, Zannetti A. Recruitment of stromal cells into tumour microenvironment promote the metastatic spread of breast cancer. Semin Cancer Biol. (2019) 60:202–13. doi: 10.1016/j.semcancer.2019.07.028
14. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. (2018) 24:541–50. doi: 10.1038/s41591-018-0014-x
15. Liguori M, Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages as incessant builders and destroyers of the cancer stroma. Cancers (Basel). (2011) 3:3740–61. doi: 10.3390/cancers3043740
16. Afik R, Zigmond E, Vugman M, Klepfish M, Shimshoni E, Pasmanik-Chor M, et al. Tumor macrophages are pivotal constructors of tumor collagenous matrix. J Exp Med. (2016) 213:2315–31. doi: 10.1084/jem.20151193
17. Ireland LV, Mielgo A. Macrophages and fibroblasts, key players in cancer chemoresistance. Front Cell Dev Biol. (2018) 6:131. doi: 10.3389/fcell.2018.00131
18. LeBleu VS, Kalluri R. A peek into cancer-associated fibroblasts: origins, functions and translational impact. Dis Models Mech. (2018) 11:dmm029447. doi: 10.1242/dmm.029447
19. De Jaeghere EA, Denys HG, De Wever O. Fibroblasts fuel immune escape in the tumor microenvironment. Trends Cancer. (2019) 5:704–23. doi: 10.1016/j.trecan.2019.09.009
20. Monteran L, Erez N. The dark side of fibroblasts: cancer-associated fibroblasts as mediators of immunosuppression in the tumor microenvironment. Front Immunol. (2019) 10:1835. doi: 10.3389/fimmu.2019.01835
21. Rockey DC, Weymouth N, Shi Z. Smooth muscle α actin (Acta2) and myofibroblast function during hepatic wound healing. PLoS ONE. (2013) 8:e77166. doi: 10.1371/journal.pone.0077166
22. Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell. (2018) 33:463–79.e10. doi: 10.1016/j.ccell.2018.01.011
23. Givel A-M, Kieffer Y, Scholer-Dahirel A, Sirven P, Cardon M, Pelon F, et al. miR200-regulated CXCL12β promotes fibroblast heterogeneity and immunosuppression in ovarian cancers. Nat Commun. (2018) 9:1056. doi: 10.1038/s41467-018-03348-z
24. Bernard V, Semaan A, Huang J, San Lucas FA, Mulu FC, Stephens BM, et al. Single-cell transcriptomics of pancreatic cancer precursors demonstrates epithelial and microenvironmental heterogeneity as an early event in neoplastic progression. Clin Cancer Res. (2019) 25:2194–205. doi: 10.1158/1078-0432.CCR-18-1955
25. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. (2009) 119:1420–8. doi: 10.1172/JCI39104
26. Nair N, Calle AS, Zahra MH, Prieto-Vila M, Oo AKK, Hurley L, et al. A cancer stem cell model as the point of origin of cancer-associated fibroblasts in tumor microenvironment. Sci Rep. (2017) 7:6838. doi: 10.1038/s41598-017-07144-5
27. Rønnov-Jessen L, Petersen OW, Koteliansky VE, Bissell MJ. The origin of the myofibroblasts in breast cancer. Recapitulation of tumor environment in culture unravels diversity and implicates converted fibroblasts and recruited smooth muscle cells. J Clin Invest. (1995) 95:859–73. doi: 10.1172/JCI117736
28. Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. (2007) 67:10123–8. doi: 10.1158/0008-5472.CAN-07-3127
29. Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, et al. IL-1-induced JAK/STAT signaling is antagonized by TGF-beta to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. (2018) CD-18-0710. doi: 10.1158/2159-8290.CD-18-0710
30. Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. (2017) 214:579–96. doi: 10.1084/jem.20162024
31. Ligorio M, Sil S, Malagon-Lopez J, Nieman LT, Misale S, Di Pilato M, et al. Stromal microenvironment shapes the intratumoral architecture of pancreatic cancer. Cell. (2019) 178:160–75.e27. doi: 10.1016/j.cell.2019.05.012
32. Bordignon P, Bottoni G, Xu X, Popescu AS, Truan Z, Guenova E, et al. Dualism of FGF and TGF-β signaling in heterogeneous cancer-associated fibroblast activation with ETV1 as a critical determinant. Cell Rep. (2019) 28:2358–72.e6. doi: 10.1016/j.celrep.2019.07.092
33. Sugimoto H, Mundel TM, Kieran MW, Kalluri R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol Ther. (2006) 5:1640–6. doi: 10.4161/cbt.5.12.3354
34. Löhr M, Schmidt C, Ringel J, Kluth M, Müller P, Nizze H, et al. Transforming growth factor-β1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. (2001) 61:550–5.
35. Meng X-M, Nikolic-Paterson DJ, Lan HY. TGF-β: the master regulator of fibrosis. Nat Rev Nephrol. (2016) 12:325–38. doi: 10.1038/nrneph.2016.48
36. Poltavets V, Kochetkova M, Pitson SM, Samuel MS. The role of the extracellular matrix and its molecular and cellular regulators in cancer cell plasticity. Front Oncol. (2018) 8:431. doi: 10.3389/fonc.2018.00431
37. Roberts AB, McCune BK, Sporn MB. TGF-β: regulation of extracellular matrix. Kidney Int. (1992) 41:557–9. doi: 10.1038/ki.1992.81
38. Qin X, Yan M, Zhang J, Wang X, Shen Z, Lv Z, et al. TGFβ3-mediated induction of periostin facilitates head and neck cancer growth and is associated with metastasis. Sci Rep. (2016) 6:20587. doi: 10.1038/srep20587
39. Calon A, Tauriello DVF, Batlle E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin Cancer Biol. (2014) 25:15–22. doi: 10.1016/j.semcancer.2013.12.008
40. Shao Z-M, Nguyen M, Barsky SH. Human breast carcinoma desmoplasia is PDGF initiated. Oncogene. (2000) 19:4337–45. doi: 10.1038/sj.onc.1203785
41. Bishen KA, Radhakrishnan R, Satyamoorthy K. The role of basic fibroblast growth factor in oral submucous fibrosis pathogenesis. J Oral Pathol Med. (2008) 37:402–11. doi: 10.1111/j.1600-0714.2008.00649.x
