 Jingyu Wang
Jingyu Wang Fang Shen
Fang Shen Ying Yao
Ying Yao Lin-lin Wang
Lin-lin Wang Yongjian Zhu
Yongjian Zhu Jue Hu
Jue Hu- 1School of Basic Medical Sciences and Forensic Medicine, Hangzhou Medical College, Hangzhou, China
- 2Department of Neurosurgery, Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
- 3Department of Orthopaedic Surgery's Spine Division, The Affiliated Hospital of Medical School of Ningbo University, Ningbo, China
- 4Department of Basic Medicine Sciences, School of Medicine, Zhejiang University, Hangzhou, China
Glioblastoma multiforme (GBM) is the most common primary brain tumor in adults with very poor prognosis and few advances in its treatment. Recently, fast-growing cancer immunotherapy provides a glimmer of hope for GBM treatment. Adoptive cell therapy (ACT) aims at infusing immune cells with direct anti-tumor activity, including tumor-infiltrating lymphocyte (TIL) transfer and genetically engineered T cells transfer. For example, complete regressions in patients with melanoma and refractory lymphoma have been shown by using naturally tumor-reactive T cells and genetically engineered T cells expressing the chimeric anti-CD19 receptor, respectively. Recently, the administration of ACT showed therapeutic potentials for GBM treatment as well. In this review, we summarize the success of ACT in the treatment of cancer and provide approaches to overcome some challenges of ACT to allow its adoption for GBM treatment.
Introduction
Gliomas, including astrocytoma, oligodendroglioma, mixed glioma, medulloblastoma, and ependymoma, are the most common primary central nervous system (CNS) tumors that arise from glial or its precursor cells (1). Glioblastoma multiforme (GBM), the highest grade (WHO IV) astrocytoma, is the most prevalent type in adults. It has been investigated that more than 11,000 individuals suffered from GBM each year in the United States. In the last 30 years, survival rates for patients with GBM have improved very little. Despite aggressive standard therapies (maximal safe surgical resection, radiation, and temozolomide), outcomes for patients with newly diagnosed GBM remain dismal. The median survival of GBM is fewer than 20 months and a 5-year survival rate is merely 4–5% (2–5). Moreover, treatments for GBM are among the costliest with the least return, bringing a significant burden to society.
Over the last decade, emerging immunotherapy aimed at improving specific immune response against tumor cells has brought a glimmer of hope to patients with GBM. Generally, immunotherapy can be divided into four aspects, including monoclonal antibodies (mAb) to the inhibitory immune checkpoint molecules, oncolytic virus therapy, adoptive cell therapy (ACT), and cellular vaccines therapy (6–9).
The immune inhibitory molecules such as cytotoxic T lymphocyte-4 (CTLA4) and programmed death 1 (PD-1) are expressed on the surfaces of T cells. When bounding by their ligands expressed on tumor cells or macrophages, these molecules inhibit T cell's activation and proliferation, resulting in tumor immune escape (10). Nowadays, anti-PD-1/PD-L1 therapy has become a routine treatment option for patients with tumors highly expressing PD-L1, such as lung cancer and melanoma. High expression of PD-L1 has also been identified in GBM, which accounts for approximately 50% of newly diagnosed GBM and 45% of recurrent GBM, respectively. Patients with PD-L1 expression are predicted to have a worse prognosis, suggesting anti-PD1/PDL-1 is a potential GBM therapy target (11, 12). However, in a phase 3 clinical trial (NCT02017717), patients with recurrent GBM received nivolumab (anti-PD1 immunotherapy) showed no notably difference in overall survival (OS) compared with another group who treated with bevacizumab (an anti-VEGF therapy) (13). It may be due to the relatively low mutant load, few T cells' infiltration, and severe immunosuppressive microenvironment in GBM. Additionally, exclusively using anti-PD-1/PDL-1 will cause the activation of other inhibitory signals such as T cell immunoglobulin mucin-domain containing-3 (Tim3), lymphocyte activation gene 3 (LAG3), and CTLA4, becoming another approach of immune escape (14). A combination of immune checkpoint inhibition has shown anti-tumor response and promoted survival in animal models with GBM, whereas more clinical trials are needed to prove the efficacy and safety of immune checkpoint inhibitors treatment (15, 16). Certainly, blood-brain barrier (BBB) obstructed antibodies entry into brain, which should be further resolved.
Oncolytic Viruses (OVs) are a group of viruses with the ability to specifically infecting tumor cells and inducing tumor lysis. Recent clinical trials revealed OVs therapy, including using recombinant adenovirus DNX-2401, polio-rhinovirus chimera, and parvovirus H-1, was able to prolong the survival of patients with GBM (>30 months of survival after treatment) (17). However, valid viral spread and replication can be resisted via cancer stem cells and innate immune cells that occur in the GBM microenvironment (18).
Tumor vaccines therapy is aimed at stimulating patients' immune systems to produce tumor-specific immune cells by transferring tumor-associated antigens. Dendritic cells (DCs) can be pulsed with a wide variety of tumor-specific antigen sources (synthetic peptides or autologous tumor lysate). After binding with MHC molecules, these antigens can be presented on DCs' surfaced to stimulate the response of T cells. Injection of DCs-based vaccine into patients with GBM can induce intracranial T-cell infiltration and anti-tumor effects (19). A clinical trial revealed 41% of patients suffered from GBM exhibited cytokine responses and survived at least 2 years after injecting autologous DC pulsed with tumor lysate (20). Moreover, vaccines combined with an adjuvant such as toll-like receptor agonists can boost continuous immune responses (21).
Adoptive cell therapy (ACT), including tumor-infiltrates lymphocytes (TILs) transfer and genetically engineered T cells transfer, is one of the most significant breakthroughs in the field of immune-oncology. Chimeric antigen receptor (CAR) engineered autologous T cells have produced sustained remissions in refractory lymphomas, but it needs further study in the treatment of solid tumors (22–24). Adoptive transfer of mutation-reactive TILs has led to durable regression in cancer such as breast cancer, lung cancer, and melanoma (25–27). In this review, we will review and focus on GBM targeted ACT.
Current Therapeutic Strategy for Glioblastoma
Over the last 15 years, the Stupp protocol, which is maximum safe tumor resection, followed by radiotherapy (RT) and temozolomide (TMZ) chemotherapy, has become the current standard first-line therapy for adult patients with newly diagnosed GBM. It has been shown that this regimen led to a median OS of 14.6 months in the combined therapy group (RT-TMZ) vs. 12.1 months in the RT only group. Methylation of the MGMT promoter was the predictor for a better outcome (28).
In addition, a new electric-physical cancer treatment modality (TTFields) was proved by the FDA in 2015 for GBM patients. A phase III trial has indicated that a better median OS occurs in GBM patients who received TTFields on the basis of standard therapy (29). However, the incremental cost-effectiveness ratio for these patients with tumor-treating fields (TTF) therapy is e549 909 per life-years gained, bringing a significant burden to the family (30). Unfortunately, the great majority of trials have revealed that more than 100 different molecularly targeted drugs showed little efficacy but increased risks of adverse events for newly diagnosed or recurrent GBM patients (31, 32). In view of the limited efficacy of standard therapy and a lack of effective molecularly targeted drugs, there is a strong interest in the development of personalized immunotherapy such as ACT.
Adoptive Cell Therapy
TIL Transfer
Adoptive cell therapy was initially pointed out by Rosenberg in 1982. He discovered that the administration of immune lymphocytes expanded in IL-2 was able to cure mice with subcutaneous lymphomas (33). Meanwhile, it has been demonstrated that the administration of a high dose of IL-2 after lymphocytes' transfer enhanced the therapeutic efficacy of ACT (34). In addition, the sustainable regression of established liver and lung tumors was mediated via TILs transfer (35). In melanoma, researches revealed that human TILs could recognize autologous tumors, and the administration of these TILs led to the regression of melanoma (36). A subsequent trial has shown that 21 of 43 patients underwent objective regressions of metastatic melanoma (37). Moreover, the regressions of brain metastases (13 of 17 patients) have also been observed, suggesting that TILs can pass the blood-brain barrier and infiltrate into brain tumors (38). In these trials, 22% of patients had complete regressions of melanoma and 20% of patients did not have recurrences 5–10 years after TILs transfer (25). In addition, 55% of patients from the NCI, 48% of patients from the MD Anderson Cancer Center, 38% of patients from the Moffitt Cancer Center, and 40% of patients from the Ella Cancer Institute have shown an objective response to TILs transfer (39–41).
Theoretically, whole-exome sequencing has confirmed that TILs amplified from melanoma tissues could recognize non-synonymous cancer gene mutation products. For example, 25-amino acid polypeptides containing mutated amino acid in the middle were established and presented on the antigen-presenting cell (APC) surface to identify the immunogenic mutations (42). But just little mutations were recognized by TILs, especially in gastrointestinal and breast cancers (27, 42, 43). A trail has identified and purified KRAS (G12D)-reactive T cells from TILs cultured from one patient with metastatic colorectal cancer. And objective regression of all lung metastases was observed after the infusion of these T cells, but progression occurred in a lesion 9 months after treatment, which was due to the absence of HLA-C*08:02 (26). Moreover, a recent trial has revealed complete durable regression of chemorefractory hormone receptor (HR)-positive metastatic breast cancer in a patient after adoptive transfer of mutant-specific TILs (reactive against SLC3A2, KIAA0368, CADPS2, and CTSB) combined with IL-2 and PD-1 treatment, which provides a new individualized immunotherapy approach for cancer (27).
