
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol., 06 March 2020
Sec. Pediatric Oncology
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.00302
This article is part of the Research TopicNovel Molecular Targets and Therapies for Pediatric Solid TumorsView all 14 articles
 Robyn T. Sussman1†
Robyn T. Sussman1† Jo Lynne Rokita1,2
Jo Lynne Rokita1,2 Kevin Huang1
Kevin Huang1 Pichai Raman1,2Komal S. Rathi1,2Daniel Martinez3
Pichai Raman1,2Komal S. Rathi1,2Daniel Martinez3 Kristopher R. Bosse1,4Maria Lane1Lori S. Hart1Tricia Bhatti3Bruce Pawel4
Kristopher R. Bosse1,4Maria Lane1Lori S. Hart1Tricia Bhatti3Bruce Pawel4 John M. Maris1,3*
John M. Maris1,3*We developed a computational pipeline designed to use RNA sequencing (n = 136) and gene expression profiling (n = 250) data from neuroblastoma tumors to identify cell surface proteins predicted to be highly expressed in MYCN amplified neuroblastomas and with little or no expression in normal human tissues. We then performed ChIP-seq in the MYCN amplified cell lines KELLY, NB-1643, and NGP to identify gene promoters that are occupied by MYCN protein to define the intersection with the differentially-expressed gene list. We initially identified 116 putative immunotherapy targets with predicted transmembrane domains, with the most significant differentially-expressed of these being the calmodulin kinase-like vesicle-associated gene (CAMKV, p = 2 × 10−6). CAMKV encodes a protein that binds calmodulin in the presence of calcium, but lacks the kinase activity of other calmodulin kinase family members. We confirmed that CAMKV is selectively expressed in 7/7 MYCN amplified neuroblastoma cell lines and showed that the transcription of CAMKV is directly controlled by MYCN. From membrane fractionation and immunohistochemistry, we verified that CAMKV is membranous in MYCN amplified neuroblastoma cell lines and patient-derived xenografts. Finally, immunohistochemistry showed that CAMKV is not expressed on normal tissues outside of the central nervous system. Together, these data demonstrate that CAMKV is a differentially-expressed cell surface protein that is transcriptionally regulated by MYCN, making it a candidate for targeting with antibodies or antibody-drug conjugates that do not cross the blood brain barrier.
Neuroblastoma is an embryonal tumor that accounts for 12% of childhood cancer-related mortality (1, 2). Prognosis for low- and intermediate-risk neuroblastoma is outstanding with the majority of patients cured, but there has been minimal progress for patients with high-risk disease. About half of all high-risk neuroblastoma patients die despite intensive multimodal chemoradiotherapy (2). Recently, clinical trials of GD2-targeting monoclonal antibody therapy have demonstrated significantly prolonged relapse free survival when used after achieving a first response to standard therapy, leading to regulatory approval in the United States and Europe (3). However, patients suffer significant pain during dinutuximab infusions due to GD2 expression on pain fibers, limiting dosing and in part explaining minimal improvements in overall survival with this immunotherapy. Our understanding of how to develop successful immunotherapies has advanced significantly in the last few years, but the limiting factor remains identification of cell surface molecules uniquely and uniformly expressed on tumor cells.
Focal amplification of MYCN occurs in roughly 40–50% of high-risk neuroblastoma cases (4–6) and is associated with an aggressive phenotype and poor prognosis (2, 7). MYCN encodes a basic helix-loop-helix transcription factor that functions in transcription activation when heterodimerized with MAX, or transcriptional repression when heterodimerized with MNT, MXI, MAD, or other negative co-factors by binding to E-boxes within gene promoters (8, 9). Gene-expression profiling has revealed a large cohort of genes involved in cell cycle, proliferation, signaling, adhesion, differentiation, and migration to be regulated by MYCN (10–12). However, while MYC family genes are known to transcriptionally regulate a very large number of genes via enhancer invasion (13), surprisingly little is known about direct MYCN target genes.
While MYCN amplification is prevalent in high-risk neuroblastoma and some other pediatric cancers, and is an important biomarker for patient outcomes, it remains an elusive drug target. While direct targeting of the MYCN transcription factor is not yet possible, several indirect methods have been proposed such as depleting MYCN protein levels with BET or AURKA inhibitors (14–17), but these appear to be with limited anti-tumor efficacy. Here, we pursue another indirect strategy, identification of direct MYCN transcriptional targets that are located in the plasma membrane and thus amendable to new immunotherapeutic strategies.
Cell lines were grown and STR validated as described (18–20). Cell lines were tested for mycoplasma when thawed and only grown for 20 passages following thaw. SHEP-2 MYCN-ER, and SK-N-AS MYCN-ER cells were obtained from the laboratory of Dr. Michael Hogarty at the Children's Hospital of Philadelphia. Cells were treated with 1 uM tamoxifen (Sigma h7904) to induce MYCN-ER nuclear translocation.
Lentiviral preparation was carried out as described (21). Briefly, using the clone TRCN0000020695 to deplete MYCN (Sigma), plasmids encoding shRNA along with the envelope encoding plasmid pMD2.G and packaging plasmid psPAX2 were transfected into 293T cells with Fugene 6 (Roche). Supernatant was collected 48 and 72 h later, filtered and added to IMR-05 cells in the presence of 8 ug/ml polybrene (Sigma). Puromycin (Sigma) was used to select for infected cells.
Total RNA was isolated from neuroblastoma cells utilizing RNeasy mini spin kits (Qiagen) and mRNAs were converted to cDNA using SuperScript II First Strand Synthesis kits (Life Technologies). CAMKV expression was detected using a Taqman probe (Hs01062060_g1, ThermoFisher) and MYCN was detected using (Hs00232074_m1, ThermoFisher), according to the methods previously described (19, 21).
Chromatin immunoprecipitation was performed as previously described (22) using anti-MYCN (Santa Cruz Biotechnology, Inc., clone B8.4B, sc-53993), anti-MAX (Santa Cruz Biotechnology, Inc., clone H-2, sc-8011) and anti-mouse IgG (Santa Cruz Biotechnology, Inc., sc-2025). Primer sequences are as follows: CAMKV TSS Forward: 5′-GGGCAGAATCCGCTCCGA-3′;
CAMKV TSS Reverse: 5′-GCGATGCTGGAGGTTCGCTA-3′;
CAMKV 5′ Forward: 5′-CAAAGTCTCCTATCCCACCCC-3′;
CAMKV 5′ Reverse: 5′-TTTGGGAAAGACTCTGGGCTT-3′.
