 Bruno Fattizzo
Bruno Fattizzo Wilma Barcellini
Wilma Barcellini- 1Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, University of Milan, Milan, Italy
- 2Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy
Autoimmune cytopenias, particularly autoimmune hemolytic anemia (AIHA) and immune thrombocytopenia (ITP), complicate up to 25% of chronic lymphocytic leukemia (CLL) cases. Their occurrence correlates with a more aggressive disease with unmutated VHIG status and unfavorable cytogenetics (17p and 11q deletions). CLL lymphocytes are thought to be responsible of a number of pathogenic mechanisms, including aberrant antigen presentation and cytokine production. Moreover, pathogenic B-cell lymphocytes may induce T-cell subsets imbalance that favors the emergence of autoreactive B-cells producing anti-red blood cells and anti-platelets autoantibodies. In the last 15 years, molecular insights into the pathogenesis of both primary and secondary AIHA/ITP has shown that autoreactive B-cells often display stereotyped B-cell receptor and that the autoantibodies themselves have restricted phenotypes. Moreover, a skewed T-cell repertoire and clonal T cells (mainly CD8+) may be present. In addition, an imbalance of T regulatory-/T helper 17-cells ratio has been involved in AIHA and ITP development, and correlates with various cytokine genes polymorphisms. Finally, altered miRNA and lnRNA profiles have been found in autoimmune cytopenias and seem to correlate with disease phase. Genomic studies are limited in these forms, except for recurrent mutations of KMT2D and CARD11 in cold agglutinin disease, which is considered a clonal B-cell lymphoproliferative disorder resulting in AIHA. In this manuscript, we review the most recent literature on AIHA and ITP secondary to CLL, focusing on available molecular evidences of pathogenic, clinical, and prognostic relevance.
Introduction
The impact of autoimmune cytopenias (AIC) complicating chronic lymphocytic leukemia (CLL), particularly autoimmune hemolytic anemia (AIHA) and immune thrombocytopenia (ITP) is variable, ranging from mild asymptomatic cytopenias case without indication to CLL treatment, to severe transfusion dependent patients with abrupt onset and CLL progression. Each patient needs to be carefully evaluated, since the different pictures require a specific approach. Given this heterogeneity, the variability of response to immune-suppression, and the possible association/development of clonal diseases (lymphoproliferation or myelodysplasia), the genomic landscape of AIC is of particular interest.
In this manuscript, we will review the most recent literature on AIHA and ITP secondary to CLL with a brief summary of their clinical management. In particular we will focus on available molecular evidences of pathogenic, clinical, and prognostic relevance.
Epidemiology and Pathogenesis
AIC may complicate CLL course at any time, from diagnosis to disease progression (Figure 1) (1). AIHA are the most frequent form (7–10% of cases), followed by ITP (1–5%), and rarer entities such as pure red cell aplasia (PRCA, <1%) and autoimmune granulocytopenia (AIG 0.17%). From a pathogenic point of view, CLL associated AIC are mediated by a complex orchestration of humoral, cellular, and innate immunity: (1) IgG auto-antibodies coat erythrocytes, platelets, and neutrophils with consequent antibody-dependent cellular cytotoxicity and complement-mediated destruction in the reticuloendothelial system (spleen and liver) or in the blood stream. (2) Anti-erythroblast and megakaryocyte autoantibodies can impair bone marrow compensatory response. (3) Autoreactive T-cells produce inflammatory cytokines and further inhibit myelopoiesis. (4) Natural killer cells have been shown to destroy erythroblasts from CLL patients in vitro, confirming a role for innate immunity.
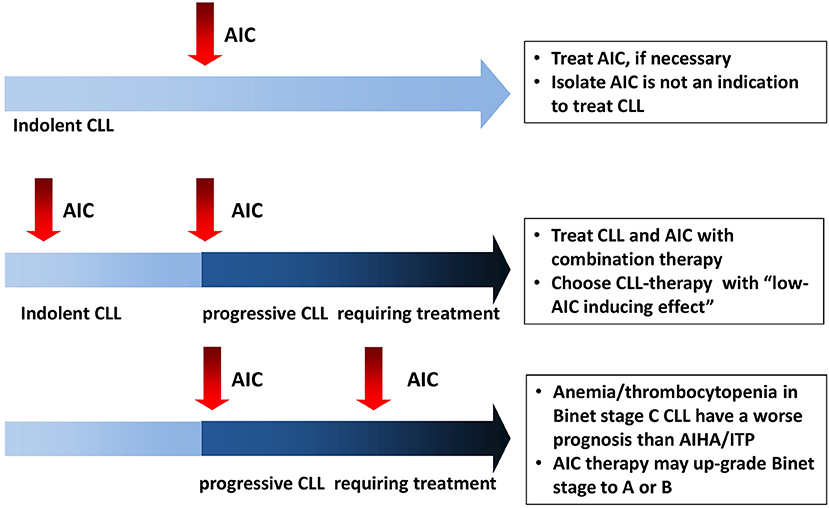
Figure 1. Autoimmune cytopenias (AIC) in chronic lymphocytic leukemia (CLL): the heterogeneity of onset imposes different management in each context.
As regards autoantibodies, they are polyclonal high-affinity IgG produced by non-malignant self-reactive B-cells in 90% of cases. CLL cells may also produce autoantibodies (mainly IgM) in <10% of cases (2–5), and have been shown to secrete soluble factors inducing a dysregulation of bone marrow microenvironment (6, 7). Further pathogenic mechanisms, are the direct antigen presentation by CLL cells that may induce self-reactive T helper cells, and the production of non-functional T regulatory cells (T-regs) (8–10). The latter become unable to eliminate non-neoplastic autoreactive T- and B-cells leading to autoimmune phenomena (11–14). In addition, an increased incidence of autoimmune cytopenias in CLL is associated to an imbalance in the ratio between Th17 cells and T-regs (15). Finally, CLL patients developing autoimmune phenomena displayed a reduction of Toll-like receptors (TLR)-4, an important player of the innate immunity, together with a lower expression of TLR2, and an increase of TLR7, TLR9, and TLR10 (16–18).
Influence of CLL Therapy on the Development of AIC
The influence of CLL therapy on the development of AIC deserves special consideration: single-agent purine analogs (i.e., fludarabine) may induce CLL-AIHA (19, 20) possibly worsening the imbalance between Th17 and T-regs (21). FC and FCR combination schemes (fludarabine, cyclophosphamide, and rituximab) in the CLL8 trial (22) showed very low incidence (<1%) of hemolytic anemia, as did bendamustine rituximab (BR) association (even if anecdotic PRCA cases have been described) (23). Alemtuzumab led to treatment-emergent ITP in 9% of CLL cases (24), again possibly due to T-cell dysregulation. Concerning small molecules, the most interesting data are available for Bruton's tyrosine kinase inhibitor ibrutinib: new-onset AIC was rarely reported in the largest studies performed so far (25–27). Moreover, AIC resolution occurred in about a half of CLL-AIC patients (N = 13) (26) and most CLL-AIC cases were able to discontinue AIC-therapy after a median of 4.7 months (N = 301 of whom 7% with ongoing AIC therapy) (27). Similar data were reported in a more recent study of 193 patients: 67% of 29 cases with AIC pre-ibrutinib could discontinue/taper AIC treatment and new-onset AIC occurred in 6% (all with unmutated IGHV) (28). Recent evidences suggest an inhibitory role of ibrutinib on autoreactive T cells, through interleukin-2-inducible kinase (ITK)suppression, leading the way for its use in T-cell mediated autoimmune conditions (i.e., graft vs. host disease) (29). Regarding other small molecules, limited data are available for idelalisib (that targets phosphoinositide 3-kinase), and venetoclax (a BCL-2 antagonist), although the presence of autoimmune phenomena was an exclusion criteria in various trials. Concerning venetoclax, it has been reported to be associated to the occurrence, although rarely, of AIHA in large CLL registrative trials (30). Interestingly, increased incidence of autoimmune complications (hepatitis, colitis, and pneumonitis) has been reported for idelalisib (31, 32).
Management of Autoimmune Hemolytic Anemia Secondary to CLL
Diagnosis
Management of AIHA in CLL requires the evaluation and exclusion of the other possible causes of anemia, including bone marrow infiltration/failure, bleeding, vitamin or iron deficiencies, and renal disease. As previously suggested, a diagnosis of AIHA can be established in the presence of Hb <11 g/dL, no chemotherapy in the previous month, variable alteration of hemolytic markers (increased unconjugated bilirubin, elevated lactate dehydrogenase, consumption of haptoglobin, increased absolute reticulocyte counts), and the positivity of the direct antiglobulin test (DAT) (1, 33). The latter allow to distinguish warm (wAIHA: DAT positive for IgG or IgG+C3d at low titer and negative autoagglutination at 20°C) from cold (cAIHA) cases (DAT positive for C3d and positive autoagglutination at 20°C). Of note, CLL itself may be a confounder in the differential diagnosis, since LDH may be elevated during disease progression, haptoglobin increased due to chronic/acute inflammation, and reticulocytosis may be absent or inadequate due to bone marrow infiltration or suppression by cytokine storm and/or anti-erythroblasts antibodies (1). The latter, demonstrated in a proportion of CLL cases through the mitogen-stimulated DAT, were associated to increased IL-4 and IFN-γ production, and may contribute to ineffective erythropoiesis (34). Furthermore, DAT positivity does not necessarily mean AIHA and in a longitudinal study of DAT+CLL cases only one third developed clinically overt hemolysis (35). Conversely, DAT negative AIHA cases may also be present (36), possibly due to the low-affinity or to the very small number of autoantibodies. In this context, the use of more sensitive techniques (microcolumn and solid-phase tests, or mitogen-stimulated DAT) may be useful (34). Finally, Bone marrow biopsy is usually necessary to document CLL infiltration and to rule out other causes (including bone marrow failure).