There was a pilot study demonstrated that the delivery of autologous TILs and IL-2 was effective for GBM patients (44). In this research, six patients underwent surgery followed by infusion of TILs and IL-2 in conjunction with chemotherapy. Three of these patients were diagnosed with anaplastic astrocytoma, 1 of these patients had a complete regression after 45 months, and 2 had a partial regression. Of other patients diagnosed with GBM, 2 had a partial regression. Dillman's group demonstrated that GBM patients who treated with lymphokine-activated killer (LAK) cells had a better prognosis compared with contemporary GBM patients (45). Subsequently, a further clinical trial (NCT00331526) was performed to explore the effect of LAK for GBM. In this study, 33 patients were received LAK cells with surgery and radiation therapy. The median survival of these patients from diagnosis of GBM was 20.5 months (46). These results supported the potential of TIL immunotherapy in GBM. But the outcome of these trials was not optimistic, which may be due to the therapeutic cells that were amplified from PBMCs. Thus, there may be low ratios of therapeutic cells that can attack tumor. Identification and purification of tumor-reactive TILs may be the key to the therapeutic success of TILs transfer in GBM. Theoretically, TILs isolated from GBM specimens possess the characteristics of diversity, divergence, and exhaustion, which indicate GBM might be recognized by these TILs (47, 48). And further studies have demonstrated that TILs cultured from patients' GBM could react against tumor cells (49, 50). In addition, exhausted molecules appear to be highly expressed in amplified TILs, and immune checkpoint therapy may promote the antitumor efficacy of TIL transfer therapy (27).
The latest protocol for transferring tumor-specific human TILs is shown in Figure 1A. Briefly, the resected tumor specimen is divided into ~1–2 mm fragments and then cultures individually in a high concentration of IL-2 (and IL-15, IL-21 if necessary). After 2-4 weeks of culture, lymphocytes overgrow, and are tested for reactivity against tumor-associated mutations. Then these selected TILs were sorted and rapidly expanded using irradiated allogenic PBMC and OKT3 antibody. After 1–2 weeks' amplification, up to 1011 TILs are collected for infusing into patients (51). lymphodepleting preparative regimen is necessary for patients before TILs transfer, which promotes cell persistence and durable immunoreaction. The general lymphodepleting preparative regimen includes the administration of 5 mg/m2 fludarabine for 5 days and 60 mg/kg cyclophosphamide for 2 days before cell infusion. After infusion, IL-2 is given at the dose of 720,000 IU/kg to induce tolerance (37). The administration of immune checkpoint agents may also promote cell persistence. However, the above protocol is time- and money-consuming, which needs a 2–3 months processing time after tumor resection with a low success rate. Some patients may die during the production of tumor-specific TILs. Therefore, simplification of processing protocol may be the research priority in the future. Recent researches have shown that CD39+ CD8+ T cells in the tumor microenvironment are tumor-reactive (52, 53). Therefore, we hypothesize that directly amplifying CD39+ CD8+ T cells sorted from solid tumors and infusing these T cells into patients may be a valid and more timesaving approach. The modified process is shown in Figure 1B, and, of course, it needs further study to test its efficacy and safety. Additionally, more molecular markers such as CD39 should be unveiled to facilitate the identification of tumor-reactive TILs.
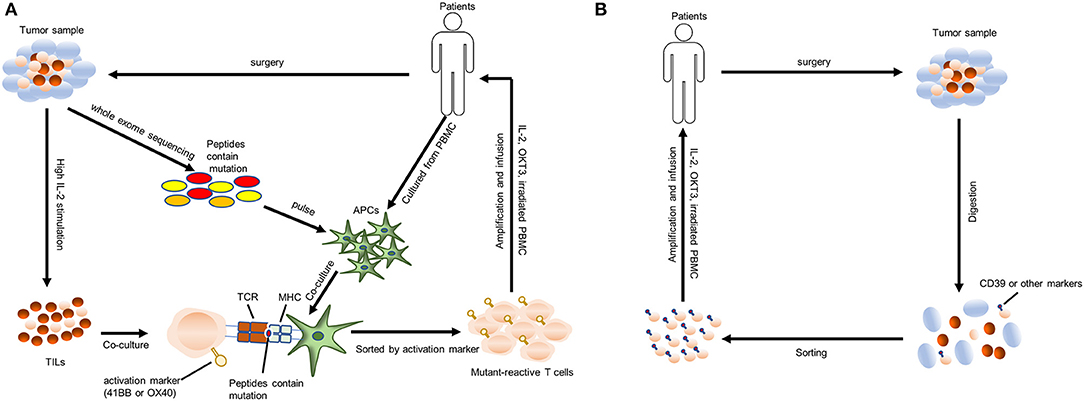
Figure 1. Protocols for identifying and transferring tumor-reactive T cells. (A) A “blueprint” for the therapy of patients using autologous T cells that recognize tumor-specific mutations. TILs, tumor-infiltrating lymphocytes; APCs, antigen-presenting cells; MHC, major histocompatibility complex; TCR, T cell receptor; PBMC, peripheral blood mononuclear cell. (B) A “blueprint” for the therapy of patients with autologous T cells recognizing tumor cells that were sorted by chronic activation markers such as CD39.
Genetically Engineered T Cells Transfer
CAR-T Cell Therapy
CARs are generally composed of ectodomain (antigen recognition domain), transmembrane (TM) domain and an intracellular domain (signaling domain). And they are genetically expressed on the surface of T cells, allowing these T cells to directly recognize the tumor-associated antigen (TAA) independent of major histocompatibility complex (MHC) molecules. The ectodomain consists of a single-chain variable fragment (scFv) of a monoclonal antibody (MAb) that is specific for TAA or a ligand if the TAA is a cell surface receptor. The intracellular domain of first-generation CARs incorporates CD3ζ chain to mediate the activation of T cells. Second-generation CARs contain an additional intracellular domain of CD28, a co-stimulatory molecule. And third-generation CARs contain both CD28 and a tumor necrosis factor receptor family member such as CD 27, CD137 (4-1BB), ICOS, CD134 (OX40), or CD244, promoting the proliferation and persistence of T cells. More recently, fourth-generation CAR-T cells, also called “TRUCK” T cells, provided with stimulatory cytokines including IL-12, IL-15, IL-18 that antagonize the immunosuppressive tumor environment (Figure 2A) (54–58). Nowadays, CD28 combined with CD137 is the most commonly used co-stimulatory domain (59). CD28 signaling induces effector memory differentiation and CD137 signaling maintains a central memory phenotype. However, there is no direct clinical evidence supporting the superiority of CD28 combined with CD137 over other combinations.
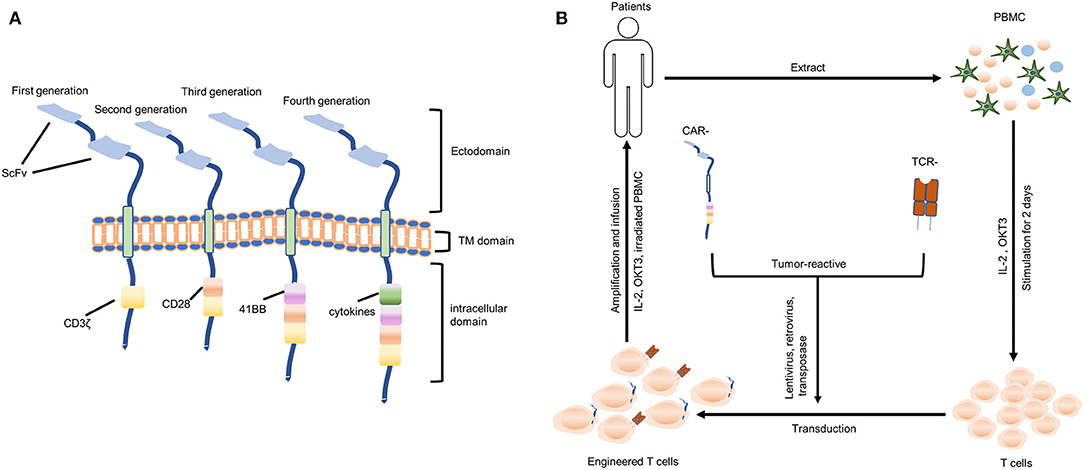
Figure 2. Genetically engineered T cells (CAR- and TCR-) transfer. (A) Structures of four generations of CAR-T cells. ScFv, single-chain variable fragment; TM, transmembrane. (B) A “blueprint” for the therapy of patients using autologous genetically engineered T cells (CAR T-cells or TCR T-cells).
Clinical trials involved administration of anti-CD19 CAR gene therapy in humans have achieved unprecedented success in refractory lymphoma. Infusion of anti-CD19 autologous CAR T-cells produced complete regression in patients with refractory lymphoma who maintains progression-free 4 years after two cycles of therapy (60). Anti-CD19 CAR T-cell therapy can lead to B cell exhaustion, which can be overcome via the periodic immunoglobulin infusions. However, solid tumors typically lack shared or specific surface antigens and therefore, there is the limited application of CAR T-cell therapy in them.
