
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Oncol., 21 April 2020
Sec. Hematologic Malignancies
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.00514
This article is part of the Research TopicThe Molecular Biology of T Cell Lymphomas/LeukemiaView all 5 articles
 Abu-Sayeef Mirza1*
Abu-Sayeef Mirza1* Pedro Horna2
Pedro Horna2 Jamie K. Teer3
Jamie K. Teer3 Jinming Song4Ratilal Akabari5
Jinming Song4Ratilal Akabari5 Mohammad Hussaini4†
Mohammad Hussaini4† Lubomir Sokol6†
Lubomir Sokol6†Sézary syndrome (SS) is a genetically and clinically distinct entity among cutaneous T-cell lymphomas (CTCL). SS is characterized by more aggressive disease compared to the most common indolent type of CTCL, mycosis fungoides. However, there are limited available genomic data regarding SS. To characterize and expand current mappings of the genomic landscape of CTCL, whole exome sequencing (WES) was performed on peripheral blood samples from seven patients with SS. We detected 21,784 variants, of which 21,140 were novel and 644 were previously described. Filtering revealed 551 nonsynonymous variants among 525 mutated genes−25 recurrent mutations and 1 recurrent variant. Several recurrently mutated genes crucial to pathogenesis pathways, including Janus kinase (JAK)/signal transducers and activators of transcription (STAT), peroxisome proliferator-activated receptors (PPAR), PI3K-serine/threonine protein kinases (AKT), and fibroblast growth factor receptors (FGFR), were identified. Furthermore, genetic mutations spanned both known and novel genes, supporting the idea of a long-tail distribution of mutations in lymphoma. Acknowledging these genetic variants and their affected pathways may inspire future targeted therapies. WES of a limited number of SS patients revealed both novel findings and corroborated complexities of the “long-tail” distribution of previously reported mutations.
Cutaneous T-cell lymphoma (CTCL) consists of a rare heterogeneous group of clonal T-cell lymphoproliferative disorders, including mycosis fungoides (MF) and Sézary syndrome (SS). Whereas MF is the most common indolent type of CTCL, SS exhibits more aggressive disease, which characteristically manifests with generalized erythroderma and circulating malignant cells in peripheral blood (1). Several studies have shown that there are distinct molecular pathogeneses and gene mutations between MF and SS (2, 3). In MF, gains in anti-apoptotic proteins and loss of cell cycle inhibitors result in increased cell survival. In SS, chromosomal alterations resulting in the dysregulation of the MYC oncogene and IL-2 receptor signaling pathway, the activation of cytokine pathways, and the inhibition of P53 accounts for the increased cell proliferation and leukemic behavior observed in patients with this disease (3, 4).
There are limited studies on whole exome sequencing (WES) of CTCL, yielding varying results. In one study, WES analyses of 42 CTCL cases, including 25 SS and 8 MF cases, showed highly prevalent chromosomal deletions involving the TP53, RB1, PTEN, DNMT3A, and CDKN1B tumor suppressors, which broadly implicates epigenetic regulation and signaling (5). In another study, whole genome and transcriptome next-generation sequencing analyses of nine patient samples showed copy variations in 8q (MYC, TOX), 17p (TP53, NCOR1), 10q (PTEN, FAS), 2p (DNMT3A), 11q (USP28), and 9p (CAAP1), but no recurrent rearrangements were identified (6). The largest retrospective WES analysis of CTCL to date included 220 patients with CTCL (including 186 SS patients and 25 MF patients) and used publicly available sequencing data across nine studies (7). This study identified 55 putative driver genes and implicated 17 novel gene mutations involving pathways that affect chromatin remodeling (BCOR, KDM6A, SMARCB1, TRRAP), immune surveillance (CD58, RFXAP), MAPK signaling (MAP2K1, NF1), NF-κB signaling (PRKCB, CSNK1A1), PI-3-kinase signaling (PIK3R1, VAV1), RHOA/cytoskeleton remodeling (ARHGEF3), RNA splicing (U2AF1), T-cell receptor signaling (PTPRN2, RLTPR), and T-cell differentiation (RARA) (7). Point mutations, single gene alterations, and copy number alterations in SS represent genomic diversity involving multiple pathways, such as T-cell receptor signaling, NF-kB and JAK/STAT pathways, apoptosis control, chromatin remodeling, and DNA damage response (8). Therefore, the clinical heterogeneity of MF and SS cannot be solely explained by known mutations.
