 Brita Singers Sørensen
Brita Singers Sørensen Michael R. Horsman
Michael R. Horsman
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 21 April 2020
Sec. Radiation Oncology
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.00562
This article is part of the Research Topic Cell Signaling Mediating Critical Radiation Responses View all 15 articles
Tumor hypoxia is a common feature of the microenvironment in solid tumors, primarily due to an inadequate, and heterogeneous vascular network. It is associated with resistance to radiotherapy and results in a poorer clinical outcome. The presence of hypoxia in tumors can be identified by various invasive and non-invasive techniques, and there are a number of approaches by which hypoxia can be modified to improve outcome. However, despite these factors and the ongoing extensive pre-clinical studies, the clinical focus on hypoxia is still to a large extent lacking. Hypoxia is a major cellular stress factor and affects a wide range of molecular pathways, and further understanding of the molecular processes involved may lead to greater clinical applicability of hypoxic modifiers. This review is a discussion of the characteristics of tumor hypoxia, hypoxia-related molecular pathways, and the role of hypoxia in treatment resistance. Understanding the molecular aspects of hypoxia will improve our ability to clinically monitor hypoxia and to predict and modify the therapeutic response.
Normal tissues require a regular supply of oxygen and nutrients to maintain viability, and a means for eliminating the waste products of metabolism (1, 2). These processes are achieved through a functional blood supply. Most solid tumors have the same metabolic requirements and to achieve this tumors initially utilize the blood supply of the host organ in which the tumor arises. Eventually that supply becomes inadequate in meeting the demands of the growing tumor mass (1, 2). To compensate, tumors develop their own functional vascular supply from the normal host vascular network by the process of angiogenesis (3, 4). However, despite the significance of this tumor neo-vasculature, the system formed is chaotic and primitive, suffering from numerous structural, and functional abnormalities (1, 2) (Figure 1). Consequently, it is actually unable to meet the metabolic demands of the developing tumor. Micro-regional areas are thus formed within the tumor that are characterized by glucose and energy deprivation, high lactate levels and extracellular acidity, and oxygen deficiency (1, 2).
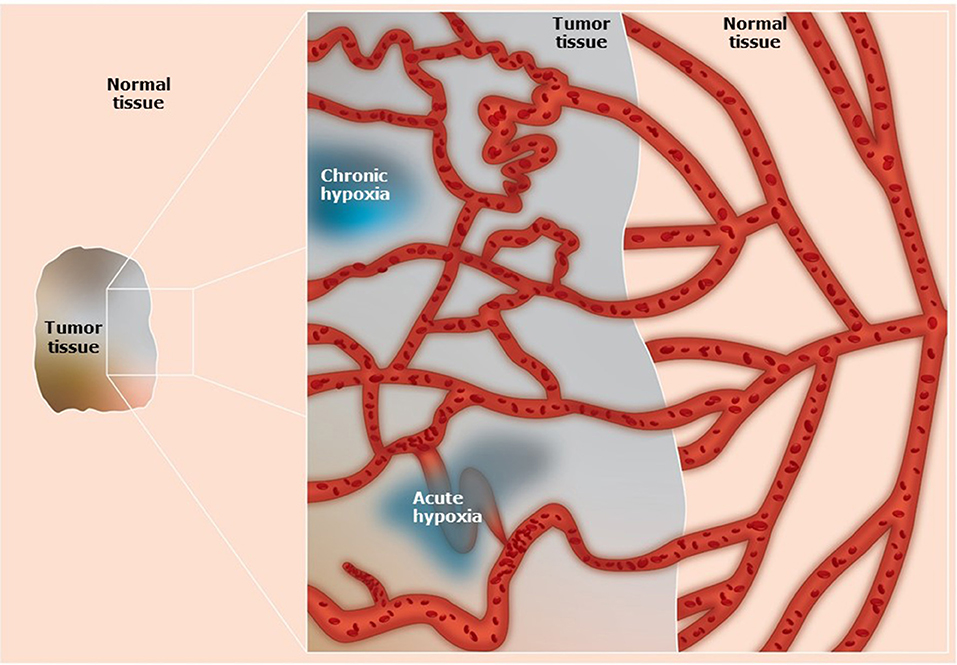
Figure 1. Schematic illustration of the vascular networks in tumors and associated normal tissues. Compared to the well-organized blood supply of normal tissues, in tumors the system is primitive and chaotic. The tumor vascular supply shows abnormal vascular density, contour irregularities, enlarged vessels, vessels with blind ends, and transiently blocked vessels. In addition, there is a loss of hierarchy, a lack of regulatory control mechanisms, and the vessel walls can be structurally defective causing increased vascular permeability. These factors result in the development of diffusion limited chronic hypoxia and perfusion limited acute hypoxia.
The most extensively studied micro-environmental parameter is hypoxia. Hypoxia is a characteristic feature of most solid tumors and is generally defined as a state of reduced oxygenation that influences biological function (5). As such, it is usually considered as a single entity, which it most definitely is not. From histological data from patients with carcinoma of the bronchus, Thomlinson & Gray suggested that hypoxia could be present in tumors due to a diffusion limit of oxygen (6). Such hypoxia would be chronic in nature (Figure 1). Later it was proposed (7) and demonstrated (8) that a form of acute/transient hypoxia could occur, resulting from periodic fluctuations in blood flow (9) (Figure 1). This acute hypoxia can result from a complete shut-down in tumor blood flow thus causing ischemic hypoxia, or from a partial shut-down sufficient to induce hypoxia by preventing red blood cell flow yet allowing plasma flow to continue to supply nutrients. The cause of chronic hypoxia can also be multi-factorial. Although the result of a diffusion limitation, the actual distance from the blood vessel can be highly variable due to several factors. These include the oxygen carrying capacity of the blood, which can be “normal” or reduced as in anemic patients or smokers and the ability of hemoglobin to release oxygen (10). It also involves the intravascular oxygen partial pressure gradient (from the arterial to the venous end of the micro-vessels), and the level of oxygen consumption by the tumor cells and the tumor growth fraction, both of which can vary within and between tumors (10). One also has to consider the degree of oxygenation, which can vary from reasonably well-oxygenated through intermediate levels of hypoxia to severely hypoxic (11).
Hypoxia is a major cellular stress factor and in response to this condition, cells undergo a wide range of molecular changes. A number of cellular pathways are affected, including increased glycolysis, decrease of cell proliferation, and enhancement processes involved in angiogenesis and erythropoiesis (Figure 2). The hypoxia-mediated intracellular signaling pathways are pre-dominantly orchestrated by intracellular signaling, mainly under control by a family of transcription factors, the hypoxia inducible factors (HIFs) (12, 13). HIF upregulates target gene expression through binding at the hypoxia responsive elements (HREs) in the enhancer and promotor regions of the target genes (14). HIF binds to the DNA as a heterodimer consisting of a alpha (α) subunit (HIF-1α, HIF-2α, or HIF-3α) and a HIF-1β subunit (15). HIF-1β is constitutively expressed, while regulation of HIFα is controlled by tissue oxygenation status, through hydroxylation of two proline residues by prolyl hydroxylase domain proteins (PHD) 1-3 (16, 17) (Figure 2). Hydroxylation, occurring only in the presence of oxygen, promotes interaction with the von Hippel-Lindau tumor suppressor protein (pVHL), which targets HIFα for ubiquitination and subsequent proteasomal degradation (18, 19). At oxygen concentrations around 2% O2 and below this hydroxylation is suppressed leading HIFα to not be degraded (20, 21), and form the active transcription complex with HIF-1β, which induce transcriptional upregulation of a broad range of target genes (22–24). The regulation of HIF-α is not only affected by PHD1-3, since a large plethora of kinases are also involved in the regulation, either directly, or indirectly (15). The major HIF complexes are comprised of HIF-1β, and one of either HIF-1α or HIF-2α, which constitutes the transcription factors referred to as HIF1 and HIF2 (25). HIF1 and HIF2 have structural similarities and identical DNA recognition motifs, but binds to different cell-specific sites across the genome (26, 27). HIF-3α has a structural difference, in that it lacks the C-terminal TAD, and as such is not able to induce the expression of hypoxia-inducible target genes to the same extent as HIF-1α and HIF-2α. HIF-3α competes with HIF-1α or HIF-2α to bind HIF-1β, and can thereby act as a suppressor of HIF-dependent gene expression (25, 28). The HIFs have been shown to influence a large range of cellular functions (Figure 2), such as angiogenesis, invasion and metastasis, apoptosis and autophagy, metabolism, intracellular acidosis, and tumor immunity.
