
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 26 February 2020
Sec. Gastrointestinal Cancers
Volume 10 - 2020 | https://doi.org/10.3389/fonc.2020.00115
 Dong-Dong Yang1,2†
Dong-Dong Yang1,2† Zhan-Hong Chen1,3†
Zhan-Hong Chen1,3† Kai Yu1†
Kai Yu1† Jia-Huan Lu1
Jia-Huan Lu1 Qi-Nian Wu1
Qi-Nian Wu1 Yun Wang1Huai-Qiang Ju1
Yun Wang1Huai-Qiang Ju1 Rui-Hua Xu1
Rui-Hua Xu1 Ze-Xian Liu1*
Ze-Xian Liu1* Zhao-Lei Zeng1*
Zhao-Lei Zeng1*Methyltransferase-like 3 (METTL3), a major component of the N6-methyladenosine (m6A) methyltransferase complex, has been suggested to function as an oncogene in several cancers. However, its biological mechanism and the involved pathways in gastric cancer (GC) remain unknown. Here, we reported that frequent upregulation of METTL3 was responsible for the aberrant m6A levels in gastric carcinoma. On the other hand, a high level of METTL3 was significantly associated with several clinicopathological features and poor survival in patients with GC. The knockdown of METTL3 effectively inhibited cell proliferation and migration and invasion capacity. Moreover, overexpression of METTL3 considerably augmented its oncogenic function. Integrated RNA-seq and m6A-seq analysis first indicated that several component molecules (e.g., MCM5, MCM6, etc.) of MYC target genes were mediated by METTL3 via altered m6A modification. Our work uncovers the oncogenic roles of METTL3 in GC and suggests a critical mechanism of GC progression.
Gastric cancer (GC) is the fifth most common malignancy and is a great burden to patients and health-care systems worldwide. Despite an overall decline in incidence and mortality rates in the past few decades, GC still kills more than 300,000 people per year in China. Although a multidisciplinary treatment (MDT) for GC patients has been developed, trastuzumab has been approved for human epidermal growth factor receptor 2 (HER2)-positive metastatic GC patients, and immunocheckpoint inhibitors have been approved for the third-line treatment of metastatic GC patients, the prognosis of most patients with metastatic GC is far from satisfactory (1–5). Thus, it is necessary to clarify the detailed molecular mechanism underlying GC progression.
N6-methyladenosine (m6A) modification is the most prominent and conservative RNA modification in both prokaryotes and eukaryotes. It is extensively involved in messenger RNA (mRNA) metabolism, including alternative splicing, stability, translation initiation, transportation, and degradation processes. Recent studies have shown that m6A modifications in mRNA play critical roles in cell meiosis, circadian clock control, and cell fate decisions, even in viral infections and immune responses (6–14). M6A modifications are dynamic and reversible processes catalyzed by methyltransferases (writers) and demethylases (erasers). Together with methyltransferase-like 14 (METTL14) and Wilms tumor 1 associated protein (WTAP), methyltransferase-like 3 (METTL3) forms a stable methyltransferase complex that can add a methyl group to RNA. Fat mass and obesity-associated protein (FTO) and alkB homolog 5 (ALKBH5) can function as erasers to deplete m6A modification (15–17). Accumulating evidence has demonstrated that m6A-related enzymes (e.g., METTL3, METTL14, FTO, etc.) can participate in carcinogenesis in several malignancies by mediating m6A modification of target RNAs (18–24). Although several studies have clarified a critical role for METTL3 in GC, the absence of the detailed working mechanism of METTL3 was the most obvious limitation of these studies (25–28). Therefore, in this research, we attempted to investigate the workflow of METTL3-m6A-target RNAs in GC.
In this study, we identified aberrant METTL3 expression and its prognostic value in GC. Then, we confirmed the functional role of METTL3 in vitro and in vivo. The most interesting thing was the regulation of several key components (e.g., MCM5, MCM6, etc.) of the MYC pathway by METTL3 through mediating m6A modification in their mRNAs. Thus, the findings of our research have provided a fresh view of m6A modification in tumor progression.
The Cancer Genome Atlas (TCGA) mRNA-seq data of 32 normal gastric tissues and 375 primary tumor tissues were downloaded from Firehose (http://gdac.broadinstitute.org/). We calculated the fold change and adjusted p-value for all m6A modification-related enzymes via the DESeq2 package, in which an adjusted p < 0.05 and fold change >2 was considered to denote a differentially expressed gene.
Patients enrolled in this study received primary radical or palliative resection without preoperative chemotherapy or radiotherapy at Sun Yat-Sen University Cancer Center (SYSUCC) between January 2007 and February 2013. The patients who met the following criteria were included: (1) pathologically confirmed gastric cancer, (2) patients received radical surgery or palliative surgery, and (3) patients with available clinicopathological information and complete follow-up information. We excluded patients who met the following criteria: (1) patients with synchronous malignant tumors, and (2) patients with incomplete baseline clinicopathological factor information. The median age of all patients was 56 (interquartile range, 50–65). Clinicopathological characteristics including gender, age, pathological tumor–node–metastasis (pTNM) stage, tumor size and grade, invasion depth, neural/vessel invasion, and survival status were described in electronic medical records. All patients were pathologically diagnosed and classified by experienced pathologist according to the 7th edition of the American Joint Committee on Cancer (AJCC) staging system (29). The patients were regularly followed up every 3–6 months until death or dropout, with a median follow-up duration of 41 months [interquartile range (IQR), 21–84 months].
GES-1, MKN45, MKN74, HGC27, SGC7901, MGC803, and pGCC (primary GC cells) were cultured in Roswell Park Memorial Institute (RPMI)-1640 (Gibco) supplemented with 10% fetal bovine serum (FBS) (Gibco), and 1% antibiotics (penicillin/streptomycin) (Gibco). AGS was maintained in F-12 (Gibco) with 10% FBS and 1% antibiotics. Cells were grown in a 5% CO2 incubator at 37°C. Lentiviral vectors expressing non-targeting control RNA (sh#nc and oe#nc), two short hairpin RNAs (shRNAs) (sh#1 and sh#2) targeting METTL3 and oe#METTL3 (overexpression of METTL3) were purchased from Gene Pharma. AGS and SGC7901 cells were incubated with lentivirus and 4 μg/ml polybrene. After 48 h of transfection, 2 μg/ml puromycin was added to the culture medium for the selection of infected cells.
Cell proliferation, colony formation, migration, and invasion assays were performed as follows. Briefly, cells were seeded at a density of 1 × 103 cells per well in 96-well plates on day 0. Then, 20 μl [3-(4,5-dimethylthiazol-2-yl)-5- (3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] (MTS) was added to each well and incubated with cells at 37°C for 2 h on days 1–4. Then, the absorption values at 490 nm were used to measure cell proliferation. For colony formation assays, cells were digested and seeded in six-well plates at 500 cells per well. After 3 weeks of growth, the cells were fixed with methanol and stained with 0.3% crystal violet. Cell migration and invasion were measured using a BioCoat Matrigel Invasion Chamber (Corning) following the manufacturer's instructions.
Four to six week-old female BALB/c nude mice (Vital River) were purchased for the construction of subcutaneous tumor xenografts. A total of 2 × 106 GC cells were injected into the flank of nude mice in a 1:1 suspension of BD Matrigel (BD Biosciences) in phosphate-buffered saline (PBS) solution. Vernier calipers were used to detect the formation of xenograft tumors every 4 days. Three weeks after injection, we euthanized nude mice for the measurement of tumor volume and tumor weights.