Other WES analyses, such as those performed by Choi et al. (9) further confirmed that CTCL genomic diversity involves multiple pathways, including T-cell receptor signaling, NF-kB and JAK/STAT pathways, apoptosis control, chromatin remodeling, and DNA damage response.
Given these limited studies and varying results, we used WES to further expand and characterize the genomic landscape of SS. The main aim of the present study was to validate current understandings of SS genomics and identify previously unreported novel mutations.
Eight patients with SS were identified through institutional query following scientific review committee and institutional review board approval (MCC17922). Patient samples were collected from peripheral blood and cryopreserved. Neoplastic cells from all samples were CD3+ and accounted for >50% of mononuclear cells, as assessed by flow cytometry. CD3− mononuclear cells sorted by flow cytometry were used as germline controls. DNA was extracted in accordance with standard protocols of the diagnostic molecular laboratory at H. Lee Moffitt Cancer Center and Research Institution (Tampa, FL, USA). WES analyses were performed at Hudson Alpha Institute for Biotechnology (Huntsville, AL, USA) on samples from eight patients. One patient lacked a paired normal sample and was excluded from the analysis.
Library prep was performed by using NimbleGen SeqCap EZ Exome Library v3.0. Sequencing was performed on HiSeq X sequencers (Illumina, San Diego, CA) at 100× for tumor sample and 30× for paired normal sample. A combined pipeline using Strelka (Illumina, San Diego, CA) and Mutect (Broad Institute, Cambridge, MA) was used to perform bioinformatics analyses. Variants with a minor allele frequency >1% in germline databases (1,000 Genomes Project) were excluded. Intronic, untranslated regions, and synonymous variants were also excluded. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt) was used to perform enrichment analyses, and Kyoto Encyclopedia of Genes and Genomes (KEGG), Reactome, and Wikipathways were used to perform pathway analyses.
WES was finalized on paired tumor/normal samples from seven unique patients (clinical characteristics are described in Table 1). After filtering, mutations that were present in both total mononuclear cells and CD3− mononuclear cells were assumed to be germline and were excluded. WES analyses detected 21,784 somatic mutations across seven samples. Twenty-six percent of variants detected were found to have no protein changes (synonymous mutations).
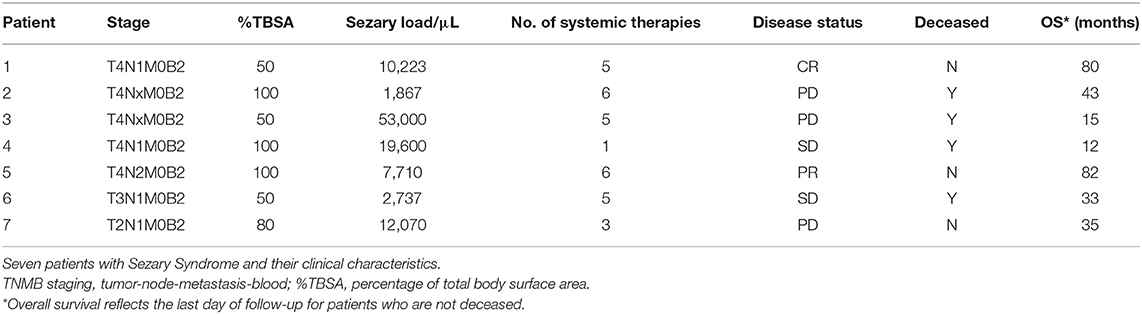
Table 1. Clinical characteristics.
Further filters were applied to exclude synonymous variants, variants that were present at >1% in 1,000 genomes, and variants with a quality score <3. WES revealed 551 non-synonymous variants distributed across 525 genes, including 478 missense mutations and 73 nonsense, splicing, or frameshift mutations. The following recurrent variants were detected: C7orf42 p.T187P (two patient samples), PTPN4 splicing variant c.1814-2A (one patient sample), and ANKRD46 splicing variant c.312-2A>T (one patient sample). The PTPN4 and ANKRD46 variants were considered to be sequencing artifacts because of their presence in a homopolymer region and presence in control specimens. The C7orf42 p.T187P variant was also suspected to be a technical artifact but was retained for having met predetermined quality criteria.
