 Stefania Oliva
Stefania Oliva Mattia D'Agostino†
Mattia D'Agostino† Mario Boccadoro
Mario Boccadoro Alessandra Larocca
Alessandra Larocca- Myeloma Unit, Division of Hematology, University of Torino, Azienda Ospedaliero-Universitaria Cittá Della Salute e Della Scienza di Torino, Turin, Italy
In the last years, the life expectancy of multiple myeloma (MM) patients has substantially improved thanks to the availability of many new drugs. Our ability to induce deep responses has improved as well, and the treatment goal in patients tolerating treatment moved from the delay of progression to the induction of the deepest possible response. As a result of these advances, a great scientific effort has been made to redefine response monitoring, resulting in the development and validation of high-sensitivity techniques to detect minimal residual disease (MRD). In 2016, the International Myeloma Working Group (IMWG) updated MM response categories defining MRD-negative responses both in the bone marrow (assessed by next-generation flow cytometry or next-generation sequencing) and outside the bone marrow. MRD is an important factor independently predicting prognosis during MM treatment. Moreover, using novel combination therapies, MRD-negative status can be achieved in a fairly high percentage of patients. However, many questions regarding the clinical use of MRD status remain unanswered. MRD monitoring can guide treatment intensity, although well-designed clinical trials are needed to demonstrate this potential. This mini-review will focus on currently available techniques and data on MRD testing and their potential future applications.
Introduction
The treatment course of multiple myeloma (MM) has been strongly improved during the last 20 years: the introduction of modern 3-drug regimen therapies combined with transplantation increased the achievement of deeper responses and the acquisition of minimal residual disease (MRD) negativity in up to 40/50% of patients enrolled in clinical trials (1). Consistently, a large number of studies showed that, among patients achieving a complete response (CR), those with detectable MRD had inferior progression-free survival (PFS), and overall survival (OS) compared to those with undetectable MRD. Moreover, among patients in CR, improved PFS and OS have been significantly associated with undetectable MRD, regardless of disease stage, prior transplant, or cytogenetic risk (2).
Therefore, the International Myeloma Working Group (IMWG) recently revised the response criteria and introduced the definition of MRD in CR patients as the persistence or re-emergence of very low levels of cancer cells, equal to about 1 tumor cell in at least 105 normal cells (3). These response criteria are the direct result of the progressive evolution of both imaging and bone marrow MRD techniques in the last 15 years (Figure 1). However, a precise knowledge of when and how to perform MRD detection is required. This review aims to examine the currently available MRD techniques recommended by IMWG and data from different clinical trials, in order to outline a possible future perspective on the role of MRD testing as a tool for decision making in standard clinical practice.
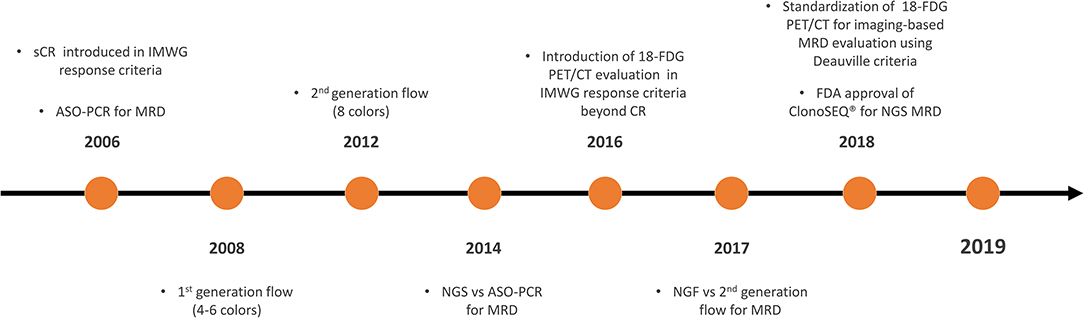
Figure 1. Timeline on the development and validation of MRD techniques in MM. MRD, minimal residual disease; MM, multiple myeloma; CR, complete response; sCR, stringent CR; IMWG, International Myeloma Working Group; ASO-PCR, allele-specific oligonucleotide polymerase chain reaction; NGS, next-generation sequencing; 18-FDG PET/CT, 18-fluorodeoxyglucose positron emission tomography/computed tomography; ClonoSEQ®, ClonoSEQ® Assay (Adaptive Biotechnologies, Seattle, US-WA); FDA, Food and Drug Administration.
MRD Techniques and Practical Considerations
Bone Marrow Techniques: NGF and NGS
There are two techniques commonly used to detect MRD in the bone marrow (BM): multiparameter flow cytometry (MFC) and next-generation sequencing (NGS) molecular technology. Both techniques show positive and negative aspects (Table 1).
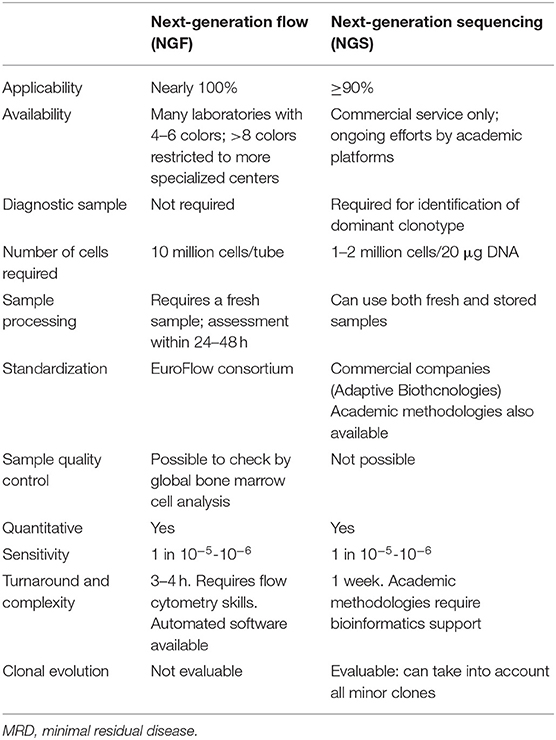
Table 1. Bone marrow techniques for MRD in myeloma: pros and cons.
MFC can detect and quantify tumor vs. normal plasma cells using cell surface and cytoplasmic markers. For the identification of plasma cells, the combined use of CD38 and CD138 is recommended even if they are also expressed on other BM cells. In particular, the aberrant expression patterns of CD19, CD56, CD45, CD38, CD27, CD20, CD28, CD33, CD117 and surface membrane immunoglobulin can characterize the phenotype of monoclonal plasma cells (4). However, antigenic expression can vary on plasma cells and should be considered when interpreting flow data.
Older conventional 4- to 7-color flow cytometry assays have now been replaced by advanced 8-color 2-tube or 10-color 1-tube assays. In this sense, the increased sensitivity of MFC (between 10−4 and 10−5) is due to the simultaneous assessment of ≥8 markers in a single tube. In this way, if sufficient cell numbers are evaluated (e.g., ≥5 × 106), it is possible to promptly identify aberrant PC phenotypes at MRD levels (5).