Treatment
As regards therapy (Table 1), the acuteness of onset, the severity of the anemia and the degree of hemolysis should be considered, together with patient' symptoms, age and comorbidities. Blood transfusions are usually indicated if Hb < 6 g/dL or higher in elderly comorbid patients. Over-transfusion should be avoided since it carries high risk of allo-immunization. In CLL-cases, given underlying bone marrow impairment and inadequate reticulocytosis, transfusion requirement may be higher than in primary cases. Moreover, the evaluation of endogenous erythropoietin (to be performed before repeated transfusions that may confound the picture) could suggest the use of recombinant erythropoietin. For warm AIHA, steroid therapy is considered the first line (usually prednisone at 1 mg/kg day for 3–4 weeks, followed by a slow tapering in a total of 6 months). Methylprednisolone boli (2–10 mg/Kg day for 3 days) may be considered, with or without intravenous immunoglobulins (0.4 g/kg for 5 days or 1 g/kg for 2 days), in patients with acute hemolysis and slow response to steroid therapy (1, 37). The fewer patients with cAIHA may have a milder clinical presentation with Hb levels >9 g/dL and cold agglutinin associated symptoms (acrocyanosis, itch, urticarial, etc.) and may require a watchful waiting approach. Treatment should be reserved for transfusion-dependent cases, active hemolysis (even if increase of LDH is difficult to judge in CLL), and invalidating cAIHA symptoms. Corticosteroids are usually effective only at high doses, and are a useful tool only in the acute setting. Prompt rituximab treatment should be considered, together with a quick steroid tapering after Hb stabilization. Rituximab is currently considered the first therapy line in cAIHA at standard dose of 375 mg/sm weekly for 4 weeks, with an overall response in up to 70–100% of patients (1, 39, 44). Considering patients refractory to first-line treatment (both wAIHA and cAIHA), current guidelines advice the introduction of a CLL directed therapy. The choice between chemoimmunotherapy and small molecules should be made according to current guidelines (patient age/comorbidities and CLL molecular characteristics) and considering potentially hemolytic side effects (avoid fludarabine single agent). As regards published studies specifically addressing refractory CLL-AIHA, rituximab in various combinations was able to induce high (>80%) and durable response rates: 89% (N = 8) with cyclophosphamide and dexamethasone (RCD) (54, 55), 95% (N = 20) with cyclophosphamide, vincristine, and prednisone (R-CVP) (56), and 80% with bendamustine (N = 26), with a median relapse free survival of 28 months (45, 46). Good results have also been reported in association with oral fludarabine, even if mainly in primary cAIHA cases (47). The only exception to this aggressive approach regards steroid-refractory wAIHA with no signs of CLL progression. In this setting, a possible strategy is to administer rituximab single agent with a reported efficacy in 72% of cases, of whom 40% sustained responses at 17 months (38, 39). Alemtuzumab has been abandoned because of serious infectious and autoimmune complications, as also happened for splenectomy (41–43). Cytotoxic immunesuppressors showed heterogeneous and weak efficacy in primary AIHA and are usually not administered in CLL secondary cases (40, 58). New generation monoclonal antibodies, such as of atumumab and obinutuzumab, may also be useful in secondary AIHA (59). As cited above, ibrutinib seems to be safe in patients with CLL-AIHA and progressive disease, and a phase II trial of ibrutinib combined to rituximab is ongoing in CLL-wAIHA [NCT03827603]. Regarding venetoclax, case reports of successful treatment have been published (60, 61).
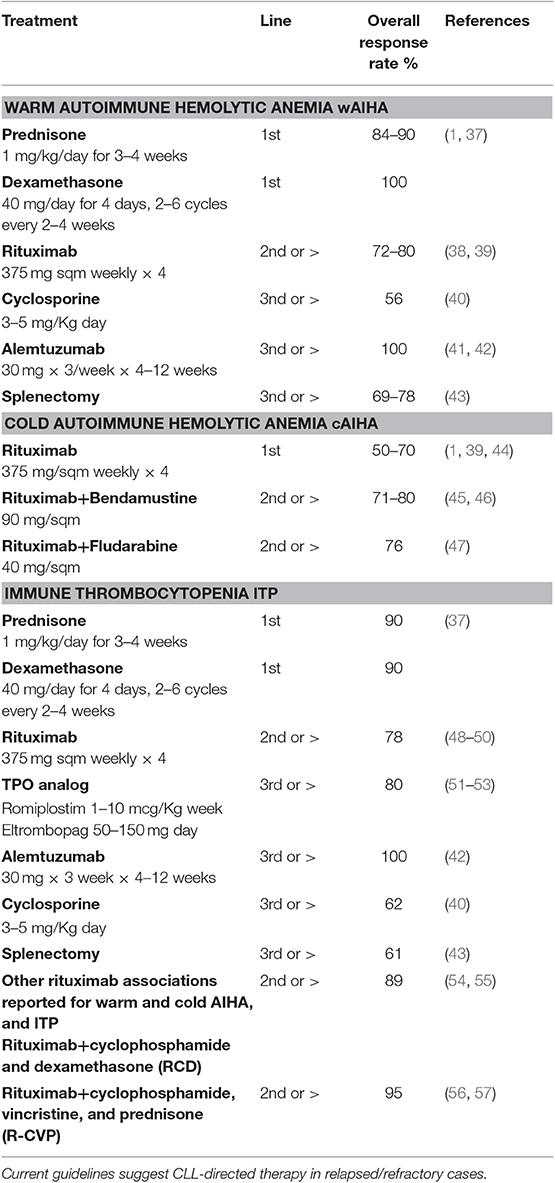
Table 1. Specific therapies and relative outcomes for warm and cold autoimmune hemolytic anemia and immune thrombocytopenia secondary to chronic lymphocytic leukemia (CLL).
Management of Immune Thrombocytopenia Secondary to CLL
Diagnosis
The same diagnostic caveats mentioned for CLL-AIHA have to be considered in the thrombocytopenic patient. ITP should be suspected in a CLL patient with <100 × 109/L platelets, with no chemotherapy in the previous month; moreover signs of CLL progression should be excluded (progressive splenomegaly, concomitant anemia, significant bone marrow CLL infiltrate, evidence of bone marrow failure/dysplasia). Other secondary causes (infections, drug-induced thrombocytopenia, thrombotic microangiopathies, and heparin-induced thrombocytopenia) should also be ruled out. Antiplatelet antibodies are of little aid due to the low sensitivity and specificity of the test, and usually not performed (1).
Treatment
ITP should be treated only in case of severe thrombocytopenia (Plt < 30 × 109/L) or bleeding. First-line therapy with steroids (prednisone at 1 mg/kg day for 1 month, followed by a slow tapering, or dexamethasone 40 mg/day × 4 days 1–3 cycles) is the standard approach, with about 50% responders. Intravenous immunoglobulin can be added in case of bleeding or slow response to steroids, again with 50% response rate [(27)]. Platelet transfusion may be required in case of life-threatening hemorrhage. Similarly to CLL-AIHA, steroid refractory cases would deserve CLL-directed therapy evaluation. Rituximab monotherapy was shown effective in 86% of CLL-ITP cases (57% complete response) (48), with 21 months response duration (49, 50). Rituximab combined to cyclophosphamide and dexamethasone or to cyclophosphamide, vincristine and prednisone had a high rate of durable responses in published experiences (55, 57). Splenectomy is usually discouraged given the increased infectious risk, older age and comorbidities of CLL patients. Finally, thrombopoietin mimetics (romiplostin and eltrombopag), indicated in refractory primary ITP, have shown high (up to 80%) and durable responses in patients with CLL-ITP (51–53, 62).
Molecular Aspects in Primary and Secondary AIHA
Table 2 shows available studies addressing molecular aspects of warm and cold AIHA, both primary and secondary to lymphoproliferative disorders.
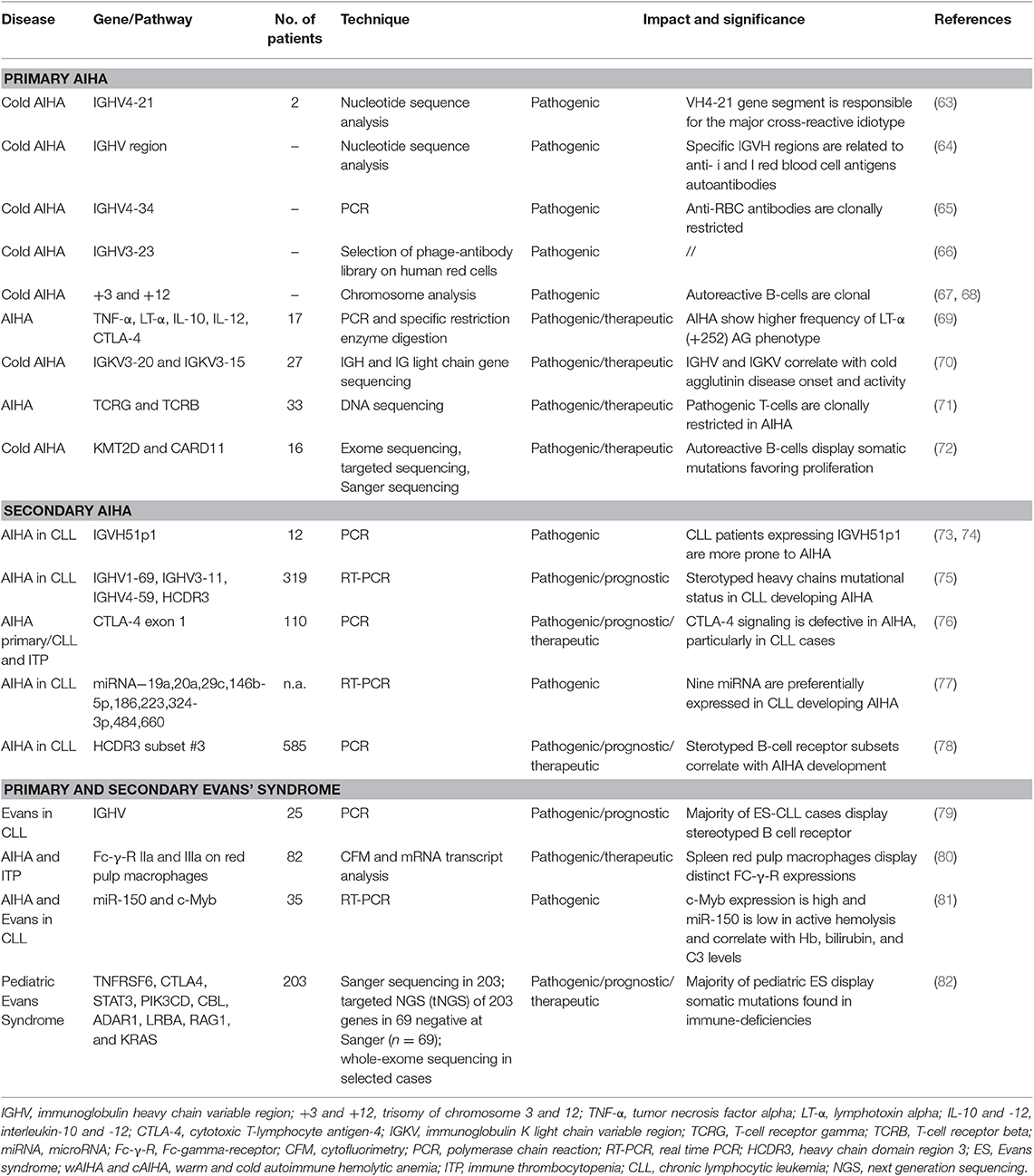
Table 2. Molecular findings in primary and secondary autoimmune hemolytic anemia (AIHA) and Evans' syndrome.
