 Christine M. Heske
Christine M. Heske- Pediatric Oncology Branch, National Cancer Institute, National Institutes of Health, Bethesda, MD, United States
Tumor cells have increased requirements for NAD+. Thus, many cancers exhibit an increased reliance on NAD+ production pathways. This dependence may be exploited therapeutically through pharmacological targeting of NAMPT, the rate-limiting enzyme in the NAD+ salvage pathway. Despite promising preclinical data using NAMPT inhibitors in cancer models, early NAMPT inhibitors showed limited efficacy in several early phase clinical trials, necessitating the identification of strategies, such as drug combinations, to enhance their efficacy. While the effect of NAMPT inhibitors on impairment of energy metabolism in cancer cells has been well-described, more recent insights have uncovered a number of additional targetable cellular processes that are impacted by inhibition of NAMPT. These include sirtuin function, DNA repair machinery, redox homeostasis, molecular signaling, cellular stemness, and immune processes. This review highlights the recent findings describing the effects of NAMPT inhibitors on the non-metabolic functions of malignant cells, with a focus on how this information can be leveraged clinically. Combining NAMPT inhibitors with other therapies that target NAD+-dependent processes or selecting tumors with specific vulnerabilities that can be co-targeted with NAMPT inhibitors may represent opportunities to exploit the multiple functions of this enzyme for greater therapeutic benefit.
Introduction
Cancer cells have altered metabolic needs, including an accelerated rate of nicotinamide adenine dinucleotide (NAD+) cycling relative to normal cells (1). To maintain this, NAD+ metabolism is altered in cancer cells, many of which have an increased dependence on certain NAD+ production enzymes (2, 3) (Figure 1). Several redundant NAD+ production pathways exist. In the de novo pathway, tryptophan is first converted to quinolinic acid (QA) through a series of steps; QA is converted to nicotinic acid mononucleotide (NAMN) via quinolinate phosphoribosyltransferase (QPRT) and is then converted to NAD+ via nicotinamide nucleotide adenylyltransferase (NMNAT) and NAD synthetase (NADS). In normal cells, QPRT expression follows a tissue-specific distribution; more recent insights have revealed that QPRT expression is altered in some cancer cells (4–7). The Preiss-Handler pathway converts nicotinic acid (NA) to NAMN through nicotinate phosphoribosyltransferase (NAPRT), an enzyme that is widely expressed in normal tissues but variably expressed in cancer cells (8–11). NAMN is then converted to NAD+ through the activity of NMNAT and NADS, as in the de novo pathway. The salvage pathway, of which nicotinamide phosphoribosyltransferase (NAMPT) is the rate-limiting enzyme, converts nicotinamide (NAM) to nicotinamide mononucleotide (NMN), which is then converted to NAD+ through NMNAT. This pathway is of major importance to cancer cells, as it recycles NAM, the product of NAD+-consuming enzymes, back to NAD+. In fact, many types of cancer cells have been shown to highly express NAMPT, reflecting potentially increased dependence on this pathway due to high NAD+ utilization and in some cases, loss of expression of other key NAD+ biosynthetic enzymes (3, 9, 12, 13). Among the types of cancers reported to have high NAMPT expression are colorectal cancer (CRC), breast cancer, osteosarcoma, chondrosarcoma, pancreatic ductal adenocarcinoma, oral squamous cell carcinoma, prostate cancer, rhabdomyosarcoma, leiomyosarcoma, esophagogastric junction adenocarcinomas, thyroid cancer, leukemia, lymphoma, ovarian cancer, and some renal cancer, and in many of these, higher expression correlated with worse outcomes (14–30). Of note, NMN may also be produced from nicotinamide riboside via nicotinamide riboside kinase (9). Currently, however, NAMPT is the only NAD+ production enzyme that has been targeted in the clinic (2, 31, 32).
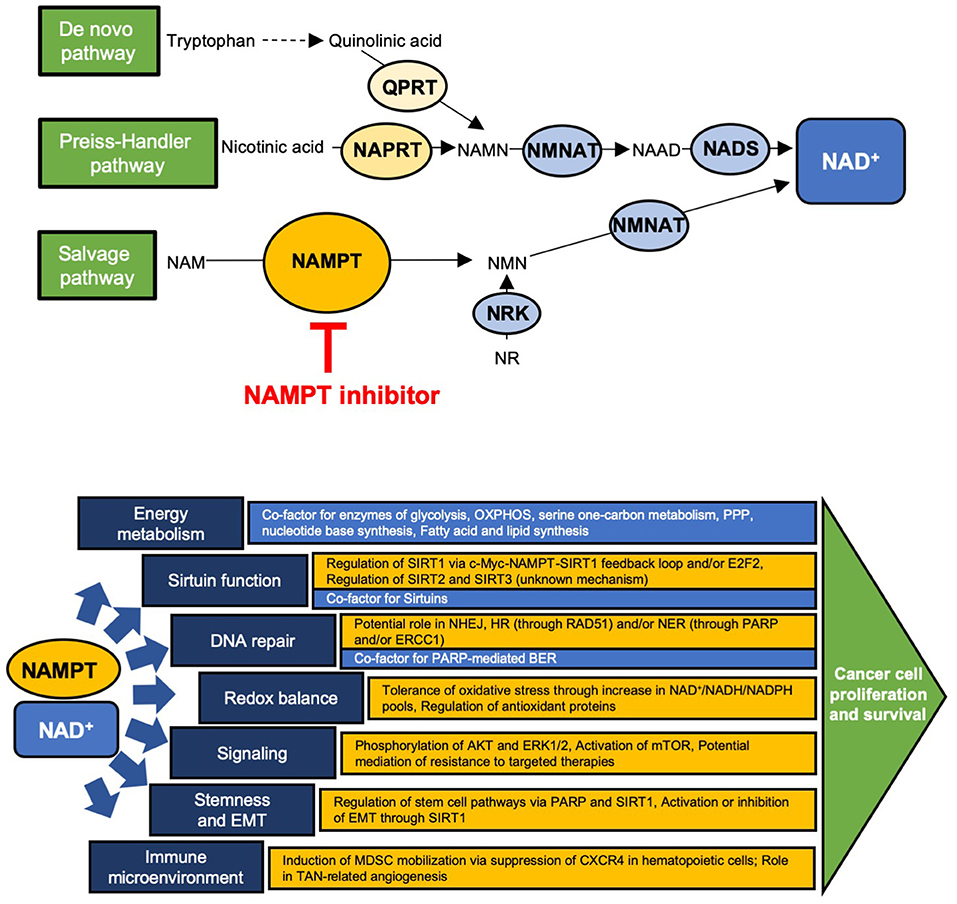
Figure 1. Schematic of the NAD+ production pathways and key enzymes and site of action of NAMPT inhibitors (Top) and the major downstream cellular functions of NAD+ (blue) and NAMPT (yellow) (Bottom). (Top) QPRT, quinolinate phosphoribosyltransferase; NAPRT, nicotinate phosphoribosyltransferase; NAMPT, nicotinamide phosphoribosyltransferase; NMNAT, nicotinamide nucleotide adenylyltransferase; NRK, nicotinamide riboside kinase; NADS, NAD+ synthetase; NAMN, nicotinic acid mononucleotide; NAAD, nicotinic acid adenine dinucleotide; NAM, nicotinamide; NMN, nicotinamide mononucleotide; NR, nicotinamide riboside. (Bottom) OXPHOS, oxidative phosphorylation; PPP, pentose phosphate pathway; E2F2, E2F family member 2; NHEJ, non-homologous end joining; HR, homologous recombination; NER, nucleotide excision repair; BER, base excision repair; PARP, poly-ADP ribose polymerase; MDSC, myeloid-derived suppressor cell; TAN, tumor associated neutrophil.
Clinical NAMPT inhibitors have investigated in a number of early phase clinical trials (Table 1). Published results on the early phase experience with first generation clinical NAMPT inhibitors describe a disease control rate of ~25% and few objective responses (33–38). Given the limited efficacy seen in these small studies, efforts to optimize the use of NAMPT inhibitors in the clinic are necessary. These include strategies such as drug combinations or selection of specific patient subsets more likely to be sensitive to these agents. Several new NAMPT inhibitors have recently entered early phase testing and preclinical efforts are focusing on use of these potential strategies to enhance activity and minimize toxicities (2, 3, 31, 32, 39).