A consensus methodology has been recently proposed by the International Myeloma Foundation's Black Swan Research Initiative, which formed the EuroFlow Next-Generation Flow (NGF) panel in order to increase sensitivity and standardization of MFC (6, 7). This panel includes two 8-color tubes (tube 1: CD138, CD27, CD38, CD56, CD45, CD19, CD117, CD81; tube 2: CD138, CD27, CD38, CD56, CD45, CD19, cIgκ, cIgλ). In this protocol, a bulk-lysis procedure was determined and the acquisition of ≥107 cells/sample was recommended. Importantly, new softwares were developed for automatic plasma cell gating in order to avoid individual assessments.
According to this consensus methodology, it is important to evaluate the limit of quantitation (LOQ) and the limit of detection (LOD) of the NGF-MRD method. The LOQ is calculated as 50 among 107 nucleated cells (based on the identification of ≥50 clonal plasma cells); the LOD as 20 among 107 nucleated cells (based on the identification of ≥20 clonal plasma cells). This evaluation allows to discriminate between positive and negative samples. Interestingly, a baseline sample is not mandatory for MRD evaluation. After the multicenter evaluation of patients with very good partial response (VGPR) or CR, 110 follow-up bone marrows showed a higher sensitivity for NGF-MRD, as compared to conventional 8-color flow-MRD: MRD-positive rates were 47 vs. 34% (P = 0.003), respectively. Thus, 25% of patients who were categorized as MRD-negative by conventional 8-color flow were categorized as MRD-positive by NGF. This translated into a significantly longer PFS using NGF to discriminate between MRD-negative and MRD-positive CR patients (P = 0.02). Importantly, NGF can also provide a qualitative assessment of the patient sample by allowing the complete analysis of the normal B-cell compartment and the detection of a significantly decreased number of non-PC BM cells (e.g., mast cells, nucleated red blood cells, myeloid precursors, B-cell precursors, and CD19– normal PC) revealing potentially hemodiluted BM samples. Finally, treatment with CD38 antibodies such as daratumumab and isatuximab can alter the antigen expression in MM cells. This sets a limit for the use of CD38 as a marker for the detection of plasma cells during MRD assessments at follow-up. The use of multi-epitope CD38 antibody in an advanced flow cytometry panel can solve this problem, since this conjugate can bind to a specific site (not covered by daratumumab) of the CD38 antigen. Nonetheless, in case of CD38 surface downregulation, the solution is the analysis of intracellular CD38 through the same protocol used for intracellular k- and λ-chain staining (7).
Allele-specific oligonucleotide polymerase chain reaction (ASO-PCR) was first explored to evaluate molecular MRD in MM, but even if its prognostic role was confirmed, different issues limited its use in favor of the NGS technique. First, its applicability ranged from 40 to 60% due to the low rate of diagnostic marker identification, since this technique does not take into account the somatic hypermutation rate of immunoglobulin loci and this translates into sequencing problems. Moreover, patient-specific reagents raised the complexity of this technique (8–11).
NGS was developed to overcome all these disadvantages. ClonoSEQ® Assay (Adaptive Biotechnologies, Seattle, US-WA) is the most frequently adopted commercial platform in the United States. In this test, DNA is extracted from patient's BM, a multiplex PCR amplifies VDJ, IgK, and IgL gene sequences and a common PCR prepares DNA for sequencing and creates a sequencing library. At the end of the process, a bioinformatic tool is essential to extrapolate and analyze all NGS data.
Using this assay, we can define as “clonotypes” two identical sequencing reads. A clonotype with frequency >5% at diagnosis is considered a clonality (clonal gene rearrangements), thus becoming a target for the detection of MRD in follow-up samples (12, 13). In lymphoid malignancies, NGS and ASO-PCR have been compared, showing similar sensitivities and results (13).
In the IFM2009 clinical trial, a comparison between NGS and 7-color MFC has been made, showing that the higher sensitivity with NGS at 10−6 allowed to predict the best outcomes in MRD-positive vs. -negative patients (3-year PFS: 53 vs. 83%, p < 0.001).
Ongoing clinical trials are evaluating NGS vs. NGF and their correlation: in the CASSIOPEIA trial, a good concordance (83.5% in paired samples) was observed using the same sensitivity (10−5) regardless of response in patients achieving ≥CR, indicating that both techniques performed similarly in evaluating MRD (14). As illustrated in Table 1, some characteristics can affect the clinician's preference of choosing NGS vs. NGF, such as the higher cost for NGS (~1,500 $ per sample vs. ~300 $ for NGF), and the required time and skills (at least 1 week for NGS vs. 3–4 h for NGF and commercial service available only for NGS).
In this regard, ongoing studies are evaluating ‘in-house’ NGS techniques: recently, Martinez-Lopez et al. described a NGS method starting from 1 μg of DNA and amplified IGH or IGK sequences. The sequencing data were analyzed by specific mathematical and bioinformatic tools to identify and quantify the clonotype present on each sample. A clonotype was identified when at least 400 identical sequencing reads were obtained, or when it was present at a frequency of >1% with a sensitivity of at least 10−5 (15).
Imaging Techniques: PET/CT
MM is a patchy disease and BM infiltration may often be heterogeneous. Indeed, ~60% of MM patients show focal lesions that represent the local accumulation of plasma cells (16). Therefore, the IMWG incorporated imaging in addition to BM evaluation to better characterize MM residual disease (3).
Different studies showed the role of imaging techniques in evaluating focal lesions: magnetic resonance imaging (MRI) is a sensitive, non-invasive imaging technique available to detect the bone involvement in the spine and to provide details regarding the soft tissue disease and the pattern of marrow infiltration (normal, focal, diffuse, or heterogeneous).
Fluorodeoxyglucose positron emission tomography/computed tomography (FDG PET/CT) can be used to analyze the vitality of the focal lesions and is therefore the current standard of care to evaluate the post-therapeutic residual infiltration (17–19).
Different studies showed the prognostic and predictive role of FDG PET/CT (20–22). Interestingly, Moreau et al. compared PET/CT with MRI. Although at diagnosis both the techniques performed similarly in the detection of bone lesions, the normalization after therapy of PET/CT, but not of MRI, was predictive of PFS and OS (20). In both responding and non-responding patients, focal lesions can still remain positive for many months. As a consequence, conventional MRI is probably not the best technique to evaluate MRD (22–24). On the other hand, functional MRI techniques based on the measurement of the movement of water molecules in the tissue (Diffusion-Weighted MRI, DWI) could be informative on the residual cellularity and the microcirculation of the focal lesions (25). No standardization of the diagnostic technique and no interpretation of results in MM after therapy are still available and no prospective comparison between PET/CT and DWI in a meaningful number of patients has been done. In a small number of MM patients, DWI seemed to be more sensitive in the detection of residual lesions. However, if this could be an advantage or could lead to an increased number of false-positive cases, still needs to be elucidated (26, 27).
