 Rohan Garje
Rohan Garje Raju Kumar Vaddepally
Raju Kumar Vaddepally Yousef Zakharia
Yousef Zakharia- 1Division of Hematology, Oncology, and Blood and Marrow Transplant, Holden Comprehensive Cancer Center, University of Iowa, Iowa City, IA, United States
- 2Yuma Regional Medical Center, Yuma, AZ, United States
Poly(ADP-ribose) polymerase (PARP) inhibitors targeting DNA repair gene mutations have shown significant clinical benefit in patients with ovarian and breast cancers. In metastatic prostate cancers, the prevalence of DNA repair gene mutations is up to 20%, and early phase studies have shown clinical activity of PARP inhibitors. Numerous clinical trials with either PARP monotherapy or in combination with other therapeutic agents are ongoing in prostate cancer. In this comprehensive review, we provide the rationale, efficacy, and safety data of PARP inhibitors in prostate as well as urothelial cancers.
Introduction
In the current era of precision medicine, the discovery of targeted agents has led to significant development in the armamentarium of cancer therapeutics (1–3). Many studies have identified new mechanisms to counter cancer cellular mechanisms (4). Targeting tumors with defects in DNA damage repair genes by inhibition of poly(ADP-ribose) polymerase (PARP) enzymes is one such breakthrough discovery (5). Multiple PARP inhibitors for the treatment of ovarian and breast cancers that harbor pathogenic BRCA mutations have been developed. PARP inhibitors are currently being explored in other cancers, including prostate, bladder, pancreas, and biliary tree (6).
Parp Inhibitor Mechanism and the Concept of Synthetic Lethality
Cellular DNA is subject to continuous damage from intrinsic and extrinsic mutagens, which gives rise to single-strand (ss) and double-strand (ds) DNA breaks (7). DNA damage response pathways (DDR) ensure the conformity of the DNA sequence, and cells respond to this perception by arresting cell cycle progression and attempting repair (8). Depending on the type of DNA damage, several repair pathways such as base excision repair (BER), mismatch repair (MMR), and nucleotide excision repair (NER) aid in ssDNA breaks (9). For dsDNA breaks, there are two main mechanisms for DNA repair-homologous recombination (HR) and non-homologous end-joining (NHEJ)—where HR matches the original DNA in a seamless repair, and NHEJ introduces deletions (10).
PARP enzyme proteins play a vital role in DNA repair, promoting ss- and dsDNA repair (11–13). PARP-1 functions as a transcription modulator and regulates the oncogenes, tumor suppressor genes, and inflammatory genes involved in chromatin modulation and gene transcription (14, 15). One of the notable dsDNA break repair (DSBR) mechanisms is the HR repair (HRR) pathway, which facilitates seamless repair of dsDNA breaks. Genes involved in HRR include BRCA2, BRCA1, PALB2, and ATM (16, 17).
The concept of synthetic lethality applies when mutation or decreased expression of two genes results in cell death, whereas mutation of one gene alone leads to viability (18, 19). Synthetic lethality with PARP inhibitor is produced by conditional drug sensitivity in HRR-deficient cells. BRCA1 and BRCA2 are tumor suppressor genes, and defective tumors with loss of the copy of either gene are shown to be intrinsically sensitive to PARP inhibitors in both pre-clinical and clinical models (20, 21). Thus, this makes the loss of a gene essential for HRR to result in synthetic lethality from PARP inhibition, in which two pathway defects that alone are non-toxic but when combined become lethal (22).
Parp Inhibitors in Prostate Cancer
Prostate cancer is the most common malignancy in men with an estimated incidence of 174,650 new cases in the United States in 2019 (23). The prevalence of mutations in the DNA repair genes involved in HRR in men with prostate cancer irrespective of age or family history is around 11–23%, with most common mutations noted in BRCA2 (24–26). The other common mutated genes include ATM, CHEK2, BRCA1, RAD51D, FANCA, CDK12, and PALB2. These mutations are more prevalent in metastatic cancer than localized disease (24–26). Additionally, these mutations have been noted in high frequency with intraductal adenocarcinoma histology, lower PSA levels at diagnosis, and tumors with lymphovascular invasion (27). Current National Comprehensive Cancer Network (NCCN) guidelines recommend germline testing for HRR genes in patients with regional and metastatic prostate cancer (25). Multiple clinical trials are now evaluating the potential role of PARP inhibitors in metastatic prostate cancer, which has opened the door for targeted therapeutics (Tables 1, 2).