42. Strutz F, Zeisberg M, Ziyadeh FN, Yang C-Q, Kalluri R, Müller GA, et al. Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation. Kidney Int. (2002) 61:1714–28. doi: 10.1046/j.1523-1755.2002.00333.x
43. Ishiguro S, Akasaka Y, Kiguchi H, Suzuki T, Imaizumi R, Ishikawa Y, et al. Basic fibroblast growth factor induces down-regulation of α-smooth muscle actin and reduction of myofibroblast areas in open skin wounds. Wound Repair Regen. (2009) 17:617–25. doi: 10.1111/j.1524-475X.2009.00511.x
44. Procopio M-G, Laszlo C, Al Labban D, Kim DE, Bordignon P, Jo S-H, et al. Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation. Nat Cell Biol. (2015) 17:1193–204. doi: 10.1038/ncb3228
45. Kartha VK, Stawski L, Han R, Haines P, Gallagher G, Noonan V, et al. PDGFRβ is a novel marker of stromal activation in oral squamous cell carcinomas. PLoS ONE. (2016) 11:e0154645. doi: 10.1371/journal.pone.0154645
46. Siedlecki J, Asani B, Wertheimer C, Hillenmayer A, Ohlmann A, Priglinger C, et al. Combined VEGF/PDGF inhibition using axitinib induces αSMA expression and a pro-fibrotic phenotype in human pericytes. Graefes Arch Clin Exp Ophthalmol. (2018) 256:1141–9. doi: 10.1007/s00417-018-3987-8
47. Costa A, Scholer-Dahirel A, Mechta-Grigoriou F. The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin Cancer Biol. (2014) 25:23–32. doi: 10.1016/j.semcancer.2013.12.007
48. Yang X, Li Y, Zou L, Zhu Z. Role of exosomes in crosstalk between cancer-associated fibroblasts and cancer cells. Front Oncol. (2019) 9:356. doi: 10.3389/fonc.2019.00356
49. Egeblad M, Rasch MG, Weaver VM. Dynamic interplay between the collagen scaffold and tumor evolution. Curr Opin Cell Biol. (2010) 22:697–706. doi: 10.1016/j.ceb.2010.08.015
50. Malik R, Lelkes PI, Cukierman E. Biomechanical and biochemical remodeling of stromal extracellular matrix in cancer. Trends Biotechnol. (2015) 33:230–6. doi: 10.1016/j.tibtech.2015.01.004
51. Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. (2006) 4:38. doi: 10.1186/1741-7015-4-38
52. Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, et al. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am J Pathol. (2011) 178:1221–32. doi: 10.1016/j.ajpath.2010.11.076
53. Sottile J, Hocking DC. Fibronectin polymerization regulates the composition and stability of extracellular matrix fibrils and cell-matrix adhesions. MBoC. (2002) 13:3546–59. doi: 10.1091/mbc.e02-01-0048
54. Velling T, Risteli J, Wennerberg K, Mosher DF, Johansson S. Polymerization of type I and III collagens is dependent on fibronectin and enhanced by integrins α11β1 and α2β1. J Biol Chem. (2002) 277:37377–81. doi: 10.1074/jbc.M206286200
55. Gopal S, Veracini L, Grall D, Butori C, Schaub S, Audebert S, et al. Fibronectin-guided migration of carcinoma collectives. Nat Commun. (2017) 8:14105. doi: 10.1038/ncomms14105
56. Lu Y, Kamel-El Sayed SA, Wang K, Tiede-Lewis LM, Grillo MA, Veno PA, et al. Live imaging of type I collagen assembly dynamics in osteoblasts stably expressing GFP and mCherry-tagged collagen constructs: live imaging of type I collagen assembly. J Bone Miner Res. (2018) 33:1166–82. doi: 10.1002/jbmr.3409
57. Barker TH, Engler AJ. The provisional matrix: setting the stage for tissue repair outcomes. Matrix Biol. (2017) 60–61:1–4. doi: 10.1016/j.matbio.2017.04.003
58. Dallas SL, Sivakumar P, Jones CJP, Chen Q, Peters DM, Mosher DF, et al. Fibronectin regulates latent transforming growth factor-β (TGFβ) by controlling matrix assembly of latent TGFβ-binding protein-1. J Biol Chem. (2005) 280:18871–80. doi: 10.1074/jbc.M410762200
59. Morrison PR, Edsall JT, Miller SG. Preparation and properties of serum and plasma proteins. XVIII. The separation of purified fibrinogen from fraction I of human plasma. J Am Chem Soc. (1948) 70:3103–8. doi: 10.1021/ja01189a080
60. Engel J, Odermatt E, Engel A, Madri JA, Furthmayr H, Rohde H, et al. Shapes, domain organizations and flexibility of laminin and fibronectin, two multifunctional proteins of the extracellular matrix. J Mol Biol. (1981) 150:97–120. doi: 10.1016/0022-2836(81)90326-0
61. Erickson HP, Carrell N, McDonagh J. Fibronectin molecule visualized in electron microscopy: a long, thin, flexible strand. J Cell Biol. (1981) 91:673–8. doi: 10.1083/jcb.91.3.673
62. Xu J, Mosher D. Fibronectin and other adhesive glycoproteins. In: Mecham RP, editor. The Extracellular Matrix: An Overview. Berlin; Heidelberg: Springer Berlin Heidelberg (2011). p. 41–75. doi: 10.1007/978-3-642-16555-9_2
63. Van Obberghen-Schilling E, Tucker RP, Saupe F, Gasser I, Cseh B, Orend G. Fibronectin and tenascin-C: accomplices in vascular morphogenesis during development and tumor growth. Int J Dev Biol. (2011) 55:511–25. doi: 10.1387/ijdb.103243eo
65. Schwarzbauer JE, DeSimone DW. Fibronectins, their fibrillogenesis, and in vivo functions. Cold Spring Harbor Perspect Biol. (2011) 3:a005041. doi: 10.1101/cshperspect.a005041
66. Baron M, Norman D, Willis A, Campbell ID. Structure of the fibronectin type 1 module. Nature. (1990) 345:642–6. doi: 10.1038/345642a0
67. Potts JR, Bright JR, Bolton D, Pickford AR, Campbell ID. Solution structure of the N-terminal F1 module pair from human fibronectin. Biochemistry. (1999) 38:8304–12. doi: 10.1021/bi990202b
68. Dickinson CD, Veerapandian B, Dai X-P, Hamlin RC, Xuong N, Ruoslahti E, et al. Crystal structure of the tenth type III cell adhesion module of human fibronectin. J Mol Biol. (1994) 236:1079–92. doi: 10.1016/0022-2836(94)90013-2
69. Dickinson CD, Gay DA, Parello J, Ruoslahti E, Ely KR. Crystals of the cell-binding module of fibronectin obtained from a series of recombinant fragments differing in length. J Mol Biol. (1994) 238:123–7. doi: 10.1006/jmbi.1994.1272
