 Luca Di Leo
Luca Di Leo Valérie Bodemeyer
Valérie Bodemeyer Daniela De Zio
Daniela De Zio
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 10 January 2020
Sec. Molecular and Cellular Oncology
Volume 9 - 2019 | https://doi.org/10.3389/fonc.2019.01506
Despite tremendous efforts in the last decade to improve treatments, melanoma still represents a major therapeutic challenge and overall survival of patients remains poor. Therefore, identifying new targets to counteract melanoma is needed. In this scenario, autophagy, the “self-eating” process of the cell, has recently arisen as new potential candidate in melanoma. Alongside its role as a recycling mechanism for dysfunctional and damaged cell components, autophagy also clearly sits at a crossroad with metabolism, thereby orchestrating cell proliferation, bioenergetics and metabolic rewiring, all hallmarks of cancer cells. In this regard, autophagy, both in tumor and host, has been flagged as an essential player in melanomagenesis and progression. To pave the way to a better understanding of such a complex interplay, the use of genetically engineered mouse models (GEMMs), as well as syngeneic mouse models, has been undoubtedly crucial. Herein, we will explore the latest discoveries in the field, with particular focus on the potential of these models in unraveling the contribution of autophagy in melanoma, along with the therapeutic advantages that may arise.
Melanoma arises from the malignant transformation of melanocytes, the melanin-producing cells mainly found in the skin's epidermis (1). Due to its mutational burden and heterogeneity, melanoma is a particularly aggressive cancer and still remains a clinical challenge (2, 3). In recent years however, a better understanding of melanoma biology and the identification of key genetic alterations causing imbalances in cell proliferation signaling (e.g., BRAF, NRAS, PTEN, CDKN2A) have revealed novel targets and therapeutic strategies for this disease (1–4). In particular, this knowledge has led to the development of the BRAF/MEK inhibitors currently used for subsets of patients harboring specific mutations and immunotherapies that aim at reactivating immune T cells through the use of immune checkpoint inhibitors (antibodies against CTLA-4 and PD-1/PD-L1) (1, 3, 5).
Macroautophagy (hereafter referred to as autophagy) is a highly-conserved self-degradative process that allows the cell to deliver unwanted cargo, such as damaged proteins and organelles, to the lysosome for degradation (6–8). So-called “bulk” autophagy virtually occurs in all cells as a general homeostatic process. Notably, in response to certain molecular triggers, cells are also able to dispose of specific cargo by selective autophagy (e.g., mitophagy, the selective removal of damaged mitochondria) (8, 9). This selectivity is mostly determined by specific protein receptors, such as SQSTM1/p62 or OPTN (9). Regardless of cargo specificity, autophagic flux is tightly regulated by two major molecular sensors, mTORC1, which acts as a nutrient sensor, mostly for amino acids, (10), therefore negatively regulating autophagy, and AMPK, which detects shifts in the AMP/ATP ratio in the cell, consequently promoting autophagy (6). In order to restore balance in cell energy supply and demand, AMPK, in an mTORC1-dependent or independent fashion, initiates an intricate signaling cascade (6). All these signals converge to the ATG proteins, which assemble in four molecular complexes to form the core autophagic machinery: the ULK1 kinase complex (ULK1, ATG13, ATG101, and FIP200), the PI3KIII complex (VPS34, VPS15, ATG14, and BECLIN1), the ubiquitin-like conjugation system (ATG3, ATG4, ATG5, ATG7, ATG10, ATG12, ATG16, and LC3/GABARAP) and transmembrane protein like ATG9 (11). Altogether, these complexes promote the initiation, elongation, formation, and maturation of autophagosomes (Figure 1A) (11, 12).
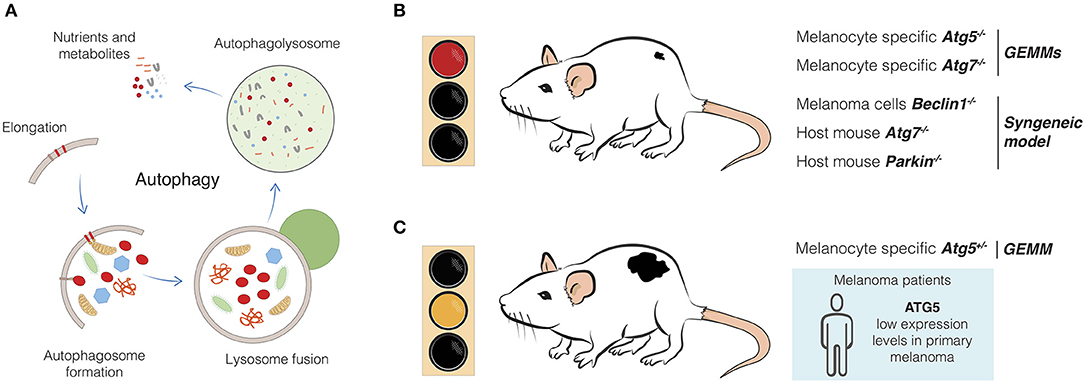
Figure 1. Effects of autophagy genes alteration in melanoma development. (A) Autophagy is a complex, dynamic, and well-conserved degradative process which involves several molecular players and steps. Starting from the elongation of the phagophore, which leads to the autophagosome formation, the cytoplasmic material is eventually engulfed and degraded in the autophagolysosome after fusion with the lysosome (11). New building blocks and metabolites are now released and available for the cell. (B) Melanomagenesis from melanocytic nevi is blocked upon melanocytic-specific depletion of autophagy genes, such as Atg5 and Atg7 in GEMMs (64, 65). Syngeneic models displayed similar evidence, as in the case of host mice injected with Beclin1-engineered B16-F10 melanoma cells (67) or upon injection of melanoma cells in Atg7- and Parkin- genetically engineered mice (68, 69). (C) Melanoma development is favored upon single-copy loss of Atg5 in GEMM (65). Primary tumors from melanoma patients have been found to have reduced expression of ATG5, if compared to melanocytic nevi (17). Therefore, impaired autophagy, as well as putative additional functions of Atg5 can induce melanoma.
In the aim of discovering novel anti-tumor therapies, autophagy has, over the past years, been investigated with great interest as a process that could potentially be modulated in tumor cells for the benefit of cancer patients (13). In melanoma, autophagy seems to play a complex and dynamic role which highly depends on the progression stage of the disease, the metabolic demand of the tumor as well as intrinsic (tissue microenvironment -TME, immunity) and extrinsic aspects (therapies) of the disease (6, 7, 14). To address this level of complexity in a clinically relevant system, syngeneic and genetically engineered mouse models (GEMMs) have been developed to fully recreate tumor progression from initiation to invasion and metastasis and to better characterize tumor-host interactions (15, 16). In this review, we will discuss how the different roles of autophagy can contribute to melanoma initiation and progression and delineate the precious insights that GEMMs and syngeneic mouse models have been able to provide to this field.