Studies on Immunoglobulin Genes
Since the autoantibody is the major pathogenic player, the larger and older experiments focused on the configuration of the genes of the variable region of the immunoglobulin heavy chains (IGHV) encoding AIHA autoantibodies and demonstrated that some rearrangements are preferentially involved. Almost all patients with cAIHA displayed monoclonal antibodies encoded by the IGHV4-34 gene, responsible for I antigen binding (63–65). Rarely, IGHV3 family genes may also encode anti-I cold agglutinins, in particular IGHV3-23 and IGKV3-20 (66, 70, 83). Concerning Ig light chain genes, the IGKV3-20 gene and the IGHV3-15 gene are used in most cAIHA patients and contribute to I antigen binding. From a clinical perspective, mutations in the complementarity determining region (CDR)2 and in the framework region 3 (FR3) of IGHV4-34 correlated with lower hemoglobin levels (70), whilst those in the IGKV3-20 CDR3 correlated with younger age at diagnosis. These findings are in line with the clonal nature of cAIHA that is currently considered a distinct lymphoproliferative disorder, with some level of bone marrow infiltration morphologically different from other non-Hodgkin lymphomas. The presence of stereotyped light chains of cAIHA may be of therapeutic interest, since anti-light chain vaccinations with IGKV3-20 are under investigation for lymphoproliferative diseases (84).
Other studies focused on B-cell receptor configuration and its contribution to AIC development. It is known that unmutated IGHV carries a strong prognostic impact on CLL course and correlates with a higher incidence of AIC (78, 85–89). The binding of auto-antigens to unmutated CLL cells activates a signal transduction (i.e., phosphorylation of SYK and ZAP-70) promoting survival and proliferation (90). More recently, a high recurrence of stereotyped IGHV aminoacid sequences has been observed in CLL patients developing AIC (91–95). Efremov et al. (73) reported an over-representation of the 51p1 VH gene; in other two large studies (N = 319 and N = 585), patients developing AIHA showed a more frequent expression of unmutated IGHV1-69, IGHV3-11, IGHV4-59, IGHV4–30, IGHD2-2, and IGHJ6 genes, unfavorable [del(17)(p13) and del(11)(q23)] cytogenetics, and stereotyped HCDR3 sequences (75, 78). Finally, stereotyped B cell receptor configuration was found in 66% of CLL secondary Evans syndrome, a known severe complication defined by the association of AIHA and ITP (79).
Studies on Cell-Mediated Immunity
Since a T-cell imbalance is known to play a part in AIC development (higher Th17/T regulatory ratio, Th1 to Th2 cytokine shift, increased APC activity), other studies focused on T-cell compartment. They showed the presence of clonal T-cell populations, mainly CD8+, in about 50% of AIHA patients (N = 33), higher than in controls (71). Another study (76) evaluated cytotoxic T-lymphocyte antigen-4 (CTLA-4) gene status in patients with primary or secondary AIC (20 primary AIHA, 30 CLL-AIHA, and 60 ITP). CTLA-4 is a negative regulator of T-cell responses and has been implicated in various autoimmune diseases (96, 97). A high prevalence of an A to G polymorphism at position 49 was found among AIHA cases, particularly in the CLL-AIHA group (73% vs. 47% in the control group), suggesting CTLA-4 mediated T-cell imbalance in these cases. A more recent study found a significant higher frequency of lymphotoxin-α (LT-α) (+252) AG phenotype in 17 AIHA cases compared to controls (41% vs. 13%) (69). LT-α (also known as TNF-β), is involved in the regulation of cell survival, proliferation, differentiation, and apoptosis, and plays an important role in innate immune regulation and immune-surveillance (98).
Finally, it has been reckoned that AIHA clinical picture also depends on the level of the monocyte-macrophage system activation and some Authors studied FcγR subtypes expressions in various tissues in 82 AIHA cases. They found that red pulp macrophages predominantly expressed the low-affinity receptors FcγRIIa and FcγRIIIa, did not express the inhibitory FcγRIIb, and expressed very low levels of the high-affinity receptor FcγRI, compared to blood monocytes (80). This may be of therapeutic interest, given that FcγR and its signaling have recently become a target in autoimmune diseases.
Genomic Studies
The use of advanced target and non-target sequencing assays offered further insights in AIHA pathogenesis. In particular, in a study of 16 primary cAIHA, next generation sequencing of bone marrow B-cells allowed the identification of recurrent mutations of KMT2D and CARD11 in 69% and 31% of cases, respectively (72). Similar mutations have also been reported in lymphomas as well as in Kabuki syndrome, a congenital disorder characterized by malformations, immune-deficiency, and development of autoimmune diseases. Loss of KMT2D function increases B cell proliferation, impedes class switch recombination (99), and may concur to survival of autoreactive B cells synergizing with IGHV4-34-encoded immunoglobulin receptor stimulation (72). CARD11 mutations were shown to induce constitutive activation of the NF-kB pathway, similarly to what observed in diffuse large B-cell lymphoma. Evaluation of KMT2D and CARD11 might be of diagnostic utility in cAIHA, and would help to distinguish it from MYD88 mutated lymphoplasmacytic lymphoma. Genomic studies may give hints for novel therapeutic approach. In fact, histone deacetylase inhibitors, that have been used in lymphoma, myeloma and Kabuki syndrome, might have a therapeutic potential in cAIHA with KMT2D mutations (72, 100). Similarly, therapies targeting CARD11 gain-of-function mutations are under investigation for B cell lymphomas and may be studied also in cAIHA (101).
Another very recent study evaluated a large series of pediatric patients with Evans syndrome by Sanger sequencing, targeted NGS, and whole exome sequencing (N = 80): 65% received a genetic diagnosis, 49 had a germline mutation, and 3 somatic variants. Pathogenic mutations in genes involved in primary immunodeficiencies (TNFRSF6, CTLA4, STAT3, PIK3CD, CBL, ADAR1, LRBA, RAG1, and KRAS) were found in 40% of cases, and probable pathogenic variants in 16 genes not previously reported in autoimmune disease were detected in 25%. It was already known that children with primary immunodeficiency are more prone to develop immune cytopenia, whilst in adult Evans' syndrome a primary immunodeficiency was identified in 9% of cases only (102). In the pediatric study, mutated patients showed more severe disease with higher treatment requirement (>number of therapy lines) and mortality. These data confirm that a higher genomic burden is probably involved in pediatric cases, and that it seems to have prognostic and therapeutic significance (82). For instance, patients with autoimmune lymphoproliferative syndrome (ALPS), caused by germline and somatic TNFRSF6 mutations, are more prone to develop severe persistent hypogammaglobulinemia after rituximab treatment, and splenectomy is contraindicated. Since rituximab is highly effective and broadly used in Evans syndrome, a prompt diagnosis of such cases is of great importance. Moreover, 36% of cases had potentially targetable mutations that will be suitable for new therapeutic approaches including rapamycin inhibitors (in ALPS or a PIK3d activation syndrome) (103, 104), CTLA-4 fusion protein (in CTLA-4 and LRBA deficiency) (105, 106), JAK inhibitors (in patients with JAK1 or JAK2 mutations) (107), and calcineurin inhibitors (in patients with NFATC1 variants) (108).
Studies on MicroRNAs
MicroRNAs (miRNAs) are small single strain RNAs mainly implied in gene expression regulation at transcriptional and post-transcriptional level. They have been associated with different clinical-biological forms of CLL and are also known to play a substantial role in autoimmunity (77). In a recent study evaluating malignant B-cells from CLL-AIHA patients, nine down-regulated miRNAs were identified (i.e., miR-19a, miR-20a, miR-29c, miR-146b-5p, miR-186, miR-223, miR-324-3p, miR-484, and miR-660), of whom two (i.e., miR-20a and miR-146b-5p) known to be involved in autoimmune phenomena. Interestingly, miR-146b-5p was shown to modulate the expression of CD80, a molecule involved in the B-T cell synapse formation and in restoring the APC capacity of CLL cells. Another miRNA, miR-150, was recently studied in 35 patients with AIHA/Evans syndrome and was found low in patients with active hemolysis compared to those in remission or with CLL-AIHA. MiR-150 negatively correlated with bilirubin values and positively with Hb and complement levels, suggesting the role of miRNAs in predicting CLL evolution and treatment response (81).
Molecular Aspects in Primary and Secondary ITP
Studies on Immunoglobulin Genes
Similarly to AIHA, first molecular studies on primary ITP showed the presence of recurrent IGHV gene rearrangements in autoreactive B cells (Table 3) (109). Roark and Colleagues, found an association with rearrangements of IGHV3-30, and further reports showed that IGHV30 encoded IgM and IgG anti-GPIIb autoantibodies (122–125). Interestingly, IGHV3-30 is highly employed also in AIHA, CLL, and immunodeficiencies and this may explain the association with ITP (74, 126). In CLL patients, it has been shown that the risk of developing ITP was higher among patients with stereotyped subset #1 (IGHV1–5-7/IGHD6–19/IGHJ4) and #7 (IGHV1–69 or IGHV3–30/IGHD3-3/IGHJ6) in HCDR3 region (78). Other IGHV involved in anti-platelets autoantibodies are VH1-02, VH1-46, VH3-21, and VH4-59. Interestingly, a specific heavy- and light-chain pairing seems to be necessary to enable antibody pathogenicity (127–131). Anti-platelets autoantibodies appear to share single heavy-chain VHDJH and have undergone isotype switching (hallmark of a T-cell-dependent, antigen-driven response). These aspects are not observed in naturally occurring anti-platelet antibodies that are polyreactive IgM with little or no somatic mutation of their variable regions, and are responsible for platelets turnover. The presence of stereotyped IGHV asset could be of therapeutic interest in ITP, since IGHV3-30-targeted reagents, such as anti-idiotypic antibodies derived from mice (132, 133) or humans (125) are under evaluation (134–137).