70. Leahy DJ, Aukhil I, Erickson HP. 2.0 Å crystal structure of a four-domain segment of human fibronectin encompassing the RGD loop and synergy region. Cell. (1996) 84:155–64. doi: 10.1016/S0092-8674(00)81002-8
71. Main AL, Harvey TS, Baron M, Boyd J, Campbell ID. The three-dimensional structure of the tenth type III module of fibronectin: An insight into RGD-mediated interactions. Cell. (1992) 71:671–8. doi: 10.1016/0092-8674(92)90600-H
72. Krammer A, Lu H, Isralewitz B, Schulten K, Vogel V. Forced unfolding of the fibronectin type III module reveals a tensile molecular recognition switch. Proc Natl Acad Sci USA. (1999) 96:1351–6. doi: 10.1073/pnas.96.4.1351
73. Hynes RO. Integrins bidirectional, allosteric signaling machines. Cell. (2002) 110:673–87. doi: 10.1016/S0092-8674(02)00971-6
74. Pytela R, Pierschbacher MD, Ruoslahti E. Identification and isolation of a 140 kd cell surface glycoprotein with properties expected of a fibronectin receptor. Cell. (1985) 40:191–8. doi: 10.1016/0092-8674(85)90322-8
75. Pierschbacher MD, Ruoslahti E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature. (1984) 309:30–3. doi: 10.1038/309030a0
76. Pankov R, Yamada KM. Fibronectin at a glance. J Cell Sci. (2002) 115:3861–3. doi: 10.1242/jcs.00059
77. Danen EHJ, Sonneveld P, Brakebusch C, Fässler R, Sonnenberg A. The fibronectin-binding integrins α5β1 and αvβ3 differentially modulate RhoA-GTP loading, organization of cell matrix adhesions, and fibronectin fibrillogenesis. J Cell Biol. (2002) 159:1071–86. doi: 10.1083/jcb.200205014
78. Zhong C, Chrzanowska-Wodnicka M, Brown J, Shaub A, Belkin AM, Burridge K. Rho-mediated contractility exposes a cryptic site in fibronectin and induces fibronectin matrix assembly. J Cell Biol. (1998) 141:539–51. doi: 10.1083/jcb.141.2.539
79. Wennerberg K, Lohikangas L, Gullberg D, Pfaff M, Johansson S, Fiissler R. 31 Integrin-dependent and -independent polymerization of fibronectin. J Cell Biol. (1996) 132:12. doi: 10.1083/jcb.132.1.227
80. Mao Y, Schwarzbauer JE. Fibronectin fibrillogenesis, a cell-mediated matrix assembly process. Matrix Biol. (2005) 24:389–99. doi: 10.1016/j.matbio.2005.06.008
81. Moyano JV, Carnemolla B, Dominguez-Jimenez C, Garcia-Gila M, Albar JP, Sanchez-Aparicio P, et al. Fibronectin type III5 repeat contains a novel cell adhesion sequence, KLDAPT, which binds activated α4β1 and α4β7 integrins. J Biol Chem. (1997) 272:24832–6. doi: 10.1074/jbc.272.40.24832
82. Komoriya A, Green L, Mervic M, Yamada SS, Yamada KM, Humphries MJ. The minimal essential sequence for a major cell type-specific adhesion site (CS1)within the alternatively spliced type I11 connecting segment domain of fibronectiins leucine-aspartic acid- valine. J Biol Chem. (1991) 266:15075–9.
83. Mould AP, Komoriya A, Yamada KM, Humphries MJ. The CS5 peptide is a second site in the IIICS region of fibronectin recognized by the integrin alpha 4 beta 1. Inhibition of alpha 4 beta 1 function by RGD peptide homologues. J Biol Chem. (1991) 266:3579–85.
84. Humphries JD. Integrin ligands at a glance. J Cell Sci. (2006) 119:3901–3. doi: 10.1242/jcs.03098
85. Dallas SL, Chen Q, Sivakumar P. Dynamics of assembly and reorganization of extracellular matrix proteins. In: Current Topics in Developmental Biology. Elsevier (2006). p. 1–24. Available online at: https://linkinghub.elsevier.com/retrieve/pii/S0070215306750013
86. Lin T-C, Yang C-H, Cheng L-H, Chang W-T, Lin Y-R, Cheng H-C. Fibronectin in cancer: friend or foe. Cells. (2019) 9:27. doi: 10.3390/cells9010027
87. Aguirre KM, McCormick R, Schwarzbauer JE. Fibronectin self-association is mediated by complementary sites within the amino-terminal one-third of the molecule. J Biol Chem. (1994) 269:27863–8.
88. Baneyx G, Baugh L, Vogel V. Fibronectin extension and unfolding within cell matrix fibrils controlled by cytoskeletal tension. Proc Natl Acad Sci USA. (2002) 99:5139–43. doi: 10.1073/pnas.072650799
89. Vogel V. Unraveling the mechanobiology of extracellular matrix. Annu Rev Physiol. (2018) 80:353–87. doi: 10.1146/annurev-physiol-021317-121312
90. Morgan MR, Humphries MJ, Bass MD. Synergistic control of cell adhesion by integrins and syndecans. Nat Rev Mol Cell Biol. (2007) 8:957–69. doi: 10.1038/nrm2289
91. Monaghan E, Gueorguiev V, Wilkins-Port C, McKeown-Longo PJ. The receptor for urokinase-type plasminogen activator regulates fibronectin matrix assembly in human skin fibroblasts. J Biol Chem. (2004) 279:1400–7. doi: 10.1074/jbc.M310374200
92. Saoncella S, Echtermeyer F, Denhez F, Nowlen JK, Mosher DF, Robinson SD, et al. Syndecan-4 signals cooperatively with integrins in a rhodependent manner in the assembly of focal adhesions and actin stress fibers. Proc Natl Acad Sci USA. (1999) 96:2805–10. doi: 10.1073/pnas.96.6.2805
93. Wilcox-Adelman SA, Denhez F, Goetinck PF. Syndecan-4 modulates focal adhesion kinase phosphorylation. J Biol Chem. (2002) 277:32970–7. doi: 10.1074/jbc.M201283200
94. Langenbach KJ, Sottile J. Identification of protein-disulfide isomerase activity in fibronectin. J Biol Chem. (1999) 274:7032–8. doi: 10.1074/jbc.274.11.7032
95. Yamada KM, Kennedy DW, Kimata K, Pratt RM. Characterization of fibronectin interactions with glycosaminoglycans and identification of active proteolytic fragments. J Biol Chem. (1980) 255:6055–63.
96. Barkalow FJB, Schwarzbauer JE. Localization of the major heparin-binding site in fibronectin. J Biol Chem. (1991) 266:7812–8.