The first studies aiming at understanding the contribution of autophagy to melanomagenesis and melanoma development revealed that, when impaired in melanocytes, autophagy can promote BRAFV600E-driven tumorigenesis, thus pointing to a tumor suppressor function for autophagy (17, 18). Importantly, autophagy seems to support BRAFV600E-dependent oncogene-induced senescence in melanocytes (17, 18). In particular, Liu et al. have found that a key autophagy gene (ATG5) is frequently down-regulated in primary melanomas when compared to benign nevi, strongly correlating with reduced progression-free survival in a cohort of early stage cutaneous melanoma patients (17). Other works have also shown that the expression of autophagy markers, such as LC3 and BECLIN1, was altered in melanocytic neoplasms, in a way that resulted in severely blunted autophagy during the early stages of melanocyte malignant transformation (19–22). This tumor suppressor role of autophagy at the onset of melanoma, as for other cancer types, has often been explained by the common view that autophagy-mediated removal of damaged organelles and redox-active protein aggregates might prevent the accumulation of detrimental and dysfunctional material, eventually preserving bioenergetics and redox metabolism (23, 24). Indeed, loss of autophagy has been often associated with increased oxidative stress, mostly as a consequence of accumulation of damaged mitochondria (see below), genomic instability and alteration of cell growth pathways, all circumstances leading to malignant transformation (23, 25). However, the mechanistic studies unraveling the real cause-effect relation between autophagy loss and melanoma initiation are still incomplete and in need of further investigation. Interestingly, Li and collaborators have recently shown that autophagy is restrained in melanoma cells at a transcriptional level in a BRAFV600E-dependent fashion in a syngeneic mouse model of melanoma (26, 27). Briefly, they have found that the master regulator of the expression of autophagy/lysosomal genes, TFEB, is under the control of the BRAF-oncogenic pathway, via its ERK-mediated phosphorylation and inactivation. This has been associated with TGFβ-signaling activation and induction of a metastatic phenotype that can be rescued by autophagy activation through the block of TFEB ERK-mediated phosphorylation and activation. This phenomenon was reverted (back to the metastatic phenotype) upon BRAF inhibition (26). Of note, BRAFV600E activity has also been shown to inhibit AMPK by compromising its interaction with LKB1, thereby promoting melanoma cells proliferation (28). Therefore, the oncogenic pathway orchestrated by BRAFV600E seems to be tightly involved in repressing autophagy, supporting the initial concept that loss of autophagy promotes melanomagenesis. The importance of understanding the intricate crosstalk between autophagy (and lysosomal-associated functions) and the oncogenic pathways activated during the onset of melanoma is supported by the evidence that melanoma cells highly depend on lysosome-associated vesicular trafficking in the very early stages of melanoma development (29). Moreover, it is noteworthy that, although the master regulators of melanocyte development MiTF, TFEB, and TFE3 belong to the same MiT/TFE transcription factor family (30), their activity correlates with the expression of diverse lysosomal and autophagy genes (31). Indeed, while it is known that MAPK/ERK signaling is involved in MiTF turnover and activation, with a crucial contribution of BRAF-mediated transcriptional and post-transcriptional regulation (32, 33), the recently discovered ERK-mediated signaling pathway that controls TFEB in BRAFV600E-driven melanoma does not work in the same way for either MiTF or TFE3 (26). To further complicate this scenario, the MiT/TFE transcription factor family has been also reported to finely regulate the lysosomal activity of mTORC1, the master regulator of cell growth as well as autophagy inhibitor (10), through direct regulation of RagD GTPase expression (34). Di Malta and colleagues elegantly demonstrated that the growth of melanoma, as for other cancer types, is deeply affected by the activity of MiT/TFE transcription factors which, besides regulating autophagy, lysosomal, and melanosome biogenesis, are specifically deputed to positively control lysosomal recruitment of mTORC1, thus enabling its activation and promoting cancer growth (34). In relation to mTORC1 activation, it is also worth mentioning that Bosenberg's group thoroughly demonstrated that mTORC1 activation, through loss of the Lkb1/AMPK pathway, is not sufficient to induce melanomagenesis in a BrafV600E-driven melanoma model (35). Indeed, melanoma development requires the concomitant activation of mTORC1 and 2 in a GEMM carrying the BrafV600E mutation uniquely in melanocytes (35). However, the link to autophagy function has not been unraveled yet in this specific context. Indeed, though providing possible clues, all these discoveries still puzzle the intricate scenario of the signaling cascades activated to control autophagy during melanomagenesis.
That said, a growing body of evidence has been pointing out a controversial oncogenic function to autophagy during melanomagenesis. Herein, we will dissect the possible explanations of such a contradictory view and how the application of GEMMs and syngeneic models (15, 16) have emerged to elucidate this complex function of autophagy in melanoma.
It is worth underlining that autophagy is principally meant as a key survival mechanism for the cell. Indeed, autophagy enables cells to recycle building blocks and metabolic substrates (primarily carbohydrates, fatty acids -FAs, amino acids, and nucleosides/nucleotides) needed for continuous growth and for sustaining the adaptive high metabolic demand cells require upon diverse stress conditions (23). This places autophagy at a crossroad with cell metabolic rewiring, a strategy adopted by melanoma cells to sustain a constant growth and metastatic progression (36). In this section, we will sum up the latest findings emphasizing the essential role of autophagy in supporting melanoma growth and metastasis, pointing out autophagy as an oncogenic/metabolic machinery in melanoma.
Metabolic reprogramming is considered one of the hallmarks of cancer, being involved in cancer initiation, maintenance, and progression (37). Historically, glycolysis represents the central metabolic pathway implicated in melanoma evolution, with the Warburg effect, i.e., the preferential use of aerobic glycolysis to oxidative phosphorylation (OxPHOS) for ATP production, having a predominant role (38–41). The glycolytic pathway of melanoma cells intrinsically relies on the activation of signaling pathways, such as (i) the MAPK pathway that, hyperactivated in BRAFV600E-driven melanomas, controls HIF-1α and MYC activities (42, 43) and (ii) the PI3K/AKT/mTOR pathway which is hyperactivated upon loss of PTEN [another common alteration found in melanoma patients (44)] or upon AKT and PI3K activating mutations (45, 46). Importantly, mitochondrial OxPHOS can also be hyperactivated in melanoma cells, mainly through the master regulator of energy metabolism PGC-1α, which was found highly expressed in a subset of melanomas (39, 41, 47, 48).