Chromatin immunoprecipitation was performed on the neuroblastoma cell lines Kelly, NB-1643 and NGPs as described (19, 23, 24). Briefly, using anti-MYCN (Santa Cruz B8.4B, sc-53993). Cells were grown to 80% confluence on 150 mm tissue culture plates in 20 mL of medium. Four hundred and fifteen microliters of 37% formaldehyde (final concentration 0.75% w/v) were added to the medium and the plate was rocked for 10 min to fix cells. 1.5 mL of 2.5 M glycine (final concentration 0.18 M) was added to quench the formaldehyde and the plate was rocked for an additional 5 min. Cells were lysed in 5 pellet volumes of FA lysis buffer (50 mM HEPES pH 7.5, 140 mM NaCl, 1 mM EDTA pH 8.0, 1.0% v/v Triton-X-100, 0.1% w/v SDS, 0.1% w/v sodium deoxycholate), supplemented with fresh protease inhibitors (Thermo Scientific, 88666) and 1 mM DTT. Beads were washed 3X in ChIP Wash Buffer (0.1% w/vSDS, 1.0% v/v Triton-X-100, 2 mM EDTA pH 8.0, 150 mM NaCl, 20 mM Tris-HCL pH 8.0) and once with Final Wash Buffer (0.1% w/v SDS, 1.0% v/v Triton-X-100, 2 mM EDTA pH 8.0, 500 mM NaCl, 20 mM Tris-HCL pH 8.0). Libraries were constructed using NEB ultra kit according to the manufacturer's instructions and sequenced on a MiSeq to a depth of ~50 M reads.
The neuroblastoma cell lines COG-N-415, LA-N-5, NB-1643, NB-69 and SK-N-SH were grown to 60–80% confluence in a 150 mm dish, fixed with 11% w/v formaldehyde for 15 min, and quenched with 2.5 M glycine for 5 min at room temperature. Cells were scraped, washed with 10 mL of chilled PBS with 0.5% v/v Igepal (Sigma #1-8896), and pelleted. Supernatant was removed and cells were washed with 10 mL of chilled PBS, 0.5% Igepal, and 100 uL 100 mM PMSF. Cells were pelleted, supernatant removed, and sent to Active Motif on dry ice for chromatin immunoprecipitation. N-Myc and c-Myc ChIP reactions were performed using 30 μg of cell line chromatin and 6 μg of N-Myc antibody (Active Motif, cat # 61185) or 4 μg of c-Myc antibody (Santa Cruz N262, cat # sc-764). Libraries were prepared according to www.activemotif.com/services and sequenced on a NextSeq 500 to a depth of ~50 M reads.
Phred sequencing scores (25) were calculated for each sample using a perl script (https://raw.githubusercontent.com/douglasgscofield/bioinfo/master/scripts/phredDetector.pl) for automated input into the alignment algorithm. Quality was assessed using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and sequences were adapter- and quality-trimmed using Trim Galore (26, 27). Following QC, bwa (28) was used to align the reads to hg 19 reference genome and Picard tools (29) was used to remove duplicates. Fragment sizes were estimated using MaSC (30) and these were used as input into MACS2 (31) for peak calling. Peaks were called significant using a q-value (minimum False Discovery Rate) cut off of 0.05. Results were returned in units of signal per million reads to get normalized peak values.
Finally, repetitive centromeric, telomeric, and satellite regions known to have low sequencing confidence were removed using the blacklisted regions defined by UCSC.
Kelly and NGP MYCN ChIP-Seq have been published under GEO accession number GSE94782 and the remaining MYCN and MYC ChIP-Seq data are available under GEO accession number GSE138295.
Cells were washed with cold PBS and fixed with a 50% Acetone/ 50% Methanol mixture for 5 min at −20°C. Cells were blocked with 0.3% v/v Triton X-100, 2% v/v goat serum in PBS for 5 min and then incubated with primary antibody in TBS-BGT (25 mM Tris, pH 8.0, 137 mM NaCl, 3 mM KCL, 1.5 mM MgCl2, 5 mg/ml BSA, 1 mg/ml Glycine, 0.05% v/v Tween 20, 0.02% v/v sodium azide) for 90 min at room temperature. Primary antibodies included m906IgG (32) and anti-CAMKV (Santa Cruz Biotechnology, Inc., clone S-17, sc-102408). Cells were then washed with TBS-BGT and incubated with secondary antibody in TBS-BGT. Cells were washed again in TBS-BGT and mounted to slides with prolong gold with DAPI (ThermoFisher Scientific, P36931).
10–20 × 106 live neuroblastoma cells were suspended in 50 uL of medium. A 1% w/v agarose in PBS solution was boiled and cooled to 50°C in a water bath. The cell suspension was mixed with 0.5 mL of the agarose and transferred into an inverted capped Eppendorf tube with the conical bottom cut off, on ice. Once solidified, the plug was removed, cut in half, and fixed in 10% v/v buffered formalin (ThermoFisher Scientific).
CAMKV staining was performed using the CAMKV S-17 antibody (Santa Cruz Biotechnology, Inc.). Each sample was scored by the same pathologist and was designated as “1” when <10% of cells stained positively, “2” when 10–90% of tumors stained positively and “3” when >90% of cells stained positively. Only cells of targeted tissue type were evaluated for NCAM staining.
Cells were lysed and western blotting was performed as previously described (18) using the following antibodies: anti-CAMKV (Santa Cruz Biotechnology, Inc., sc-102408) was used at a 1:250 dilution, anti-ACTIN (Cell Signaling, 4967s) was used at a 1:5,000 dilution, anti-ADAM12 (abcam, ab28225) was used at 1:1,000 and anti-BCL-2 (Cell Signaling, 2870S) was used at a 1:2,000 dilution.
Membrane fractionation was performed using the Proteoextract Native Membrane Protein Extraction Kit (Millipore, 444810) according to the manufacturer's instructions.
RNA sequencing data from 150 primary neuroblastoma tumors (136 high risk) was generated by the Therapeutically Applicable Research to Generate Effective Treatments project (TARGET data matrix, http://ocg.programs/target/data-matrix). RNA sequencing data were obtained from The Cancer Genome Atlas Program (TCGA), the Children's Brain Tumor Tissue Consortium (CBTTC), the Pacific Pediatric Neuro-Oncology Consortium (PNOC), the GEO database (GSE60052), and the Genotype-Tissue Expression (GTEx) project. Gene level data was generated using STAR alignment and RSEM normalization using hg 38 as a reference genome and GENCODE V23 gene annotation (33). The voom procedure was used to normalize the RSEM generated expected counts followed by differential expression testing using the R package limma to obtain adjusted p-values and Log-fold changes (LogFCs). Tumors were stratified by MYCN expression and sequencing data was queried for genes that had a LogFC >1 in the MYCN amplified tumors compared to non-amplified tumors with an adjusted p-value of 0.05 by the Benhanini-Hochberg procedure.
Gene expression data generated using Human Exon arrays (Affymetrix) from 250 primary tumors were obtained from the TARGET data matrix. These data were processed with Robust Mutlichip Average (RMA) normalization analysis implemented in the Affymetrix Power Tools (Affymetrix, Inc.). Samples were stratified by MYCN expression and the top and bottom 15% from this stratification were then used to query for the most differentially overexpressed genes in the MYCN-high subset of tumors.
The COMPARTMENTS database (http://compartments.jensenlab.org) was used to determine association with the plasma membrane (34). We designated only those genes to be potentially associated with the plasma membrane where the max confidence score across all GO categories was associated with Plasma Membrane or Cell Surface and was ≥3.