97. Moyano JV, Carnemolla B, Albar JP, Leprini A, Gaggero B, Zardi L, et al. Identification of a novel heparin and cell binding sequence in repeat III5. J Biol Chem. (1999) 274:135–42. doi: 10.1074/jbc.274.1.135
98. Mostafavi-Pour Z, Askari JA, Whittard JD, Humphries MJ. Identification of a novel heparin-binding site in the alternatively spliced IIICS region of fibronectin: roles of integrins and proteoglycans in cell adhesion to fibronectin splice variants. Matrix Biol. (2001) 20:63–73. doi: 10.1016/S0945-053X(00)00131-1
99. Woods A. Syndecans: transmembrane modulators of adhesion and matrix assembly. J Clin Invest. (2001) 107:935–41. doi: 10.1172/JCI12802
100. Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. (2010) 123:4195–200. doi: 10.1242/jcs.023820
101. Hynes RO. The extracellular matrix: not just pretty fibrils. Science. (2009) 326:1216–9. doi: 10.1126/science.1176009
102. Umezawa K, Kornblihtt AR, Baralle FE. Isolation and characterization of cDNA clones for human liver fibronectin. FEBS Lett. (1985) 186:31–4. doi: 10.1016/0014-5793(85)81333-8
103. Schwarzbauer JE, Tamkun JW, Lemischka IR, Hynes RO. Three different fibronectin mRNAs arise by alternative splicing within the coding region. Cell. (1983) 35:421–31. doi: 10.1016/0092-8674(83)90175-7
104. Kornblihtt AR, Vibe-Pedersen K, Baralle FE. Human fibronectin: molecular cloning evidence for two mRNA species differing by an internal segment coding for a structural domain. EMBO J. (1984) 3:221–6. doi: 10.1002/j.1460-2075.1984.tb01787.x
105. Gutman A, Kornblihtt AR. Identification of a third region of cell-specific alternative splicing in human fibronectin mRNA. Proc Natl Acad Sci USA. (1987) 84:7179–82. doi: 10.1073/pnas.84.20.7179
106. Schwarzbauer JE, Patel RS, Fonda D, Hynes RO. Multiple sites of alternative splicing of the rat fibronectin gene transcript. EMBO J. (1987) 6:2573–80. doi: 10.1002/j.1460-2075.1987.tb02547.x
107. White E, Baralle F, Muro A. New insights into form and function of fibronectin splice variants. J Pathol. (2008) 216:1–14. doi: 10.1002/path.2388
108. Mamuya W, Chobanian A, Brecher P. Age-related changes in fibronectin expression in spontaneously hypertensive, Wistar-Kyoto, and Wistar rat hearts. Circ Res. (1992) 71:1341–50. doi: 10.1161/01.RES.71.6.1341
109. Wang J, Yin L, Chen Z. New insights into the altered fibronectin matrix and extrasynaptic transmission in the aging brain. J Clin Gerontol Geriatr. (2011) 2:35–41. doi: 10.1016/j.jcgg.2010.12.002
110. Negishi H, Yamada H, Okuyama K, Sagawa T, Makinoda S, Fujimoto S. Fetal fibronectin concentration in amniotic fluid decreases with advancing gestational age. Int J Gynecol Obstet. (1995) 49:17–20. doi: 10.1016/0020-7292(95)02341-9
111. Chauhan AK, Iaconcig A, Baralle FE, Muro AF. Alternative splicing of fibronectin: a mouse model demonstrates the identity of in vitro and in vivo systems and the processing autonomy of regulated exons in adult mice. Gene. (2004) 324:55–63. doi: 10.1016/j.gene.2003.09.026
112. Pagani F. Tissue-specific splicing pattern of fibronectin messenger RNA precursor during development and aging in rat. J Cell Biol. (1991) 113:1223–9. doi: 10.1083/jcb.113.5.1223
113. Magnuson VL, Young M, Schattenberg DG, Mancini MA, Chen D, Steffensen B, et al. The alternative splicing of fibronectin pre-mRNAIs altered during aging andin response to growth factors. J Biol Chem. (1991) 266:14654–62.
114. Ffrench-Constant C. Reappearance of an embryonic pattern of fibronectin splicing during wound healing in the adult rat. J Cell Biol. (1989) 109:903–14. doi: 10.1083/jcb.109.2.903
115. Jarnagin WR. Expression of variant fibronectins in wound healing: cellular source and biological activity of the EIIIA segment in rat hepatic fibrogenesis. J Cell Biol. (1994) 127:2037–48. doi: 10.1083/jcb.127.6.2037
116. Du K, Peng Y, Greenbaum LE, Haber BA, Taub R. HRS/SRp40-mediated inclusion of the fibronectin EIIIB exon, a possible cause of increased EIIIB expression in proliferating liver. Mol Cell Biol. (1997) 17:4096–104. doi: 10.1128/MCB.17.7.4096
117. Ffrench-Constant C. Alternative splicing of fibronectin–many different proteins but few different functions. Exp Cell Res. (1995) 221:261–71. doi: 10.1006/excr.1995.1374
118. Chen Y, Huang Q, Liu W, Zhu Q, Cui C-P, Xu L, et al. Mutually exclusive acetylation and ubiquitylation of the splicing factor SRSF5 control tumor growth. Nat Commun. (2018) 9:2464. doi: 10.1038/s41467-018-04815-3
119. Yang S, Jia R, Bian Z. SRSF5 functions as a novel oncogenic splicing factor and is upregulated by oncogene SRSF3 in oral squamous cell carcinoma. Biochim Biophys Acta. (2018) 1865:1161–72. doi: 10.1016/j.bbamcr.2018.05.017
120. Hallgren O, Malmström J, Malmström L, Andersson-Sjöland A, Wildt M, Tufvesson E, et al. Splicosomal and serine and arginine-rich splicing factors as targets for TGF-β. Fibrogen Tissue Repair. (2012) 5:6. doi: 10.1186/1755-1536-5-6
121. Balza E, Borsi L, Allemanni G, Zardi L. Transforming growth factor β regulates the levels of different fibronectin isoforms in normal human cultured fibroblasts. FEBS Lett. (1988) 228:42–4. doi: 10.1016/0014-5793(88)80580-5
122. Borsi L, Castellani P, Risso AM, Leprini A, Zardi L. Transforming growth factor-β regulates the splicing pattern of fibronectin messenger RNA precursor. FEBS Letters. (1990) 261:175-8. doi: 10.1016/0014-5793(90)80664-5
123. Viedt C, Bürger A, Hänsch GM. Fibronectin synthesis in tubular epithelial cells: up-regulation of the EDA splice variant by transforming growth factor β. Kidney Int. (1995) 48:1810–7. doi: 10.1038/ki.1995.479
124. Muro AF, Moretti FA, Moore BB, Yan M, Atrasz RG, Wilke CA, et al. An essential role for fibronectin extra type III domain A in pulmonary fibrosis. Am J Respir Crit Care Med. (2008) 177:638–45. doi: 10.1164/rccm.200708-1291OC
125. To WS, Midwood KS. Plasma and cellular fibronectin: distinct and independent functions during tissue repair. Fibrogen Tissue Repair. (2011) 4:21. doi: 10.1186/1755-1536-4-21
126. Muro AF, Chauhan AK, Gajovic S, Iaconcig A, Porro F, Stanta G, et al. Regulated splicing of the fibronectin EDA exon is essential for proper skin wound healing and normal lifespan. J Cell Biol. (2003) 162:149–60. doi: 10.1083/jcb.200212079
127. Chauhan AK, Moretti FA, Iaconcig A, Baralle FE, Muro AF. Impaired motor coordination in mice lacking the EDA exon of the fibronectin gene. Behav Brain Res. (2005) 161:31–8. doi: 10.1016/j.bbr.2005.02.020
128. Fukuda T, Yoshida N, Kataoka Y, Manabe R, Mizuno-Horikawa Y, Sato M, et al. Mice lacking the EDB segment of fibronectin develop normally but exhibit reduced cell growth and fibronectin matrix assembly in vitro. Cancer Res. (2002) 62:5603–10.