Nevertheless, there have been multiple trials of CAR T-cell therapy for GBM. Human epidermal growth factor receptor 2 (HER2) is overexpressed in some GBM. And a phase I trial (NCT01109095) has revealed that 17 patients with progressive GBM have no severe adverse events after the administration of autologous HER2-CAR T cells. And 8 of which patients had a clinical benefit with a median OS of 24.5 months (61). Moreover, it has been shown that there is an epidermal growth factor receptor (EGFR) overexpression in GBM. And EGFR variant 3 (EGFRvIII) is a frequently occurring mutation in GBM (62). Administration of CAR-T cells targeting EGFRvIII led to regression of GBM in the animal model, while it has been failed in a pilot trial (NCT01454596) (63–65). The loss of EGFRvIII in recurrent tumors may account for this failure, and EGFRvIII may not be the driven mutation in GBM (64). In addition, a recent clinical trial (NCT02208362) showed that the regression of all intracranial and spinal lesions was observed in a patient with recurrent multifocal GBM who received CAR-T cells targeting the interleukin-13 receptor alpha 2 (IL13Rα2) (66). The clinical response continued for 7.5 months until tumors eventually recurred at four new locations in the spinal cord, and there was low IL13Rα2 expression in these new lesions. Above trials have demonstrated the limited clinical benefits obtained from single-epitope CAR-T cell therapy. Multivalent CAR T-cells may overcome the problem of antigenic variability and immune escape of GBM. Recently, a trivalent CAR T-cell targeting 3 targetable glioma antigens (IL13Rα2, HER2, and ephrin-A2 [EphA2]) was designed, which could recognize almost 100% GBM (67). Moreover, podoplanin (PDPN), CD70, and IL7 Receptor are emerging targets of CAR T-cell therapy for GBM (68–70). In diffuse intrinsic pontine glioma (DIPG) with mutated histone H3 K27M (H3-K27M), disialoganglioside 2 (GD2) is highly expressed, and GD2-targeted CAR T-cells have shown a robust generation of antigen-dependent cytokine, killing of DIPG cells in vitro, and complete regression of DIPG in a mouse model (71). Driven antigens or receptors that are not shared with essential organ/tissue cells are required to be discovered in GBM.
TCR-T Cell Therapy
The T cell receptor (TCR) expressed on the surface of T cells recognizes antigenic peptides with MHC restriction. TCR heterodimer contains two-chains, either αβ or γδ, and αβ TCR is the most commonly used molecule for genetic engineering. The cDNAs for the TCR α and β chains can be sequenced and molecularly cloned from tumor-reactive T cells. The genetic transfer of this TCR is able to produce tumor antigen-specific T cells from autologous T cells. However, direct transfer of α- and β- chains can result in mispairing between endogenous and transgenic TCR-derived α- and β- chains. The mispairment of TCR will not recognize tumor antigens and may even target normal cells. Multiple strategies have been developed to resolve this problem, including replacing human regions with murine C regions, application of small interfering RNA constructs that inhibit the expression of endogenous TCR, TCR chain leucine zipper fusions, and substituting the extra cysteine residues with a disulfide bridge (72–74). It has been discovered that the murine-human hybrid TCRs were able to promote engineered T cells to release cytokine and kill tumor cells compared with pure human TCR (75).
The first application of TCR-transduced T cells targeting MART-1 in patients with metastatic melanoma has been performed in 2006, and 2 of 15 treated patients showed tumor regression (76). And a clinical trial has demonstrated objective cancer regressions occur in 19 and 30% of patients with metastatic melanoma who received the gp100 or MART-1 TCR, respectively, while there was on-target toxicity presented in the eyes, ears, or skin where there was an abundance of normal melanocytes (77). Therefore, the application of TCR T-cell therapy should be careful. Unknown cross-reactive antigens may present in healthy important organs. Meanwhile, even when MAGE-A3 is not previously found in any normal organs, targeting this antigen led to 2 deaths because this TCR recognized a related epitope in MAGE-A12, which is expressed in the brain, and induced necrotizing leukoencephalopathy (78). The available strategy to avoid this reaction was the identification and evaluation of mutant-reactive TCR. A human leukocyte antigen (HLA)-C*08:02-restricted TCR from CD8+ TILs targeting the KRAS(G12D) hotspot driver mutation presented in many different types of human cancers such as lung, pancreatic, and gastrointestinal cancer has been identified (43).
Unfortunately, there is no clinical trial with TCR T-cell therapy for GBM in the present. But recent research has identified a TCR that targeted histone 3 variant 3 K27M (H3.3K27M) mutation that was frequently expressed in DIPG with HLA-A2 restriction. Transfer of this mutant-reactive TCR-transgenic T cells notably inhibited the progression of DIPG xenografts in mice and this provided laboratory evidence for evaluating TCR T-cell therapy targeting specific epitope in GBM, while brain inflammation during treatment should be concerned when translating into clinic (79). The general process of genetically engineered T-cell therapy (CAR- and TCR-) is shown in Figure 2B. And the existing clinical trials relating to ACT for the treatment of GBM are summarized in Table 1.
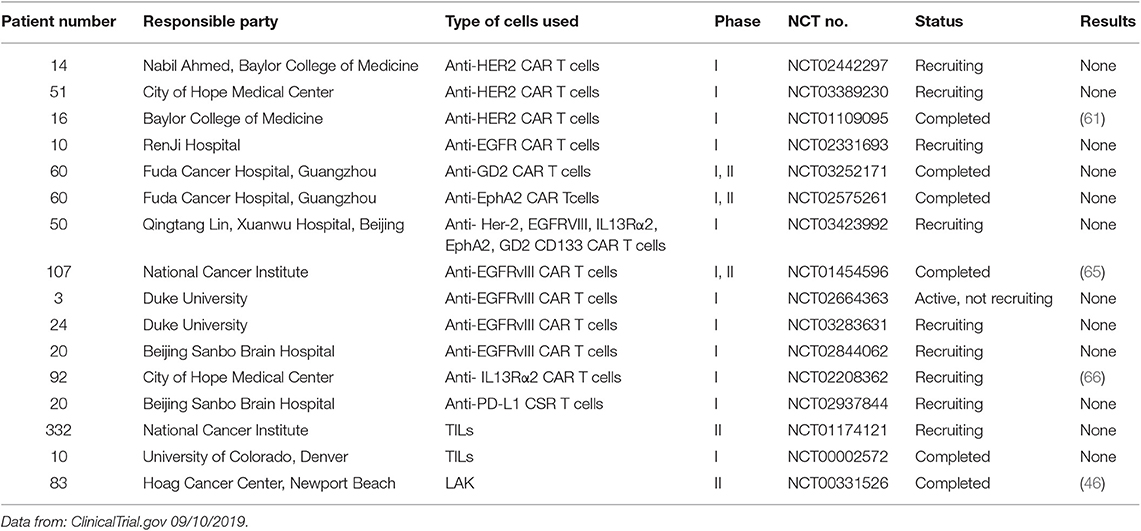
Table 1. Clinical trials of adoptive cell therapy in patients with glioblastoma.
Challenges
ACT depends on the identification of common and specific tumor antigens and has already exhibited prominent anti-tumor activity in cancer patients. The ACT has been successful in the treatment of hematologic malignancies and certain solid cancers such as melanoma, lung cancer, and breast cancer. However, there remain some critical challenges for the application of ACT in GBM.
Recruitment of Infused T Cells Into GBM
Immune cells can be prevented from entering the brain parenchyma via the blood-brain barrier (BBB) under physiological conditions. Only little activated T cells are allowed to reach the brain parenchyma (80, 81). BBB consists of an endothelial basement membrane and a parenchymal basement membrane (81). Unique endothelial cells connected through tight junctions are localized on the endothelial basement membrane, where some embedded pericytes are able to found. Nevertheless, the parenchymal basement membrane is formed via astrocytic end-feet, which is also called glia limitans perivascular.
The process of T cells crossing the inflamed BBB is coordinating, sequential, and complicated, as shown in Figure 3. Briefly, activated T cells initially arresting on the endothelium is mediated by the lymphocyte-associated antigen-1 (LFA-1) and α4β1-integrin expressed on the T cells, respectively binding to the intracellular cell adhesion molecule 1 (ICAM1) and adhesion molecules vascular cell adhesion molecule 1 (VCAM1) on brain endothelial cells (82). Subsequently, the T cell crawling and polarization exclusively involve LFA-1 and ICAM1/2 interactions (83). After arriving at sites where are rich in the laminin isoform α4 but not laminin α5, the T cells use α6β1-integrin to traverse the endothelial basement membrane (81). Matrix metalloproteinases (MMP)-2 and MMP-9 existing in the perivascular space promote T cells to finally cross the glia limitans into the brain parenchyma, which cleave the β-dystroglycan (β-DG, an e extracellular matrix receptor) expressed on the astrocyte end-feed (81).