While metabolic rewiring allows cancer cells to find alternative sources of energy to adapt to nutrient and oxygen limitation, tumor cells may also control nutrient demand indirectly by modulating protein synthesis. This strategy, extensively reviewed by García-Jiménez and Goding, is present in many cancer types including melanoma (49). Indeed, recent findings suggest that translation reprogramming in melanoma can be initiated by different stress kinases and is mainly orchestrated via phosphorylation of the eukaryotic translation initiation factor eIF2α (50–52). This reprogramming can be triggered by various molecular cues such as oncogenic BRAF-induced ER stress (53–55), amino acid limitation (50, 51) or inflammation (51), that ultimately converge toward reducing global protein translation and inducing autophagy (49, 50, 53, 54). In this context, the downstream autophagic response seems to be an essential pro-survival mechanism that not only allows melanoma cells to thrive but also promotes an invasive phenotype (50, 52, 55). Of note, ER stress induced autophagy can also occur in an eIF2α-independent fashion via JNK signaling (53, 54).
Recently, a key role has emerged also for lipid metabolism in melanoma. As lipids are a source for membranes and a reservoir for ATP and acetyl-CoA, it is not surprising that FAs synthesis and oxidation have been entangled in melanoma growth and progression (39, 56). Interestingly, Zhang and colleagues have recently demonstrated that fatty acid transporters, such as FATP1, are highly expressed in melanoma (56). In this way, melanoma cells metabolism was considerably affected and melanoma progression sustained by increased uptake of FAs from TME-resident adipocytes, hence revisiting FATPs as new possible targets in melanoma therapy (56). Moreover, beyond cell energetic requirements, lipid alterations can also affect lipid signaling in melanoma (57, 58). Indeed, FAs can bind and activate specific nuclear receptors, i.e., PPARs, thus controlling the expression of genes involved in lipid homeostasis, oxidative stress, and inflammation (59, 60). Unfortunately, poor knowledge exists on this matter as the only evidence for a role of lipid signaling in melanoma biology comes from two recent papers in which the authors independently remarked an anti-tumor effect of PPARβ/δ and PPARγ in melanoma progression and metastasis (61, 62).
By making available different sources of energy and by directly controlling mitochondria homeostasis through selective removal of mitochondria (see below), autophagy is unquestionably a central player in tumor metabolism (36). Indeed, recent evidence demonstrated a tight interplay between autophagy and lipid metabolism, proposing autophagy as a mechanism regulating both β-oxidation and production of ketone bodies (63).
The most emerging discoveries posing the question on autophagy as promoting or suppressive mechanism during melanoma evolution come from studies performed in GEMMs (Table 1). Results from a very sophisticated BrafV600E-mutation and Pten-deletion tamoxifen-inducible mouse model of melanoma, which develops cutaneous melanoma resembling the human disease (70, 71), have mainly flagged an oncogenic role for autophagy in melanoma development, highlighting the complexity of this process in the malignant transformation of melanocytes. In particular, melanocyte-specific Atg7 or Atg5 ablation can prevent melanoma formation and drive melanocytic senescence in the same GEMM model (Figure 1B) (64, 65), being the Atg7-related phenotype associated with increased oxidative stress and extended survival rate (64). In this context, autophagy clearly seems to harbor an oncogenic role. Strikingly, hemizygous deletion of Atg5 enhances melanoma growth (compared to Atg5+/+ or Atg5−/−), metastatic power and resistance to targeted therapy, such as BRAF inhibitor (Figure 1C) (65). Of note, as for the Atg7-deficient BrafV600E; Pten−/− driven melanomas (64), complete loss of Atg5 reduced tumor formation (65). Importantly, single-copy loss of ATG5 has been identified as a distinctive feature of advanced melanomas, regardless of the mutational status of melanoma associated-oncogenes (e.g., BRAF, RAS), and associated with poor overall patient survival (Figure 1C) (65). These very interesting findings add another layer of complexity to the general knowledge about the impact of autophagy on melanoma evolution, even arguing for a dose-dependent contribution of autophagy genes, as seen for Atg5, and opening new hypotheses on additional autophagy-unrelated function(s) of Atg5 (72). In line with this idea, a very recent work indicates that the well-known autophagy receptor Sqstm1/p62 is involved in fueling melanoma progression through a distinct non-autophagy pathway (66). In particular, the authors demonstrated that p62 can positively regulate the mRNA stability of a cluster of pro-tumorigenic factors in melanomas, thus arguing for p62, an alternative oncogenic role which seems to be far from its autophagy function, at least in this type of tumor (66).
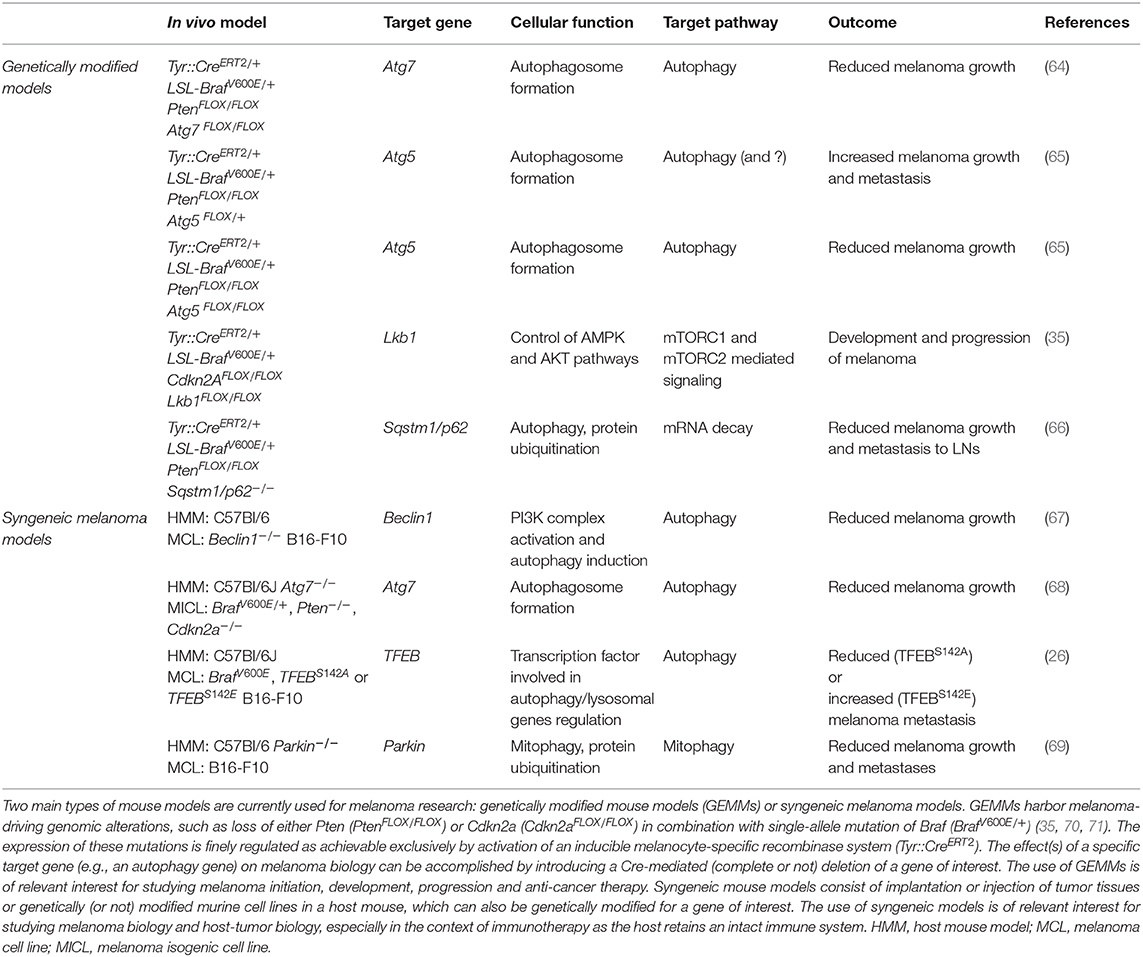
Table 1. Melanoma mouse models.