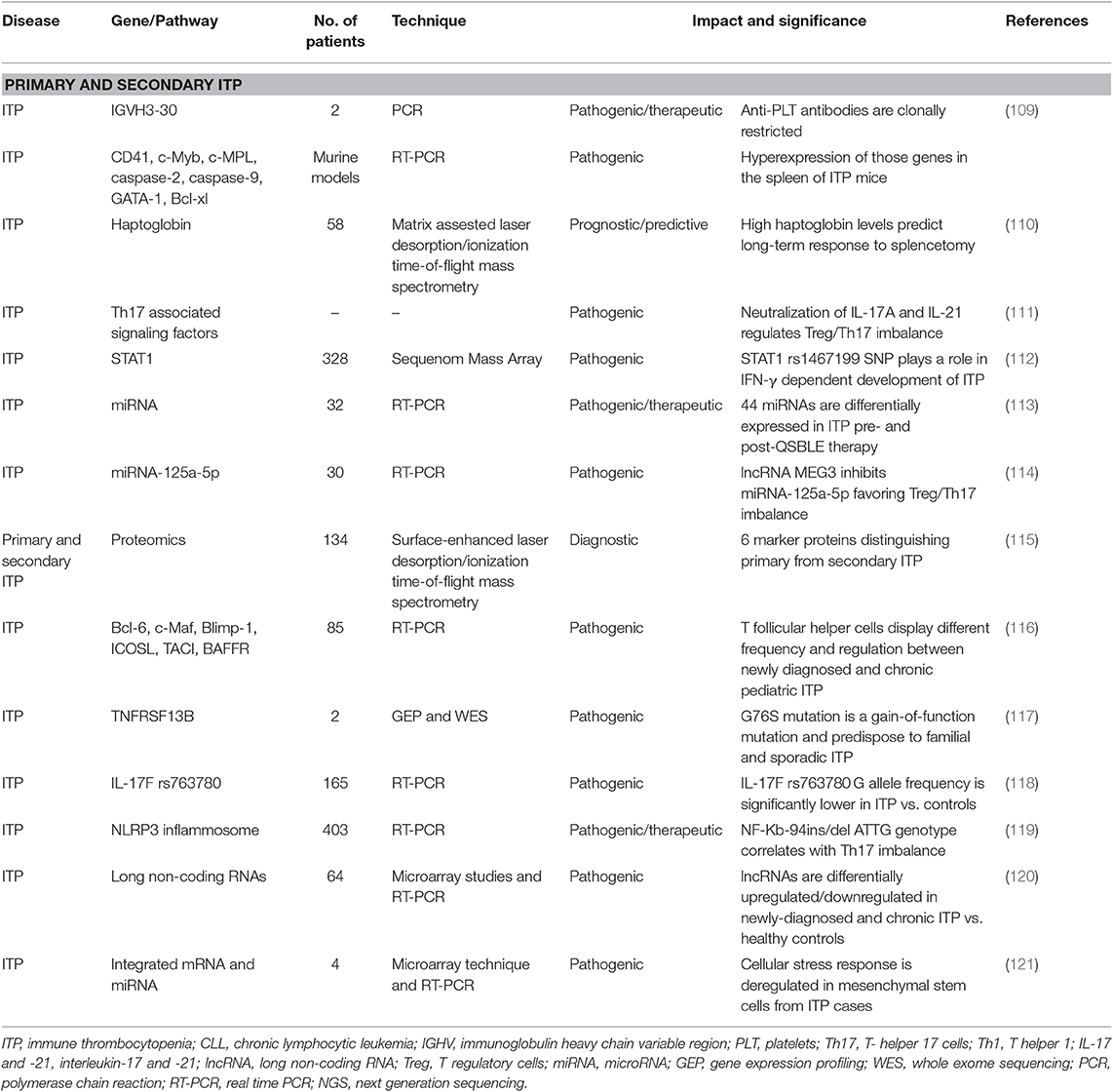
Table 3. Molecular findings in primary and secondary immune thrombocytopenia (ITP).
Studies on Cell-Mediated Immunity
Th17 are known to mediate autoimmunity through the release of pro-inflammatory cytokines (IL-2/IL-17). Th17 cells response, together with Th2 (anti-inflammatory), regulatory B (Breg), and Treg cells inhibition (with decrease in IL-10/TGF-β), favor ITP persistent/chronic phase. As a matter of fact, therapy with corticosteroids, rituximab, and thrombopoietin receptor agonists have all be shown to increase Tregs and TGF-β levels (TPO agonists also increase Breg). Given the importance of these cytokine dysregulation, some Authors focused on Treg/Th17 imbalance and on cytokine genes polymorphisms. In a recent study, it has been shown that NF-κB-94ins/del ATTG genotype (involved in the NLRP3 inflammasome) contributes to ITP development and to imbalanced Th17 cell response (119). Another study on IL-17F rs763780 polymorphism, that has been associated with IL-17 expression and activity, showed a lower prevalence in ITP cases (N = 165) compared to healthy controls (118). Finally, Hu et al. demonstrated that IL-17A and IL-21 are able to upregulate STAT-1, STAT-3, STAT-5 or RAR-related orphan receptor C (RORC), resulting in decreased Treg/Th17 balance in newly diagnosed ITP cases. This imbalance recovered after ITP remission and was reversed by the neutralization of IL-17A or IL-21 through targeting antibodies (111). IL-21 levels, together with IL-4, were also found to be abnormal in pediatric ITP (N = 85), and to affect T follicular helper cells levels and regulation (116). IL-17A or IL-21 blockade could be a novel target for ITP.
Studies on Inflammatory Cytokines
Interferon (IFN)-γ signaling and tumor necrosis factor (TNF) are highly implicated in ITP pathogenesis and provides a link between autoimmunity, inflammation, and bone marrow failure. A polymorphism in the signal transducer and activator of transcription 1 protein (STAT1) rs1467199 SNP, the main target of IFN-γ down-stream emerged in a study of 328 ITP children, and was differentially found between newly diagnosed and chronic patients (112). More recently, microarray studies showed that a huge number of long non-coding RNAs (lncRNAs) were significantly up-regulated or down-regulated in newly diagnosed and chronic ITP patients vs. healthy individuals. TNF and granulocyte macrophage colony-stimulating factor signaling were the most interested pathways. Interestingly, lncRNAs ENST00000440492, ENST00000528366, NR_038920, and ENST00000552576 were able to distinguish newly diagnosed from chronic ITP (120). Finally, Peng et al. used gene expression profiling analysis and whole-exome sequencing on samples from family members with ITP, sporadic ITP cases and healthy individuals and identified a potential pathologic p.G76S heterozygous mutation on the TNFRSF13B gene. Mutated cases had upregulated cytokine-cytokine receptor interaction, increased serum TNFα, IL-17α, IFNγ, and BAFF levels, and enhanced binding capacity of APRIL ligand to B cells. Moreover, B cells transfected with the G76S mutation could induce human megakaryocyte apoptosis in vitro (117).
Studies on MicroRNAs
MiRNAs expression was also evaluated in ITP in various reports: molecular studies of bone marrow mesenchymal stem cells from ITP patients showed that 740 genes and 32 miRNAs were differentially expressed compared to controls and correlated with the presence of cellular growth defects and functional abnormalities. The latter seem to be due to impaired cellular stress response, unfolded protein response, and reduced DNA transcription (121). Burenbatu and Colleagues, identified 44 miRNAs that are differentially expressed in ITP patients before and after treatment with the Mongolian medicine Qishunbaolier (QSBLE). Interestingly, 25 from these 44 miRNAs are downregulated in ITP as compared to controls, and are restored after QSBLE exposure (113). Finally, reduced miR-125a-5p expression has been linked Treg/Th17 imbalance. Li et al. demonstrated that miR-125a-5p expression is inhibited by MEG3 overexpression in ITP patients (N = 30). Interestingly, dexamethasone was able to reduce MEG3 expression in vitro, thus restoring Treg/Th17 ratio (114).
Proteomics
Proteomic studies found some clinical implications: screen of 64 primary and 70 secondary ITP cases using surface-enhanced laser desorption/ionization time-of-flight mass spectrometry (SELDI-TOF-MS) allowed the identification of 6 proteins able to distinguish primary from secondary cases with high sensitivity (115). Another proteomic study identified higher haptoglobin levels as a favorable serum biomarker for predicting long-term response to splenectomy in ITP, with a positive correlation with postoperative platelet count (110).
Discussion and Future Perspectives
AIC secondary to CLL are a nice model of close intersection between cancer and autoimmunity. Both are the result of uncontrolled and dysregulated homeostatic mechanisms leading to aberrant proliferation and activity of specific cellular subsets with heterogeneous epiphenomena. Leukemic B-cells show impaired apoptosis, are unable to efficiently produce immunoglobulins, may function as antigen presenting cells, and release a variety of inflammatory cytokines leading to three main immune-related complications: infections, autoimmune diseases, and decreased immune-surveillance on secondary malignancies. These complications seem to correlate with advanced stage CLL and with poor prognostic markers. Moreover, CLL therapy may have an impact on their development.
The genomic landscape of primary and secondary AIC is of particular interest, since the type and the depth of the immune response is likely under genetic control and it could be hypothesized that a predisposing genetic background correlates with a more profound immune dysregulation. Molecular studies performed so far, mainly focused on B-cell/autoantibodies characteristics and functioning, and on T cell aberrations: sterotyped B cells with specific IGHV and light chain configuration are involved in AIC development, clonal T cells, specifically CD8+ ones are present, and various cytokine genes polymorphisms may correlate with Treg/Th17 imbalance. Other experiences showed a dysregulation at the gene expression level as demonstrated by altered miRNA and lnRNA profiles in AIC cases compared to healthy subjects, but also in newly-diagnosed vs. chronic patients, and in the same patients in different tissues. Finally, proteomic studies reported differentially translated proteins in primary vs. secondary cases. In this regard, all the guidelines on AIC state that secondary causes should always be excluded. However, current workup relies mainly on laboratory, morphologic and imaging techniques that could be unable to disclose the presence of clonal disorders (Figure 2). In this context, the genetic/molecular characterization of AIC patients will probably increase our sensitivity in diagnosing secondary cases. This has been demonstrated in the recent paper on a pediatric Evans' population, where NGS/WES techniques revealed the presence of an underlying disease in 65% of cases, with important clinical/therapeutic implications. No data are available for adults, but for cAIHA, where a clonal lymphoid infiltrate is almost invariably present. This form is particularly difficult to distinguish from secondary cases. Berentsen and Colleagues proposed to differentiate cold agglutinin “disease” from “syndrome” basing on the absence or presence of a secondary cause. The demonstration that MYD88 mutation is always absent and that KMT2D and CARD11 ones are present in a proportion of cases, carry diagnostic, prognostic and therapeutic impact, further stressing the utility of molecular studies in AIC. Finally, there is growing evidence that AIC may evolve to overt clonal diseases of myeloid or lymphoid lineages and no predictors are available (138–141). This tempts to speculate about a model of “double clonality” unique for these forms, where either myeloid or lymphoid populations may undergo clonal expansion/selection. As a matter of fact, clonality and malignancy are distinct although overlapping concepts, and the evolution of a clonal disorder into an overt malignancy may require a long time, even longer than human lifespan. The immune system has a role in this process. However, it is not always clear whether it acts as an effector or spectator, and the exact molecular/genetic mechanisms and therapeutic implications have still to be disclosed.
