 Rebecca Epperly
Rebecca Epperly Stephen Gottschalk
Stephen Gottschalk M. Paulina Velasquez
M. Paulina Velasquez
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 28 February 2020
Sec. Pediatric Oncology
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.00262
This article is part of the Research Topic Microenvironment and Therapy-Resistance in Leukemias View all 8 articles
Chimeric antigen receptor (CAR) T cells targeting CD19 have been successful treating patients with relapsed/refractory B cell acute lymphoblastic leukemia (ALL) and B cell lymphomas. However, relapse after CAR T cell therapy is still a challenge. In addition, preclinical and early clinical studies targeting acute myeloid leukemia (AML) have not been as successful. This can be attributed in part to the presence of an AML microenvironment that has a dampening effect on the antitumor activity of CAR T cells. The AML microenvironment includes cellular interactions, soluble environmental factors, and structural components. Suppressive immune cells including myeloid derived suppressor cells and regulatory T cells are known to inhibit T cell function. Environmental factors contributing to T cell exhaustion, including immune checkpoints, anti-inflammatory cytokines, chemokines, and metabolic alterations, impact T cell activity, persistence, and localization. Lastly, structural factors of the bone marrow niche, secondary lymphoid organs, and extramedullary sites provide opportunities for CAR T cell evasion by AML blasts, contributing to treatment resistance and relapse. In this review we discuss the effect of the AML microenvironment on CAR T cell function. We highlight opportunities to enhance CAR T cell efficacy for AML through manipulating, targeting, and evading the anti-inflammatory leukemic microenvironment.
Chimeric Antigen Receptors (CARs) are a novel immunotherapeutic strategy that incorporates the antigen specificity of an antibody's single chain variable fragment (scFv) with the transmembrane and intracellular signaling domains of the CD3ζ chain and costimulatory molecules (1, 2). CD19 targeted CAR T cell therapy has proven successful for the treatment of relapsed/refractory B cell acute lymphoblastic leukemia (ALL) (3–7). Efforts to expand adoptive immunotherapy strategies to acute myeloid leukemia (AML) are complicated by antigen overlap between normal hematopoietic progenitor cells (HPCs) and leukemic blasts (8). Early clinical studies of CAR T cells for AML are ongoing, exploring several targets including CD123 (9, 10), CD33 (11), C-type lectin-like molecule 1 (CLL-1) (12, 13), and Lewis-Y (14). Additional targets under preclinical investigation include CD135 (FLT-3 receptor) (15–17), Folate receptor β (18), CD44v6 (19), WT1 (20), B7-H3 (21–23), CD70 (24, 25), and CD7 (26). While clinical experience thus far has shown feasibility and safety of CAR T cells for AML, efficacy has been limited in comparison to CD19-CAR T cell therapy for ALL (11, 14).
In addition to these challenges, the AML microenvironment is highly immunosuppressive. Data on the direct impact of this aspect on CAR T cells in AML is still emerging. However, we can extrapolate from the well-established role of the microenvironment in CAR T cell therapy for solid tumors (27, 28), clinical experience with CD19-CAR T cells for B-ALL, and evidence detailing the dynamics of the AML microenvironment. This includes complex interactions between native immune cells, secreted factors, and structural components (Figure 1). In this review we discuss how each of these aspects can impact CAR T cell therapy and highlight opportunities to improve CAR T cell therapy for AML.
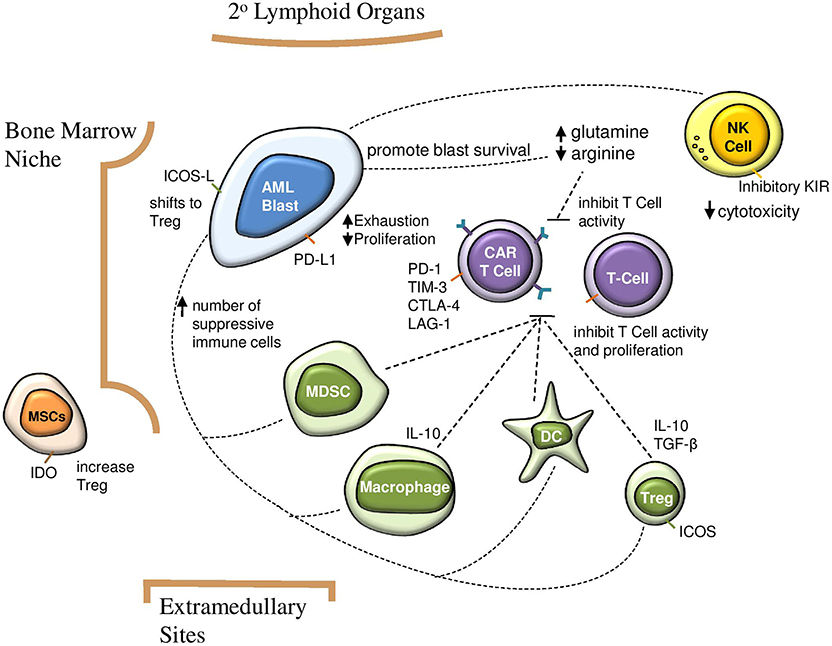
Figure 1. Impact of suppressive AML microenvironment on immunosurveillance and CAR T cell efficacy. AML blasts increase number of suppressive immune cells, which in turn inhibit CAR T cell activity and proliferation. Direct interactions between AML blasts and CAR T cells contribute to T cell exhaustion and decreased proliferation. A glutamine rich and arginine low environment contributes to AML blast survival and impairs CAR T cell function. AML, acute myeloid leukemia; CAR, chimeric antigen receptor; NK, natural killer; Treg, regulatory T cell; MDSC, myeloid derived suppressor cell; MSC, mesenchymal stem cell; DC, dendritic cell; IDO, indoleamine 2,3-dioxygenase; ICOS, inducible T cell costimulator.
The AML microenvironment contains cell types which can dampen T cell responses. These include AML blasts, myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), macrophages, and dendritic cells. Counteracting these cellular interactions which promote leukemic survival may bolster the efficacy of CAR T cell therapy in AML.
In order to escape immune surveillance, AML blasts downregulate major histocompatibility complex (MHC) class I and II expression (29–32) and express inhibitory ligands such as programmed death-ligand 1(PDL-1), B7-H3 (CD276), and Galectin 9 (Gal-9) (33–35). Immune checkpoint ligands expressed on cells in the AML microenvironment interact with receptors including programmed death receptor 1 (PD-1), cytotoxic T-lymphocyte associated protein 4 (CTLA-4), lymphocyte-activation gene 3 (LAG3), and T cell immunoglobulin and mucin-containing-3 (TIM3) on T cells, leading to T cell exhaustion (34, 36, 37). Despite success of antibody-based immune checkpoint blockade for solid tumors, only modest response has been demonstrated in early trials for AML (38–40). While the mechanism for limited efficacy of immune checkpoint inhibitors as monotherapy in AML is not well-understood, disease burden is most likely one contributing factor. Thus, there is still potential for application of checkpoint inhibitors as combination therapies. In the case of combination with CAR T cells, there is the additional potential benefit that checkpoint inhibitors may improve T cell persistence and enhance response.
CAR T cells have been designed to intrinsically block PD-1 through secreted single chain variable fragments (scFv) or antibodies, shRNA, dominant-negative receptors, and CRISPR/cas9 mediated knockout (41–44). Intrinsically blocking PD-1 has similar preclinical efficacy in preventing PD-1 mediated T cell exhaustion as co-administration of PD-1/PDL-1 antibodies, with the added benefit of localizing the effect to the site of T cell activity.