Finally, different researchers confirmed the complementarity of PET/CT and BM techniques. Rasche et al. showed how patients who were both Flow-MRD- and PET/CT-negative had the best PFS outcome when compared with those who were Flow-MRD-negative but PET/CT-positive (28). Paiva et al. demonstrated that, even if NGF-negative patients had a long PFS, there was a proportion of subjects who relapsed with extramedullary disease in the presence of a previous negative BM sample, confirming the importance of combining BM and imaging analyses (29).
PET/CT has some limitations, some of which are linked to the tracer used (FDG). Indeed, a low expression of the enzymes responsible for the glycolysis process (e.g., hexokinase 2 gene) in MM cells could lead to false-negative cases with FDG PET/CT (30). Alternative tracers could overcome these limitations. For instance, 11C-Methionine uptake correlates with protein synthesis, a very active mechanism in malignant plasma cells, and can be used as an alternative PET/CT tracer in MM (31).
In a head-to-head prospective comparison in a heterogeneous MM patient population, 11C-Methionine PET/CT was more sensitive than FDG PET/CT in the detection of focal lesions, both within and outside the bone. More data are needed in a homogenous patient population to understand whether this tracer could be an alternative to FDG in the detection of residual disease after treatment. Currently, other tracers targeting lipid membrane (e.g., Choline, Acetate) and CXCR4 are also under study (32).
MRD Results in the Clinical Setting: Relevant Questions
In this section we focus on clinically relevant questions regarding MRD, reviewing available data on newly diagnosed MM (NDMM) patients. Single studies are summarized in Table 2. Data on MRD evaluation in relapsed and/or refractory MM patients (59) and high-risk smoldering MM (60) are beginning to emerge as well, and have been recently reviewed elsewhere (61).
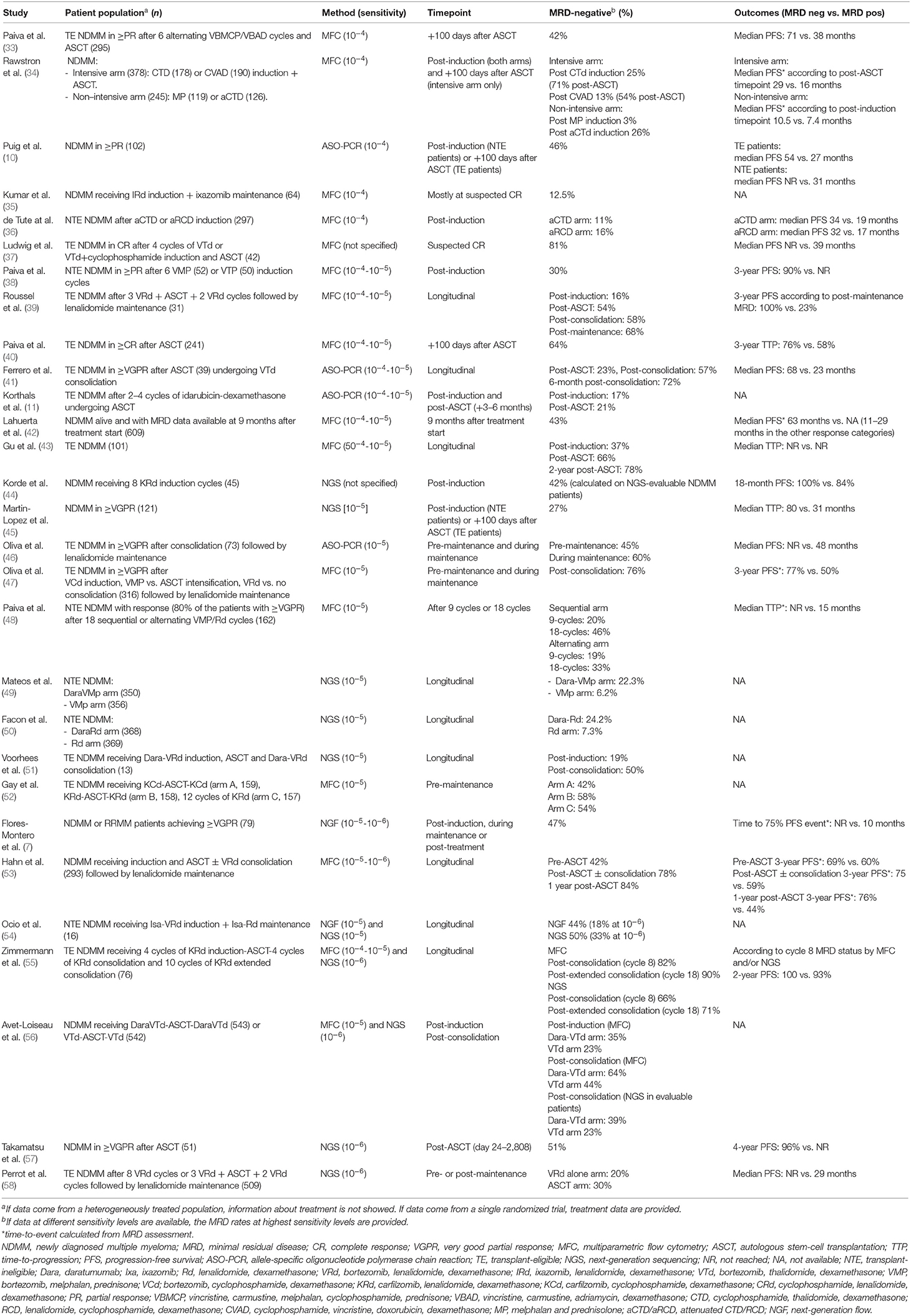
Table 2. Selected trials on NDMM patients reporting MRD data.
In the MM field, a major question concerned the prognostic role of MRD and its ability to perform better than conventionally defined response criteria. As already discussed, there is now compelling evidence coming from multiple studies (Table 2) and two meta-analyses (2, 62) confirming that MRD-negative patients have a significantly better PFS and OS compared to MRD-positive patients. The beneficial effect of MRD negativity was confirmed also focusing on CR patients (2). Using MFC with a sensitivity of 10−4-10−5, Lahuerta et al. nicely demonstrated that MRD-negative patients with a conventionally defined CR had better PFS (median, 63 vs. 27 months, p < 0.001) and OS (median, not reached vs. 59 months, p < 0.001) than MRD-positive CR patients (42). Moreover, MRD-positive CR patients had similar outcomes compared to patients achieving a partial response (PR) (median PFS, 27 vs. 29 months; median OS, 59 vs. 65 months, respectively) showing that the prognostic advantage of conventionally defined CR over PR resided in the MRD-negative patient population (42).
The best timing for MRD measurement is another important unanswered question. Usually, MRD is measured at specific timepoints during therapy [e.g., post-induction (39), +100 days post-ASCT (33), post-consolidation (41), pre-maintenance, and during maintenance (46)]. If treatment does not provide for a phase-specific timepoint (as in the case of the continuous treatment strategy commonly adopted for transplant-ineligible patients), MRD testing is usually done at unconfirmed CR/sCR and at fixed timepoints thereafter (50).