Total RNA from tissues or cell cultures was extracted using TRIzol (Invitrogen) following the manufacturer's protocol. Then, we used an m6A RNA methylation quantification kit (P-9005-48, EpiGentek) to measure the m6A levels in global RNAs. In brief, 200 ng of total RNA from the samples was added to each well and coated at 37°C for 90 min. Then, capture antibody, detection antibody, and enhancer solution were sequentially added to assay wells according to the user guide. We added a developer solution to wells for color development and measured the absorption value at 450 nm. Then, the m6A levels of each well were calculated by standard curve. Quantitative reverse transcription PCR (qRT-PCR) was performed using PrimeScript RTTM Master Mix (RR036Q, TAKARA) and Go Taq® qPCR Master Mix (A6002, Promega). The primers used in this study are listed in Supplementary Table 1.
The procedures of Western blot analysis were conducted as described previously (30). The antibodies used for Western blotting in this research were as follows: METTL3 (ab195352, Abcam), MYC (ab32072, Abcam), MCM5 (11703-1-AP, Proteintech), MCM6 (13347-2-AP, Proteintech), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (ab181602, Abcam).
A dozen tissue microarrays (TMAs) were prepared from 196 paraffin-embedded primary tumor blocks, corresponding adjacent normal mucosa and metastatic lesions. METTL3 immunostaining was performed as previously described (31). The estimation of METTL3 expression was determined by two independent pathologists who were blinded to the clinical data. The staining intensity was scored as 0 (negative), 1 (weak), 2 (moderate), or 3 (strong). The final score of each tissue block was the mean of the products of positive staining rate (0–100%) and intensity score (0–3), ranging from 0 to 300. We applied receiver operating characterizing (ROC) curve analysis to define the optimal cut-off value. The best threshold of METTL3 expression in primary tumors was 145, which meant that patients whose primary tumor score was above that value were grouped into the high expression group (n = 69); otherwise, they were grouped into the low expression group (n = 127).
Characterization of the cell methylome was carried out by m6A-seq following procedures described previously (30, 32). Briefly, a total of ~100 μg of global cellular RNAs was isolated by a poly(A) mRNA with poly(T) oligo-attached magnetic beads (Invitrogen) (Supplementary Figure 1A). Then, purified poly(A) mRNA samples were fragmented into ~100 nt oligonucleotides under the divalent cations with elevated temperature. Fragmented mRNA was incubated with m6A-specific antibody (No. 202003, Synaptic Systems, Germany) in immunoprecipitation (IP) buffer (50 mM Tris–HCl, 750 mM NaCl, and 0.5% Igepal CA-630) for 2 h at 4°C. The solution was mixed with protein-A beads for m6A immunoprecipitation, and the beads were eluted by elution buffer (1 × IP buffer and 6.7 mM m6A) three times. Both input and m6A IP samples were prepared according to the manufacturer's instructions for sequencing on an Illumina NovaSeq 6000 platform at the LC-BIO Biotech Ltd.
For the data analysis, the enriched m6A peaks were identified by exomePeak for further analysis. The m6A peaks were then annotated by HOMER, and consensus sequence motifs were identified using MEME with the default parameters. Differentially methylated m6A peaks were calculated by limma. The gene expression matrix was quantified and normalized using the RSEM software, and the “DESeq” package was used to find differentially expressed genes. Gene set enrichment analysis (GSEA) was performed to evaluate the pathway differences in expression and m6A modification patterns between the sh#METTL3 and control groups.
Each cellular experiment was repeated at three biological replicates. The cut-off value of METTL3 was defined using ROC curve analysis. We compared differences of continuous variables by Student's t-tests. We compared differences in categorical factors between groups by chi-square test and Fisher's exact test. We compared median values between different groups using the Mann–Whitney test.
In univariate analyses, we estimated the survival difference of distinct variables using the Kaplan–Meier method (log-rank test), and we subsequently identified independent prognostic factors by Cox proportional hazards regression models in multivariate analysis.
All tests were two-tailed, and P < 0.05 was considered statistically significant. Statistical analysis was performed using GraphPad Prism 6.0 (GraphPad Software Inc., La Jolla, CA, United States) and R statistical package (R software version 3.4.1; R Foundation for Statistical Computing, Vienna, Austria).
We retrieved m6A methyltransferase (METTL3, METTL14, and WTAP) and demethylase (FTO, ALKBH5) expression in GC and normal tissues using online TCGA mRNA-seq data in Firehose. Compared with normal tissues, all three core components of the methyltransferase complex were upregulated, and the obvious alteration of demethylases was not observed in cancerous tissues (Figure 1A). Then, we quantitatively examined m6A levels in the total RNAs of 13 paired cancerous and adjacent tissues. Consistent with the results of the TCGA dataset, we found that m6A levels were increased in the cancerous group compared with their corresponding paratumor group (Figure 1B). Because m6A modification are primarily regulated by the five enzymes above, we explored the mRNA levels of these genes in the corresponding 13 paired tissues. As shown in Figure 1C, the mRNA expression of METTL3 and METTL14 was significantly elevated in GC tissues. In contrast, the expression of WTAP, FTO, or ALKBH5 was not significantly altered in tumor tissues. Next, we further estimated METTL3 and METTL14 expression in a larger cohort containing 136 paired tumor and paratumor tissues. In this sample cohort, METTL3 expression was markedly elevated in 66.18% (90/136) of patients with GC (Figure 1D). However, elevation of METTL14 was detected in 44.11% (60/136) of patients (Supplementary Figure 1B). Considering the higher percentage of METTL3 elevation in GC and its catalytic roles in the methyltransferase complex, we selected METTL3 as the candidate molecule for aberrant m6A modification in GC.
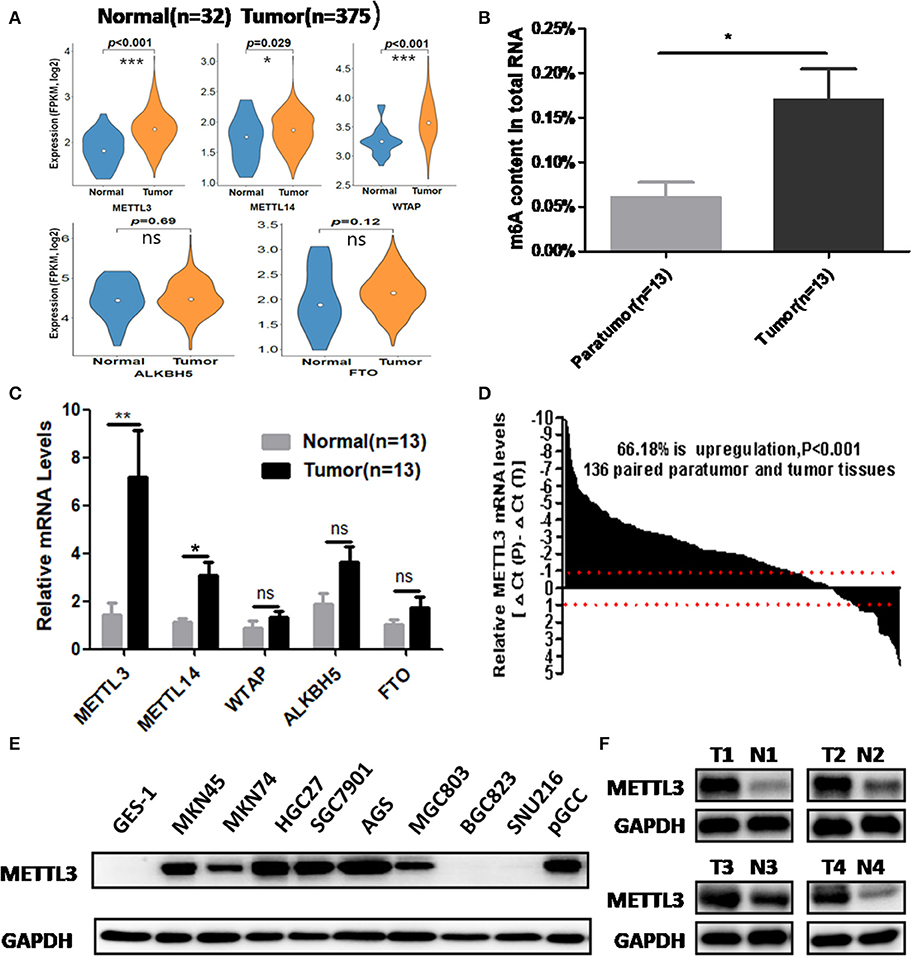
Figure 1. The association between methyltransferase-like 3 (METTL3) upregulation and abnormal m6A levels in gastric cancer (GC). (A) Relative expression pf writers and erasers in the The Cancer Genome Atlas (TCGA) database (normal mucosa = 32, tumor tissue = 375). (B) Total RNA m6A levels in 13 paired gastric carcinoma tissues and adjacent tissues. (C) Relative messenger RNA (mRNA) expression of writers and erasers in the above 13 paired tissues. (D) The mRNA expression of METTL3 in 136 paired gastric carcinoma tissues and adjacent tissues. (E) METTL3 expression in GES-1, pGCC, and cancer cell lines at the protein level. (F) METTL3 protein expression in four paired tumor tissues and adjacent tissues. *p < 0.05, **p < 0.01, ***p < 0.001. ns, non-significant.