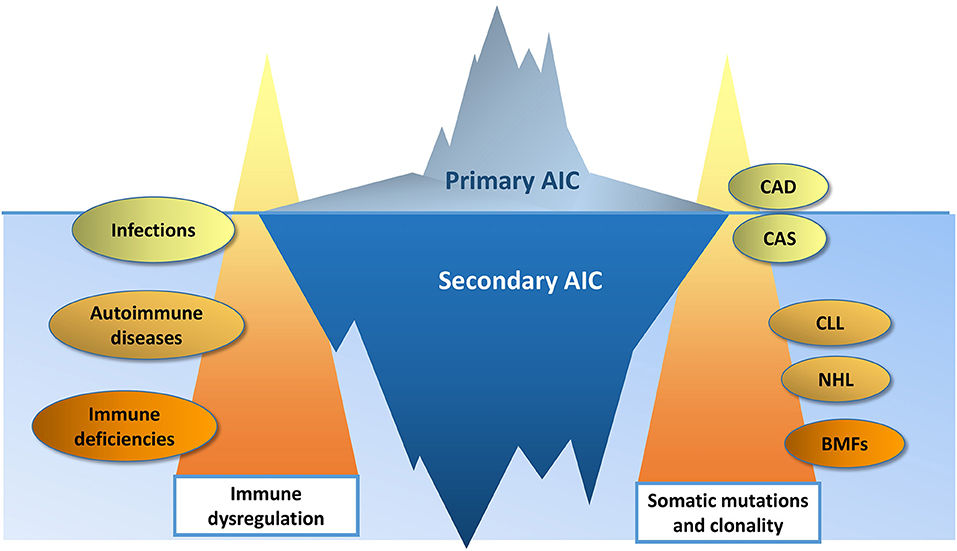
Figure 2. The changing border between primary and secondary autoimmune cytopenias (AIC). Immune dysregulation is more profound in AIC secondary to systemic autoimmune diseases and immune deficiencies, than in AIC secondary to infections. Likewise, a higher burden of somatic mutations is more typical of bone marrow failures (BMF) and lymphoproliferative disorders (chronic lymphocytic leukemia, CLL; non-Hodgkin lymphomas, NHL), than in cold agglutinin disease (CAD) and syndrome (CAS). The increasing availability of genomic testing will improve the diagnostic sensitivity, moving upward the border between primary and secondary AIC.
Author Contributions
BF and WB designed and wrote the review and participated to the final revision. All authors participated to the design of the review, literature revision, manuscript writing, and final revision for important intellectual content.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Visco C, Barcellini W, Maura F, Neri A, Cortelezzi A, Rodeghiero F. Autoimmune cytopenias in chronic lymphocytic leukemia. Am J Hematol. (2014) 89:1055–62. doi: 10.1002/ajh.23785
2. Hamblin TJ, Oscier DG, Young BJ. Autoimmunity in chronic lymphocytic leukaemia. J Clin Pathol. (1986) 39:713–6. doi: 10.1136/jcp.39.7.713
3. Beaume A, Brizard A, Dreyfus B, Preud'homme JL. High incidence of serum monoclonal Igs detected by a sensitive immunoblotting technique in B-cell chronic lymphocytic leukemia. Blood. (1994) 84:1216–9. doi: 10.1182/blood.V84.4.1216.1216
4. Broker BM, Klajman A, Youinou P, Jouquan J, Worman CP, Murphy J, et al. Chronic lymphocytic leukemic (CLL) cells secrete multispecific autoantibodies. J Autoimmun. (1998) 1:469–81. doi: 10.1016/0896-8411(88)90068-6
5. Stevenson FK, Hamblin TJ, Stevenson GT, Tutt AL. Extracellular idiotypic immunoglobulin arising from human leukemic B lymphocytes. J Exp Med. (1980) 152:1484–96. doi: 10.1084/jem.152.6.1484
6. Diehl LF, Ketchum LH. Autoimmune disease and chronic lymphocytic leukemia: autoimmune hemolytic anemia, pure red cell aplasia, and autoimmune thrombocytopenia. Semin Oncol. (1998) 25:80–97.
7. Hodgson K, Ferrer G, Pereira A, Moreno C, Montserrat E. Autoimmune cytopenia in chronic lymphocytic leukaemia: diagnosis and treatment. Br J Haematol. (2011) 154:14–22. doi: 10.1111/j.1365-2141.2011.08707.x
8. Galletti J, Canones C, Morande P, Borge M, Oppezzo P, Geffner J, et al. Chronic lymphocytic leukemia cells bind and present the erythrocyte protein band 3: possible role as initiators of autoimmune hemolytic anemia. J Immunol. (2008) 181:3674–83. doi: 10.4049/jimmunol.181.5.3674
9. Hall AM, Vickers MA, McLeod E, Barker RN. Rh autoantigen presentation to helper T cells in chronic lymphocytic leukemia by malignant B cells. Blood. (2005) 105:2007–15. doi: 10.1182/blood-2003-10-3563
10. Strati P, Caligaris-Cappio F. A matter of debate in chronic lymphocytic leukemia: is the occurrence of autoimmune disorders an indicator of chronic lymphocytic leukemia therapy? Curr Opin Oncol. (2011) 23:455–60. doi: 10.1097/CCO.0b013e328348c683
11. Gorgun G, Holderried TA, Zahrieh D, Neuberg D, Gribben JG. Chronic lymphocytic leukemia cells induce changes in gene expression of CD4 and CD8 T cells. J Clin Invest. (2005) 115:1797–805. doi: 10.1172/JCI24176
12. Beyer M, Kochanek M, Darabi K, Popov A, Jensen M, Endl E, et al. Reduced frequencies and suppressive function of CD4+CD25hi regulatory T cells in patients with chronic lymphocytic leukemia after therapy with fludarabine. Blood. (2005) 106:2018–25. doi: 10.1182/blood-2005-02-0642
13. Ward FJ, Hall AM, Cairns LS, Leggat AS, Urbaniak SJ, Vickers MA, et al. Clonal regulatory T cells specific for a red blood cell autoantigen in human autoimmune hemolytic anemia. Blood. (2008) 111:680–7. doi: 10.1182/blood-2007-07-101345
14. Wright GP, Ehrenstein MR, Stauss HJ. Regulatory T-cell adoptive immunotherapy: Potential for treatment of autoimmunity. Expert Rev Clin Immunol. (2011) 7:213–25. doi: 10.1586/eci.10.96
15. Lad D, Varma S, Varma N, Sachdeva MU, Bose P, Malhotra P. Regulatory T-cell and T-helper 17 balance in chronic lymphocytic leukemia progression and autoimmune cytopenias. Leuk Lymphoma. (2015) 56:2424–8. doi: 10.3109/10428194.2014.986479
16. Grandjenette C, Kennel A, Faure GC, Béné MC, Feugier P. Expression of functional toll-like receptors by B-chronic lymphocytic leukemia cells. Haematologica. (2007) 92:1279–81. doi: 10.3324/haematol.10975
17. Muzio M, Bertilaccio MT, Simonetti G, Frenquelli M, Caligaris-Cappio F. The role of toll-like receptors in chronic B-cell malignancies. Leuk Lymphoma. (2009) 50:1573–80. doi: 10.1080/10428190903115410
18. Barcellini W, Imperiali FG, Zaninoni A, Reda G, Consonni D, Fattizzo B, et al. Toll-like receptor 4 and 9 expression in B-chronic lymphocytic leukemia: relationship with infections, autoimmunity and disease progression. Leuk Lymphoma. (2014) 55:1768–73. doi: 10.3109/10428194.2013.856426
19. Weiss R, Freiman J, Kweder SL, Diehl LF, Byrd JC. Haemolytic anaemia after fludarabine therapy for chronic lymphocytic leukemia. J Clin Oncol. (1998) 16:1885–9. doi: 10.1200/JCO.1998.16.5.1885
20. Gonazalez H, Leblond V, Azar N, Sutton L, Gabarre J, Binet JL, et al. Severe autoimmune haemolytic anaemia in eight patients treated with fludarabine. Hematol Cell Ther. (1998) 40:113–8.
21. Molica S, Polliack A. Autoimmune hemolytic anemia (AIHA) associated with chronic lymphocytic leukemia in the current era of targeted therapy. Leuk Res. (2016) 50:31–6. doi: 10.1016/j.leukres.2016.09.002
22. Fischer K, Bahlo J, Fink AM, Goede V, Herling CD, Cramer P, et al. Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: updated results of the CLL8 trial. Blood. (2016) 127:208–15. doi: 10.1182/blood-2015-06-651125
23. Tsang M, Chaffee KR, Call TG, Ding W, Leis J, Chanan-Khan A, et al. Pure red cell aplasia (PRCA) in chronic lymphocytic leukemia (CLL): etiology, therapy, and outcomes. Blood. (2015) 126:4169. doi: 10.1182/blood.V126.23.4169.4169
24. Reda G, Maura F, Gritti G, Gregorini A, Binda F, Guidotti F, et al. Low-dose alemtuzumab-associated immune thrombocytopenia in chronic lymphocytic leukemia. Am J Hematol. (2012) 87:936–7. doi: 10.1002/ajh.23268
25. Montillo M, O'Brien S, Tedeschi A, Byrd JC, Dearden C, Gill D, et al. Ibrutinib in previously treated chronic lymphocytic leukemia patients with autoimmune cytopenias in the RESONATE study. Blood Cancer J. (2017) 7:e524. doi: 10.1038/bcj.2017.5
26. Vitale C, Ahn IE, Sivina M, Ferrajoli A, Wierda WG, Estrov Z, et al. Autoimmune cytopenias in patients with chronic lymphocytic leukemia treated with ibrutinib. Haematologica. (2016) 102:e254–8. doi: 10.3324/haematol.2015.138289
27. Rogers K, Ruppert AS, Bingman A, Andritsos LA, Awan FT, Blum KA, et al. Incidence and description of autoimmune cytopenias during treatment with ibrutinib for chronic lymphocytic leukemia autoimmune cytopenias during ibrutinib treatment. Leukemia. (2016) 30:346–50. doi: 10.1038/leu.2015.273
28. Hampel PJ, Larson MC, Kabat B, Call TG, Ding W, Kenderian SS. Autoimmune cytopenias in patients with chronic lymphocytic leukaemia treated with ibrutinib in routine clinical practice at an academic medical centre. Br J Haematol. (2018) 183:421–7. doi: 10.1111/bjh.15545
29. Jaglowski SM, Blazar BR. How ibrutinib, a B-cell malignancy drug, became an FDA-approved second-line therapy for steroid-resistant chronic GVHD. Blood Adv. (2018) 2:2012–9. doi: 10.1182/bloodadvances.2018013060
30. Stilgenbauer S, Eichhorst B, Schetelig J, Coutre S, Seymour JF, Munir T, et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol. (2016) 17:768–78. doi: 10.1016/S1470-2045(16)30019-5
31. Lampson B, Kasar SN, Matos TR, Morgan EA, Rassenti L, Davids M, et al. Idelalisib given front-line for treatment of chronic lymphocytic leukemia causes frequent immune-mediated hepatotoxicity. Blood. (2016) 128:195–203. doi: 10.1182/blood-2016-03-707133
32. Furman R, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. (2014) 370:997–1007. doi: 10.1056/NEJMoa1315226
33. Tsang M, Parikh SA. A concise review of autoimmune cytopenias in chronic lymphocytic leukemia. Curr Hematol Malig Rep. (2017) 12:29–38. doi: 10.1007/s11899-017-0366-1
34. Barcellini W, Montesano R, Clerici G, Zaninoni A, Imperiali FG, Calori R, et al. In vitro production of anti-RBC antibodies and cytokines in chronic lymphocytic leukemia. Am J Hematol. (2002) 71:177–83. doi: 10.1002/ajh.10210
35. Quinquenel A, Al Nawakil C, Baran-Marszak F, Eclache V, Letestu R, Khalloufi M, et al. Old DAT and new data: positive direct antiglobulin test identifies a subgroup with poor outcome among chronic lymphocytic leukemia stage A patients. Am J Hematol. (2015) 90:E5–8. doi: 10.1002/ajh.23861
36. Mauro F, Foa R, Cerretti R, Giannarelli D, Coluzzi S, Mandelli F, et al. Autoimmune hemolytic anemia in chronic lymphocytic leukemia: clinical, therapeutic, and prognostic features. Blood. (2000) 95:2786–92. doi: 10.1182/blood.V95.9.2786.009k30_2786_2792
37. Rogers K, Woyach JA. Secondary autoimmune cytopenias in chronic lymphocytic leukemia. Semin Oncol. (2016) 43:300–10. doi: 10.1053/j.seminoncol.2016.02.011
38. Narat S, Gandla J, Hoffbrand AV, Hughes RG, Mehta AB. Rituximab in the treatment of refractory autoimmune cytopenias in adults. Haematologica. (2005) 90:1273–4.