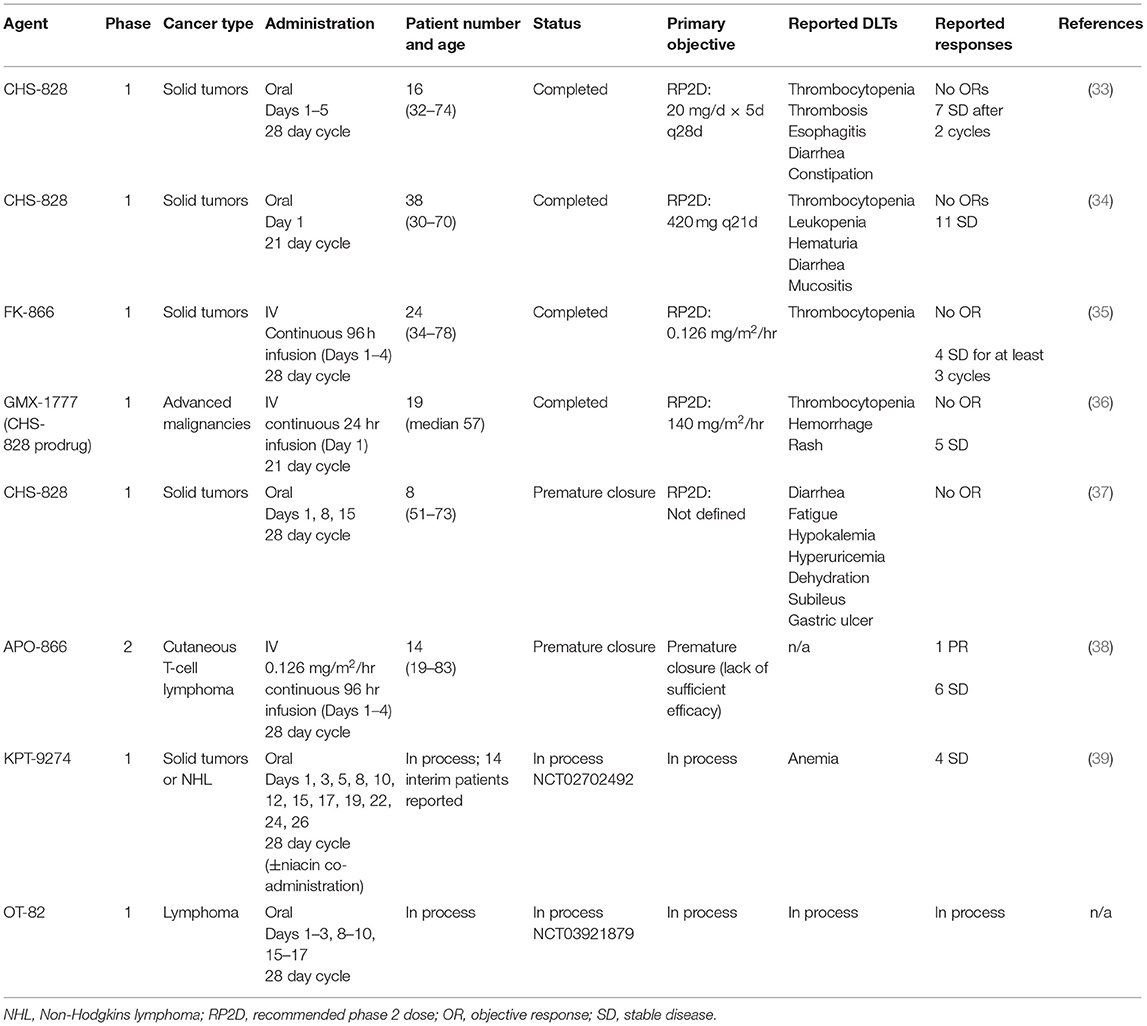
Table 1. Summary of clinical trials testing NAMPT inhibitors.
The role of NAD+ in supporting the energy metabolism of cancer cells has been well-established as NAD+ is an important co-factor for a number of metabolic enzymes (10). Accordingly, NAMPT inhibitors have been shown to impair energy metabolism through disruption of specific metabolic pathways, decreased ATP production, and increased energetic stress (40–44). In cancer cells, NAMPT inhibitors affect glycolysis (40, 45–55), oxidative phosphorylation (45, 49, 53, 56–59), serine biosynthesis and one-carbon metabolism (60), the pentose phosphate pathway (40, 53, 61), amino acid metabolism (53), purine and pyrimidine metabolism (53), and fatty acid and lipid synthesis (45, 62). In addition, cancer types harboring mutations in metabolic pathways, such as isocitrate dehydrogenase (IDH), have been shown to be exquisitely sensitive to loss of NAMPT activity (63–65).
Beyond its role in energy metabolism, NAD+ plays a vital role in many other cellular functions which may similarly be targeted with NAMPT inhibitors (13, 32, 66–70). An understanding of the non-metabolic implications of NAMPT inhibition may uncover additional targetable vulnerabilities, providing combinatorial opportunities for therapeutic intervention in cancer. The purpose of this review is to describe the impact of NAMPT and NAMPT inhibition on the non-energetic cellular functions of NAD+ in cancer cells.
Sirtuin Function
The sirtuins are a family of NAD+-dependent deacylases and ADP-ribosyltransferases that are responsible for a significant amount of cellular NAD+ consumption (71). The role of sirtuins in cancer is increasingly being described and consequently, there is a growing understanding of how targeting sirtuin function may be beneficial in certain cancer types (72). Among these insights are an appreciation of the role of NAMPT in sirtuin expression (73). In CRC models, several groups have reported that regulation of SIRT1 activity is mediated by NAMPT (74–77). In some cases, NAMPT was found to be a direct transcriptional target of c-Myc, resulting in a positive feedback loop between c-Myc, NAMPT, and SIRT1 that drove tumor cell proliferation and progression (74–76). These studies showed that the use of NAMPT inhibitors resulted in a loss of SIRT1 expression, de-repression of TP53, and decreased tumor cell growth in CRC models (74, 75, 77). A similar effect has been observed in prostate (19) and gastric cancer models (78).
Regulation of the sirtuins by NAMPT has additionally been reported in other cancer types. In melanoma cells, NAMPT regulation of the E2F family member 2 was shown to impact transcription and translation of SIRT1, and genetic or pharmacologic loss of NAMPT activity resulted in activation of TP53 and apoptotic cell death (79). A similar regulatory network has been described in breast cancer cells where increased SIRT1 activity and induction of deacetylation of TP53 were observed upon exposure to the extracellular form of NAMPT (80).
In cancers where a direct regulatory link between NAMPT and the sirtuins has not yet been elucidated, experimental induction of NAMPT has been shown to have a corresponding effect on SIRT1 activity (81), while genetic or pharmacologic inhibition of NAMPT has resulted in decreased SIRT1 (82–85), SIRT2 (86), and SIRT3 (87). Interestingly, the functional significance of NAMPT on sirtuins is likely cancer cell type specific, since changes in SIRT1 expression with NAMPT inhibition have not been observed in all NAMPT inhibitor-sensitive cells (88). These differences may be important for clinical translation of NAMPT inhibitors, as they may play a role in determining which cancer types are more susceptible to NAMPT inhibitors (89).
DNA Damage Repair Response
Poly-ADP ribose polymerases (PARPs) represent another group of NAD+-dependent proteins that consume a large proportion of cellular NAD+ (90). Among their functions, PARPs play a key role in DNA damage detection and repair (91). Thus, an expected consequence of NAD+ depletion is impaired DNA damage repair. Decreased PARP activity with NAMPT inhibitor use has been reported in several cancer models, forming the basis for preclinical testing of NAMPT inhibitors plus PARP inhibitors in Ewing sarcoma and triple-negative breast cancer (92, 93). In both studies, synergy between NAMPT inhibitors and PARP inhibitors was observed. In the breast cancer study, the effect was noted to be greatest in BRCA-deficient models, suggesting that underlying defects in homologous recombination (HR) may further enhance the efficacy of NAMPT inhibition (93). This is supported by data indicating that NAMPT inhibition impairs non-homologous end joining and increases cellular dependence on HR (94). In ovarian cancer models, a regulatory relationship between BRCA1 and NAMPT has been described (95). Although it is not BRCA-deficient, Ewing sarcoma is also characterized by defective HR (96), further supporting the idea that cancers with defective DNA repair mechanisms may have increased susceptibility to NAMPT inhibition. Interestingly, results from a recent study in preclinical leukemia models revealed functional antagonism between NAMPT and PARP inhibitors, suggesting that cell type specific differences in how these pathways interact may be present (97).
Other cancer types with DNA repair deficiencies have been identified as selectively sensitive to NAMPT inhibitors. Non-small cell lung cancers (NSCLC) with excision repair cross-complementation group 1 (ERCC1) deficiency were exquisitely sensitive to NAMPT inhibitors, in vitro and in vivo (98). ERCC1 deficiency is also associated with mitochondrial defects, suggesting that additional factors may contribute to NAMPT inhibitor sensitivity in this cancer subtype. In ovarian cancer, expression of NAPRT, the key enzyme in the Preiss-Handler pathway, correlated with a BRCA-ness gene expression signature, and cells carrying these features were more sensitive to NAMPT inhibitors (99). Mechanistic studies of the downstream consequences of NAMPT inhibitors on DNA damage repair have described a variety of effects including decreased PARylation (92, 99) decreased RAD51, and impaired double-strand break repair by the HR pathway (100).
Given these insights, a number of studies have sought to determine the efficacy of NAMPT inhibitors when combined with DNA damaging agents. Enhanced antitumor activity has been reported across multiple malignancies when genetic or pharmacological inhibition of NAMPT has been combined with radiation (101), DNA alkylating agents (63, 99, 100, 102, 103), topoisomerase inhibitors (19, 46, 86), or other classes of chemotherapy known to augment the effects of impaired DNA repair (19, 20, 43, 46, 104–106). Surprisingly, in some cancers, an improvement in efficacy was restricted to combinations with only certain chemotherapeutic agents, as in preclinical studies in pancreatic cancer models which revealed that gemcitabine, but not 5-fluorouracil (5-FU) or oxaliplatin, enhanced the antiproliferative effect of NAMPT inhibitors (107). In other studies, the combinatorial effects of NAMPT inhibitors with drugs from across chemotherapeutic classes was similar (46). The mechanisms for these differences are not known. Lastly, a study characterizing the effects of resistance to NAMPT inhibitors in CRC cell lines reported changes in expression of genes involved in DNA repair and an increased sensitivity to DNA damaging agents, further suggesting an intimate connection between NAMPT dependency and DNA damage repair (108). Taken together, these insights have clinical implications as they suggest that tumors with certain defects in DNA repair mechanisms may be selectively sensitive to NAMPT inhibitors, and that rational combinations with chemotherapies may enhance the efficacy of this class of agents, particularly in selected tumor types.