Additionally, AML blasts release reactive oxygen species and indolamine 2, 3 dioxygenase (IDO). IDO in turn catabolizes tryptophan degradation, which interferes with T cell proliferation and effector function (45, 46). Leukemic blasts recruit other immunosuppressive cells such as MDSCs and Tregs to the tumor microenvironment, as well as altering metabolite availability.
MDSCs are immature myeloid cells that arise in the bone marrow from myeloid progenitors (47, 48). Their presence has been documented in cancer patients regardless of tumor, and their potent immunosuppressive function is widely recognized (49). An increase in inflammatory mediators including interleukin (IL) 6, IL1β, granulocyte-macrophage colony-stimulating factor (GMCSF) and granulocyte-colony stimulating factor (GCSF) drive the accumulation and inhibitory function of MDSCs. MDSCs localize to the tumor microenvironment through chemokines such as VEGF (50). MDSCs polarize macrophages, inhibit natural killer (NK) cell-mediated tumor cell lysis and recruit regulatory T cells (Tregs). They prevent T cell activation by sequestering essential amino acids, generating reactive oxygen species (ROS), and downregulating the expression of CD62L on circulating T cells (51, 52).
An expansion in the number of MDSCs has been reported in AML patients, where they inhibit T cell responses (53–56). Expansion of MDSC decreases CAR T cell efficacy in solid tumors (57), and interventions that aim to inhibit MDSC function have had preclinical success in improving CAR T cell function (58). Given this inhibitory role on T cell proliferation and activity, MDSCs have the potential to contribute to resistance to CAR T cell therapy. Thus, strategies that are specific for antigens such as CD33 which are also present on MDSCs have antitumor activity not only through T-cell mediated direct cytotoxicity of CD33+ blasts, but also through inhibition of CD33+ MDSC (55). This is the case for a CD33xCD3 bispecific T-cell engaging antibody (BiTE)55 and potentially of CD33-CAR T cells (59–61).
Tregs limit activation and proliferation of cytolytic T cells. They act both directly through anti-inflammatory cytokine secretion and contact dependent suppression and indirectly, by interfering with the activation status of antigen presenting cells (62). The AML microenvironment favors the expansion of Tregs (37, 63–65). Expression of inducible T cell co-stimulator ligand (ICOSL) on AML blasts stimulates T cells through inducible T cell co-stimulator (ICOS), leading to differentiation to Treg phenotype and expansion of the Treg subset (66). Additionally, AML blasts and bone marrow mesenchymal cells overexpress IDO, which promotes the emergence of a Treg phenotype and limits T cell proliferation (67).
The depletion of Tregs in a murine AML model led to an increase in proliferation and activity of adoptively transferred tumor reactive cytotoxic T cells, highlighting the immunosuppressive capabilities of Tregs in the AML microenvironment (68). In the clinical setting a lower frequency of Tregs has proven predictive of better antitumor response to the CD19xCD3 bispecific BiTE blinatumomab for ALL (69).
Several strategies can potentially circumvent the inhibitory effects of Tregs on CAR T cells. The integration of co-stimulatory domains in second or third generation CAR T cells allows CAR T cells to proliferate despite the inhibitory effects of Treg cells (70). Genetic modifications in the PYAP Lck-binding motif of CD28 costimulatory domain of a CD28.4-1BB CAR have resulted in disruption of the IL2 signaling pathway, blocking Treg activity with modest enhancement of efficacy in preclinical solid tumor models (71). In addition, transgenic expression of IL15 in CAR T cells favors proliferation of cytotoxic T cells over Tregs (72). Additionally, the administration of lymphodepleting chemotherapy prior to CAR T cell infusion depletes Tregs in the tumor microenvironment and allows for expansion of adoptively transferred CAR T cells (71).
NK cells that are present in the AML microenvironment are often dysfunctional since it promotes the expression of inhibitory Killer-Cell Immunoglobulin-like Receptors (KIRs) resulting in decreased interferon (IFN)-γ secretion and cytolytic capacity (73). Additionally, downregulation of micro-RNAs (MIRs), single stranded non-coding RNAs that play a role in gene expression, leads to a downregulation of IL2 and IL15 cytokine receptors, NKG2D, and transcription factors such as c-myb in NK cells (74–76). Downregulation of transcription factors inhibits the activity of naturally occurring NK cells in the leukemic microenvironment. Despite this, the infusion of unmodified donor-derived NK cells in post-transplant patients has proven beneficial in controlling leukemia relapse by contributing to Graft-vs.-Leukemia (GVL) effects (77–81). This antitumor response can be enhanced by generation of a cytokine-induced memory-like NK cell subset (82). Genetic modifications to NK cells including introduction of CARs have been investigated as a platform for cellular immunotherapy in preclinical leukemia models (83–86). CD19-CAR NK cells expressing IL15 have shown improved persistence, with encouraging results in an ongoing clinical study (NCT03056339) (86). CAR NK cells have potential benefit for use as an allogeneic product given a lower risk of graft-vs.-host disease than T cell-based therapies as well as the presence of native activating receptors such as NKG2D, which may amplify antitumor activity (77, 87–89).
Macrophages can perform both inhibitory and stimulatory functions. In the AML microenvironment, blasts and MDSCs skew macrophage differentiation to an inhibitory phenotype (90). Inhibitory macrophages contribute to a hostile environment for CAR T cells in AML. In patients receiving CD19-CAR T cells for B cell lymphoma, infiltration with tumor associated macrophages diminishes response to CAR T cells (91). Macrophages have been implicated in the pathogenesis of cytokine release syndrome associated with CAR T cell therapy (92), evidence that modulation of macrophage function has significance for titrating clinical response.
Dendritic cells also play an important role in modulating the immune microenvironment. AML blasts can cause arrest of dendritic cell maturation, promoting immune tolerance and inducing Treg development (93). Chemotherapy induced cell death contributes to production of tolerogenic dendritic cells, potentially impacting immunotherapy efforts when used in combination (94). Driving dendritic cells toward a T cell stimulatory phenotype could improve CAR T cell therapy. For example, CAR T cells genetically modified to constitutively express CD40 ligand promote secretion of the pro-inflammatory cytokine IL12 by dendritic cells, which enhances antitumor efficacy (95).
Many suppressive effects of the AML microenvironment are mediated through soluble environmental factors. AML blasts influence cells in the microenvironment to secrete anti-inflammatory cytokines and alter chemokine-mediated trafficking. Metabolic changes drive the microenvironment to support leukemic cell growth and survival while limiting immune responses.
Both exogenous and endogenous cytokines are integral in promoting the expansion and effector function of CAR T cells (96, 97). Specific cytokine signatures have been linked to efficacy, for example increased IL6 levels observed in responders to CD19-directed CAR T therapy for ALL (98). While the presence of AML blasts can stimulate monocytes to secrete pro-inflammatory cytokines including tumor necrosis factor-α (TNF-α), IL1β, and IL6, they also cause increased production of the anti-inflammatory cytokine IL10 (93). Additionally, increased Treg subsets contribute to production of IL10 and transforming growth factor β (TGF-β), which can limit the effector function of CAR T cells (66). In analysis of CD123-CAR T cells targeting AML, TNF-α and IFN-γ upregulate CD123 expression on endothelial cells, increasing risk for capillary leak. Production of IL6 and IL1 by monocytes and macrophages mediates cytokine release syndrome and is associated with neurotoxicity in the CD19-CAR clinical experience (99, 100). This illustrates that the cytokine milieu in the leukemic microenvironment impacts not only antitumor activity of CAR T cells, but also toxicities.