To determine the subset of genes which were differentially overexpressed in MYCN amplified neuroblastoma, we first used RNA-sequencing data from 150 primary tumors (136 of which were high-risk) and stratified them by MYCN expression (Figure 1A). We then filtered for genes that had a log fold change over 1 in the MYCN amplified tumors compared to non-amplified tumors with an adjusted p-value of 0.05. We similarly analyzed microarray gene expression data from 250 primary neuroblastomas stratified by MYCN expression (Figure 1B). The top and bottom 15% from this stratification were then used to query for the most differentially overexpressed genes in the MYCN-high subset. We intersected these data with a list of genes predicted to encode proteins located in the plasma membrane by the COMPARTMENTS database (34) and found 14 to be consistently differentially overexpressed in the MYCN amplified tumors (Figure 1C, Table 1, Table S1).
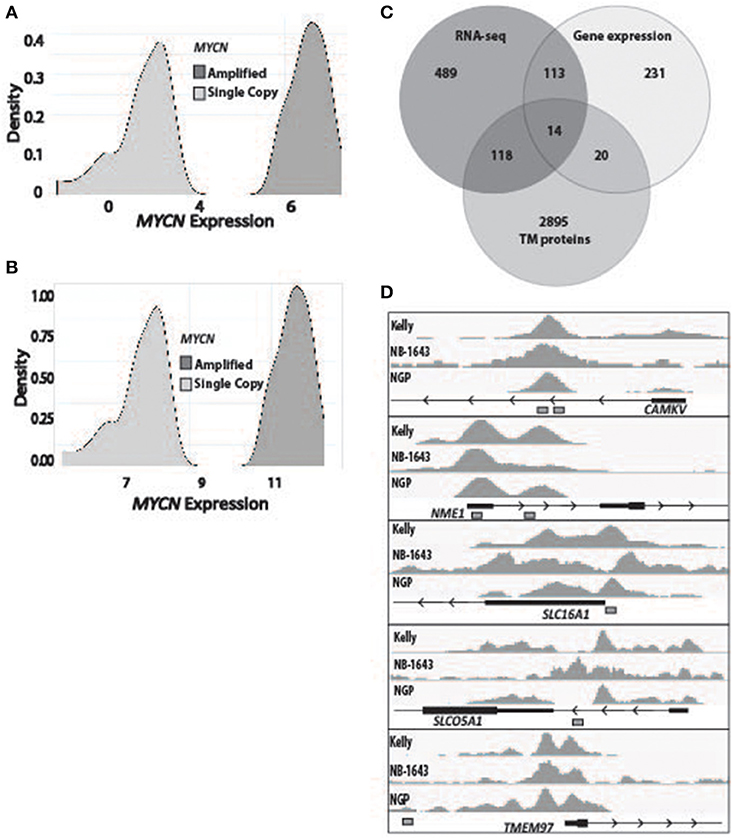
Figure 1. Identification of transmembrane-domain containing MYCN target genes in MYCN amplified neuroblastoma. (A) RNA sequencing data from 150 primary neuroblastoma tumors were stratified by MYCN expression and queried for the most differentially overexpressed genes (Log fold change >1, adjusted p-value of 0.05) in the MYCN high tumors. (B) Gene expression data from 250 primary neuroblastoma tumors were stratified by MYCN expression and queried for the most differentially overexpressed genes in the MYCN high tumors. (C) The most differentially overexpressed genes from the MYCN high tumors from RNA-sequencing data and gene expression data were intersected with a list of genes that produce transmembrane proteins (34). (D) Chromatin Immunoprecipitation sequencing tracks from five targets that were bound by MYCN protein at transcription start site-proximal E-boxes (depicted by white boxes) in the MYCN amplified Kelly, NB-1643 and NGP cell lines.
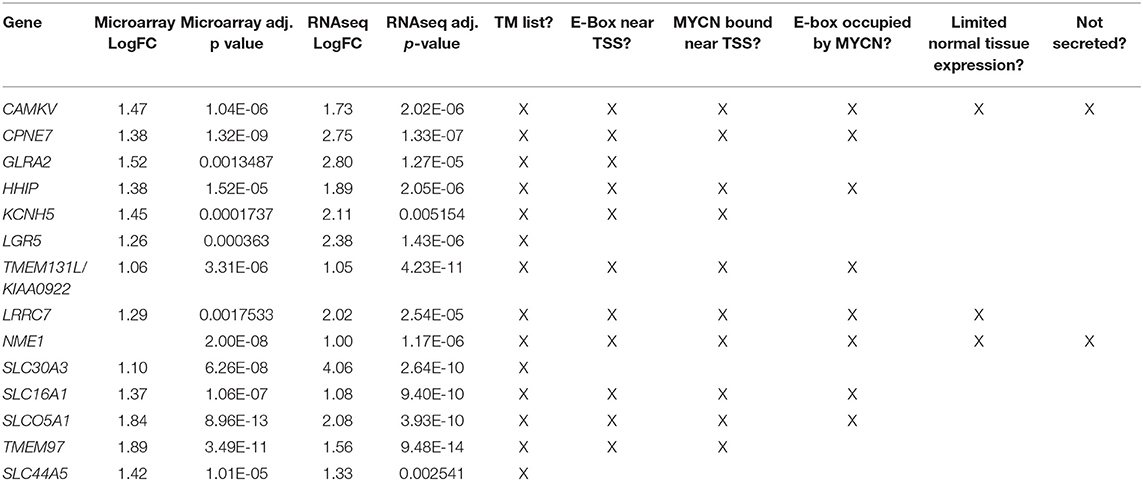
Table 1. Differentially overexpressed genes in MYCN high tumors with transmembrane domains.
To prioritize these 14 candidate immunotherapeutic targets, we first surveyed for transcription start site (TSS)-proximal E-box motifs, and showed that these were present in 11 (Table 1). To determine which of these genes are transcriptionally dependent on MYCN being bound to their promoters, we performed ChIP-seq in the MYCN amplified cell lines NB-1643, NGP, and Kelly (discovery cohort). We later performed additional MYCN ChIP-seq in the MYCN amplified cell lines COG-N-415, LA-N-5, and repeated NB-1643, as well as the non-amplified cell line NB-69 (validation cohort). Ten of these 11 differentially overexpressed gene loci were occupied by MYCN, 8 at transcription start site-proximal E-boxes (Figure 1D, Figure S1, Table 1). We next looked at the normal expression of these genes and eliminated any with widespread normal tissue expression. For this analysis, we included genes that had normal tissue expression limited to the central nervous system, considering larger biologics such as antibodies would not cross the blood-brain barrier (35), reducing our gene list to 3 (Table 1). To ensure that the protein products of these genes were not only expressed at the membrane, but were membrane bound and able to be targeted with a therapeutic, we eliminated genes with secreted products. Of the remaining two genes, NME1 has been described for its prognostic value in neuroblastoma and mutations in NME1 correlate with an aggressive phenotype (36, 37). While NME1 is somewhat differentially overexpressed in MYCN amplified neuroblastoma (logFC = 1, adjusted p-value = 1.17 × 10−6, Table S1) the protein product of NME1, nm23-H1, has diffuse sub-cellular localization that does not always include the plasma membrane (38) and mostly consists of the cytosol and endoplasmic reticulum (39), suggesting it would not be an ideal candidate for targeted immunotherapy. Calmodulin kinase-like vesicle associated (CAMKV) is more significantly differentially overexpressed in MYCN amplified tumors than NME1 (logFC = 1.73, adjusted p-value = 2.02 × 10−6). CAMKV has limited normal tissue expression in the brain (Figure S2) and has a TSS-proximal E-box that is occupied by MYCN protein. While we had intended to nominate candidate immunotherapy targets for MYCN amplified neuroblastoma that lacked normal tissue expression, the analysis did not yield any targets. Thus, we expanded our analysis to include normal expression on CNS tissues with the goal of using an antibody-based therapeutic that wouldn't cross the blood-brain barrier. From this analysis, we nominated the unstudied protein CAMKV as a putative immunotherapeutic target for MYCN amplified neuroblastoma.