Out of the 21,784 somatic variants detected, 21,140 (97%) were novel variants and 644 were previously described variants based on Ensemble analyses; 86.8% of mutations were missense and 13.3% of mutations were truncating. The 525 genes affected by nonsynonymous somatic changes (single nucleotide variants and indels), along with genes affected by copy number loss or gain (i.e., TP53, STK11, MYC, MAP2K4, GNA11), were further analyzed (Supplementary Table 1). Recurrently mutated genes (mutated in >2 patient samples) included ANK3, CAMSAP1, C7orf42, CSMD1, DH11, FAT1, FLAD1, FLNB, FRAS1, GLUD2, GRIA2, ITGB8, KCND2, LRP1B, LRP2, MYH4, NRCAM, OR2L2, PAPPA2, PCLO, PKHD1L1, UNC13C, VWA3B, and XIRP2. LRP2 was mutated in three patient samples. Certain genes were not mutated across patient samples but harbored multiple mutations in the same patient sample.
Among the genes that were mutated more than once (regardless of whether the mutation occurred twice in the same patient), the most frequently mutated genes were GLUD2, LRP2, and PABPC3 (Table 2). Copy number variant analyses showed three samples with TP53 loss, three samples with STK11 loss, three samples with MYC gain, three samples with MAP2K4 loss, and three samples with GNA11 loss (Supplementary Figure 1).

Table 2. Recurrent mutations.
WEB-based GEne SeT AnaLysis Toolkit (WebGestalt) gene ontology functional database was used to investigate the role of the identified altered genes. GoSlim summary divided genes into biological processes, cellular components, and molecular function categories. Four hundred eighty-seven genes and 42 IDs were excluded from the analysis, as they were inadequately mapped to Entrez Gene IDs. A minimum of five genes per category were required for binning. Genes in the biological processes category were most commonly involved in biological regulation, metabolic processes, and response to stimuli. The molecular functions of the genes involved were most commonly protein binding, iron binding, and nucleic acid binding.
WebGestalt enrichment analyses showed enrichment of genes in the glutamate receptor signaling pathway (enrichment ratio = 4.81, P = 9.83 × 10−5). Similar overrepresentation analyses (ORA) performed using KEGG demonstrated enrichment of genes in the PI3K-AKT signaling pathway (R = 1.98; P = 5.43 × 10−3) (Supplementary Figure 2). This finding was confirmed by Wikipathways analyses. Overall mapping of mutated genes to cancer pathways showed that pathways, including the PPAR and JAK/STAT pathways, were mainly involved in providing proliferation signals. KEGG gene mapping further confirmed the involvement of the PPAR and JAK/STAT pathways. Reactome pathway analyses reconfirmed that the PI3K pathway and signal transduction particularly involved the fibroblast growth factor receptor (FGFR; Supplementary Figure 3).
WES analyses were used to elucidate the molecular biology of SS and its genomic landscape. Despite having a limited sample size, this study validated the genomic diversity of SS, characterized by the disease's long-tail distribution of genomic mutations. By focusing on recurrent gene mutations in multiple samples from seven SS patients, we highlighted both novel and known mutations and pathways.
Multiply mutated genes included LRP2, GLUD2, and PABC3. LRP2 is a member of the LDLR family and an endocytic receptor. LRP2 is expressed on the apical surface of absorptive epithelial cells and facilitates internalization of different ligands, such as lipoproteins, sterols, vitamin-binding proteins, hormones, signaling molecules, and extracellular matrix proteins (10). Once internalized, these ligands undergo lysosomal degradation or transcytosis (10). LRP2 can also form complexes with cubilin, which can be inhibited by sodium maleate (11, 12). LRP2 expression has been shown to be crucial for cell maintenance in malignant melanoma, and siRNA-mediated reduction of LRP2 in melanoma cells significantly decreased melanoma cell proliferation and survival rates (12). LRP2 gene polymorphisms have also been studied in regards to prostate cancer given the influence of steroid hormone uptake by endocytic receptors in prostate epithelial cells (13).
GLUD2 mutations have not been previously mentioned in regards to SS. GLUD2 is a housekeeping gene that is widely expressed and plays a crucial role in glutamate metabolism (14). RNA sequencing of triple-negative breast cancer samples has shown GLUD2 variant mutations (15).