Chemokines play an important role in trafficking of T cells to the lymphoid compartment and toward malignant cells in other sites. The serum chemokine profile in AML patients differs from healthy controls including levels of CCL3, CCL4, CCL5, CCL17, and CXCL10 (101, 102). Variations in systemic chemokine levels and expression of chemokine receptors in patients with AML have been linked with prognosis and treatment response (103, 104). Chemokine-mediated trafficking has been exploited to enhance CAR T cell activity. For example, forced co-expression of the chemokine receptor CCR4 with CAR increases accumulation of CD8+ effector CAR T cells in the lymphoid compartment in a Hodgkin Lymphoma model (105). Expression of chemokine ligand CCL19 together with IL7, which are typically produced by lymphoid T-cell zones, enhances CAR T infiltration into solid tumors (106). These strategies are particularly relevant to targeting CAR T cells toward AML disease burden in chloromas or extramedullary sites.
The immunosuppressive capabilities of the AML microenvironment are potentiated by metabolic alterations including changes in amino acid and nucleotide concentrations. The AML microenvironment rich in glutamine contributes to immunosuppression as higher concentrations of this amino acid inhibit T cells by contributing to T cell exhaustion (107). Inhibiting glutamine metabolism using L-asparaginase, a chemotherapeutic agent that also has glutaminase activity, has proven effective treating AML (108–111). In addition, culturing T cells in glutamine restricted media can improve antitumor activity of CD8+ T cells by reducing T cell exhaustion and allowing for enrichment of an effector memory T cell subset (107). Thus, modulating glutamine concentrations in culture media has the potential to improve CAR T cell activity in a similar fashion.
Conversely, AML blasts promote a low arginine microenvironment mediated through the expression of arginase II. Limiting arginine availability steers monocytes toward a suppressive phenotype and acts as a metabolic brake for T cells, evidenced by lower IFN-γ production and increased expression of checkpoint inhibitors leading to decreased proliferation (112). Inhibiting arginine metabolism enhances antitumor activity of CD33-CAR T cells for AML in preclinical studies (113), demonstrating the importance of metabolic dynamics on efficacy of CAR T cell therapy.
Increases in adenosine concentration also inhibit T cell activity in the leukemia microenvironment. Adenosine is metabolized by CD73 and CD39 from extracellular ATP (114, 115). CD73 is expressed on tumor cells, MDSCs and Tregs (114), while CD39 has been described on CD8+ T cells. Increased adenosine leads to signaling through adenosine receptors, such as adenosine 2A (A2A), which in turn results in T cell suppression. Altering adenosine metabolism by targeting CD73 or CD39 with monoclonal antibodies or inhibitory drugs result in more effective antitumor activity (116). Targeting downstream adenosine metabolism by blocking A2A receptors with pharmacological agents has resulted in enhanced CAR T cell therapy for solid tumors preclinically (114), but its role in hematologic malignancies is less established.
The physical spaces where AML blasts reside include the bone marrow niche, secondary lymphoid organs, and extramedullary sites. For CAR T cells to be effective, they not only have to penetrate the complex bone marrow environment, but also sanctuary sites such as the central nervous system, which can harbor blasts and allow for immune escape.
The bone marrow microenvironment includes hematopoietic, endothelial, osteoblastic, and stromal components. Mesenchymal stem cells (MSCs) are stromal cells which define the bone marrow microenvironment and give rise to other supporting cells. MSCs highly express IDO, which correlates with expansion of Tregs and could inhibit CAR T cell effector function (117).
MSCs from AML patients have a higher propensity to differentiate into adipocytes, and the interactions between AML blasts and adipocytes in the bone marrow niche impact cellular metabolism (118). AML blasts induce lipolysis in bone marrow adipocytes, shifting toward fatty acid oxidation and an environment favorable for leukemic survival (119). These AML-adipocyte interactions have been linked to chemotherapeutic resistance (120, 121). The ability of substrates for fatty acid oxidation can impact not only AML blast survival, but also T cell persistence. Specifically, fatty acid oxidation is key in development of memory CD8+ T cells (122, 123). Signaling through a 4-1BB costimulatory domain is associated with a shift toward fatty acid oxidation rather than glycolysis (124), which is proposed as a potential mechanism for improved persistence of 41-BB containing CAR constructs (124).
Interactions through the chemokine receptor CXCR4/CXCL12 pathway are integral in leukocyte trafficking in the bone marrow niche, involving both the endothelium and leukocytes. CXCR4 expression dictates AML blast migration, is implicated in prognosis, and is being explored as a therapeutic target (125). The CXCR4 pathway can also be involved in migration of CAR T cells to the bone marrow niche, demonstrated by improvement in bone marrow localization when CAR T cells are co-transduced with CXCR4 in preclinical studies (126).
Inflammatory responses impact the interaction between HPCs and the bone marrow niche, prompting quiescent HPCs to actively proliferate. This has been demonstrated secondary to viral infections (127), chemotherapy, and mediated through cytokines including the IFN-α signaling pathway (128). This IFN-α based shift in HSC populations can sensitize leukemic stem cells to cytotoxic therapies and has justified the use of IFN-α in management of chronic myelogenous leukemia. IFN-α supports proliferation of T cells, and co-administration of IFN-α with CD19-CAR T cells has shown enhanced activity in vitro for treatment of B cell lymphoma (129). The impact on myeloid progenitors in the bone marrow niche and enhanced T cell proliferation suggests a potential benefit for combining IFN-α with CAR T cell therapy to enhance anti-leukemic effect in AML.
Clinical trials with CD30-CAR T cells in Hodgkin lymphoma and CD19-CAR T cells in non-Hodgkin lymphoma have shown that CAR T cells do penetrate into lymph nodes and have persistent antitumor activity (130, 131). While lymphoid tissues have an important role to enhance antigen presentation and selective T cell proliferation, fibroblastic reticular cells (FRC) can attenuate T cell expansion through immune suppressive mediators including IDO, A2A receptor, prostaglandins, and TGFβ (132, 133). This suppressive effect has been demonstrated on native T cells both in murine models and humanized in vitro systems, however there is some evidence that activated effector CAR T cells may be resistant to this suppression (133).
AML demonstrates a variety of extramedullary manifestations, either in isolation or associated with bone marrow disease (134, 135). Chloromas are noted both at the time of initial diagnosis and relapse. The central nervous system and reproductive organs are particularly vulnerable to relapse, including after allogeneic hematopoietic stem cell transplant, as they can act as sanctuary sites to harbor leukemic cells through physical barriers (136). In order for CAR T cell therapy to be effective in treating refractory or relapsed AML, CAR T cells must be able to penetrate and persist in these sites. In clinical studies, CD19-CAR T cells have been shown to infiltrate, expand, and have antitumor activity in the CNS (137) and reproductive sites (138).