Considering the intricate role played by mitochondria in maintaining metabolism and cellular homeostasis, it is not surprising that alterations in mitochondria may occur and be involved in melanoma features. Indeed, disruption of mitochondrial capacity has been implicated in reduced melanoma cell growth in an in vivo mouse xenograft model (73).
Even though mitochondrial bioenergetics largely sustains both the high proliferation rate and the energetic demand of cancer cells (74), mitochondrial metabolism can also act as a double-edged sword as mitochondria are primary producers and targets of reactive oxygen species (ROS). Although enhanced ROS production can activate signaling cascades that are favorable for tumorigenesis (75), the role of ROS in melanoma biology is still controversial and widely debated. A recent study revealed that scavenging ROS ensures maintenance of melanoma cells proliferation and migration both in vitro and in an in vivo xenograft tumor growth model (76). Bagati and colleagues shed further light on such a complex role by suggesting a double and stage-specific role for ROS in melanoma. Indeed, while mechanistically demonstrating that ablation of Klf9, a pro-oxidant transcriptional regulator, promotes metastases in a BrafV600E; Pten−/− GEMM of melanoma without affecting primary tumor growth, the authors also reasoned that Klf9 deficiency inhibits premalignant melanocytic hyperplasia in a BrafV600E-induced hyperplasia model (77).
On the other hand, massive mitochondrial oxidative damage can occur upon excessive ROS production and culminate in the activation of mitophagy, the autophagy-mediated mechanism that, tightly coordinating with the mitochondrial fission/fusion machinery, controls the clearance of dysfunctional mitochondria (78). As such, mitophagy can be exploited by cancer cells to isolate and degrade damaged mitochondria to ensure qualitatively functional organelles. Indeed, a reduced proliferative rate has been associated with compromised fission machinery and retention of dysfunctional mitochondria in melanoma cell lines (69, 73). Also, increased hyper-activation of the fission machinery, which has been positively correlated to the BRAFV600E mutation in human patients, has been implicated in high proliferation of melanoma cells (79).
Unfortunately, little is known about the role of mitophagy regulators in melanoma biology. However, some evidence speaks in favor of an oncogenic role of these proteins in melanoma. As a matter of fact, the activity of BNIP3 has been discovered to crucially regulate removal of ROS-producing mitochondria. Doing so, BNIP3 favors mitochondrial fitness and maintains survival, bioenergetics, growth and aggressiveness of melanoma cells (80, 81). In parallel, Lee et al. have recently used an elaborate syngeneic model of B16-F10 melanoma cells injected in a transgenic Parkin knock-out mouse and assessed that Parkin favors melanoma growth and distal metastases by preventing the activation of the apoptotic cascade (Figure 1B) (69).
Over the last years, several researchers have attempted to address the interesting contribution of TME and systemic non-cell autonomous autophagy to tumor growth. Here, we report the most relevant publications on this matter that are using GEMMs or other genetically modified organism-based studies (Table 1).
White's group has recently demonstrated that, once autophagy is systemically depleted in a mouse model with conditional whole-body Atg7 deficiency or when autophagy-proficient melanoma cells are subcutaneously injected into mice, melanoma growth significantly slows down (Figure 1B) (68). The authors neatly proved that, in the context of arginine auxotrophic melanoma (82), host autophagy is necessary to sustain tumor growth by systemically replenishing arginine. According to the authors, autophagy-deficient hosts release hepatic ARG1 to process circulating arginine. Even though the mechanism still needs to be elucidated, this interplay is essential for tumor growth (68). This important discovery further underpins the relevance of autophagy in supporting tumor growth: though cancer cells are autophagy-proficient, maintenance of their growth requires an autophagy-competent environment. Moreover, the whole scenario suggests that also restricting the availability of essential tumor nutrients deserves to be exploited as a strategy for improving melanoma therapy.
Of note, this discovery has shed light on the non-cell-autonomous contribution of autophagy to tumor growth, which was already supported by the elegant work of Katheder and colleagues that demonstrated the extremely relevant role of TME autophagy in promoting tumor development (83). Briefly, they generated sophisticated transgenic models of Drosophila where different autophagy depletion assets were applied, in order to distinguish the contribution of tumor-only, TME-only (or the combination of both), and of host autophagy to tumor growth. Their interesting results displayed a more significant reduction of tumor growth and invasion in the neighboring tissue when local- and distal-autophagy deficiency were induced, giving enormous relevance to the microenvironmental signaling component (e.g., chemotactic cytokines) and to the metabolic addiction of the tumor to the microenvironment (83).
The last breakthroughs in dissecting the role of autophagy in melanomagenesis and melanoma progression have brought the metabolic aspects of autophagy to our attention, revealing autophagy as a fundamental machinery that sustains tumor metabolism. This implies the autophagic process to be pro-tumorigenic in the evolution of melanoma, especially supporting tumor growth (Figure 1B). It becomes even more relevant when considering that autophagy activation in both tumor cells and host can all affect melanoma development. The application of genetically engineered or syngeneic melanoma mouse models improved our understanding of these phenomena (Figure 1) and, most importantly, allowed researchers to test the therapeutic advantages of blocking autophagy in the treatment of melanoma, the beneficial effects of which have already been emphasized (13, 84, 85).