129. Astrof S, Crowley D, Hynes RO. Multiple cardiovascular defects caused by the absence of alternatively spliced segments of fibronectin. Dev Biol. (2007) 311:11–24. doi: 10.1016/j.ydbio.2007.07.005
130. Serini G, Bochaton-Piallat M-L, Ropraz P, Geinoz A, Borsi L, Zardi L, et al. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-β1. J Cell Biol. (1998) 142:9. doi: 10.1083/jcb.142.3.873
131. Saito S, Yamaji N, Yasunaga K, Saito T, Matsumoto S, Katoh M, et al. The fibronectin extra domain A activates matrix metalloproteinase gene expression by an interleukin-1-dependent mechanism. J Biol Chem. (1999) 274:30756–63. doi: 10.1074/jbc.274.43.30756
132. Glukhova MA, Frid MG, Shekhonin BV, Vasilevskaya TD, Grunwald J, Saginati M, et al. Expression of extra domain A fibronectin sequence in vascular smooth muscle cells is phenotype dependent. J Cell Biol. (1989) 109:357–66. doi: 10.1083/jcb.109.1.357
133. Castellani P, Viale G, Dorcaratto A, Nicolo G, Kaczmarek J, Querze G, et al. The fibronectin isoform containing the ED-B oncofetal domain: a marker of angiogenesis. Int J Cancer. (1994) 59:612–8. doi: 10.1002/ijc.2910590507
134. Manabe R, Oh-e N, Sekiguchi K. Alternatively spliced EDA segment regulates fibronectin-dependent cell cycle progression and mitogenic signal transduction. J Biol Chem. (1999) 274:5919–24. doi: 10.1074/jbc.274.9.5919
135. Liao YF, Gotwals PJ, Koteliansky VE, Sheppard D, Van De Water L. The EIIIA segment of fibronectin is a ligand for integrins α9β1 and α4β1 providing a novel mechanism for regulating cell adhesion by alternative splicing. J Biol Chem. (2002) 277:14467–74. doi: 10.1074/jbc.M201100200
136. Shinde AV, Bystroff C, Wang C, Vogelezang MG, Vincent PA, Hynes RO, et al. Identification of the peptide sequences within the EIIIA (EDA) segment of fibronectin that mediate integrin α9β1-dependent cellular activities. J Biol Chem. (2008) 283:2858–70. doi: 10.1074/jbc.M708306200
137. Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, et al. The extra domain A of fibronectin activates toll-like receptor 4. J Biol Chem. (2001) 276:10229–33. doi: 10.1074/jbc.M100099200
138. Sens C, Huck K, Pettera S, Uebel S, Wabnitz G, Moser M, et al. Fibronectins containing extradomain A or B enhance osteoblast differentiation via distinct integrins. J Biol Chem. (2017) 292:7745–60. doi: 10.1074/jbc.M116.739987
139. Ruoslahti E, Vaheri A. Novel human serum protein from fibroblast plasma membrane. Nature. (1974) 248:789–91. doi: 10.1038/248789a0
140. Keski-Oja J, Vaheri A, Ruoslahti E. Fibroblast surface antigen (SF): THE external glycoprotein lost in proteolytic stimulation and malignant transformation. Int J Cancer. (1976) 17:261–9. doi: 10.1002/ijc.2910170215
141. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. (2000) 100:57–70. doi: 10.1016/S0092-8674(00)81683-9
142. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
143. George EL, Georges-Labouesse EN, Patel-King RS, Rayburn H, Hynes RO. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development. (1993) 119:1079–91.
144. Lenselink EA. Role of fibronectin in normal wound healing: role of fibronectin in normal wound healing. Int Wound J. (2015) 12:313–6. doi: 10.1111/iwj.12109
145. Liao Y-X, Zhang Z-P, Zhao J, Liu J-P. Effects of fibronectin 1 on cell proliferation, senescence and apoptosis of human glioma cells through the PI3K/AKT signaling pathway. Cell Physiol Biochem. (2018) 48:1382–96. doi: 10.1159/000492096
146. Ou Y-C, Li J-R, Wang J-D, Chang C-Y, Wu C-C, Chen W-Y, et al. Fibronectin promotes cell growth and migration in human renal cell carcinoma cells. IJMS. (2019) 20:2792. doi: 10.3390/ijms20112792
147. Cao Y, Liu X, Lu W, Chen Y, Wu X, Li M, et al. Fibronectin promotes cell proliferation and invasion through mTOR signaling pathway activation in gallbladder cancer. Cancer Lett. (2015) 360:141–50. doi: 10.1016/j.canlet.2015.01.041
148. von Au A, Vasel M, Kraft S, Sens C, Hackl N, Marx A, et al. Circulating fibronectin controls tumor growth. Neoplasia. (2013) 15:925–38. doi: 10.1593/neo.13762
149. Kohan M, Muro AF, White ES, Berkman N. EDA-containing cellular fibronectin induces fibroblast differentiation through binding to α 4 β 7 integrin receptor and MAPK/Erk 1/2-dependent signaling. FASEB J. (2010) 24:4503–12. doi: 10.1096/fj.10-154435
150. Nam J-M, Onodera Y, Bissell MJ, Park CC. Breast cancer cells in three-dimensional culture display an enhanced radioresponse after coordinate targeting of integrin alpha5beta1 and fibronectin. Cancer Res. (2010) 70:5238–48. doi: 10.1158/0008-5472.CAN-09-2319
151. Manabe R, Oh-e N, Maeda T, Fukuda T, Sekiguchi K. Modulation of cell-adhesive activity of fibronectin by the alternatively spliced EDA segment. J Cell Biol. (1997) 139:295–307. doi: 10.1083/jcb.139.1.295
152. Lv W-Q, Wang H-C, Peng J, Wang Y-X, Jiang J-H, Li C-Y. Gene editing of the extra domain A positive fibronectin in various tumors, amplified the effects of CRISPR/Cas system on the inhibition of tumor progression. Oncotarget. (2017) 8:105020–36. doi: 10.18632/oncotarget.21136
153. Kenny HA, Chiang C-Y, White EA, Schryver EM, Habis M, Romero IL, et al. Mesothelial cells promote early ovarian cancer metastasis through fibronectin secretion. J Clin Invest. (2014) 124:4614–28. doi: 10.1172/JCI74778
154. Khan ZA, Chan BM, Uniyal S, Barbin YP, Farhangkhoee H, Chen S, et al. EDB fibronectin and angiogenesis–a novel mechanistic pathway. Angiogenesis. (2005) 8:183–96. doi: 10.1007/s10456-005-9017-6
155. Cseh B, Fernandez-Sauze S, Grall D, Schaub S, Doma E, Van Obberghen-Schilling E. Autocrine fibronectin directs matrix assembly and crosstalk between cell-matrix and cell-cell adhesion in vascular endothelial cells. J Cell Sci. (2010) 123:3989–99. doi: 10.1242/jcs.073346
156. Chu L-H, Rivera CG, Popel AS, Bader JS. Constructing the angiome: a global angiogenesis protein interaction network. Physiol Genom. (2012) 44:915–24. doi: 10.1152/physiolgenomics.00181.2011
157. Turner CJ, Badu-Nkansah K, Hynes RO. Endothelium-derived fibronectin regulates neonatal vascular morphogenesis in an autocrine fashion. Angiogenesis. (2017) 20:519–31. doi: 10.1007/s10456-017-9563-8
158. Zhou X, Rowe RG, Hiraoka N, George JP, Wirtz D, Mosher DF, et al. Fibronectin fibrillogenesis regulates three-dimensional neovessel formation. Genes Dev. (2008) 22:1231–43. doi: 10.1101/gad.1643308
159. Murphy PA, Begum S, Hynes RO. Tumor angiogenesis in the absence of fibronectin or its cognate integrin receptors. PLoS ONE. (2015) 10:e0120872. doi: 10.1371/journal.pone.0120872