The hostile AML microenvironment has a notable role in dampening T cell effector function. The cellular interactions, soluble environmental factors, and structural components of the AML microenvironment have potential to limit antitumor efficacy of CAR T cells. Investigating complex interactions between the AML microenvironment, CAR T cell therapy, and other novel anti-leukemic therapies allows the opportunity to improve upon our current regimens. Targeting antigens shared between AML blasts and suppressive immune cells such as CD33 and B7-H3 present the opportunity to modulate the microenvironment while targeting tumor cells. Designing CAR T cells capable of modulating the microenvironment's cytokine and chemokine milieu have the potential to enhance T cell effector function, leading to increased antileukemic activity. In addition, exploring combinatorial therapies with antibodies and other pharmacological compounds, such as checkpoint inhibitors or adenosine receptor blockers may improve CAR T cell efficacy and persistence. In our opinion, incorporation of combination therapies would tackle antigen escape and bypass limitations regarding the number of additional CAR modifications that can be performed with current technologies. Current clinical experience has stemmed predominantly from autologous CAR T cells. The use of allogeneic CAR T cells could overcome limitations of autologous T cell production including logistics and reduced T cell quality in heavily pretreated patients. However, most allogeneic CAR T cell products require additional genetic engineering to reduce the risk for graft-vs.-host effect; in addition their in vivo expansion and persistence may be limited in comparison to autologous products. As we gain insights into the intricate dynamics that affect modulation of immune cells, there is an opportunity to convert an immunosuppressive microenvironment into one that favors CAR T cell effector function and persistence.
RE and MV conceptualized the manuscript. RE, SG, and MV provided content. All authors reviewed, edited, and approved the final manuscript.
SG and MV hold patent applications in the field of gene and cell therapy.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors' AML research was supported by grants from the Leukemia and Lymphoma Society, the Cancer Prevention Research Institute of Texas (RP160693), Alex Lemonade Stand Foundation, St. Baldrick's Foundation, Assisi Foundation of Memphis and American Lebanese Syrian Associated Charities (ALSAC).
1. Riddell SR, Jensen MC, June CH. Chimeric antigen receptor–modified T cells: clinical translation in stem cell transplantation and beyond. Biol Blood Marrow Transplant. (2013) 19:S2–5. doi: 10.1016/j.bbmt.2012.10.021
2. Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discov. (2013) 3:388–98. doi: 10.1158/2159-8290.CD-12-0548
3. Davila M, Riviere I, Wang X, Vartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. (2014) 19:224. doi: 10.1126/scitranslmed.3008226
4. Maude S, Frey N, Shaw P, Aplenc R, Barrett D, Bunin N, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
5. Lee D, Kochenderfer J, Stetler-Stevenson M, Cui Y, Delbrook C, Feldman S, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. (2015) 385:517–28. doi: 10.1016/S0140-6736(14)61403-3
6. Gardner R, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. (2017) 129:3322–31. doi: 10.1182/blood-2017-02-769208
7. Maude S, Laetsch T, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
8. Perna F, Berman S, Soni R, Mansilla-Soto J, Eyguem J, Hamieh M, et al. Integrating proteomics and transcriptomics for systematic combinatorial chimeric antigen receptor therapy of AML. Cancer Cell. (2017) 32:506–19. doi: 10.1016/j.ccell.2017.09.004
9. Budde L, Song J, Kim Y, Blanchard S, Wagner J, Stein A, et al. Remissions of acute myeloid leukemia and blastic plasmacytoid dendritic cell neoplasm following treatment with CD123-specific CAR T cells: a first-in-human clinical trial. Blood. (2017) 130:811. doi: 10.1182/blood.V130.Suppl_1.811.811
10. Cummins K, Frey N, Nelson A, Schmidt A, Luger S, Isaacs R, et al. Treating relapsed/refractory (RR) AML with biodegradable anti-CD123 CAR modified T cells. Blood. (2017) 130:1359. doi: 10.1111/bjh.12282
11. Wang Q, Wang Y, Lv H, Han Q, Fan H, Guo B, et al. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol Ther. (2015) 23:184–91. doi: 10.1038/mt.2014.164
12. Laborda E, Mazagova M, Shao S, Wang X, Quirino H, Woods A, et al. Development of a chimeric antigen receptor targeting C-type lectin-like molecule-1 for human acute myeloid leukemia. Int J Mol Sci. (2017) 18:2259. doi: 10.3390/ijms18112259
13. Tashiro H, Sauer T, Shum T, Parikh K, Mamonkin M, Omer B. Treatment of acute myeloid leukemia with T cells expressing chimeric antigen receptors directed to C-type lectin-like molecule 1. Mol Ther. (2017) 6:2202–13. doi: 10.1016/j.ymthe.2017.05.024
14. Ritchie D, Neeson P, Khot A, Peinert S, Tai T, Tainton K, et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol Ther. (2013) 21:2122–9. doi: 10.1038/mt.2013.154
15. Chen L, Mao H, Zhang J, Chu J, Devine S, Caligiuri MA, et al. Targeting FLT3 by chimeric antigen receptor T cells for the treatment of acute myeloid leukemia. Leukemia. (2017) 31:1830–4. doi: 10.1038/leu.2017.147
16. Jetani H, Garcia-Cadenas I, Nerreter T, Thomas S, Rydzek J, Meijide JB, et al. CAR T-cells targeting FLT3 have potent activity against FLT3(-)ITD(+) AML and act synergistically with the FLT3-inhibitor crenolanib. Leukemia. (2018) 32:1168–79. doi: 10.1038/s41375-018-0009-0
17. Wang Y, Xu Y, Li S, Liu J, Xing Y, Xing H, et al. Targeting FLT3 in acute myeloid leukemia using ligand-based chimeric antigen receptor-engineered T cells. J Hematol Oncol. (2018) 11:60. doi: 10.1186/s13045-018-0603-7
18. Lynn RC, Poussin M, Kalota A, Feng Y, Low PS, Dimitrov DS, et al. Targeting of folate receptor beta on acute myeloid leukemia blasts with chimeric antigen receptor-expressing T cells. Blood. (2015) 125:3466–76. doi: 10.1182/blood-2014-11-612721
19. Casucci M, Nicolis Di Robilant B, Falcone L, Camisa B, Norelli M, Genovese P, et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood. (2013) 122:3461–72. doi: 10.1182/blood-2013-04-493361
20. Rafiq S, Purdon TJ, Daniyan AF, Koneru M, Dao T, Liu C, et al. Optimized T-cell receptor-mimic chimeric antigen receptor T cells directed toward the intracellular Wilms Tumor 1 antigen. Leukemia. (2017) 31:1788–97. doi: 10.1038/leu.2016.373
21. Guery T, Roumier C, Berthon C, Renneville A, Preudhomme C, Quesnel B. B7-H3 protein expression in acute myeloid leukemia. Cancer Med. (2015) 4:1879–83. doi: 10.1002/cam4.522