Redox Homeostasis
Maintenance of intracellular redox homeostasis is a critical cellular process requiring a balance between reactive oxygen species (ROS) generation and elimination, as excessive levels of ROS can result in cell death (109). NAD+ is an important regulator of cellular ROS levels which can accumulate upon depletion of NAD+ (110). This is particularly true in cancer cells which generally have increased ROS production and require very tight control of ROS balance (111). Hence, an additional consequence of NAMPT inhibition is disruption of ROS homeostasis.
NAMPT has been shown to contribute to the cellular capacity to tolerate oxidative stress in a number of studies. In CRC cell lines, NAMPT functioned to increase NADH pools, protecting cells against oxidative stress (112). In breast cancer models, NAMPT increased the pool of NAD+ that could be converted to NADPH through the pentose phosphate pathway, thus maintaining glutathione in the reduced state. This was of particular importance in cells undergoing glucose-deprivation, for which high levels of NAMPT decreased mitochondrial ROS levels (61). Additionally, in several studies, NAMPT inhibition enhanced cancer cell susceptibility to oxidative stress through a reduction in antioxidative capacity via downregulation of antioxidant proteins (102, 113).
Depletion or inhibition of NAMPT has been shown to increase ROS in models of NSCLC (40), leukemia (43, 97), prostate cancer (19), breast cancer (61), glioblastoma (102), CRC (112), and others (85). In all cases, this was associated with a loss of cancer cell viability. In addition, there was a differential effect noted between the induction of ROS in cancer cells compared to normal cells treated with NAMPT inhibitors, suggesting the existence of a therapeutic window for ROS induction with these agents (114). Interestingly, not all cancer cells exhibit an increase in ROS upon inhibition of NAMPT. In Ewing sarcoma and some NSCLC models, cells were able to maintain ROS balance in the presence of a NAMPT inhibitor, suggesting that there may be cell-type dependent differences in these effects (40, 92). This may be the result of active compensatory NAD+ production pathways, such as the Preiss-Handler pathway, in certain cancers (114). A comprehensive understanding of these differences will be important to clinical translation of this class of agents as they may have implications for patient selection.
Finally, several studies have reported on the efficacy of combining NAMPT inhibitors with ROS inducing agents. Use of NAMPT inhibitors plus β-lapachone, an agent that targets the NADPH quinone oxidoreductase-1 (NQO1) and generates ROS, resulted in excessive ROS production and had enhanced efficacy against growth of pancreatic adenocarcinoma cells, particularly those which overexpressed NQO1 (52, 115). A similar effect was seen with this combination in NSCLC models (83). In addition, adding a NAMPT inhibitor to ROS-containing plasma-activated medium resulted in increased ROS production, decreased intracellular reduced glutathione, and cell death of breast cancer cells (116).
Oncogenic Signaling
Crosstalk between NAMPT and oncogenic signaling pathways has been reported in several cancer models. From a clinical perspective, co-targeting NAMPT with these pathways may be a beneficial strategy. In some cases, oncogenic factors regulate expression and activity of NAMPT, such as in Ewing sarcoma, where the oncogenic transcription factor EWS-FLI1 has been shown to regulate NAMPT expression (49) and in breast cancer, where FOXO1, a tumor suppressor, negatively regulates the expression of NAMPT while AKT positively regulates it (117). In other cases, NAMPT regulates the activity of oncogenic signaling pathways. For example, NAMPT overexpression in breast cancer cells and extracellular NAMPT (eNAMPT) released by melanoma cell have both been associated with AKT phosphorylation (118, 119). Exogenous eNAMPT was also found to induce phosphorylation of AKT and ERK1/2 and increase proliferation of breast cancer cells, and the use of AKT and ERK1/2 inhibitors could abrogate these effects (120). In multiple cancer models, a decrease in phospho-ERK1/2 was observed with NAMPT inhibition (44, 121, 122) and combining NAMPT inhibitors with ERK1/2 blockade enhanced cell death (121).
An interaction between NAMPT and mTOR has also been described in a number of malignancies. In hepatocellular carcinoma cells, NAMPT inhibition was associated with loss of activation of mTOR and its downstream targets. A corresponding increase in AMPKα activation was also noted (41). A similar effect was observed in leukemia cells (42), pancreatic ductal adenocarcinoma cells (46), and pancreatic neuroendocrine tumor cells (123). In both pancreatic cancer subtypes, the antiproliferative effect of NAMPT inhibition could be potentiated with concurrent mTOR inhibitor treatment (46, 123). In multiple myeloma models, NAMPT inhibition was also associated with loss of mTOR activation which was thought to contribute to autophagic death (121). In contrast, changes in AMPKα and mTOR were not observed in studies of non-cancerous cells treated with NAMPT inhibitors (41).
A number of studies have investigated changes in NAMPT expression that occur with development of drug resistance to targeted therapies. Both in clinical samples and experimental models, BRAF inhibitor resistant melanoma cells expressed higher levels of both intra- and extracellular NAMPT than their sensitive counterparts (57, 124). Remarkably, BRAF inhibitor resistance could be overcome with addition of NAMPT inhibitors (57). In addition, induced expression of NAMPT was able to render melanoma cells resistant to BRAF inhibitors while BRAF inhibition in sensitive cells resulted in transcriptional downregulation of NAMPT (125).
In addition to the oncogenic signaling molecules already described, correlative studies have also proposed a link between NAMPT expression and EGFR (44, 126), HER2, and estrogen receptor positivity (127). Furthermore, based on the data supporting crosstalk between oncogenic signaling and NAMPT, co-targeting NAMPT along with other signaling pathway molecules, as has been described with the BTK inhibitor ibrutinib in Waldenstrom macroglobulinemia cells, could be a promising therapeutic strategy (128).
Epithelial-Mesenchymal Transformation and Stemness
NAMPT has been described as a mediator of cancer cell stemness (129). Studies in clinical CRC tumor samples revealed that high NAMPT expression was associated with the presence of a high proportion of cancer-initiating cells. Mechanistically, this was the result of the influence of NAMPT on transcriptional regulation of stem cell signaling pathways and was mediated by SIRT1 and PARP (126, 130). In glioblastoma tumors and patient-derived stem-like cells, high NAMPT expression was observed (131). NAMPT overexpression in experimental models of glioblastoma resulted in a cellular phenotype consistent with that of a cancer stem-like cell (126), while pharmacological and genetic inhibition of NAMPT decreased the ability of glioblastoma stem cells to self-renew and form in vivo tumors (131). In one study, the loss of cancer stem cell pluripotency upon inhibition of NAMPT was the result of an excess of autophagy, a well-described consequence of NAMPT inhibition (15, 58, 64, 121, 132–135), which disrupted the maintenance of cancer cell stemness (136). NAMPT inhibition has also been shown to reverse the ability of cancer cells to dedifferentiate (137).
NAMPT inhibition also affects epithelial-mesenchymal transition (EMT) in cancer cells. In hepatocellular carcinoma cells, pharmacological NAMPT inhibition resulted in changes in EMT marker proteins indicating a reversal of EMT, as well as a reduction in cellular capacity for invasion and metastasis formation, through a decrease in SIRT1 (84). Similarly, data showing that both NAMPT overexpression and exogenous eNAMPT induced EMT in breast cancer cell lines (127), and that eNAMPT promoted osteosarcoma cell migration and invasion (138). Furthermore, NAMPT inhibition diminished motility in glioma cells (54). In contrast, in lung cancer cell lines, NAMPT inhibition activated EMT and increased cellular invasiveness also through decreased SIRT1 (85), and in breast cancer models, NAMPT inhibition enhanced metastatic behavior (139), suggesting the impact of NAMPT inhibition on EMT may be cell-type specific. Interestingly, expression of NAPRT, which differs across cancer cell lines, correlates with EMT status (140), and may be related to the differential effects of NAMPT inhibition on EMT. Thus, an understanding of the effect of NAMPT inhibition on metastasis is important as it may differ for different malignancies, impacting optimal clinical translation of these agents.
Immune Regulation of Tumor Microenvironment
In addition to the effect of NAMPT on primary tumor cells, recent insights have begun to elucidate the impact of NAMPT on the immune suppressive characteristics of the tumor microenvironment (26, 141). In murine cancer models, macrophage colony stimulating factor was shown to increase NAMPT expression in myeloid cells which, in turn, negatively regulated CXCR4 expression in hematopoietic cells in the bone marrow. Consequently, low CXCR4 resulted in mobilization of immature myeloid-derived suppressor cells (MDSCs), contributing to tumor immunosuppression. Importantly, pharmacologic inhibition of NAMPT resulted in a decrease in MDSC mobilization, reversing the immunosuppression and re-sensitizing tumor cells to immunotherapeutic agents in preclinical models (142).
NAMPT has also been shown to be upregulated in tumor associated neutrophils (TANs) in patients with melanoma and head and neck cancer, and in murine cancer models. Inhibition of NAMPT in ex vivo TANs followed by adoptive transfer of the TANs into tumor-bearing mice reduced tumor angiogenesis and proliferation through suppression of SIRT1 and resultant transcriptional blockade of pro-angiogenic genes (143). While more studies are required to better understand the role NAMPT inhibition plays in the microenvironmental immune milieu, these and other preliminary reports suggest NAMPT inhibitors could be used to enhance immunotherapies in the clinic (144). In addition, correlative studies describing the effects of NAMPT inhibition on tumor microenvironmental factors are currently lacking in the clinical literature but would be informative to further clinical development of this class of agents and should be pursued in future studies.