Simultaneously, we determined METTL3 expression in nine GC cell lines and GES-1 (an immortalized gastric epithelial cell line) by Western blot analysis. The results indicated that METTL3 protein levels in most cancer cell lines (except for BGC823 and SUN216) were higher than those in GES-1 (Figure 1E). In addition, METTL3 expression was also elevated in four cases of gastric carcinoma tissues (Figure 1F). Based on the above evidence, we deduced that the upregulation of METTL3, a leading cause of higher m6A levels in GC, occurred frequently in patients who suffered from this deadly disease.
We enrolled 196 patients diagnosed with GC in our hospital to determine the prognostic value of METTL3 in these patients. In total, we examined METTL3 expression in 455 paraffin-embedded blocks (paratumor, n = 146; primary tumor, n = 196; lymph node metastasis, n = 92; distant metastasis, n = 21). The immunohistochemistry (IHC) scores of METTL3 in the primary tumor group were significantly higher than those in the paratumor group (Figure 2A, p < 0.001). Meanwhile, METTL3 expression in lymph node metastatic and distant metastatic tissues is significantly higher than that of primary tumor tissues (Figure 2A). Representative IHC images of different tissues are shown in Figure 2B. Furthermore, matched normal and cancerous tissues from this cohort also showed similar differences (Supplementary Figures 1C,D).
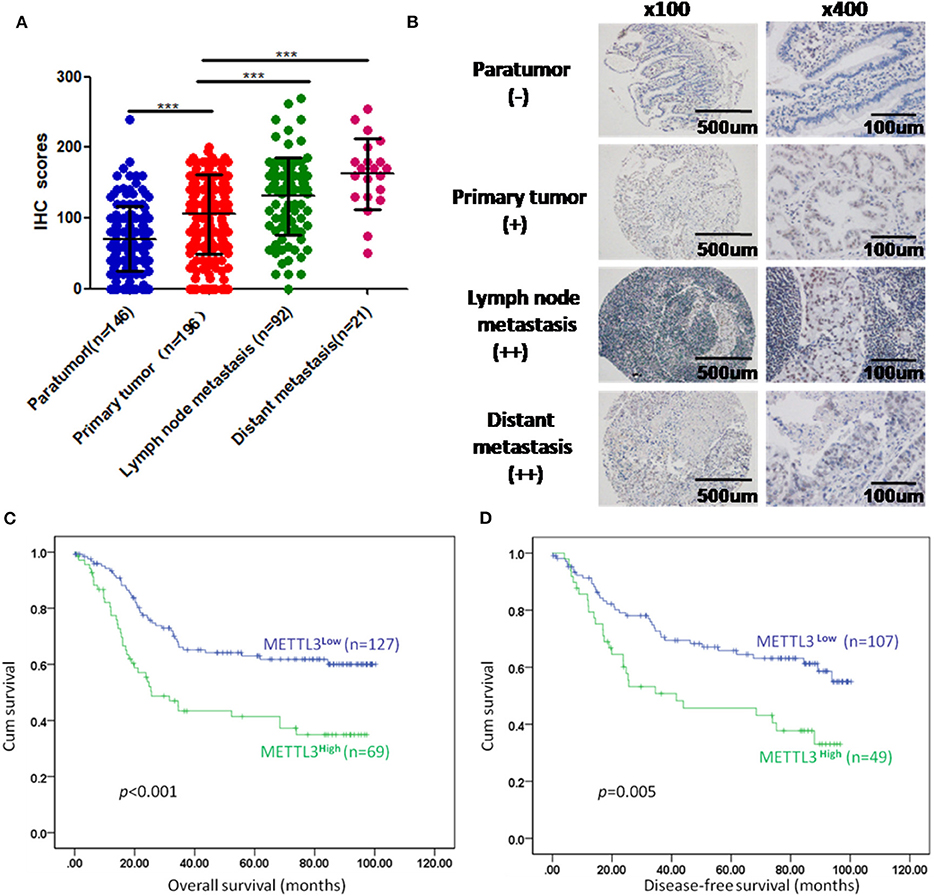
Figure 2. Immunohistochemistry (IHC) scores of methyltransferase-like 3 (METTL3) in different types of tissues and Kaplan–Meier survival curves in gastric cancer (GC) patients. (A) IHC scores of METTL3 in four different types of tissues from 196 patients with GC (paratumor, mean ± SD = 70.74 ± 46.29; primary tumor, mean ± SD = 105.60 ± 56.16; lymph node metastasis, mean ± SD = 130.80 ± 55.31; distant metastasis: mean ± SD = 162.6 ± 50.19). (B) Immunohistochemical staining of METTL3 in four different types of tissues from the same patient. (C) Overall survival analysis in all GC patients (n = 196). (D) Disease-free survival analysis in GC patients who were first diagnosed with tumor–node–metastasis (TNM) stage I, II, or III (n = 156). ***p < 0.001.
Subsequently, we further assessed the relationship between METTL3 expression and other clinicopathological variables. As shown in Table 1, METTL3 expression was significantly associated with tumor size (p = 0.011) and distant metastasis (p = 0.040) and TNM stage (p = 0.035). Other remaining factors had no statistical correlation with METTL3 (Table 1). Moreover, METTL3 expression in the primary tumor was elevated with increased M stage and TNM stage (Supplementary Figures 1E,F). Then, we performed Kaplan–Meier survival curve analysis (log-rank test) to confirm the prognostic value of METTL3 expression in GC. As shown in Figures 2C,D, GC patients with higher METTL3 expression had significantly shorter overall survival (OS) (p < 0.001) and disease-free survival (DFS) (p = 0.005). In addition, in the tumor size ≤5 cm group and grade III group, high METTL3 expression was associated with worse OS and DFS (Supplementary Figure 2).