39. D'Arena G, Laurenti L, Capalbo S, D'Arco AM, De Filippi R, Marcacci G, et al. Rituximab therapy for chronic lymphocytic leukemia-associated autoimmune hemolytic anemia. Am J Hematol. (2006) 81:598–602. doi: 10.1002/ajh.20665
40. Cortes J, O'Brien S, Loscertales J, Kantarjian H, Giles F, Thomas D, et al. Cyclosporin A for the treatment of cytopenia associated with chronic lymphocytic leukemia. Cancer. (2001) 92:2016–22. doi: 10.1002/1097-0142(20011015)92:8<2016::AID-CNCR1539>3.0.CO;2-E
41. Karlsson C, Hansson L, Celsing F, Lundin J. Treatment of severe refractory autoimmune hemolytic anemia in B-cell chronic lymphocytic leukemia with alemtuzumab (humanized CD52 monoclonal antibody) Leukemia. (2007) 21:511–4. doi: 10.1038/sj.leu.2404512
42. Osterborg A, Karlsson C, Lundin J. Alemtuzumab to treat refractory autoimmune hemolytic anemia or thrombocytopenia in chronic lymphocytic leukemia. Curr Hematol Malig Rep. (2009) 4:47–53. doi: 10.1007/s11899-009-0007-4
43. Cusack JC Jr, Seymour JF, Lerner S, Keating MJ, Pollock RE. Role of splenectomy in chronic lymphocytic leukemia. J Am Coll Surg. (1997) 185:237–43. doi: 10.1016/S1072-7515(97)00057-4
44. Pamuk G, Turgut B, Demir M, Tezcan F, Vural O. The successful treatment of refractory autoimmune hemolytic anemia with rituximab in a patient with chronic lymphocytic leukemia. Am J Hematol. (2006) 81:631–3. doi: 10.1002/ajh.20671
45. Berentsen S, Randen U, Oksman M, Birgens H, Tvedt THA, Dalgaard J, et al. Bendamustine plus rituximab for chronic cold agglutinin disease: results of a Nordic prospective multicenter trial. Blood. (2017) 130:537–41. doi: 10.1182/blood-2017-04-778175
46. Quinquenel A, Willekens C, Dupuis J, Royer B, Ysebaert L, De Guibert S, et al. Bendamustine and rituximab combination in the management of chronic lymphocytic leukemia-associated autoimmune hemolytic anemia: a multicentric retrospective study of the French CLL intergroup (GCFLLC/MW and GOELAMS). Am J Hematol. (2015) 90:204–7. doi: 10.1002/ajh.23909
47. Berentsen S, Randen U, Vågan AM, Hjorth-Hansen H, Vik A, Dalgaard J, et al. High response rate and durable remissions following fludarabine and rituximab combination therapy for chronic cold agglutinin disease. Blood. (2010) 116:3180–4. doi: 10.1182/blood-2010-06-288647
48. D'Arena G, Capalbo S, Laurenti L, Del Poeta G, Nunziata G, Deaglio S, et al. Chronic lymphocytic leukemia-associated immune thrombocytopenia treated with rituximab: a retrospective study of 21 patients. Eur J Haematol. (2010) 85:502–7. doi: 10.1111/j.1600-0609.2010.01527.x
49. Hegde UP, Wilson WH, White T, Cheson BD. Rituximab treatment of refractory fludarabine-associated immune thrombocytopenia in chronic lymphocytic leukemia. Blood. (2002) 100:2260–2. doi: 10.1182/blood.V100.6.2260
50. Fernandez M, Llopis I, Pastor E, Real E, Grau E. Immune thrombocytoepnia induced by fludarabine successfully treated with rituximab. Haematologica. (2003) 88:ELT02.
51. Jolliffe E, Romeril K. Eltrombopag for resistant immune thrombocytopenia secondary to secondary lymphocytic leukemia. Intern Med J. (2014) 44:697–9. doi: 10.1111/imj.12468
52. Koehrer S, Keating MJ, Wierda WG. Eltrombopag, a second-generation thrombopoietin receptor agonist, for chronic lymphocytic leukemia-associated ITP. Leukemia. (2010) 24:1096–8. doi: 10.1038/leu.2010.45
53. Paul S, Jain N, Ferrajoli A, O'Brien S, Burger J, Keating M, et al. A phase II trial of eltrombopag for patients with chronic lymphocytic leukaemia (CLL) and thrombocytopenia. Br J Haematol. (2019) 185:606–8. doi: 10.1111/bjh.15581
54. Gupta N, Kavuru S, Patel D, Janson D, Driscoll N, Ahmed S, et al. Rituximab-based chemotheray for steroid-refractory autoimmune hemolytic anemia of chronic lymphocytic leukemia. Leukemia. (2002) 16:2092–5. doi: 10.1038/sj.leu.2402676
55. Michallet A, Rossignol J, Cazin B, Ysebaert L. Rituximab–cyclophosphamide–dexamethasone combination in management of autoimmune cytopenias associated with chronic lymphocytic leukemia. Leuk Lymphoma. (2011) 52:1401–3. doi: 10.3109/10428194.2011.591005
56. Bowen DA, Call TG, Shanafelt TD, Kay NE, Schwager SM, Reinalda MS, et al. Treatment of autoimmune cytopenia complicating progressive chronic lymphocytic leukemia/small lymphocytic lymphoma with rituximab, cyclophosphamide, vincristine, and prednisone. Leuk Lymphoma. (2010) 51:620–7. doi: 10.3109/10428191003682767
57. Kaufman M, Limaye SA, Driscoll N, Johnson C, Caramanica A, Lebowicz Y, et al. A combination of rituximab, cyclophosphamide, and dexamethasone effectively treats immune cytopenias of chronic lymphocytic leukemia. Leuk Lymphoma. (2009) 50:892–9. doi: 10.1080/10428190902887563
58. Borthakur G, O'Brien S, Wierda WG, Thomas DA, Cortes JE, Giles FJ, et al. Immune anaemias in patients with chronic lymphocytic leukaemia treated with fludrabine, cyclophosphamide and rituximab–incidence and predictors. Br J Haematol. (2007) 136:800–5. doi: 10.1111/j.1365-2141.2007.06513.x
59. Church A, VanDerMeid KR, Baig NA, Baran AM, Witzig TE, Nowakowski GS, et al. Anti-CD20 monoclonal antibody-dependent phagocytosis of chronic lymphocytic leukaemia cells by autologous macrophages. Clin Exp Immunol. (2016) 183:90–101. doi: 10.1111/cei.12697
60. Lacerda MP, Guedes NR, Yamakawa PE, Pereira AD, Fonseca ARBMD, Chauffaille MLLF, et al. Treatment of refractory autoimmune hemolytic anemia with venetoclax in relapsed chronic lymphocytic leukemia with del(17p). Ann Hematol. (2017) 96:1577–8. doi: 10.1007/s00277-017-3039-1
61. Gordon MJ, Maldonado E, Danilov AV. Refractory autoimmune cytopenias treated with venetoclax. Hemasphere. (2019) 3:e202. doi: 10.1097/HS9.0000000000000202
62. Visco C, Rodeghiero F, Romano A, Valeri F, Merli M, Quaresimini G, et al. Eltrombopag for immune thrombocytopenia secondary to chronic lymphoproliferative disorders: a phase 2 multicenter study. Blood. (2019) 134:1708–11. doi: 10.1182/blood.2019001617
63. Pascual V, Victor K, Lelsz D, Spellerberg MB, Hamblin TJ, Thompson KM, et al. Nucleotide sequence analysis of the V regions of two IgM cold agglutinins. Evidence that the VH4-21 gene segment is responsible for the major cross-reactive idiotype. J Immunol. (1991) 146:4385–91.
64. Silberstein LE, Jefferies LC, Goldman J, Friedman D, Moore JS, Nowell PC, et al. Variable region gene analysis of pathologic human autoantibodies to the related i and I red blood cell antigens. Blood. (1991) 78:2372–86. doi: 10.1182/blood.V78.9.2372.2372
65. Potter KN, Hobby P, Klijn S, Stevenson FK, Sutton BJ. Evidence for involvement of a hydrophobic patch in framework region 1 of human V4-34-encoded Igs in recognition of the red blood cell I antigen. J Immunol. (2002) 169:3777–82. doi: 10.4049/jimmunol.169.7.3777
66. Marks JD, Ouwehand WH, Bye JM, Finnern R, Gorick BD, Voak D, et al. Human antibody fragments specific for human blood group antigens from a phage display library. Biotechnology. (1993) 11:1145–9. doi: 10.1038/nbt1093-1145
67. Michaux L, Dierlamm J, Wlodarska I, Stul M, Bosly A, Delannoy A, et al. Trisomy 3 is a consistent chromosome change in malignant lymphoproliferative disorders preceded by cold agglutinin disease. Br J Haematol. (1995) 91:421–4. doi: 10.1111/j.1365-2141.1995.tb05315.x
68. Silberstein LE, Robertson GA, Harris AC, Moreau L, Besa E, Nowell PC. Etiologic aspects of cold agglutinin disease: evidence for cytogenetically defined clones of lymphoid cells and the demonstration that an anti-Pr cold autoantibody is derived from a chromosomally aberrant B cell clone. Blood. (1986) 67:1705–9.