Discussion
Given the critical role that NAD+ plays in the growth and survival of malignant cells, NAMPT is an attractive therapeutic target in cancer. In addition to its function in cellular energy metabolism, NAMPT is involved in sirtuin function, support of DNA repair mechanisms, maintenance of redox balance, molecular signaling, determination of cellular states, and tumor-related immune suppression. Depending on the cancer cell type, NAMPT inhibitors may be able to impair many of these additional functions. Furthermore, combination therapies with agents that target these functions in a complementary manner have the potential to dramatically improve the efficacy of NAMPT inhibitors. There are an increasing number of reports describing additive or synergistic effects of NAMPT inhibitors being used in combination with other agents in the preclinical setting. With newer generation NAMPT inhibitors currently undergoing phase 1 evaluation, clinical translation of these rational combinations is a logical next step.
In addition to developing rational combination regimens using NAMPT inhibitors, careful patient selection represents an additional opportunity to maximize the efficacy of these agents. For example, IDH mutant cancers have been shown to have exquisite sensitivity to NAMPT inhibitors (63–65), as have tumors deficient in NAPRT (9, 145–151). Patient selection may also be guided by recognition of specific vulnerabilities in the non-metabolic pathways supported by NAMPT, such as HR-deficiency or EMT targeting for metastatic disease. In conclusion, it is critical to understand the impact of NAMPT and NAMPT inhibitors on both the energetic and the non-energetic cellular functions of NAD+ in cancer as these insights may be key to future development of this class of agents.
Author Contributions
CH researched, wrote, and edited the manuscript.
Funding
The author was supported by grants from the Intramural Research Program of the National Institutes of Health (NIH), National Cancer Institute, Center for Cancer Research.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author would like to thank Mr. Choh Yeung, Dr. Marielle Yohe, and Dr. Sameer Issaq for critical reading of the manuscript.
References
1. Chiarugi A, Dolle C, Felici R, Ziegler M. The NAD metabolome–a key determinant of cancer cell biology. Nat Rev Cancer. (2012) 12:741–52. doi: 10.1038/nrc3340
2. Roulston A, Shore GC. New strategies to maximize therapeutic opportunities for NAMPT inhibitors in oncology. Mol Cell Oncol. (2016) 3:e1052180. doi: 10.1080/23723556.2015.1052180
3. Sampath D, Zabka TS, Misner DL, O'Brien T, Dragovich PS. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) as a therapeutic strategy in cancer. Pharmacol Therap. (2015) 151:16–31. doi: 10.1016/j.pharmthera.2015.02.004
4. Ishidoh K, Kamemura N, Imagawa T, Oda M, Sakurai J, Katunuma N. Quinolinate phosphoribosyl transferase, a key enzyme in de novo NAD(+) synthesis, suppresses spontaneous cell death by inhibiting overproduction of active-caspase-3. Biochim Biophys Acta. (2010) 1803:527–33. doi: 10.1016/j.bbamcr.2010.02.007
5. Hinsch N, Frank M, Doring C, Vorlander C, Hansmann ML. QPRT: a potential marker for follicular thyroid carcinoma including minimal invasive variant; a gene expression, RNA and immunohistochemical study. BMC Cancer. (2009) 9:93. doi: 10.1186/1471-2407-9-93
6. Ullmark T, Montano G, Jarvstrat L, Jernmark Nilsson H, Hakansson E, Drott K, et al. Anti-apoptotic quinolinate phosphoribosyltransferase (QPRT) is a target gene of Wilms' tumor gene 1 (WT1) protein in leukemic cells. Biochem Biophys Res Commun. (2017) 482:802–7. doi: 10.1016/j.bbrc.2016.11.114
7. Haslinger D, Waltes R, Yousaf A, Lindlar S, Schneider I, Lim CK, et al. Loss of the Chr16p11.2 ASD candidate gene QPRT leads to aberrant neuronal differentiation in the SH-SY5Y neuronal cell model. Mol Autism. (2018) 9:56. doi: 10.1186/s13229-018-0239-z
8. Olesen UH, Thougaard AV, Jensen PB, Sehested M. A preclinical study on the rescue of normal tissue by nicotinic acid in high-dose treatment with APO866, a specific nicotinamide phosphoribosyltransferase inhibitor. Mol Cancer Ther. (2010) 9:1609–17. doi: 10.1158/1535-7163.MCT-09-1130
9. Chowdhry S, Zanca C, Rajkumar U, Koga T, Diao Y, Raviram R, et al. NAD metabolic dependency in cancer is shaped by gene amplification and enhancer remodelling. Nature. (2019) 569:570. doi: 10.1038/s41586-019-1150-2
10. Garten A, Schuster S, Penke M, Gorski T, de Giorgis T, Kiess W. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat Rev Endocrinol. (2015) 11:535–46. doi: 10.1038/nrendo.2015.117
11. Duarte-Pereira S, Pereira-Castro I, Silva SS, Correia MG, Neto C, da Costa LT, et al. Extensive regulation of nicotinate phosphoribosyltransferase (NAPRT) expression in human tissues and tumors. Oncotarget. (2016) 7:1973–83. doi: 10.18632/oncotarget.6538
12. Xiao Y, Elkins K, Durieux JK, Lee L, Oeh J, Yang LX, et al. Dependence of tumor cell lines and patient-derived tumors on the NAD salvage pathway renders them sensitive to NAMPT inhibition with GNE-618. Neoplasia. (2013) 15:1151–60. doi: 10.1593/neo.131304
13. Shackelford RE, Mayhall K, Maxwell NM, Kandil E, Coppola D. Nicotinamide phosphoribosyltransferase in malignancy: a review. Genes Cancer. (2013) 4:447–56. doi: 10.1177/1947601913507576
14. Li XQ, Lei J, Mao LH, Wang QL, Xu F, Ran T, et al. NAMPT and NAPRT, key enzymes in NAD salvage synthesis pathway, are of negative prognostic value in colorectal cancer. Front Oncol. (2019) 9:736. doi: 10.3389/fonc.2019.00736
15. Zhang H, Zhang N, Liu Y, Su P, Liang Y, Li Y, et al. Epigenetic regulation of NAMPT by NAMPT-AS drives metastatic progression in triple-negative breast cancer. Cancer Res. (2019) 79:3347–59. doi: 10.1158/0008-5472.CAN-18-3418
16. Meram AT, Alzubaidi Y, Cotelingam J, Ghali G, Lopez L, Coppola D, et al. Nicotinamide phosphoribosyl transferase is increased in osteosarcomas and chondrosarcomas compared to benign bone and cartilage. Anticancer Res. (2019) 39:1761–5. doi: 10.21873/anticanres.13282
17. Davis K, Dunseth CD, Mott SL, Cramer-Morales KL, Miller AM, Ear PH, et al. Nicotinamide phosphoribosyltransferase expression and clinical outcome of resected stage I/II pancreatic ductal adenocarcinoma. PLoS ONE. (2019) 4:e0213576. doi: 10.1371/journal.pone.0213576
18. Zhou SJ, Bi TQ, Qin CX, Yang XQ, Pang K. Expression of NAMPT is associated with breast invasive ductal carcinoma development and prognosis. Oncol Lett. (2018) 15:6648–54. doi: 10.3892/ol.2018.8164
19. Wang B, Hasan MK, Alvarado E, Yuan H, Wu H, Chen WY. NAMPT overexpression in prostate cancer and its contribution to tumor cell survival and stress response. Oncogene. (2011) 30:907–21. doi: 10.1038/onc.2010.468
20. Wang XY, Wang JZ, Gao L, Zhang FY, Wang Q, Liu KJ, et al. Inhibition of nicotinamide phosphoribosyltransferase and depletion of nicotinamide adenine dinucleotide contribute to arsenic trioxide suppression of oral squamous cell carcinoma. Toxicol Appl Pharmacol. (2017) 331:54–61. doi: 10.1016/j.taap.2017.05.008
21. Vora M, Ansari J, Shanti RM, Veillon D, Cotelingam J, Coppola D, et al. Increased nicotinamide phosphoribosyltransferase in rhabdomyosarcomas and leiomyosarcomas compared to skeletal and smooth muscle tissue. Anticancer Res. (2016) 36:503–7.
22. Li H, Bai E, Zhang Y, Jia Z, He S, Fu J. Role of nampt and visceral adiposity in esophagogastric junction adenocarcinoma. J Immunol Res. (2017) 2017:3970605. doi: 10.1155/2017/3970605
23. Zhu Y, Guo M, Zhang L, Xu T, Wang L, Xu G. Biomarker triplet NAMPT/VEGF/HER2 as a de novo detection panel for the diagnosis and prognosis of human breast cancer. Oncol Rep. (2016) 35:454–62. doi: 10.3892/or.2015.4391
24. Lv X, Zhang L, Zhu Y, Said HM, Shi J, Xu G. Regulative effect of nampt on tumor progression and cell viability in human colorectal cancer. J Cancer. (2015) 6:849–58. doi: 10.7150/jca.12341
25. Sawicka-Gutaj N, Waligorska-Stachura J, Andrusiewicz M, Biczysko M, Sowinski J, Skrobisz J, et al. Nicotinamide phosphorybosiltransferase overexpression in thyroid malignancies and its correlation with tumor stage and with survivin/survivin DEx3 expression. Tumour Biol. (2015) 36:7859–63. doi: 10.1007/s13277-015-3506-z
26. Audrito V, Serra S, Brusa D, Mazzola F, Arruga F, Vaisitti T, et al. Extracellular nicotinamide phosphoribosyltransferase (NAMPT) promotes M2 macrophage polarization in chronic lymphocytic leukemia. Blood. (2015) 125:111–23. doi: 10.1182/blood-2014-07-589069
27. Shackelford R, Hirsh S, Henry K, Abdel-Mageed A, Kandil E, Coppola D. Nicotinamide phosphoribosyltransferase and SIRT3 expression are increased in well-differentiated thyroid carcinomas. Anticancer Res. (2013) 33:3047–52. doi: 10.1093/ajcp/140.suppl1.011
28. Olesen UH, Hastrup N, Sehested M. Expression patterns of nicotinamide phosphoribosyltransferase and nicotinic acid phosphoribosyltransferase in human malignant lymphomas. APMIS. (2011) 119:296–303. doi: 10.1111/j.1600-0463.2011.02733.x
29. Shackelford RE, Bui MM, Coppola D, Hakam A. Over-expression of nicotinamide phosphoribosyltransferase in ovarian cancers. Int J Clin Exp Pathol. (2010) 3:522–7.