One issue, that has yet to be mentioned, is the contribution of autophagy to the regulation of the antitumor immunity and how this can influence the development of melanoma. As many other cancer types, melanoma can provoke an intricate and multifaceted immune response which can be more or less deleterious for the tumor, depending on several factors (86, 87). The involvement of autophagy in this context can very much depend on where it is exerting a major function in regulating the immune response and which immune cells are sensing and then responding to the different levels of autophagy (88–91). For instance, the group of Verginis demonstrated that autophagy inhibition in myeloid-derived suppressor cells (MDSCs), a population of immune cells accumulating within the tumors with the function of suppressing the immune response (92), can redirect these cells toward an antitumor immune reaction (93). In particular, they observed that MDSCs in both melanoma patients and mouse models displayed high autophagy levels. Therefore, by interfering with the autophagy status specifically in MDSCs, they successfully re-activated the tumor-specific CD4 positive T-cells and repressed the MDSCs suppressive function toward the tumor, with a resulting remarkable delay in tumor growth (93). Another interesting example is the study of Mgrditchian and co-workers, where the authors thoroughly demonstrated that inhibition of autophagy in melanoma cells negatively affected tumor growth by favoring the infiltration of natural killer (NK) cells into the tumor (67, 94–96). Placing autophagy again as an oncogenic process, Mgrditchian et al. demonstrated that syngeneic melanoma models depleted of Beclin1 or other autophagy genes (Atg5, Sqtsm1/p62) or with pharmacologically-inhibited autophagy (e.g., with chloroquine), showed significantly decreased melanoma development due to an enhanced NK cells-mediated immune response (Figure 1B) (67, 94). Taken together, these findings provide further insights into the oncogenic role of autophagy and how it can influence tumor growth by modulating the immune response, a highly relevant matter considering that melanoma is a great candidate for immunotherapy application (3, 5). This other layer of regulation by autophagy therefore undoubtedly bears a high clinical relevance for the future and could open new venues for autophagy-targeted therapy in melanoma.
DD, LD, and VB wrote the manuscript. LD and DD drafted the figure and table. DD provided senior supervision and critically revised the manuscript.
DD was supported by grants from the Danish Cancer Society (KBVU 204-A12424), the LEO foundation (LF-OC-19-000004), the Melanoma Research Alliance (620385). DD, LD, and VB are part of the Center of Excellence for Autophagy, Recycling and Disease (CARD), funded by the Danmarks Grundforskningsfond (DNRF125).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors are grateful to Per Guldberg for the fruitful discussions, Shona Willis for helping with manuscript editing, Mette Vixø Vistesen for the autophagy artwork in Figure 1, and to Laila Fischer for secretarial work.
AKT, AKT serine/threonine kinase 1; AMPK, 5′ adenosine monophosphate-activated protein kinase; ARG1, arginase 1; ATG, autophagy-related gene; BNIP3, BCL2-interacting protein 3; BRAF, v-raf murine sarcoma viral oncogene homolog; CD4, cluster of differentiation 4; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; eIF2α, eukaryotic initiation factor 2 alpha; ER, endoplasmic reticulum; ERK, extracellular signal-regulated kinase; FA, fatty acid; FATP1, fatty acid transfer protein 1; FIP200, focal adhesion kinase-interacting protein 200 kDa; GABARAP, GABA type A receptor-associated protein; GEMM, genetically engineered mouse model; HIF-1α, hypoxia inducing factor 1 alpha; JNK, jun N-terminal kinase; Klf9, Krueppel-like factor 9; LC3, light chain 3; LKB1, liver kinase B1; MAPK, mitogen-activated protein kinase; MDSCs, myeloid-derived suppressor cells; MEK, MAPK/ERK kinase 1; MiTF, melanocyte inducing transcription factor; mTORC1, mammalian target of rapamycin complex 1; MYC, myelocytomatosis oncogene cellular homolog; NK, natural killer; NRAS, neuroblastoma RAS viral oncogene homolog; OPTN, optineurin; OxPHOS, oxidative phosphorylation; PD-1, programmed cell death protein 1; PD-L1, programmed cell death ligand 1; PGC-1α, peroxisome proliferator-activated receptor-gamma coactivator 1 alpha; PI3K, phosphatidylinositol-3-kinase; PI3KIII, class III phosphatidylinositol-3-kinase; PPAR, peroxisome proliferator-activated receptor; PTEN, phosphatase and tensin homolog; RagD, Ras-related GTP-binding protein D; RAS, rat sarcoma; ROS, reactive oxygen species; SQSTM1, sequestosome 1; TFEB, transcription factor EB; TFE3, Transcription Factor Binding To IGHM Enhancer 3; TGF-β, tumor growth factor beta; TME, tissue microenvironment; ULK1, unc-51 like autophagy activating kinase 1; Vps 15 34, vacuolar protein sorting 15 34.
1. Shain HA, Bastian BC. From melanocytes to melanomas. Nat Rev Cancer. (2016) 16:345–58. doi: 10.1038/nrc.2016.37
2. Leonardi GC, Falzone L, Salemi R, Zanghì A, Spandidos DA, McCubrey JA, et al. Cutaneous melanoma: from pathogenesis to therapy (review). Int J Oncol. (2018) 52:1071–80. doi: 10.3892/ijo.2018.4287
3. Luke JJ, Flaherty KT, Ribas A, Long GV. Targeted agents and immunotherapies: optimizing outcomes in melanoma. Nat Rev Clin Oncol. (2017) 14:463–82. doi: 10.1038/nrclinonc.2017.43
4. Damsky W, Bosenberg M. Melanocytic nevi and melanoma: unraveling a complex relationship. Oncogene. (2017) 36:5771–92. doi: 10.1038/onc.2017.189
5. Domingues B, Lopes J, Soares P, Pópulo H. Melanoma treatment in review. Immunotargets Ther. (2018) 7:35–49. doi: 10.2147/ITT.S134842
6. Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. (2019) 176:11–42. doi: 10.1016/j.cell.2018.09.048
7. Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Bio. (2018) 19:349–64. doi: 10.1038/s41580-018-0003-4
8. Mizushima N. A brief history of autophagy from cell biology to physiology and disease. Nat Cell Biol. (2018) 20:521–7. doi: 10.1038/s41556-018-0092-5
9. Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. (2018) 20:233–42. doi: 10.1038/s41556-018-0037-z