Another multiply mutated gene was PABPC3, which is known to be an important RNA-binding protein in the translational regulation of mRNAs in spermatogenesis (16). WES analyses of six follicular thyroid cancer cell lines revealed PABPC3 to be a recurrently mutated cancer driver gene (17). These findings support the idea of a potential pathogenic role of these mutations in SS.
Our study confirmed the dysregulation of the PI3K/AKT pathway in SS, as previously reported (18). The PI3K/AKT pathway is implicated in multiple malignancies and is involved with tumor suppression when antagonized by PTEN. PI3K overexpression is an oncogenic factor in squamous cell carcinomas and is considered to be a therapeutic target (19). Interestingly, the cytokine IL-31R was found to be involved with the PI3K/AKT pathway in relation to the pathogenesis of intense pruritis among MF/SS patients (20, 21). Furthermore, AKT activation as a proxy for hyperproliferation and growth was more often found in SS skin cells than in circulating SS cells, suggesting a molecular pathogenesis of cutaneous manifestations (22).
Activated AKT is considered to be a survival factor for inhibiting apoptosis via phosphorylation of several key targets, including FOXO transcription factors (23). Inactivation of various elements of the FOXO family has been reported in the development of multiple myeloid leukemias and oncogenesis in pre-B acute lymphoblastic leukemia (24). In SS patients, FOXO1A was found to be downregulated, resulting in loss of control mechanisms for cell cycle, cell death, cell metabolism, and oxidative stress (25).
Cristofoletti et al. discovered PTEN to be deleted in 36% of patients with SS and downregulated in almost all SS samples (n = 44) (22). The PTEN gene (locus at 10q23) may enhance resistance to apoptosis in SS cells by downregulating FOXO3a, thereby contributing to malignant expansion (22). In mice studies, T-cell PTEN deletions have resulted in the development of CD4+ T-cell lymphomas (26). In MF and SS, PI3K inhibition has been shown to potentiate HDAC-inhibitor antitumor activity (27). However, the role of PIK3 inhibition as a single agent or adjuvant therapy remains to be elucidated.
Reactome analyses showed FGFR signaling involvement. Mutations in the FGFR family of proteins have been reported in regards to several malignancies (28). In certain malignancies, FGFR signaling inhibition can result in antiproliferative and/or proapoptotic effects, and FGFRs can activate several oncogenic pathways, such as STAT-dependent signaling, Ras-dependent MAPK, and Ras-independent PI3K/AKT signaling pathways (28). Several targeted therapies against FGFR, ranging from monoclonal antibodies to specific inhibitors, are currently being studied in phase 1/2 clinical trials for the treatment of several types of cancer (28, 29). As anti-FGFR therapy for CTCL has not been studied, our data provide the basis for further therapeutic investigation of this therapy.
We also detected a 17p deletion in three out of seven SS patients. This finding was consistent with that of Prasad et al. who reported a TP53 gene deletion and/or mutation in 58% of SS patients (30). Given the positive regulatory relationship between TP53 and PTEN, the combined dysfunction of PTEN and TP53 is suspected to contribute to the genetic instability of SS cells. This genetic instability facilitates chromosomal alterations, namely losses, gains, and rearrangements (22). The recurrent nature of TP53 aberrations in SS patients may constitute a distinct clinical subtype (8). Pharmacological activation of P53 may be considered in the future if it is combined with traditional chemotherapies (31).
Several studies have shown large regions of chromosomes affected by recurrent copy number variations in regions of known oncogenes (4, 32). Our copy number variant analyses showed that several genes had losses in three tumor samples (TP53, STK11, MAP2K4, GNA11), and one gene had a gain in three tumor samples (MYC).
STK11 loss was found in three out of seven patients in our study. STK11 is a tumor suppressor gene that plays a crucial role in cell growth regulation and apoptosis (33). STK11 kinase activity elimination is associated with the Peutz-Jeghers syndrome and an elevated cancer risk (34). In a genotypically and phenotypically distinct subset of lung adenocarcinoma cell lines, STK11 inactivation was found to be common and to attenuate the PI3K/AKT pathway (35). In one case report, a patient with triple-negative breast cancer with a point mutation in STK11 with loss of heterozygosity had a near-complete response with everolimus therapy. This response may be explained by the relationship between STK11 and the PI3K/AKT/mTOR signaling pathway (36).