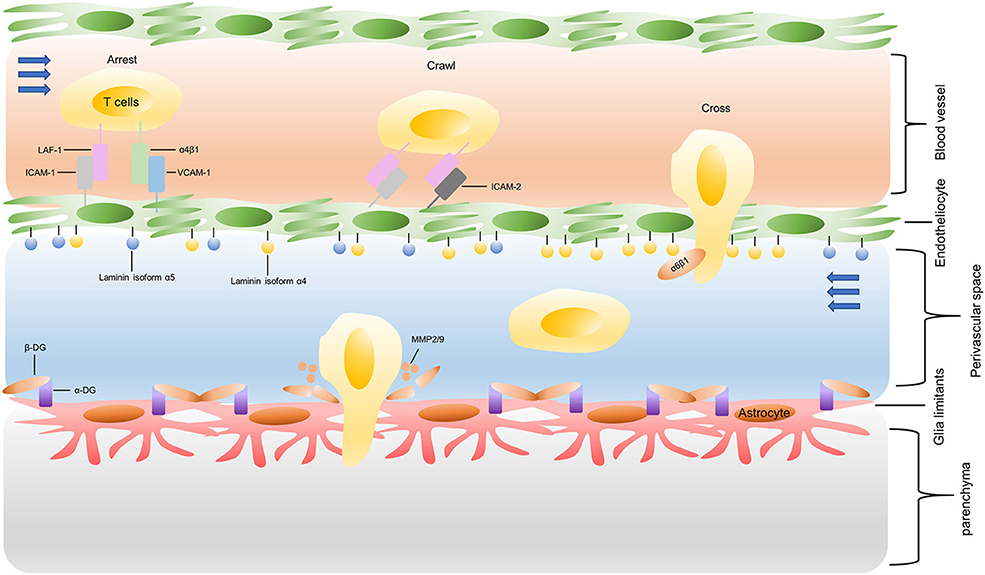
Figure 3. Recruitment of infused T cells into GBM. LAF-1, lymphocyte-associated antigen-1; ICAM-1, intracellular cell adhesion molecule 1; VCAM1, vascular cell adhesion molecule 1; MMP: matrix metalloproteinases; β-DG: β-dystroglycan.
Fortunately, contrast-enhanced MRI of GBM has confirmed BBB was disrupted by these tumors, while BBB is complete in the position where GBM infiltrates into the normal brain parenchyma (84). All of the above illustrate that infused T cells can arrive at GBM, while these T cells need to be activated by APCs and the efficiency of the recruitment is unclear. The route of T cell administration may affect the recruitment of infused T cells into GBM. Safety intracavitary or intratumoral delivery of T cells has been established by several investigators in clinical trials to alleviate systemic toxicity and increase T-cell homing (45, 66). Nevertheless, the intratumoral infusion has technical limitations, especially in the multi-nodulose or deep regional GBM. Additionally, intracavitary delivery of T cells does not equate to effective recruitment into the GBM and transferred T cells still must cross the BBB (85). Importantly, it is indistinct for the persistence of infused T cells about these local injections. The approach of intravenous injection promotes the persistence of transferred T cells (61, 65). A phase 1 clinical trial has shown CAR-modified T cells still exist in the peripheral blood 1 year after the infusion (61). However, it has been found that adoptive T cells initially migrate into irrelevant organs like spleen, lung, and liver, without preferential migration in tumor tissues after intravenous injection (86). Therefore, it is necessary to develop novel strategies assisting the efficient infiltration of infused T cells.
Preferential migration of T cells to tumor tissues can be induced by tumor-associated chemoattractants such as CXCL 9, 10, 11, and CCL2. Adoptive T cells can be transduced with the chemokine receptor for this chemoattractant to promote the preferential migration (87–89). However, these chemoattractants could be inhibited in highly vascular tumors such as GBM due to its capability of overproducing vascular endothelial growth factor (VEGF) (90, 91). Due to CCL7 and CCL22 that attract T regulatory cells are overexpressed in GBM, the transgene of CCR4 into adoptive T cells may improve the infiltration (92, 93). In addition, cancer endothelium overexpresses activated leukocyte cell adhesion molecule (ALCAM) in GBM. And a research group has created an ALCAM-restricted homing system (HS) to support brain cancers capture T cells (94). On the other hand, strategies designed to destroy the BBB need to be tested in the ACT, such as Focused Ultrasound (FUS), HAV6 peptide, and glutamate (95–97).
Identification of GBM-Specific Antigens, TILs, and TCRs
For designing CAR-T cells, it is critical to select antigens shared with nonessential normal organs or those specific for GBM. IL13Rα2, HER2, EphA2, and EGFR have been identified as targets for GBM (Figure 4A). Although clinical trials have shown there are no severe adverse events after the administration of above CAR-T therapies, these markers may be expressed in vital organs such as lung and heart (61, 66). To overcome this potential risk, mutational antigens can be a good option, but the GBM as a type of “cold cancer” lacks pervasive mutations and it is difficult to design a CAR targeting mutations with high affinity. Importantly, some critical mutations such as isocitrate dehydrogenase (IDH) 1 (R132C/G) and BRAF (V600E) located in the cytoplasm are unable to be designed as a target for CAR-T cell therapy, which is able to be recognized by TCRs. Exploring these TCRs restricted by common HLA can be the focus of future research. Of cause, driven mutations in GBM should be detected. On the other hand, directly sorting and amplifying tumor-reactive TILs facilitate the process of ACT. Despite CD39 has been proposed as a tumor-reactive marker in CD8+ TILs, it needs to be further confirmed in GBM. And other markers that indicate tumor reactivity in TILs also need to be discovered. Nevertheless, the lack of tumor-reactive TILs in GBM hinders the application of TILs transfer and identification of tumor-specific TCRs. This may be resolved by combining it with dendritic vaccine therapy that can stimulate the host to produce tumor-reactive T cells (98, 99).
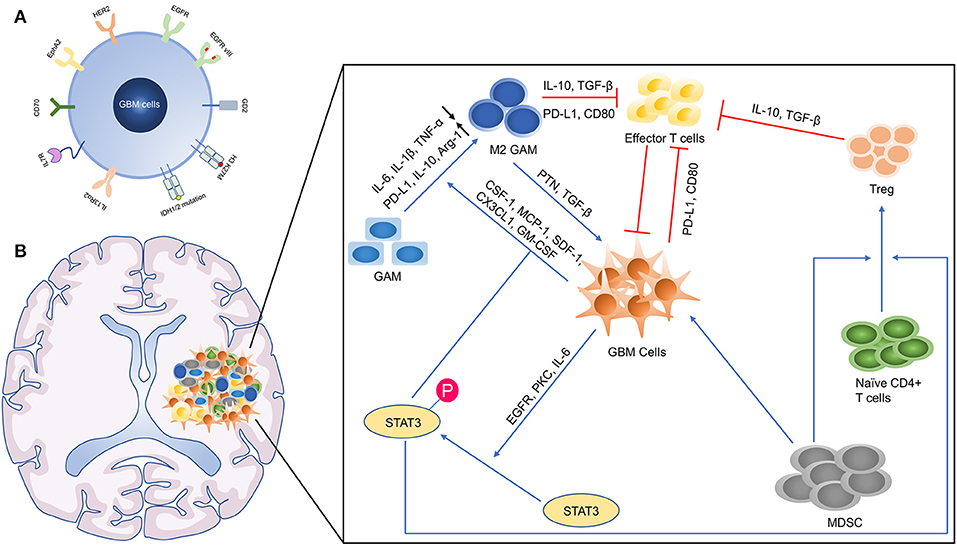
Figure 4. The microenvironmental landscape of GBM. (A) The GBM associated antigens. HER2, Human epidermal growth factor receptor 2; EGFR, epidermal growth factor receptor; GD2, disialoganglioside 2; H3 K27M, mutated histone H3 K27M; IDH, isocitrate dehydrogenase; IL13Rα2, interleukin-13 receptor alpha 2; EphA2, ephrin-A2. (B) The Immunosuppressive GBM microenvironment. TNF-α, tumor necrosis factor α; PD-1, programmed death 1; TGF-β, transforming growth factor-β; Arg-1, arginase 1; GM-CSF, granulocyte-macrophage colony-stimulating factor; MCP-1, monocyte chemoattractant protein 1; SDF-1, stromal cell-derived factor 1; PKC, protein kinase C.
Immunosuppressive GBM Microenvironment
The GBM microenvironment is developed to prevent the cancer cells from the immune attack and promote the growth of them (Figure 4B). Macrophages form the largest part of the GBM infiltrated immune cells (100). Differing remarkably from other types of cancer, macrophages possess of vary origin, including bone marrow-derived monocytes/macrophages (BMDMs) (85%) and microglia (15%). Microglia is prominent in peritumoral regions, while BMDMs recruited from peripheral blood are gathered in perivascular areas. RNA-sequencing display that BMDMs in GBM highly express genes associated with “cellular migration,” while microglia upregulate genes related to “pro-inflammatory cytokines,” which show these two subsets assume different function (101). Similarly, BMDMs and microglia are close to M2 phenotypes, producing anti-inflammatory cytokines and lacking expression of CD40, CD86, and CD80 (T cell co-stimulated molecules) (102, 103). Considering that macrophages are essential accomplices in immunosuppress, it is possible to develop effective therapies to inhibit or switch them. The administration of colony-stimulating factor 1 receptor (CSF-1R) inhibitors aimed at this goal needs to be seen, despite around 50% of animals showed resistance to these inhibitors after treatment (104). The failure of CSF-1R in the treatment of recurrent GBM patients suggests us to pay attention to the heterogeneity of macrophage (105).