10. Laplante M, Sabatini DM. Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci. (2013) 126:1713–9. doi: 10.1242/jcs.125773
11. Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Cell Dev Biol. (2011) 27:107–32. doi: 10.1146/annurev-cellbio-092910-154005
12. Klionsky DJ, Baehrecke EH, Brumell JH, Chu CT, Codogno P, Cuervo A, et al. A comprehensive glossary of autophagy-related molecules and processes (2nd edition). Autophagy. (2011) 7:1273–94. doi: 10.4161/auto.7.11.17661
13. Amaravadi RK, Kimmelman AC, Debnath J. Targeting autophagy in cancer: recent advances and future directions. Cancer Discov. (2019) 9:1167–81. doi: 10.1158/2159-8290.CD-19-0292
14. Rosenfeldt MT, Ryan KM. The multiple roles of autophagy in cancer. Carcinogenesis. (2011) 32:955–63. doi: 10.1093/carcin/bgr031
15. Day C-P, Merlino G, Van Dyke T. Preclinical mouse cancer models: a maze of opportunities and challenges. Cell. (2015) 163:39–53. doi: 10.1016/j.cell.2015.08.068
16. Walker GJ, Soyer H, Terzian T, Box NF. Modelling melanoma in mice. Pigm Cell Melanoma R. (2011) 24:1158–76. doi: 10.1111/j.1755-148X.2011.00923.x
17. Liu H, He Z, von Rütte T, Yousefi S, Hunger RE, Simon H-U. Down-regulation of autophagy-related protein 5 (ATG5) contributes to the pathogenesis of early-stage cutaneous melanoma. Sci Transl Med. (2013) 5:202ra123. doi: 10.1126/scitranslmed.3005864
18. Liu H, He Z, Simon H-U. Autophagy suppresses melanoma tumorigenesis by inducing senescence. Autophagy. (2013) 10:372–3. doi: 10.4161/auto.27163
19. Maes H, Agostinis P. Autophagy and mitophagy interplay in melanoma progression. Mitochondrion. (2014) 19:58–68. doi: 10.1016/j.mito.2014.07.003
20. Hara Y, Nakamura M. Overexpression of autophagy-related beclin-1 in advanced malignant melanoma and its low expression in melanoma-in-situ. Eur J Dermatol. (2012) 22:128–9. doi: 10.1684/ejd.2011.1562
21. Miracco C, Cevenini G, Franchi A, Luzi P, Cosci E, Mourmouras V, et al. Beclin 1 and LC3 autophagic gene expression in cutaneous melanocytic lesions. Hum Pathol. (2010) 41:503–12. doi: 10.1016/j.humpath.2009.09.004
22. Maes H, Martin S, Verfaillie T, Agostinis P. Dynamic interplay between autophagic flux and Akt during melanoma progression in vitro. Exp Dermatol. (2014) 23:101–6. doi: 10.1111/exd.12298
23. Rybstein MD, Pedro J, Kroemer G, Galluzzi L. The autophagic network and cancer. Nat Cell Biol. (2018) 20:243–51. doi: 10.1038/s41556-018-0042-2
24. Poillet-Perez L, White E. Role of tumor and host autophagy in cancer metabolism. Gene Dev. (2019) 33:610–9. doi: 10.1101/gad.325514.119
25. Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. (2015) 22:377. doi: 10.1038/cdd.2014.150
26. Li S, Song Y, Quach C, Guo H, Jang G-B, Maazi H, et al. Transcriptional regulation of autophagy-lysosomal function in BRAF-driven melanoma progression and chemoresistance. Nat Commun. (2019) 10:1693. doi: 10.1038/s41467-019-09634-8
27. Li S, Song Y, Quach C, Nemecio D, Liang C. Revisiting the role of autophagy in melanoma. Autophagy. (2019) 15:1843–4. doi: 10.1080/15548627.2019.1635386
28. Zheng B, Jeong JH, Asara JM, Yuan Y-Y, Granter SR, Chin L, et al. Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Mol Cell. (2009) 33:237–47. doi: 10.1016/j.molcel.2008.12.026
29. Alonso-Curbelo D, Riveiro-Falkenbach E, Pérez-Guijarro E, Cifdaloz M, Karras P, Osterloh L, et al. RAB7 controls melanoma progression by exploiting a lineage-specific wiring of the endolysosomal pathway. Cancer Cell. (2014) 26:61–76. doi: 10.1016/j.ccr.2014.04.030
30. Kawakami A, Fisher DE. The master role of microphthalmia-associated transcription factor in melanocyte and melanoma biology. Lab Invest. (2017) 97:649–56. doi: 10.1038/labinvest.2017.9
31. Möller K, Sigurbjornsdottir S, Arnthorsson AO, Pogenberg V, Dilshat R, Fock V, et al. MITF has a central role in regulating starvation-induced autophagy in melanoma. Sci Rep. (2019) 9:1055. doi: 10.1038/s41598-018-37522-6
32. Wellbrock C, Rana S, Paterson H, Pickersgill H, Brummelkamp T, Marais R. Oncogenic BRAF regulates melanoma proliferation through the lineage specific factor MITF. PLoS ONE. (2008) 3:e2734. doi: 10.1371/journal.pone.0002734
33. Wellbrock C, Arozarena I. Microphthalmia-associated transcription factor in melanoma development and MAP-kinase pathway targeted therapy. Pigment Cell Melanoma Res. (2015) 28:390–406. doi: 10.1111/pcmr.12370
34. Malta C, Siciliano D, Calcagni A, Monfregola J, Punzi S, Pastore N, et al. Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science. (2017) 356:1188–92. doi: 10.1126/science.aag2553
35. Damsky W, Micevic G, Meeth K, Muthusamy V, Curley DP, Santhanakrishnan M, et al. mTORC1 activation blocks Braf V600E -induced growth arrest but is insufficient for melanoma formation. Cancer Cell. (2015) 27:41–56. doi: 10.1016/j.ccell.2014.11.014
36. Rabinowitz JD, White E. Autophagy and metabolism. Science. (2010) 330:1344–8. doi: 10.1126/science.1193497
37. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
38. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. (2016) 23:27–47. doi: 10.1016/j.cmet.2015.12.006
39. Fischer GM, Gopal VY, McQuade JL, Peng W, DeBerardinis RJ, Davies MA. Metabolic strategies of melanoma cells: mechanisms, interactions with the tumor microenvironment, and therapeutic implications. Pigment Cell Melanoma Res. (2018) 31:11–30. doi: 10.1111/pcmr.12661
40. Smith LK, Rao AD, McArthur GA. Targeting metabolic reprogramming as a potential therapeutic strategy in melanoma. Pharmacol Res. (2016) 107:42–7. doi: 10.1016/j.phrs.2016.02.009
41. Abildgaard C, Guldberg P. Molecular drivers of cellular metabolic reprogramming in melanoma. Trends Mol Med. (2015) 21:164–71. doi: 10.1016/j.molmed.2014.12.007
42. Parmenter TJ, Kleinschmidt M, Kinross KM, Bond ST, Li J, Kaadige MR, et al. Response of BRAF-mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cancer Discov. (2014) 4:423–33. doi: 10.1158/2159-8290.CD-13-0440
43. Kumar SM, Yu H, Edwards R, Chen L, Kazianis S, Brafford P, et al. Mutant V600E BRAF increases hypoxia inducible factor-1α expression in melanoma. Cancer Res. (2007) 67:3177–84. doi: 10.1158/0008-5472.CAN-06-3312
44. Guldberg P, Thor Straten P, Birck A, Ahrenkiel V, Kirkin A, Zeuthen J. Disruption of the MMAC1/PTEN gene by deletion or mutation is a frequent event in malignant melanoma. Cancer Res. (1997) 57:3660–3.