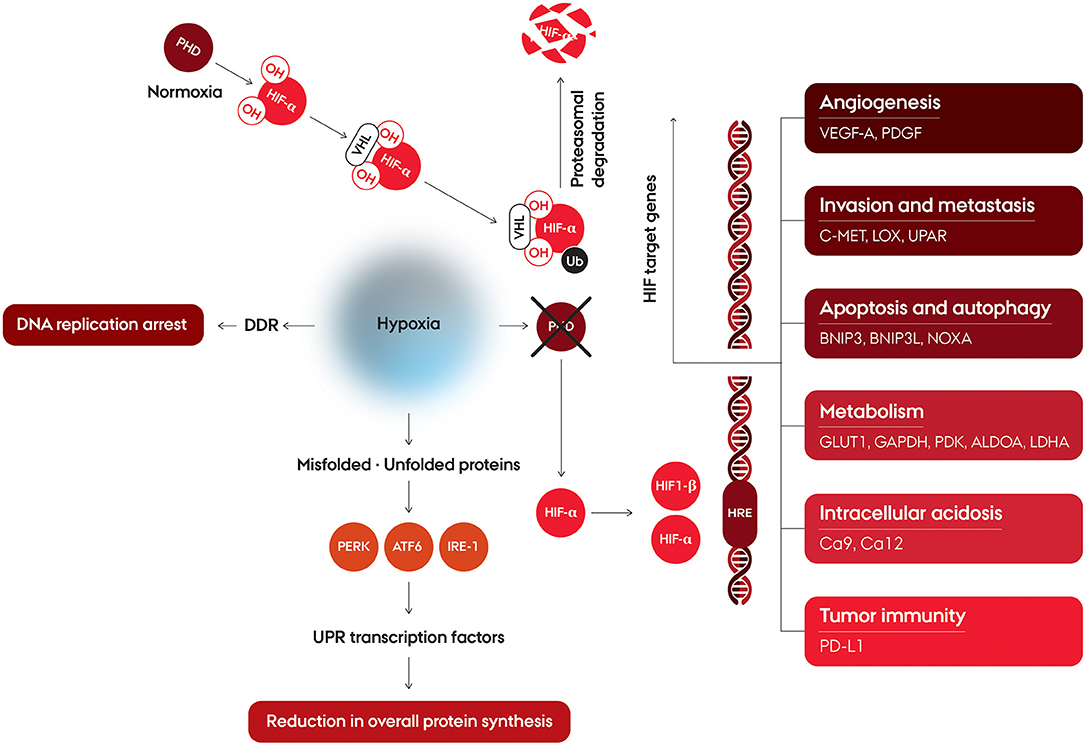
Figure 2. Schematic illustration of cellular pathways affected by hypoxia. Hypoxia affects regulation of hypoxia-inducible factors and induction of HIF target. HIF is a heterodimer consisting of an alpha (α) subunit (HIF-1α, HIF-2α or HIF-3α) and a HIF-1β subunit. Under normoxic conditions, HIF-α is rapidly degraded due to hydroxylation by prolyl hydroxylase domain (PHD) protein. The proline-hydroxylated HIF-α interacts with the von Hippel-Lindau protein (VHL), which targets HIF-α for ubiquitination and degradation via the proteasome. Under hypoxia, HIF-α is stable, and forms the active transcription complex with HIF-1β. After translocation to the nucleus the HIF heterodimer binds at the hypoxia response element (HRE) of target genes thereby initiating the transcription of the HIF target genes. At severe hypoxia, the cellular response also affects the DNA Damage Response (DDR), which leads to DNA replication arrest. Exposure to very low oxygen concentrations also leads to an reduction in mRNA translation initiation and overall protein synthesis, through an activation of the Untranslated Protein Response (UPR).
Cancer cells adapt to hypoxia by a number of stress responses, mediated by the intracellular signaling aimed at facilitating the cells ability to cope with the microenvironment, and to alter the energy requirements as necessary. One of the stress responses is the unfolded protein response (UPR) activated in response to ER stress, endoplasmic reticulum stress, and leads to a downstream activation of adaptive mechanisms. ER stress is the result of an accumulation of unfolded or misfolded proteins, as oxygen depletion can interfere with protein folding (29, 30) Unlike HIF, which is activated at oxygen concentrations below 2%, UPR is activated at exposure to more severe hypoxia (<0.02% O2) (31). The UPR is a complex of intracellular signaling pathways which are mediated by three independent ER transmembrane proteins: PKR-like ER kinase (PERK), Activating Transcription Factor 6 (ATF6) and inositol-requiring enzyme 1(IRE-1) (32). Exposure to severe hypoxia leads to a reduction in mRNA translation initiation and overall protein synthesis, through a activation of PERK which subsequently phosphorylates eIF2α (33) (Figure 2). Activated ATF6 and IRE-11 directly modulates transcriptional induction of UPR target genes. Activation of IRE-1 leads to expression of a panel of genes maintaining metabolic homeostasis and ER through activation of a transcription factor, spliced XBP1 (XBP1s) (34–36). ATF6 is cleaved in the Golgi apparatus, where after the active transcriptional form, ATF6f, translocate to the nucleus and induce transcription of the UPR target genes (29, 37, 38).
Translation of the majority of genes is inhibited under these conditions, but due to regulatory sequences in the 5′ untranslated regions, some gene transcripts are able to escape this inhibition, resulting in an alteration in differential protein expression during hypoxia due to the change in translational efficiency (33, 39).
UPR has been suggested to induce autophagy, an intracellular self-degradation process which can both induce or protect from cell death, through the PERK and BNIP3 pathways (40, 41). The impact of hypoxia on autophagy pathways in malignant cells, and the balance of autophagy in survival and death pathways under hypoxia has shown to be complex. It is susceptible to the genetic background of the cells, as well as the severity of the oxygen deprivation and of other tumor microenvironmental factors (40, 41).
The cellular response to hypoxia also affects the DNA Damage Response (DDR) at very low oxygen concentrations, which includes DNA replication arrest and rapid accumulation of replication stress (Figure 2). This is thought to be due to the enzyme responsible for nucleotide production, ribonucleotide reductase, being dependent on cellular oxygen for its function and, therefore, compromised in hypoxic conditions (32, 42). The DDR involves a complex collaboration between signaling pathways activated due to different types of DNA damaging stresses, and the hypoxia induced effects includes both ATR- and ATM-mediated signaling, despite the absence of detectable DNA damage. This results in cellular protection of the replication forks, minimizing the risk of further genomic instability (42–44). Activation of p53 is a consequence of the hypoxia induced DDR, by phosphorylation at a number of residues (45, 46).
The tumor microenvironment is characterized by factors other than tumor hypoxia, such as low pH. Lactic acid accumulation can cause acidosis in solid tumors. In order to compensate for reduced mitochondrial ATP, low oxygen concentrations leads to anaerobic energy production and the formation of lactic acid production, referred to as the Pasteur effect (47). Significant disparities in the temporal and spatial distribution of areas in tumors with low oxygenation level and high level of acidosis results from tumor cells maintaining a high rate of glycolysis even in the presence of oxygen, a which is referred to as aerobic glycolysis or the Warburg effect (48–50). The cellular response in terms of DNA repair and gene transcription and translation succeeding combination of low oxygen concentration and low extracellular pH in combination has shown to be very different compared to the response to either hypoxia or acidosis alone (51, 52). While both hypoxia and acidosis greatly effects the cellular response, simultaneous hypoxia and acidosis in vitro suppresses metabolic rate and protein synthesis to a greater extent than each of the factors on their own (53).