69. D'Abronzo LS, Barros MM, Bordin JO, Figueiredo MS. Analysis of polymorphisms of TNF-α, LT-α, IL-10, IL-12 and CTLA-4 in patients with warm autoimmune haemolytic anaemia. Int J Lab Hematol. (2012) 34:356–61. doi: 10.1111/j.1751-553X.2012.01400.x
70. Malecka A, Trøen G, Tierens A, Østlie I, Małecki J, Randen U, et al. Immunoglobulin heavy and light chain gene features are correlated with primary cold agglutinin disease onset and activity. Haematologica. (2016) 101:e361–4. doi: 10.3324/haematol.2016.146126
71. Smirnova SJ, Sidorova JV, Tsvetaeva NV, Nikulina OF, Biderman BV, Nikulina EE, et al. Expansion of CD8+ cells in autoimmune hemolytic anemia. Autoimmunity. (2016) 49:147–54. doi: 10.3109/08916934.2016.1138219
72. Malecka A, Troen G, Tierens A, Østlie I, Małecki J, Randen U, et al. Frequent somatic mutations of KMT2D (MLL2) and CARD11 genes in primary cold agglutinin disease. Br J Haematol. (2018) 183:838–42. doi: 10.1111/bjh.15063
73. Efremov DG, Ivanovski M, Burrone OR. The pathologic significance of the immunoglobulins expressed by chronic lymphocytic leukemia B-cells in the development of autoimmune hemolytic anemia. Leuk Lymphoma. (1998) 28:285–93. doi: 10.3109/10428199809092684
74. Efremov DG, Ivanovski M, Siljanovski N, Pozzato G, Cevreska L, Fais F, et al. Restricted immunoglobulin VH region repertoire in chronic lymphocytic leukemia patients with autoimmune hemolytic anemia. Blood. (1996) 87:3869–76. doi: 10.1182/blood.V87.9.3869.bloodjournal8793869
75. Kryachok I, Abramenko I, Bilous N, Chumak A, Martina Z, Filonenko I. IGHV gene rearrangements as outcome predictors for CLL patients: experience of Ukrainian group. Med Oncol. (2012) 29:1093–101. doi: 10.1007/s12032-011-9872-5
76. Pavkovic M, Georgievski B, Cevreska L, Spiroski M, Efremov DG. CTLA-4 exon 1 polymorphism in patients with autoimmune blood disorders. Am J Hematol. (2003) 72:147–9. doi: 10.1002/ajh.10278
77. Ferrer G, Navarro A, Hodgson K, Aymerich M, Pereira A, Baumann T, et al. MicroRNA expression in chronic lymphocytic leukemia developing autoimmune hemolytic anemia. Leuk Lymphoma. (2013) 54:2016–22. doi: 10.3109/10428194.2012.763123
78. Maura F, Visco C, Falisi E, Reda G, Fabris S, Agnelli L, et al. B-cell receptor configuration and adverse cytogenetics are associated with autoimmune hemolytic anemia in chronic lymphocytic leukemia. Am J Hematol. (2013) 88:32–6. doi: 10.1002/ajh.23342
79. Carli G, Visco C, Falisi E, Perbellini O, Novella E, Giaretta I, et al. Evans syndrome secondary to chronic lymphocytic leukaemia: presentation, treatment, and outcome. Ann Hematol. (2016) 95:863–70. doi: 10.1007/s00277-016-2642-x
80. Nagelkerke SQ, Bruggeman CW, den Haan JMM, Mul EPJ, van den Berg TK, van Bruggen R, et al. Red pulp macrophages in the human spleen are a distinct cell population with a unique expression of Fc-γ receptors. Blood Adv. (2018) 2:941–53. doi: 10.1182/bloodadvances.2017015008
81. Xing L, Xu W, Qu Y, Zhao M, Zhu H, Liu H, et al. miR-150 regulates B lymphocyte in autoimmune hemolytic anemia/Evans syndrome by c-Myb. Int J Hematol. (2018) 107:666–72. doi: 10.1007/s12185-018-2429-z
82. Hadjadj J, Aladjidi N, Fernandes H, Leverger G, Magérus-Chatinet A, Mazerolles F. Pediatric Evans syndrome is associated with a high frequency of potentially damaging variants in immune genes. Blood. (2019) 134:9–21. doi: 10.1182/blood-2018-11-887141
83. Jefferies LC, Carchidi CM, Silberstein LE. Naturally occurring anti-i/I cold agglutinins may be encoded by different VH3 genes as well as the VH4.21 gene segment. J Clin Invest. (1993) 92:2821–33. doi: 10.1172/JCI116902
84. Martorelli D, Guidoboni M, De Re V, Muraro E, Turrini R, Merlo A, et al. IGKV3 proteins as candidate “off-the-shelf” vaccines for kappa-light chain-restricted B-cell non-Hodgkin lymphomas. Clin Cancer Res. (2012) 18:4080–91. doi: 10.1158/1078-0432.CCR-12-0763
85. Herve M, Xu K, Ng YS, Wardemann H, Albesiano E, Messmer BT, et al. Unmutated and mutated chronic lymphocytic leukemias derive from self-reactive B cell precursors despite expressing different antibody reactivity. J Clin Invest. (2005) 115:1636–43. doi: 10.1172/JCI24387
86. Visco C, Giaretta I, Ruggeri M, Madeo D, Tosetto A, Rodeghiero F. Un-mutated IgVH in chronic lymphocytic leukemia is associated with a higher risk of immune thrombocytopenia. Leukemia. (2007) 21:1092–3. doi: 10.1038/sj.leu.2404592
87. Visco C, Ruggeri M, Laura EM, Stasi R, Zanotti R, Giaretta I, et al. Impact of immune thrombocytopenia on the clinical course of chronic lymphocytic leukemia. Blood. (2008) 111:1110–6. doi: 10.1182/blood-2007-09-111492
88. Visco C, Novella E, Peotta E, Paolini R, Giaretta I, Rodeghiero F. Autoimmune hemolytic anemia in patients with chronic lymphocytic leukemia is associated with IgVH status. Haematologica. (2010) 95:1230–2. doi: 10.3324/haematol.2010.022079
89. Visco C, Maura F, Tuana G, Agnelli L, Lionetti M, Fabris S, et al. Immune thrombocytopenia in patients with chronic lymphocytic leukemia is associated with stereotyped B-cell receptors. Clin Cancer Res. (2012) 18:1870–8. doi: 10.1158/1078-0432.CCR-11-3019
90. Mockridge CI, Potter KN, Wheatley I, Neville LA, Packham G, Stevenson FK. Reversible anergy of sIgM-mediated signaling in the two subsets of CLL defined by VH-gene mutational status. Blood. (2007) 109:4424–31. doi: 10.1182/blood-2006-11-056648
91. Bomben R, Dal BM, Capello D, Forconi F, Maffei R, Laurenti L, et al. Molecular and clinical features of chronic lymphocytic leukaemia with stereotyped B cell receptors: results from an Italian multicentre study. Br J Haematol. (2009) 144:492–506. doi: 10.1111/j.1365-2141.2008.07469.x
92. Maura F, Cutrona G, Fabris S, Colombo M, Tuana G, Agnelli L, et al. Relevance of stereotyped B-cell receptors in the context of the molecular, cytogenetic and clinical features of chronic lymphocytic leukemia. PLoS ONE. (2011) 6:e24313. doi: 10.1371/journal.pone.0024313
93. Stamatopoulos K, Belessi C, Moreno C, Boudjograh M, Guida G, Smilevska T, et al. Over 20% of patients with chronic lymphocytic leukemia carry stereotyped receptors: Pathogenetic implications and clinical correlations. Blood. (2007) 109:259–70. doi: 10.1182/blood-2006-03-012948
94. Tobin G, Thunberg U, Johnson A, Eriksson I, Söderberg O, Karlsson K, et al. Chronic lymphocytic leukemias utilizing the VH3–21 gene display highly restricted Vlambda2-14 gene use and homologous CDR3s: implicating recognition of a common antigen epitope. Blood. (2003) 101:4952–7. doi: 10.1182/blood-2002-11-3485
95. Widhopf GF, Rassenti LZ, Toy TL, Gribben JG, Wierda WG, Kipps TJ. Chronic lymphocytic leukemia B cells of more than 1% of patients express virtually identical immunoglobulins. Blood. (2004) 104:2499–504. doi: 10.1182/blood-2004-03-0818
96. Marron MP, Raffel LJ, Garchon HJ, Jacob CO, Serrano-Rios M, Martinez Larrad MT, et al. Insulin-dependentdiabetes mellitus (IDDM) is associated with CTLA-4 polymorphismsin multiple ethnic groups. Hum Mol Genet. (1997) 6:1275–82. doi: 10.1093/hmg/6.8.1275
97. Donner H, Braun J, Seidl C, Rau H, Finke R, Ventz M, et al. Codon 17 polymorphism of the cytotoxicT lymphocyte antigen 4 gene in Hashimoto's thyroiditis and Addison'sdisease. J Clin Endocrinol Metab. (1997) 82:4130–2. doi: 10.1210/jcem.82.12.4406
98. Shaker OG, Alnoury AM, Hegazy GA, El Haddad HE, Sayed S, Hamdy A. Methylene tetrahydrofolate reductase, transforming growth factor-β1 and lymphotoxin-α genes polymorphisms and susceptibility to rheumatoid arthritis. Rev Bras Reumatol Engl Ed. (2016) 56:414–20. doi: 10.1016/j.rbre.2016.04.002
99. Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med. (2015) 21:1199–208. doi: 10.1038/nm.3943
100. Imai Y, Maru Y, Tanaka J. Action mechanisms of histone deacetylase inhibitors in the treatment of hematological malignancies. Cancer Sci. (2016) 107:1543–9. doi: 10.1111/cas.13062
101. Young RM, Staudt LM. A new “brew” of MALT1 inhibitors. Cancer Cell. (2012) 22:706–7. doi: 10.1016/j.ccr.2012.11.011
102. Michel M, Chanet V, Dechartres A, Morin AS, Piette JC, Cirasino L, et al. The spectrum of Evans syndrome in adults:new insight into the disease based on the analysis of 68 cases. Blood. (2009) 114:3167–72. doi: 10.1182/blood-2009-04-215368
103. Klemann C, Esquivel M, Magerus-Chatinet A, Lorenz MR, Fuchs I, Neveux N, et al. Evolution of disease activity and biomarkers on and off rapamycin in 28 patients with autoimmune lymphoproliferative syndrome. Haematologica. (2017) 102:e52–6. doi: 10.3324/haematol.2016.153411
104. Rao VK, Webster S, Dalm VASH, Šedivá A, van Hagen PM, Holland S, et al. Effective “activated PI3Kδ syndrome”-targeted therapy with the PI3Kδ inhibitor leniolisib. Blood. (2017) 130:2307–16. doi: 10.1182/blood-2017-08-801191
105. Schwab C, Gabrysch A, Olbrich P, Patiño V, Warnatz K, Wolff D, et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J Allergy Clin Immunol. (2018) 142:1932–46. doi: 10.1016/j.jaci.2018.02.055
106. Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. Patients with LRBAdeficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science. (2015) 349:436–40. doi: 10.1126/science.aaa1663
107. Del Bel KL, Ragotte RJ, Saferali A, Lee S, Vercauteren SM, Mostafavi SA, et al. JAK1 gain-of-function causes an autosomal dominant immune dysregulatory and hypereosinophilic syndrome. J Allergy Clin Immunol. (2017) 139:2016–20.e5. doi: 10.1016/j.jaci.2016.12.957
108. Penel Page M, Bertrand Y, Fernandes H, Kherfellah D, Leverger G, Leblanc T, et al. Treatment with cyclosporin in autoimmune cytopenias in children: the experience from the French cohort OBS'CEREVANCE. Am J Hematol. (2018). doi: 10.1002/ajh.25137. [Epub ahead of print].