Copy number variation analyses, as reported by Lee et al. (37) also showed loss of STK11, MAP2K4, GNA11, and MYC gene gain-of-function aberrations. The observed loss in this study supports the theory that the more aggressive behavior of SS involving blood, skin, and lymph nodes may be facilitated via MYC dysregulation (4).
MAP2K4 loss was seen in three out of seven of our patients. The MAPK pathway, similar to the PI3K pathway, contributes to oncogenesis by cell proliferation and antiapoptotic activity (38). The loss-of-function mutation in MAP2K4 has been observed to be highly frequent in several cancers (37–40). Xue et al. (38) demonstrated that inactivating mutations in MAP2K4 increased cell line sensitivity to MEK inhibitor therapy, thereby enhancing response to the therapy.
GNA11 loss was observed in three out of seven patients. GNA11 is considered to be an early driver mutation in leptomeningeal and uveal melanomas (41, 42). Over 80% of uveal melanomas are known to have mutations in GNA11 (43). Using GNA11 mutated melanoma cell lines, MEK inhibitors to suppress MAPK pathways, and suppressing protein kinase C led to the synergistic inhibition of proliferation (44).
MYC gene gain-of-function aberrations from translocation, amplification, or overexpression are common in tumorigenesis (45) and are associated with TP53 loss mutation. Both MYC gain and TP53 loss are inversely related to poor 5-year overall survival rates (46). When the proto-oncogene MYC is overactivated, it triggers an antioncogenic mitosis-differentiation checkpoint in human epidermal keratinocytes, resulting in impaired cell division, and squamous differentiation (47). Our data appear to support the hypothesis that disseminated leukemic behavior of SS may be affected by MYC dysregulation (4).
Our study underscores the need to sequence more SS cases, given the genomic heterogeneity of this disease and the potential for identifying targetable therapies. To further understand the implications of the genomic alterations in SS described in this study, functional characterization of the detected genetic alterations, prospective studies using larger sample sets, therapeutic clinical trials with targeted agents, and correlation with outcomes are all needed.
All datasets generated for this study are included in the article/Supplementary Material.
The studies involving human participants were reviewed and approved by Scientific Review Committee of Moffitt Cancer Center, Institutional Review Board of Moffitt Cancer Center (MCC17922). The patients/participants provided their written informed consent to participate in this study.
A-SM: developed manuscript, developed figures, and data analysis. PH: data analysis. JT: bioinformatics, data analysis. JS: hematopathology research, reviewed paper. RA: hematopathology research, sample preparation, and quality control. MH: hematopathology research, data analysis, IRB proposal/renewal, and developed supplemental figures. LS: malignant hematology research, clinician, principal investigator, and reviewed paper.
LS received funding for this study from The Macauley and Helen Dow Whiting Foundation.
LS served as a coinvestigator on MAVORIC and ALCANZA studies and received research funding from Spectrum Pharmaceuticals, Celgene, and Seattle Genetics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Paul Fletcher and Daley Drucker (H. Lee Moffitt Cancer Center and Research Institute) for editorial assistance. They were not compensated for their assistance beyond their regular salaries.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.00514/full#supplementary-material