Data clearly show that, as we continue to intensify patient treatment, the percentage of MRD-negative patients increases (39, 43, 53, 55, 56) and even maintenance treatment can convert a significant percentage of MRD-positive patients into MRD-negative [e.g., 27–30% with lenalidomide maintenance in a pooled analysis (9, 46)]. Each timepoint can be important due to different clinical reasons. For instance, the post-induction timepoint can be used to design clinical trials addressing different intensification regimens, while pre-maintenance or during maintenance timepoints can be exploited to design clinical trials addressing the intensity and the duration of maintenance. Regarding the prognostic effect of different timepoints, in the Myeloma IX study, which used MFC with a sensitivity of 10−4, a PFS advantage was found in patients that were MRD-negative both post-induction and post-ASCT, as compared with patients that were MRD-positive post-induction and became MRD-negative post-ASCT, although this effect did not translate into an OS benefit (34). On the other hand, Hahn et al. demonstrated in a transplant-eligible population that patients who were MRD-negative pre-ASCT, pre-maintenance, and 1-year post-ASCT showed all a better PFS compared to MRD-positive patients. Only the 1-year post-ASCT timepoint was associated with better OS (3-year post-ASCT OS 96 vs. 66% for MRD-negative vs. MRD-positive patients) (53). These data suggest that the duration of MRD negativity may be important, but little data are available on sustained MRD negativity (i.e., the need to confirm MRD at different timepoints) and on its optimal duration. Gu et al. used MFC to monitor 104 MM patients post-induction and at different post-ASCT timepoints (3 to 24 months), showing that patients with persistent MRD negativity post-induction until 24 months after ASCT (n = 33) had better time to progression (median, not reached vs. 15.4 months) and OS (not reached vs. 35.2 months), as compared to patients that were MRD-negative post-induction but MRD-positive within 24 months post-ASCT (n = 5) (43). The low numbers in the latter group do not allow the exploration of different time cutoffs for sustained MRD negativity. However, 2/5 patients became MRD-positive 18 months post-ASCT, thus suggesting that long-term confirmation of sustained MRD negativity may be necessary.
Another question is whether the sensibility of the technique impacts the reliability of MRD. Using MFC with a sensibility of 10−4, Rawstron et al. demonstrated that each log depletion in MRD levels predicted a 1-year median OS advantage (5.9 years for 10−2-10−3, 6.8 years for 10−3-10−4, and more than 7.5 years for 10−4), suggesting that MRD level is a continuous rather than a discrete variable (63). Recently, several studies using both flow cytometry-based methods with a sensitivity of 10−5 (48) or 10−5-10−6 (7) and NGS-based methods with a sensitivity of 10−6 (58, 64) demonstrated that lower levels of MRD are associated with better outcomes and that the best possible sensitivity should be pursued. Indeed, in the IFM/DFCI 2009 trial, among 163 patients who were MRD-negative pre-maintenance using MFC with a sensibility of 10−4, 84 (56%) were indeed MRD-positive using NGS with a sensibility of 10−6 (3-year PFS, 86 vs. 66% in NGS-negative vs. NGS-positive among MFC-negative patients). This is especially important in clinical trials designed to explore treatment interruption based on MRD levels because a low sensibility of the technique can lead to unacceptable risk of patients' undertreatment.
This observation leads to our last question: if MRD negativity is a major prognostic determinant, do treatment administered and baseline risk stratification matter as long as MRD negativity is achieved? Many studies demonstrated that even if a more effective regimen induced MRD negativity in a higher number of patients, the prognosis of MRD-negative patients was similar independently from treatment arm (49, 58). However, we do need MRD-driven clinical trials to determine if treatment deintensification in MRD-negative patients is feasible without worsening patient prognosis (65). In this regard, in the Myeloma IX trial, MRD-negative patients (MFC at 10−4) receiving thalidomide maintenance remained in a MRD-negative state more often than patients not receiving maintenance treatment (96 vs. 68.8%, p = 0.026). Regarding MM patients who are at high risk according to baseline prognostic factors (e.g., high-risk cytogenetics or unfavorable Revised International Staging System score), MRD-negative patients at a low level of sensitivity (10−4) still showed inferior clinical outcomes than standard-risk patients (34). Conversely, reaching MRD negativity at a sensitivity of 10−5-10−6 seemed to overcome the inferior outcome observed in high-risk vs. standard-risk patients (48, 58). However, it should be noted that high-risk patients require highly intensive regimens in order to achieve a proper level of MRD negativity (47, 52, 55).
Future Perspectives
Is MRD a Surrogate Endpoint for Drug Approval?
Improving OS and quality of life is the final aim of MM treatment. In the past years, the PFS endpoint has been used as a surrogate endpoint for OS to speed up the drug approval process. However, following the achievement of long-standing and deep responses (especially in NDMM patients), PFS is inappropriately becoming a late endpoint. MRD is considered the best candidate as a PFS/OS surrogate marker for provisional drug approval by regulatory agencies. Indeed, ClonoSEQ® Assay is now authorized by FDA (66) and MRD negativity with a sensitivity of 10−5 is the most common primary endpoint of new clinical trials designed for NDMM patients. However, as discussed above, continuous efforts should be exterted to define the optimal sensitivity cut-off (10−5 vs. 10−6), the timing of evaluation and the need for a sustained MRD negativity. Moreover, safety should be closely addressed, as it was demonstrated by higher MRD (13.4 vs. 1%) but worse OS rates (HR 2.03, 95% CI 1.04–3.94) in the experimental arm of the BELLINI trial (M14-031) comparing venetoclax-Vd vs. Vd (67, 68). Moreover, in some settings, the correlation between MRD negativity rates and PFS improvement could be less clear because of technical pitfalls (e.g., early MRD evaluation after myelosuppressive treatments in hypocellular bone marrows).
How to Address Spatial Heterogeneity?
MM is a spatially heterogeneous disease and simultaneous MRD negativization inside and outside the bone marrow showed synergistic predictive values (28).
Moreover, MRD analysis within the bone marrow is done on bone marrow aspirates coming from a single random site and, in some patients, MM cells show a patchy infiltration (69). To overcome this issue and to possibly link the information on residual disease coming from both bone marrow and extramedullary sites, liquid biopsy approaches are beginning to emerge. Currently under exploration are the detections at high sensitivity levels of circulating tumor DNA (70), circulating plasma cells (71), and M protein peptides (72–74). The further optimization of the available techniques will be essential for their future success.
As an example, applying the ClonoSEQ® assay to peripheral blood ctDNA and paired BM samples, Mazzotti et al. showed that residual disease in the peripheral blood was undetectable in 69% of patients with concurrent MRD-positive bone marrow samples (70). This was mainly due to an insufficient sensibility to detect specific Ig gene rearrangements in the peripheral blood when disease burden was low in the BM (70), underlying the need to improve the technique before we can routinely exploit peripheral blood to monitor MM burden.