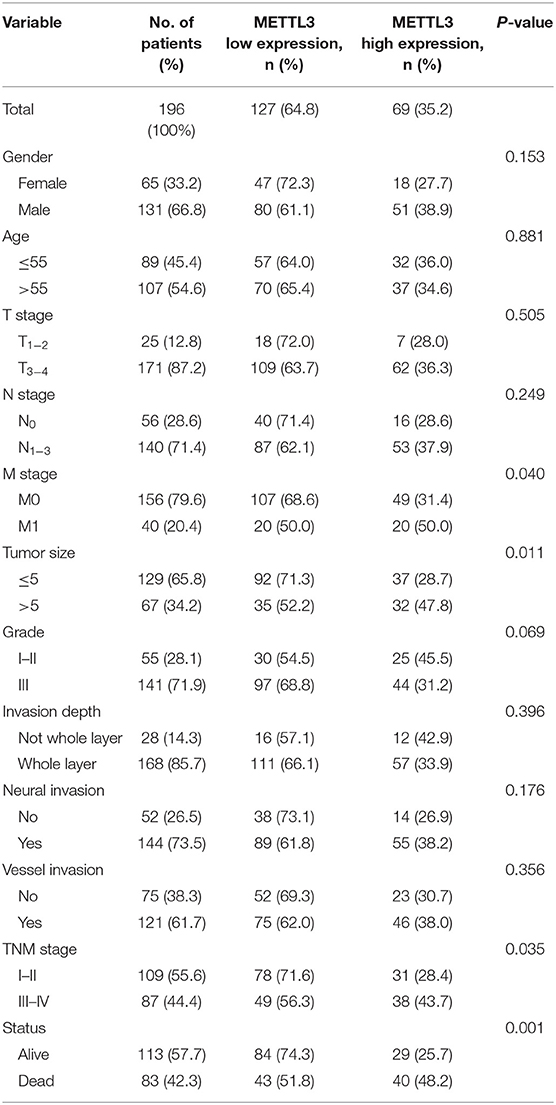
Table 1. The relationship between METTL3 expression and clinicopathological parameters.
Importantly, the results of univariate analysis revealed that well-recognized clinical prognostic parameters and METTL3 expression were significantly correlated with OS and DFS (Table 2). Multivariate analysis confirmed that METTL3 expression was an independent prognostic factor for OS [hazard ratio (HR), 1.741; 95% confidence interval (CI), 1.123–2.698; p = 0.013] and DFS (HR, 1.936; 95% CI, 1.189–3.512; p = 0.008). Collectively, these findings suggested that high METTL3 expression was an independent prognostic factor of poor overall survival in GC patients.
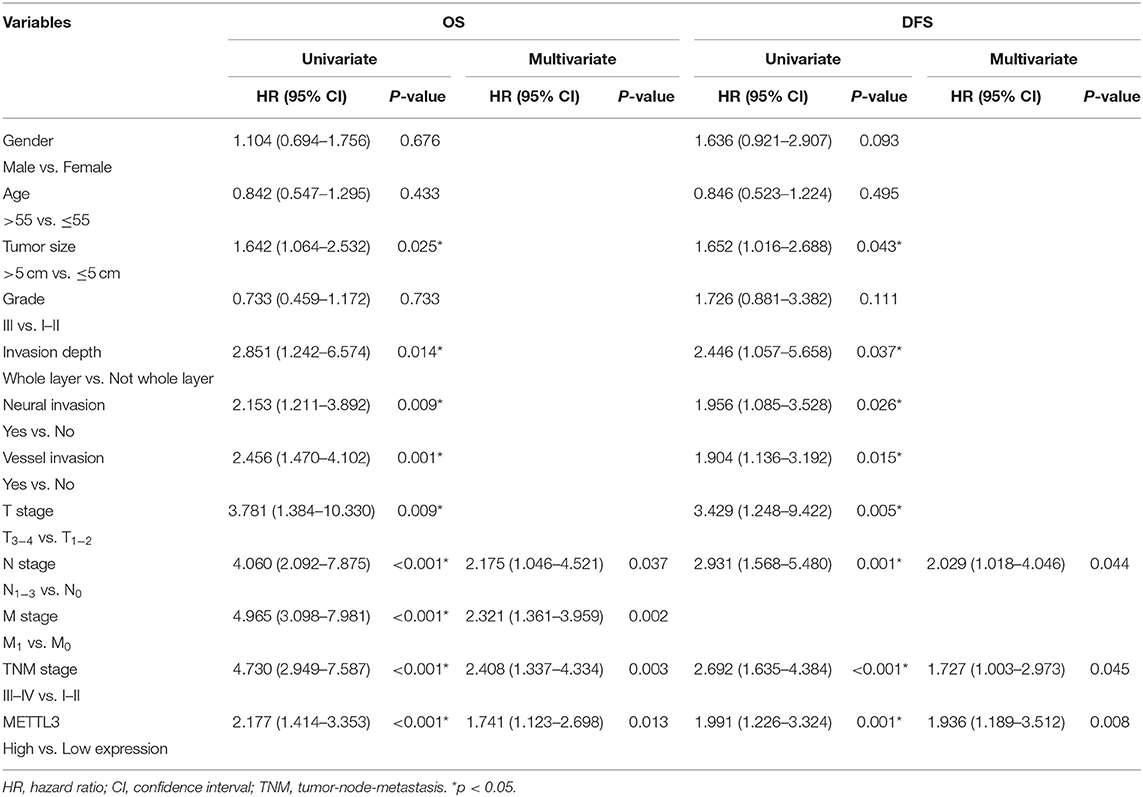
Table 2. Univariate and multivariate analysis of OS in 196 patients (TNM stage I–IV) and DFS in 156 patients (TNM stage I–III).
To explore the functional roles of METTL3 in GC, we stably knocked down METTL3 in two GC cell lines, SGC7901 and AGS, using two independent shRNAs (sh#1, sh#2). Then, qRT-PCR and Western blotting were conducted to confirm the high knocking efficiency of METTL3. As expected, once successfully knocking down METTL3, m6A levels in SGC7901 and AGS sharply reduced (Figure 3A). We performed in vitro functional assays to determine the role of METTL3 in the proliferation, colony formation, migration, and invasion of GC cells. The results of the MTS assays and colony-forming assays indicated that depletion of METTL3 could obviously suppress proliferation and colony-forming abilities of GC cells (Figures 3B,C). In the migration and invasion assays, the silencing of METTL3 significantly inhibited the migratory and invasive abilities of SGC7901 and AGS cells (Figure 3D).