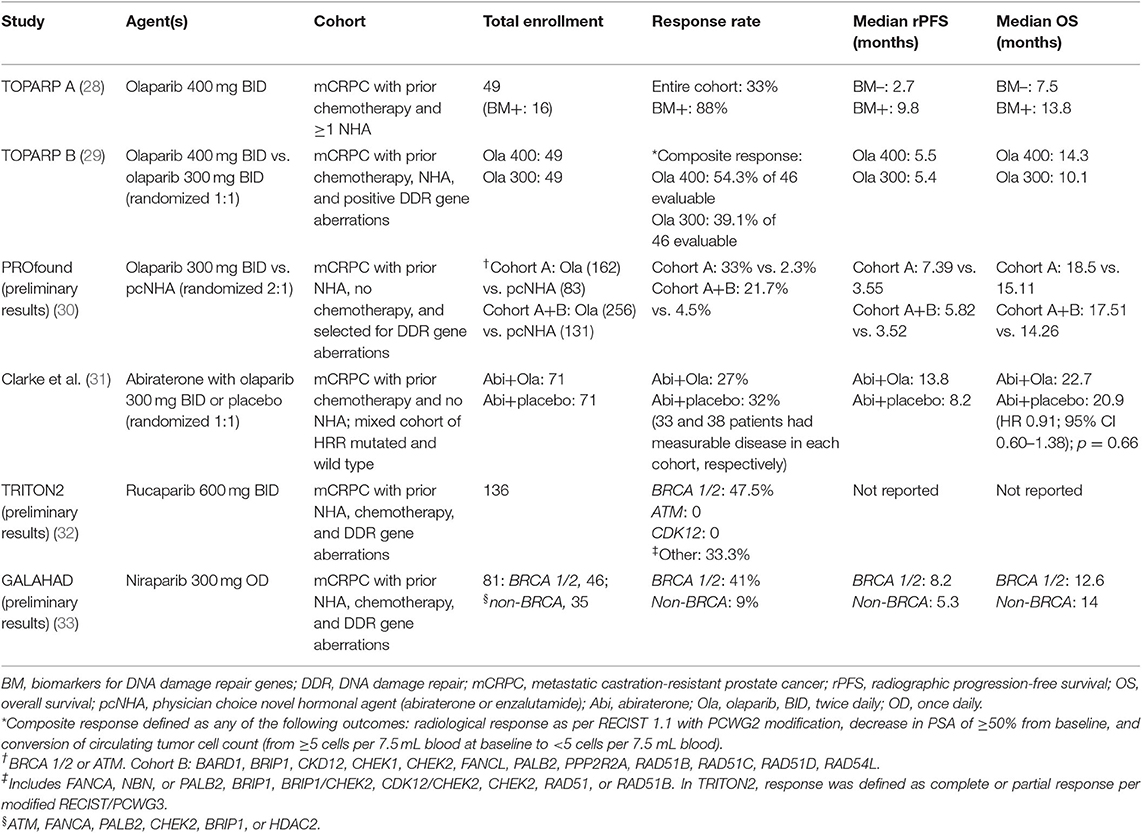
Table 1. Results of PARP inhibitors in prostate cancer.
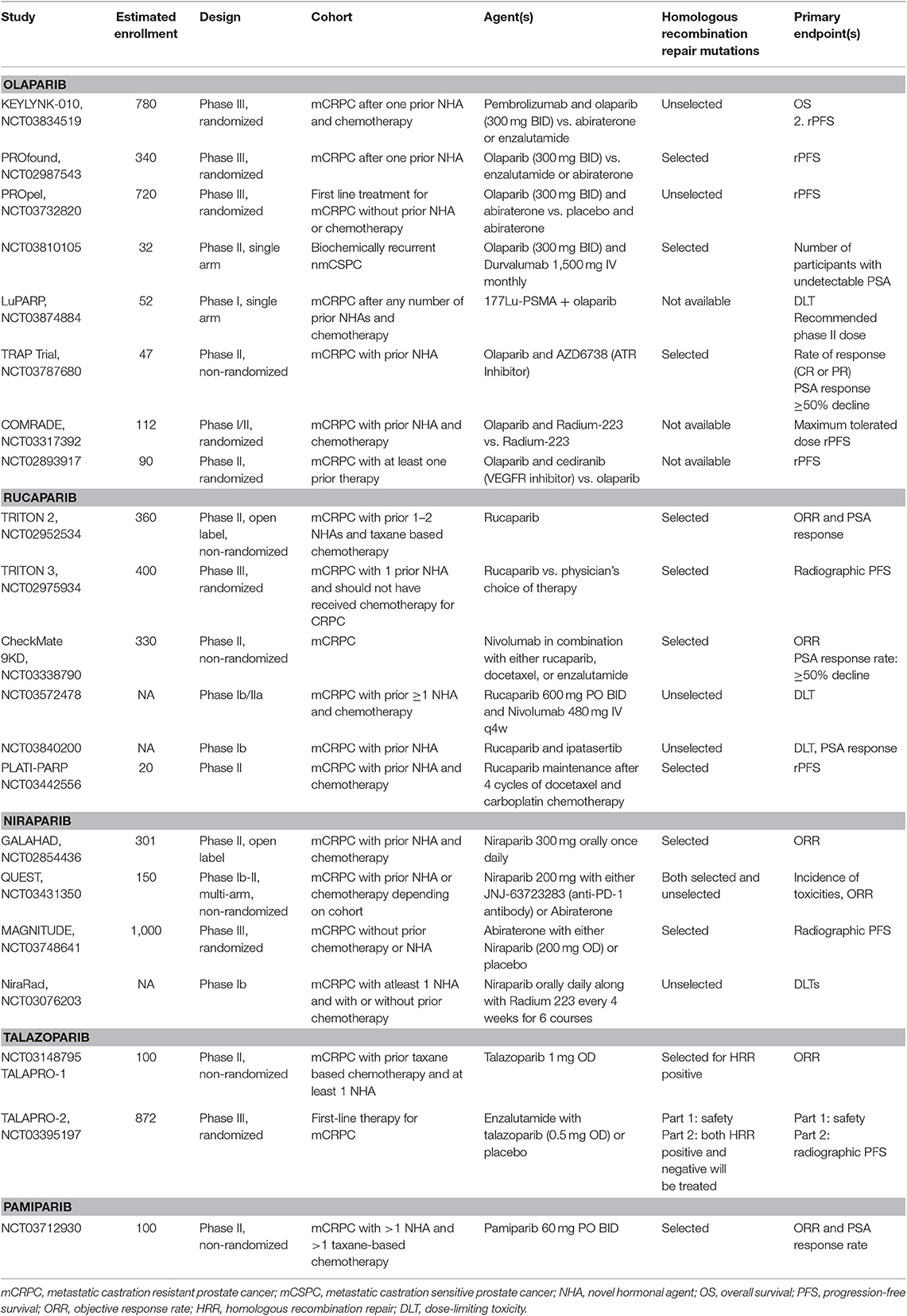
Table 2. Summary of ongoing clinical trials evaluating PARP inhibitors as monotherapy or in combination with other therapeutic agents in prostate cancer.