To further explore the association of CAMKV and MYCN co-expression, we stratified RNA-sequencing and gene expression data from primary tumors by MYCN amplification status. We performed correlation analysis in primary tumors between MYCN and CAMKV and found them to be significantly correlated (Figures 2A,C). Finally, we examined if CAMKV protein expression is selectively expressed in MYCN amplified neuroblastoma cell lines. Whole cell lysate from 7 MYCN amplified neuroblastoma cell lines was compared to two non-amplified neuroblastoma cell lines, immortalized retinal pigment epithelial cells, and three non neuroblastoma, non-neural crest origin cancer cell lines and found CAMKV protein to be exclusively expressed in the MYCN-amplified neuroblastoma cell lines (Figure 2B). Together, these data suggest that MYCN amplification is a predictive biomarker of high CAMKV mRNA and protein expression.
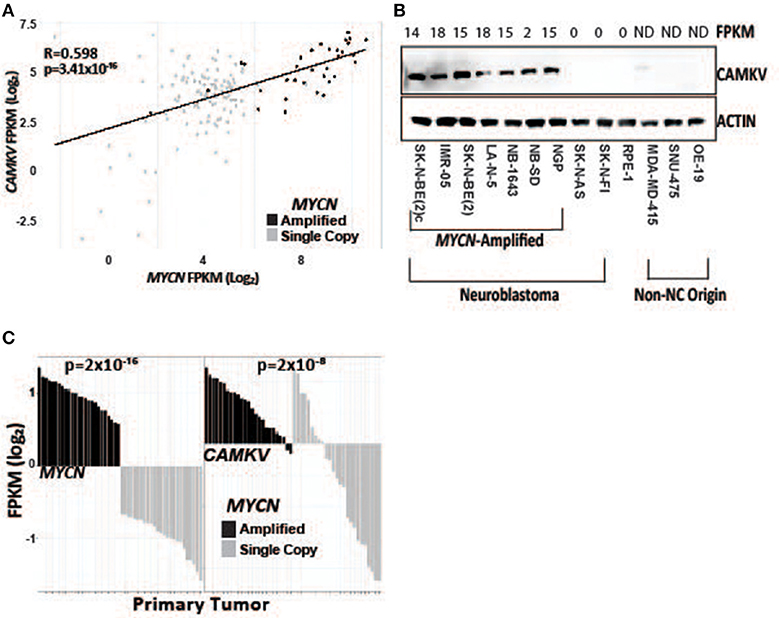
Figure 2. CAMKV expression correlates with MYCN expression. (A) MYCN and CAMKV FPKM correlate across 150 primary neuroblastoma tumors (R = 0.598, p = 3.41 × 10−16). (B) CAMKV is expressed in MYCN amplified neuroblastoma cell lines (lines 1–7) but not non-amplified lines (8, 9), retinal pigment epithelial cells (9) or non-neural-crest origin cells (11–13). FPKM for CAMKV in cell lines are shown at top, ND = not detected. (C) RNA sequencing data from 150 primary neuroblastoma tumors were stratified by MYCN expression and the FPKM for MYCN (left) and CAMKV (right) for each tumor are plotted. Tumors with amplified MYCN are colored in black and non-amplified tumors are gray. MYCN amplification correlates with MYCN FPKM (p = 2 × 10−16) and CAMKV FPKM (p = 2 × 10−8).
A limited number of non-amplified tumors appeared to have high CAMKV expression (Figures 2A,C). A recently defined subset of high-risk neuroblastoma tumors that lack MYCN amplification overexpress the MYC protein and have a poor prognosis comparable to those with amplified MYCN (40–42). While the majority of CAMKV-expressing cell lines are MYCN amplified (Figure 3A), the highest CAMKV-expressing cell line is NB-69 which does not harbor MYCN amplification, but has the highest MYC expression (Figure 3B). To further interrogate how MYCN and/or MYC protein(s) control CAMKV expression, we performed validation CHIP-sequencing for MYCN protein in the MYCN amplified neuroblastoma cell lines: COG-N-415, LA-N-5, NB-1643, and for MYC protein in the MYCN non-amplified lines: NB-69 and SK-N-SH (Figure 3C). We additionally performed MYCN ChIP-Seq on the non-amplified NB-69 cell line as a negative control. The MYCN amplified cell lines COG-N-415, LA-N-5, and NB-1643 all show MYCN occupancy at the CAMKV TSS-proximal E-boxes, similar to KELLY and NGP from the discovery cohort (top two tracks). The NB-69 cell line does not harbor MYCN amplification and has no MYCN occupancy at the TSS-proximal E-box; NB-69 is the highest MYC- and CAMKV-expressing cell line, and indeed shows MYC occupancy at the CAMKV TSS-proximal E-box (Figure 3C). The SK-N-SH cell line has the second highest MYC expression after NB-69, the second-highest CAMKV expression among non-amplified cell lines, and also has MYC occupancy at the CAMKV TSS-proximal E-box (Figure 3C). Together, these data suggest that in some high-risk neuroblastomas that lack MYCN amplification, MYC can drive CAMKV transcription.