160. van der Flier A, Badu-Nkansah K, Whittaker CA, Crowley D, Bronson RT, Lacy-Hulbert A, et al. Endothelial 5 and v integrins cooperate in remodeling of the vasculature during development. Development. (2010) 137:2439–49. doi: 10.1242/dev.049551
161. Kaspar M, Zardi L, Neri D. Fibronectin as target for tumor therapy. Int J Cancer. (2006) 118:1331–9. doi: 10.1002/ijc.21677
162. Rybak J-N, Roesli C, Kaspar M, Villa A, Neri D. The extra-domain A of fibronectin is a vascular marker of solid tumors and metastases. Cancer Res. (2007) 67:10948–57. doi: 10.1158/0008-5472.CAN-07-1436
163. Colin C, Baeza N, Bartoli C, Fina F, Eudes N, Nanni I, et al. Identification of genes differentially expressed in glioblastoma versus pilocytic astrocytoma using suppression subtractive hybridization. Oncogene. (2006) 25:2818–26. doi: 10.1038/sj.onc.1209305
164. Serres E, Debarbieux F, Stanchi F, Maggiorella L, Grall D, Turchi L, et al. Fibronectin expression in glioblastomas promotes cell cohesion, collective invasion of basement membrane in vitro and orthotopic tumor growth in mice. Oncogene. (2014) 33:3451–62. doi: 10.1038/onc.2013.305
165. Wijelath ES, Murray J, Rahman S, Patel Y, Ishida A, Strand K, et al. Novel vascular endothelial growth factor binding domains of fibronectin enhance vascular endothelial growth factor biological activity. Circ Res. (2002) 91:25–31. doi: 10.1161/01.RES.0000026420.22406.79
166. Wijelath ES, Rahman S, Namekata M, Murray J, Nishimura T, Mostafavi-Pour Z, et al. Heparin-II domain of fibronectin is a vascular endothelial growth factor-binding domain: enhancement of vegf biological activity by a singular growth factor/matrix protein synergism. Circ Res. (2006) 99:853–60. doi: 10.1161/01.RES.0000246849.17887.66
167. Stenzel D, Lundkvist A, Sauvaget D, Busse M, Graupera M, van der Flier A, et al. Integrin-dependent and -independent functions of astrocytic fibronectin in retinal angiogenesis. Development. (2011) 138:4451–63. doi: 10.1242/dev.071381
168. Franke FE, Von Georgi R, Zygmunt M, Münstedt K. Association between fibronectin expression and prognosis in ovarian carcinoma. Anticancer Res. (2003) 23:4261–7.
169. Shi K, Wang S-L, Shen B, Yu F-Q, Weng D-F, Lin J-H. Clinicopathological and prognostic values of fibronectin and integrin αvβ3 expression in primary osteosarcoma. World J Surg Oncol. (2019) 17:23. doi: 10.1186/s12957-019-1566-z
170. Wang JP, Hielscher A. Fibronectin: how its aberrant expression in tumors may improve therapeutic targeting. J Cancer. (2017) 8:674–82. doi: 10.7150/jca.16901
171. Yi W, Xiao E, Ding R, Luo P, Yang Y. High expression of fibronectin is associated with poor prognosis, cell proliferation and malignancy via the NF-κB/p53-apoptosis signaling pathway in colorectal cancer. Oncol Rep. (2016) 36:3145–53. doi: 10.3892/or.2016.5177
172. Richter P, Junker K, Franz M, Berndt A, Geyer C, Gajda M, et al. IIICS de novo glycosylated fibronectin as a marker for invasiveness in urothelial carcinoma of the urinary bladder (UBC). J Cancer Res Clin Oncol. (2008) 134:1059–65. doi: 10.1007/s00432-008-0390-6
173. Stenman S, Vaheri A. Fibronectin in human solid tumors. Int J Cancer. (1981) 27:427–35. doi: 10.1002/ijc.2910270403
174. Attieh Y, Vignjevic DM. The hallmarks of CAFs in cancer invasion. Eur J Cell Biol. (2016) 95:493–502. doi: 10.1016/j.ejcb.2016.07.004
175. Attieh Y, Clark AG, Grass C, Richon S, Pocard M, Mariani P, et al. Cancer-associated fibroblasts lead tumor invasion through integrin-β3-dependent fibronectin assembly. J Cell Biol. (2017) 216:3509–20. doi: 10.1083/jcb.201702033
176. Erdogan B, Ao M, White LM, Means AL, Brewer BM, Yang L, et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J Cell Biol. (2017) 216:3799–816. doi: 10.1083/jcb.201704053
177. Malik G, Knowles LM, Dhir R, Xu S, Yang S, Ruoslahti E, et al. Plasma fibronectin promotes lung metastasis by contributions to fibrin clots and tumor cell invasion. Cancer Res. (2010) 70:4327–34. doi: 10.1158/0008-5472.CAN-09-3312
178. Meng XN, Jin Y, Yu Y, Bai J, Liu GY, Zhu J, et al. Characterisation of fibronectin-mediated FAK signalling pathways in lung cancer cell migration and invasion. Br J Cancer. (2009) 101:327–34. doi: 10.1038/sj.bjc.6605154
179. Sengupta S, Nandi S, Hindi ES, Wainwright DA, Han Y, Lesniak MS. Short hairpin RNA-mediated fibronectin knockdown delays tumor growth in a mouse glioma model. Neoplasia. (2010) 12:837–47. doi: 10.1593/neo.10662
180. Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. (2019) 20:69–84. doi: 10.1038/s41580-018-0080-4
181. Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. (2002) 2:442–54. doi: 10.1038/nrc822
182. Duband JL, Thiery JP. Appearance and distribution of fibronectin during chick embryo gastrulation and neurulation. Dev Biol. (1982) 94:337–50. doi: 10.1016/0012-1606(82)90352-9
183. Fontana L, Chen Y, Prijatelj P, Sakai T, Fässler R, Sakai LY, et al. Fibronectin is required for integrin αvβ6-mediated activation of latent TGF-β complexes containing LTBP-1. FASEB J. (2005) 19:1798–808. doi: 10.1096/fj.05-4134com
184. Barbazán J, Alonso-Alconada L, Elkhatib N, Geraldo S, Gurchenkov V, Glentis A, et al. Liver metastasis is facilitated by the adherence of circulating tumor cells to vascular fibronectin deposits. Cancer Res. (2017) 77:3431–41. doi: 10.1158/0008-5472.CAN-16-1917
185. Kai F, Drain AP, Weaver VM. The extracellular matrix modulates the metastatic journey. Dev Cell. (2019) 49:332–46. doi: 10.1016/j.devcel.2019.03.026
186. Liu Y, Cao X. Characteristics and significance of the pre-metastatic niche. Cancer Cell. (2016) 30:668–81. doi: 10.1016/j.ccell.2016.09.011
187. Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. (2005) 438:820–7. doi: 10.1038/nature04186
188. Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. (2015) 17:816–26. doi: 10.1038/ncb3169
189. Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. (2009) 15:35–44. doi: 10.1016/j.ccr.2008.11.012
190. Wang Y, Sun Y, Li D, Zhang L, Wang K, Zuo Y, et al. Platelet P2Y12 is involved in murine pulmonary metastasis. PLoS ONE. (2013) 8:e80780. doi: 10.1371/journal.pone.0080780
191. Maquart F-X, Pasco S, Ramont L, Hornebeck W, Monboisse J-C. An introduction to matrikines: extracellular matrix-derived peptides which regulate cell activity. Crit Rev Oncol Hematol. (2004) 49:199–202. doi: 10.1016/j.critrevonc.2003.06.007
192. Kelsh RM, McKeown-Longo PJ. Topographical changes in extracellular matrix: activation of TLR4 signaling and solid tumor progression. Trends Cancer Res. (2013) 9:1–13.