Olaparib Monotherapy
Olaparib is an inhibitor of PARP1, PARP2, and PARP3 enzymes involved in DNA repair and is currently approved for the treatment of ovarian and breast cancers. In the pivotal investigator-initiated, phase II Trial of PARP Inhibition in Prostate Cancer (TOPARP-A), the antitumor activity of olaparib 400 mg twice a day was evaluated in patients with metastatic, castration-resistant prostate cancer (CRPC) previously treated with chemotherapy and novel hormonal therapy (28). A response (defined as radiological response based on RECIST v1.1, or ≥ 50% decline in PSA or a reduction in the number of circulating tumor cells from ≥5/7.5 ml blood at baseline to <5/7.5 ml) was noted in 16 of 49 evaluable patients. Among the patients who had a response, 88% (14/16) harbored homozygous deletions and/or deleterious mutations in BRCA 1/2, ATM, Fanconi anemia genes, and CHEK2. Interestingly, 100% of the patients (7/7) with BRCA2 mutation had a response. The median progression-free survival (PFS) and median overall survival in patients with genomic aberrations were 9.8 and 13.8 months, respectively (28). Myelosuppression and fatigue were the most common treatment-related adverse effects (28). It is important to note that a predominant number of patients (94%, n = 31) who did not harbor these deleterious mutations had no response to olaparib (28).
Based on TOPARP-A data, the multicenter randomized phase III clinical trial (PROfound study) evaluated the efficacy of olaparib (30). In this landmark trial, patients with metastatic CRPC who received prior novel hormonal therapy and harbored alterations in HRR genes were randomized in a 2:1 fashion to receive either olaparib (300 mg BID) or physician's choice of novel anti-androgen agents such as enzalutamide or abiraterone. Patients were enrolled in cohort A (n = 245) if the tumors harbored BRCA1, BRCA2, or ATM mutations and cohort B (n = 142) with other DNA repair gene alterations. In both cohorts, median PFS significantly improved with olaparib when compared to the novel hormonal therapy. Clinical benefit was more prominent in cohort A, where the median PFS was 7.39 months with olaparib and 3.55 months with novel hormonal therapy (HR 0.34; 95% CI 0.25–0.47; p < 0.0001). In cohort A, objective response rates were also better with olaparib (33.3%) when compared to enzalutamide/abiraterone (2.3%). The interim overall survival (OS) analysis in cohort A showed a median OS of 18.5 months with olaparib and 15.11 months with novel hormonal therapy; however, statistical significance was not reached at the time of data cutoff. The most common adverse events with olaparib therapy were anemia, nausea, decreased appetite, and fatigue. At the time of data cutoff, there were no reports of myelodysplastic syndrome or leukemia (30).
Combination Studies With Olaparib in Prostate Cancer
Preclinical data have shown potential synergy of PARP inhibitors and androgen receptor antagonists irrespective of HRR status in prostate cancer (34). One preclinical study demonstrated that reduced levels of the tumor suppressor protein CCDC6 led to sensitization of cancer cells to PARP inhibitors (35). The turnover of CCDC6 protein is regulated by the de-ubiquitinase USP7, which also controls androgen receptor (AR) stability. The combination of USP7-inhibitors and PARP inhibitors, by affecting CCDC6 stability and HRR and accelerating AR/ARv7 turnover, may provide a novel therapeutic option in advanced prostate cancer.
In a randomized, placebo-controlled, and double-blind phase II clinical trial, Clarke et al. evaluated the efficacy of olaparib and abiraterone irrespective of HRR mutations in patients with metastatic CRPC (31). Of the 142 patients enrolled, 71 received olaparib, and abiraterone and 71 received placebo and abiraterone. HRR mutation status was not known in all patients. Radiological PFS was 13.8 months with olaparib and 8.2 months with placebo. Time to second progression or death was 23.3 months vs. 18.5 months, respectively. Median OS was 22.7 months in the olaparib arm and 20.9 months in the placebo arm (not statistically significant). However, grade ≥ 3 adverse events and serious adverse events were higher in the olaparib arm when compared to the placebo arm (54% vs. 28% and 34% vs. 18%, respectively). Four treatment-related deaths were noted in olaparib arm (1 each due to pneumonitis, ischemic stroke, cardiac failure, and mediastinitis) (31). To further evaluate this combination therapy, a multicenter, randomized phase III study (NCT03732820, PROpel study) is currently recruiting patients.
Cohort A of the phase Ib Keynote-365 clinical trial is evaluating the safety and antitumor activity of the combination of pembrolizumab (200 mg IV every 3 weeks) along with olaparib (400 mg orally twice daily). Preliminary results were presented at ASCO Genitourinary symposium in 2019 (36). In 41 patients with metastatic CRPC previously treated with chemotherapy and novel hormonal agent, the combination therapy had a median PFS per prostate cancer working group 3 (PCWG-3) RECIST criteria of 5 months and a median OS of 14 months. In this cohort, none of the patients had DNA repair gene defects as detected by either by Guardant360 ctDNA or whole exome sequencing of biopsy tissue. Per RECIST criteria, partial response was seen in 7% and stable disease was noted in 46%. The disease control rate of ≥6 months was seen in 32%. Responses were observed in both PD-L1+ (combined positive score ≥1) and PDL1– patients. The most common treatment-related adverse events were anemia, fatigue, and nausea. There was one treatment-related death. A phase III clinical trial evaluating this combination therapy is currently open for enrollment in unselected metastatic CRPC patients (NCT03834519, KEYLYNK-010 study).