30. Shackelford RE, Abdulsattar J, Wei EX, Cotelingam J, Coppola D, Herrera GA. Increased nicotinamide phosphoribosyltransferase and cystathionine-β-synthase in renal oncocytomas, renal urothelial carcinoma, and renal clear cell carcinoma. Anticancer Res. (2017) 37:3423–7. doi: 10.21873/anticanres.11709
31. Montecucco F, Cea M, Bauer I, Soncini D, Caffa I, Lasiglie D, et al. Nicotinamide phosphoribosyltransferase (NAMPT) inhibitors as therapeutics: rationales, controversies, clinical experience. Curr Drug Targets. (2013) 14:637–43. doi: 10.2174/1389450111314060003
32. Dalamaga M, Christodoulatos GS, Mantzoros CS. The role of extracellular and intracellular nicotinamide phosphoribosyl-transferase in cancer: diagnostic and therapeutic perspectives and challenges. Metabolism. (2018) 82:72–87. doi: 10.1016/j.metabol.2018.01.001
33. Hovstadius P, Larsson R, Jonsson E, Skov T, Kissmeyer AM, Krasilnikoff K, et al. A Phase I study of CHS 828 in patients with solid tumor malignancy. Clin Cancer Res. (2002) 8:2843–50.
34. Ravaud A, Cerny T, Terret C, Wanders J, Bui BN, Hess D, et al. Phase I study and pharmacokinetic of CHS-828, a guanidino-containing compound, administered orally as a single dose every 3 weeks in solid tumours: an ECSG/EORTC study. Eur J Cancer. (2005) 41:702–7. doi: 10.1016/j.ejca.2004.12.023
35. Holen K, Saltz LB, Hollywood E, Burk K, Hanauske AR. The pharmacokinetics, toxicities, and biologic effects of FK866, a nicotinamide adenine dinucleotide biosynthesis inhibitor. Invest New Drugs. (2008) 26:45–51. doi: 10.1007/s10637-007-9083-2
36. Pishvaian MJ, Marshall JL, Hwang JJ, Malik S, He AR, Deeken JF, et al. A phase I trial of GMX1777, an inhibitor of nicotinamide phosphoribosyl transferase (NAMPRT), given as a 24-hour infusion. J Clin Oncol. (2009) 27:3581. doi: 10.1200/jco.2008.26.15_suppl.14568
37. von Heideman A, Berglund A, Larsson R, Nygren P. Safety and efficacy of NAD depleting cancer drugs: results of a phase I clinical trial of CHS 828 and overview of published data. Cancer Chemother Pharmacol. (2010) 65:1165–72. doi: 10.1007/s00280-009-1125-3
38. Goldinger SM, Gobbi Bischof S, Fink-Puches R, Klemke CD, Dreno B, Bagot M, et al. Efficacy and safety of APO866 in patients with refractory or relapsed cutaneous T-cell lymphoma: a phase 2 clinical trial. JAMA Dermatol. (2016) 152:837–9. doi: 10.1001/jamadermatol.2016.0401
39. Naing A, Leong S, Pishvaian MJ, Razak ARA, Mahipal A, Berlin J, et al. A first in human phase 1 study of KPT-9274, a first in class dual inhibitor of PAK4 and NAMPT, in patients with advance solid malignancies or NHL. Ann Oncol. (2017) 28(Suppl. 5):v122–41. doi: 10.1093/annonc/mdx367.008
40. Xiao Y, Kwong M, Daemen A, Belvin M, Liang X, Hatzivassiliou G, et al. Metabolic response to NAD depletion across cell lines is highly variable. PLoS ONE. (2016) 11:e0164166. doi: 10.1371/journal.pone.0164166
41. Schuster S, Penke M, Gorski T, Gebhardt R, Weiss TS, Kiess W, et al. FK866-induced NAMPT inhibition activates AMPK and downregulates mTOR signaling in hepatocarcinoma cells. Biochem Biophys Res Commun. (2015) 458:334–40. doi: 10.1016/j.bbrc.2015.01.111
42. Zucal C, D'Agostino VG, Casini A, Mantelli B, Thongon N, Soncini D, et al. EIF2A-dependent translational arrest protects leukemia cells from the energetic stress induced by NAMPT inhibition. BMC Cancer. (2015) 15:855. doi: 10.1186/s12885-015-1845-1
43. Gehrke I, Bouchard ED, Beiggi S, Poeppl AG, Johnston JB, Gibson SB, et al. On-target effect of FK866, a nicotinamide phosphoribosyl transferase inhibitor, by apoptosis-mediated death in chronic lymphocytic leukemia cells. Clin Cancer Res. (2014) 20:4861–72. doi: 10.1158/1078-0432.CCR-14-0624
44. Okumura S, Sasaki T, Minami Y, Ohsaki Y. Nicotinamide phosphoribosyltransferase: a potent therapeutic target in non-small cell lung cancer with epidermal growth factor receptor-gene mutation. J Thorac Oncol. (2012) 7:49–56. doi: 10.1097/JTO.0b013e318233d686
45. Chmielewski JP, Bowlby SC, Wheeler FB, Shi L, Sui G, Davis AL, et al. CD38 inhibits prostate cancer metabolism and proliferation by reducing cellular NAD(+) pools. Mol Cancer Res. (2018) 16:1687–700. doi: 10.1158/1541-7786.MCR-17-0526
46. Espindola-Netto JM, Chini CCS, Tarrago M, Wang E, Dutta S, Pal K, et al. Preclinical efficacy of the novel competitive NAMPT inhibitor STF-118804 in pancreatic cancer. Oncotarget. (2017) 8:85054–67. doi: 10.18632/oncotarget.18841
47. Tateishi K, Iafrate AJ, Ho Q, Curry WT, Batchelor TT, Flaherty KT, et al. Myc-Driven glycolysis is a therapeutic target in glioblastoma. Clin Cancer Res. (2016) 22:4452–65. doi: 10.1158/1078-0432.CCR-15-2274
48. Ohayon D, De Chiara A, Chapuis N, Candalh C, Mocek J, Ribeil JA, et al. Cytoplasmic proliferating cell nuclear antigen connects glycolysis and cell survival in acute myeloid leukemia. Sci Rep. (2016) 6:35561. doi: 10.1038/srep35561
49. Mutz CN, Schwentner R, Aryee DN, Bouchard ED, Mejia EM, Hatch GM, et al. EWS-FLI1 confers exquisite sensitivity to NAMPT inhibition in Ewing sarcoma cells. Oncotarget. (2017) 8:24679. doi: 10.18632/oncotarget.14976
50. Tan B, Dong S, Shepard RL, Kays L, Roth KD, Geeganage S, et al. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT), an enzyme essential for NAD+ biosynthesis, leads to altered carbohydrate metabolism in cancer cells. J Biol Chem. (2015) 290:15812–24. doi: 10.1074/jbc.M114.632141
51. Buonvicino D, Mazzola F, Zamporlini F, Resta F, Ranieri G, Camaioni E, et al. Identification of the nicotinamide salvage pathway as a new toxification route for antimetabolites. Cell Chem Biol. (2018) 25:471–82.e7. doi: 10.1016/j.chembiol.2018.01.012
52. Moore Z, Chakrabarti G, Luo X, Ali A, Hu Z, Fattah FJ, et al. NAMPT inhibition sensitizes pancreatic adenocarcinoma cells to tumor-selective, PAR-independent metabolic catastrophe and cell death induced by beta-lapachone. Cell Death Dis. (2015) 6:e1599. doi: 10.1038/cddis.2014.564
53. Tolstikov V, Nikolayev A, Dong S, Zhao G, Kuo MS. Metabolomics analysis of metabolic effects of nicotinamide phosphoribosyltransferase (NAMPT) inhibition on human cancer cells. PLoS ONE. (2014) 9:e114019. doi: 10.1371/journal.pone.0114019
54. van Horssen R, Willemse M, Haeger A, Attanasio F, Guneri T, Schwab A, et al. Intracellular NAD(H) levels control motility and invasion of glioma cells. Cell Mol Life Sci. (2013) 70:2175–90. doi: 10.1007/s00018-012-1249-1
55. Mitchell SR, Larkin K, Grieselhuber NR, Lai TH, Cannon M, Orwick S, et al. Selective targeting of NAMPT by KPT-9274 in acute myeloid leukemia. Blood Adv. (2019) 3:242–55. doi: 10.1182/bloodadvances.2018024182
56. Cagnetta A, Caffa I, Acharya C, Soncini D, Acharya P, Adamia S, et al. APO866 increases antitumor activity of cyclosporin-A by inducing mitochondrial and endoplasmic reticulum stress in leukemia cells. Clin Cancer Res. (2015) 21:3934–45. doi: 10.1158/1078-0432.CCR-14-3023
57. Audrito V, Manago A, La Vecchia S, Zamporlini F, Vitale N, Baroni G, et al. Nicotinamide phosphoribosyltransferase (NAMPT) as a therapeutic target in BRAF-mutated metastatic melanoma. J Natl Cancer Inst. (2018) 110:209–303. doi: 10.1093/jnci/djx198
58. Kozako T, Aikawa A, Ohsugi T, Uchida YI, Kato N, Sato K, et al. High expression of NAMPT in adult T-cell leukemia/lymphoma and anti-tumor activity of a NAMPT inhibitor. Eur J Pharmacol. (2019) 865:172738. doi: 10.1016/j.ejphar.2019.172738