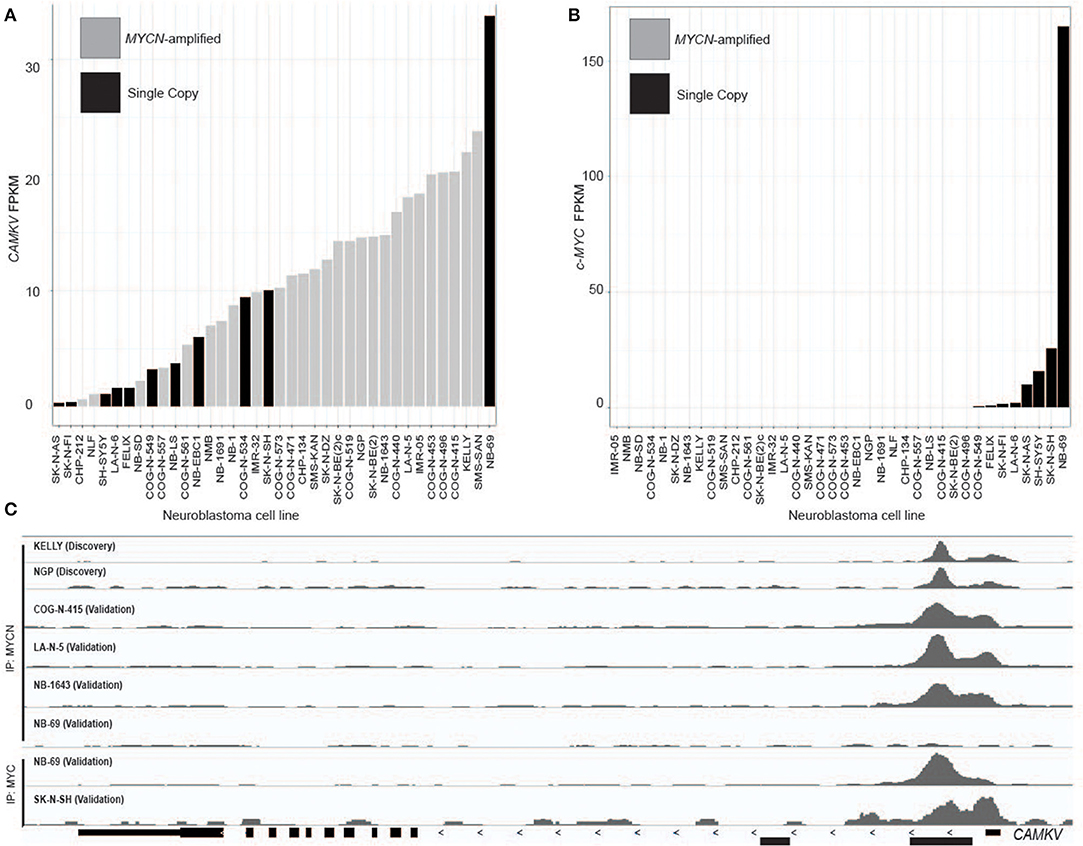
Figure 3. CAMKV is expressed in a limited number of non-amplified tumors with high MYC. (A) Neuroblastoma cell lines in rank order of CAMKV expression. MYCN amplified tumors are gray and single copy tumors are black. (B) Neuroblastoma cell lines in rank order of c-MYC expression. MYCN amplified tumors are gray and single copy tumors are black. (C) Chromatin immunoprecipitation sequencing tracks at the CAMKV locus from the MYCN amplified cell lines Kelly, NGP, COG-N-415, LA-N-5, NB-1643, and non-amplified lines NB-69 and SK-N-SH. Cells were immunoprecipitated using either an anti-MYCN antibody (top tracks) or an anti-MYC antibody (bottom tracks).
To investigate whether MYCN protein regulates CAMKV expression, we transiently depleted MYCN from MYCN amplified IMR-05 cells using shRNA and saw a significant reduction in CAMKV mRNA levels (Figure 4A, p < 0.005), suggesting that MYCN is required for CAMKV transcription in MYCN amplified neuroblastoma cells. Next, to determine if MYCN protein could drive CAMKV transcription, we used SHEP and SK-N-AS cells with a tamoxifen-inducible construct to translocate MYCN to the nucleus in cells that do not normally express MYCN. Upon MYCN translocation, we observed a significant increase in CAMKV mRNA expression in these cells, indicating that MYCN is also sufficient to drive CAMKV transcription (Figure 4B, p < 0.01). The MYCN ChIP-seq data revealed that the CAMKV TSS has a MYCN-occupied E-box motif. To confirm this occupancy, we designed primers to amplify the locus containing the E-box motif and a 5′ locus that did not have an E-box motif or an occupancy peak in the ChIP-seq data sets. We performed ChIP-qPCR in three MYCN amplified cell lines (KELLY, NGP, and IMR-05) and the non-amplified cell line, SK-N-AS. These data confirmed that the MYCN and MAX proteins occupy the E-box motif at the TSS in MYCN amplified cell lines and not in SK-N-AS cells and do not have significant binding to the 5′ locus (Figure 4C).
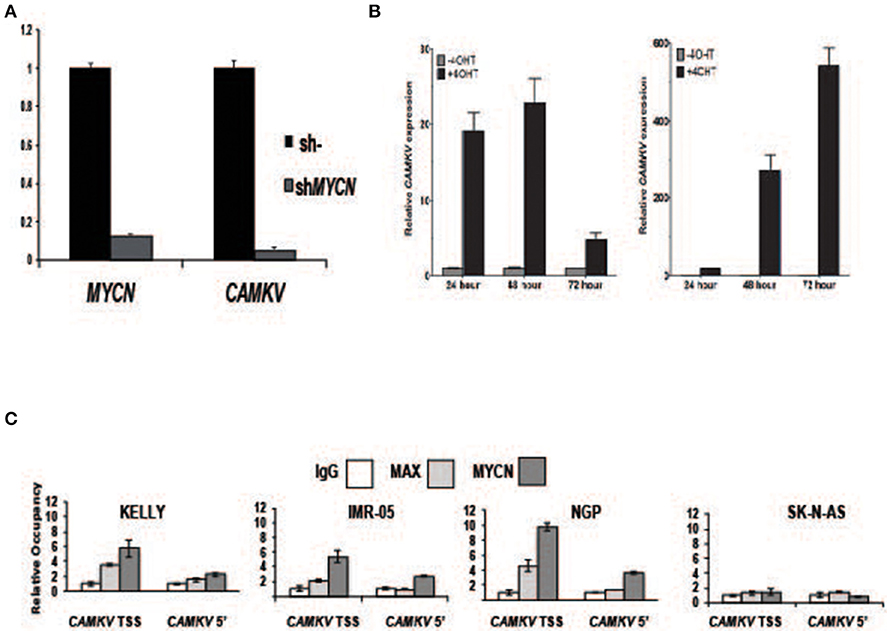
Figure 4. MYCN protein directly controls CAMKV transcription. (A) MYCN depletion by shRNA in the MYCN amplified IMR-05 cell line causes a reduction in CAMKV mRNA. Error bars represent standard deviation of technical triplicates, p = < 0.005 for both MYCN and CAMKV transcript depletions. (B) MYCN-inducible SK-N-AS (left) and SHEP (right) were treated with tamoxifen and expression of CAMKV was analyzed following 24, 48, and 72 h of treatment. Error bars represent standard deviation of technical triplicates, p < 0.01 for all changes in CAMKV expression. (C) Chromatin immunoprecipitation followed by qPCR was used to confirm the presence of MYCN and MAX proteins at the CAMKV TSS and a downstream (5') site in the CAMKV locus in three MYCN amplified cell lines (Kelly, IMR-05, and NGP) and the non-amplified cell line SK-N-AS. Error bars represent standard deviation of technical triplicates.