22. Du H, Hirabayashi K, Ahn S, Kren NP, Montgomery SA, Wang X, et al. Antitumor responses in the absence of toxicity in solid tumors by targeting B7-H3 via chimeric antigen receptor T Cells. Cancer Cell. (2019) 35:221–37 e228. doi: 10.1016/j.ccell.2019.01.002
23. Majzner RG, Theruvath JL, Nellan A, Heitzeneder S, Cui Y, Mount CW, et al. CAR T Cells targeting B7-H3, a pan-cancer antigen, demonstrate potent preclinical activity against pediatric solid tumors and brain tumors. Clin Cancer Res. (2019) 25:2560–74. doi: 10.1158/1078-0432.CCR-18-0432
24. Leick M, Maus M, Scarfo I, Larson R, Frigault M, Schmidts A, et al. Use of CD70 targeted chimeric antigen receptor (CAR) T cells for the treatment of acute myeloid leukemia (AML). Blood. (2019) 134:4443. doi: 10.1182/blood-2019-127154
25. Sauer T, Parikh K, Rooney C, Omer B, Gottschalk S, Sharma S. CD70-specific CAR T cells have potent activity against acute myeloid leukemia (AML) without HSC toxicity. Blood. (2019) 134:1932. doi: 10.1182/blood-2019-125534
26. Gomes-Silva D, Atilla E, Atilla PA, Mo F, Tashiro H, Srinivasan M, et al. CD7 CAR T Cells for the therapy of acute myeloid leukemia. Mol Ther. (2019) 27:272–80. doi: 10.1016/j.ymthe.2018.10.001
27. Derenzo C, Krenciute G, Gottschalk S. The landscape of CAR T cells beyond acute lymphoblastic leukemia for pediatric solid tumors. Am Soc Clin Oncol Educ Book. (2018) 38:830–7. doi: 10.1200/EDBK_200773
28. Martinez M, Moon EK. CAR T Cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front Immunol. (2019) 10:128. doi: 10.3389/fimmu.2019.00128
29. Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol. (2002) 3:99–1005. doi: 10.1038/ni1102-999
30. Vago L, Perna SK, Zanussi M, Mazzi B, Barlassina C, Stanghellini MT, et al. Loss of mismatched HLA in leukemia after stem-cell transplantation. N Engl J Med. (2009) 361:478–88. doi: 10.1056/NEJMoa0811036
31. Christopher MJ, Petti AA, Rettig MP, Miller CA, Chendamarai E, Duncavage EJ, et al. Immune escape of relapsed AML cells after allogeneic transplantation. N Engl J Med. (2018) 379:2330–41. doi: 10.1056/NEJMoa1808777
32. Toffalori C, Zito L, Gambacorta V, Riba M, Oliveira G, Bucci G, et al. Immune signature drives leukemia escape and relapse after hematopoietic cell transplantation. Nat Med. (2019) 25:603–11. doi: 10.1038/s41591-019-0400-z
33. Vereecque R, Buffenoir G, Gonzalez R, Cambier N, Hetuin D, Bauters F, et al. gamma-ray irradiation induces B7.1 expression in myeloid leukaemic cells. Br J Haematol. (2000) 108:825–31. doi: 10.1046/j.1365-2141.2000.01967.x
34. Zhou Q, Munger M, Veenstra R, Weigel B, Hirashima M, Munn D, et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhustion phenotype in mice with disseminated acute myelogenous leukemia. Blood. (2011) 117:4501–10. doi: 10.1182/blood-2010-10-310425
35. Zhou P, Shaffer DR, Alvarez Arias DA, Nakazaki Y, Pos W, Torres AJ, et al. In vivo discovery of immunotherapy targets in the tumour microenvironment. Nature. (2014) 506:52–7. doi: 10.1038/nature12988
36. Zhang L, Gajewski T, Kline J. PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood. (2009) 114:1545–52. doi: 10.1182/blood-2009-03-206672
37. Williams P, Basu S, Garcia-Manero G, Hourigan C, Oetjen K, Cortes J, et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patient with newly diagnosed eand relapsed acute myeloid leukemia. Cancer. (2019) 125:1470–81. doi: 10.1002/cncr.31896
38. Daver N, Basu S, Garcia-Manero G, Cortes J, Ravandi E, Jabbour E, et al. Phase IB/II study of nivolumab in combination wth azacytidine in patients with relapsed acute myeloid leukemia. Blood. (2016) 22:763. doi: 10.1182/blood.V128.22.763.763
39. Davids M, Kim H, Bachireddy P, Costello C, Liguori R, Savell A, et al. Ipilimumab for patients with relapse after allogeneic transplantation. N Engl J Med. (2016) 375:143–53. doi: 10.1056/NEJMoa1601202
40. Kadia T, Cortes J, Ghorab A, Ravandi F, Jabbour E, Daver N, et al. Nivolumab maintenance in high-risk acute myeloid leukemia patients. J Clin Oncol. (2018) 36:15. doi: 10.1200/JCO.2018.36.15_suppl.7014
41. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov D, Jones D, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. (2016) 126:3130–44. doi: 10.1172/JCI83092
42. Suarez E, Chang D, Sun J, Sui J, Freeman G, Signoretti S, et al. Chimeric antigen receptor T cells secreting antii-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget. (2016) 7:3431–55. doi: 10.18632/oncotarget.9114
43. Rupp L, Schumann K, Roybal K, Gate R, Ye C, Lim W, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. (2017) 7:737. doi: 10.1038/s41598-017-00462-8
44. Rafiq S, Yeku O, Jackson H, Purdon T, Van Leeuwen D, Drakes D, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tuor efficacy in vivo. Nat Biotechnol. (2018) 36:847–56. doi: 10.1038/nbt.4195
45. Teague RM, Kline J. Immune evasion in acute myeloid leukemia: current concepts and future directions. J Immunother Cancer. (2013) 1. doi: 10.1186/2051-1426-1-13
46. Bonifant CL, Velasquez MP, Gottschalk S. Advances in immunotherapy for pediatric acute myeloid leukemia. Expert Opin Biol Ther. (2018) 18:51–63. doi: 10.1080/14712598.2018.1384463
47. Nagaraj S, Schrum AG, Cho HI, Celis E, Gabrilovich DI. Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J Immunol. (2010) 184:3106–16. doi: 10.4049/jimmunol.0902661
48. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. (2016) 7:12150. doi: 10.1038/ncomms12150
49. Zhang S, Ma X, Zhu C, Liu L, Wang G, Yuan X. The Role of myeloid-derived suppressor cells in patients with solid tumors: a meta-analysis. PLoS ONE. (2016) 11:e0164514. doi: 10.1371/journal.pone.0164514
50. Ostrand-Rosenberg S, Fenselau C. Myeloid-derived suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol. (2018) 200:422–31. doi: 10.4049/jimmunol.1701019
51. Movahedi K, Guilliams M, Van Den Bossche J, Van Den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. (2008) 111:4233–44. doi: 10.1182/blood-2007-07-099226
52. Schouppe E, Mommer C, Movahedi K, Laoui D, Morias Y, Gysemans C, et al. Tumor-induced myeloid-derived suppressor cell subsets exert either inhibitory or stimulatory effects on distinct CD8+ T-cell activation events. Eur J Immunol. (2013) 43:2930–42. doi: 10.1002/eji.201343349
53. Sun H, Li Y, Zhang Z, Ju Y, Li L, Zhang B, et al. Increase in myeloid-derived suppressor cells (MDSCs) associated with minimal residual disease (MRD) detection in adult acute myeloid leukemia. Int J Hematol. (2015) 102:579–86. doi: 10.1007/s12185-015-1865-2
54. Pyzer A, Stroopinsky D, Rajabi H, Washington A, Tagde A, Coll M, et al. MUC1-mediated induction of myeloid-derived suppressor cells in patients with acute myeloid leukemia. Blood. (2017) 129:1791–801. doi: 10.1182/blood-2016-07-730614
55. Jitschin R, Saul D, Braun M, Tohumeken S, Volkl S, Kischel R, et al. CD33/CD3-bispecific T-cell engaging (BiTE®) antibody construct targets monocytic AML myeloid-derived suppressor cells. J Immunother Cancer. (2018) 6:116. doi: 10.1186/s40425-018-0432-9