45. Davies M, Stemke-Hale K, Tellez C, Calderone T, Deng W, Prieto V, et al. A novel AKT3 mutation in melanoma tumours and cell lines. Brit J Cancer. (2008) 99:1265. doi: 10.1038/sj.bjc.6604637
46. Kwong LN, Davies MA. Navigating the therapeutic complexity of PI3K pathway inhibition in melanoma. Clin Cancer Res. (2013) 19:5310–9. doi: 10.1158/1078-0432.CCR-13-0142
47. Scott DA, Richardson AD, Filipp FV, Knutzen CA, Chiang GG, Ronai ZA, et al. Comparative metabolic flux profiling of melanoma cell lines beyond the Warburg effect. J Biol Chem. (2011) 286:42626–34. doi: 10.1074/jbc.M111.282046
48. Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell. (2013) 23:302–15. doi: 10.1016/j.ccr.2013.02.003
49. García-Jiménez C, Goding CR. Starvation and pseudo-starvation as drivers of cancer metastasis through translation reprogramming. Cell Metab. (2019) 29:254–67. doi: 10.1016/j.cmet.2018.11.018
50. Falletta P, Sanchez-del-Campo L, Chauhan J, Effern M, Kenyon A, Kershaw CJ, et al. Translation reprogramming is an evolutionarily conserved driver of phenotypic plasticity and therapeutic resistance in melanoma. Gene Dev. (2017) 31:18–33. doi: 10.1101/gad.290940.116
51. Giglio P, Gagliardi M, Tumino N, Antunes F, Smaili S, Cotella D, et al. PKR and GCN2 stress kinases promote an ER stress-independent eIF2α phosphorylation responsible for calreticulin exposure in melanoma cells. Oncoimmunology. (2018) 7:e1466765. doi: 10.1080/2162402X.2018.1466765
52. Maida I, Zanna P, Guida S, Ferretta A, Cocco T, Palese L, et al. Translational control mechanisms in cutaneous malignant melanoma: the role of eIF2α. J Transl Med. (2019) 17:20. doi: 10.1186/s12967-019-1772-z
53. Giglio P, Fimia G, Lovat PE, Piacentini M, Corazzari M. Fateful music from a talented orchestra with a wicked conductor: connection between oncogenic BRAF, ER stress, and autophagy in human melanoma. Mol Cell Oncol. (2015) 2:e995016. doi: 10.4161/23723556.2014.995016
54. Corazzari M, Rapino F, Ciccosanti F, Giglio P, Antonioli M, Conti B, et al. Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differ. (2015) 22:946–58. doi: 10.1038/cdd.2014.183
55. Ferretta A, Maida I, Guida S, Azzariti A, Porcelli L, Tommasi S, et al. New insight into the role of metabolic reprogramming in melanoma cells harboring BRAF mutations. Biochim Biophys Acta. (2016) 1863:2710–8. doi: 10.1016/j.bbamcr.2016.08.007
56. Zhang M, Martino JS, Bowman RL, Campbell NR, Baksh SC, Simon-Vermot T, et al. Adipocyte-derived lipids mediate melanoma progression via FATP proteins. Cancer Discov. (2018) 8:1006–25. doi: 10.1158/1538-7445.AM2018-5120
57. Luo X, Zhao X, Cheng C, Li N, Liu Y, Cao Y. The implications of signaling lipids in cancer metastasis. Exp Mol Med. (2018) 50:127. doi: 10.1038/s12276-018-0150-x
58. Tímár J, Hegedüs B, Rásó E. The role of lipid signaling in the progression of malignant melanoma. Cancer Metast Rev. (2018) 37:245–55. doi: 10.1007/s10555-018-9729-x
59. Vegliante R, Di Leo L, Ciccarone F, Ciriolo M. Hints on ATGL implications in cancer: beyond bioenergetic clues. Cell Death Dis. (2018) 9:316. doi: 10.1038/s41419-018-0345-z
60. Di Leo L, Vegliante R, Ciccarone F, Salvatori I, Scimeca M, Bonanno E, et al. Forcing ATGL expression in hepatocarcinoma cells imposes glycolytic rewiring through PPAR-α/p300-mediated acetylation of p53. Oncogene. (2019) 38:1860–75. doi: 10.1038/s41388-018-0545-0
61. Lim J, Kwan Y, Tan M, Teo M, Chiba S, Wahli W, et al. The role of PPARβ/δ in melanoma metastasis. Int J Mol Sci. (2018) 19:2860. doi: 10.3390/ijms19102860
62. Borland MG, Yao P-L, Kehres EM, Lee C, Pritzlaff AM, Ola E, et al. Editor's highlight: PPARβ/δ and PPARγ inhibit melanoma tumorigenicity by modulating inflammation and apoptosis. Toxicol Sci. (2017) 159:436–48. doi: 10.1093/toxsci/kfx147
63. Saito T, Kuma A, Sugiura Y, Ichimura Y, Obata M, Kitamura H, et al. Autophagy regulates lipid metabolism through selective turnover of NCoR1. Nat Commun. (2019) 10:1567. doi: 10.1038/s41467-019-08829-3
64. Xie X, Koh J, Price S, White E, Mehnert JM. Atg7 overcomes senescence and promotes growth of BrafV600E-driven melanoma. Cancer Discov. (2015) 5:410–23. doi: 10.1158/2159-8290.CD-14-1473
65. García-Fernández M, Karras P, Checinska A, Cañón E, Calvo GT, Gómez-López G, et al. Metastatic risk and resistance to BRAF inhibitors in melanoma defined by selective allelic loss of ATG5. Autophagy. (2016) 12:1–15. doi: 10.1080/15548627.2016.1199301
66. Karras P, Riveiro-Falkenbach E, Cañón E, Tejedo C, Calvo TG, Martínez-Herranz R, et al. p62/SQSTM1 fuels melanoma progression by opposing mRNA decay of a selective set of pro-metastatic factors. Cancer Cell. (2019) 35:46–63 e10. doi: 10.1016/j.ccell.2018.11.008
67. Mgrditchian T, Arakelian T, Paggetti J, Noman M, Viry E, Moussay E, et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc Natl Acad Sci USA. (2017) 114:E9271–9. doi: 10.1073/pnas.1703921114
68. Poillet-Perez L, Xie X, Zhan L, Yang Y, Sharp DW, Hu Z, et al. Autophagy maintains tumour growth through circulating arginine. Nature. (2018) 563:569–73. doi: 10.1038/s41586-018-0697-7
69. Lee Y, Jung Y, Park M, Yeo I, Im H, Nam K, et al. Deficiency of parkin suppresses melanoma tumor development and metastasis through inhibition of MFN2 ubiquitination. Cancer Lett. (2018) 433:156–64. doi: 10.1016/j.canlet.2018.07.007