MRD-Driven Trials
MRD has not yet entered the clinical practice, but it represents an attractive tool to potentially guide treatment choices. To address this hypothesis, many MRD-driven trials are beginning to explore treatment intensification in MRD-positive patients after standard treatment (e.g., NCT03901963) or treatment deintensification in sustained MRD-negative patients (e.g., NCT03710603). Ongoing and future MRD-driven trials will contribute to solve the unanswered question: is it recommended to evaluate other induction cycles until the achievement of MRD negativity in patients who are MRD-positive after 4 induction cycles? Can we perform post-transplant consolidation on the basis of MRD status? Can we stop maintenance after 1 year of sustained MRD negativity?
Ongoing and future clinical trials will evaluate the definition and the role of sustained MRD-negativity in treatment decision-making. On the one hand, the achievement of a MRD-negative status does not necessarily mean that treatment should be stopped. Indeed, it should be noted that what we define as “MRD-negative” is a MRD undetectable with the current techniques, each one of them having a sensitivity limit. This means that we are not sure that the disease is eradicated even in MRD-negative cases. On the other hand, the achievement of a MRD-positive status after treatment brings the question of whether it is necessary to change treatment, improving the depth of response. However, before developing response-adjusted treatment strategies based on MRD status—either intensifying/changing treatment for MRD-positive patients or de-escalating treatment for MRD-negative patients—we need to understand if sustained MRD negativity should be the treatment goal and to define the most appropriate timepoint for its evaluation (after 1 year or after more years).
Author Contributions
SO, MD'A, MB, and AL: substantial contributions to the conception or design, acquisition, analysis, or interpretation of data, critical revision for important intellectual content, final approval of the version to be published, and agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. SO, MD'A, and AL: first draft. MB and AL: supervision.
Conflict of Interest
SO has received honoraria from Amgen, Celgene, and Janssen; has served on the advisory boards for Adaptive Biotechnologies, Janssen, Amgen, and Takeda. MD'A has served on the advisory board for GSK. MB has received honoraria from Sanofi, Celgene, Amgen, Janssen, Novartis, AbbVie, and Bristol-Myers Squibb; has received research funding from Celgene, Janssen, Amgen, Bristol-Myers Squibb, Mundipharma, Novartis, and Sanofi. AL has received honoraria from Amgen, Bristol-Myers Squibb, Celgene, and Janssen; has served on the advisory boards for Bristol-Myers Squibb, Celgene, Janssen, and Takeda.
References
1. Landgren O, Lu SX, Hultcrantz M. MRD testing in multiple myeloma: the main future driver for modern tailored treatment. Semin Hematol. (2018) 55:44–50. doi: 10.1053/j.seminhematol.2018.03.001
2. Munshi NC, Avet-Loiseau H, Rawstron AC, Owen RG, Child JA, Thakurta A, et al. Association of minimal residual disease with superior survival outcomes in patients with multiple myeloma: a meta-analysis. JAMA Oncol. (2017) 3:28–35. doi: 10.1001/jamaoncol.2016.3160
3. Kumar S, Paiva B, Anderson KC, Durie B, Landgren O, Moreau P, et al. International myeloma working group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. (2016) 17:e328–46. doi: 10.1016/S1470-2045(16)30206-6
4. Flores-Montero J, de Tute R, Paiva B, Perez JJ, Böttcher S, Wind H, et al. Immunophenotype of normal vs. myeloma plasma cells: toward antibody panel specifications for MRD detection in multiple myeloma. Cytometry B Clin Cytom. (2016) 90:61–72. doi: 10.1002/cyto.b.21265
5. Kalina T, Flores-Montero J, Lecrevisse Q, Pedreira CE, van der Velden VHJ, Novakova M, et al. Quality assessment program for euroflow protocols: summary results of four-year (2010-2013) quality assurance rounds. Cytom Part A. (2015) 87:145–56. doi: 10.1002/cyto.a.22581
6. Stetler-Stevenson M, Paiva B, Stoolman L, Lin P, Jorgensen JL, Orfao A, et al. Consensus guidelines for myeloma minimal residual disease sample staining and data acquisition. Cytometry B Clin Cytom. (2016) 90:26–30. doi: 10.1002/cyto.b.21249
7. Flores-Montero J, Sanoja-Flores L, Paiva B, Puig N, García-Sánchez O, Böttcher S, et al. Next Generation Flow for highly sensitive and standardized detection of minimal residual disease in multiple myeloma. Leukemia. (2017) 31:2094–103. doi: 10.1038/leu.2017.29
8. Ladetto M, Pagliano G, Ferrero S, Cavallo F, Drandi D, Santo L, et al. Major tumor shrinking and persistent molecular remissions after consolidation with bortezomib, thalidomide, and dexamethasone in patients with autografted myeloma. J Clin Oncol. (2010) 28:2077–84. doi: 10.1200/JCO.2009.23.7172
9. Gambella M, Omedé P, Spada S, Muccio VE, Gilestro M, Saraci E, et al. Minimal residual disease by flow cytometry and allelic-specific oligonucleotide real-time quantitative polymerase chain reaction in patients with myeloma receiving lenalidomide maintenance: a pooled analysis. Cancer. (2019) 125:750–60. doi: 10.1002/cncr.31854
10. Puig N, Sarasquete ME, Balanzategui A, Martínez J, Paiva B, García H, et al. Critical evaluation of ASO RQ-PCR for minimal residual disease evaluation in multiple myeloma. A comparative analysis with flow cytometry. Leukemia. (2014) 28:391–7. doi: 10.1038/leu.2013.217
11. Korthals M, Sehnke N, Kronenwett R, Bruns I, Mau J, Zohren F, et al. The level of minimal residual disease in the bone marrow of patients with multiple myeloma before high-dose therapy and autologous blood stem cell transplantation is an independent predictive parameter. Biol Blood Marrow Transplant. (2012) 18:423–31.e3. doi: 10.1016/j.bbmt.2011.07.002
12. Faham M, Zheng J, Moorhead M, Carlton VEH, Stow P, Coustan-Smith E, et al. Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood. (2012) 120:5173–80. doi: 10.1182/blood-2012-07-444042
13. Ladetto M, Brüggemann M, Monitillo L, Ferrero S, Pepin F, Drandi D, et al. Next-generation sequencing and real-time quantitative PCR for minimal residual disease detection in B-cell disorders. Leukemia. (2014) 28:1299–307. doi: 10.1038/leu.2013.375
14. Avet-Loiseau H, Bene MC, Wuilleme S, Corre J, Attal M, Arnulf B, et al. Concordance of post-consolidation minimal residual disease rates by multiparametric flow cytometry and next-generation sequencing in CASSIOPEIA. In: 17th International Myeloma Workshop Abstract Book. Boston, MA. 8 [Abstract #OAB-004]. Available online: http://imw2019boston.org/images/Abstracts/17th_IMW_Abstract_Book_FINAL_V2.pdf (accessed December 12, 2019).