193. Bhattacharyya S, Varga J. Endogenous ligands of TLR4 promote unresolving tissue fibrosis: Implications for systemic sclerosis and its targeted therapy. Immunol Lett. (2018) 195:9–17. doi: 10.1016/j.imlet.2017.09.011
194. Mishra S, Kundu K. Synthesis, characterization and applications of polyacrylamide grafted fenugreek gum (FG-g-PAM) as flocculant: microwave vs thermal synthesis approach. Int J Biol Macromol. (2019) 141:792–808. doi: 10.1016/j.ijbiomac.2019.09.033
195. Lasarte JJ, Casares N, Gorraiz M, Hervás-Stubbs S, Arribillaga L, Mansilla C, et al. The extra domain a from fibronectin targets antigens to TLR4-expressing cells and induces cytotoxic T cell responses in vivo. J Immunol. (2007) 178:748–56. doi: 10.4049/jimmunol.178.2.748
196. Malara A, Gruppi C, Abbonante V, Cattaneo D, De Marco L, Massa M, et al. EDA fibronectin-TLR4 axis sustains megakaryocyte expansion and inflammation in bone marrow fibrosis. J Exp Med. (2019) 216:587–604. doi: 10.1084/jem.20181074
197. Kelsh R, You R, Horzempa C, Zheng M, McKeown-Longo PJ. Regulation of the innate immune response by fibronectin: synergism between the III-1 and EDA domains. PLoS ONE. (2014) 9:e102974. doi: 10.1371/journal.pone.0102974
198. Smith ML, Gourdon D, Little WC, Kubow KE, Eguiluz RA, Luna-Morris S, et al. Force-induced unfolding of fibronectin in the extracellular matrix of living cells. PLoS Biol. (2007) 5:e268. doi: 10.1371/journal.pbio.0050268
199. McFadden JP, Basketter DA, Dearman RJ, Kimber IR. Extra domain A-positive fibronectin-positive feedback loops and their association with cutaneous inflammatory disease. Clin Dermatol. (2011) 29:257–65. doi: 10.1016/j.clindermatol.2010.11.003
200. Galon J. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. (2006) 313:1960–4. doi: 10.1126/science.1129139
201. Mlecnik B, Tosolini M, Kirilovsky A, Berger A, Bindea G, Meatchi T, et al. Histopathologic-based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. JCO. (2011) 29:610–8. doi: 10.1200/JCO.2010.30.5425
202. Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. (2003) 348:203–13. doi: 10.1056/NEJMoa020177
203. Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. (2012) 12:298–306. doi: 10.1038/nrc3245
204. Barnes TA, Amir E. HYPE or HOPE: the prognostic value of infiltrating immune cells in cancer. Br J Cancer. (2017) 117:451–60. doi: 10.1038/bjc.2017.220
205. Garner H, de Visser K. Immune crosstalk in cancer progression and metastatic spread: a complex conversation. Nat Rev Immunol. (in press). doi: 10.1038/s41577-019-0271-z
206. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. (2017) 541:321–30. doi: 10.1038/nature21349
207. Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean M-C, Validire P, Trautmann A, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest. (2012) 122:899–910. doi: 10.1172/JCI45817
208. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. (2015) 348:74–80. doi: 10.1126/science.aaa6204
209. Peranzoni E, Rivas-Caicedo A, Bougherara H, Salmon H, Donnadieu E. Positive and negative influence of the matrix architecture on antitumor immune surveillance. Cell Mol Life Sci. (2013) 70:4431–48. doi: 10.1007/s00018-013-1339-8
210. Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc Natl Acad Sci USA. (2018) 115:E4041–50. doi: 10.1073/pnas.1720948115
211. Varol C, Sagi I. Phagocyte-extracellular matrix crosstalk empowers tumor development and dissemination. FEBS J. (2018) 285:734–51. doi: 10.1111/febs.14317
212. Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. (2009) 8:3984–4001. doi: 10.4161/cc.8.23.10238
213. Sullivan WJ, Mullen PJ, Schmid EW, Flores A, Momcilovic M, Sharpley MS, et al. Extracellular matrix remodeling regulates glucose metabolism through TXNIP destabilization. Cell. (2018) 175:117–32.e21. doi: 10.1016/j.cell.2018.08.017
214. Morris BA, Burkel B, Ponik SM, Fan J, Condeelis JS, Aguirre-Ghiso JA, et al. Collagen matrix density drives the metabolic shift in breast cancer cells. EBio Med. (2016) 13:146–56. doi: 10.1016/j.ebiom.2016.10.012
215. Xu S, Xu H, Wang W, Li S, Li H, Li T, et al. The role of collagen in cancer: from bench to bedside. J Transl Med. (2019) 17:309. doi: 10.1186/s12967-019-2058-1
216. Kato Y, Ozawa S, Miyamoto C, Maehata Y, Suzuki A, Maeda T, et al. Acidic extracellular microenvironment and cancer. Cancer Cell Int. (2013) 13:89. doi: 10.1186/1475-2867-13-89
217. Gortan Cappellari G, Barazzoni R, Cattin L, Muro AF, Zanetti M. Lack of fibronectin extra domain A alternative splicing exacerbates endothelial dysfunction in diabetes. Sci Rep. (2016) 6:37965. doi: 10.1038/srep37965
218. Du H, Wang Y, Zhang Z, Yang J, Zhang J, Zhang Y. Fibronectin overexpression modulates formation of macrophage foam cells by activating SREBP2 involved in endoplasmic reticulum stress. Cell Physiol Biochem. (2015) 36:1821–34. doi: 10.1159/000430153
219. Rick JW, Chandra A, Dalle Ore C, Nguyen AT, Yagnik G, Aghi MK. Fibronectin in malignancy: cancer-specific alterations, protumoral effects, and therapeutic implications. Semin Oncol. (2019) 46:284–90. doi: 10.1053/j.seminoncol.2019.08.002
220. Kumra H, Reinhardt DP. Fibronectin-targeted drug delivery in cancer. Adv Drug Deliv Rev. (2016) 97:101–10. doi: 10.1016/j.addr.2015.11.014
221. Johannsen M, Spitaleri G, Curigliano G, Roigas J, Weikert S, Kempkensteffen C, et al. The tumour-targeting human L19-IL2 immunocytokine: preclinical safety studies, phase I clinical trial in patients with solid tumours and expansion into patients with advanced renal cell carcinoma. Eur J Cancer. (2010) 46:2926–35. doi: 10.1016/j.ejca.2010.07.033