59. Akhavan D, Pourzia AL, Nourian AA, Williams KJ, Nathanson D, Babic I, et al. De-repression of PDGFRbeta transcription promotes acquired resistance to EGFR tyrosine kinase inhibitors in glioblastoma patients. Cancer Discov. (2013) 3:534–47. doi: 10.1158/2159-8290.CD-12-0502
60. Murphy JP, Giacomantonio MA, Paulo JA, Everley RA, Kennedy BE, Pathak GP, et al. The NAD(+) salvage pathway supports PHGDH-driven serine biosynthesis. Cell Rep. (2018) 24:2381–91.e5. doi: 10.1016/j.celrep.2018.07.086
61. Hong SM, Park CW, Kim SW, Nam YJ, Yu JH, Shin JH, et al. NAMPT suppresses glucose deprivation-induced oxidative stress by increasing NADPH levels in breast cancer. Oncogene. (2016) 35:3544–54. doi: 10.1038/onc.2015.415
62. Bowlby SC, Thomas MJ, D'Agostino RB Jr, Kridel SJ. Nicotinamide phosphoribosyl transferase (Nampt) is required for de novo lipogenesis in tumor cells. PLoS ONE. (2012) 7:e40195. doi: 10.1371/journal.pone.0040195
63. Tateishi K, Higuchi F, Miller JJ, Koerner MVA, Lelic N, Shankar GM, et al. The alkylating chemotherapeutic temozolomide induces metabolic stress in IDH1-mutant cancers and potentiates NAD(+) depletion-mediated cytotoxicity. Cancer Res. (2017) 77:4102–15. doi: 10.1158/0008-5472.CAN-16-2263
64. Tateishi K, Wakimoto H, Iafrate AJ, Tanaka S, Loebel F, Lelic N, et al. Extreme vulnerability of IDH1 mutant cancers to NAD+ depletion. Cancer Cell. (2015) 28:773–84. doi: 10.1016/j.ccell.2015.11.006
65. Shankar GM, Kirtane AR, Miller JJ, Mazdiyasni H, Rogner J, Tai T, et al. Genotype-targeted local therapy of glioma. Proc Natl Acad Sci USA. (2018) 115:E8388–94. doi: 10.1073/pnas.1805751115
66. Yaku K, Okabe K, Hikosaka K, Nakagawa T. NAD metabolism in cancer therapeutics. Front Oncol. (2018) 8:622. doi: 10.3389/fonc.2018.00622
67. Chen H, Wang S, Zhang H, Nice EC, Huang C. Nicotinamide phosphoribosyltransferase (Nampt) in carcinogenesis: new clinical opportunities. Expert Rev Anticancer Ther. (2016) 16:827–38. doi: 10.1080/14737140.2016.1190649
68. Zhu Y, Liu J, Park J, Rai P, Zhai RG. Subcellular compartmentalization of NAD(+) and its role in cancer: a sereNADe of metabolic melodies. Pharmacol Ther. (2019) 200:27–41. doi: 10.1016/j.pharmthera.2019.04.002
69. Jieyu H, Chao T, Mengjun L, Shalong W, Xiaomei G, Jianfeng L, et al. Nampt/Visfatin/PBEF: a functionally multi-faceted protein with a pivotal role in malignant tumors. Curr Pharm Des. (2012) 18:6123–32. doi: 10.2174/138161212803582531
70. Galli M, Van Gool F, Rongvaux A, Andris F, Leo O. The nicotinamide phosphoribosyltransferase: a molecular link between metabolism, inflammation, and cancer. Cancer Res. (2010) 70:8–11. doi: 10.1158/0008-5472.CAN-09-2465
71. Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. (2014) 24:464–71. doi: 10.1016/j.tcb.2014.04.002
72. Chalkiadaki A, Guarente L. The multifaceted functions of sirtuins in cancer. Nat Rev Cancer. (2015) 15:608–24. doi: 10.1038/nrc3985
73. Zhang T, Kraus WL. SIRT1-dependent regulation of chromatin and transcription: linking NAD(+) metabolism and signaling to the control of cellular functions. Biochim Biophys Acta. (2010) 1804:1666–75. doi: 10.1016/j.bbapap.2009.10.022
74. Brandl L, Kirstein N, Neumann J, Sendelhofert A, Vieth M, Kirchner T, et al. The c-MYC/NAMPT/SIRT1 feedback loop is activated in early classical and serrated route colorectal cancer and represents a therapeutic target. Med Oncol. (2018) 36:5. doi: 10.1007/s12032-018-1225-1
75. Brandl L, Zhang Y, Kirstein N, Sendelhofert A, Boos SL, Jung P, et al. Targeting c-MYC through Interference with NAMPT and SIRT1 and their association to oncogenic drivers in murine serrated intestinal tumorigenesis. Neoplasia. (2019) 21:974–88. doi: 10.1016/j.neo.2019.07.009
76. Menssen A, Hydbring P, Kapelle K, Vervoorts J, Diebold J, Luscher B, et al. The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop. Proc Natl Acad Sci USA. (2012) 109:E187–96. doi: 10.1073/pnas.1105304109
77. Pan JH, Zhou H, Zhu SB, Huang JL, Zhao XX, Ding H, et al. Nicotinamide phosphoribosyl transferase regulates cell growth via the Sirt1/P53 signaling pathway and is a prognosis marker in colorectal cancer. J Cell Physiol. (2019) 234:4385–95. doi: 10.1002/jcp.27228
78. Liu H, Liu N, Zhao Y, Zhu X, Wang C, Liu Q, et al. Oncogenic USP22 supports gastric cancer growth and metastasis by activating c-Myc/NAMPT/SIRT1-dependent FOXO1 and YAP signaling. Aging. (2019) 11:9643–60. doi: 10.18632/aging.102410
79. Zhao H, Tang W, Chen X, Wang S, Wang X, Xu H, et al. The NAMPT/E2F2/SIRT1 axis promotes proliferation and inhibits p53-dependent apoptosis in human melanoma cells. Biochem Biophys Res Commun. (2017) 493:77–84. doi: 10.1016/j.bbrc.2017.09.071
80. Behrouzfar K, Alaee M, Nourbakhsh M, Gholinejad Z, Golestani A. Extracellular NAMPT/visfatin causes p53 deacetylation via NAD production and SIRT1 activation in breast cancer cells. Cell Biochem Funct. (2017) 35:327–33. doi: 10.1002/cbf.3279
81. Ma R, Wu Y, Zhai Y, Hu B, Ma W, Yang W, et al. Exogenous pyruvate represses histone gene expression and inhibits cancer cell proliferation via the NAMPT-NAD+-SIRT1 pathway. Nucleic Acids Res. (2019) 47:11132–50. doi: 10.1093/nar/gkz864
82. Venkateshaiah SU, Khan S, Ling W, Bam R, Li X, van Rhee F, et al. NAMPT/PBEF1 enzymatic activity is indispensable for myeloma cell growth and osteoclast activity. Exp Hematol. (2013) 41:547–57.e2. doi: 10.1016/j.exphem.2013.02.008
83. Liu HY, Li QR, Cheng XF, Wang GJ, Hao HP. NAMPT inhibition synergizes with NQO1-targeting agents in inducing apoptotic cell death in non-small cell lung cancer cells. Chin J Nat Med. (2016) 14:582–9. doi: 10.1016/S1875-5364(16)30068-1
84. Zhang B, Shi D, Zhang X, Liang G, Liu W, Qiao S. FK866 inhibits the epithelial-mesenchymal transition of hepatocarcinoma MHCC97-H cells. Oncol Lett. (2018) 16:7231–8. doi: 10.3892/ol.2018.9541
85. Wang W, Hu Y, Yang C, Zhu S, Wang X, Zhang Z, et al. Decreased NAD activates STAT3 and integrin pathways to drive epithelial-mesenchymal transition. Mol Cell Proteomics. (2018) 17:2005–17. doi: 10.1074/mcp.RA118.000882
86. Grohmann T, Penke M, Petzold-Quinque S, Schuster S, Richter S, Kiess W, et al. Inhibition of NAMPT sensitizes MOLT4 leukemia cells for etoposide treatment through the SIRT2-p53 pathway. Leuk Res. (2018) 69:39–46. doi: 10.1016/j.leukres.2018.04.004
87. Bergaggio E, Riganti C, Garaffo G, Vitale N, Mereu E, Bandini C, et al. IDH2 inhibition enhances proteasome inhibitor responsiveness in hematological malignancies. Blood. (2019) 133:156–67. doi: 10.1182/blood-2018-05-850826
88. Chini CC, Guerrico AM, Nin V, Camacho-Pereira J, Escande C, Barbosa MT, et al. Targeting of NAD metabolism in pancreatic cancer cells: potential novel therapy for pancreatic tumors. Clin Cancer Res. (2014) 20:120–30. doi: 10.1158/1078-0432.CCR-13-0150
89. Thakur BK, Dittrich T, Chandra P, Becker A, Kuehnau W, Klusmann JH, et al. Involvement of p53 in the cytotoxic activity of the NAMPT inhibitor FK866 in myeloid leukemic cells. Int J Cancer. (2013) 132:766–74. doi: 10.1002/ijc.27726
90. Ying W. NAD+ and NADH in cellular functions and cell death. Front Biosci. (2006) 11:3129–48. doi: 10.2741/2038
91. Kim MY, Zhang T, Kraus WL. Poly(ADP-ribosyl)ation by PARP-1: 'PAR-laying' NAD+ into a nuclear signal. Genes Dev. (2005) 19:1951–67. doi: 10.1101/gad.1331805
92. Heske CM, Davis MI, Baumgart JT, Wilson KM, Gormally MV, Chen L, et al. Matrix screen identifies synergistic combination of PARP inhibitors and nicotinamide phosphoribosyltransferase (NAMPT) inhibitors in Ewing sarcoma. Clin Cancer Res. (2017) 23:7301–11. doi: 10.1158/1078-0432.CCR-17-1121