15. Martinez-Lopez J, Sanchez-Vega B, Barrio S, Cuenca I, Ruiz-Heredia Y, Alonso R, et al. Analytical and clinical validation of a novel in-house deep-sequencing method for minimal residual disease monitoring in a phase II trial for multiple myeloma. Leukemia. (2017) 31:1446–9. doi: 10.1038/leu.2017.58
16. Hillengass J, Landgren O. Challenges and opportunities of novel imaging techniques in monoclonal plasma cell disorders: imaging “early myeloma.” Leuk Lymphoma. (2013) 54:1355–63. doi: 10.3109/10428194.2012.740559
17. Bladé J, Fernández De Larrea C, Rosiñol L, Cibeira MT, Jiménez R, Powles R. Soft-tissue plasmacytomas in multiple myeloma: incidence, mechanisms of extramedullary spread, and treatment approach. J Clin Oncol. (2011) 29:3805–12. doi: 10.1200/JCO.2011.34.9290
18. Hillengass J, Bäuerle T, Bartl R, Andrulis M, Mcclanahan F, Laun FB, et al. Diffusion-weighted imaging for non-invasive and quantitative monitoring of bone marrow infiltration in patients with monoclonal plasma cell disease: a comparative study with histology. Br J Haematol. (2011) 153:721–8. doi: 10.1111/j.1365-2141.2011.08658.x
19. Moreau P. PET-CT in MM: a new definition of CR. Blood. (2011) 118:5984–5. doi: 10.1182/blood-2011-09-379818
20. Moreau P, Attal M, Caillot D, Macro M, Karlin L, Garderet L, et al. Prospective evaluation of magnetic resonance imaging and [18F]fluorodeoxyglucose positron emission tomography-computed tomography at diagnosis and before maintenance therapy in symptomatic patients with multiple myeloma included in the IFM/DFCI 2009 trial. J Clin Oncol. (2017) 35:2911–8. doi: 10.1200/JCO.2017.72.2975
21. Zamagni E, Patriarca F, Nanni C, Zannetti B, Englaro E, Pezzi A, et al. Prognostic relevance of 18-F FDG PET/CT in newly diagnosed multiple myeloma patients treated with up-front autologous transplantation. Blood. (2011) 118:5989–95. doi: 10.1182/blood-2011-06-361386
22. Bartel TB, Haessler J, Brown TLY, Shaughnessy JD, van Rhee F, Anaissie E, et al. F18-fluorodeoxyglucose positron emission tomography in the context of other imaging techniques and prognostic factors in multiple myeloma. Blood. (2009) 114:2068–76. doi: 10.1182/blood-2009-03-213280
23. Hillengass J, Ayyaz S, Kilk K, Weber MA, Hielscher T, Shah R, et al. Changes in magnetic resonance imaging before and after autologous stem cell transplantation correlate with response and survival in multiple myeloma. Haematologica. (2012) 97:1757–60. doi: 10.3324/haematol.2012.065359
24. Zamagni E, Cavo M. The role of imaging techniques in the management of multiple myeloma. Br J Haematol. (2012) 159:499–513. doi: 10.1111/bjh.12007
25. Hillengass J, Merz M, Delorme S. Minimal residual disease in multiple myeloma: use of magnetic resonance imaging. Semin Hematol. (2018) 55:19–21. doi: 10.1053/j.seminhematol.2018.02.001
26. Sachpekidis C, Mosebach J, Freitag MT, Wilhelm T, Mai EK, Goldschmidt H, et al. Application of (18)F-FDG PET and diffusion weighted imaging (DWI) in multiple myeloma: comparison of functional imaging modalities. Am J Nucl Med Mol Imaging. (2015) 5:479–92.
27. Pawlyn C, Fowkes L, Otero S, Jones JR, Boyd KD, Davies FE, et al. Whole-body diffusion-weighted MRI: A new gold standard for assessing disease burden in patients with multiple myeloma? Leukemia. (2016) 30:1446–8. doi: 10.1038/leu.2015.338
28. Rasche L, Alapat D, Kumar M, Gershner G, McDonald J, Wardell CP, et al. Combination of flow cytometry and functional imaging for monitoring of residual disease in myeloma. Leukemia. (2019) 33:1713–22. doi: 10.1038/s41375-018-0329-0
29. Paiva B, Puig N, Cedena MT, Cordon L, Vidriales M-B, Burgos L, et al. Impact of next-generation flow (NGF) Minimal Residual Disease (MRD) monitoring in multiple myeloma (MM): results from the Pethema/GEM2012 trial. Blood. (2017) 130: Abstract #905 [ASH 2017 58th Meeting]. doi: 10.1182/BLOOD.V130.SUPPL_1.905.905
30. Rasche L, Angtuaco E, McDonald JE, Buros A, Stein C, Pawlyn C, et al. Low expression of hexokinase-2 is associated with false-negative FDG–positron emission tomography in multiple myeloma. Blood. (2017) 130:30–4. doi: 10.1182/blood-2017-03-774422
31. Lapa C, Garcia-Velloso MJ, Lückerath K, Samnick S, Schreder M, Otero PR, et al. 11C-methionine-PET in multiple myeloma: a combined study from two different institutions. Theranostics. (2017) 7:2956–64. doi: 10.7150/thno.20491
32. Jamet B, Bailly C, Carlier T, Touzeau C, Nanni C, Zamagni E, et al. Interest of pet imaging in multiple myeloma. Front Med. (2019) 6:69. doi: 10.3389/fmed.2019.00069
33. Paiva B, Vidriales M-B, Cerveró J, Mateo G, Pérez JJ, Montalbán MA, et al. Multiparameter flow cytometric remission is the most relevant prognostic factor for multiple myeloma patients who undergo autologous stem cell transplantation. Blood. (2008) 112:4017–23. doi: 10.1182/blood-2008-05-159624
34. Rawstron AC, Child JA, de Tute RM, Davies FE, Gregory WM, Bell SE, et al. Minimal residual disease assessed by multiparameter flow cytometry in multiple myeloma: impact on outcome in the medical research council myeloma IX study. J Clin Oncol. (2013) 31:2540–7. doi: 10.1200/JCO.2012.46.2119
35. Kumar SK, Berdeja JG, Niesvizky R, Lonial S, Laubach JP, Hamadani M, et al. Ixazomib, lenalidomide, and dexamethasone in patients with newly diagnosed multiple myeloma: long-term follow-up including ixazomib maintenance. Leukemia. (2019) 33:1736–46. doi: 10.1038/s41375-019-0384-1
36. de Tute RM, Rawstron AC, Cairns DA, Pawlyn C, Davies FE, Collett C, et al. Impact of minimal residual disease in transplant ineligible myeloma patients: results from the UK NCRI myeloma XI trial. Blood. (2016) 128: Abstract #245 [ASH 2016 58th Meeting]. doi: 10.1182/blood.V128.22.245.245
37. Ludwig H, Greil R, Masszi T, Spicka I, Shpilberg O, Hajek R, et al. Bortezomib, thalidomide and dexamethasone, with or without cyclophosphamide, for patients with previously untreated multiple myeloma: 5-year follow-up. Br J Haematol. (2015) 171:344–54. doi: 10.1111/bjh.13582