70. Hooijkaas AI, Gadiot J, van der Valk M, Mooi WJ, Blank CU. Targeting BRAF V600E in an inducible murine model of melanoma. Am J Pathol. (2012) 181:785–94. doi: 10.1016/j.ajpath.2012.06.002
71. Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE Jr, et al. BrafV600E cooperates with Pten loss to induce metastatic melanoma. Nat Genet. (2009) 41:544–52. doi: 10.1038/ng.356
72. Galluzzi L, Green DR. Autophagy-independent functions of the autophagy machinery. Cell. (2019) 177:1682–99. doi: 10.1016/j.cell.2019.05.026
73. Pal HC, Prasad R, Katiyar SK. Cryptolepine inhibits melanoma cell growth through coordinated changes in mitochondrial biogenesis, dynamics and metabolic tumor suppressor AMPKα1/2-LKB1. Sci Rep. (2017) 7:1498. doi: 10.1038/s41598-017-01659-7
74. Ciccarone F, Vegliante R, Di Leo L, Ciriolo M. The TCA cycle as a bridge between oncometabolism and DNA transactions in cancer. Semin Cancer Biol. (2017) 47:50–6. doi: 10.1016/j.semcancer.2017.06.008
75. del Idelchik M, Begley U, Begley TJ, Melendez AJ. Mitochondrial ROS control of cancer. Semin Cancer Biol. (2017) 47:57–66. doi: 10.1016/j.semcancer.2017.04.005
76. Zhang X, Gibhardt CS, Will T, Stanisz H, Körbel C, Mitkovski M, et al. Redox signals at the ER–mitochondria interface control melanoma progression. Embo J. (2019) 38:e100871. doi: 10.15252/embj.2018100871
77. Bagati A, Moparthy S, Fink EE, Bianchi-Smiraglia A, Yun D, Kolesnikova M, et al. KLF9-dependent ROS regulate melanoma progression in stage-specific manner. Oncogene. (2019) 38:3585–97. doi: 10.1038/s41388-019-0689-6
78. Vara-Perez M, Felipe-Abrio B, Agostinis P. Mitophagy in cancer: a tale of adaptation. Cells. (2019) 8:493. doi: 10.3390/cells8050493
79. Wieder SY, Serasinghe MN, Sung JC, Choi DC, Birge MB, Yao JL, et al. Activation of the mitochondrial fragmentation protein DRP1 correlates with BRAFV600E melanoma. J Invest Dermatol. (2015) 135:2544–7. doi: 10.1038/jid.2015.196
80. Maes H, Eygen VS, Krysko D, Vandenabeele P, Nys K, Rillaerts K, et al. BNIP3 supports melanoma cell migration and vasculogenic mimicry by orchestrating the actin cytoskeleton. Cell Death Dis. (2017) 5:e1127. doi: 10.1038/cddis.2014.94
81. Vara-Perez M, Maes H, Dingenen S, Agostinis P. BNIP3 contributes to the glutamine-driven aggressive behavior of melanoma cells. Biol Chem. (2019) 400:187–93. doi: 10.1515/hsz-2018-0208
82. Dillon BJ, Prieto VG, Curley SA, Ensor MC, Holtsberg FW, Bomalaski JS, et al. Incidence and distribution of argininosuccinate synthetase deficiency in human cancers. Cancer. (2004) 100:826–33. doi: 10.1002/cncr.20057
83. Katheder NS, Khezri R, O'Farrell F, Schultz SW, Jain A, Rahman MM, et al. Microenvironmental autophagy promotes tumour growth. Nature. (2017) 541:417. doi: 10.1038/nature20815
84. Ndoye A, Weeraratna AT. Autophagy- an emerging target for melanoma therapy. F1000research. (2016) 5:F1000 Faculty Rev-1888. doi: 10.12688/f1000research.8347.1
85. Galluzzi L, Pedro J, Levine B, Green DR, Kroemer G. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov. (2017) 16:nrd.2017.22. doi: 10.1038/nrd.2017.22
86. Barnes TA, Amir E. HYPE or HOPE: the prognostic value of infiltrating immune cells in cancer. Brit J Cancer. (2017) 117:451–60. doi: 10.1038/bjc.2017.220
87. O'Donnell JS, Teng MW, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. (2018) 16:1. doi: 10.1038/s41571-018-0142-8
88. Jiang G-M, Tan Y, Wang H, Peng L, Chen H-T, Meng X-J, et al. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol Cancer. (2019) 18:17. doi: 10.1186/s12943-019-0944-z
89. Li Y-Y, Feun LG, Thongkum A, Tu C-H, Chen S-M, Wangpaichitr M, et al. Autophagic mechanism in anti-cancer immunity: its pros and cons for cancer therapy. Int J Mol Sci. (2017) 18:1297. doi: 10.3390/ijms18061297
90. Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, inflammation, and immunity: a troika governing cancer and its treatment. Cell. (2016) 166:288–98. doi: 10.1016/j.cell.2016.05.051
91. León-Letelier RA, Bonifaz LC, Fuentes-Pananá EM. OMIC signatures to understand cancer immunosurveillance and immunoediting: melanoma and immune cells interplay in immunotherapy. J Leukocyte Biol. (2019) 105:915–33. doi: 10.1002/JLB.MR0618-241RR
92. Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. (2017) 5:3–8. doi: 10.1158/2326-6066.CIR-16-0297
93. Alissafi T, Hatzioannou A, Mintzas K, Barouni R, Banos A, Sormendi S, et al. Autophagy orchestrates the regulatory program of tumor-associated myeloid-derived suppressor cells. J Clin Invest. (2018) 128:3840–52. doi: 10.1172/JCI120888
94. Wennerberg E, Galluzzi L. Born to kill: NK cells go to war against cancer. Trends Cancer. (2019) 5:143–5. doi: 10.1016/j.trecan.2018.12.005
95. Baginska J, Viry E, Berchem G, Poli A, Noman M, van Moer K, et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci USA. (2013) 110:17450–5. doi: 10.1073/pnas.1304790110
Keywords: melanoma, autophagy, metabolism, GEMM, syngeneic mouse model
Citation: Di Leo L, Bodemeyer V and De Zio D (2020) The Complex Role of Autophagy in Melanoma Evolution: New Perspectives From Mouse Models. Front. Oncol. 9:1506. doi: 10.3389/fonc.2019.01506
Received: 03 October 2019; Accepted: 16 December 2019;
Published: 10 January 2020.
Edited by:
Geert Bultynck, KU Leuven, BelgiumReviewed by:
Gabriella Guida, University of Bari Aldo Moro, ItalyCopyright © 2020 Di Leo, Bodemeyer and De Zio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniela De Zio, ZHppb0BjYW5jZXIuZGs=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.