Additional studies evaluating the safety and antitumor activity of olaparib in combination with radioligands such as radium 223 (COMRADE, NCT03317392) and 177Lu-PSMA (NCT03874884), cediranib (VEGFR inhibitor, NCT02893917) and AZD6738 (ATR inhibitor, NCT03787680) are ongoing (Table 2).
Rucaparib
Rucaparib camsylate is an oral, highly selective small molecule inhibitor of PARP1, PARP2, and PARP3. It is currently FDA-approved for the treatment of recurrent epithelial ovarian, fallopian tube, and primary peritoneal cancers (37). Multiple studies are evaluating its role in prostate cancer.
The preliminary results of the industry-funded multicenter, phase II clinical trial (TRITON2) evaluating the antitumor activity of rucaparib were presented at the European Society for Medical Oncology (ESMO) Congress in 2019 (38). In this clinical trial, patients with metastatic CRPC who were previously treated with chemotherapy, androgen receptor antagonists, and had HRR genomic alterations (somatic or germline) were treated with rucaparib 600 mg orally twice daily. An objective response (defined as complete or partial response) of 47.5% (19/40) and a PSA response (≥50% decrease) rate of 53.6% (37/69) was seen in patients with BRCA 1/2 mutations. However, no objective responses were noted in patients with ATM or CDK12 gene alterations. The most common grade ≥3 toxicities were anemia, fatigue, and elevated liver enzymes. No treatment-related deaths were reported. A randomized phase III clinical trial (TRITON3, NCT02975934) is currently comparing the efficacy of rucaparib to either novel hormonal therapy or docetaxel chemotherapy in meteastatic CRPC with HRR deficiency. Another pilot study of 30 patients (TRIUMP, NCT03413995) is evaluating the role of upfront rucaparib as an alternative to androgen deprivation therapy in patients with metastatic castration sensitive prostate cancer who have germline HRR mutations. The combination of rucaparib with the checkpoint inhibitor nivolumab is also currently being evaluated in a phase II clinical trial (CheckMate 9KD, NCT03338790).
Niraparib
Niraparib is another PARP inhibitor currently being explored in the management of prostate cancer. It is a selective inhibitor of PARP1 and PARP2 enzymes involved in DNA repair with a long half-life of 36 h, permitting once-daily dosing. Additionally, niraparib traps PARP1 and PARP2 enzymes with higher potency when compared to other PARP inhibitors, and the trapped PARP-DNA complexes have been demonstrated to be more cytotoxic than unrepaired ssDNA breaks (37). It is the only PARP inhibitor approved by the FDA for maintenance treatment of patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer irrespective of BRCA mutation status (38).
The preliminary results of the phase II, open-label, multicenter, and industry sponsored GALAHAD clinical trial (NCT02854436) were presented at the ESMO congress in 2019 (33). This study is evaluating the safety and efficacy of niraparib 300 mg once daily in patients with metastatic CRPC who have HRR gene mutations and were previously treated with taxane-based chemotherapy and at least one androgen receptor antagonist. To date, of the 81 patients with biallelic HRR gene alterations, 46 have BRCA 1/2 and the rest have other HRR mutations (biallelic mutations in ATM, FANCA, PALB2, CHEK2, BRIP1, or HDAC2). In the BRCA 1/2 cohort, the median radiographic PFS reported to date is 8.2 months, and median OS is 12.6 months. An objective response has been noted in 41% of patients. In the non-BRCA cohort, the median radiologic PFS to date is 5.3 months, and median OS is 14 months. The most common grade 3–4 adverse events are myelosuppression, asthenia, and back pain.
The combination of niraparib and abiraterone has also been studied and niraparib dosing of 200 mg once daily was deemed safe to study in further trials (39). Efficacy data or HRR status not yet been reported. A phase III randomized, blinded study of abiraterone with niraparib (200 mg once daily) or placebo is also currently enrolling patients (MAGNITUDE, NCT03748641). HRR gene mutation status will be used for randomization.
Veliparib
Veliparib, a PARP1 and PARP2 inhibitor, was one of the earliest PARP inhibitors evaluated in combination with abiraterone in patients with metastatic CRPC (40). In a phase II multicenter study, 148 patients stratified by ETS status were randomly assigned to abiraterone alone or abiraterone with veliparib. The study was negative, with no difference between both arms either for PSA response or radiological response. On exploratory biomarker analysis, response rates and radiologic PFS were better in both arms in patients with HRR gene mutations when compared to HRR wild type. Currently, no further studies are planned with veliparib in prostate cancer.
Talazoparib
Talazoparib is an inhibitor of PARP1 and PARP2 enzymes currently FDA-approved for germline BRCA mutated locally advanced, metastatic breast cancer. TALAPRO-1 and−2 studies are currently evaluating the efficacy of talazoparib in metastatic CRPC.