38. Paiva B, Martinez-Lopez J, Vidriales MB, Mateos MV, Montalban MA, Fernandez-Redondo E, et al. Comparison of immunofixation, serum free light chain, and immunophenotyping for response evaluation and prognostication in multiple myeloma. J Clin Oncol. (2011) 29:1627–33. doi: 10.1200/JCO.2010.33.1967
39. Roussel M, Lauwers-Cances V, Robillard N, Hulin C, Leleu X, Benboubker L, et al. Front-line transplantation program with lenalidomide, bortezomib, and dexamethasone combination as induction and consolidation followed by lenalidomide maintenance in patients with multiple myeloma: a phase II study by the Intergroupe Francophone du Myélo. J Clin Oncol. (2014) 32:2712–7. doi: 10.1200/JCO.2013.54.8164
40. Paiva B, Gutiérrez NC, Rosiñol L, Vídriales M-B, Montalbán M-Á, Martínez-López J, et al. High-risk cytogenetics and persistent minimal residual disease by multiparameter flow cytometry predict unsustained complete response after autologous stem cell transplantation in multiple myeloma. Blood. (2012) 119:687–91. doi: 10.1182/blood-2011-07-370460
41. Ferrero S, Ladetto M, Drandi D, Cavallo F, Genuardi E, Urbano M, et al. Long-term results of the GIMEMA VEL-03-096 trial in MM patients receiving VTD consolidation after ASCT: MRD kinetics' impact on survival. Leukemia. (2015) 29:689–95. doi: 10.1038/leu.2014.219
42. Lahuerta J-J, Paiva B, Vidriales M-B, Cordón L, Cedena M-T, Puig N, et al. Depth of response in multiple myeloma: a pooled analysis of three PETHEMA/GEM clinical trials. J Clin Oncol. (2017) 35:2900–10. doi: 10.1200/JCO.2016.69.2517
43. Gu J, Liu J, Chen M, Huang B, Li J. Longitudinal flow cytometry identified “minimal residual disease” (MRD) evolution patterns for predicting the prognosis of patients with transplant-eligible multiple myeloma. Biol Blood Marrow Transplant. (2018) 24:2568–74. doi: 10.1016/j.bbmt.2018.07.040
44. Korde N, Roschewski M, Zingone A, Kwok M, Manasanch EE, Bhutani M, et al. Treatment with carfilzomib-lenalidomide-dexamethasone with lenalidomide extension in patients with smoldering or newly diagnosed multiple myeloma. JAMA Oncol. (2015) 1:746–54. doi: 10.1001/jamaoncol.2015.2010
45. Martinez-Lopez J, Lahuerta JJ, Pepin F, González M, Barrio S, Ayala R, et al. Prognostic value of deep sequencing method for minimal residual disease detection in multiple myeloma. Blood. (2014) 123:3073–9. doi: 10.1182/blood-2014-01-550020
46. Oliva S, Gambella M, Larocca A, Spada S, Marzanati E, Mantoan B, et al. Prognostic impact of minimal residual disease by ASO-RQ-PCR in multiple myeloma: a pooled analysis of 2 phase III studies in patients treated with lenalidomide after front-line therapy. Blood. (2016) 128: Abstract #4409 [ASH 2016 58th Meeting]. doi: 10.1182/blood.V128.22.4409.4409
47. Oliva S, Op Bruinink DH, RÍhová L, Spada S, van der Holt B, Troia R, et al. Minimal residual disease (MRD) monitoring by multiparameter flow cytometry (MFC) in newly diagnosed transplant eligible multiple myeloma (MM) patients: results from the EMN02/HO95 phase 3 trial. J Clin Oncol. (2017) 35 (15_suppl): Abstract #8011 [ASCO 2017 Annual Meeting]. doi: 10.1200/JCO.2017.35.15_suppl.8011
48. Paiva B, Cedena MT, Puig N, Arana P, Vidriales MB, Cordon L, et al. Minimal residual disease monitoring and immune profiling in multiple myeloma in elderly patients. Blood. (2016) 127:3165–74. doi: 10.1182/blood-2016-03-705319
49. Mateos M-V, Dimopoulos MA, Cavo M, Suzuki K, Jakubowiak A, Knop S, et al. Daratumumab plus bortezomib, melphalan, and prednisone for untreated myeloma. N Engl J Med. (2018) 378:518–28. doi: 10.1056/NEJMoa1714678
50. Facon T, Kumar S, Plesner T, Orlowski RZ, Moreau P, Bahlis N, et al. Daratumumab plus lenalidomide and dexamethasone for untreated myeloma. N Engl J Med. (2019) 380:2104–15. doi: 10.1056/NEJMoa1817249
51. Voorhees PM, Rodriguez C, Reeves B, Nathwani N, Costa LJ, Lutska Y, et al. Efficacy and updated safety analysis of a safety run-in cohort from griffin, a phase 2 randomized study of daratumumab (Dara), Bortezomib (V), Lenalidomide (R), and Dexamethasone (D; Dara-Vrd) Vs. Vrd in Patients (Pts) with Newly Diagnosed (ND) Multiple M. Blood. (2018) 132: Abstract #151 [ASH 2018 59th Meeting]. doi: 10.1182/blood-2018-151
52. Gay F, Cerrato C, Petrucci MT, Zambello R, Gamberi B, Ballanti S, et al. Efficacy of carfilzomib lenalidomide dexamethasone (KRd) with or without transplantation in newly diagnosed myeloma according to risk status: results from the forte trial. J Clin Oncol. (2019) 37: Abstract #8002 [ASCO 2019 Annual Meeting]. doi: 10.1200/JCO.2019.37.15_suppl.8002
53. Hahn TE, Wallace PK, Fraser R, Fei M, Tario JD, Howard A, et al. Minimal residual disease (MRD) assessment before and after autologous hematopoietic cell transplantation (AutoHCT) and maintenance for multiple myeloma (MM): results of the prognostic immunophenotyping for myeloma response (PRIMeR) study. Biol Blood Marrow Transplant. (2019) 25:S4–6. doi: 10.1016/j.bbmt.2018.12.687
54. Ocio EM, Otero PR, Bringhen S, Oliva S, Nogai A, Attal M, et al. Preliminary Results from a Phase I Study of Isatuximab (ISA) in Combination with Bortezomib, Lenalidomide, Dexamethasone (VRd) in patients with newly diagnosed multiple myeloma (NDMM) non-eligible for transplant. Blood. (2018) 132 (Suppl. 1): Abstract #595 [ASH 2018 60th Meeting]. doi: 10.1182/blood-2018-99-111244
55. Zimmerman T, Raje NS, Vij R, Reece D, Berdeja JG, Stephens LA, et al. Final results of a phase 2 trial of extended treatment (tx) with carfilzomib (CFZ), lenalidomide (LEN), and dexamethasone (KRd) plus autologous stem cell transplantation (ASCT) in newly diagnosed multiple myeloma (NDMM). Blood. (2016) 128: Abstract #675 [ASH 2016 58th Meeting]. doi: 10.1182/blood.V128.22.675.675