Cancer immunotherapy has resulted in unprecedented improvements in outcome in patients with a spectrum of solid tumors, and has established itself as the fourth modality in cancer treatment. This is primarily the result of development of vaccines and agents targeting immune regulatory checkpoints, namely the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), or programmed death 1 (PD-1) and programmed death 1 ligand (PD-L1) (54). Despite positive results, many patients show little or no response to vaccines and checkpoint inhibitors (55). The immune response to tumors is a complex balance between antitumor mechanisms, where infiltrating lymphocytes recognize tumor specific antigens on the surface of cancer cells and eliminate the cancer cells thereby decrease tumor growth, and the protumor inflammatory response, which increases immune tolerance, cell survival, and proliferation (56–58). There is evidence that radiation alone can induce an innate immune response, and recent studies have shown that the combination of radiotherapy with immunotherapy has the potential to be an effective treatment modality (59–61).
Hypoxia seems to play a significant role in influencing anti-cancer immune responses (62, 63). It promotes an immunosuppressive microenvironment by regulating the recruitment of T-cells, myeloid-derived suppressor cells (MDSCs), macrophages, and neutrophils (64, 65). In addition, hypoxia can have a negative effect on immunogenicity by altering the function of immune cells and/or increasing resistance of tumor cells to the cytolytic activity of immune effectors (66, 67). There is also evidence that hypoxia can influence immune checkpoints. A rapid and selective up-regulation of PD-L1 is induced by hypoxia on MDSCs, and significant increased expression of PD-L1 on macrophages, dendritic cells and tumor cells, all due to HIF1 binding directly to the HRE in the PD-L1 proximal promoter (68). Hypoxia has also been shown to regulate the CTLA-4 receptor, again potentially via HIF1 (69). Apart from direct immune suppressive effects, hypoxia can also indirectly affect immune response since it causes an increased accumulation of adenosine, drives the expression of vascular endothelial growth factor, and is associated with higher levels of lactate, all of which can inhibit anti-tumor immunity (62, 70). Interestingly, one pre-clinical study using a variety of tumor models showed that by allowing tumor-bearing mice to breathe high oxygen content gas (60% oxygen) rather than the normal 21% oxygen, resulted in an inhibition of tumor progression, a decrease in metastatic disease, and prolonged animal survival (67). This hyperoxia decreased tumor hypoxia, increased pro-inflammatory cytokines, decreased the levels of immunosuppressive molecules, and weakened immunosuppression by regulatory T-cells.
Clearly, there is a need to investigate role of hypoxia on immune response and understand how modifiers of hypoxia influence that response. Non-invasive imaging may be helpful in this context. Substantial pre-clinical and clinical effort has been made in finding clinically relevant approaches that can non-invasively identify hypoxia in tumors (71). The techniques include positron emission tomography (PET), magnetic resonance imaging, and computed tomography. Using these techniques, especially the PET-based approaches, one not only identifies tumor hypoxia, but also shows its relationship to patient outcome following radiotherapy (71). More recently, a PET based approach has also been developed for non-invasively imaging immunotherapy. It involves radiolabeling various monoclonal antibodies with 89-Zirconium (89Zr). Pre-clinically, these conjugates have included CD4 and CD8 antibodies (72), or an anti-PD-L1 antibody (73). Both approaches allowed for whole body visualization and evaluation of tumor response. Such approaches have even undergone clinical evaluation using 89Zr-labeled atezolizumab, an antibody against PD-L1, and the images obtained in cancer patients was able to assess response to PD-L1 blockade (74). Combining PET-hypoxia markers with immunotherapy based PET markers should allow us to investigate the interaction between both parameters and how that influences patient outcome.
Estimates of tumor hypoxia obtained using electrodes, exogenous marker expression, or the upregulation of endogenous hypoxia-associated molecules, have not only demonstrated hypoxia to be a common feature of animal solid tumors, human tumor xenografts and human cancers (49, 75), but also a major negative factor influencing tumor radiation response. Pre-clinical studies in the early 1950s demonstrated that when the partial pressure of oxygen was reduced below about 20 mmHg at the time of irradiation cells became resistant to the radiation damage (76). When radiation is absorbed in biological material, highly reactive free radicals are produced either directly or indirectly in the target. These radicals are unstable and will react with oxygen to change the chemical composition of the target, ultimately causing damage. However, under hypoxic conditions the target can be chemically restored to its original form. Typically, under hypoxia one requires 2.5–3.0 fold higher radiation doses to induce the same level of damage as seen under normoxic conditions (77). The type of hypoxia (i.e., chronic or acute) is irrelevant for the initial radioprotection. However, while chronically hypoxic cells are generally also nutrient deprived, acutely hypoxic cells are hypoxic for only a short period (78) and as such are less likely to be nutrient deprived, and this could play a role in influencing the cells ability to repair the radiation damage, thus making acute hypoxia a more resistant factor.
Regardless of whether one type of hypoxia is more of a negative factor, there is good clinical evidence that hypoxia significantly impacts patient outcome following radiation therapy (71). Consequently, substantial effort has been made in the last 50 years to identify approaches that can overcome hypoxia-induced radiation resistance (1, 71). These have involved using agents that either increase oxygen delivery, radiosensitize the hypoxic cells, or preferentially kill them. Attempts have also been made to use dose painting, whereby the hypoxic areas are identified and the radiation dose to these areas is increased, or the use of high LET (linear energy transfer) radiation where hypoxia is less of an issue (79). However, despite the pre-clinical and even clinical demonstrations of the benefit of several of these approaches, only one approach has become established in routine clinical practice and that is the hypoxic cell radiosensitizer nimorazole, and only in head & neck squamous cell carcinoma and only in Denmark (80) and Norway (81).
A principal factor controlling the tumor microenvironment, and thus the degree of hypoxia, is its vascular supply. As a result, any treatment that modifies this tumor vascular supply can consequently change the level of hypoxia. One such group are the so-called vascular targeting agents (VTAs). These include angiogenesis inhibitors (AIs) that inhibit the development of the tumor neo-vasculature, and vascular disrupting agents (VDAs) that damage the already establish tumor vascular supply (82, 83).
With VDAs, the vascular damage induced causes a reduction in tumor blood flow and this increases the adverse microenvironmental conditions within tumors leading to substantial cell killing and subsequent increase in necrosis (82, 84, 85). The overall result is a reduction in tumor volume. AIs also inhibit tumor growth, but their effects on the tumor vascular supply and microenvironment are more complex and somewhat controversial. Some years ago it was suggested that rather than AIs simply stopping the angiogenesis process and thus decreasing vessel density they could also actually reduce or abolish the vascular abnormalities of the remaining vessels, causing vessel stabilization resulting in a more efficient vasculature similar to that seen in normal tissues. This stabilization process was termed “normalization” (86) and the more stable, organized vasculature that resulted would likely lead to a better delivery of oxygen and nutrients to the tumor, thus reducing the degree of tumor hypoxia. Numerous studies have since reported that treatment with a range of AIs can indeed give rise to an apparent decrease in tumor hypoxia (82, 84). The first study that demonstrated an improvement in oxygenation status that was associated with vessel normalization was that of Winkler and colleagues (87), using the anti-VEGF (vascular endothelial growth factor) monoclonal antibody DC101. Using a human glioblastoma xenograft grown orthotopically in the mouse brain they found that during treatment with DC101 there was a significant decrease in the level of binding of the hypoxic marker, pimonidazole, and a similar increase in radiation sensitivity, an affect that was clearly associated with pericyte recruitment. They also found that when pericyte coverage was maximal there was an upregulation of human angiopoietin-1 (Ang-1) and Ephrin B2. Ang-1 is associated with pericyte recruitment and additional studies showed that an increased synthesis of Ang-1 mRNA resulted in an increased Ang-1 protein deposition close to its receptor Tie2 on the endothelial cells (87). Furthermore, when using a Tie2-blocking antibody or peptide to block Ang-1/Tie2 signaling, DC101 was unable to increase pericyte coverage of vessels. However, the reported improvements in oxygenation by AIs are not all due to vessel normalization. Using SU5416, an antagonist of the VEGF receptor, the increase in tumor oxygenation resulted from an inhibition of mitochondrial respiration, thereby decreasing hypoxia by increasing the oxygen diffusion distance (88).