CAMKV is known to localize to neurite outgrowths in the brain and thus is predicted to be a membrane-bound protein. Furthermore, CAMKV is predicted to have a transmembrane domain (43). To confirm transmembrane localization in neuroblastoma, we first performed a membrane extraction in four MYCN amplified neuroblastoma cell lines [KELLY, CHP-134, LA-N-5, and SK-N-BE (2)c], three non-amplified neuroblastoma cell lines (NB-Ebc1, SK-N-AS, SK-N-FI) and RPE-1 cells. We used ADAM12 as a marker for the membrane fraction (44) and BCL-2 as a marker for the soluble fraction of cells (45). CAMKV protein was present in the membrane fraction of the four MYCN amplified neuroblastoma cell lines and NB-EBc1 cells [which have known low basal MYCN protein expression without locus amplification (18)] (Figure 5A). Next, we created cell plugs from both MYCN amplified and non-amplified neuroblastoma cell lines and performed immunocytochemistry for CAMKV. This revealed CAMKV protein to be expressed and membrane-bound in MYCN amplified SK-N-BE (2)c cells but not expressed in non-amplified SK-N-AS cells (Figure 5B). Finally, the membrane localization of CAMKV was confirmed in MYCN amplified NGP neuroblastoma cells by immunofluorescence (Figure 5C). We used NCAM1 (CD56), an established cell surface molecule expressed in neuroblastoma as a marker for the plasma membrane (32, 46) and found NCAM1 and CAMKV proteins co-localize on the plasma membrane of NGP cells. Together, these data show that CAMKV is a membrane-bound protein in MYCN amplified neuroblastoma with the potential to be targeted using a CAMKV-specific biologic.
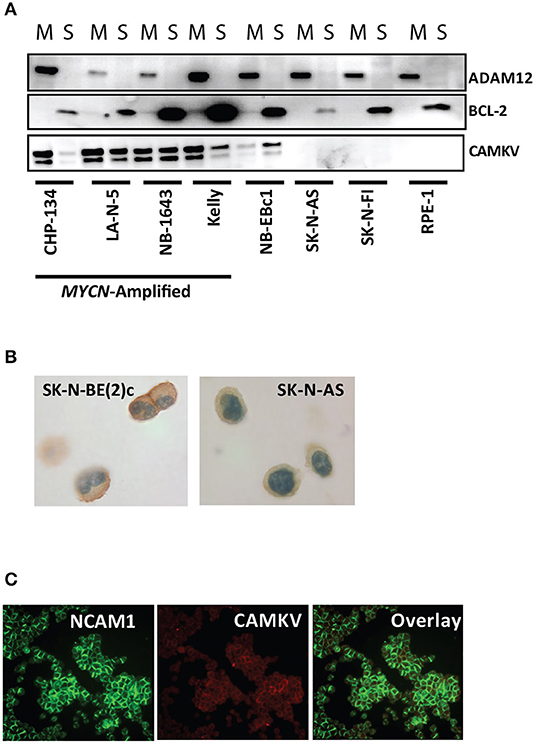
Figure 5. CAMKV protein is expressed on the plasma membrane. (A) Membrane fractionation of the MYCN amplified neuroblastoma cell lines CHP-134, LA-N-5, NB-1643, and KELLY, the non-amplified cell lines NB-Ebc1, SK-N-AS, and SK-N-FI, and immortalized retinal pigment epithelial cells (RPE-1). ADAM12 is a marker for the membrane fraction (M) and BCL-2 is a marker for the soluble fraction (S). (B) CAMKV protein expression detected by immunohistochemistry on cell plugs from the MYCN amplified SK-N-BE (2)c and non-amplified SK-N-AS neuroblastoma cell lines. (C) CAMKV protein expression detected by immunofluorescence on the MYCN amplified NGP neuroblastoma cell line. NCAM1 is used as a marker for the plasma membrane.
Here we present a computational pipeline to discover cell-surface immunotherapeutic targets in MYCN amplified neuroblastoma. MYCN amplification status is routinely tested for in all newly diagnosed neuroblastoma patients (1), and we sought to discover therapeutic targets that could utilize MYCN amplification as a biomarker for efficacy. The intersection of the most differentially overexpressed genes in tumors with the highest MYCN expression and genes predicted to be located in the plasma membrane resulted in 14 potential cell-surface targets in MYCN amplified neuroblastoma tumors, 11 of which have TSS-proximal E-boxes. Chromatin immunoprecipitation with sequencing (ChIP-seq) in MYCN amplified cell lines revealed that 4 of these TSS-proximal E-boxes were bound by MYCN protein. Two of these genes had low or absent expression in normal tissues outside of the brain, and CAMKV had the highest differential expression in MYCN amplified tumors and correlated with MYCN expression across all tumors. Perhaps not surprisingly due to the neural lineage specificity of MYCN expression, all candidate immunotherapeutic targets identified here showed expression in several brain tissues, obviating cellular immunotherapeutic or small molecule targeting approaches.
CAMKV encodes a pseudokinase that has sequence similarity to the calmodulin kinase family of proteins but lacks the essential residues required for kinase activity. CAMKV is present post-natally in the mammalian forebrain and is associated with vesicles in axons and dendrites. Recombinant CAMKV was found to bind calmodulin in the presence of calcium but lacks kinase activity with a sample substrate (47). CAMKV appears to be a substrate of CDK5 in the developing brain and functions in regulating dendritic spine maintenance (48). Remarkably, CAMKV is one of a very limited number of MYCN target genes whose protein product is localized in the plasma membrane and shows significant differential expression compared to normal non-central nervous system tissues. We previously reported that MYCN mediates GPC2 expression in neuroblastoma and that GPC2 was overexpressed in neuroblastomas that have 7q gains, where the GPC2 locus is. Due to high GPC2 expression in neuroblastomas with 7q gain and diploid MYCN, GPC2 was not one of the genes to be differentially overexpressed when comparing MYCN amplified to non-amplified tumors. Here, we nominate CAMKV as an immunotherapeutic target for MYCN amplified neuroblastoma, and suggest pursuing the identification of specific human scFv binders to this protein for the creation of antibody-drug conjugate therapeutics that should not cross the blood-brain barrier.
The datasets generated for this study can be found in the NCBI Gene Expression Omnibus (GEO) database (GSE138295 and GSE94782).
RS, PR, and JM: conceptualization. RS, JR, PR, and JM: methodology. JR, PR, and KR: formal analysis. RS, KH, JR, PR, KB, ML, and LH: investigation. DM, TB, and BP: pathology. RS, JR, and JM: funding acquisition and writing. RS and JM: supervision.
This work was supported by a St. Baldrick's-Stand Up to Cancer Dream Team Translational Research Grant (SU2C-AACR-DT-27-17). Stand Up to Cancer is a division of the Entertainment Industry Foundation. Research Grants are administered by the American Association for Cancer Research, the Scientific Partner of SU2C. This work was also supported by NIH grants R35220500 (JM), U54232568 (JM), U54 CA232568 (JM), the Damon Runyon Cancer Research Foundation PST-07-16 (KB), the Alex's Lemonade Stand Foundation with Northwestern Mutual Foundation (RS, JR, and KB), and the Giulio D'Angio Endowed Chair (JM). Some of the data in this manuscript were previously presented at conferences in the following presentations (49–52).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.00302/full#supplementary-material
Figure S1. Additional MYCN-target genes identified in the validation Chromatin immunoprecipitation experiments. ChIP-Seq tracks from 5 genes bound by MYCN at transcription start site-proximal E-boxes in the MYCN amplified COG-N-415, LA-N-5 and NB-1643 cell lines.