In addition, another major subset of immunosuppressive cells is T regulatory (Treg) cells. Circulating T helper cells were decreased in GBM patients, whereas Treg cells were increased in the proportion of Th cells and prominently infiltrate into GBM tissues (106). In a preclinical model, the systemic application of anti-CD25 hinders Treg cell's function but not results in absolute elimination, which promotes the immune response (107). However, oxidative stress-induced apoptotic Treg cells maintain even amplify their immunosuppressive ability, explaining that the sole utilization of anti-CD25 maybe not enough (108). Moreover, immune checkpoint molecules such as CTLA-4 and PD-1 are upregulated as a result of T cell activation, which also occurs in adoptive T cells (109, 110). The combination of ACT and immune checkpoint inhibitors may promote the survival of these T cells, and adoptive T cells can be transduced with a PD-1-CD28 switch receptor that can overcome the inhibitory signaling by checkpoint activation (111).
Furthermore, a mass of abnormal molecular signaling activation involved in immunosuppression occurs in GBM, especially the activator of transcription 3 (STAT3) protein signaling. The phosphorylation of STAT3 at Ser-727 or Tyr-705 has been discovered in up to 90% GBM tissues, relating to the high histopathological grade and worse prognosis (112–114). This activation of STAT3 may not be induced by its intrinsic mutations but by upstream signaling molecules such as EGFR mutations, overexpression of interleukin 6 (IL-6) and protein kinase C (PKC) (115–117). There are certain mechanisms by which STAT3 activation can induce immune tolerance: (i) by increase of M2 like macrophages and microglia (118); (ii) through the recruitment and accumulation of Treg cells (119, 120); and (iii) via the suppression of DCs' maturation (121). The administration of WP1066 (a molecule inhibitor of p-STAT3) reverses immunosuppression in GBM patients and a clinical trial of phase I is on the way (NCT01904123) (122).
Immune-Related Complications Associated With ACT
The ACT is correlated with life-threatening side effects, notably immune effector cell-associated neurotoxicity syndrome (ICANS) and cytokine release syndrome (CRS), which require urgent attention (123). CRS is a systemic and excessive inflammatory response initially caused by adoptive T cells through generating and releasing proinflammatory cytokines (124). These cytokines later activate endotheliocytes and bystander immune cells, formatting the vicious circle of a cytokine storm in a waterfall manner. IL-6 primarily released by macrophages seems to play a central role in CRS (125). Additionally, other cytokines like granulocyte-macrophage colony-stimulating factor (GM-CSF), monocyte chemoattractant protein 1 (MCP-1), tumor necrosis factor (TNF), interferon (IFN)-γ, and IL-1,2,8,10 participate in CRS (124).
Clinically, the symptoms of CRS may be light and limiting, or serious that require vasopressor or ventilator support. Fever is a symbolic symptom, occurring in the early phase with other mild discomforts such as myalgia, rash, headache, and arthralgia (123). Seriously, symptoms will progress to vascular leakage, hypotension, disseminated intravascular coagulation (DIC), and even multiple organ failure (MOF).
The severity of CRS is divided into 4 grades according to the American Society for Transplantation and Cellular Therapy (ASTCT) consensus guidelines (126). CRS with grade 1 is defined as a patient undergoes fevers only. Hypotension without the utilization of vasopressors and/or hypoxia with the administration of low flow oxygen occurs in grade 2 CRS patients. Grade 3 CRS requires the existence of hypotension requiring the support of one vasopressor with/without the administration of vasopressin and/or hypoxia with the use of high flow oxygen. Grade 4 CRS entails the presence of hypotension that needs multiple vasopressors excepting vasopressin and hypoxia that needs positive pressure ventilation systems, representing a life-threatening complication. It is critical to exclude infections and sepsis for the diagnosis of CRS, notably bacterial infections, and the identification of elements predicting severe CRS is developing (124, 127). Tocilizumab (an antibody against humanized IL-6 receptor) that is recommended with a maximum of 800 mg per dose and a maximum of 3 doses in 24 h and corticosteroids are the core of CRS therapy (123).
ICANS is another one of the most frequent complications in patients treated with ACT. Presently, the pathophysiology of ICANS is barely understood, while it is demonstrated that the severity of ICANS may be aggravated by a severe CRS or a high cancer burden (128). In addition, the activation of endotheliocytes and the disruption of BBB possibly lead to ICANS (129). High cytokines' levels such as GM-CSF, IL-6 present in the cerebrospinal fluid (CSF) of patients treated with ACT, resulting in brain inflammation (128). The symptoms of ICANS are more diverse than CRS, including headache, dysgraphia, aphasia, tremor, lethargy, impaired attention, and apraxia. Relatively, expressive aphasia is possibly one of the most specific symptoms in patients with ICANS (128, 130).
Similarly, the symptoms of ICANS are divided into 4 grades as well by ASTCT depending on Immune Effector Cell-Associated Encephalopathy (ICE) score (126). The preferred therapy for patients with ICANS is corticosteroids relying on the ICANS Consensus Grading. The recommended dose of dexamethasone is 10 mg per 6 h until clinical cure. However, patients with grade 4 ICANS require 1,000 mg methylprednisolone per 24 h (123, 128). Additionally, tocilizumab is absolutely recommended for patients with concurrent CRS (128).
Fortunately, present clinical trials confirm that there is no severe side-effect during the administration of ACT for GBM patients (44, 45, 61, 65, 66). It is a consensus that lymphodepletion is required before the begin of ACT by using fludarabine and cyclophosphamide to promote the persistence of infused T cells. Nevertheless, TMZ is a portion of the present therapeutic strategy for patients with GBM, which can also induce lymphopenia (131). Therefore, TMZ is supposed to be used as an inducer of lymphodepleting replacing fludarabine and cyclophosphamide (132). The pretreatment of dose-intensified TMZ induces durable lymphodepleting, whereas standard dose was transient (132). This replacement reduces the drug burden for patients who receive ACT, which entails cautious trails to interpret the dose. In addition, a high dose of IL-2 is needed for the persistence of T cells. However, the administration of IL-2 appears to lead to discomfort and even patients cannot tolerate it. It has been revealed that adoptive T cells engineered to express IL-12 were able to survive in the absence of IL-2 while they cannot stay long-term in the host (133).
Conclusion
To conclude, ACT is a highly personalized therapy that possesses a significant potential for the treatment of different types of cancers. GBM is unique in terms of its characteristics like low immunogenicity, immunosuppression, and exclusive location, all of which make it particularly difficult to treat. ACT can be a potential therapy for the treatment of GBM. Further researches are necessary to solve the above challenges and a combination of several immunotherapies may prove to be a solution.
Author Contributions
All authors participated in the preparation of this manuscript. JH and YZ were responsible for determining the topic. YZ, JW, FS, and YY collected data. JW, FS, and JH contributed to writing the first draft of this article. LW was responsible for furtherly editing the manuscript.
Funding
This review was supported by the Provincial Administration of Traditional Chinese Medicine of Zhejiang (No. 2018ZZ015), Natural Science Foundation of Zhejiang (No. Y19H170008), and National Natural Science Foundation of China (No. 81673777).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
GBM, glioblastoma multiforme; ACT, adoptive cell therapy; TIL, tumor-infiltrating lymphocyte; CNS, central nervous system; TTF, tumor-treating fields; mAB, monoclonal antibodies; CTLA4, cytotoxic T lymphocyte-4; PD-1, programmed death 1; OVs, oncolytic viruses; DCs, dendritic cells; CAR, chimeric antigen receptor; RT, radiotherapy; TMZ, temozolomide; HR, hormone receptor; PBMC, peripheral blood mononuclear cell; TM, transmembrane; TAA, tumor-associated antigen; MHC, major histocompatibility complex; scFv, single-chain variable fragment; EGFR, epidermal growth factor receptor; IL13Rα2, interleukin-13 receptor alpha 2; HER2, human epidermal growth factor receptor 2; EphA2, ephrin-A2; PDPN, podoplanin; DIPG, diffuse intrinsic pontine glioma; H3-K27M, mutated histone H3 K27M; GD2, disialoganglioside 2; TCR, T cell receptor; HLA, human leukocyte antigen; VEGF, vascular endothelial growth factor; BBB, blood-brain barrier; LFA-1, lymphocyte- associated antigen-1; ICAM1, intracellular cell adhesion molecule 1; VCAM1, vascular cell adhesion molecule 1; MMP, matrix metalloproteinase; β-DG, β-dystroglycan; ALCAM, activated leukocyte cell adhesion molecule; HS, homing system; FUS, Focused Ultrasound; IDH, isocitrate dehydrogenase; CSF-1R, colony stimulating factor 1 receptor; STAT3, activator of transcription 3; PKC, protein kinase C; ICANS, immune effector cell-associated neurotoxicity syndrome; CRS, cytokine release syndrome; GM-CSF, granulocyte-macrophage colony-stimulating factor; MCP-1, monocyte chemoattractant protein 1; TNF, tumor necrosis factor; DIC, disseminated intravascular coagulation; MOF, multiple organ failure; ASTCT, American Society for Transplantation and Cellular Therapy; CSF, cerebrospinal fluid; TGF-β, transforming growth factor-β; IL-10, interleukin 10; TAMs, tumor-associated macrophages; MDSCs, myeloid-derived suppressor cells.