56. Wang L, Jia B, Claxton D, Ehmann W, Rybka W, Mineishi S, et al. VISTA is highly expressed on MDSCs and mediates an inhibition of T cell response in patients with AML. Oncoimmunology. (2018) 7:e1469594. doi: 10.1080/2162402X.2018.1469594
57. Burga R, Thorn M, Point G, Guha P, Nguyen C, Licata L, et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol Immunother. (2015) 64:817–29. doi: 10.1007/s00262-015-1692-6
58. Long A, Highfill S, Cui Y, Smith J, Walker A, Ramakrishna S, et al. Reduction of MDSCs with all-trans retinoic acid improves CAR therapy efficacy for sarcomas. Cancer Immunol Res. (2016) 4:869–80. doi: 10.1158/2326-6066.CIR-15-0230
59. Pizzitola I, Anjos-Afonso F, Rouault-Pierre K, Lassailly F, Tettamanti S, Spinelli O, et al. Chimeric antigen receptors against CD33/CD123 antigens efficiently target pirmary acute myeloid leukemia cells in vivo. Leukemia. (2014) 28:1596–605. doi: 10.1038/leu.2014.62
60. Kenderian S, Ruella M, Shestova O, Klichinsky M, Aikawa V, Morrissette J, et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia. (2015) 29:1637–47. doi: 10.1038/leu.2015.52
61. O'hear C, Heiber J, Schubert I, Fey G, Geiger T. Anti-CD33 chimeric antigen receptor targeting of acute myeloid leukemia. Haematologica. (2015) 100:336–44. doi: 10.3324/haematol.2014.112748
62. Schmidt A, Oberle N, Krammer PH. Molecular mechanisms of treg-mediated T cell suppression. Front Immunol. (2012) 3:51. doi: 10.3389/fimmu.2012.00051
63. Szczepanski M, Szajnik M, Czystowska M, Mandapathil M, Strauss L, Welsh A, et al. Increased frequency and suppression by regulatory T cells in patients with acute myelogenous leukemia. Clin Cancer Rev. (2009) 15:3325–32. doi: 10.1158/1078-0432.CCR-08-3010
64. Wang M, Zhang C, Tian T, Zhang T, Wang R, Han F, et al. Increased regulatory T cells in peripheral blood of acute myeloid leukemia patients rely on tumor necrosis factor (TNF)-α-receptor 2 pathway. Front Immunol. (2018) 5:1274. doi: 10.3389/fimmu.2018.01274
65. Zahran A, Mohammed Saleh M, Sayed M, Rayan A, Ali A, Hetta H. Up-regulation of regulatory T cells, CD200 and TIM3 expression in cytogenetically normal acute myeloid leukemia. Cancer Biomark. (2018) 22:587–95. doi: 10.3233/CBM-181368
66. Han Y, Dong Y, Yang Q, Xu W, Jiang S, Yu Z, et al. Acute myeloid leukemia cells express ICOS ligand to promote the expansion of regulatory T cells. Front Immunol. (2018) 9:2227. doi: 10.3389/fimmu.2018.02227
67. Arandi NR, Ramzi M, Safaei F, Monabati A. Overexpression of indoleamine 2,3-dioxygenase correlates with regulatory T cell phenotype in acute myeloid leukemia patients with normal karyotype. Blood Res. (2018) 53:294–8. doi: 10.5045/br.2018.53.4.294
68. Zhou Q, Bucher C, Munger M, Highfill S, Tolar J, Munn D, et al. Depletion of endogenous tumor-associated regulatory T cells improves the efficacy of adoptive cytotoxic T-cell immunotherapy in murine acute myeloid leukemia. Blood. (2009) 114:3793–802. doi: 10.1182/blood-2009-03-208181
69. Duell J, Dittrich M, Bedke T, Mueller T, Eisele F, Rosenwald A, et al. Frequency of regulatory T cells determines the outcome of the T-cell-engaging antibody blinatumomab in patients with B-precursor ALL. Leukemia. (2017) 31:2181–90. doi: 10.1038/leu.2017.41
70. Loskog A, Giandomenico V, Rossig C, Pule M, Dotti G, Brenner M. Addition of the CD28 signaling domain to chimeric T-cell receptors enhances chimeric T-cell resistance to T regulatory cells. Leukemia. (2006) 20:1819–28. doi: 10.1038/sj.leu.2404366
71. Suryadevara C, Desai R, Farber S, Choi B, Swartz A, Shen S, et al. Preventing Lck activation in CAR T cells confers Treg resistance but requires 4-1BB signaling for them to persist and treat solid tumors in nonlymphodepleted hosts. Clin Cancer Res. (2019) 25:358–68. doi: 10.1158/1078-0432.CCR-18-1211
72. Perna SK, De Angelis B, Pagliara D, Hasan ST, Zhang L, Mahendravada A, et al. Interleukin 15 provides relief to CTLs from regulatory T cell-mediated inhibition: implications for adoptive T cell-based therapies for lymphoma. Clin Cancer Res. (2013) 19:106–17. doi: 10.1158/1078-0432.CCR-12-2143
73. Lion E, Willemen Y, Berneman ZN, Van Tendeloo VF, Smits EL. Natural killer cell immune escape in acute myeloid leukemia. Leukemia. (2012) 26:2019–26. doi: 10.1038/leu.2012.87
74. Gao S, Xing C, Chen C, Lin S, Dong P, Yu F. miR-15a and miR-16-1 inhibit the proliferation of leukemic cells by down-regulating WT1 protein level. J Exp Clin Cancer Res. (2011) 1:110. doi: 10.1186/1756-9966-30-110
75. Liu X, Wang Y, Sun Q, Yan J, Huang J, Zhu S, et al. Identification of microRNA transcriptome involved in human natural killer cell activation. Immunol Lett. (2012) 143:208–17. doi: 10.1016/j.imlet.2012.02.014
76. Hassani S, Rezaeeyan H, Ghodsi A, Saki N. Restoration of natural killer cell cytotoxicity in the suppressive tumor microenvironment: novel approaches to treat AML. J Hematopathol. (2017) 10:109–16. doi: 10.1007/s12308-017-0306-y
77. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. (2002) 295:2097–100. doi: 10.1126/science.1068440
78. Miller J, Soignier Y, Panoskaltsis-Mortari A, Mcnearney S, Yun G, Fautsch S, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. (2005) 105:3051–7. doi: 10.1182/blood-2004-07-2974
79. Rubnitz J, Inaba H, Ribeiro R, Pounds S, Rooney B, Bell T, et al. NKAML: a pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J Clin Oncol. (2010) 28: 955–9. doi: 10.1200/JCO.2009.24.4590
80. Curti A, Ruggeri L, D'addio A, Bontadini A, Dan E, Motta M, et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood. (2011) 118:3273–9. doi: 10.1182/blood-2011-01-329508
81. Rubnitz J, Inaba H, Kang G, Gan K, Hartford C, Triplett B, et al. Natural killer cell therapy in children with relapsed leukemia. Pediatr Blood Cancer. (2015) 62:1468–72. doi: 10.1002/pbc.25555
82. Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. (2016) 8:357ra123. doi: 10.1126/scitranslmed.aaf2341
83. Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. (2005) 106:376–83. doi: 10.1182/blood-2004-12-4797
84. Muller T, Uherek C, Maki G, Chow K, Schimpf A, Kllingemann H, et al. Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol Immunother. (2008) 57:411–23. doi: 10.1007/s00262-007-0383-3
85. Chang Y, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. (2013) 73:1777–86. doi: 10.1158/0008-5472.CAN-12-3558
86. Liu E, Tong Y, Dotti G, Shaim H, Salvoldo B, Mukherjee M, et al. Cord blood derived natural killer cells engineered with a chimeric antigen receptor targeting CD19 and expressing IL-15 have long term persistence and exert potent anti-leukemia activity. Leukemia. (2018) 32:520–31. doi: 10.1038/leu.2017.226