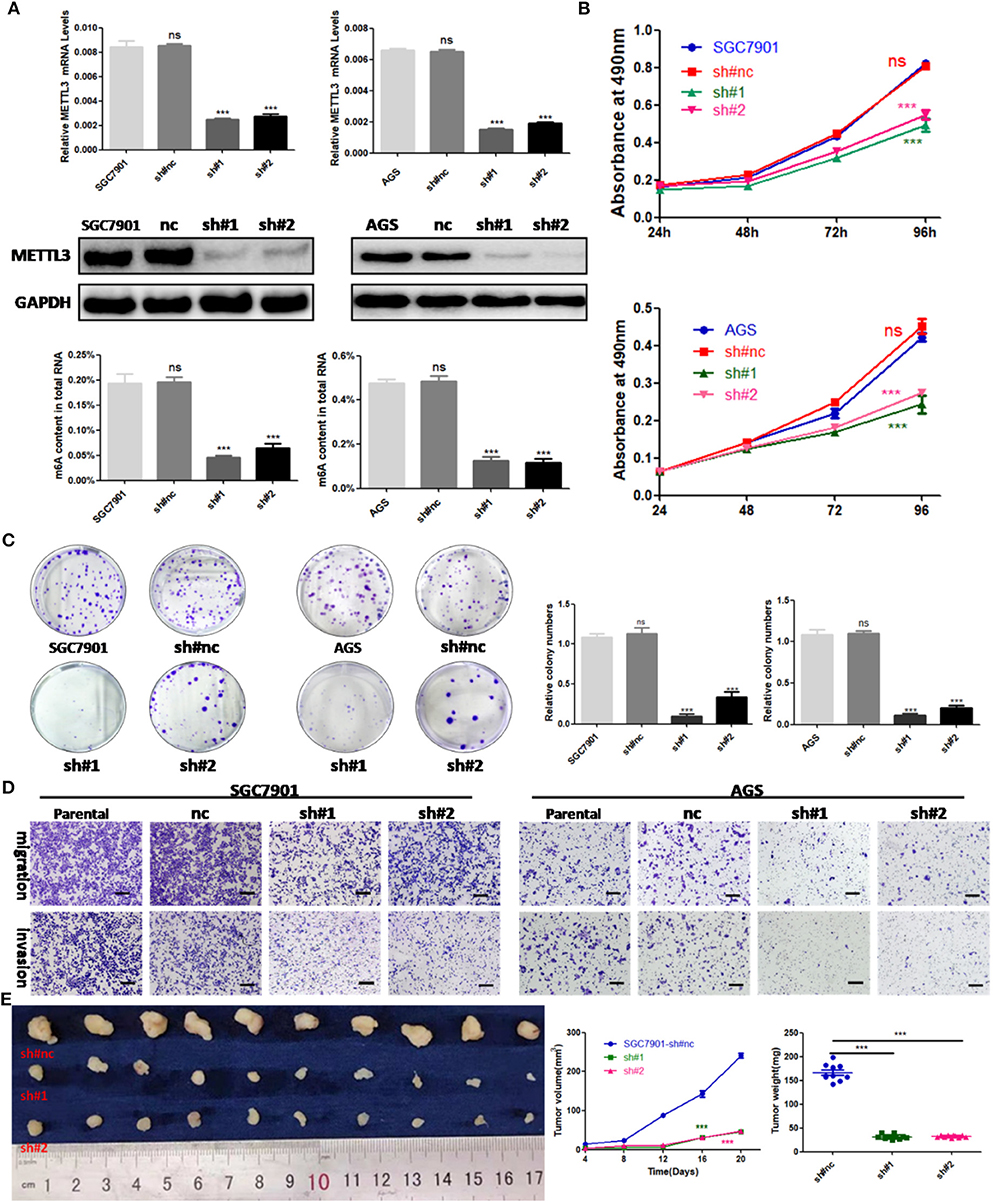
Figure 3. The effects of methyltransferase-like 3 (METTL3) knockdown on cell function in vitro and in vivo. (A) METTL3 knockdown efficiency and m6A levels of total RNAs in SGC7901 and AGS. (B) Cell proliferation assays after METTL3 knockdown in SGC7901 and AGS cells. (C) Colony formation assays of METTL3 knockdown in SGC7901 and AGS cells. (D) Transwell migration and invasion assays after METTL3 knockdown in SGC7901 and AGS. (E) The nude mice xenograft models (N = 5) of METTL3 knockdown SGC7901 cells. ***p < 0.001. ns, non-significant. All experiments were repeated three times. Scale bars = 50 μm.
SGC7901 infected with sh#METTL3 and control lentiviruses were used to establish a subcutaneous tumor xenograft in nude mice. We observed xenograft growth for 3 weeks and found that METTL3 silencing could suppress tumor growth in vivo (Figure 3E). All in all, these findings in vitro and in vivo revealed the oncogenic roles of METTL3 in GC.
Considering the findings of the functional assays above, we were motivated to determine if overexpression of METTL3 could promote GC progression. Then, control lentivirus and lentivirus for overexpressing endogenous METTL3 were used to transfect SGC7901 and AGS cells. The upregulation of METTL3 was verified by qRT-PCR and Western blot analysis. In addition, we observed elevated m6A levels in cells overexpressing METTL3 (Figure 4A). Similarly, cell growth and colony-forming abilities were increased upon METTL3 overexpression as well as migratory and invasive capacities (Figures 4B–D). In nude mouse xenograft models, tumors induced by METTL3-overexpression cells showed significantly larger tumor volumes and heavier tumor weights than those in the control group (Figure 4E). Taken together, METTL3 may act as an oncogene in GC by potentiating cellular proliferation and metastasis.
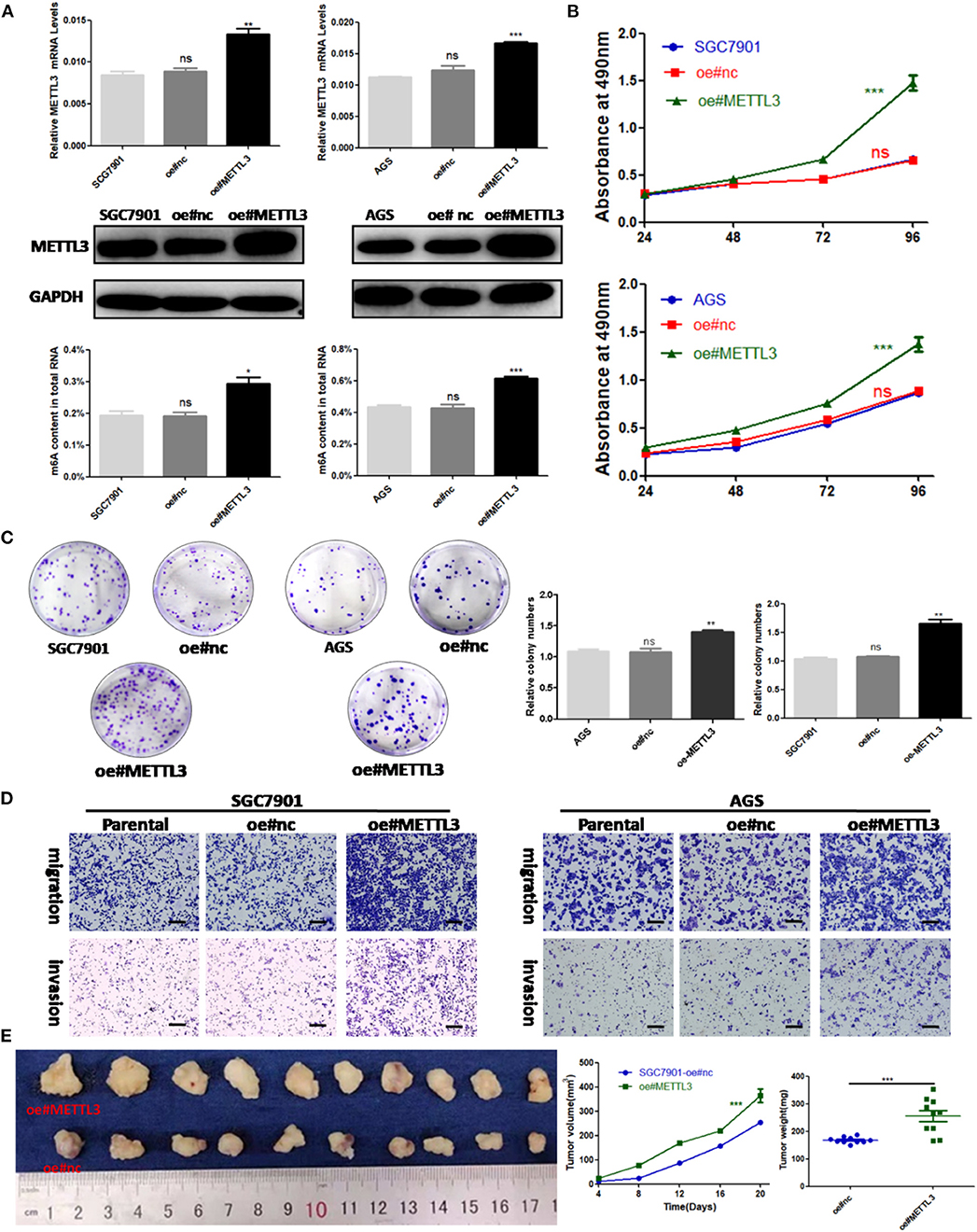
Figure 4. Methyltransferase-like 3 (METTL3) overexpression enhances migration and invasion in vitro and promotes proliferation in vivo. (A) METTL3 overexpression efficiency and m6A levels of total RNA in SGC7901 and AGS. (B) Cell proliferation assays of METTL3-overexpressing SGC7901 and AGS cells. (C) Colony formation assays of METTL3 overexpressing SGC7901 and AGS cells. (D) Transwell migration and invasion assays of METTL3-overexpressing SGC7901 and AGS cells. (E) The nude mice xenograft models (N = 5) of METTL3 overexpressing SGC7901 cells. *p < 0.05, **p < 0.01, ***p < 0.001. ns, non-significant. All experiments were repeated three times. Scale bars = 50 μm.