93. Bajrami I, Kigozi A, Van Weverwijk A, Brough R, Frankum J, Lord CJ, et al. Synthetic lethality of PARP and NAMPT inhibition in triple-negative breast cancer cells. EMBO Mol Med. (2012) 4:1087–96. doi: 10.1002/emmm.201201250
94. Zhu B, Deng X, Sun Y, Bai L, Xiahou Z, Cong Y, et al. Nampt is involved in DNA double-strand break repair. Chin J Cancer. (2012) 31:392–8. doi: 10.5732/cjc.012.10089
95. Li D, Chen NN, Cao JM, Sun WP, Zhou YM, Li CY, et al. BRCA1 as a nicotinamide adenine dinucleotide (NAD)-dependent metabolic switch in ovarian cancer. Cell Cycle. (2014) 13:2564–71. doi: 10.4161/15384101.2015.942208
96. Gorthi A, Romero JC, Loranc E, Cao L, Lawrence LA, Goodale E, et al. EWS-FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma. Nature. (2018) 555:387–91. doi: 10.1038/nature25748
97. Cloux AJ, Aubry D, Heulot M, Widmann C, ElMokh O, Piacente F, et al. Reactive oxygen/nitrogen species contribute substantially to the antileukemia effect of APO866, a NAD lowering agent. Oncotarget. (2019) 10:6723–38. doi: 10.18632/oncotarget.27336
98. Touat M, Sourisseau T, Dorvault N, Chabanon RM, Garrido M, Morel D, et al. DNA repair deficiency sensitizes lung cancer cells to NAD+ biosynthesis blockade. J Clin Invest. (2018) 128:1671–87. doi: 10.1172/JCI90277
99. Piacente F, Caffa I, Ravera S, Sociali G, Passalacqua M, Vellone VG, et al. Nicotinic acid phosphoribosyltransferase regulates cancer cell metabolism, susceptibility to NAMPT inhibitors, and DNA repair. Cancer Res. (2017) 77:3857–69. doi: 10.1158/0008-5472.CAN-16-3079
100. Li N, Lopez MA, Linares M, Kumar S, Oliva S, Martinez-Lopez J, et al. Dual PAK4-NAMPT inhibition impacts growth and survival, and increases sensitivity to DNA-damaging agents in waldenstrom macroglobulinemia. Clin Cancer Res. (2019) 25:369–77. doi: 10.1158/1078-0432.CCR-18-1776
101. Zerp SF, Vens C, Floot B, Verheij M, van Triest B. NAD(+) depletion by APO866 in combination with radiation in a prostate cancer model, results from an in vitro and in vivo study. Radiother Oncol. (2014) 110:348–54. doi: 10.1016/j.radonc.2013.10.039
102. Feng J, Yan PF, Zhao HY, Zhang FC, Zhao WH, Feng M. Inhibitor of nicotinamide phosphoribosyltransferase sensitizes glioblastoma cells to temozolomide via activating ROS/JNK signaling pathway. Biomed Res Int. (2016) 2016:1450843. doi: 10.1155/2016/1450843
103. Sun T, Zhao Q, Zhang C, Cao L, Song M, Maimela NR, et al. Screening common signaling pathways associated with drug resistance in non-small cell lung cancer via gene expression profile analysis. Cancer Med. (2019) 8:3059–71. doi: 10.1002/cam4.2190
104. Chan M, Gravel M, Bramoulle A, Bridon G, Avizonis D, Shore GC, et al. Synergy between the NAMPT inhibitor GMX1777(8) and pemetrexed in non-small cell lung cancer cells is mediated by PARP activation and enhanced NAD consumption. Cancer Res. (2014) 74:5948–54. doi: 10.1158/0008-5472.CAN-14-0809
105. Ju HQ, Zhuang ZN, Li H, Tian T, Lu YX, Fan XQ, et al. Regulation of the nampt-mediated NAD salvage pathway and its therapeutic implications in pancreatic cancer. Cancer Lett. (2016) 379:1–11. doi: 10.1016/j.canlet.2016.05.024
106. Bi TQ, Che XM, Liao XH, Zhang DJ, Long HL, Li HJ, et al. Overexpression of Nampt in gastric cancer and chemopotentiating effects of the Nampt inhibitor FK866 in combination with fluorouracil. Oncol Rep. (2011) 26:1251–7. doi: 10.3892/or.2011.1378
107. Barraud M, Garnier J, Loncle C, Gayet O, Lequeue C, Vasseur S, et al. A pancreatic ductal adenocarcinoma subpopulation is sensitive to FK866, an inhibitor of NAMPT. Oncotarget. (2016) 7:53783–96. doi: 10.18632/oncotarget.10776
108. Ogino Y, Sato A, Uchiumi F, Tanuma SI. Genomic and tumor biological aspects of the anticancer nicotinamide phosphoribosyltransferase inhibitor FK866 in resistant human colorectal cancer cells. Genomics. (2018) 111:1889–95. doi: 10.1016/j.ygeno.2018.12.012
109. Wondrak GT. Redox-directed cancer therapeutics: molecular mechanisms and opportunities. Antioxid Redox Signal. (2009) 11:3013–69. doi: 10.1089/ars.2009.2541
110. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science. (2009) 324:1029–33. doi: 10.1126/science.1160809
111. Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. (2009) 8:579–91. doi: 10.1038/nrd2803
112. Hong SM, Hwang SW, Wang T, Park CW, Ryu YM, Jung JH, et al. Increased nicotinamide adenine dinucleotide pool promotes colon cancer progression by suppressing reactive oxygen species level. Cancer Sci. (2019) 110:629–38. doi: 10.1111/cas.13886
113. Xu R, Yuan Z, Yang L, Li L, Li D, Lv C. Inhibition of NAMPT decreases cell growth and enhances susceptibility to oxidative stress. Oncol Rep. (2017) 38:1767–73. doi: 10.3892/or.2017.5793
114. Cerna D, Li H, Flaherty S, Takebe N, Coleman CN, Yoo SS. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) activity by small molecule GMX1778 regulates reactive oxygen species (ROS)-mediated cytotoxicity in a p53- and nicotinic acid phosphoribosyltransferase1 (NAPRT1)-dependent manner. J Biol Chem. (2012) 287:22408–17. doi: 10.1074/jbc.M112.357301
115. Breton CS, Aubry D, Ginet V, Puyal J, Heulot M, Widmann C, et al. Combinative effects of beta-Lapachone and APO866 on pancreatic cancer cell death through reactive oxygen species production and PARP-1 activation. Biochimie. (2015) 116:141–53. doi: 10.1016/j.biochi.2015.07.012
116. Nagaya M, Hara H, Kamiya T, Adachi T. Inhibition of NAMPT markedly enhances plasma-activated medium-induced cell death in human breast cancer MDA-MB-231 cells. Arch Biochem Biophys. (2019) 2019:108155. doi: 10.1016/j.abb.2019.108155
117. Jeong B, Park JW, Kim JG, Lee BJ. FOXO1 functions in the regulation of nicotinamide phosphoribosyltransferase (Nampt) expression. Biochem Biophys Res Commun. (2019) 511:398–403. doi: 10.1016/j.bbrc.2019.02.069
118. Ge X, Zhao Y, Dong L, Seng J, Zhang X, Dou D. NAMPT regulates PKM2 nuclear location through 14–3-3zeta: conferring resistance to tamoxifen in breast cancer. J Cell Physiol. (2019) 234:23409–20. doi: 10.1002/jcp.28910
119. Grolla AA, Torretta S, Gnemmi I, Amoruso A, Orsomando G, Gatti M, et al. Nicotinamide phosphoribosyltransferase (NAMPT/PBEF/visfatin) is a tumoural cytokine released from melanoma. Pigment Cell Melanoma Res. (2015) 28:718–29. doi: 10.1111/pcmr.12420
120. Gholinejad Z, Kheiripour N, Nourbakhsh M, Ilbeigi D, Behroozfar K, Hesari Z, et al. Extracellular NAMPT/Visfatin induces proliferation through ERK1/2 and AKT and inhibits apoptosis in breast cancer cells. Peptides. (2017) 92:9–15. doi: 10.1016/j.peptides.2017.04.007
121. Cea M, Cagnetta A, Fulciniti M, Tai YT, Hideshima T, Chauhan D, et al. Targeting NAD+ salvage pathway induces autophagy in multiple myeloma cells via mTORC1 and extracellular signal-regulated kinase (ERK1/2) inhibition. Blood. (2012) 120:3519–29. doi: 10.1182/blood-2012-03-416776
122. Zhang LY, Liu LY, Qie LL, Ling KN, Xu LH, Wang F, et al. Anti-proliferation effect of APO866 on C6 glioblastoma cells by inhibiting nicotinamide phosphoribosyltransferase. Eur J Pharmacol. (2012) 674:163–70. doi: 10.1016/j.ejphar.2011.11.017
123. Mpilla G, Aboukameel A, Muqbil I, Kim S, Beydoun R, Philip PA, et al. PAK4-NAMPT dual inhibition as a novel strategy for therapy resistant pancreatic neuroendocrine tumors. Cancers. (2019) 11:1902. doi: 10.3390/cancers11121902
124. Audrito V, Manago A, Zamporlini F, Rulli E, Gaudino F, Madonna G, et al. Extracellular nicotinamide phosphoribosyltransferase (eNAMPT) is a novel marker for patients with BRAF-mutated metastatic melanoma. Oncotarget. (2018) 9:18997–9005. doi: 10.18632/oncotarget.24871