222. Eigentler TK, Weide B, de Braud F, Spitaleri G, Romanini A, Pflugfelder A, et al. A dose-escalation and signal-generating study of the immunocytokine L19-IL2 in combination with dacarbazine for the therapy of patients with metastatic melanoma. Clin Cancer Res. (2011) 17:7732–42. doi: 10.1158/1078-0432.CCR-11-1203
223. Weide B, Eigentler T, Catania C, Ascierto PA, Cascinu S, Becker JC, et al. A phase II study of the L19IL2 immunocytokine in combination with dacarbazine in advanced metastatic melanoma patients. Cancer Immunol Immunother. (2019) 68:1547–59. doi: 10.1007/s00262-019-02383-z
224. Rekers NH, Zegers CM, Germeraad WT, Dubois L, Lambin P. Long-lasting antitumor effects provided by radiotherapy combined with the immunocytokine L19-IL2. Oncoimmunology. (2015) 4:e1021541. doi: 10.1080/2162402X.2015.1021541
225. Arribillaga L, Durantez M, Lozano T, Rudilla F, Rehberger F, Casares N, et al. A fusion protein between streptavidin and the endogenous TLR4 ligand EDA targets biotinylated antigens to dendritic cells and induces T cell responses in vivo. BioMed Res Int. (2013) 2013:1–9. doi: 10.1155/2013/864720
226. Mansilla C, Berraondo P, Durantez M, Martínez M, Casares N, Arribillaga L, et al. Eradication of large tumors expressing human papillomavirus E7 protein by therapeutic vaccination with E7 fused to the extra domain a from fibronectin. Int J Cancer. (2012) 131:641–51. doi: 10.1002/ijc.26412
227. Rudilla F, Fayolle C, Casares N, Durantez M, Arribillaga L, Lozano T, et al. Combination of a TLR4 ligand and anaphylatoxin C5a for the induction of antigen-specific cytotoxic T cell responses. Vaccine. (2012) 30:2848–58. doi: 10.1016/j.vaccine.2012.02.052
228. Julier Z, Martino MM, de Titta A, Jeanbart L, Hubbell JA. The TLR4 agonist fibronectin extra domain A is cryptic, exposed by elastase-2; use in a fibrin matrix cancer vaccine. Sci Rep. (2015) 5:8569. doi: 10.1038/srep08569
229. Schaffner F, Ray A, Dontenwill M. Integrin α5β1, the fibronectin receptor, as a pertinent therapeutic target in solid tumors. Cancers. (2013) 5:27–47. doi: 10.3390/cancers5010027
230. Bell-McGuinn KM, Matthews CM, Ho SN, Barve M, Gilbert L, Penson RT, et al. A phase II, single-arm study of the anti-α5β1 integrin antibody volociximab as monotherapy in patients with platinum-resistant advanced epithelial ovarian or primary peritoneal cancer. Gynecol Oncol. (2011) 121:273–9. doi: 10.1016/j.ygyno.2010.12.362
231. Jin H, Su J, Garmy-Susini B, Kleeman J, Varner J. Integrin α4β1 promotes monocyte trafficking and angiogenesis in tumors. Cancer Res. (2006) 66:2146–52. doi: 10.1158/0008-5472.CAN-05-2704
232. Oh J, Magnuson A, Benoist C, Pittet MJ, Weissleder R. Age-related tumor growth in mice is related to integrin α4 in CD8+ T cells. JCI Insight. (2018) 3:e122961. doi: 10.1172/jci.insight.122961
233. Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. (2010) 10:9–22. doi: 10.1038/nrc2748
234. Batlle E, Massagué J. Transforming Growth factor-β signaling in immunity and cancer. Immunity. (2019) 50:924–40. doi: 10.1016/j.immuni.2019.03.024
235. Sethi T, Rintoul RC, Moore SM, MacKinnon AC, Salter D, Choo C, et al. Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: a mechanism for small cell lung cancer growth and drug resistance in vivo. Nat Med. (1999) 5:662–8. doi: 10.1038/9511
236. Hazlehurst L, Damiano J, Buyuksal I, Pledger W, Dalton W. Adhesion to fibronectin via β1 integrins regulates p27kip1 levels and contributes to cell adhesion mediated drug resistance (CAM-DR). Oncogene. (2000) 19:4319–27. doi: 10.1038/sj.onc.1203782
237. Eke I, Storch K, Krause M, Cordes N. Cetuximab attenuates its cytotoxic and radiosensitizing potential by inducing fibronectin biosynthesis. Cancer Res. (2013) 73:5869–79. doi: 10.1158/0008-5472.CAN-13-0344
238. Matsunaga T, Takemoto N, Sato T, Takimoto R, Tanaka I, Fujimi A, et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med. (2003) 9:1158–65. doi: 10.1038/nm909
239. Eke I, Sandfort V, Storch K, Baumann M, Röper B, Cordes N. Pharmacological inhibition of EGFR tyrosine kinase affects ILK-mediated cellular radiosensitization in vitro. Int J Radiat Biol. (2007) 83:793–802. doi: 10.1080/09553000701727549
240. Hehlgans S, Haase M, Cordes N. Signalling via integrins: Implications for cell survival and anticancer strategies. Biochim Biophys Acta. (2007) 1775:163–80. doi: 10.1016/j.bbcan.2006.09.001
241. Estrugo D, Fischer A, Hess F, Scherthan H, Belka C, Cordes N. Ligand bound β1 integrins inhibit procaspase-8 for mediating cell adhesion-mediated drug and radiation resistance in human leukemia cells. PLoS ONE. (2007) 2:e269. doi: 10.1371/journal.pone.0000269
242. Senthebane D, Jonker T, Rowe A, Thomford N, Munro D, Dandara C, et al. The role of tumor microenvironment in chemoresistance: 3D extracellular matrices as accomplices. IJMS. (2018) 19:2861. doi: 10.3390/ijms19102861
Keywords: tumor, extracellular matrix, fibronectin, oncofetal variants, cancer hallmarks
Citation: Efthymiou G, Saint A, Ruff M, Rekad Z, Ciais D and Van Obberghen-Schilling E (2020) Shaping Up the Tumor Microenvironment With Cellular Fibronectin. Front. Oncol. 10:641. doi: 10.3389/fonc.2020.00641
Received: 24 February 2020; Accepted: 06 April 2020;
Published: 30 April 2020.
Edited by:
Vasiliki Gkretsi, European University Cyprus, CyprusReviewed by:
Oliver Kepp, Institut Gustave Roussy, FranceGiuseppina Comito, University of Florence, Italy
Copyright © 2020 Efthymiou, Saint, Ruff, Rekad, Ciais and Van Obberghen-Schilling. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ellen Van Obberghen-Schilling, dmFub2JiZXJAdW5pY2UuZnI=
†These authors have contributed equally to this work