References
1. Weller M, Wick W, Aldape K, Brada M, Berger M, Pfister SM, et al. Glioma. Nat Rev Dis Primers. (2015) 1:15017. doi: 10.1038/nrdp.2015.17
2. McLendon RE, Halperin EC. Is the long-term survival of patients with intracranial glioblastoma multiforme overstated? Cancer. (2003) 98:1745–8. doi: 10.1002/cncr.11666
3. Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. (2017) 18:1373–85. doi: 10.1016/S1470-2045(17)30517-X
4. Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2011-2015. Neuro Oncol. (2018) 20(Suppl. 4):iv1–86. doi: 10.1093/neuonc/noy131
5. Gittleman H, Cioffi G, Vecchione-Koval T, Ostrom QT, Kruchko C, Osorio DS, et al. Descriptive epidemiology of germ cell tumors of the central nervous system diagnosed in the United States from 2006 to 2015. J Neurooncol. (2019) 143:251–60. doi: 10.1007/s11060-019-03173-4
6. Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest. (2015) 125:3335–7. doi: 10.1172/JCI83871
7. Bilusic M, Madan RA, Gulley JL. Immunotherapy of prostate cancer: facts and hopes. Clin Cancer Res. (2017) 23:6764–70. doi: 10.1158/1078-0432.CCR-17-0019
8. Filley AC, Henriquez M, Dey M. CART immunotherapy: development, success, and translation to malignant gliomas and other solid tumors. Front Oncol. (2018) 8:453. doi: 10.3389/fonc.2018.00453
9. Lynes J, Sanchez V, Dominah G, Nwankwo A, Nduom E. Current options and future directions in immune therapy for glioblastoma. Front Oncol. (2018) 8:578. doi: 10.3389/fonc.2018.00578
10. Yang B, Liu T, Qu Y, Liu H, Zheng SG, Cheng B, et al. Progresses and perspectives of anti-PD-1/PD-L1 antibody therapy in head and neck cancers. Front Oncol. (2018) 8:563. doi: 10.3389/fonc.2018.00563
11. Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wohrer A, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol. (2015) 17:1064–75. doi: 10.1093/neuonc/nou307
12. Wang Z, Zhang C, Liu X, Wang Z, Sun L, Li G, et al. Molecular and clinical characterization of PD-L1 expression at transcriptional level via 976 samples of brain glioma. Oncoimmunology. (2016) 5:e1196310. doi: 10.1080/2162402X.2016.1196310
13. Reardon DA, Omuro A, Brandes AA, Rieger J, Wick A, Sepulveda J, et al. OS10.3 Randomized phase 3 study evaluating the efficacy and safety of Nivolumab vs Bevacizumab in patients with recurrent glioblastoma: checkmate 143. Neuro-Oncology. (2017) 19(Suppl. 3):iii21. doi: 10.1093/neuonc/nox036.071
14. Herter-Sprie GS, Koyama S, Korideck H, Hai J, Deng J, Li YY, et al. Synergy of radiotherapy and PD-1 blockade in Kras-mutant lung cancer. JCI Insight. (2016) 1:e87415. doi: 10.1172/jci.insight.87415
15. Jahan N, Talat H, Alonso A, Saha D, Curry WT. Triple combination immunotherapy with GVAX, anti-PD-1 monoclonal antibody, and agonist anti-OX40 monoclonal antibody is highly effective against murine intracranial glioma. Oncoimmunology. (2019) 8:e1577108. doi: 10.1080/2162402X.2019.1577108
16. Wu A, Maxwell R, Xia Y, Cardarelli P, Oyasu M, Belcaid Z, et al. Combination anti-CXCR4 and anti-PD-1 immunotherapy provides survival benefit in glioblastoma through immune cell modulation of tumor microenvironment. J Neurooncol. (2019) 143:241–9. doi: 10.1007/s11060-019-03172-5
17. Stepanenko AA, Chekhonin VP. Recent advances in oncolytic virotherapy and immunotherapy for glioblastoma: a glimmer of hope in the search for an effective therapy? Cancers. (2018) 10:E492. doi: 10.3390/cancers10120492
18. Martikainen M, Essand M. Virus-based immunotherapy of glioblastoma. Cancers. (2019) 11:E186. doi: 10.3390/cancers11020186
19. Yu JS, Wheeler CJ, Zeltzer PM, Ying H, Finger DN, Lee PK, et al. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T-cell infiltration. Cancer Res. (2001) 61:842–7.
20. Wheeler CJ, Black KL, Liu G, Mazer M, Zhang XX, Pepkowitz S, et al. Vaccination elicits correlated immune and clinical responses in glioblastoma multiforme patients. Cancer Res. (2008) 68:5955–64. doi: 10.1158/0008-5472.CAN-07-5973
21. Zhu S, Lv X, Zhang X, Li T, Zang G, Yang N, et al. An effective dendritic cell-based vaccine containing glioma stem-like cell lysate and CpG adjuvant for an orthotopic mouse model of glioma. Int J Cancer. (2019) 144:2867–79. doi: 10.1002/ijc.32008
22. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
23. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. (2015) 7:303ra139. doi: 10.1126/scitranslmed.aac5415
24. Hu Y, Wu Z, Luo Y, Shi J, Yu J, Pu C, et al. Potent anti-leukemia activities of chimeric antigen receptor-modified T Cells against CD19 in Chinese patients with relapsed/refractory acute lymphocytic leukemia. Clin Cancer Res. (2017) 23:3297–306. doi: 10.1158/1078-0432.CCR-16-1799
25. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. (2015) 348:62–8. doi: 10.1126/science.aaa4967
26. Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, et al. T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med. (2016) 375:2255–62. doi: 10.1056/NEJMoa1609279
27. Zacharakis N, Chinnasamy H, Black M, Xu H, Lu YC, Zheng Z, et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med. (2018) 24:724–30. doi: 10.1038/s41591-018-0040-8
28. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. (2009) 10:459–66. doi: 10.1016/S1470-2045(09)70025-7
29. Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA. (2017) 318:2306–16. doi: 10.1001/jama.2017.18718
30. Bernard-Arnoux F, Lamure M, Ducray F, Aulagner G, Honnorat J, Armoiry X. The cost-effectiveness of tumor-treating fields therapy in patients with newly diagnosed glioblastoma. Neuro Oncol. (2016) 18:1129–36. doi: 10.1093/neuonc/now102
31. dos Santos MA, Pignon JP, Blanchard P, Lefeuvre D, Levy A, Touat M, et al. Systematic review and meta-analysis of phase I/II targeted therapy combined with radiotherapy in patients with glioblastoma multiforme: quality of report, toxicity, and survival. J Neurooncol. (2015) 123:307–14. doi: 10.1007/s11060-015-1802-5
32. Su J, Cai M, Li W, Hou B, He H, Ling C, et al. Molecularly targeted drugs plus radiotherapy and temozolomide treatment for newly diagnosed glioblastoma: a meta-analysis and systematic review. Oncol Res. (2016) 24:117–28. doi: 10.3727/096504016X14612603423511
33. Eberlein TJ, Rosenstein M, Rosenberg SA. Regression of a disseminated syngeneic solid tumor by systemic transfer of lymphoid cells expanded in interleukin 2. J Exp Med. (1982) 156:385–97.
34. Donohue JH, Rosenstein M, Chang AE, Lotze MT, Robb RJ, Rosenberg SA. The systemic administration of purified interleukin 2 enhances the ability of sensitized murine lymphocytes to cure a disseminated syngeneic lymphoma. J Immunol. (1984) 132:2123–8.
35. Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. (1986) 233:1318–21. doi: 10.1126/science.3489291
36. Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. (1988) 319:1676–80. doi: 10.1056/NEJM198812223192527
37. Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. (2002) 298:850–4. doi: 10.1126/science.1076514
38. Hong JJ, Rosenberg SA, Dudley ME, Yang JC, White DE, Butman JA, et al. Successful treatment of melanoma brain metastases with adoptive cell therapy. Clin Cancer Res. (2010) 16:4892–8. doi: 10.1158/1078-0432.CCR-10-1507
39. Pilon-Thomas S, Kuhn L, Ellwanger S, Janssen W, Royster E, Marzban S, et al. Efficacy of adoptive cell transfer of tumor-infiltrating lymphocytes after lymphopenia induction for metastatic melanoma. J Immunother. (2012) 35:615–20. doi: 10.1097/CJI.0b013e31826e8f5f
40. Radvanyi LG, Bernatchez C, Zhang M, Fox PS, Miller P, Chacon J, et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin Cancer Res. (2012) 18:6758–70. doi: 10.1158/1078-0432.CCR-12-1177
41. Besser MJ, Shapira-Frommer R, Itzhaki O, Treves AJ, Zippel DB, Levy D, et al. Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin Cancer Res. (2013) 19:4792–800. doi: 10.1158/1078-0432.CCR-13-0380
42. Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. (2013) 19:747–52. doi: 10.1038/nm.3161
43. Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. (2015) 350:1387–90. doi: 10.1126/science.aad1253
44. Quattrocchi KB, Miller CH, Cush S, Bernard SA, Dull ST, Smith M, et al. Pilot study of local autologous tumor infiltrating lymphocytes for the treatment of recurrent malignant gliomas. J Neurooncol. (1999) 45:141–57. doi: 10.1023/A:1006293606710
45. Dillman RO, Duma CM, Schiltz PM, DePriest C, Ellis RA, Okamoto K, et al. Intracavitary placement of autologous lymphokine-activated killer (LAK) cells after resection of recurrent glioblastoma. J Immunother. (2004) 27:398–404. doi: 10.1097/00002371-200409000-00009
46. Dillman RO, Duma CM, Ellis RA, Cornforth AN, Schiltz PM, Sharp SL, et al. Intralesional lymphokine-activated killer cells as adjuvant therapy for primary glioblastoma. J Immunother. (2009) 32:914–9. doi: 10.1097/CJI.0b013e3181b2910f
47. Sims JS, Grinshpun B, Feng Y, Ung TH, Neira JA, Samanamud JL, et al. Diversity and divergence of the glioma-infiltrating T-cell receptor repertoire. Proc Natl Acad Sci USA. (2016) 113:E3529–37. doi: 10.1073/pnas.1601012113
48. Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, et al. T-Cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res. (2018) 24:4175–86. doi: 10.1158/1078-0432.CCR-17-1846
49. Liu Z, Meng Q, Bartek JJr, Poiret T, Persson O, Rane L, et al. Tumor-infiltrating lymphocytes (TILs) from patients with glioma. Oncoimmunology. (2017) 6:e1252894. doi: 10.1080/2162402X.2016.1252894
50. Romani M, Pistillo MP, Carosio R, Morabito A, Banelli B. Immune checkpoints and innovative therapies in glioblastoma. Front Oncol. (2018) 8:464. doi: 10.3389/fonc.2018.00464
51. Parkhurst MR, Robbins PF, Tran E, Prickett TD, Gartner JJ, Jia L, et al. Unique neoantigens arise from somatic mutations in patients with gastrointestinal cancers. Cancer Discov. (2019) 9:1022–35. doi: 10.1158/2159-8290.CD-18-1494
52. Duhen T, Duhen R, Montler R, Moses J, Moudgil T, de Miranda NF, et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat Commun. (2018) 9:2724. doi: 10.1038/s41467-018-05072-0
53. Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. (2018) 557:575–9. doi: 10.1038/s41586-018-0130-2
54. Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. (2012) 119:4133–41. doi: 10.1182/blood-2011-12-400044
55. Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood. (2012) 119:3940–50. doi: 10.1182/blood-2011-10-387969
56. Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. (2015) 15:1145–54. doi: 10.1517/14712598.2015.1046430
57. Maus MV, June CH. Making better chimeric antigen receptors for adoptive T-cell therapy. Clin Cancer Res. (2016) 22:1875–84. doi: 10.1158/1078-0432.CCR-15-1433
58. Hu B, Ren J, Luo Y, Keith B, Young RM, Scholler J, et al. Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL-18. Cell Rep. (2017) 20:3025–33. doi: 10.1016/j.celrep.2017.09.002
59. Prinzing BL, Gottschalk SM, Krenciute G. CAR T-cell therapy for glioblastoma: ready for the next round of clinical testing? Expert Rev Anticancer Ther. (2018) 18:451–61. doi: 10.1080/14737140.2018.1451749
60. Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. (2010) 116:4099–102. doi: 10.1182/blood-2010-04-281931
61. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. (2017) 3:1094–101. doi: 10.1001/jamaoncol.2017.0184
62. Fan QW, Cheng CK, Gustafson WC, Charron E, Zipper P, Wong RA, et al. EGFR phosphorylates tumor-derived EGFRvIII driving STAT3/5 and progression in glioblastoma. Cancer Cell. (2013) 24:438–49. doi: 10.1016/j.ccr.2013.09.004
63. Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med. (2015) 7:275ra222. doi: 10.1126/scitranslmed.aaa4963
64. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. (2017) 9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984
65. Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, et al. Pilot trial of adoptive transfer of chimeric antigen receptor-transduced T cells targeting EGFRvIII in patients with glioblastoma. J Immunother. (2019) 42:126–35. doi: 10.1097/CJI.0000000000000260
66. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
67. Bielamowicz K, Fousek K, Byrd TT, Samaha H, Mukherjee M, Aware N, et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. (2018) 20:506–18. doi: 10.1093/neuonc/nox182
68. Shiina S, Ohno M, Ohka F, Kuramitsu S, Yamamichi A, Kato A, et al. CAR T cells targeting podoplanin reduce orthotopic glioblastomas in mouse brains. Cancer Immunol Res. (2016) 4:259–68. doi: 10.1158/2326-6066.CIR-15-0060
69. Shum T, Omer B, Tashiro H, Kruse RL, Wagner DL, Parikh K, et al. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Discov. (2017) 7:1238–47. doi: 10.1158/2159-8290.CD-17-0538
70. Jin L, Ge H, Long Y, Yang C, Chang YE, Mu L, et al. CD70, a novel target of CAR T-cell therapy for gliomas. Neuro Oncol. (2018) 20:55–65. doi: 10.1093/neuonc/nox116
71. Mount CW, Majzner RG, Sundaresh S, Arnold EP, Kadapakkam M, Haile S, et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M(+) diffuse midline gliomas. Nat Med. (2018) 24:572–9. doi: 10.1038/s41591-018-0006-x
72. Okamoto S, Mineno J, Ikeda H, Fujiwara H, Yasukawa M, Shiku H, et al. Improved expression and reactivity of transduced tumor-specific TCRs in human lymphocytes by specific silencing of endogenous TCR. Cancer Res. (2009) 69:9003–11. doi: 10.1158/0008-5472.CAN-09-1450
73. Zhang L, Morgan RA. Genetic engineering with T cell receptors. Adv Drug Deliv Rev. (2012) 64:756–62. doi: 10.1016/j.addr.2011.11.009
74. Lin Y, Okada H. Cellular immunotherapy for malignant gliomas. Expert Opin Biol Ther. (2016) 16:1265–75. doi: 10.1080/14712598.2016.1214266
75. Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. (2006) 66:8878–86. doi: 10.1158/0008-5472.CAN-06-1450
76. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. (2006) 314:126–9. doi: 10.1126/science.1129003
77. Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. (2009) 114:535–46. doi: 10.1182/blood-2009-03-211714
78. Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. (2013) 36:133–51. doi: 10.1097/CJI.0b013e3182829903
79. Chheda ZS, Kohanbash G, Okada K, Jahan N, Sidney J, Pecoraro M, et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J Exp Med. (2018) 215:141–57. doi: 10.1084/jem.20171046
80. Engelhardt B. Regulation of immune cell entry into the central nervous system. Results Probl Cell Differ. (2006) 43:259–80. doi: 10.1007/400_020
81. Engelhardt B, Ransohoff RM. Capture, crawl, cross: the T cell code to breach the blood-brain barriers. Trends Immunol. (2012) 33:579–89. doi: 10.1016/j.it.2012.07.004
82. Steffen BJ, Butcher EC, Engelhardt B. Evidence for involvement of ICAM-1 and VCAM-1 in lymphocyte interaction with endothelium in experimental autoimmune encephalomyelitis in the central nervous system in the SJL/J mouse. Am J Pathol. (1994) 145:189–201.