125. Ohanna M, Cerezo M, Nottet N, Bille K, Didier R, Beranger G, et al. Pivotal role of NAMPT in the switch of melanoma cells toward an invasive and drug-resistant phenotype. Genes Dev. (2018) 32:448–61. doi: 10.1101/gad.305854.117
126. Lucena-Cacace A, Otero-Albiol D, Jimenez-Garcia MP, Peinado-Serrano J, Carnero A. NAMPT overexpression induces cancer stemness and defines a novel tumor signature for glioma prognosis. Oncotarget. (2017) 8:99514–30. doi: 10.18632/oncotarget.20577
127. Soncini D, Caffa I, Zoppoli G, Cea M, Cagnetta A, Passalacqua M, et al. Nicotinamide phosphoribosyltransferase promotes epithelial-to-mesenchymal transition as a soluble factor independent of its enzymatic activity. J Biol Chem. (2014) 289:34189–204. doi: 10.1074/jbc.M114.594721
128. Cea M, Cagnetta A, Acharya C, Acharya P, Tai YT, Yang C, et al. Dual NAMPT and BTK targeting leads to synergistic killing of waldenstrom macroglobulinemia cells regardless of MYD88 and CXCR4 somatic mutation status. Clin Cancer Res. (2016) 22:6099–109. doi: 10.1158/1078-0432.CCR-16-0630
129. Lucena-Cacace A, Umeda M, Navas LE, Carnero A. NAMPT as a dedifferentiation-inducer gene: NAD(+) as core axis for glioma cancer stem-like cells maintenance. Front Oncol. (2019) 9:292. doi: 10.3389/fonc.2019.00292
130. Lucena-Cacace A, Otero-Albiol D, Jimenez-Garcia MP, Munoz-Galvan S, Carnero A. NAMPT is a potent oncogene in colon cancer progression that modulates cancer stem cell properties and resistance to therapy through sirt1 and PARP. Clin Cancer Res. (2018) 24:1202–15. doi: 10.1158/1078-0432.CCR-17-2575
131. Gujar AD, Le S, Mao DD, Dadey DY, Turski A, Sasaki Y, et al. An NAD+-dependent transcriptional program governs self-renewal and radiation resistance in glioblastoma. Proc Natl Acad Sci USA. (2016) 113:E8247–56. doi: 10.1073/pnas.1610921114
132. Cea M, Cagnetta A, Patrone F, Nencioni A, Gobbi M, Anderson KC. Intracellular NAD(+) depletion induces autophagic death in multiple myeloma cells. Autophagy. (2013) 9:410–2. doi: 10.4161/auto.22866
133. Sharif T, Ahn DG, Liu RZ, Pringle E, Martell E, Dai C, et al. The NAD(+) salvage pathway modulates cancer cell viability via p73. Cell Death Differ. (2016) 23:669–80. doi: 10.1038/cdd.2015.134
134. Yang P, Zhang L, Shi QJ, Lu YB, Wu M, Wei EQ, et al. Nicotinamide phosphoribosyltransferase inhibitor APO866 induces C6 glioblastoma cell death via autophagy. Pharmazie. (2015) 70:650–5. doi: 10.1691/ph.2015.5614
135. Zoppoli G, Cea M, Soncini D, Fruscione F, Rudner J, Moran E, et al. Potent synergistic interaction between the nampt inhibitor APO866 and the apoptosis activator TRAIL in human leukemia cells. Exp Hematol. (2010) 38:979–88. doi: 10.1016/j.exphem.2010.07.013
136. Sharif T, Martell E, Dai C, Kennedy BE, Murphy P, Clements DR, et al. Autophagic homeostasis is required for the pluripotency of cancer stem cells. Autophagy. (2017) 13:264–84. doi: 10.1080/15548627.2016.1260808
137. Ostrakhovitch EA, Akakura S, Sanokawa-Akakura R, Goodwin S, Tabibzadeh S. Dedifferentiation of cancer cells following recovery from a potentially lethal damage is mediated by H2S-Nampt. Exp Cell Res. (2015) 330:135–50. doi: 10.1016/j.yexcr.2014.09.027
138. Cheng G, Liu C, Sun X, Zhang L, Liu L, Ouyang J, et al. Visfatin promotes osteosarcoma cell migration and invasion via induction of epithelial-mesenchymal transition. Oncol Rep. (2015) 34:987–94. doi: 10.3892/or.2015.4053
139. Santidrian AF, LeBoeuf SE, Wold ED, Ritland M, Forsyth JS, Felding BH. Nicotinamide phosphoribosyltransferase can affect metastatic activity and cell adhesive functions by regulating integrins in breast cancer. DNA Repair. (2014) 23:79–87. doi: 10.1016/j.dnarep.2014.08.006
140. Lee J, Kim H, Lee JE, Shin SJ, Oh S, Kwon G, et al. Selective Cytotoxicity of the NAMPT Inhibitor FK866 toward gastric cancer cells with markers of the epithelial-mesenchymal transition, due to loss of NAPRT. Gastroenterology. (2018) 155:799–814.e13. doi: 10.1053/j.gastro.2018.05.024
141. Chaker M, Minden A, Chen S, Weiss RH, Chini EN, Mahipal A, et al. Rho GTPase effectors and NAD metabolism in cancer immune suppression. Expert Opin Ther Targets. (2018) 22:9–17. doi: 10.1080/14728222.2018.1413091
142. Travelli C, Consonni FM, Sangaletti S, Storto M, Morlacchi S, Grolla AA, et al. Nicotinamide phosphoribosyltransferase acts as a metabolic gate for mobilization of myeloid-derived suppressor cells. Cancer Res. (2019) 79:1938–51. doi: 10.1158/0008-5472.CAN-18-1544
143. Pylaeva E, Harati MD, Spyra I, Bordbari S, Strachan S, Thakur BK, et al. NAMPT signaling is critical for the proangiogenic activity of tumor-associated neutrophils. Int J Cancer. (2019) 144:136–49. doi: 10.1002/ijc.31808
144. Yang HJ, Yen MC, Lin CC, Lin CM, Chen YL, Weng TY, et al. A combination of the metabolic enzyme inhibitor APO866 and the immune adjuvant L-1-methyl tryptophan induces additive antitumor activity. Exp Biol Med. (2010) 235:869–76. doi: 10.1258/ebm.2010.010001
145. Fons NR, Sundaram RK, Breuer GA, Peng S, McLean RL, Kalathil AN, et al. PPM1D mutations silence NAPRT gene expression and confer NAMPT inhibitor sensitivity in glioma. Nat Commun. (2019) 10:3790. doi: 10.1038/s41467-019-11732-6
146. Abu Aboud O, Chen CH, Senapedis W, Baloglu E, Argueta C, Weiss RH. Dual and specific inhibition of NAMPT and PAK4 By KPT-9274 decreases kidney cancer growth. Mol Cancer Ther. (2016) 15:2119–29. doi: 10.1158/1535-7163.MCT-16-0197
147. Cole J, Guiot MC, Gravel M, Bernier C, Shore GC, Roulston A. Novel NAPRT specific antibody identifies small cell lung cancer and neuronal cancers as promising clinical indications for a NAMPT inhibitor/niacin co-administration strategy. Oncotarget. (2017) 8:77846–59. doi: 10.18632/oncotarget.20840
148. Zhao G, Green CF, Hui YH, Prieto L, Shepard R, Dong S, et al. Discovery of a highly selective NAMPT inhibitor that demonstrates robust efficacy and improved retinal toxicity with nicotinic acid coadministration. Mol Cancer Ther. (2017) 16:2677–88. doi: 10.1158/1535-7163.MCT-16-0674
149. Duarte-Pereira S, Silva SS, Azevedo L, Castro L, Amorim A, Silva RM. NAMPT and NAPRT1: novel polymorphisms and distribution of variants between normal tissues and tumor samples. Sci Rep. (2014) 4:6311. doi: 10.1038/srep06311
150. Shames DS, Elkins K, Walter K, Holcomb T, Du P, Mohl D, et al. Loss of NAPRT1 expression by tumor-specific promoter methylation provides a novel predictive biomarker for NAMPT inhibitors. Clin Cancer Res. (2013) 19:6912–23. doi: 10.1158/1078-0432.CCR-13-1186
151. Watson M, Roulston A, Belec L, Billot X, Marcellus R, Bedard D, et al. The small molecule GMX1778 is a potent inhibitor of NAD+ biosynthesis: strategy for enhanced therapy in nicotinic acid phosphoribosyltransferase 1-deficient tumors. Mol Cell Biol. (2009) 29:5872–88. doi: 10.1128/MCB.00112-09
Keywords: NAD+, NAMPT, sirtuins, PARP, ROS, cancer
Citation: Heske CM (2020) Beyond Energy Metabolism: Exploiting the Additional Roles of NAMPT for Cancer Therapy. Front. Oncol. 9:1514. doi: 10.3389/fonc.2019.01514
Received: 29 October 2019; Accepted: 16 December 2019;
Published: 17 January 2020.
Edited by:
Paolo E. Porporato, University of Turin, ItalyReviewed by:
Valentina Audrito, University of Turin, ItalyCyril Corbet, Catholic University of Louvain, Belgium
Copyright © 2020 Heske. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christine M. Heske, Y2hyaXN0aW5lLmhlc2tlQG5paC5nb3Y=