109. Roark JH, Bussel JB, Cines DB, Siegel DL. Genetic analysis of autoantibodies in idiopathic thrombocytopenic purpura reveals evidence of clonal expansion and somatic mutation. Blood. (2002) 100:1388–98. doi: 10.1182/blood.V100.4.1388.h81602001388_1388_1398
110. Zheng CX, Ji ZQ, Zhang LJ, Wen Q, Chen LH, Yu JF, et al. Proteomics-based identification of haptoglobin as a favourable serum biomarker for predicting long-term response to splenectomy in patients with primary immune thrombocytopenia. J Transl Med. (2012) 10:208. doi: 10.1186/1479-5876-10-208
111. Hu Y, Wang X, Yu S, Hou Y, Ma D, Hou M. Neutralizations of IL-17A and IL-21 regulate regulatory T cell/T-helper 17 imbalance via T-helper 17-associated signaling pathway in immune thrombocytopenia. Expert Opin Ther Targets. (2015) 19:723–32. doi: 10.1517/14728222.2015.1016499
112. Chen Z, Guo Z, Ma J, Liu F, Gao C, Liu S, et al. STAT1 single nucleotide polymorphisms and susceptibility to immune thrombocytopenia Autoimmunity. (2015) 48:305–12. doi: 10.3109/08916934.2015.1016218
113. Burenbatu, Borjigin M, Eerdunduleng, Huo W, Gong C, Hasengaowa, et al. Profiling of miRNA expression in immune thrombocytopenia patients before and after Qishunbaolier (QSBLE) treatment. Biomed Pharmacother. (2015) 75:196–204. doi: 10.1016/j.biopha.2015.07.022
114. Li JQ, Hu SY, Wang ZY, Lin J, Jian S, Dong YC. Long non-coding RNA MEG3 inhibits microRNA-125a-5p expression and induces immune imbalance of Treg/Th17 in immune thrombocytopenic purpura. Biomed Pharmacother. (2016) 83:905–11. doi: 10.1016/j.biopha.2016.07.057
115. Zhang HW, Zhou P, Wang KZ, Liu JB, Huang YS, Tu YT, et al. Platelet proteomics in diagnostic differentiation of primary immune thrombocytopenia using SELDI-TOF-MS. Clin Chim Acta. (2016) 455:75–79. doi: 10.1016/j.cca.2016.01.028
116. Yao X, Li C, Yang J, Wang G, Li C, Xia Y. Differences in frequency and regulation of T follicular helper cells between newly diagnosed and chronic pediatric immune thrombocytopenia. Blood Cells Mol Dis. (2016) 61:26–36. doi: 10.1016/j.bcmd.2016.06.006
117. Peng HL, Zhang Y, Sun NN, Yin YF, Wang YW, Cheng Z, et al. A gain-of-function mutation in TNFRSF13B is a candidate for predisposition to familial or sporadic immune thrombocytopenia. J Thromb Haemost. (2017) 15:2259–69. doi: 10.1111/jth.13806
118. Li H, Zhou Z, Tai W, Feng W, Zhang D, Gu X, et al. Decreased frequency of IL-17F rs763780 site allele G is associated with genetic susceptibility to immune thrombocytopenia in a Chinese population. Clin Appl Thromb Hemost. (2017) 23:466–71. doi: 10.1177/1076029615618022
119. Yu J, Hua M, Zhao X, Wang R, Zhong C, Zhang C, et al. NF-κB-94ins/del ATTG genotype contributes to the susceptibility and imbalanced Th17 cells in patients with immune thrombocytopenia. J Immunol Res. (2018) 2018:8170436. doi: 10.1155/2018/8170436
120. Li T, Gu M, Liu P, Liu Y, Guo J, Zhang W, et al. Abnormal expression of long noncoding RNAs in primary immune thrombocytopenia: a microarray related study. Cell Physiol Biochem. (2018) 48:618–32. doi: 10.1159/000491890
121. Zhang JM, Zhu XL, Xue J, Liu X, Long Zheng X, Chang YJ, et al. Integrated mRNA and miRNA profiling revealed deregulation of cellular stress response in bone marrow mesenchymal stem cells derived from patients with immune thrombocytopenia. Funct Integr Genomics. (2018) 18:287–99. doi: 10.1007/s10142-018-0591-2
122. Kunicki TJ, Annis DS, Gorski J, Nugent DJ. Nucleotide sequence of the human autoantibody 2E7 specific for the platelet integrin IIb heavy chain. J Autoimmun. (1991) 4:433–46. doi: 10.1016/0896-8411(91)90157-8
123. Kunicki TJ, Plow EF, Kekomaki R, Nugent DJ. Human monoclonal autoantibody 2E7 is specific for a peptide sequence of platelet glycoprotein IIb: localization of the epitope to IIb231–238 with an immunodominant Trp235. J Autoimmun. (1991) 4:415–31. doi: 10.1016/0896-8411(91)90156-7
124. Jendreyko N, Uttenreuther-Fischer MM, Lerch H, Gaedicke G, Fischer P. Genetic origin of IgG antibodies cloned by phage display and anti-idiotypic panning from three patients with autoimmune thrombocytopenia. Eur J Immunol. (1998) 28:4236–47. doi: 10.1002/(SICI)1521-4141(199812)28:12<4236::AID-IMMU4236>3.0.CO;2-R
125. Fischer P, Jendreyko N, Hoffmann M, Lerch H, Uttenreuther-Fischer MM, Chen PP, et al. Platelet-reactive IgG antibodies cloned by phage display and panning with IVIG from three patients with autoimmune thrombocytopenia. Br J Haematol. (1999) 105:626–40. doi: 10.1046/j.1365-2141.1999.01407.x
126. Bettaieb A, Oksenhendler E, Duedari N, Bierling P. Cross-reactive antibodies between HIV-gp120 and platelet gpIIIa (CD61) in HIV-related immune thrombocytopenic purpura. Clin Exp Immunol. (1996) 103:19–23. doi: 10.1046/j.1365-2249.1996.917606.x
127. van der Harst D, de Jong D, Limpens J, Kluin PM, Rozier Y, van Ommen GJ, et al. Clonal B-cell populations in patients with idiopathic thrombocytopenic purpura. Blood. (1990) 76:2321–6. doi: 10.1182/blood.V76.11.2321.bloodjournal76112321
128. Christie DJ, Sauro SC, Fairbanks KD, Kay NE. Detection of clonal platelet antibodies in immunologically-mediated thrombocytopenias: association with circulating clonal/oligoclonal B cells. Br J Haematol. (1993) 85:277–84. doi: 10.1111/j.1365-2141.1993.tb03167.x
129. Stockelberg D, Hou M, Jacobsson S, Kutti J, Wadenvik H. Evidence for a light chain restriction of glycoprotein Ib/IX and IIb/IIIa reactive antibodies in chronic idiopathic thrombocytopenic purpura (ITP). Br J Haematol. (1995) 90:175–9. doi: 10.1111/j.1365-2141.1995.tb03397.x
130. Stockelberg D, Hou M, Jacobsson S, Kutti J, Wadenvik H. Light chain-restricted autoantibodies in chronic idiopathic thrombocytopenic purpura, but no evidence for circulating clone B-lymphocytes. Ann Hematol. (1996) 72:29–34. doi: 10.1007/BF00663013
131. McMillan R, Lopez-Dee J, Bowditch R. Clonal restriction of platelet-associated anti-GPIIb/IIIa autoantibodies in patients with chronic ITP. Thromb Haemost. (2001) 85:821–3. doi: 10.1055/s-0037-1615754
132. Crowley JJ, Mageed RA, Silverman GJ, Chen PP, Kozin F, Erger RA, et al. The incidence of a new human cross-reactive idiotype linked to subgroup VHIII heavy chains. Mol Immunol. (1990) 27:87–94. doi: 10.1016/0161-5890(90)90063-6
133. Shokri F, Mageed RA, Maziak BR, Jefferis R. Expression of VHIII-associated cross-reactive idiotype on human B lymphocytes: association with staphylococcal protein A binding and Staphylococcus aureus Cowan I stimulation. J Immunol. (1991) 146:936–40.
134. Silverman G. B cell superantigens: possible roles in immunodeficiency and autoimmunity. Semin Immunol. (1998) 10:43–55. doi: 10.1006/smim.1997.0104
135. Graille M, Stura EA, Corper AL, Sutton BJ, Taussig MJ, Charbonnier JB, et al. Crystal structure of a Staphylococcus aureus protein A domain complexed with the Fab fragment of a human IgM antibody: structural basis for recognition of B-cell receptors and superantigen activity. Proc Natl Acad Sci USA. (2000) 97:5399–404. doi: 10.1073/pnas.97.10.5399
136. Silverman G, Cary S, Dwyer D, Luo L, Wagenknecht R, Curtiss V. A B-cell superantigen induced persistent “hole” in the B-1 repertoire. J Exp Med. (2000) 192:87–98. doi: 10.1084/jem.192.1.87
137. Goodyear CS, Silverman GJ. Evidence of a novel immunomodulatory mechanism of action of Prosorba therapy: release of staphylococcal protein A induces VH region targeted apoptotic death of B lymphocytes. Arthritis Rheum. (2001) 44:S296. Available online at: https://mafiadoc.com/arthritis-rheumatism-2001-annual-scientific_5c17b804097c47b3388b469e.html
138. Barcellini W, Fattizzo B, Zaninoni A, Valli V, Ferri V, Gianelli U, et al. Clinical evolution of autoimmune cytopenias to idiopathic cytopenias/dysplasias of uncertain significance (ICUS/IDUS) and bone marrow failure syndromes. Am J Hematol. (2017) 92:E26–9. doi: 10.1002/ajh.24618
139. Fattizzo B, Zaninoni A, Consonni D, Zanella A, Gianelli U, Cortelezzi A, et al. Is chronic neutropenia always a benign disease? Evidences from a 5-year prospective study. Eur J Intern Med. (2015) 26:611–5. doi: 10.1016/j.ejim.2015.05.019
140. Fattizzo B, Zaninoni A, Gianelli U, Zanella A, Cortelezzi A, Kulasekararaj AG, et al. Prognostic impact of bone marrow fibrosis and dyserythropoiesis in autoimmune hemolytic anemia. Am J Hematol. (2018) 93:E88–E91. doi: 10.1002/ajh.25020
Keywords: autoimmune hemolytic anemia, immune thrombocytopenia, chronic lymphocytic leukemia, Evans' syndrome, molecular
Citation: Fattizzo B and Barcellini W (2020) Autoimmune Cytopenias in Chronic Lymphocytic Leukemia: Focus on Molecular Aspects. Front. Oncol. 9:1435. doi: 10.3389/fonc.2019.01435
Received: 21 October 2019; Accepted: 02 December 2019;
Published: 10 January 2020.
Edited by:
Francesco Maura, Memorial Sloan Kettering Cancer Center, United StatesCopyright © 2020 Fattizzo and Barcellini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bruno Fattizzo, YnJ1bm8uZmF0dGl6em9AcG9saWNsaW5pY28ubWkuaXQ=