To identify the potential targets methylated by METTL3 in GC, we performed RNA-seq and m6A-seq in SGC7901 cell lines. Then, we applied two algorithms widely used in m6A-seq analysis (masc2 and exomePeak) to confirm the profiles of m6A peaks (Figure 5A). In line with previously published works, we identified 24,195 peaks in 13,322 genes. The distribution of the identified m6A peaks was the highest enriched near the stop codon, with a higher percentage in the mRNA intron and mRNA exon (Figures 5B,C). Consistent with other cell types, the consensus motif of all m6A sites was GGACT (Figure 5C).
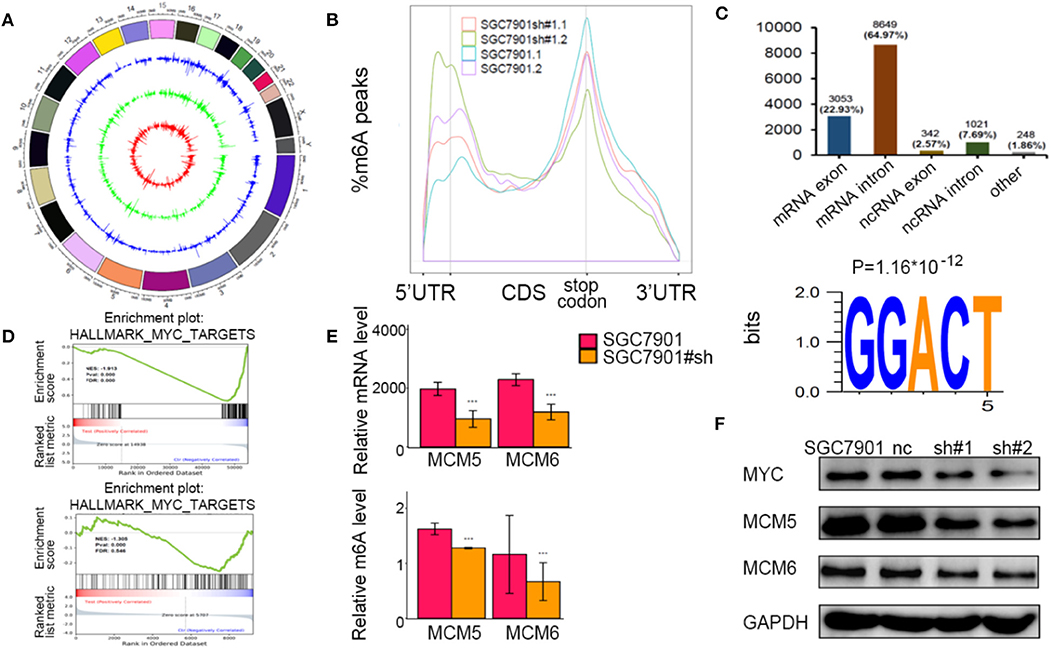
Figure 5. Identification of the potential m6A-dependent targets of methyltransferase-like 3 (METTL3) in GC. (A) Identification of m6A peaks by two algorithms. The layers from outer to inner represent the m6A peaks identified by exomePeak, masc2, and the overlap from both algorithms. (B) The density distribution of m6A peaks in messenger RNA (mRNA) transcripts. (C) Distribution of the m6A peaks in exonic or intronic regions of protein-coding and non-coding genes and in other regions. The consensus motif from all identified m6A peaks. (D) Gene set enrichment analysis determined by both RNA-seq and m6A-seq selected MYC pathway as downstream of METTL3. (E) Relative m6A levels of MCM5 and MCM6 upon depletion of METTL3. (F) The protein levels of MCM5, MCM6, and MYC upon depletion of METTL3. *p < 0.05, **p < 0.01, ***p < 0.001. ns, non-significant.
To explore the METTL3-m6A-target genes, we performed GSEA using the hypo-methylation site lists combined with differentially expressed genes upon METTL3 depletion. There were multiple signaling pathways that were positively/negatively associated with METTL3 depletion in this merged analysis (Supplementary Table 2). MYC target genes had a significant decrease at both the m6A and mRNA levels after METTL3 knockdown (Figure 5D). Here, we showed decreases in MCM5 and MCM6 (key components of MYC target genes) at epigenetic levels (Figure 5E). In addition, the protein levels of MYC, MCM5, and MCM6 were decreased with METTL3 knockdown in SGC7901 cells (Figure 5F). These findings strongly indicated that MYC target genes were downstream of METTL3 in GC.
In the past few years, METTL3, with other writers and erasers, has been reported to extensively take part in tumorigenesis (33–35). It was reported that METTL3 could participate in glioblastoma tumorigenesis by enhancing glioma stem-like cell (GSC) maintenance and radioresistance. In contrast, another study showed that METTL3 could inhibit GSC growth and self-renewal by decreasing the expression of a series of oncogenes (ADAM19, EPHA3, and KLF4). Several studies have identified that METTL3 can promote cancer cell proliferation and invasion by upregulating downstream key oncogenes via post-transcriptional modification. However, the results from an American research team indicated that METTL3 can increase the invasiveness of lung cancer cells by initiating the translation of oncogenes (EGFR, TAZ, and MAPKAPK2) (36–40).
The roles METTL3 have been identified in several studies of GC. Zhang et al. showed that reduced m6A levels could activate Wnt/phosphatidylinositol-3-kinase (PI3K)-protein kinase B (AKT) signaling and promote malignant phenotypes in GC cells. Lin and collaborators found that METTL3 could promote the proliferation and mobility of GC cells by activating the AKT pathway. The results of Liu and coworkers indicated that METTL3 knockdown reduces α-smooth muscle actin and that METTL3 is an adverse factor in patients with GC (25–27). However, these studies explored the METTL3 regulatory network in GC simply by bioinformatics analysis. In Yue's research, the results of m6A-seq indicated that METTL3 could enhance ZMYM1 mRNA stability, which facilitates the EMT program and metastasis of GC (41). Similarly, in our research, we explored the downstream targets of METTL3 by high-throughput m6A-seq and in-depth bioinformatics analysis in GC cell lines and comprehensive experiments. Specifically, we first observed that frequent upregulation of METTL3 rather than METTL14 or WTAP was responsible for elevated m6A levels and poor survival prognosis. Then, we confirmed the carcinogenesis of METTL3 by silencing or overexpressing METTL3 in cells. The novelty of this study was the identification of MYC and its target genes downstream of METTL3.