1. Mao X, Lillington D, Scarisbrick JJ, Mitchell T, Czepulkowski B, Russell-Jones R, et al. Molecular cytogenetic analysis of cutaneous T-cell lymphomas: identification of common genetic alterations in Sézary syndrome and mycosis fungoides. Br J Dermatol. (2002) 147:464–75. doi: 10.1046/j.1365-2133.2002.04966.x
2. Kamarashev J, Burg G, Kempf W, Hess Schmid M, Dummer R. Comparative analysis of histological and immunohistological features in mycosis fungoides and Sézary syndrome. J Cutan Pathol. (1998) 25:407–12. doi: 10.1111/j.1600-0560.1998.tb01766.x
3. Van Doorn R, Van Kester MS, Dijkman R, Vermeer MH, Mulde, AA, Szuhai K, et al. Oncogenomic analysis of mycosis fungoides reveals major differences with Sézary syndrome. Blood. (2009) 113:127–36. doi: 10.1182/blood-2008-04-153031
4. Vermeer MH, Van Doorn R, Dijkman R, Mao X, Whittaker S, Van Voorst Vader PC, et al. Novel and highly recurrent chromosomal alterations in Sézary syndrome. Cancer Res. (2008) 68:2689–98. doi: 10.1158/0008-5472.CAN-07-6398
5. Da Silva Almeida AC, Abate F, Khiabanian H, Martinez-Escala E, Guitart J, Tensen CP, et al. The mutational landscape of cutaneous t cell lymphoma and Sézary syndrome. Nat Genet. (2015) 47:1465–70. doi: 10.1038/ng.3442
6. Izykowska K, Przybylski GK, Gand C, Braun FC, Grabarczyk P, Kuss AW, et al. Genetic rearrangements result in altered gene expression and novel fusion transcripts in Sézary syndrome. Oncotarget. (2017) 8:39627–39. doi: 10.18632/oncotarget.17383
7. Park J, Yang J, Wenzel AT, Ramachandran A, Lee WJ, Daniels JC, et al. Genomic analysis of 220 CTCLs identifies a novel recurrent gain-of-function alteration in RLTPR (P.Q575E). Blood. (2017) 130:1430–40. doi: 10.1182/blood-2017-02-768234
8. Chevret E, Merlio JP. Sézary syndrome: translating genetic diversity into personalized medicine. J Invest Dermatol. (2016) 136:1319–24. doi: 10.1016/j.jid.2016.04.027
9. Choi J, Goh G, Walradt T, Hong BS, Bunick CG, Chen K, et al. Genomic landscape of cutaneous t cell lymphoma. Nat Genet. (2015) 47:1011–9. doi: 10.1038/ng.3356
10. Marzolo MP, Farfan P. New insights into the roles of megalin/LRP2 and the regulation of its functional expression. Biol Res. (2011) 44:89–105. doi: 10.4067/S0716-97602011000100012
11. De S, Kuwahara S, Saito A. The endocytic receptor megalin and its associated proteins in proximal tubule epithelial cells. Membranes (Basel). (2014) 4:333–55. doi: 10.3390/membranes4030333
12. Andersen RK, Hammer K, Hager H, Christensen JN, Ludvigsen M, Honoré B, et al. Melanoma tumors frequently acquire lRP2/megalin expression, which modulates melanoma cell proliferation and survival rates. Pigment Cell Melanoma Res. (2015) 28:267–80. doi: 10.1111/pcmr.12352
13. Holt SK, Karyadi DM, Kwon EM, Stanford JL, Nelson PS, Ostrander EA. Association of megalin genetic polymorphisms with prostate cancer risk and prognosis. Clin Cancer Res. (2008) 14:3823–31. doi: 10.1158/1078-0432.CCR-07-4566
14. Zaganas I, Kanavouras K, Mastorodemos V, Latsoudis H, Spanaki C, Plaitakis A. The human GLUD2 glutamate dehydrogenase: localization and functional aspects. Neurochem Int. (2009) 55:52–63. doi: 10.1016/j.neuint.2009.03.001
15. Horvath A, Pakala SB, Mudvari P, Reddy SD, Ohshiro K, Casimiro S, et al. Novel insights into breast cancer genetic variance through RNA sequencing. Sci Rep. (2013) 3:2256. doi: 10.1038/srep02256
16. Ozturk S, Sozen B, Uysal F, Bassorgun IC, Usta MF, Akkoyunlu G, et al. The poly(A)-binding protein genes, EPAB, PABPC1, and PABPC3 are differentially expressed in infertile men with non-obstructive azoospermia. J Assist Reprod Genet. (2016) 33:335–48. doi: 10.1007/s10815-016-0654-z
17. Erinjeri NJ, Nicolson NG, Deyholos C, Korah R, Carling T. Whole-exome sequencing identifies two discrete druggable signaling pathways in follicular thyroid cancer. J Am Coll Surg. (2018) 226:950–9.e5. doi: 10.1016/j.jamcollsurg.2018.01.059