Figure S2. CAMKV expression in cancers and normal tissues. (A) RNA sequencing data from 150 neuroblastoma tumors and normal tissues in the GTEx project were queried for CAMKV (B) RNA sequencing data obtained from normal tissues in the GTEx project, and tumors from the TARGET program (including 150 neuroblastomas), PNOC, CBTTC, TCGA and the GEO database (GSE60052, small cell lung cancer) were queried for CAMKV RNA expression. For both plots, RNA expression values were FPKM-normalized and log2-transformed. Data are represented by boxplots of the median plus the first and third quartiles. Dots represent outliers.
Table S1. Gene lists from microarray or RNAseq analysis for MYCN high tumors and transmembrane domain-containing gene products.
1. Maris JM. Recent advances in neuroblastoma. N Engl J Med. (2010) 362:2202–11. doi: 10.1056/NEJMra0804577
2. Matthay KK, Maris JM, Schleiermacher G, Nakagawara A, Mackall CL, Diller L, et al. Neuroblastoma. Nat Rev Dis Primers. (2016) 2:16078. doi: 10.1038/nrdp.2016.78
3. Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. (2010) 363:1324–34. doi: 10.1056/NEJMoa0911123
4. Schwab M, Alitalo K, Klempnauer KH, Varmus HE, Bishop JM, Gilbert F, et al. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature. (1983) 305:245–8. doi: 10.1038/305245a0
5. Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. (1984) 224:1121–4. doi: 10.1126/science.6719137
6. Seeger RC, Brodeur GM, Sather H, Dalton A, Siegel S, Wong KY, et al. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N Engl J Med. (1985) 313:1111–6. doi: 10.1056/NEJM198510313131802
7. Deyell RJ, Attiyeh EF. Advances in the understanding of constitutional and somatic genomic alterations in neuroblastoma. Cancer Genet. (2011) 204:113–21. doi: 10.1016/j.cancergen.2011.03.001
8. Sakamuro D, Prendergast GC. New Myc-interacting proteins: a second Myc network emerges. Oncogene. (1999) 18:2942–54. doi: 10.1038/sj.onc.1202725
9. Grandori C, Cowley SM, James LP, Eisenman R. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu Rev Cell Dev Biol. (2000) 16:653–99. doi: 10.1146/annurev.cellbio.16.1.653
10. Wakamatsu Y, Watanabe Y, Nakamura H, Kondoh H. Regulation of the neural crest cell fate by N-myc: promotion of ventral migration and neuronal differentiation. Development. (1997) 124:1953–62.
11. Oliver TG, Grasfeder LL, Carroll AL, Kaiser C, Gillingham CL, Lin SM, et al. Transcriptional profiling of the Sonic hedgehog response: a critical role for N-myc in proliferation of neuronal precursors. Proc Natl Acad Sci USA. (2003) 100:7331–6. doi: 10.1073/pnas.0832317100
12. Kenney AM, Widlund HR, Rowitch DH. Hedgehog and PI-3 kinase signaling converge on Nmyc1 to promote cell cycle progression in cerebellar neuronal precursors. Development. (2004) 131:217–28. doi: 10.1242/dev.00891
13. Zeid R, Lawlor MA, Poon E, Reyes JM, Fulciniti M, Lopez MA, et al. Enhancer invasion shapes MYCN-dependent transcriptional amplification in neuroblastoma. Nat Genet. (2018) 50:515–23. doi: 10.1038/s41588-018-0044-9
14. Faisal A, Vaughan L, Bavetsias V, Sun C, Atrash B, Avery S, et al. The aurora kinase inhibitor CCT137690 downregulates MYCN and sensitizes MYCN-amplified neuroblastoma in vivo. Mol Cancer Ther. (2011) 10:2115–23. doi: 10.1158/1535-7163.MCT-11-0333
15. Valentijn LJ, Koster J, Haneveld F, Aissa RA, van Sluis P, Broekmans ME, et al. Functional MYCN signature predicts outcome of neuroblastoma irrespective of MYCN amplification. Proc Natl Acad Sci USA. (2012) 109:19190–5. doi: 10.1073/pnas.1208215109
16. Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. (2013) 3:308–23. doi: 10.1158/2159-8290.CD-12-0418
17. Gustafson WC, Meyerowitz JG, Nekritz EA, Chen J, Benes C, Charron E, et al. Drugging MYCN through an allosteric transition in Aurora kinase A. Cancer Cell. (2014) 26:414–27. doi: 10.1016/j.ccr.2014.07.015
18. Hart LS, Rader J, Raman P, Batra V, Russell MR, Tsang M, et al. Preclinical therapeutic synergy of MEK1/2 and CDK4/6 inhibition in neuroblastoma. Clin Cancer Res. (2017) 23:1785–96. doi: 10.1158/1078-0432.CCR-16-1131
19. Bosse KR, Raman P, Zhu Z, Lane M, Martinez D, Heitzeneder S, et al. Identification of GPC2 as an oncoprotein and candidate immunotherapeutic target in high-risk neuroblastoma. Cancer Cell. (2017) 32:295–309 e212. doi: 10.1016/j.ccell.2017.08.003
20. Harenza JL, Diamond MA, Adams RN, Song M, Davidson HL, Hart LS, et al. Transcriptomic profiling of 39 commonly-used neuroblastoma cell lines. Sci Data. (2017) 4:170033. doi: 10.1038/sdata.2017.33
21. Schnepp RW, Khurana P, Attiyeh EF, Raman P, Chodosh SE, Oldridge DA, et al. A LIN28B-RAN-AURKA signaling network promotes neuroblastoma tumorigenesis. Cancer Cell. (2015) 28:599–609. doi: 10.1016/j.ccell.2015.09.012
22. Sussman RT, Stanek TJ, Esteso P, Gearhart JD, Knudsen KE, McMahon SB. The epigenetic modifier ubiquitin-specific protease 22 (USP22) regulates embryonic stem cell differentiation via transcriptional repression of sex-determining region Y-box 2 (SOX2). J Biol Chem. (2013) 288:24234–46. doi: 10.1074/jbc.M113.469783
23. Lee TI, Johnstone SE, Young RA. Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat Protoc. (2006) 1:729–48. doi: 10.1038/nprot.2006.98
24. Upton K, Modi A, Patel K, Conkrite KL, Sussman R, Way GP, et al. Epigenomic profiling of neuroblastoma cell lines. bioRxiv [Preprint]. (2019). doi: 10.1101/829754
25. Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. accuracy assessment. Genome Res. (1998) 8:175–85. doi: 10.1101/gr.8.3.175
26. Lindgreen S. AdapterRemoval: easy cleaning of next-generation sequencing reads. BMC Res Notes. (2012) 5:337. doi: 10.1186/1756-0500-5-337
27. Krueger F. Trim Galore: A Wrapper Tool Around Cutadapt and FastQC to Consistently Apply Quality and Adapter Trimming to FastQ files, with some Extra Functionality for MspI-Digested RRBS-Type (Reduced Representation Bisufite-Seq) Libraries. (2015). Available online at: http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/
28. Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. (2009) 25:1754–60. doi: 10.1093/bioinformatics/btp324
29. Alec Wysoker KT, Fennell T. Picard Tools. (2013). Available online at: http://broadinstitute.github.io/picard/
30. Ramachandran P, Palidwor GA, Porter CJ, Perkins T. MaSC: mappability-sensitive cross-correlation for estimating mean fragment length of single-end short-read sequencing data. Bioinformatics. (2013) 29:444–50. doi: 10.1093/bioinformatics/btt001
31. Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson D, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. (2008) 9:R137. doi: 10.1186/gb-2008-9-9-r137
32. Feng Y, Wang Y, Zhu Z, Li W, Sussman RT, Randall M, et al. Differential killing of CD56-expressing cells by drug-conjugated human antibodies targeting membrane-distal and membrane-proximal non-overlapping epitopes. MAbs. (2016) 8:799–810. doi: 10.1080/19420862.2016.1155014
33. Vivian J, Rao AA, Nothaft FA, Ketchum C, Armstrong J, Novak A, et al. Toil enables reproducible, open source, big biomedical data analyses. Nat Biotechnol. (2017) 35:314–6. doi: 10.1038/nbt.3772
34. Binder JX, Pletscher-Frankild S, Tsafou K, Stolte C, O'Donoghue SI, Schneider R, et al. COMPARTMENTS: unification and visualization of protein subcellular localization evidence. Database. (2014) 2014:bau012. doi: 10.1093/database/bau012
35. Pardridge WM. Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab. (2012) 32:1959–72. doi: 10.1038/jcbfm.2012.126
36. Almgren MA, Henriksson KC, Fujimoto J, Chang CL. Nucleoside diphosphate kinase A/nm23-H1 promotes metastasis of NB69-derived human neuroblastoma. Mol Cancer Res. (2004) 2:387–94. Available online at: https://mcr.aacrjournals.org/content/2/7/387.long
37. Garcia I, Mayol G, Rios J, Domenech G, Cheung NK, Oberthuer A, et al. A three-gene expression signature model for risk stratification of patients with neuroblastoma. Clin Cancer Res. (2012) 18:2012–23. doi: 10.1158/1078-0432.CCR-11-2483
38. Marioni G, Ottaviano G, Lionello M, Lora L, Lovato A, Staffieri C, et al. Nm23-H1 nuclear expression is associated with a more favourable prognosis in laryngeal carcinoma: univariate and multivariate analysis. Histopathology. (2012) 61:1057–64. doi: 10.1111/j.1365-2559.2012.04331.x
39. Bosnar MH, De Gunzburg J, Bago R, Brecevic L, Weber I, Pavelic J. Subcellular localization of A and B Nm23/NDPK subunits. Exp Cell Res. (2004) 298:275–84. doi: 10.1016/j.yexcr.2004.04.018
40. Wang LL, Suganuma R, Ikegaki N, Tang X, Naranjo A, McGrady P, et al. Neuroblastoma of undifferentiated subtype, prognostic significance of prominent nucleolar formation, and MYC/MYCN protein expression: a report from the Children's Oncology Group. Cancer. (2013) 119:3718–26. doi: 10.1002/cncr.28251
41. Wang LL, Teshiba R, Ikegaki N, Tang XX, Naranjo A, London WB, et al. Augmented expression of MYC and/or MYCN protein defines highly aggressive MYC-driven neuroblastoma: a Children's Oncology Group study. Br J Cancer. (2015) 113:57–63. doi: 10.1038/bjc.2015.188
42. Zimmerman MW, Liu Y, He S, Durbin AD, Abraham B, Easton J, et al. MYC drives a subset of high-risk pediatric neuroblastomas and is activated through mechanisms including enhancer hijacking and focal enhancer amplification. Cancer Discov. (2018) 8:320–35. doi: 10.1158/2159-8290.CD-17-0993
43. Hofmann F, James P, Vorherr T, Carafoli E. The C-terminal domain of the plasma membrane Ca2+ pump contains three high affinity Ca2+ binding sites. J Biol Chem. (1993) 268:10252–9.
44. Stautz D, Leyme A, Grandal MV, Albrechtsen R, van Deurs B, Wewer U, et al. Cell-surface metalloprotease ADAM12 is internalized by a clathrin- and Grb2-dependent mechanism. Traffic. (2012) 13:1532–46. doi: 10.1111/j.1600-0854.2012.01405.x
45. Danial NN. BCL-2 family proteins: critical checkpoints of apoptotic cell death. Clin Cancer Res. (2007) 13:7254–63. doi: 10.1158/1078-0432.CCR-07-1598
46. Cunningham BA, Hemperly JJ, Murray BA, Prediger EA, Brackenbury R, Edelman GM. Neural cell adhesion molecule: structure, immunoglobulin-like domains, cell surface modulation, and alternative RNA splicing. Science. (1987) 236:799–806. doi: 10.1126/science.3576199
47. Godbout M, Erlander MG, Hasel KW, Danielson P, Wong KK, Battenberg EL, et al. 1G5: a calmodulin-binding, vesicle-associated, protein kinase-like protein enriched in forebrain neurites. J Neurosci. (1994) 14:1–13. doi: 10.1523/JNEUROSCI.14-01-00001.1994
48. Liang Z, Zhan Y, Shen Y, Wong CC, Yates JR III, Plattner F, et al. The pseudokinase CaMKv is required for the activity-dependent maintenance of dendritic spines. Nat Commun. (2016) 7:13282. doi: 10.1038/ncomms13282
49. Sussman RT, Huang K, Raman P, Maris JM. CAMKV as an immunotherapy target in MYCN-amplified neuroblastoma. Cancer Res. (2015) 75(15 Suppl): 1712. doi: 10.1158/1538-7445.AM2015-1712
50. Sussman RT. CAMKV as a Target for Immunotherapy in MYCN-Amplified Neuroblastoma. Oral Presentation. Philadelphia, PA: Alex's Lemonade Stand Foundation Young Investigator Summit (2015).
Keywords: CAMKV, MYCN, immunotherapy, neuroblastoma, ChIP-Seq
Citation: Sussman RT, Rokita JL, Huang K, Raman P, Rathi KS, Martinez D, Bosse KR, Lane M, Hart LS, Bhatti T, Pawel B and Maris JM (2020) CAMKV Is a Candidate Immunotherapeutic Target in MYCN Amplified Neuroblastoma. Front. Oncol. 10:302. doi: 10.3389/fonc.2020.00302
Received: 23 October 2019; Accepted: 20 February 2020;
Published: 06 March 2020.
Edited by:
Marianna Szemes, University of Bristol, United KingdomReviewed by:
Ken Lieuw, Walter Reed National Military Medical Center, United StatesCopyright © 2020 Sussman, Rokita, Huang, Raman, Rathi, Martinez, Bosse, Lane, Hart, Bhatti, Pawel and Maris. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John M. Maris, TWFyaXNAZW1haWwuY2hvcC5lZHU=
†Present address: Robyn T. Sussman, Department of Pathology and Laboratory Medicine, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.