83. Steiner O, Coisne C, Cecchelli R, Boscacci R, Deutsch U, Engelhardt B, et al. Differential roles for endothelial ICAM-1, ICAM-2, and VCAM-1 in shear-resistant T cell arrest, polarization, and directed crawling on blood-brain barrier endothelium. J Immunol. (2010) 185:4846–55. doi: 10.4049/jimmunol.0903732
84. Sarkaria JN, Hu LS, Parney IF, Pafundi DH, Brinkmann DH, Laack NN, et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-oncology. (2018) 20:184–91. doi: 10.1093/neuonc/nox175
85. Chuntova P, Downey KM, Hegde B, Almeida ND, Okada H. Genetically engineered T-cells for malignant glioma: overcoming the barriers to effective immunotherapy. Front Immunol. (2019) 9:3062. doi: 10.3389/fimmu.2018.03062
86. Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. (2006) 12(20 Pt 1):6106–15. doi: 10.1158/1078-0432.CCR-06-1183
87. Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. (2009) 113:6392–402. doi: 10.1182/blood-2009-03-209650
88. Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. (2010) 33:780–8. doi: 10.1097/CJI.0b013e3181ee6675
89. Moon EK, Carpenito C, Sun J, Wang LC, Kapoor V, Predina J, et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res. (2011) 17:4719–30. doi: 10.1158/1078-0432.CCR-11-0351
90. Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. (2009) 69:3077–85. doi: 10.1158/0008-5472.CAN-08-2281
91. Slaney CY, Kershaw MH, Darcy PK. Trafficking of T cells into tumors. Cancer Res. (2014) 74:7168–74. doi: 10.1158/0008-5472.CAN-14-2458
92. Oelkrug C, Ramage JM. Enhancement of T cell recruitment and infiltration into tumours. Clin Exp Immunol. (2014) 178:1–8. doi: 10.1111/cei.12382
93. Jung Y, Ahn SH, Park H, Park SH, Choi K, Choi C, et al. MCP-1 and MIP-3alpha secreted from necrotic cell-treated glioblastoma cells promote migration/infiltration of microglia. Cell Physiol Biochem. (2018) 48:1332–46. doi: 10.1159/000492092
94. Samaha H, Pignata A, Fousek K, Ren J, Lam FW, Stossi F, et al. A homing system targets therapeutic T cells to brain cancer. Nature. (2018) 561:331–7. doi: 10.1038/s41586-018-0499-y
95. Chen PY, Hsieh HY, Huang CY, Lin CY, Wei KC, Liu HL. Focused ultrasound-induced blood-brain barrier opening to enhance interleukin-12 delivery for brain tumor immunotherapy: a preclinical feasibility study. J Transl Med. (2015) 13:93. doi: 10.1186/s12967-015-0451-y
96. Vazana U, Veksler R, Pell GS, Prager O, Fassler M, Chassidim Y, et al. Glutamate-mediated blood-brain barrier opening: implications for neuroprotection and drug delivery. J Neurosci. (2016) 36:7727–39. doi: 10.1523/JNEUROSCI.0587-16.2016
97. Sajesh BV, On NH, Omar R, Alrushaid S, Kopec BM, Wang W-G, et al. Validation of cadherin HAV6 peptide in the transient modulation of the blood-brain barrier for the treatment of brain tumors. Pharmaceutics. (2019) 11:481. doi: 10.3390/pharmaceutics11090481
98. Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanovic S, Gouttefangeas C, et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature. (2019) 565:240–5. doi: 10.1038/s41586-018-0810-y
99. Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. (2019) 565:234–9. doi: 10.1038/s41586-018-0792-9
100. Quail DF, Joyce JA. The microenvironmental landscape of brain tumors. Cancer Cell. (2017) 31:326–41. doi: 10.1016/j.ccell.2017.02.009
101. Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res. (2017) 77:2266–78. doi: 10.1158/0008-5472.CAN-16-2310
102. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. (2002) 23:549–55. doi: 10.1016/S1471-4906(02)02302-5
103. Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. (2006) 8:261–79. doi: 10.1215/15228517-2006-008
104. Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT, et al. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science. (2016) 352:aad3018. doi: 10.1126/science.aad3018
105. Butowski N, Colman H, De Groot JF, Omuro AM, Nayak L, Wen PY, et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro Oncol. (2016) 18:557–64. doi: 10.1093/neuonc/nov245
106. Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. (2006) 66:3294–302. doi: 10.1158/0008-5472.CAN-05-3773
107. Fecci PE, Sweeney AE, Grossi PM, Nair SK, Learn CA, Mitchell DA, et al. Systemic anti-CD25 monoclonal antibody administration safely enhances immunity in murine glioma without eliminating regulatory T cells. Clin Cancer Res. (2006) 12:4294–305. doi: 10.1158/1078-0432.CCR-06-0053
108. Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat Immunol. (2017) 18:1332–41. doi: 10.1038/ni.3868
109. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331
110. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. (2016) 126:3130–44. doi: 10.1172/JCI83092
111. Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, et al. A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res. (2016) 76:1578–90. doi: 10.1158/0008-5472.CAN-15-2524
112. Brantley EC, Nabors LB, Gillespie GY, Choi YH, Palmer CA, Harrison K, et al. Loss of protein inhibitors of activated STAT-3 expression in glioblastoma multiforme tumors: implications for STAT-3 activation and gene expression. Clin Cancer Res. (2008) 14:4694–704. doi: 10.1158/1078-0432.CCR-08-0618
113. Lin GS, Chen YP, Lin ZX, Wang XF, Zheng ZQ, Chen L. STAT3 serine 727 phosphorylation influences clinical outcome in glioblastoma. Int J Clin Exp Pathol. (2014) 7:3141–9.
114. Piperi C, Papavassiliou KA, Papavassiliou AG. Pivotal role of STAT3 in shaping glioblastoma immune microenvironment. Cells. (2019) 8:E1398. doi: 10.3390/cells8111398
115. Wang H, Lathia JD, Wu Q, Wang J, Li Z, Heddleston JM, et al. Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem Cells. (2009) 27:2393–404. doi: 10.1002/stem.188
116. Puram SV, Yeung CM, Jahani-Asl A, Lin C, de la Iglesia N, Konopka G, et al. STAT3-iNOS signaling mediates EGFRvIII-induced glial proliferation and transformation. J Neurosci. (2012) 32:7806–18. doi: 10.1523/JNEUROSCI.3243-11.2012
117. Xu Y, Li Z, Zhang C, Zhang S, Ji Y, Chen F. Knockdown of PKCepsilon expression inhibits growth, induces apoptosis and decreases invasiveness of human glioma cells partially through Stat3. J Mol Neurosci. (2015) 55:21–31. doi: 10.1007/s12031-014-0341-4
118. Su Y-L, Banerjee S, White SV, Kortylewski M. STAT3 in tumor-associated myeloid cells: multitasking to disrupt immunity. Int J Mol Sci. (2018) 19:1803. doi: 10.3390/ijms19061803
119. Wei J, Barr J, Kong LY, Wang Y, Wu A, Sharma AK, et al. Glioblastoma cancer-initiating cells inhibit T-cell proliferation and effector responses by the signal transducers and activators of transcription 3 pathway. Mol Cancer Ther. (2010) 9:67–78. doi: 10.1158/1535-7163.MCT-09-0734
120. Gieryng A, Pszczolkowska D, Walentynowicz KA, Rajan WD, Kaminska B. Immune microenvironment of gliomas. Lab Invest. (2017) 97:498–518. doi: 10.1038/labinvest.2017.19
121. Alshamsan A, Hamdy S, Das S, Lavasanifar A, Samuel J, El-Kadi AO. Validation of bone marrow derived dendritic cells as an appropriate model to study tumor-mediated suppression of DC maturation through STAT3 hyperactivation. J Pharm Pharm Sci. (2010) 13:21–6. doi: 10.18433/J37598
122. Hussain SF, Kong LY, Jordan J, Conrad C, Madden T, Fokt I, et al. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res. (2007) 67:9630–6. doi: 10.1158/0008-5472.CAN-07-1243
123. Garcia Borrega J, Godel P, Ruger MA, Onur OA, Shimabukuro-Vornhagen A, Kochanek M, et al. In the eye of the storm: immune-mediated toxicities associated with CAR-T cell therapy. Hemasphere. (2019) 3:e191. doi: 10.1097/HS9.0000000000000191
124. Murthy H, Iqbal M, Chavez JC, Kharfan-Dabaja MA. Cytokine release syndrome: current perspectives. ImmunoTargets Therap. (2019) 8:43–52. doi: 10.2147/ITT.s202015
125. Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. (2018) 24:739–48. doi: 10.1038/s41591-018-0036-4
126. Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. (2019) 25:625–38. doi: 10.1016/j.bbmt.2018.12.758
127. Hill JA, Li D, Hay KA, Green ML, Cherian S, Chen X, et al. Infectious complications of CD19-targeted chimeric antigen receptor-modified T-cell immunotherapy. Blood. (2018) 131:121–30. doi: 10.1182/blood-2017-07-793760
128. Santomasso BD, Park JH, Salloum D, Riviere I, Flynn J, Mead E, et al. Clinical and biological correlates of neurotoxicity associated with CAR T-cell therapy in patients with B-cell acute lymphoblastic leukemia. Cancer Discov. (2018) 8:958–71. doi: 10.1158/2159-8290.CD-17-1319
129. Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T Cells. Cancer Discov. (2017) 7:1404–19. doi: 10.1158/2159-8290.CD-17-0698
130. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
131. Mrugala MM, Chamberlain MC. Mechanisms of disease: temozolomide and glioblastoma–look to the future. Nat Clin Pract Oncol. (2008) 5:476–86. doi: 10.1038/ncponc1155
132. Suryadevara CM, Desai R, Abel ML, Riccione KA, Batich KA, Shen SH, et al. Temozolomide lymphodepletion enhances CAR abundance and correlates with antitumor efficacy against established glioblastoma. Oncoimmunology. (2018) 7:e1434464. doi: 10.1080/2162402X.2018.1434464
Keywords: adoptive cell therapy, Glioblastoma multiforme, chimeric antigen receptor, T cell receptor, tumor-infiltrating lymphocyte
Citation: Wang J, Shen F, Yao Y, Wang L, Zhu Y and Hu J (2020) Adoptive Cell Therapy: A Novel and Potential Immunotherapy for Glioblastoma. Front. Oncol. 10:59. doi: 10.3389/fonc.2020.00059
Received: 28 September 2019; Accepted: 13 January 2020;
Published: 31 January 2020.
Edited by:
Haotian Zhao, New York Institute of Technology, United StatesReviewed by:
Yong-Chen Lu, National Institutes of Health (NIH), United StatesAmorette Barber, Longwood University, United States
Copyright © 2020 Wang, Shen, Yao, Wang, Zhu and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jue Hu, aGptaWNlQDEyNi5jb20=; Yongjian Zhu, bmV1cm9zdXJnZXJ5QHpqdS5lZHUuY24=
†These authors have contributed equally to this work