18. Lee CS, Ungewickell A, Bhaduri A, Qu K, Webster DE, Armstrong R, et al. Transcriptome sequencing in Sezary syndrome identifies Sezary cell and mycosis fungoides-associated lncRNAs and novel transcripts. Blood. (2012) 120:3288–97. doi: 10.1182/blood-2012-04-423061
19. Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. (2005) 4:988–1004. doi: 10.1038/nrd1902
20. Black GE, Sokol KK, Moe DM, Simmons JD, Muscat D, Pastukh V, et al. Impact of a novel phosphoinositol-3 kinase inhibitor in preventing mitochondrial DNA damage and damage-associated molecular pattern accumulation: results from the biochronicity project. J Trauma Acute Care Surg. (2017) 83:683–9. doi: 10.1097/TA.0000000000001593
21. Ferretti E, Corcione A, Pistoia V. The IL-31/IL-31 receptor axis: general features and role in tumor microenvironment. J Leukoc Biol. (2017) 102:711–7. doi: 10.1189/jlb.3MR0117-033R
22. Cristofoletti C, Picchio MC, Lazzeri C, Tocco V, Pagani E, Bresin A, et al. Comprehensive analysis of PEN status in Sézary syndrome. Blood. (2013) 122:3511–20. doi: 10.1182/blood-2013-06-510578
23. Serrano J, Serrano-Lopez J, Sanchez Garcia J, Herrera C, Torres Gomez AP. Analysis of the PI3/Akt/Survivin pathway in acute myelogenous leukemia. Blood. (2009) 114:2645. doi: 10.1182/blood.V114.22.2645.2645
24. Köhrer S, Havranek O, Seyfried F, Hurtz C, Coffey GP, Kim E, et al. Pre-BCR signaling in precursor b-cell acute lymphoblastic leukemia regulates PI3K/AKT, FOXO1, and MYC, and can be targeted by SYK inhibition. Leukemia. (2016) 30:1246–54. doi: 10.1038/leu.2016.9
25. Van Doorn R, Dijkman R, Vermeer MH, Out-Luiting JJ, Van Der Raaij-Helmer EMH Willemze R, et al. Aberrant expression of the tyrosine kinase receptor EphA4 and the transcription factor twist in Sézary syndrome identified by gene expression analysis. Cancer Res. (2004) 64:5578–86. doi: 10.1158/0008-5472.CAN-04-1253
26. Suzuki A, Yamaguchi MT, Ohteki T, Sasaki T, Kaisho T, Kimura Y, et al. T cell-specific loss of Pten leads to defects in central and peripheral tolerance. Immunity. (2001) 14:523–34. doi: 10.1016/S1074-7613(01)00134-0
27. Ai W, Yang C-Y, Faraj R, Rakhshandhroo T, Afghani S, Pincus L, et al. Pathway-Directed high throughput drug screen identifies PI3K inhibitors that synergistically potentiate anti-tumor activity of HDAC inhibitors in mycosis fungoides and Sézary syndrome. Blood. (2015) 126:2755. doi: 10.1182/blood.V126.23.2755.2755
28. Brooks AN, Kilgour E, Smith PD. Molecular pathways: fibroblast growth factor signaling: a new therapeutic opportunity in cancer. Clin Cancer Res. (2012) 18:1855–62. doi: 10.1158/1078-0432.CCR-11-0699
29. Porta R, Borea R, Coelho A, Khan S, Araujo A, Reclusa P, et al. FGFR a promising druggable target in cancer: molecular biology and new drugs. Crit Rev Oncol Hematol. (2017) 113:256–67. doi: 10.1016/j.critrevonc.2017.02.018
30. Prasad A, Rabionet R, Espinet B, Zapata L, Puiggros A, Melero C, et al. Identification of gene mutations and fusion genes in patients with Sézary syndrome. J Invest Dermatol. (2016) 136:1490–9. doi: 10.1016/j.jid.2016.03.024
31. Lamprecht B, Kreher S, Mobs M, Sterry W, Dorken B, Janz M, et al. The tumour suppressor P53 is frequently nonfunctional in Sézary syndrome. Br J Dermatol. (2012) 167:240–6. doi: 10.1111/j.1365-2133.2012.10918.x
32. Cristofoletti C, Narducci MG, Russo G. Sézary syndrome, recent biomarkers and new drugs. Chin Clin Oncol. (2018) 8:2. doi: 10.21037/cco.2018.11.02
33. Lopus M, Paul DM, Rajasekaran R. Unraveling the deleterious effects of cancer-Driven STK11 mutants through conformational sampling approach. Cancer Inform. (2016) 15:35–44. doi: 10.4137/CIN.S38044