Mechanism of Resistance to Parp Inhibitors
Results of ongoing studies with PARP inhibitors are encouraging, especially with better objective response rates and PFS data. However, despite good initial benefit, these responses are short-term, and patients eventually experience disease progression. It is crucial to understand resistance mechanisms, which will help formulate subsequent treatment strategies. One such mechanism is acquired BRCA2 reversion mutations, where previously BRCA-2-deficient tumor cells are able to achieve BRCA2 proficiency due to constant selection pressure of PARP inhibition (41). Serial sequencing of circulating cell-free (cf) DNA can help monitor these reversion mutations in a non-invasive manner. In addition to this mechanism, the loss of MLL3/4 complex protein and PTIP helps stabilize the replication fork and, in turn, protect DNA from degradation (42). It is also prudent to note the differential response to PARP inhibitors in tumors that harbor BRCA vs. non-BRCA HRR genomic aberrations. In a retrospective study of 23 patients with BRCA 1/2 and ATM mutations, none of the 6 patients with ATM mutations responded to olaparib (43). In this study, the median PFS in patients with BRCA 1/2 was 12.3 months as compared to 2.4 months with ATM mutations. It is postulated that ATM is a DNA damage sensor rather than a mediator of DNA repair—a potential explanation for this differential response. Further studies are needed to understand the primary refractoriness and acquired resistance mechanisms to PARP inhibitors.
Parp Inhibitors in Urothelial Cancer
Bladder cancer is the most common malignancy of urinary system, and urothelial histopathology is the most common subtype in the United States (23). Platinum-based chemotherapy and checkpoint inhibitors are the most common modalities of treatment for advanced, unresectable urothelial cancer. Recently, enfortumab vedotin was granted accelerated approval by the FDA in third-line setting (44). Additionally, for a subset of patients with fibroblast growth factor receptor (FGFR2/3) alterations, erdafitinib was also granted accelerated approval (45).
Molecular characterization of 412 tissue specimens with muscle-invasive bladder cancer in the cancer genome atlas (TCGA) program revealed the frequency of genomic aberrations in DNA repair genes such as ATM, ERCC2, RAD51B to be in the range of 2–14% (46). In another review of 81 muscle-invasive bladder cancer specimens, the somatic mutations in the BRCA 1/2, PALB2, FANCD2, ERCC2, ATM genes were in the range of 3.7–12.3% (47). Additionally, the presence of DDR and repair gene mutations was associated with improved response to platinum-based chemotherapy in metastatic urothelial cancer (48). These findings led to the fruition of several studies evaluating the efficacy of PARP inhibitors in metastatic urothelial carcinoma.
In a phase II, open-label, industry-sponsored study, patients with advanced urothelial cancer who progressed on one or two lines of systemic therapy were treated with rucaparib 600 mg twice daily (49). This study enrolled patients with both HRR deficient and proficient tumors. However, the study was terminated after preliminary review by an independent data monitoring committee did not show adequate objective response rate and met the criteria for study discontinuation. Additional studies evaluating the efficacy of PARP inhibitors in a cohort of urothelial cancers that is selected for HRR deficiency are ongoing (Table 3).
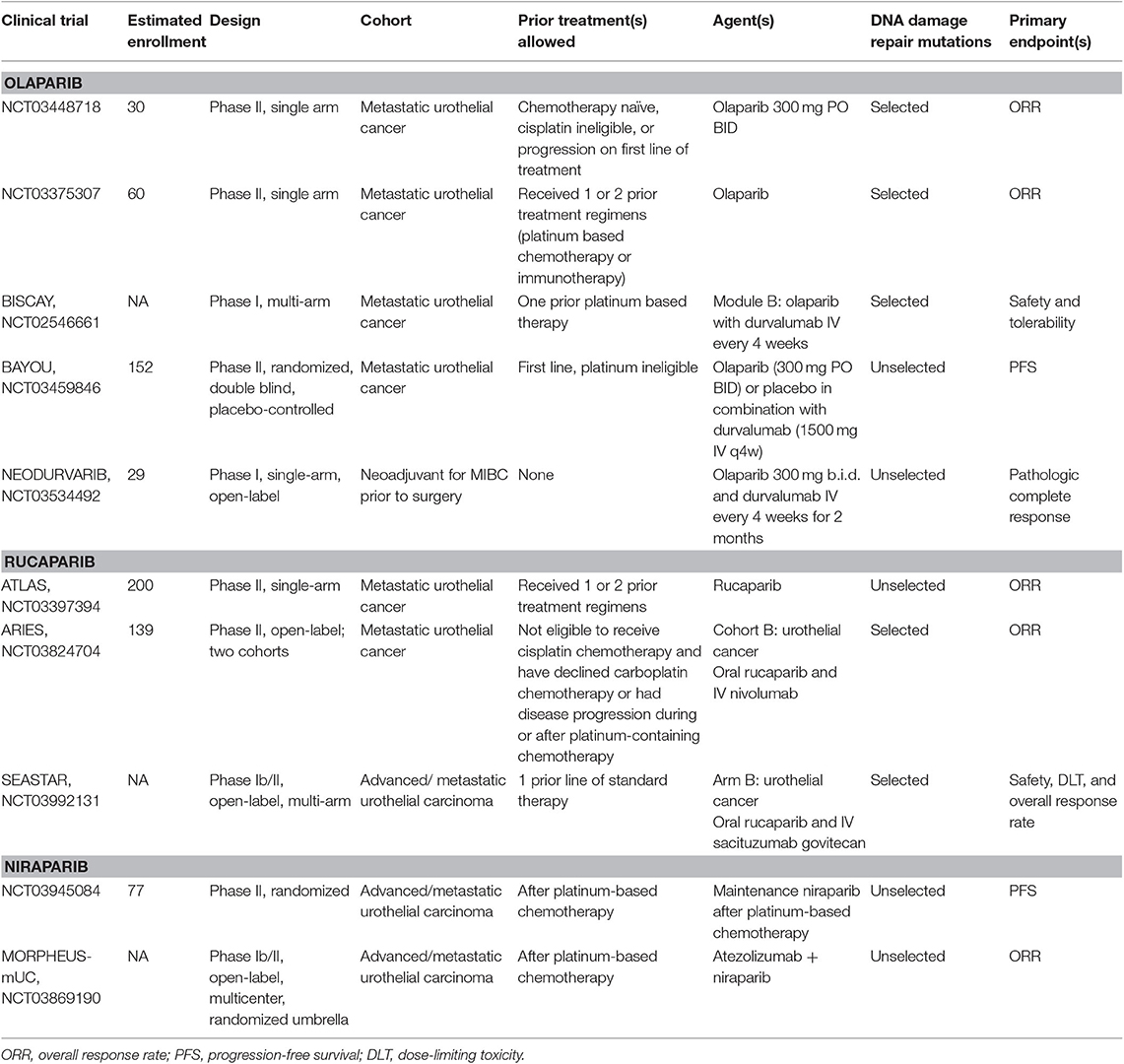
Table 3. Summary of ongoing clinical trials evaluating PARP inhibitors as monotherapy or in combination with other therapeutic agents in urothelial cancer.