Regardless of the mechanisms for these decreases in tumor hypoxia, the improved oxygenation in both these studies was somewhat transient and only lasted for a period of a few days despite the AI treatment being continued. This “narrow window” of improved oxygenation has also been seen with thalidomide (89, 90), a nucleolin antagonist (91), and bevacizumab (92–95), regardless of the technique used to monitor the changes in oxygenation/hypoxia. The transient nature of this effect would suggest that the timing of hypoxia measurement is critical. In fact, two studies reported both a decrease and increase in hypoxia depending on the time of measurement after treatment with either DC101 (96) or bevacizumab (95).
Although at least one clinical study suggested an apparent improvement in oxygenation with AI therapy (97), several pre-clinical studies reported no change in tumor oxygenation status despite the AIs causing a decrease in vascular density and blood perfusion (1, 98). More significantly, in the majority of reported pre-clinical studies these AI-induced anti-vascular effects actually led to an increase in hypoxia, in line with what one would expect (1, 82). It could be argued that these different effects on tumor oxygenation status could be the result of using different drugs, doses, scheduling, or the time of hypoxia assessment. However, it seems more likely that the effects are a tumor dependent phenomena. This is probably best illustrated using DC101, where one study showed that 2 days after treating animals with DC101 (3 × 40 mg/kg), U87 gliomas were significantly better oxygenated when measured using a hypoxic specific marker (87). Yet another study using the same drug, almost identical dose schedule (3 × 45 mg/kg), and similar hypoxic specific marker, found that 2 days after treatment, MCa4/MCa35 mammary carcinomas were significantly more hypoxic (99). This same controversy was seen in the limited clinical studies in which both a decrease (100) and an increase (101, 102) in tumor hypoxia have been reported. Such findings clearly argue against making sweeping statements about the effects of AIs on tumor hypoxia and that either measurements of the oxygenation status need to be routinely made when AIs are administered or that they be given in such a way as to avoid any negative influence on the conventional treatment with which they are combined.
To take advantage of the cellular response to hypoxia, the use of expression levels of hypoxia induced genes as biomarkers for tumor hypoxia has been widely investigated. Initially, single genes such as HIF-1, Ca9, and Glut1 measured either at the protein level, with for example immunohistochemistry, or on the mRNA level with for instance qPCR, was used in a range of studies (103–106). The use of single gene expression markers for tumor hypoxia has often led to conflicting reports, due to the genes being influenced by factors other than hypoxia, such as extracellular pH or glucose concentrations (107, 108). Ca9 expression was one such factor proposed as a hypoxia marker in a number of studies, however other experimental studies clearly demonstrated that hypoxia and Ca9 expression did not exclusively correlate (109). Certain microRNAs (miRNAs) have also demonstrated to be inducible by hypoxia (110, 111), as for example hsa-mir-210 which has shown to be hypoxia related and to have prognostic significance in several tumor types, e.g., in cervical cancer (112), in breast cancer (113), and in bladder cancer (114).
Progresses in gene expression profiling have let to a higher level of understanding of the biology of hypoxia, and development of hypoxic signatures based on a number of genes rather than on single genes as biomarkers for tumor hypoxia (115–122). These have typically been developed by determining global gene expression levels by gene expression arrays, and identifying genes preferentially upregulated by hypoxia based on either in vitro or clinically derived gene expression data sets. There is no consensus to the optimal way to develop gene expression signatures, and the currently published hypoxia gene expression signatures are at different stages in respect to clinical usability and validation (123).
The Toustrup 15-gene-classifier was developed from a panel of genes, identified in an in vitro study in a panel of Head and Neck Squamous Cell Carcinoma (HNSCC) cell lines as upregulated by low oxygen concentration, independent of pH. It was developed in a training cohort of 58 HNSCC patients with the oxygenation status measured using an oxygen electrode. The classifier was validated in the DAHANCA 5 cohort, which is a Danish study where patients were randomized to receive either the previous mentioned hypoxic cell radiosensitizer nimorazole, or placebo, with radiotherapy. The classifier was in this cohort demonstrated to be both prognostic and have predictive impact for hypoxic modification (124). The 26-gene classifier by Eustace et al. (121), is another hypoxia signature in HNSCC. This signature is based on a metagene signature developed for patients with breast, lung and head and neck cancers. In the Dutch ARCON trial, which compared treatment with radiotherapy combined with carbogen and nicotinamide, two hypoxia modifying agents, compared to radiotherapy alone in patients with laryngeal cancer, the patients classified as “more hypoxic” according to the 26-gene classifier showed a significantly improved locoregional control in when treated with the modifying agents (123, 125).
Several studies have aimed at comparing the published gene signatures (126–129), but with the constraint that common analyzing methods have been used, such as the two-class k-means clustering, and not the validated analysis method, which for some of the gene signatures include cutoff values.
To utilize the biological knowledge, studies have been focused on combining gene signatures for hypoxia with other factors known to affect cellular factors influencing the response to radiotherapy, such as markers for cancer stem cells (129), and for proliferation and DNA repair (119). Currently, for all signatures there is a need for a continued validation, both at the technical and clinical level (130, 131), especially to be able to advance from retrospective to prospective classification of the hypoxic status of patients and subsequently the assignment to hypoxia-modifying therapies in the clinic.
Tumor hypoxia mediates intercellular signaling through the regulation of many cytokines and angiogenic factors (CAF), and serum or plasma levels of hypoxia associated proteins have also been suggested as markers for hypoxia (132–134). One of the proteins which have been intensively studied is osteopontin (OPN). OPN has both in vitro and in vivo shown to be upregulated by hypoxia (108, 135), and clinical studies have found a high level of OPN to be associated with a poor prognosis, both in HNSCC (136, 137) and small cell lung cancer (138). The findings of a correlation of OPN levels and hypoxia is not consistent across studies (139), and it has been demonstrated that the measured level of OPN is sensitive to the choice of analysis platform (140). Nonetheless, hypoxia associated circulating proteins could add prognostic information on patient outcome.
In the age of targeted therapies, hypoxia has to be considered the ultimate target. Hypoxia exists in virtually all solid tumor types, it influences patient response to radio-, chemo-, and immune-therapy, and plays a major role in malignant progression. Its presence in tumors can be identified by various invasive and non-invasive techniques, and there are a number of approaches by which hypoxia can be modified to improve outcome. However, despite these factors and the ongoing extensive pre-clinical studies, the clinical focus on hypoxia is still to a large extent lacking. Molecular pathways are the fundamental background for the cellular response to hypoxia, and further understanding of the molecular processes involved may help overcome this limitation.
MH and BS formulated the topic of the review and drafted and approved the manuscript.
BS was a co-inventor on a patent on a method (gene expression profile) for determining clinically relevant hypoxia in cancer (WO2012146259 A1) that is owned by Aarhus University, Aarhus, Denmark.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Siemann DW, Horsman MR. Modulation of the tumor vasculature and oxygenation to improve therapy. Pharmacol Ther. (2015) 153:107–24. doi: 10.1016/j.pharmthera.2015.06.006
2. Horsman MR, Vaupel P. Pathophysiological basis for the formation of the tumor microenvironment. Front Oncol. (2016) 6:66. doi: 10.3389/fonc.2016.00066
3. Folkman J. How is blood vessel growth regulated in normal and neoplastic tissue? G.H.A. clowes memorial award lecture. Cancer Res. (1986) 46:467–73.
4. Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. (2003) 3:401–10. doi: 10.1038/nrc1093
5. Hockel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. JNCI J Natl Cancer Inst. (2001) 93:266–76. doi: 10.1093/jnci/93.4.266
6. Thomlinson RH, Gray LH. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br J Cancer. (1955) 9:539–49. doi: 10.1038/bjc.1955.55
7. Brown JM. Evidence for acutely hypoxic cells in mouse tumours, and a possible mechanism of reoxygenation. Br J Radiol. (1979) 52:650–6. doi: 10.1259/0007-1285-52-620-650
8. Chaplin DJ, Olive PL, Durand RE. Intermittent blood flow in a murine tumor: radiobiological effects. Cancer Res. (1987) 47:597–601.
9. Kimura H, Braun RD, Ong ET, Hsu R, Secomb TW, Papahadjopoulos D, et al. Fluctuations in red cell flux in tumor microvessels can lead to transient hypoxia and reoxygenation in tumor parenchyma. Cancer Res. (1996) 56:5522–8.