34. Zhao ZY, Jiang YL, Li BR, Yang F, Li J, Jin XW, et al. Sanger sequencing in exonic regions of sTK11 gene uncovers a novel de-novo germline mutation (c.962_963delCC) associated with Peutz-Jeghers syndrome and elevated cancer risk: case report of a chinese patient. BMC Med Genet. (2017) 18:130. doi: 10.1186/s12881-017-0471-y
35. Kaufman JM, Amann JM, Park K, Arasada RR, Li H, Shyr Y, et al. LKB1 loss induces characteristic patterns of gene expression in human tumors associated with NRF2 activation and attenuation of PI3K-AKT. J Thorac Oncol. (2014) 9:794–804. doi: 10.1097/JTO.0000000000000173
36. Parachoniak CA, Rankin A, Gaffney B, Hartmaier R, Spritz D, Erlich RL, et al. Exceptional durable response to everolimus in a patient with biphenotypic breast cancer harboring an STK11 variant. Cold Spring Harb Mol Case Stud. (2017) 3:a000778. doi: 10.1101/mcs.a000778
37. Lee SH, Jung SH, Kim TM, Rhee JK, Park HC, Kim MS, et al. Whole-exome sequencing identified mutational profiles of high-grade colon adenomas. Oncotarget. (2017) 8:6579–88. doi: 10.18632/oncotarget.14172
38. Xue Z, Vis DJ, Bruna A, Sustic T, Van Wageningen S, Batra AS, et al. MAP3K1 and MAP2K4 mutations are associated with sensitivity to MEK inhibitors in multiple cancer models. Cell Res. (2018) 28:719–29. doi: 10.1038/s41422-018-0044-4
39. Xin W, Yun KJ, Ricci F, Zahurak M, Qiu W, Su GH, et al. MAP2K4/MKK4 expression in pancreatic cancer: genetic validation of immunohistochemistry and relationship to disease course. Clin Cancer Res. (2004) 10:8516–8520. doi: 10.1158/1078-0432.CCR-04-0885
40. Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. (2012) 486:353–60. doi: 10.1038/nature11143
41. Costa S, Byrne M, Pissaloux D, Haddad V, Paindavoine S, Thomas L, et al. Melanomas associated with blue nevi or mimicking cellular blue nevi: clinical, pathologic, and molecular study of 11 cases displaying a high frequency of GNA11 mutations, BAP1 expression loss, and a predilection for the scalp. Am J Surg Pathol. (2016) 40:368–77. doi: 10.1097/PAS.0000000000000568
42. Staby KM, Gravdal K, Mork SJ, Heegaard S, Vintermyr OK, Krohn J. Prognostic impact of chromosomal aberrations and GNAQ, GNA11 and BAP1 mutations in uveal melanoma. Acta Ophthalmol. (2018) 96:31–8. doi: 10.1111/aos.13452
43. Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O'brien JM, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. (2009) 457:599–602. doi: 10.1038/nature07586
44. Chen X, Wu Q, Tan L, Porter D, Jager MJ, Emery C, et al. Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene. (2014) 33:4724–34. doi: 10.1038/onc.2013.418
45. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
46. Tessoulin B, Eveillard M, Lok A, Chiron D, Moreau P, Amiot M, et al. p53 dysregulation in B-cell malignancies: more than a single gene in the pathway to hell. Blood Rev. (2017) 31:251–9. doi: 10.1016/j.blre.2017.03.001
Keywords: cutaneous T-cell lymphoma, Sézary syndrome, whole exome sequencing, genomics, translational oncology
Citation: Mirza A-S, Horna P, Teer JK, Song J, Akabari R, Hussaini M and Sokol L (2020) New Insights Into the Complex Mutational Landscape of Sézary Syndrome. Front. Oncol. 10:514. doi: 10.3389/fonc.2020.00514
Received: 01 September 2019; Accepted: 23 March 2020;
Published: 21 April 2020.
Edited by:
Onder Alpdogan, Thomas Jefferson University, United StatesReviewed by:
Guru Prasad Maiti, Oklahoma Medical Research Foundation, United StatesCopyright © 2020 Mirza, Horna, Teer, Song, Akabari, Hussaini and Sokol. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Abu-Sayeef Mirza, bWlyemFhQG1haWwudXNmLmVkdQ==
†These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.