56. Avet-Loiseau H, Moreau P, Attal M, Hulin C, Arnulf B, Corre J, et al. Efficacy of daratumumab. (DARA) + bortezomib/thalidomide/dexamethasone (D-VTd) in transplant-eligible newly diagnosed multiple myeloma (TE NDMM) based on minimal residual disease (MRD) status: analysis of the CASSIOPEIA trial. J Clin Oncol. (2019) 37: Abstract #8017 [ASCO 2019 International Meeting]. doi: 10.1200/JCO.2019.37.15_suppl.8017
57. Takamatsu H, Takezako N, Zheng J, Moorhead M, Carlton VEH, Kong KA, et al. Prognostic value of sequencing-based minimal residual disease detection in patients with multiple myeloma who underwent autologous stem-cell transplantation. Ann Oncol. (2017) 28:2503–10. doi: 10.1093/annonc/mdx340
58. Perrot A, Lauwers-Cances V, Corre J, Robillard N, Hulin C, Chretien ML, et al. Minimal residual disease negativity using deep sequencing is a major prognostic factor in multiple myeloma. Blood. (2018) 132:2456–64. doi: 10.1182/blood-2018-06-858613
59. Dimopoulos MA, San-Miguel J, Belch A, White D, Benboubker L, Cook G, et al. Daratumumab plus lenalidomide and dexamethasone versus lenalidomide and dexamethasone in relapsed or refractory multiple myeloma: updated analysis of POLLUX. Haematologica. (2018) 103:2088–96. doi: 10.3324/haematol.2018.194282
60. Mateos M-V, Martínez-López J, Rodríguez-Otero P, de la Calle GV, González M-S, Oriol A, et al. Curative strategy (gem-cesar) for high-risk smoldering myeloma: carfilzomib, lenalidomide and dexamethasone (krd) as induction followed by hdt-asct, consolidation with krd and maintenance with rd. HemaSphere. (2019) 3: 390 [Abstract #S871, EHA 2019 24th Congress]. doi: 10.1097/01.HS9.0000561764.53887.c0
61. Kunacheewa C, Lin P, Manasanch EE. Minimal residual disease in multiple myeloma 2019. Adv Cell Gene Ther. (2019) e71:1–15. doi: 10.1002/acg2.71
62. Landgren O, Devlin S, Boulad M, Mailankody S. Role of MRD status in relation to clinical outcomes in newly diagnosed multiple myeloma patients: a meta-analysis. Bone Marrow Transplant. (2016) 51:1565–8. doi: 10.1038/bmt.2016.222
63. Rawstron AC, Gregory WM, de Tute RM, Davies FE, Bell SE, Drayson MT, et al. Minimal residual disease in myeloma by flow cytometry: independent prediction of survival benefit per log reduction. Blood. (2015) 125:1932–5. doi: 10.1182/blood-2014-07-590166
64. Avet-Loiseau H, Corre J, Lauwers-Cances V, Chretien M-L, Robillard N, Leleu X, et al. Evaluation of minimal residual disease (MRD) by next generation sequencing (NGS) is highly predictive of progression free survival in the IFM/DFCI 2009 trial. Blood. (2015) 126: Abstract #191 [ASH 2015 57th Meeting]. doi: 10.1182/blood.V126.23.191.191
65. D'Agostino M, De Paoli L, Conticello C, Offidani M, Ria R, Petrucci MT, et al. Continuous therapy in standard- and high-risk newly-diagnosed multiple myeloma: a pooled analysis of 2 phase III trials. Crit Rev Oncol Hematol. (2018) 132:9–16. doi: 10.1016/j.critrevonc.2018.09.008
66. FDA Authorizes First Next Generation Sequencing-Based Test to Detect Very Low Levels of Remaining Cancer Cells in Patients With Acute Lymphoblastic Leukemia or Multiple Myeloma. (2018) Available online at: https://www.fda.gov/news-events/press-announcements/fda-authorizes-first-next-generation-sequencing-based-test-detect-very-low-levels-remaining-cancer (accessed October 23, 2019).
67. NCT02755597—A Study Evaluating Venetoclax (ABT-199) in Multiple Myeloma Subjects Who Are Receiving Bortezomib Dexamethasone as Standard Therapy - Full Text View - ClinicalTrials,.gov. (2016) Available online at: https://clinicaltrials.gov/ct2/show/NCT02755597 (accessed October 23, 2019).
68. Kumar S, Rajkumar SV. Surrogate endpoints in randomised controlled trials: a reality check. Lancet. (2019) 394:281–3. doi: 10.1016/S0140-6736(19)31711-8
69. Lee N, Moon SY, Lee J-H, Park H-K, Kong S-Y, Bang S-M, et al. Discrepancies between the percentage of plasma cells in bone marrow aspiration and BM biopsy: impact on the revised IMWG diagnostic criteria of multiple myeloma. Blood Cancer J. (2017) 7:e530. doi: 10.1038/bcj.2017.14
70. Mazzotti C, Buisson L, Maheo S, Perrot A, Chretien M-L, Leleu X, et al. Myeloma MRD by deep sequencing from circulating tumor DNA does not correlate with results obtained in the bone marrow. Blood Adv. (2018) 2:2811–3. doi: 10.1182/bloodadvances.2018025197
71. Thiele JA, Pitule P, Hicks J, Kuhn P. Single-cell analysis of circulating tumor cells. In: Murray S, editor. Methods in Molecular Biology (New York, NY: Humana Press Inc.). p. 243–64. doi: 10.1007/978-1-4939-9004-7_17
72. Thoren KL. Mass spectrometry methods for detecting monoclonal immunoglobulins in multiple myeloma minimal residual disease. Semin Hematol. (2018) 55:41–3. doi: 10.1053/j.seminhematol.2018.02.008
73. Bergen HR, Dasari S, Dispenzieri A, Mills JR, Ramirez-Alvarado M, Tschumper RC, et al. Clonotypic light chain peptides identified for monitoring minimal residual disease in multiple myeloma without bone marrow aspiration. Clin Chem. (2016) 62:243–51. doi: 10.1373/clinchem.2015.242651
Keywords: multiple myeloma (MM), minimal residual disease (MRD), clinical practice, next-generation flow (NGF), next-generation sequencing (NGS), PET/CT
Citation: Oliva S, D'Agostino M, Boccadoro M and Larocca A (2020) Clinical Applications and Future Directions of Minimal Residual Disease Testing in Multiple Myeloma. Front. Oncol. 10:1. doi: 10.3389/fonc.2020.00001
Received: 29 October 2019; Accepted: 02 January 2020;
Published: 31 January 2020.
Edited by:
Francesco Maura, Memorial Sloan Kettering Cancer Center, United StatesReviewed by:
Cirino Botta, Cosenza Hospital, ItalyMatteo Claudio Da Vià, University of Milan, Italy
Dickran Kazandjian, National Cancer Institute, National Institutes of Health (NIH), United States
Copyright © 2020 Oliva, D'Agostino, Boccadoro and Larocca. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandra Larocca, YWxlbGFyb2NjYUBob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work and share first authorship