10. Bayer C, Shi K, Astner ST, Maftei C-A, Vaupel P. Acute versus chronic hypoxia: why a simplified classification is simply not enough. Int J Radiat Oncol. (2011) 80:965–8. doi: 10.1016/j.ijrobp.2011.02.049
11. Wouters BG, Brown JM. Cells at intermediate oxygen levels can be more important than the “Hypoxic Fraction” in determining tumor response to fractionated radiotherapy. Radiat Res. (1997) 147:541. doi: 10.2307/3579620
12. Semenza GL. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol. (2000) 88:1474–80. doi: 10.1152/jappl.2000.88.4.1474
13. Pugh CW, Ratcliffe PJ. New horizons in hypoxia signaling pathways. Exp Cell Res. (2017) 356:116–21. doi: 10.1016/j.yexcr.2017.03.008
14. Wang GL, Semenza GL. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc Natl Acad Sci USA. (1993) 90:4304–8. doi: 10.1073/pnas.90.9.4304
15. Kietzmann T, Mennerich D, Dimova EY. Hypoxia-Inducible Factors (HIFs) and phosphorylation: impact on stability, localization, and transactivity. Front Cell Dev Biol. (2016) 4:11. doi: 10.3389/fcell.2016.00011
16. Bruick R., McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. (2001) 294:1337–40. doi: 10.1126/science.1066373
17. Epstein ACR, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR, et al. C. Elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. (2001) 107:43–54. doi: 10.1016/S0092-8674(01)00507-4
18. Pugh CW, Ratcliffe PJ. The von Hippel–Lindau tumor suppressor, hypoxia-inducible factor-1 (HIF-1) degradation, and cancer pathogenesis. Semin Cancer Biol. (2003) 13:83–9. doi: 10.1016/S1044-579X(02)00103-7
19. Cockman ME, Masson N, Mole DR, Jaakkola P, Chang GW, Clifford SC, et al. Hypoxia inducible factor-α binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J Biol Chem. (2000) 275:25733–41. doi: 10.1074/jbc.M002740200
20. Bracken CP, Fedele AO, Linke S, Balrak W, Lisy K, Whitelaw ML, et al. Cell-specific regulation of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha stabilization and transactivation in a graded oxygen environment. J Biol Chem. (2006) 281:22575–85. doi: 10.1074/jbc.M600288200
21. Jiang BH, Semenza GL, Bauer C, Marti HH. Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am J Physiol. (1996) 271:C1172–80. doi: 10.1152/ajpcell.1996.271.4.C1172
22. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. (2001) 292:468–72. doi: 10.1126/science.1059796
23. Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. (2001) 292:464–8. doi: 10.1126/science.1059817
24. Maxwell PH, Wiesener MS, Chang G-W, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. (1999) 399:271–5. doi: 10.1038/20459
25. Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. (2012) 148:399–408. doi: 10.1016/j.cell.2012.01.021
26. Smythies JA, Sun M, Masson N, Salama R, Simpson PD, Murray E, et al. Inherent DNA-binding specificities of the HI-1α and HIF-2α transcription factors in chromatin. EMBO Rep. (2019) 20:e46401. doi: 10.15252/embr.201846401
27. Loboda A, Jozkowicz A, Dulak J. HIF-1 versus HIF-2 — Is one more important than the other? Vascul Pharmacol. (2012) 56:245–51. doi: 10.1016/j.vph.2012.02.006
28. Gu YZ, Moran SM, Hogenesch JB, Wartman L, Bradfield CA. Molecular characterization and chromosomal localization of a third alpha-class hypoxia inducible factor subunit, HIF3alpha. Gene Expr. (1998) 7:205–13.
29. Lee P, Chandel NS, Simon MC. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol. (2020) doi: 10.1038/s41580-020-0227-y
30. Feldman DE, Chauhan V, Koong AC. The unfolded protein response: a novel component of the hypoxic stress response in tumors. Mol Cancer Res. (2005) 3:597–605. doi: 10.1158/1541-7786.MCR-05-0221
31. Koumenis C, Wouters BG. “Translating” Tumor Hypoxia: Unfolded Protein Response (UPR)-dependent and UPR-independent pathways. Mol Cancer Res. (2006). 4:423–36. doi: 10.1158/1541-7786.MCR-06-0150
32. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. (2007) 8:519–29. doi: 10.1038/nrm2199
33. Koritzinsky M, Magagnin MG, van den Beucken T, Seigneuric R, Savelkouls K, Dostie J, et al. Gene expression during acute and prolonged hypoxia is regulated by distinct mechanisms of translational control. EMBO J. (2006) 25:1114–25. doi: 10.1038/sj.emboj.7600998
34. Xie H, Tang C-HA, Song JH, Mancuso A, Del Valle JR, Cao J, et al. IRE1α RNase–dependent lipid homeostasis promotes survival in Myc-transformed cancers. J Clin Invest. (2018) 128:1300–16. doi: 10.1172/JCI95864
35. Liu Y, Adachi M, Zhao S, Hareyama M, Koong AC, Luo D, et al. Preventing oxidative stress: a new role for XBP1. Cell Death Differ. (2009) 16:847–57. doi: 10.1038/cdd.2009.14
36. Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature. (2014) 508:103–7. doi: 10.1038/nature13119
37. Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 Is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. (1999) 10:3787–99. doi: 10.1091/mbc.10.11.3787
38. Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. (2001) 107:881–91. doi: 10.1016/S0092-8674(01)00611-0
39. Chipurupalli S, Kannan E, Tergaonkar V, D'Andrea R, Robinson N. Hypoxia induced ER stress response as an adaptive mechanism in Cancer. Int J Mol Sci. (2019) 20. doi: 10.3390/ijms20030749
40. Fang Y, Tan J, Zhang Q. Signaling pathways and mechanisms of hypoxia-induced autophagy in the animal cells. Cell Biol Int. (2015) 39:891–8. doi: 10.1002/cbin.10463
41. Mazure NM, Pouysségur J. Hypoxia-induced autophagy: cell death or cell survival? Curr Opin Cell Biol. (2010) 22:177–80. doi: 10.1016/j.ceb.2009.11.015
42. Olcina M, Lecane PS, Hammond EM. Targeting hypoxic cells through the DNA damage response. Clin Cancer Res. (2010) 16:5624–9. doi: 10.1158/1078-0432.CCR-10-0286
43. Ng N, Purshouse K, Foskolou IP, Olcina MM, Hammond EM. Challenges to DNA replication in hypoxic conditions. FEBS J. (2018) 285:1563–71. doi: 10.1111/febs.14377
44. Bristow RG, Hill RP. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer. (2008) 8:180–92. doi: 10.1038/nrc2344
45. Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. (2009) 458:1127–30. doi: 10.1038/nature07986
46. Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. (2008) 9:402–12. doi: 10.1038/nrm2395
47. Barnett JA. A history of research on yeasts 2: Louis Pasteur and his contemporaries, 1850-1880. Yeast. (2000) 16:755–71. doi: 10.1002/1097-0061(20000615)16:8<755::AID-YEA587>3.0.CO;2-4
48. Koppenol WH, Bounds PL, Dang C V. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer. (2011) 11:325–37. doi: 10.1038/nrc3038
49. Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res. (1989) 49:6449–65.