87. Olson J, Leveson-Gower D, Gill S, Baker J, Beilhack A, Negrin R. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood. (2010) 115:4293–301. doi: 10.1182/blood-2009-05-222190
88. Sivori S, Carlomagno S, Falco M, Romeo E, Moretta L, Moretta A. Natural killer cells expressing the KIR2DS1-activating receptor efficiently kill T-cell blasts and dendritic cells: implications in haploidentical HSCT. Blood. (2011) 117:4284–92. doi: 10.1182/blood-2010-10-316125
89. Meinhardt K, Kroeger I, Bauer R, Ganss F, Ovsiy I, Rothamer J, et al. Identification and characterization of the specific murine NK cell subset supporting graft-versus-leukemia-and reducing graft-versus-host-effects. Oncoimmunology. (2015) 3:e981483. doi: 10.4161/2162402X.2014.981483
90. Al-Matary Y, Botezatu L, Opalka B, Hones J, Lams R, Thivakaram A, et al. Acute myeloid leukemia cells polarize macrophages towards a leukemia supporting state in a growth factor indepnedence 1 dependent manner. Haematologica. (2016) 101:1216–27. doi: 10.3324/haematol.2016.143180
91. Yan Z, Li L, Wang W, Ouyang B, Cheng S, Wang L, et al. Clinical efficacy and tumor microenvironment influence in a dose-escalation study of anti-CD19 chimeric antigen receptor T cells in refractory B-cell non-Hodgkin's lymphoma. Clin Cancer Res. (2019) 25:6995–7003. doi: 10.1158/1078-0432.CCR-19-0101
92. Giavridis T, Van Der Stegen S, Eyquen J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induuced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. (2018) 24:731–8. doi: 10.1038/s41591-018-0041-7
93. Rickmann M, Macke L, Sundarasetty B, Stamer K, Figueiredo C, Blasczyk R, et al. Monitoring dendritic cell and cytokine biomarkersduring remission prior to relapse in patients with FLT3-ITD acute myeloid leukemia. Ann Hematol. (2013) 92:1079–90. doi: 10.1007/s00277-013-1744-y
94. Lecciso M, Ocadlikova D, Sangaletti S, Trabanelli S, De Marchi E, Orioli E, et al. ATP release from chemotherapy-treated dying leukemia cells elicits an immune suppressive effect by increasing regulatory T cells and tolerogenic dendritic cells. Front Immunol. (2017) 8:1918. doi: 10.3389/fimmu.2017.01918
95. Curran K, Seinstra B, Nikhamin Y, Yeh R, Usachenko Y, Van Leeuwen D, et al. Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol Ther. (2015) 23:769–78. doi: 10.1038/mt.2015.4
96. Textor A, Listopad JJ, Wuhrmann LL, Perez C, Kruschinski A, Chmielewski M, et al. Efficacy of CAR T-cell therapy in large tumors relies upon stromal targeting by IFNgamma. Cancer Res. (2014) 74:6796–805. doi: 10.1158/0008-5472.CAN-14-0079
97. Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood. (2014) 123:3750–9. doi: 10.1182/blood-2014-01-552174
98. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. (2018) 24:563–71. doi: 10.1038/s41591-018-0010-1
99. Norelli M, Camisa B, Barbiera G, Falcone L, Purevdori A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. (2018) 24:739–48. doi: 10.1038/s41591-018-0036-4
100. Santomasso B, Park J, Salloum D, Riviere I, Flynn J, Mead E, et al. Clinical and biological correlates of neurotoxicity associated with CAR T-cell therapy in patients with B-cell acute lymphoblastic leukemia. Cancer Discov. (2018) 8:958–71. doi: 10.1158/2159-8290.CD-17-1319
101. Olsnes AM, Motorin D, Ryningen A, Zaritskey AY, Bruserud O. T lymphocyte chemotactic chemokines in acute myelogenous leukemia (AML): local release by native human AML blasts and systemic levels of CXCL10 (IP-10), CCL5 (RANTES) and CCL17 (TARC). Cancer Immunol Immunother. (2006) 55:830–40. doi: 10.1007/s00262-005-0080-z
102. Yazdani Z, Mousavi Z, Ghasemimehr N, Kalantary Khandany B, Nikbakht R, Jafari E, et al. Differential regulatory effects of chemotherapeutic protocol on CCL3_CCL4_CCL5/CCR5 axes in acute myeloid leukemia patients with monocytic lineage. Life Sci. (2020) 240:117071. doi: 10.1016/j.lfs.2019.117071
103. Faaij CM, Willemze AJ, Revesz T, Balzarolo M, Tensen CP, Hoogeboom M, et al. Chemokine/chemokine receptor interactions in extramedullary leukaemia of the skin in childhood AML: differential roles for CCR2, CCR5, CXCR4 and CXCR7. Pediatr Blood Cancer. (2010) 55:344–8. doi: 10.1002/pbc.22500
104. Merle M, Fischbacher D, Liepert A, Grabrucker C, Kroell T, Kremser A, et al. Serum chemokine-release profiles in AML-patients might contribute to predict the clinical course of the disease. Immunol Invest. (2019) 1–21. doi: 10.1080/08820139.2019.1661429
105. Di Stasi A, De Angelis B, Rooney C, Zhang L, Mahendravada A, Foster A, et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. (2009) 113:6392–402. doi: 10.1182/blood-2009-03-209650
106. Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotech. (2018) 36:346–51. doi: 10.1038/nbt.4086
107. Nabe S, Yamada T, Suzuki J, Toriyama K, Yasuoka T, Kuwahara M, et al. Reinforce the antitumor activity of CD8+ T cells via glutamine restriction. Cancer Sci. (2018) 12:3737–50. doi: 10.1111/cas.13827
108. Willems L, Jacque N, Jacquel A, Neveux N, Maciel T, Lambert M, et al. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood. (2013) 122:3521–32. doi: 10.1182/blood-2013-03-493163
109. Goto M, Miwa H, Shikami M, Tsunekawa-Imai N, Suganuma K, Mizuno S, et al. Importance of glutamine metabolism in leukemia cells by energy production through TCA cycle and by redox homeostasis. Cancer Invest. (2014) 32:241–7. doi: 10.3109/07357907.2014.907419
110. Jacque N, Ronchetti A, Larrue C, Meunier G, Birsen R, Willems L, et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood. (2015) 126:1346–56. doi: 10.1182/blood-2015-01-621870
111. Matre P, Velez J, Jacamo R, Qi Y, Su X, Cai T, et al. Inhibiting glutaminase in acute myeloid leukemia: metabolic dependency of selected AML subtypes. Oncotarget. (2016) 29:79722–35. doi: 10.18632/oncotarget.12944
112. Mussai F, De Santo C, Abu-Dayyeh I, Booth S, Quek L, Mcewen-Smith R, et al. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood. (2013) 122:749–58. doi: 10.1182/blood-2013-01-480129
113. Mussai F, Wheat R, Sarrou E, Booth S, Stavrou V, Fultang L, et al. Targeting the arginine metabolic brake enhances immunotherapy for leukaemia. Int J Cancer. (2019) 145:2201–8. doi: 10.1002/ijc.32028
114. Beavis P, Henderson M, Giuffrida L, Mills J, Sek K, Cross R, et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J Clin Invest. (2017) 127:929–41. doi: 10.1172/JCI89455
115. Canale F, Ramello M, Nunez N, Araujo Furlan C, Bossio S, Piaggio E, et al. CD39 Expression defines cell exhaustion in tumor-infiltrating CD8+ T cells. Cancer Res. (2018) 78:115–28. doi: 10.1158/0008-5472.CAN-16-2684
116. Hausler S, Del Barrio I, Diessner J, Stein R, Strohschein J, Honig A, et al. Anti-CD39 and anti-CD73 antibodies A1 and 7G2 improve targeted therapy in ovarian cancer by blocking adenosine-dependent immune evasion. Am J Transl Res. (2014) 6:129–39.