Combination Strategies in Urothelial Cancer
In addition to enhancing sensitization to platinum chemotherapy, the presence of DNA repair gene aberrations is also associated with an increase in tumor mutation load and infiltration of lymphocytes in the tumor microenvironment (50, 51). Also, there is an activation of the stimulator of interferon genes (STING) pathway, which leads to enhanced antitumor immune response and PDL1 expression on cancer cells (52, 53). In a retrospective review of patients with urothelial cancer harboring known and unknown deleterious HRR gene mutations, monotherapy with PD1/PDL1 inhibitors showed higher response rates (54). Based on this observation, it is hypothesized that the combination of PARP inhibitors with PD1/PDL1 inhibitors in urothelial cancer can improve antitumor activity and clinical outcomes (55). The preliminary results of a multi-arm, biomarker-driven, adaptive study of durvalumab (anti-PDL1 antibody) in combination with either olaparib or an FGFR1-3 inhibitor (AZD4547) or a TORC 1 and 2 inhibitor (vistusertib) were presented at ESMO congress in 2019 (56). Of 391 screened patients, 54 (14%) had HRR genomic alterations and were treated with olaparib combination therapy. This group had a high tumor mutation burden and a confirmed objective response rate of 35.7%. The 6-month PFS rate was 42%, and 1 year OS rate was 54%. Grades 3/4 treatment-related adverse events were seen in 21.4%.
Summary
In the management of advanced prostate cancer harboring DDR genomic aberrations, multiple PARP inhibitors are currently under study with encouraging results. This is a giant leap toward precision oncology for a cancer that over the past several decades was devoid of predictive therapeutic biomarkers. Similarly, multiple studies evaluating the safety and efficacy of PARP inhibitors in urothelial cancers are up and coming. Precise biomarkers that can accurately predict response to these agents is pivotal for the success of these agents as differential responses are noted in BRCA vs. non-BRCA mutations. Finally, it is prudent to explore the resistance mechanisms to PARP inhibitors by utilizing non-invasive tools such as cfDNA, as this would help the development of subsequent treatment strategies.
Author Contributions
RG: project development and research, manuscript writing, and editing. RV: manuscript writing and editing. YZ: manuscript editing.
Funding
The authors from University of Iowa are supported by National Institutes of Health grant P30 CA086862.
Conflict of Interest
YZ: Advisory Board: Amgen, Roche Diagnostics, Novartis, Jansen, Eisai, Exelixis, Castle Bioscience, Array, Bayer, Pfizer, Clovis, EMD Serono. Grant/research support: institutional clinical trial support from NewLink Genetics, Pfizer, Exelixis, Eisai. DSMC: Jansen.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Abrahams E, Eck SL. Molecular medicine: precision oncology is not an illusion. Nature. (2016) 539:357. doi: 10.1038/539357e
2. Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER, et al. Next-generation characterization of the cancer cell line encyclopedia. Nature. (2019) 569:503–8. doi: 10.1038/s41586-019-1186-3
4. Palmbos PL, Hussain MH. Targeting PARP in prostate cancer: novelty, pitfalls, and promise. Oncology. (2016) 30:377–85.
5. Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. (2012) 481:287–94. doi: 10.1038/nature10760
6. Sachdev E, Tabatabai R, Roy V, Rimel BJ, Mita MM. PARP inhibition in cancer: an update on clinical development. Target Oncol. (2019) 14:657–79. doi: 10.1007/s11523-019-00680-2
7. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. (2009) 461:1071. doi: 10.1038/nature08467
8. d'Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer. (2008) 8:512. doi: 10.1038/nrc2440
9. Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol. (2008) 9:297. doi: 10.1038/nrm2351
10. Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, Utsumi H, et al. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. (1998) 17:5497–508. doi: 10.1093/emboj/17.18.5497
11. de Murcia G, Ménissier-de Murcia J, Schreiber V. Poly(ADP-ribose) polymerase: molecular biological aspects. Bioessays. (1991) 13:455–62. doi: 10.1002/bies.950130905
12. Pellegrini L, Yu DS, Lo T, Anand S, Lee M, Blundell TL, et al. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature. (2002) 420:287–93. doi: 10.1038/nature01230
13. Amé JC, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays. (2004) 26:882–93. doi: 10.1002/bies.20085
14. Feng FY, de Bono JS, Rubin MA, Knudsen KE. Chromatin to clinic: the molecular rationale for PARP1 inhibitor function. Mol Cell. (2015) 58:925–34. doi: 10.1016/j.molcel.2015.04.016
15. Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly (ADP-ribose) and PARPs. Nat Rev Mol Cell Biol. (2012) 13:411. doi: 10.1038/nrm3376
16. O'Connor MJ. Targeting the DNA damage response in cancer. Mol Cell. (2015) 60:547–60. doi: 10.1016/j.molcel.2015.10.040
17. Bouchard VJ, Rouleau M, Poirier GG. PARP-1, a determinant of cell survival in response to DNA damage. Exp Hematol. (2003) 31:446–54. doi: 10.1016/S0301-472X(03)00083-3
18. Hartwell L. Theoretical biology: a robust view of biochemical pathways. Nature. (1997) 387:855. doi: 10.1038/43072
19. Kaelin WG. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. (2005) 5:689. doi: 10.1038/nrc1691
20. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. (2005) 434:917–21. doi: 10.1038/nature03445
21. Ashworth A, Lord CJ. Synthetic lethal therapies for cancer: what's next after PARP inhibitors? Nat Rev Clin Oncol. (2018) 15:564–76. doi: 10.1038/s41571-018-0055-6
22. Nombela P, Lozano R, Aytes A, Mateo J, Olmos D, Castro E. BRCA2 and other DDR genes in prostate cancer. Cancers. (2019) 11:E352. doi: 10.3390/cancers11030352
23. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. (2019) 69:7–34. doi: 10.3322/caac.21551
24. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. (2015) 161:1215–28. doi: 10.1016/j.cell.2015.06.053
25. Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. New Engl J Med. (2016) 375:443–53. doi: 10.1056/NEJMoa1603144
26. Armenia J, Wankowicz SAM, Liu D, Gao J, Kundra R, Reznik E, et al. The long tail of oncogenic drivers in prostate cancer. Nat Genet. (2018) 50:645–51. doi: 10.1038/s41588-018-0078-z
27. Isaacsson Velho P, Silberstein JL, Markowski MC, Luo J, Lotan TL, Isaacs WB, et al. Intraductal/ductal histology and lymphovascular invasion are associated with germline DNA-repair gene mutations in prostate cancer. Prostate. (2018) 78:401–7. doi: 10.1002/pros.23484
28. Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-repair defects and olaparib in metastatic prostate cancer. New Engl J Med. (2015) 373:1697–708. doi: 10.1056/NEJMoa1506859
29. Mateo J, Porta N, Bianchini D, McGovern U, Elliott T, Jones R, et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. (2020) 21:162–74. doi: 10.1016/S1470-2045(19)30684-9
30. Hussain M, Mateo J, Fizazi K, Saad F, Shore ND, Sandhu S, et al. LBA12_PRPROfound: phase III study of olaparib versus enzalutamide or abiraterone for metastatic castration-resistant prostate cancer (mCRPC) with homologous recombination repair (HRR) gene alterations. Ann Oncol. (2019) 30(Suppl_5):mdz394.039. doi: 10.1093/annonc/mdz394.039
31. Clarke N, Wiechno P, Alekseev B, Sala N, Jones R, Kocak I, et al. Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. (2018) 19:975–86. doi: 10.1016/S1470-2045(18)30365-6
32. Abida W, Campbell D, Patnaik A, Sautois B, Shapiro J, Vogelzang NJ, et al. 846PD Preliminary results from the TRITON2 study of rucaparib in patients (pts) with DNA damage repair (DDR)-deficient metastatic castration-resistant prostate cancer (mCRPC): Updated analyses. Ann Oncol. (2019) 30(Suppl_5):mdz248.003. doi: 10.1093/annonc/mdz248.003
33. Smith MR, Sandhu SK, Kelly WK, Scher HI, Efstathiou E, Lara P, et al. LBA50Pre-specified interim analysis of GALAHAD: a phase II study of niraparib in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) and biallelic DNA-repair gene defects (DRD). Ann Oncol. (2019) 30(Suppl_5):mdz394.043. doi: 10.1093/annonc/mdz394.043
34. Asim M, Tarish F, Zecchini HI, Sanjiv K, Gelali E, Massie CE, et al. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat Commun. (2017) 8:374. doi: 10.1038/s41467-017-00393-y
35. Morra F, Merolla F, Napolitano V, Ilardi G, Miro C, Paladino S, et al. The combined effect of USP7 inhibitors and PARP inhibitors in hormone-sensitive and castration-resistant prostate cancer cells. Oncotarget. (2017) 8:31815–29. doi: 10.18632/oncotarget.16463
36. Yu EY, Massard C, Retz M, Tafreshi A, Galceran JC, Hammerer P, et al. Keynote-365 cohort a: pembrolizumab (pembro) plus olaparib in docetaxel-pretreated patients (pts) with metastatic castrate-resistant prostate cancer (mCRPC). J Clin Oncol. (2019) 37(7_suppl):145. doi: 10.1200/JCO.2019.37.7_suppl.145
37. Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. (2012) 72:5588–99. doi: 10.1158/0008-5472.CAN-12-2753
38. Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. (2016) 375:2154–64. doi: 10.1056/NEJMoa1611310
39. Saad F, Zhao X, Rezazadeh A, Chi KN, Shore N, Graff JN, et al. 834PInterim results of a phase Ib study of niraparib plus androgen receptor-targeted therapy in men with metastatic castration-resistant prostate cancer. Ann Oncol. (2018) 29(suppl_8):mdy284.043. doi: 10.1093/annonc/mdy284.043
40. Hussain M, Daignault-Newton S, Twardowski PW, Albany C, Stein MN, Kunju LP, et al. Targeting androgen receptor and DNA repair in metastatic castration-resistant prostate cancer: results from NCI 9012. J Clin Oncol. (2017) 36:991–9. doi: 10.1200/JCO.2017.75.7310
41. Quigley D, Alumkal JJ, Wyatt AW, Kothari V, Foye A, Lloyd P, et al. Analysis of circulating cell-free DNA identifies multiclonal heterogeneity of BRCA2 reversion mutations associated with resistance to PARP inhibitors. Cancer Disc. (2017) 7:999–1005. doi: 10.1158/2159-8290.CD-17-0146
42. Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. (2016) 535:382–7. doi: 10.1038/nature18325
43. Marshall CH, Sokolova AO, McNatty AL, Cheng HH, Eisenberger MA, Bryce AH, et al. Differential response to olaparib treatment among men with metastatic castration-resistant prostate cancer harboring BRCA1 or BRCA2 versus ATM mutations. Eur Urol. (2019) 76:452–8. doi: 10.1016/j.eururo.2019.02.002
44. Petrylak DP, Balar AV, O'Donnell PH, McGregor BA, Heath EI, Yu EY, et al. EV-201: results of enfortumab vedotin monotherapy for locally advanced or metastatic urothelial cancer previously treated with platinum and immune checkpoint inhibitors. J Clin Oncol. (2019) 37(18_suppl):4505. doi: 10.1200/JCO.2019.37.18_suppl.LBA4505
45. Loriot Y, Necchi A, Park SH, Garcia-Donas J, Huddart R, Burgess E, et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N Engl J Med. (2019) 381:338–48. doi: 10.1056/NEJMoa1817323
46. Robertson AG, Kim J, Al-Ahmadie H, Bellmunt J, Guo G, Cherniack AD, et al. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell. (2017) 171:540–56.e525. doi: 10.1016/j.cell.2017.09.007
47. Yap KL, Kiyotani K, Tamura K, Antic T, Jang M, Montoya M, et al. Whole-exome sequencing of muscle-invasive bladder cancer identifies recurrent mutations of UNC5C and prognostic importance of DNA repair gene mutations on survival. Clin Cancer Res. (2014) 20:6605–17. doi: 10.1158/1078-0432.CCR-14-0257
48. Teo MY, Bambury RM, Zabor EC, Jordan E, Al-Ahmadie H, Boyd ME, et al. DNA damage response and repair gene alterations are associated with improved survival in patients with platinum-treated advanced urothelial carcinoma. Clin Cancer Res. (2017) 23:3610–8. doi: 10.1158/1078-0432.CCR-16-2520
49. Clovis Oncology, Inc. Available online at: https://www.sec.gov/Archives/edgar/data/1466301/000119312519105182/d719637d8k.htm
50. Galsky MD, Uzilov AV, McBride RB, Wang H, Patel VG, Sfakianos J, et al. DNA damage response (DDR) gene mutations (mut), mut load, and sensitivity to chemotherapy plus immune checkpoint blockade in urothelial cancer (UC). J Clin Oncol. (2017) 35(6_suppl):300. doi: 10.1200/JCO.2017.35.6_suppl.300
51. McBride RB, Patel VG, Lorduy AC, Castillo-Martin M, Cha EK, Berger MF, et al. Prognostic significance of DNA damage repair (DDR) mutations in patients with urothelial carcinoma (UC) and associations with tumor infiltrating lymphocytes (TILs). J Clin Oncol. (2016) 34(15_suppl):4538. doi: 10.1200/JCO.2016.34.15_suppl.4538
52. Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. (2015) 15:760–70. doi: 10.1038/nri3921
53. Parkes EE, Walker SM, Taggart LE, McCabe N, Knight LA, Wilkinson R, et al. Activation of STING-dependent innate immune signaling by S-Phase-Specific DNA damage in breast cancer. J Natl Cancer Inst. (2016) 109. doi: 10.1093/jnci/djw199
54. Teo MY, Seier K, Ostrovnaya I, Regazzi AM, Kania BE, Moran MM, et al. Alterations in DNA damage response and repair genes as potential marker of clinical benefit from PD-1/PD-L1 blockade in advanced urothelial cancers. J Clin Oncol. (2018) 36:1685–94. doi: 10.1200/JCO.2017.75.7740
55. Criscuolo D, Morra F, Giannella R, Visconti R, Cerrato A, Celetti A. New combinatorial strategies to improve the PARP inhibitors efficacy in the urothelial bladder Cancer treatment. J Exp Clin Cancer Res. (2019) 38:91. doi: 10.1186/s13046-019-1089-z
56. Michael Lattanzi M. ESMO 2019: An Adaptive, Biomarker-Directed Platform Study With Durvalumab in Combination With Targeted Therapies in Metastatic Urothelial Cancer (BISCAY). (2019). Available online at: https://www.urotoday.com/conference-highlights/esmo-2019/esmo-2019-bladder-cancer/115240-esmo-2019-an-adaptive-biomarker-directed-platform-study-with-durvalumab-in-combination-with-targeted-therapies-in-metastatic-urothelial-cancer-biscay.html (accessed October 9, 2019).
Keywords: prosate cancer, urothelial cancer (UC), PARP inhibitor, precision oncology, DNA damage repair genes
Citation: Garje R, Vaddepally RK and Zakharia Y (2020) PARP Inhibitors in Prostate and Urothelial Cancers. Front. Oncol. 10:114. doi: 10.3389/fonc.2020.00114
Received: 13 November 2019; Accepted: 22 January 2020;
Published: 07 February 2020.
Edited by:
Marijo Bilusic, National Institutes of Health (NIH), United StatesReviewed by:
Joseph W. Kim, Yale University, United StatesAngela Celetti, Italian National Research Council, Italy
Copyright © 2020 Garje, Vaddepally and Zakharia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rohan Garje, cm9oYW4tZ2FyamVAdWlvd2EuZWR1