The MYC oncogene and its target genes contribute to the genesis of many human cancers. The upstream and downstream of MYC were extensively involved in cell growth and proliferation as well as nutrient metabolism. Indeed, antitumor strategies targeting MYC have offered new cancer therapeutic opportunities (42). Several studies have indicated that METTL3, METTL14, or FTO could affect the stability of MYC mRNA via the alteration of m6A abundance (39, 43, 44). However, in the current research, we did not observe influences on the relative m6A abundance of MYC transcripts by METTL3 depletion. Indeed, MYC and its downstream proteins were degraded upon METTL3 depletion. The precise mechanism of how METTL3 manipulates MYC expression remains unknown in GC and needs to be further explored.
More importantly, we identified the key components of the MYC pathway as direct target genes of METTL3 in GC. MCM5 and MCM6, which belong to the minichromosome maintenance (MCM) family, assembled into the prereplication complex for the initiation of eukaryotic genome replication. It was reported that high MCM5 expression was associated with poor clinicopathological parameters and poor survival in GC. According to a previous report, MCM6 can serve as a prognostic predictor and promote metastasis in hepatocellular carcinoma (HCC) (45–47). In our research, m6A abundances of MCM5 and MCM6 transcripts were sharply decreased upon the depletion of METTL3 in cells. Some published studies have shown that certain gene transcripts are degraded upon the downregulation of m6A modification in several cancers. Thus, we presumed that the possible regulatory mechanisms were a decrease in the stability of MCM5 and MCM6 transcripts mediated by METTL3 depletion. The exact mechanism requires further experiments.
With the continuous improvement of m6A-seq and other related detection technologies, we could further detect the precise modality of METTL3 in cancers (48). Currently, three FTO inhibitors (Rhein, MA2, and R-2HG) have effective anticancer potential (34, 36, 49). Future efforts could be made to develop inhibitors targeting METTL3.
In conclusion, our study highlights a new pathway of METTL3-mediated m6A modification in the epigenetic silencing of MYC downstream molecules in GC, which can provide a new strategy for more careful surveillance and aggressive therapeutic intervention.
The RNA-seq and m6A-seq data from METTL3 knockdown or non-treated SGC7901 have been deposited in BioProject under accession PRJNA595769. Other data generated during this study are not publicly available, and we are willing to share it upon reasonable request.
The studies involving human participants were reviewed and approved by Institute Research Ethics Committee of Cancer Center of Sun Yat-sen University. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Institute Research Ethics Committee of Cancer Center of Sun Yat-sen University.
D-DY, Z-HC, and Z-LZ designed the study, acquired the data, and wrote the manuscript. D-DY, Q-NW, YW, Z-LZ, and J-HL collected cell samples from the Pathology Department and did the experiments in vitro and in vivo. KY and Z-XL interpreted and analyzed the data. R-HX, H-QJ, and Z-LZ revised and approved the final version of the manuscript.
This research was supported by the National Natural Science Foundation of China (81572392, 31501069, 81872011, 81972239, 91953123), Tip-Top Scientific and Technical Innovative Youth Talents of Guangdong special support program (2016TQ03R614), the National Key R&D Program of China (2018YFC1313300, 2016YFC1201704, 2017YFC1308900), Natural Science Foundation of Guangdong Province (2018A0303130282, 2018B030306049, 2017A030313485, 2014A030312015, 2019A1515010634), Science and Technology Program of Guangdong (2015B020232008, 2019B020227002), Science and Technology Program of Guangzhou (201904020046, 201803040019, 201704020228), Health & Medical Collaborative Innovation Program of Guangzhou (201803040019), Program for Guangdong Introducing Innovative and Entrepreneurial Teams (2017ZT07S096), and Pearl River S&T Nova Program of Guangzhou (201906010088).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the patients and the investigators who participated in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.00115/full#supplementary-material
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
2. Ferro A, Peleteiro B, Malvezzi M, Bosetti C, Bertuccio P, Levi F, et al. Worldwide trends in gastric cancer mortality (1980-2011), with predictions to 2015, and incidence by subtype. Eur J Cancer. (2014) 50:1330–44. doi: 10.1016/j.ejca.2014.01.029
3. Ferro A, Peleteiro B, Malvezzi M, Bosetti C, Bertuccio P, Levi F, et al. Risk factors for gastric cancer: a systematic review. Asian Pac J Cancer Prev. (2018) 19:591–603. doi: 10.22034/APJCP.2018.19.3.591
5. Takenoya T, Yabe N, Morishige S, Tamura E, Sakimoto Y, Tajima Y, et al. Trastuzumab combination chemotherapy followed by R0 resection for locally advanced human epidermal growth factor receptor 2-positive gastric cancer-a case report. Gan To Kagaku Ryoho. (2019) 46:121–23.
6. Cao G, Li HB, Yin Z, Flavell RA. Recent advances in dynamic m6A RNA modification. Open Biol. (2016) 6:160003. doi: 10.1098/rsob.160003
7. Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. (2014) 505:117–20. doi: 10.1038/nature12730
8. Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, et al. Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science. (2015) 347:1002–6. doi: 10.1126/science.1261417
9. Hastings MH. m(6)A mRNA methylation: a new circadian pacesetter. Cell. (2013) 155:740–1. doi: 10.1016/j.cell.2013.10.028
10. Gokhale NS, McIntyre ABR, McFadden MJ, Roder AE, Kennedy EM, Gandara JA, et al. N6-Methyladenosine in flaviviridae viral RNA genomes regulates infection. Cell Host Microbe. (2016) 20:654–65. doi: 10.1016/j.chom.2016.09.015
11. Li HB, Tong J, Zhu S, Batista PJ, Duffy EE, Zhao J, et al. m(6)A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature. (2017) 548:338–42. doi: 10.1038/nature23450
12. Pereira-Montecinos C, Valiente-Echeverría F, Soto-Rifo R. Epitranscriptomic regulation of viral replication. Biochim Biophys Acta Gene Regul Mech. (2017) 1860:460–71. doi: 10.1016/j.bbagrm.2017.02.002
13. Lichinchi G, Zhao BS, Wu Y, Lu Z, Qin Y, He C, et al. Dynamics of human and viral RNA methylation during Zika virus infection. Cell Host Microbe. (2016) 20:666–73. doi: 10.1016/j.chom.2016.10.002
14. Wei W, Ji X, Guo X, Ji S. Regulatory role of N(6) -methyladenosine (m(6) A) methylation in RNA processing and human diseases. J Cell Biochem. (2017) 118:2534–43. doi: 10.1002/jcb.25967
15. Yang Y, Hsu PJ, Chen YS, Yang YG. Dynamic transcriptomic m(6)A decoration: writers, erasers, readers and function sinRNA metabolism. Cell Res. (2018) 28:616–24. doi: 10.1038/s41422-018-0040-8
16. Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. (2014) 10:93–5. doi: 10.1038/nchembio.1432