50. Helmlinger G, Yuan F, Dellian M, Jain RK. Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. NatMed. (1997) 3:177–82. doi: 10.1038/nm0297-177
51. Sørensen BS, Toustrup K, Horsman MR, Overgaard J, Alsner J. Identifying pH independent hypoxia induced genes in human squamous cell carcinomas in vitro. Acta Oncol. (2010) 49:895–905. doi: 10.3109/02841861003614343
52. Chen JL, Lucas JE, Schroeder T, Mori S, Wu J, Nevins J, et al. The genomic analysis of lactic acidosis and acidosis response in human cancers. PLoS Genet. (2008) 4:e1000293. doi: 10.1371/journal.pgen.1000293
53. Sørensen BS, Busk M, Overgaard J, Horsman MR, Alsner J. Simultaneous hypoxia and low extracellular pH suppress overall metabolic rate and protein synthesis in vitro. PLoS ONE. (2015) 10:e0134955. doi: 10.1371/journal.pone.0134955
54. Drake CG, Lipson EJ, Brahmer JR. Breathing new life into immunotherapy: review of melanoma, lung and kidney cancer. Nat Rev Clin Oncol. (2014) 11:24–37. doi: 10.1038/nrclinonc.2013.208
55. Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol. (2015) 33:1974–82. doi: 10.1200/JCO.2014.59.4358
56. Coussens LM, Werb Z. Inflammation and cancer. Nature. (2002) 420:860–7. doi: 10.1038/nature01322
57. Qu X, Tang Y, Hua S. Immunological approaches towards cancer and inflammation: a cross talk. Front Immunol. (2018) 9:563. doi: 10.3389/fimmu.2018.00563
58. Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. (2018) 32:1267–84. doi: 10.1101/gad.314617.118
59. van Limbergen EJ, De Ruysscher DK, Olivo Pimentel V, Marcus D, Berbee M, Hoeben A, et al. Combining radiotherapy with immunotherapy: the past, the present and the future. Br J Radiol. (2017) 90:20170157. doi: 10.1259/bjr.20170157
60. Bernier J. Immuno-oncology: allying forces of radio- and immuno-therapy to enhance cancer cell killing. Crit Rev Oncol Hematol. (2016) 108:97–108. doi: 10.1016/j.critrevonc.2016.11.001
61. Durante M, Reppingen N, Held KD. Immunologically augmented cancer treatment using modern radiotherapy. Trends Mol Med. (2013) 19:565–82. doi: 10.1016/j.molmed.2013.05.007
62. Vaupel P, Multhoff G. Accomplices of the hypoxic tumor microenvironment compromising antitumor immunity: adenosine, lactate, acidosis, vascular endothelial growth factor, potassium ions, and phosphatidylserine. Front Immunol. (2017) 8:1–6. doi: 10.3389/fimmu.2017.01887
63. Multhoff G, Vaupel P. Hypoxia compromises anti-cancer immune responses. Adv Exp Med Biol vol. (2020) 1232:131–43. doi: 10.1007/978-3-030-34461-0_18
64. Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Science. (2016) 352:175–80. doi: 10.1126/science.aaf4405
65. Triner D, Shah YM. Hypoxia-inducible factors: a central link between inflammation and cancer. J Clin Invest. (2016) 126:3689–98. doi: 10.1172/JCI84430
66. Barsoum IB, Koti M, Siemens DR, Graham CH. Mechanisms of hypoxia-mediated immune escape in cancer. Cancer Res. (2014) 74:7185–90. doi: 10.1158/0008-5472.CAN-14-2598
67. Hatfield SM, Kjaergaard J, Lukashev D, Schreiber TH, Belikoff B, Abbott R, et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci Transl Med. (2015) 7:277ra30. doi: 10.1126/scitranslmed.aaa1260
68. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. (2014) 211:781–90. doi: 10.1084/jem.20131916
69. Petrova V, Annicchiarico-Petruzzelli M, Melino G, Amelio I. The hypoxic tumour microenvironment. Oncogenesis. (2018) 7:10. doi: 10.1038/s41389-017-0011-9
70. Vaupel P, Multhoff G. Fatal alliance of Hypoxia-/HIF-1α-driven microenvironmental traits promoting cancer progression. Adv Exp Med Biol. (2020) 1232:169–76. doi: 10.1007/978-3-030-34461-0_21
71. Horsman MR, Mortensen LS, Petersen JB, Busk M, Overgaard J. Imaging hypoxia to improve radiotherapy outcome. Nat Rev Clin Oncol. (2012) 9:674–87. doi: 10.1038/nrclinonc.2012.171
72. Kristensen LK, Fröhlich C, Christensen C, Melander MC, Poulsen TT, Galler GR, et al. CD4+ and CD8a+ PET imaging predicts response to novel PD-1 checkpoint inhibitor: studies of Sym021 in syngeneic mouse cancer models. Theranostics. (2019) 9:8221–38. doi: 10.7150/thno.37513
73. Christensen C, Kristensen LK, Alfsen MZ, Nielsen CH, Kjaer A. Quantitative PET imaging of PD-L1 expression in xenograft and syngeneic tumour models using a site-specifically labelled PD-L1 antibody. Eur J Nucl Med Mol Imaging. (2019) doi: 10.1158/1538-7445.AM2018-3030
74. Bensch F, van der Veen EL, Lub-de Hooge MN, Jorritsma-Smit A, Boellaard R, Kok IC, et al. 89Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer. Nat Med. (2018). 24:1852–8. doi: 10.1038/s41591-018-0255-8
75. Moulder JE, Rockwell S. Hypoxic fractions of solid tumors: experimental techniques, methods of analysis, and a survey of existing data. Int J Radiat Oncol. (1984) 10:695–712. doi: 10.1016/0360-3016(84)90301-8
76. Gray LH, Conger AD, Ebert M, Hornsey S, Scott OCA. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br J Radiol. (1953) 26:638–48. doi: 10.1259/0007-1285-26-312-638
77. Horsman MR, Wouters BG, Joiner M, Overgaard J. The oxygen effect and fractionated radiotherapy. In: Joiner M, van der Kogel AJ, editors. Basic Clinical Radiobiology 4th edit. London: Hodder Arnold. (2009) p. 207–16. doi: 10.1201/b13224-16
78. Chaplin DJ, Trotter MJ. Chemical modifiers of tumour blood flow. In Vaupel P, Jain RK, editors. Tumour blood supply Metabolism Microenvironment, Stuttgart: Gustav Fischer. (1991) p. 65–85.
79. Bassler N, Toftegaard J, Lühr A, Sørensen BS, Scifoni E, Krämer M, et al. LET-painting increases tumour control probability in hypoxic tumours. Acta Oncol. (2014) 53:25–32. doi: 10.3109/0284186X.2013.832835
80. Overgaard J, Hansen HS, Overgaard M, Bastholt L, Berthelsen A, Specht L, et al. A randomized double-blind phase III study of nimorazole as a hypoxic radiosensitizer of primary radiotherapy in supraglottic larynx and pharynx carcinoma. results of the Danish Head and Neck Cancer Study (DAHANCA) protocol 5-85. RadiotherOncol. (1998) 46:135–46. doi: 10.1016/S0167-8140(97)00220-X
81. www.ous-research.no/hn/ n.d.
82. Horsman MR, Siemann DW. Pathophysiologic effects of vascular-targeting agents and the implications for combination with conventional therapies. Cancer Res. (2006) 66:11520–39. doi: 10.1158/0008-5472.CAN-06-2848
83. Siemann DW, Bibby MC, Dark GG, Dicker AP, Eskens FALM, Horsman MR, et al. Differentiation and definition of vascular-targeted therapies. Clin Cancer Res. (2005) 11:416–20.
84. Siemann DW, Chaplin DJ, Horsman MR. Realizing the potential of vascular targeted therapy: the rationale for combining vascular disrupting agents and anti-angiogenic agents to treat cancer. Cancer Invest. (2017) 35:519–34. doi: 10.1080/07357907.2017.1364745
85. Tozer GM, Kanthou C, Baguley BC. Disrupting tumour blood vessels. Nat Rev Cancer. (2005) 5:423–35. doi: 10.1038/nrc1628
86. Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med. (2001) 7:987–9. doi: 10.1038/nm0901-987
87. Winkler F, Kozin S V, Tong RT, Chae S-S, Booth MF, Garkavtsev I, et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell. (2004) 6:553–63. doi: 10.1016/S1535-6108(04)00305-8
88. Ansiaux R, Baudelet C, Jordan BF, Crokart N, Martinive P, DeWever J, et al. Mechanism of reoxygenation after antiangiogenic therapy using SU5416 and its importance for guiding combined antitumor therapy. Cancer Res. (2006) 66:9698–704. doi: 10.1158/0008-5472.CAN-06-1854
89. Segers J, Di Fazio V, Ansiaux R, Martinive P, Feron O, Wallemacq P, et al. Potentiation of cyclophosphamide chemotherapy using the anti-angiogenic drug thalidomide: importance of optimal scheduling to exploit the “normalization” window of the tumor vasculature. Cancer Lett. (2006) 244:129–35. doi: 10.1016/j.canlet.2005.12.017
90. Ansiaux R, Baudelet C, Jordan BF, Beghein N, Sonveaux P, De Wever J, et al. Thalidomide radiosensitizes tumors through early changes in the tumor microenvironment. Clin Cancer Res. (2005) 11:743–50.