117. Mansour I, Zyed R, Said F, La L. Indoleamine 2,3-dioxygenase and regulatory T cells in acute myeloid leukemia. Hematology. (2016) 21:447–53. doi: 10.1080/10245332.2015.1106814
118. Azadniv M, Myers J, Mcmurray H, Guo N, Rock P, Coppage M, et al. Bone marrow mesenchymal stromal cells from acute myelogenous leukemia patients demonstrate adipogenic differentiation propensity with implications for leukemia cell support. Leukemia. (2020) 34:391–403. doi: 10.1038/s41375-019-0568-8
119. Shafat M, Oellerich T, Mohr S, Robinson S, Edwards D, Marlein C, et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood. (2017) 129:1320–32. doi: 10.1182/blood-2016-08-734798
120. Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M, et al. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell. (2016) 19:23–37. doi: 10.1016/j.stem.2016.06.001
121. Tabe Y, Yamamoto S, Saitoh K, Sekihara K, Monma N, Ikeo K, et al. Bone marrow adipocytes facilitate fatty acid oxidation activating AMPK and a transcriptional network supporting survival of acute monocytic leukemia cells. Cancer Res. (2017) 77:1453–64. doi: 10.1158/0008-5472.CAN-16-1645
122. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. (2009) 460:103–7. doi: 10.1038/nature08097
123. Van Der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. (2012) 36:68–78. doi: 10.1016/j.immuni.2011.12.007
124. Kawalekar O, O'connor R, Fraietta J, Guo L, Mcgettigan S, Posey AJ, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. (2016) 44:380–90. doi: 10.1016/j.immuni.2016.01.021
125. Drury L, Ziarek J, Gravel S, Veldkamp C, Takekoshi T, Hwang S, et al. Monomeric and dimeric CXCL12 inhibit metastasis through distinct CXCR4 interactions and signaling pathways. PNAS. (2011) 108:17655–60. doi: 10.1073/pnas.1101133108
126. Arai Y, Choi U, Corsino CI, Koontz SM, Tajima M, Sweeney CL, et al. Myeloid conditioning with c-kit-Targeted CAR-T cells enables donor stem cell engraftment. Mol Ther. (2018) 26:1181–97. doi: 10.1016/j.ymthe.2018.03.003
127. Hirche C, Frenz T, Haas S, Doring M, Borst K, Tegtmeyer P, et al. Systemic virus infections differentially moduate cell cycle state and functionality of long-term hematopoietic stem cells in vivo. Cell Rep. (2017) 19:2345–56. doi: 10.1016/j.celrep.2017.05.063
128. Essers M, Offner S, Blanco-Bose W, Waibler Z, Kalinke U, Duchosal M, et al. IFNalpha activates dormant ahematopoietic stem cells in vivo. Nature. (2009) 458:904–8. doi: 10.1038/nature07815
129. Young P, Yamada R, Trinh K, Vasuthasawat A, De Oliveira S, Yamada D, et al. Activity of anti-CD19 chimeric antigen receptor T cells against B cell lymphoma is enhanced by antibody-targeted interferon-alpha. J Interferon Cytokine Res. (2018) 38:239–54. doi: 10.1089/jir.2018.0030
130. Kochenderfer J, Dudley M, Kassim S, Somerville R, Carpenter R, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma an dindolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. (2015) 33:540–9. doi: 10.1200/JCO.2014.56.2025
131. Wang CM, Wu ZQ, Wang Y, Guo YL, Dai HR, Wang XH, et al. Autologous T cells expressing CD30 chimeric antigen receptors for relapsed or refractory Hodgkin lymphoma: an open-label phase I trial. Clin Cancer Res. (2017) 2017:1156–66. doi: 10.1158/1078-0432.CCR-16-1365
132. Siegert S, Huang H, Yang C, Scarpellino L, Carrie L, Essex S, et al. Fibroblastic reticular cells from lymph nodes attenuate T cell expansion by producing nitric oxide. PLoS ONE. (2011) 6:e27618. doi: 10.1371/journal.pone.0027618
133. Knoblich K, Migoni S, Siew S, Jinks E, Kaul B, Jeffery H, et al. The human lymph node microenvironment unilaterally regulates T-cell activation and differentiation. PLoS Biol. (2018) 16:e2005046. doi: 10.1371/journal.pbio.2005046
134. Tsimberidou AM, Kantarjian H, Wen S, Keating M, O'brien S, et al. Myeloid sarcoma is associated with superior event-free survival and overall survival compared with acute myeloid leukemia. Cancer. (2008) 113:1370–8. doi: 10.1002/cncr.23691
135. Campidelli C, Agostinelli C, Stitson R, Pileri S. Myeloid sarcoma: extramedullary manifestation of myeloid disorders. Am J Clin Path. (2009) 132:426–37. doi: 10.1309/AJCP1ZA7HYZKAZHS
136. Solh M, Defor T, Weisdorf D, Kaufman D. Extramedullary relapse of acute myelogenous leukemia after allogeneic hematopoietic stem cell transplantation: better prognosis than systemic relapse. Biol Blood Marrow Transplant. (2012) 18:106–12. doi: 10.1016/j.bbmt.2011.05.023
137. Frigault M, Dietrich J, Martinez-Lage M, Leick M, Choi B, Defilipp Z, et al. Tisagenlecleucel CAR-T cell therapy in secondary CNS lymphoma. Blood. (2019) 134:860–6. doi: 10.1182/blood.2019001694
Keywords: chimeric antigen receptor, cellular therapy, immunotherapy, acute myeloid leukemia, microenvironment
Citation: Epperly R, Gottschalk S and Velasquez MP (2020) A Bump in the Road: How the Hostile AML Microenvironment Affects CAR T Cell Therapy. Front. Oncol. 10:262. doi: 10.3389/fonc.2020.00262
Received: 06 December 2019; Accepted: 14 February 2020;
Published: 28 February 2020.
Edited by:
Yong-mi Kim, Children's Hospital of Los Angeles, United StatesReviewed by:
Saar Gill, University of Pennsylvania, United StatesCopyright © 2020 Epperly, Gottschalk and Velasquez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: M. Paulina Velasquez, cGF1bGluYS52ZWxhc3F1ZXpAc3RqdWRlLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.