17. Chen K, Zhao BS, He C. Nucleic acid modifications in regulation of gene expression. Cell Chem Biol. (2016) 23:74–85. doi: 10.1016/j.chembiol.2015.11.007
18. Maity A, Das B. N6-methyladenosine modification in mRNA: machinery, function and implications for health and diseases. FEBS J. (2016) 283:1607–30. doi: 10.1111/febs.13614
19. Deng X, Su R, Weng H, Huang H, Li Z, Chen J. RNA N(6)-methyladenosine modification incancers: current status and perspectives. Cell Res. (2018) 28:507–17. doi: 10.1038/s41422-018-0034-6
20. Cai X, Wang X, Cao C, Gao Y, Zhang S, Yang Z, et al. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer Lett. (2018) 415:11–9. doi: 10.1016/j.canlet.2017.11.018
21. Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millán-Zambrano G, Robson SC, et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature. (2017) 552:126–31. doi: 10.1038/nature24678
22. Singh B, Kinne HE, Milligan RD, Washburn LJ, Olsen M, Lucci A. Important role of FTO in the survival of rare panresistant triple-negative inflammatory breast cancer cells facing a severe metabolic challenge. PLoS ONE. (2016) 11:e0159072. doi: 10.1371/journal.pone.0159072
23. Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, et al. FTO plays an oncogenic role in acute myeloid leukemia as a N(6)-methyladenosine RNA demethylase. Cancer Cell. (2017) 31:127–41. doi: 10.1016/j.ccell.2016.11.017
24. Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m(6)A-demethylation of NANOG mRNA. Proc Natl Acad Sci USA. (2016) 113:E2047–56. doi: 10.1073/pnas.1602883113
25. Zhang C, Zhang M, Ge S, Huang W, Lin X, Gao J, et al. Reduced m6A modification predicts malignant phenotypes and augmented Wnt/PI3K-Akt signaling in gastric cancer. Cancer Med. (2019) 8:4766–81. doi: 10.1002/cam4.2360
26. Lin S, Liu J, Jiang W, Wang P, Sun C, Wang X, et al. METTL3 promotes the proliferation and mobility of gastric cancer cells. Open Med. (2019) 14:25–31. doi: 10.1515/med-2019-0005
27. Liu T, Yang S, Sui J, Xu SY, Cheng YP, Shen B, et al. Dysregulated N6-methyladenosine methylation writer METTL3 contributes to the proliferation and migration of gastric cancer. J Cell Physiol. (2019) 235:1–15. doi: 10.1002/jcp.28994
28. Zhu L, Zhu Y, Han S, Chen M, Song P, Dai D, et al. Impaired autophagic degradation of lncRNA ARHGAP5-AS1 promotes chemoresistance in gastric cancer. Cell Death Dis. (2019) 10:383. doi: 10.1038/s41419-019-1585-2
29. Edge SB, Compton CC. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol. (2010) 17:1471–4. doi: 10.1245/s10434-010-0985-4
30. Li T, Hu PS, Zuo Z, Lin JF, Li X, Wu QN, et al. METTL3 facilitates tumor progression via anm6A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol Cancer. (2019) 18:112–26. doi: 10.1186/s12943-019-1038-7
31. Shaoyan X, Juanjuan Y, Yalan T, Ping H, Jianzhong L, Qinian W. Downregulation of EIF4A2 in non-small-cell lung cancer associates with poor prognosis. Clin Lung Cancer. (2013) 14:658–65. doi: 10.1016/j.cllc.2013.04.011
32. Dominissini D, Moshitch-Moshkovitz S, Salmon-Divon M, Amariglio N, Rechavi G. Mapping of Transcriptome-wide N6-methyladenosine by m6A-seq based on immunocapturing and massively parallel sequencing. Nat Protoc. (2013) 8:176–89. doi: 10.1038/nprot.2012.148
33. Wang S, Sun C, Li J, Zhang E, Ma Z, Xu W, et al. Roles of RNA methylation by means of N(6)-methyladenosine (m(6)A) in human cancers. Cancer Lett. (2017) 408:112–20. doi: 10.1016/j.canlet.2017.08.030
34. Jaffrey SR, Kharas MG. Emerging links between m(6)A and mRNA methylation in cancer. Genome Med. (2017) 9:2. doi: 10.1186/s13073-016-0395-8
35. Tong J, Flavell RA, Li HB. RNA m(6)A modification and its function in diseases. Front Med. (2018) 12:481–9. doi: 10.1007/s11684-018-0654-8
36. Visvanathan A, Patil V, Arora A, Hegde AS, Arivazhagan A, Santosh V, et al. Essential role of METTL3-mediatedm(6)A modification in glioma stem-like cells maintenance and radioresistance. Oncogene. (2018) 37:522–33. doi: 10.1038/onc.2017.351
37. Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, et al. m(6)A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. (2017) 18:2622–34. doi: 10.1016/j.celrep.2017.02.059
38. Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. (2018) 67:2254–70. doi: 10.1002/hep.29683
39. Cheng M, Sheng L, Gao Q, Xiong Q, Zhang H, Wu M, et al. The m(6)A methyltransferase METTL3 promotes bladder cancer progression via AFF4/NF-κB/MYC signaling network. Oncogene. (2019) 38:3667–80. doi: 10.1038/s41388-019-0683-z
40. Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell. (2016) 62:335–45. doi: 10.1016/j.molcel.2016.03.021
41. Yue B, Song C, Yang L, Cui R, Cheng X, Zhang Z, et al. METTL3-mediated N6-methyladenosinemodification is critical for epithelial mesenchymal transition and metastasis of gastric cancer. Mol Cancer. (2019) 18:142. doi: 10.1186/s12943-019-1065-4
43. Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell. (2018) 22:191–205.e9. doi: 10.1016/j.stem.2017.11.016
44. Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, et al. R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell. (2018) 172:90–105.e123. doi: 10.1016/j.cell.2017.11.031
45. Gou K, Liu J, Feng X, Li H, Yuan Y, Xing C. Expression of minichromosome maintenance proteins (MCM) and cancer prognosis: a meta-analysis. J Cancer. (2018) 9:1518–26. doi: 10.7150/jca.22691
46. Giaginis C, Giagini A, Tsourouflis G, Gatzidou E, Agapitos E, Kouraklis G, et al. MCM-2 and MCM-5 expression in gastric adenocarcinoma: clinical significance and comparison with Ki-67 proliferative marker. Dig Dis Sci. (2010) 56:777–85. doi: 10.1007/s10620-010-1348-5
47. Liu M, Hu Q, Tu M, Wang X, Yang Z, Yang G, et al. MCM6 promotes metastasis of hepatocellular carcinoma via MEK/ERK pathway and serves as a novel serum biomarker for early recurrence. J Exp Clin Cancer Res. (2018) 37:10. doi: 10.1186/s13046-017-0669-z
48. Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. Single-nucleotide resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. (2015) 12:767–72. doi: 10.1038/nmeth.3453
Keywords: METTL3, gastric cancer, prognostic factor, MYC target genes, minichromosome maintenance complex component 5, minichromosome maintenance complex component 6
Citation: Yang D-D, Chen Z-H, Yu K, Lu J-H, Wu Q-N, Wang Y, Ju H-Q, Xu R-H, Liu Z-X and Zeng Z-L (2020) METTL3 Promotes the Progression of Gastric Cancer via Targeting the MYC Pathway. Front. Oncol. 10:115. doi: 10.3389/fonc.2020.00115
Received: 14 September 2019; Accepted: 22 January 2020;
Published: 26 February 2020.
Edited by:
Yun Dai, Peking University First Hospital, ChinaReviewed by:
Sean Porazinski, The Kinghorn Cancer Centre, AustraliaCopyright © 2020 Yang, Chen, Yu, Lu, Wu, Wang, Ju, Xu, Liu and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ze-Xian Liu, bGl1enhAc3lzdWNjLm9yZy5jbg==; Zhao-Lei Zeng, emVuZ3pobEBzeXN1Y2Mub3JnLmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.