91. Fogal V, Sugahara KN, Ruoslahti E, Christian S. Cell surface nucleolin antagonist causes endothelial cell apoptosis and normalization of tumor vasculature. Angiogenesis. (2009) 12:91–100. doi: 10.1007/s10456-009-9137-5
92. Vangestel C, Van de Wiele C, Mees G, Mertens K, Staelens S, Reutelingsperger C, et al. Single-photon emission computed tomographic imaging of the early time course of therapy-induced cell death using technetium 99m tricarbonyl His-annexin A5 in a colorectal cancer xenograft model. Mol Imaging. (2012) 11:135–47. doi: 10.2310/7290.2011.00034
93. Myers AL, Williams RF, Ng CY, Hartwich JE, Davidoff AM. Bevacizumab-induced tumor vessel remodeling in rhabdomyosarcoma xenografts increases the effectiveness of adjuvant ionizing radiation. J Pediatr Surg. (2010) 45:1080–5. doi: 10.1016/j.jpedsurg.2010.02.068
94. McGee MC, Hamner JB, Williams RF, Rosati SF, Sims TL, Ng CY, et al. Improved intratumoral oxygenation through vascular normalization increases glioma sensitivity to ionizing radiation. Int J Radiat Oncol Biol Phys. (2010) 76:1537–45. doi: 10.1016/j.ijrobp.2009.12.010
95. Dings RPM, Loren M, Heun H, McNiel E, Griffioen AW, Mayo KH, et al. Scheduling of radiation with angiogenesis inhibitors anginex and Avastin improves therapeutic outcome via vessel normalization. Clin Cancer Res. (2007) 13:3395–402. doi: 10.1158/1078-0432.CCR-06-2441
96. Hansen-Algenstaedt N, Stoll BR, Padera TP, Dolmans DE, Hicklin DJ, Fukumura D, et al. Tumor oxygenation in hormone-dependent tumors during vascular endothelial growth factor receptor-2 blockade, hormone ablation, and chemotherapy. Cancer Res. (2000) 60:4556–60.
97. Batchelor TT, Gerstner ER, Emblem KE, Duda DG, Kalpathy-Cramer J, Snuderl M, et al. Improved tumor oxygenation and survival in glioblastoma patients who show increased blood perfusion after cediranib and chemoradiation. Proc Natl Acad Sci. (2013) 110:19059–64. doi: 10.1073/pnas.1318022110
98. Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res. (1989) 49:6449–65.
99. Fenton BM, Paoni SF, Ding I. Pathophysiological effects of vascular endothelial growth factor receptor-2-blocking antibody plus fractionated radiotherapy on murine mammary tumors. Cancer Res. (2004) 64:5712–9. doi: 10.1158/0008-5472.CAN-04-0434
100. Hugonnet F, Fournier L, Medioni J, Smadja C, Hindie E, Huchet V, et al. Metastatic renal cell carcinoma: relationship between initial metastasis hypoxia, change after 1 month's sunitinib, and therapeutic response: an 18F-Fluoromisonidazole PET/CT Study. J Nucl Med. (2011) 52:1048–55. doi: 10.2967/jnumed.110.084517
101. Hattingen E, Jurcoane A, Bähr O, Rieger J, Magerkurth J, Anti S, et al. Bevacizumab impairs oxidative energy metabolism and shows antitumoral effects in recurrent glioblastomas: a 31P/1H MRSI and quantitative magnetic resonance imaging study. Neuro Oncol. (2011) 13:1349–63. doi: 10.1093/neuonc/nor132
102. Milosevic MF, Townsley CA, Chaudary N, Clarke B, Pintilie M, Fan S, et al. Sorafenib increases tumor hypoxia in cervical cancer patients treated with radiation therapy: results of a phase 1 clinical study. Int J Radiat Oncol Biol Phys. (2016) 94:111–7. doi: 10.1016/j.ijrobp.2015.09.009
103. Olive PL, Aquino-Parsons C, MacPhail SH, Liao SY, Raleigh JA, Lerman MI, et al. Carbonic anhydrase 9 as an endogenous marker for hypoxic cells in cervical cancer. Cancer Res. (2001) 61:8924–9.
104. Giatromanolaki A, Koukourakis MI, Sivridis E, Pastorek J, Wykoff CC, Gatter KC, et al. Expression of hypoxia-inducible carbonic anhydrase-9 relates to angiogenic pathways and independently to poor outcome in non-small cell lung cancer. Cancer Res. (2001) 61:7992–8.
105. Airley RE, Loncaster J, Raleigh JA, Harris AL, Davidson SE, Hunter RD, et al. GLUT-1 and CAIX as intrinsic markers of hypoxia in carcinoma of the cervix: relationship to pimonidazole binding. IntJ Cancer. (2003) 104:85–91. doi: 10.1002/ijc.10904
106. Haugland HK, Vukovic V, Pintilie M, Fyles AW, Milosevic M, Hill RP, et al. Expression of hypoxia-inducible factor-1alpha in cervical carcinomas: correlation with tumor oxygenation. Int J Radiat Oncol Biol Phys. (2002) 53:854–61. doi: 10.1016/S0360-3016(02)02815-8
107. Sørensen BS, Hao J, Overgaard J, Vorum H, Honoré B, Alsner J, et al. Influence of oxygen concentration and pH on expression of hypoxia induced genes. Radiother Oncol. (2005) 76:187–93. doi: 10.1016/j.radonc.2005.06.037
108. Sørensen BS, Alsner J, Overgaard J, Horsman MR. Hypoxia induced expression of endogenous markers in vitro is highly influenced by pH. Radiother Oncol. (2007) 83:362–6. doi: 10.1016/j.radonc.2007.04.028
109. Troost EG, Bussink J, Kaanders JH, van EJ, Peters JP, Rijken PF, et al. Comparison of different methods of CAIX quantification in relation to hypoxia in three human head and neck tumor lines. RadiotherOncol. (2005) 76:194–9. doi: 10.1016/j.radonc.2005.06.031
110. Gee HE, Ivan C, Calin GA, Ivan M. HypoxamiRs and cancer: from biology to targeted therapy. Antioxid Redox Signal. (2014) 21:1220–38. doi: 10.1089/ars.2013.5639
111. Choudhry H, Harris AL, McIntyre A. The tumour hypoxia induced non-coding transcriptome. Mol Aspects Med. (2016) 47–48:35–53. doi: 10.1016/j.mam.2016.01.003
112. Nilsen A, Jonsson M, Aarnes E-K, Kristensen GB, Lyng H. Reference microRNAs for RT-qPCR assays in cervical cancer patients and their application to studies of HPV16 and hypoxia biomarkers. Transl Oncol. (2019) 12:576–84. doi: 10.1016/j.tranon.2018.12.010
113. Camps C, Buffa FM, Colella S, Moore J, Sotiriou C, Sheldon H, et al. Hsa-miR-210 is induced by hypoxia and is an independent prognostic factor in breast cancer. Clin Cancer Res. (2008) 14:1340–8. doi: 10.1158/1078-0432.CCR-07-1755
114. Irlam-Jones JJ, Eustace A, Denley H, Choudhury A, Harris AL, Hoskin PJ, et al. Expression of miR-210 in relation to other measures of hypoxia and prediction of benefit from hypoxia modification in patients with bladder cancer. Br J Cancer. (2016) 115:571–8. doi: 10.1038/bjc.2016.218
115. Toustrup K, Sørensen BS, Nordsmark M, Busk M, Wiuf C, Alsner J, et al. Development of a hypoxia gene expression classifier with predictive impact for hypoxic modification of radiotherapy in head and neck cancer. Cancer Res. (2011) 71:5923–31. doi: 10.1158/0008-5472.CAN-11-1182
116. Seigneuric R, Starmans MHW, Fung G, Krishnapuram B, Nuyten DSA, van Erk A, et al. Impact of supervised gene signatures of early hypoxia on patient survival. Radiother Oncol. (2007) 83:374–82. doi: 10.1016/j.radonc.2007.05.002
117. Winter SC, Buffa FM, Silva P, Miller C, Valentine HR, Turley H, et al. Relation of a hypoxia metagene derived from head and neck cancer to prognosis of multiple cancers. Cancer Res. (2007) 67:3441–9. doi: 10.1158/0008-5472.CAN-06-3322
118. Buffa FM, Harris AL, West CM, Miller CJ. Large meta-analysis of multiple cancers reveals a common, compact and highly prognostic hypoxia metagene. Br J Cancer. (2010) 102:428–35. doi: 10.1038/sj.bjc.6605450
119. Ragnum HB, Vlatkovic L, Lie AK, Axcrona K, Julin CH, Frikstad KM, et al. The tumour hypoxia marker pimonidazole reflects a transcriptional programme associated with aggressive prostate cancer. Br J Cancer. (2015) 112:382–90. doi: 10.1038/bjc.2014.604
120. Yang L, Roberts D, Takhar M, Erho N, Bibby BAS, Thiruthaneeswaran N, et al. Development and validation of a 28-gene hypoxia-related prognostic signature for localized prostate cancer. EBioMedicine. (2018) 31:182–9. doi: 10.1016/j.ebiom.2018.04.019
121. Eustace A, Mani N, Span PN, Irlam JJ, Taylor J, Betts GNJ, et al. A 26-gene hypoxia signature predicts benefit from hypoxia-modifying therapy in laryngeal cancer but not bladder cancer. Clin Cancer Res. (2013) 19:4879–88. doi: 10.1158/1078-0432.CCR-13-0542
122. Fjeldbo CS, Julin CH, Lando M, Forsberg MF, Aarnes E-K, Alsner J, et al. Integrative analysis of DCE-MRI and gene expression profiles in construction of a gene classifier for assessment of hypoxia-related risk of chemoradiotherapy failure in cervical cancer. Clin Cancer Res. (2016) 22:4067–76. doi: 10.1158/1078-0432.CCR-15-2322
123. Harris BHLHL, Barberis A, West CMLML, Buffa FMM. Gene expression signatures as biomarkers of tumour hypoxia. Clin Oncol. (2015) 27:547–60. doi: 10.1016/j.clon.2015.07.004
124. Toustrup K, Sørensen BS, Alsner J, Overgaard J. Hypoxia gene expression signatures as prognostic and predictive markers in head and neck radiotherapy. Semin Radiat Oncol. (2012) 22:119–27. doi: 10.1016/j.semradonc.2011.12.006
125. Janssens GO, Rademakers SE, Terhaard CH, Doornaert PA, Bijl HP, van den Ende P, et al. Accelerated radiotherapy with carbogen and nicotinamide for laryngeal cancer: results of a phase III randomized trial. J Clin Oncol. (2012) 30:1777–83. doi: 10.1200/JCO.2011.35.9315
126. Bhandari V, Hoey C, Liu LY, Lalonde E, Ray J, Livingstone J, et al. Molecular landmarks of tumor hypoxia across cancer types. Nat Genet. (2019) 51:308–18. doi: 10.1038/s41588-018-0318-2
127. van der Heijden M, de Jong MC, Verhagen CVM, de Roest RH, Sanduleanu S, Hoebers F, et al. Acute hypoxia profile is a stronger prognostic factor than chronic hypoxia in advanced stage head and neck cancer patients. Cancers. (2019) 11:583. doi: 10.3390/cancers11040583
128. Tawk B, Schwager C, Deffaa O, Dyckhoff G, Warta R, Linge A, et al. Comparative analysis of transcriptomics based hypoxia signatures in head- and neck squamous cell carcinoma. Radiother Oncol. (2016) 118:350–8. doi: 10.1016/j.radonc.2015.11.027
129. Linge A, Löck S, Gudziol V, Nowak A, Lohaus F, von Neubeck C, et al. Low cancer stem cell marker expression and low hypoxia identify good prognosis subgroups in HPV(–) HNSCC after postoperative radiochemotherapy: a multicenter study of the DKTK-ROG. Clin Cancer Res. (2016) 22:2639–49. doi: 10.1158/1078-0432.CCR-15-1990
130. Betts GNJ, Eustace A, Patiar S, Valentine HR, Irlam J, Ramachandran A, et al. Prospective technical validation and assessment of intra-tumour heterogeneity of a low density array hypoxia gene profile in head and neck squamous cell carcinoma. Eur J Cancer. (2013) 49:156–65. doi: 10.1016/j.ejca.2012.07.028
131. Toustrup K, Sørensen BS, Metwally MAH, Tramm T, Mortensen LS, Overgaard J, et al. Validation of a 15-gene hypoxia classifier in head and neck cancer for prospective use in clinical trials. Acta Oncol. (2016) 55:1091–8. doi: 10.3109/0284186X.2016.1167959
132. Byers LA, Holsinger FC, Kies MS, William WN, El-Naggar AK, Lee JJ, et al. Serum signature of hypoxia-regulated factors is associated with progression after induction therapy in head and neck squamous cell cancer. Mol Cancer Ther. (2010) 9:1755–63. doi: 10.1158/1535-7163.MCT-09-1047
133. Brøndum L, Sørensen BS, Eriksen JG, Mortensen LS, Lønbro S, Overgaard J, et al. An evaluation of multiplex bead-based analysis of cytokines and soluble proteins in archived lithium heparin plasma, EDTA plasma and serum samples. Scand J Clin Lab Invest. (2016) 76:601–11. doi: 10.1080/00365513.2016.1230882
134. Ock C-Y, Nam A-R, Bang J-H, Kim T-Y, Lee K-H, Han S-W, et al. Signature of cytokines and angiogenic factors (CAFs) defines a clinically distinct subgroup of gastric cancer. Gastric Cancer. (2017) 20:164–74. doi: 10.1007/s10120-015-0583-z
135. Lukacova S, Khalil AA, Overgaard J, Alsner J, Horsman MR. Relationship between radiobiological hypoxia in a C3H mouse mammary carcinoma and osteopontin levels in mouse serum. IntJRadiatBiol. (2005) 81:937–44. doi: 10.1080/09553000600567616
136. Overgaard J, Eriksen JG, Nordsmark M, Alsner J, Horsman MR. Plasma osteopontin, hypoxia, and response to the hypoxia sensitiser nimorazole in radiotherapy of head and neck cancer: results from the DAHANCA 5 randomised double-blind placebo-controlled trial. Lancet Oncol. (2005) 6:757–64. doi: 10.1016/S1470-2045(05)70292-8
137. Petrik D, Lavori PW, Cao H, Zhu Y, Wong P, Christofferson E, et al. Plasma osteopontin is an independent prognostic marker for head and neck cancers. J Clin Oncol. (2006) 24:5291–7. doi: 10.1200/JCO.2006.06.8627
138. Carvalho S, Troost EGC, Bons J, Menheere P, Lambin P, Oberije C. Prognostic value of blood-biomarkers related to hypoxia, inflammation, immune response and tumour load in non-small cell lung cancer – A survival model with external validation. Radiother Oncol. (2016) 119:487–94. doi: 10.1016/j.radonc.2016.04.024
139. Hui EP, Sung FL, Yu BKH, Wong CSC, Ma BBY, Lin X, et al. Plasma osteopontin, hypoxia, and response to radiotherapy in nasopharyngeal cancer. Clin Cancer Res. (2008) 14:7080–7. doi: 10.1158/1078-0432.CCR-08-0364
Keywords: hypoxia, radiation response, gene regulation, intracellular signaling, hypoxia classifyer
Citation: Sørensen BS and Horsman MR (2020) Tumor Hypoxia: Impact on Radiation Therapy and Molecular Pathways. Front. Oncol. 10:562. doi: 10.3389/fonc.2020.00562
Received: 14 January 2020; Accepted: 30 March 2020;
Published: 21 April 2020.
Edited by:
Carsten Herskind, University of Heidelberg, GermanyReviewed by:
Chuan-Yuan Li, Duke University Medical Center, United StatesCopyright © 2020 Sørensen and Horsman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brita Singers Sørensen, YnNpbkBvbmNvbG9neS5hdS5kaw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.